
Sixantone® 30 mg Retardmikrokapseln und Suspensionsmittel
Jede Zweikammerspritze mit 352,9 mg Retardmikrokapseln und 1 ml Suspensionsmittel enthält:
30,0 mg Leuprorelinacetat, entsprechend 28,58 mg Leuprorelin.
Sonstige Bestandteile mit bekannter Wirkung:
Eine Zweikammerspritze enthält 5,0 mg Carmellose-Natrium.
Dieses Arzneimittel enthält 1 mg Polysorbat 80 pro Zweikammerspritze.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
Retardmikrokapseln und Suspensionsmittel
Sixantone wird bei erwachsenen Männern zur Therapie des fortgeschrittenen hormonabhängigen Prostatakarzinoms angewendet.
Zur Behandlung des lokal fortgeschrittenen, hormonabhängigen Prostatakarzinoms; begleitend zur und nach der Strahlentherapie.
Zur Behandlung des lokalisierten hormonabhängigen Prostatakarzinoms bei Patienten des mittleren und Hoch-Risikoprofils in Kombination mit der Strahlentherapie.
Zur Behandlung des metastasierten hormonsensitiven Prostatakarzinoms (mHSPC) in Kombination mit Androgen-Rezeptor-Inhibitoren (Apalutamid, Enzalutamid).
Zur Behandlung des metastasierten hormonsensitiven Prostatakarzinoms (mHSPC) in Kombination mit Darolutamid und Docetaxel.
Zur Behandlung des metastasierten hormonsensitiven Prostatakarzinoms (mHSPC) in Kombination mit Abirateronacetat und Prednison oder Prednisolon (nur für Patienten mit neu diagnostiziertem Hochrisiko-Prostatakarzinom).
Zur Behandlung des metastasierten hormonsensitiven Prostatakarzinoms (mHSPC) in Kombination mit Docetaxel mit oder ohne Prednison oder Prednisolon.
Zur Behandlung erwachsener Männer mit nicht-metastasiertem kastrationsresistentem Prostatakarzinom (nmCRPC), die ein hohes Risiko für die Entwicklung von Metastasen aufweisen, in Kombination mit Apalutamid, Darolutamid oder Enzalutamid.
Dosierung
Einmal sechsmonatlich 352,9 mg Retardmikrokapseln mit 30,0 mg Leuprorelinacetat suspendiert in 1 ml Suspensionsmittel s.c. applizieren.
Sixantone 30 mg kann als neoadjuvante oder adjuvante Therapie in Kombination mit Strahlentherapie bei lokal fortgeschrittenem, hormonabhängigem Prostatakarzinom sowie bei lokalisiertem Prostatakarzinom des mittleren und Hoch-Risikoprofils angewendet werden.
Kinder und Jugendliche
Zur Anwendung des 6-Monats-Depots bei Kindern liegen zurzeit keine Erfahrungen vor. Für die Anwendung bei Kindern mit Pubertas praecox stehen die 1-Monats-Depots Enantone Monats-Depot oder Enantone-Paed Montas-Depot 1,88 mg und das 3-Monats-Depot Trenantone zur Verfügung.
Art der Anwendung
Sixantone darf nur von medizinischem Fachpersonal zubereitet, rekonstituiert und verabreicht werden, das mit der sachgemäßen Handhabung vertraut ist.
Die Suspension von Sixantone ist vor der Gabe frisch zuzubereiten (Hinweise zur Vorbereitung der Zweikammerspritze vor der Anwendung, siehe Abschnitt 6.6).
Sixantone wird einmal alle sechs Monate als subkutane Injektion verabreicht. Das Applikationsintervall sollte 168 Tage bis maximal 182 Tage (24 bis 26 Wochen) betragen.
Die Injektionsstelle sollte alle 6 Monate gewechselt werden. Dabei kann die subkutane Injektion in die Bauchhaut, das Gesäß oder z. B. den Oberschenkel erfolgen.
In der Regel ist die Therapie hormonabhängiger Prostatakarzinome mit Sixantone eine Langzeitbehandlung.
Die Behandlung von Patienten, die an einem Prostatakarzinom erkrankt sind, mit einem GnRH (Gonadotropin-Releasing-Hormon)-Analogon, kann auch nach Erreichen einer Kastrationsresistenz fortgeführt werden. Die relevanten Leitlinien sind hierbei zu beachten.
Klinische Daten haben gezeigt, dass bei lokal fortgeschrittenem, hormonabhängigem Prostatakarzinom, eine begleitend zur und nach der Strahlentherapie eingesetzte 3-jährige Androgenentzugstherapie einer 6-monatigen vorzuziehen ist (siehe auch Abschnitt 5.1). In medizinischen Leitlinien wird für Patienten (T3 - T4), die eine Strahlentherapie erhalten, eine Androgenentzugstherapie mit einer Behandlungsdauer von 2 - 3 Jahren empfohlen.
Bei lokalisiertem Prostatakarzinom des mittleren Risikoprofils wird die Kombination aus Strahlentherapie und einer 4 - 6-monatigen Androgenentzugstherapie mit GnRH-Analoga und bei Tumoren des Hoch-Risikoprofils wird eine 2 - 3-jährige Androgenentzugstherapie empfohlen.
Eine versehentliche intraarterielle Injektion ist aufgrund tierexperimenteller Befunde (Thrombosierung kleiner Gefäße distal des Applikationsortes) unbedingt zu vermeiden.
- Überempfindlichkeit gegen Leuprorelin oder andere synthetische GnRH-Analoga, gegen Polymilchsäure oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile des Suspensionsmittels.
- Nachgewiesene Hormonunabhängigkeit des Karzinoms.
Sixantone ist nicht für die Anwendung bei Frauen bestimmt und ist während der Schwangerschaft und Stillzeit generell kontraindiziert.
Patienten mit Bluthochdruck sollten sorgfältig überwacht werden.
Bei Patienten, die mit GnRH-Agonisten wie Leuprorelin behandelt werden, besteht ein erhöhtes Risiko für Depressionen (die schwerwiegend sein können). Die Patienten sind über das Risiko aufzuklären und im Falle auftretender Symptomatik entsprechend zu behandeln.
Nach chirurgischer Kastration bewirkt Sixantone keine weitere Absenkung des Testosteronspiegels.
Wegen des kurzfristigen Anstiegs des Serumtestosteronspiegels zu Beginn der Therapie, der zu einer vorübergehenden Verstärkung bestimmter Krankheitssymptome führen kann, sollten Patienten mit drohenden neurologischen Komplikationen, Wirbelsäulenmetastasen sowie Harnwegsobstruktionen während der ersten Behandlungswochen engmaschig überwacht werden.
Für die Initialphase der Behandlung sollte die zusätzliche Gabe eines geeigneten Antiandrogens erwogen werden, um so die möglichen Folgeerscheinungen des anfänglichen Testosteronanstiegs und die Verschlechterung der klinischen Symptomatik abzuschwächen.
Der Therapieerfolg sollte regelmäßig (insbesondere aber bei Anzeichen für eine Progression trotz adäquater Therapie) durch klinische Untersuchungen (rektale Austastung der Prostata, Sonographie, Skelettszintigraphie, Computertomographie) und durch Überprüfung der Phosphatasen bzw. des prostataspezifischen Antigens (PSA) und des Serumtestosterons kontrolliert werden.
Eine Langzeit-Androgendeprivationstherapie mit GnRH-Analoga bzw. Orchiektomie ist mit einem erhöhten Risiko der Knochendemineralisierung assoziiert. Bei Risikopatienten kann dies zu einer Osteoporose und erhöhtem Frakturrisiko führen.
Metabolische Veränderungen und kardiovaskuläres Risiko:
Epidemiologische Daten haben gezeigt, dass die Hemmung der körpereigenen Sexualhormonproduktion, wie z.B. während einer Androgendeprivationstherapie mit GnRH-Analoga zu einer Veränderung des Stoffwechsels (Herabsetzung der Glucosetoleranz oder Verschlechterung eines bestehenden Diabetes mellitus) führt und dass ein erhöhtes Risiko für kardiovaskuläre Erkrankungen bestehen kann (siehe Abschnitt 4.8). Prospektive Daten bestätigten jedoch den Zusammenhang zwischen der Behandlung mit GnRH-Analoga und einer erhöhten kardiovaskulären Mortalität nicht. Diabetiker und Patienten mit erhöhtem Risiko für metabolische oder kardiovaskuläre Erkrankungen sollen während der Behandlung mit Sixantone angemessen überwacht werden. Zu den möglichen metabolischen Veränderungen, die im Zusammenhang mit GnRH-Agonisten auftreten können, zählt auch eine Fettlebererkrankung.
Eine Androgendeprivationstherapie kann die QT-Zeit verlängern. Bei Patienten mit einer QT-Zeitverlängerung in der Vorgeschichte oder mit einem Risiko für eine QT-Zeitverlängerung und bei Patienten, die gleichzeitig QT-zeitverlängernde Arzneimittel einnehmen (siehe Abschnitt 4.5), sollte daher vor der Anwendung von Sixantone eine sorgfältige Nutzen-Risiko-Bewertung inklusive des Risikos für die Entstehung von Torsade de pointes durchgeführt werden.
Nach der Markteinführung von Leuprorelinacetat wurden Krampfanfälle bei Kindern und Erwachsenen mit oder ohne eine Vorgeschichte von Epilepsie, Anfallsleiden oder Risikofaktoren für Krampfanfälle, beobachtet und berichtet.
Idiopathische intrakranielle Hypertension
Bei Patienten, die Leuprorelin erhalten, wurde über idiopathische intrakranielle Hypertonie (Pseudotumor cerebri) berichtet. Die Patienten sollten auf Anzeichen und Symptome einer idiopathischen intrakraniellen Hypertonie, einschließlich starker oder wiederkehrender Kopfschmerzen, Sehstörungen und Tinnitus, hingewiesen werden. Wenn eine idiopathische intrakranielle Hypertonie auftritt, sollte ein Absetzen von Leuprorelin in Betracht gezogen werden.
Schwerwiegende unerwünschte Reaktionen der Haut
Im Zusammenhang mit der Behandlung mit Leuprorelin wurden schwerwiegende unerwünschte Reaktionen der Haut (SCAR) einschließlich Stevens-Johnson-Syndrom (SJS) und Toxische epidermale Nekrolyse (TEN) berichtet, die lebensbedrohlich oder tödlich verlaufen können. Die Patienten sollten zum Zeitpunkt der Verschreibung über die Anzeichen und Symptome aufgeklärt und engmaschig auf schwerwiegende unerwünschte Reaktionen der Haut hin überwacht werden. Wenn Anzeichen und Symptome solcher Reaktionen auftreten, sollte Leuprorelin sofort abgesetzt und ggf. eine alternative Behandlung in Betracht gezogen werden.
Sofort nach der Herstellung verwenden, da sich die Suspension nach der Rekonstitution sehr schnell absetzt.
Die Anwendung von Sixantone kann bei Dopingkontrollen zu positiven Ergebnissen führen.
Sixantone enthält weniger als 1 mmol (23 mg) Natrium pro Zweikammerspritze, d.h., es ist nahezu „natriumfrei“.
Polysorbate können allergische Reaktionen hervorrufen.
Da eine Androgendeprivationstherapie zu einer Verlängerung der QT-Zeit führen kann, sollte die gleichzeitige Anwendung von Sixantone mit anderen Arzneimitteln, die bekanntermaßen zu einer QT-Zeitverlängerung führen, oder Arzneimitteln, die ein Risiko zur Entstehung von Torsade de pointes haben, wie Antiarrhythmika der Klasse IA (z. B. Chinidin, Disopyramid) oder Klasse III (z. B. Amiodaron, Sotalol, Dofetilid, Ibutilid), Methadon, Moxifloxacin, Antipsychotika und andere sorgfältig geprüft werden (siehe Abschnitt 4.4).
Sixantone ist nicht für die Anwendung bei Frauen bestimmt und ist während der Schwangerschaft und Stillzeit generell kontraindiziert.
Fertilität bei Männern: Klinische und pharmakologische Studien an Männern haben gezeigt, dass die Unterdrückung der Fertilität spätestens 24 Wochen nach Absetzen einer kontinuierlichen Leuprorelin-Applikation voll reversibel war.
Wegen der insbesondere zu Therapiebeginn häufig auftretenden Müdigkeit, welche auch durch die zugrunde liegende Tumorerkrankung bedingt sein kann, kann dieses Arzneimittel auch bei bestimmungsgemäßem Gebrauch das Reaktionsvermögen so weit verändern, dass die Fähigkeit zur aktiven Teilnahme am Straßenverkehr oder zum Bedienen von Maschinen gering oder mäßig beeinträchtigt wird. Dies gilt in verstärktem Maße im Zusammenwirken mit Alkohol.
Anfangs kommt es regelmäßig zu einem kurzfristigen Anstieg des Serumtestosteronspiegels (Flare-Phänomen), der zu einer vorübergehenden Verstärkung bestimmter Krankheitssymptome führen kann. Es kann zum Auftreten oder zu einer Zunahme von Harnwegsobstruktion und deren Folgen (beispielsweise Hämaturie) kommen. Bei Patienten mit ossären Metastasen kann es zu einer Zunahme von Knochenschmerzen kommen. Bei Patienten mit Rückenmarkskompression können Muskelschwäche in den Beinen, Lymphödeme und Parästhesie (als neurologische Symptome) auftreten. Diese Zunahme der Beschwerden geht üblicherweise spontan zurück, ohne dass Sixantone abgesetzt werden muss.
Aufgrund des Entzuges der Geschlechtshormone kann es zum Auftreten von Nebenwirkungen kommen. Bei den Häufigkeiten zu Nebenwirkungen sind folgende Kategorien zugrunde gelegt: sehr häufig (≥ 1/10), häufig (≥ 1/100 bis < 1/10), gelegentlich (≥ 1/1 000 bis < 1/100), selten (≥ 1/10 000 bis < 1/1 000), sehr selten (< 1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 1. Nebenwirkungen bei Männern
Erkrankungen des Blutes und des Lymphsystems | |
Nicht bekannt |
Anämie, Leukopenie, Thrombozytopenie |
Erkrankungen des Immunsystems | |
Gelegentlich |
Allgemeine allergische Reaktionen (Fieber, Juckreiz, Eosinophilie, Hautausschlag) |
Sehr selten |
Anaphylaktische Reaktionen |
Nicht bekannt |
Nesselsucht, Atembeschwerden, Schüttelfrost |
Stoffwechsel- und Ernährungsstörungen | |
Sehr häufig |
Gewichtszunahme |
Häufig |
Appetitabnahme/Appetitzunahme |
Selten |
Veränderung einer diabetischen Stoffwechsellage (Erhöhung oder Senkung von Blutzuckerwerten) |
Nicht bekannt |
Metabolisches Syndrom (inklusive Bluthochdruck, Dyslipidämie, Insulinresistenz, gestörte Glukosetoleranz), Steatosis hepatis |
Psychiatrische Erkrankungen | |
Häufig |
Depression, Stimmungsschwankungen (bei Langzeitanwendung von Sixantone), Schlafstörungen |
Gelegentlich |
Stimmungsschwankungen (bei kurzer Anwendung von Sixantone) |
Nicht bekannt |
Suizidgedanken, Suizidales Verhalten, Suizidversuch |
Erkrankungen des Nervensystems | |
Häufig |
Kopfschmerzen |
Gelegentlich |
Schwindel |
Selten |
Vorübergehende Geschmacksveränderungen |
Sehr selten |
Wie auch bei anderen Arzneimitteln dieser Stoffklasse wurde in sehr seltenen Fällen über eine Apoplexie der Hypophyse nach initialer Verabreichung von Leuprorelin bei Patienten mit Hypophysenadenom berichtet. Hypophysenblutungen |
Nicht bekannt |
Krampfanfälle, Idiopathische intrakranielle Hypertonie (Pseudotumor cerebri) (siehe Abschnitt 4.4) |
Augenerkrankungen | |
Nicht bekannt |
Sehstörung |
Herzerkrankungen | |
Nicht bekannt |
Herzklopfen, Verlängerung der QT-Zeit (siehe Abschnitte 4.4 und 4.5) |
Gefäßerkrankungen | |
Sehr häufig |
Hitzewallungen |
Gelegentlich |
Blutdruckveränderungen (Hypertonie, Hypotonie) |
Erkrankungen des Gastrointestinaltrakts | |
Häufig |
Übelkeit, Erbrechen |
Gelegentlich |
Diarrhöe |
Affektionen der Leber und Gallenblase | |
Häufig |
Abnorme Leberfunktion (inkl. Gelbsucht), Abnormer Leberfunktionstest, gewöhnlich vorübergehend |
Erkrankungen der Haut und des Unterhautzellgewebes | |
Sehr häufig |
Hyperhidrosis |
Gelegentlich |
Trockene Haut bzw. Schleimhaut, Nachtschweiß |
Selten |
Alopezie |
Nicht bekannt |
Stevens-Johnson-Syndrom/Toxische epidermale Nekrolyse (SJS/TEN) (siehe Abschnitt 4.4), Toxischer Hautausschlag, Erythema multiforme, Bullöse Dermatitis |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
Nicht bekannt |
Interstitielle Lungenerkrankung |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |
Sehr häufig |
Knochenschmerzen, Muskelschwäche |
Häufig |
Auftreten von Gelenk- bzw. Rückenschmerzen |
Gelegentlich |
Muskelschmerzen |
Nicht bekannt |
Knochendemineralisierung (siehe Abschnitt 4.4), Osteoporose (inklusive vertebrale Frakturen) |
Erkrankungen der Geschlechtsorgane und Brustdrüse | |
Sehr häufig |
Erektile Dysfunktion, Verminderung oder Verlust der Libido und der Potenz, Verkleinerung der Hoden |
Häufig |
Gynäkomastie |
Gelegentlich |
Testikuläre Schmerzen |
Erkrankungen der Nieren und Harnwege | |
Häufig |
Nykturie, Dysurie, Pollakisurie |
Gelegentlich |
Harnverhaltung |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Sehr häufig |
Reaktionen an der Injektionsstelle (z. B. Verhärtungen, |
Häufig |
Periphere Ödeme, Parästhesie, Schlafstörungen |
Untersuchungen | |
Sehr häufig |
Auftreten einer Gewichtszunahme |
Häufig |
Anstiege der LDH, der Transaminasen, der Gamma-GT und der alkalischen Phosphatase, die jedoch Ausdruck der Grundkrankheit sein können. |
Gelegentlich |
Gewichtsabnahme |
Hinweise:
Die Reaktion auf die Sixantone-Therapie kann durch Messung der Serumspiegel von Testosteron, saurer Phosphatase und PSA kontrolliert werden. So steigt der Testosteronspiegel bei Behandlungsbeginn zunächst an und sinkt dann während eines Zeitraumes von zwei Wochen wieder ab. Nach zwei bis vier Wochen werden Testosteronspiegel erreicht, wie sie nach einer beidseitigen Orchiektomie beobachtet werden, und die über den gesamten Behandlungszeitraum bestehen bleiben.
Ein Anstieg saurer Phosphatasespiegel kann in der Anfangsphase der Therapie erfolgen und ist vorübergehender Natur. Gewöhnlich werden nach einigen Wochen wieder Normalwerte bzw. annähernde Normalwerte erreicht.
Im Fall von sehr häufig auftretenden Spritzenabszessen sollten die Testosteronspiegel überprüft werden, da eine unzureichende Resorption von Leuprorelin aus dem Depot mit einem möglichen Wiederansteigen des Testosteronspiegels resultieren kann.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
53175 Bonn
Website: www.bfarm.de
anzuzeigen.
Intoxikationssymptome wurden bisher nicht beobachtet. Im Falle einer Überdosierung sollten die Patienten genau überwacht sowie symptomatisch und supportiv behandelt werden.
Selbst bei Verabreichung von Dosen bis zu 20 mg Leuprorelinacetat pro Tag über zwei Jahre, die bei ersten klinischen Studien Anwendung fanden, konnten keine anderen bzw. neuen Nebenwirkungen gefunden werden, die sich von denen nach täglicher Applikation von 1 mg oder einer Applikation von 30 mg alle 6 Monate unterschieden.
4.10 Missbrauch und Abhängigkeit
Das Risiko einer Abhängigkeit gegenüber Leuprorelinacetat ist sehr gering, da die Applikation nur durch medizinisches Fachpersonal erfolgt. Ein Missbrauch von Leuprorelinacetat würde die endogene Produktion von Sexualhormonen unterdrücken.
Pharmakotherapeutische Gruppe: GnRH-Analoga
ATC-Code: L02AE02
Leuprorelinacetat, der Wirkstoff von Sixantone, ist ein synthetisches Nonapeptid-Analogon des natürlich vorkommenden hypothalamischen Gonadotropin-Releasing-Hormons (GnRH). Dieses Peptid hat durch seine erhöhte Resistenz gegenüber Peptidasen eine längere Halbwertzeit und ist durch seine erhöhte Bindungsaffinität zum GnRH-Rezeptor 80- bis 100-mal potenter als natürlich vorkommendes GnRH.
Die Applikation von Leuprorelinacetat bei Tieren und Menschen führt zu einem Anstieg der gonadotropen Hormone LH (luteinisierendes Hormon) und FSH (follikelstimulierendes Hormon) aus dem Hypophysenvorderlappen. Diese Hormone stimulieren ihrerseits die gonadale Steroidsynthese.
Im Gegensatz zum physiologischen GnRH, das pulsatil vom Hypothalamus freigesetzt wird, blockiert das auch als GnRH-Agonist bezeichnete Leuprorelinacetat bei therapeutischer Daueranwendung die GnRH-Rezeptoren der Hypophyse kontinuierlich und verursacht nach einer initialen, kurzfristigen Stimulation deren Desensibilisierung ("down regulation"). Als Folge kommt es zu einer reversiblen hypophysären Suppression der Gonadotropin-Freisetzung mit nachfolgendem Abfall der Testosteron-Spiegel, und damit zu einer Beeinflussung des Wachstums des karzinomatös veränderten Prostatagewebes, das durch Dihydrotestosteron - gebildet durch Reduktion von Testosteron in den Prostatazellen - normalerweise stimuliert wird.
Die kontinuierliche Applikation von Leuprorelinacetat führt zu einer Abnahme der Anzahl und/oder der Empfindlichkeit (sogenannte "down regulation") der in der Hypophyse vorhandenen Rezeptoren und in der Folge zum Abfall der LH-, FSH- und DHT-Spiegel. Der Testosteronspiegel wird dabei innerhalb von 2 bis 4 Wochen in den Kastrationsbereich abgesenkt.
Auch in Tierversuchen konnte die hormonsenkende Wirkung und Wachstumshemmung von Prostatakarzinomen nachgewiesen werden.
Den experimentellen und klinischen Studien zufolge bewirkt die 6-monatliche Behandlung mit Sixantone nach anfänglicher Stimulation eine Hemmung der Gonadotropinfreisetzung.
Beim Mann bewirkt die subkutane Verabreichung von Sixantone einen anfänglichen Anstieg von LH und FSH, gekennzeichnet durch einen passageren Spiegelanstieg von Testosteron und Dihydrotestosteron.
Da in Einzelfällen in den ersten drei Wochen eine damit zusammenhängende kurzfristige symptomatische Verschlechterung des Krankheitsbildes beobachtet wurde, ist bei Männern mit Prostatakarzinom die zusätzliche Gabe von Antiandrogenen zu erwägen.
Die Langzeittherapie mit Sixantone bewirkt bei allen Patienten eine Senkung der LH- und FSH-Spiegel; es werden beim Mann Androgenspiegel erreicht, wie sie nach einer beidseitigen Orchiektomie vorliegen. Diese Veränderungen treten meist zwei bis drei Wochen nach Therapiebeginn auf und sind über den gesamten Behandlungszeitraum manifest. Gegebenenfalls kann eine Orchiektomie durch eine Therapie mit Sixantone, das alle 6 Monate verabreicht werden muss, ersetzt werden. In der pivotalen Zulassungsstudie verblieben nach 12-monatiger Therapie mit Sixantone die medianen Testosteronspiegel der Patienten zu 98 % im Kastrationsbereich. Dabei wurden für den Beobachtungszeitraum (Monat 1-12) mediane Testosteron-Serumspiegel von 12-15 ng/dl erreicht. Kastrationsspiegel für Testosteron konnten bisher nach kontinuierlicher Gabe von Leuprorelinacetat über fünf Jahre gehalten werden.
Klinische Wirksamkeit
In einer multizentrischen, randomisierten Phase III Studie mit Leuprorelinacetat wurden 263 Patienten mit lokal fortgeschrittenem Prostatakarzinom der Stadien T3 - T4 oder pT3, N0, M0 ausgewertet. Eine Kombination aus Strahlentherapie mit einer Langzeit-Androgenentzugstherapie über 3 Jahre erhielten 133 Patienten und eine alleinige dreijährige Androgenentzugstherapie mit Leuprorelinacetat 130 Patienten.
Basierend auf den ASTRO (Phoenix) Kriterien lag das 5-Jahres-progressionsfreie Überleben bei 60,9 % (64,7 %) in der Kombinationstherapie im Vergleich zu 8,5 % (15,4 %) in der Gruppe mit alleiniger Hormontherapie [p = 0,0001; (p = 0,0005)]. Entsprechend den ASTRO Kriterien lag das Progressionsrisiko 3,8-mal höher in der Gruppe mit alleiniger Hormontherapie (95 % KI [2,17; 6,49]). Die mediane klinische oder biochemische progressionsfreie Überlebenszeit nach ASTRO Definition lag bei 641 Tagen (95 % KI [626; 812]) in der Gruppe mit alleiniger Androgenentzugstherapie und bei 2.804 Tagen (95 % KI [2090; -]; p < 0,0001) in der Gruppe mit Kombinationstherapie. Es ergaben sich weitere statistisch signifikante Unterschiede hinsichtlich einer lokoregionalen Progression [HR 3,6 (95 % KI [1,9; 6,8]; p < 0,0001)], metastatischer Progression (p < 0,018) und metastasenfreiem Überleben (p = 0,018) für die Gruppe mit Kombinationstherapie im Vergleich zur alleinigen Androgenentzugstherapie.
Die Ergebnisse dieser Studie konnten zeigen, dass eine 3-jährige Androgenentzugstherapie mit Leuprorelinacetat in Kombination mit Strahlentherapie der alleinigen 3-jährigen Androgenentzugstherapie mit Leuprorelinacetat überlegen ist.
Die kombinierte Androgenentzugstherapie mit GnRH-Analoga war auch einer alleinigen Strahlentherapie beim lokal fortgeschrittenen Prostatakarzinom überlegen, wie die folgende Studie belegt.
In die randomisierte RTOG 85-31 Studie wurden 977 Patienten mit lokal fortgeschrittenem Prostatakarzinom der Stadien T1- T3 mit Lymphknotenmetastasen, Kapseldurchbruch der Prostata oder Penetration des Prostatakarzinoms in die Samenbläschen aufgenommen. Eine Kombination aus Strahlentherapie mit einer Langzeit-Androgenentzugstherapie mit Goserelin erhielten 488 Patienten und eine alleinige Strahlentherapie 489 Patienten. Wie die Studienergebnisse zeigen, war die kombinierte Androgenentzugstherapie mit Strahlentherapie der alleinigen Strahlentherapie überlegen. Das 10-Jahres-Progressionsfreie-Überleben lag bei 37 % gegenüber 23 % (p < 0,001), das Progressionsfreie-Überleben mit einem PSA-Wert < 1,5 ng/ml lag bei 31 % gegenüber 9 % (p < 0,0001), lokale Rezidive traten bei 23 % gegenüber 38 % auf (p < 0, 0001) und eine Metastasen-bedingte Progression trat bei 24 % gegenüber 39 % auf (p < 0,0001). Das Gesamtüberleben lag bei 49 % gegenüber 39 % (p = 0,002) und die krankheitsspezifische Mortalität lag bei 16 % gegenüber 22 % (p = 0,0052).
Die Überlegenheit für die Androgenentzugstherapie mit GnRH-Analoga in Kombination mit einer Strahlentherapie im Vergleich zu einer alleinigen Strahlentherapie bei lokalisiertem Prostatakarzinom des mittleren Risikoprofils wird durch die folgende Studie belegt.
Die randomisierte Phase III Studie RTOG 94-08 wurde bei Patienten mit lokalisiertem Prostatakarzinom im Stadium T1b, T1c, T2a oder T2b und einem PSA-Wert ≤ 10 ng/ml durchgeführt. Die Subpopulation der Patienten mit mittlerem Risikoprofil, definiert als ein Gleason-Score von 7 oder ein Gleason-Score ≤ 6 in Verbindung mit einem PSA-Wert von > 10 ng/ml bis 20 ng/ml oder das klinische Tumorstadium T2b umfasste in der Gruppe mit Kurzzeit-Androgenentzugstherapie über 4 Monate, zwei Monate vor und zwei Monate überlappend mit der Strahlentherapie 524 Patienten und in der Gruppe mit alleiniger Strahlentherapie 544 Patienten. In der Subpopulation mit mittlerem Risikoprofil war die Gruppe der kombinierten Behandlung aus Androgenentzugstherapie mit Leuprorelinacetat oder Goserelin und Strahlentherapie der Patientengruppe mit alleiniger Strahlentherapie überlegen. Das Gesamtüberleben lag nach 10 Jahren bei 61 % gegenüber 54 % [Hazard Ratio 1,23, 95 % KI (1,02 - 1,49; p = 0,03)]. Die krankheitspezifische Mortalität lag bei 3% gegenüber 10 % [Hazard Ratio 2,49, 95 % KI (1,50 - 4,11; p = 0,004)] und die biochemische Progression bei 28 % gegenüber 45 % [Hazard Ratio 1,79, 95 % KI (1,45 - 2,21; p < 0,001)].
Der Nachweis für den Einsatz bei lokalisiertem Prostatakarzinom des Hoch-Risikoprofils beruht auf veröffentlichten Studien zur Strahlentherapie in Kombination mit GnRH-Analoga einschließlich Leuprorelinacetat. Es wurden klinische Daten aus fünf veröffentlichten Studien analysiert (EORTC 22863, RTOG 85-31, RTOG 92-02, RTOG 8610 und D’Amico et al., JAMA 2004), die alle den Vorteil der Kombination von GnRH-Analoga mit Strahlentherapie zeigen. Eine klare Differenzierung zwischen den Studienpopulationen für die Indikationen lokal fortgeschrittenes Prostatakarzinom und lokales Prostatakarzinom des Hoch-Risikoprofils war in den veröffentlichten Studien nicht möglich.
Klinische Daten haben gezeigt, dass Strahlentherapie gefolgt von 3 Jahren Androgenentzugstherapie gegenüber Strahlentherapie gefolgt von 6 Monaten Androgenentzugstherapie vorzuziehen ist. Die in medizinischen Leitlinien empfohlene Dauer der Androgenentzugstherapie bei Patienten mit T3 bis T4 Tumoren, die eine Strahlentherapie erhalten, beträgt 2 bis 3 Jahre.
Metastasiertes hormonsensitives Prostatakarzinom (mHSPC)
Kombination mit Enzalutamid (ARCHES-Studie)
In die ARCHES-Studie wurden 1150 Patienten mit mHSPC eingeschlossen, welche im Verhältnis 1:1 randomisiert wurden, um eine Behandlung mit Enzalutamid plus Androgenentzugstherapie oder Placebo plus Androgenentzugstherapie (Androgenentzugstherapie war definiert als LHRH-Analogon oder bilaterale Orchiektomie) zu erhalten. Die Patienten erhielten 160 mg Enzalutamid einmal täglich (N = 574) oder Placebo (N = 576).
Das radiologisch progressionsfreie Überleben (rPFS), basierend auf einer unabhängigen zentralen Bewertung, war der primäre Endpunkt, definiert als die Zeit von der Randomisierung bis zum ersten objektiven Nachweis einer radiologischen Krankheitsprogression oder bis zum Tod (aufgrund jeglicher Ursache vom Zeitpunkt der Randomisierung bis zu 24 Wochen nach Absetzen der Studienmedikation), je nachdem, was zuerst eintrat. Enzalutamid zeigte im Vergleich zu Placebo eine statistisch signifikante Reduktion des Risikos eines rPFS-Ereignisses um 61 % [HR = 0,39 (95 %-KI: 0,30; 0,50); p < 0,0001]. Übereinstimmende rPFS-Ergebnisse wurden beobachtet bei Patienten mit hoher oder niedriger Tumorlast und Patienten mit oder ohne vorherige Chemotherapie mit Docetaxel.
Tabelle 2. Zusammenfassung der Wirksamkeitsergebnisse bei Patienten, die im Rahmen der ARCHES-Studie entweder mit Enzalutamid oder Placebo behandelt wurden (Intent-to-Treat-Analyse)
Enzalutamid plus Androgenentzugstherapie |
Placebo plus Androgenentzugstherapie |
|
Radiologisch progressionsfreies Überleben | ||
Anzahl Ereignisse (%) |
91 (15,9) |
201 (34,9) |
Median, Monate (95%-KI)1 |
nicht erreicht |
19,0 (16,6; 22,2) |
Hazard Ratio (95%-KI)2 |
0,39 (0,30; 0,50) |
|
p-Wert2 |
p < 0,0001 |
|
|
1. Berechnet unter Verwendung der Methode nach Brookmeyer und Crowley. 2. Stratifiziert nach Tumorlast (niedrig oder hoch) und vorheriger Anwendung von Docetaxel (ja oder nein). | ||
Wichtigste in der Studie beurteilte sekundäre Wirksamkeitsendpunkte waren unter anderem die Zeit bis zur PSA-Progression, die Zeit bis zum Beginn einer neuen antineoplastischen
Therapie, die Rate nicht nachweisbarer PSA-Werte (Rückgang auf < 0,2 μg/l) und die objektive Ansprechrate (RECIST 1.1 laut unabhängiger Bewertung). Für all diese sekundären Endpunkte wurden statistisch signifikante Verbesserungen für mit Enzalutamid behandelte Patienten im Vergleich zu Placebo gezeigt.
Ein weiterer wichtiger in der Studie beurteilter sekundärer Wirksamkeitsendpunkt war das Gesamtüberleben. Bei der zuvor geplanten abschließenden Analyse des Gesamtüberlebens, die nach 356 beobachteten Todesfällen durchgeführt wurde, zeigte sich eine statistisch signifikante Verringerung des Sterberisikos um 34 % in der Gruppe, die zur Behandlung mit Enzalutamid randomisiert worden war, im Vergleich zu der Gruppe, die zur Behandlung mit Placebo randomisiert worden war [HR = 0,66, (95 %-KI: 0,53; 0,81), p < 0,0001]. Die mediane Zeit für das Gesamtüberleben wurde in beiden Behandlungsgruppen nicht erreicht. Die geschätzte mediane Nachbeobachtungszeit für alle Patienten betrug 44,6 Monate.
Kombination mit Apalutamid (TITAN-Studie)
Die TITAN-Studie war eine randomisierte, doppelblinde, placebokontrollierte, multinationale, multizentrische klinische Studie, in der 1 052 mHSPC-Patienten im Verhältnis 1:1 randomisiert wurden. Sie erhielten entweder einmal täglich 240 mg Apalutamid oral (n = 525) oder einmal täglich Placebo (n = 527). Alle Patienten mussten mindestens eine Knochenmetastase in der Technetium 99 m Skelettszintigraphie aufweisen. Patienten wurden ausgeschlossen, wenn nur isolierte Lymphknoten- oder Viszeralmetastasen (z. B. Leber oder Lunge) vorlagen. Alle Patienten in der TITAN-Studie erhielten begleitend ein GnRH-Analogon oder hatten sich zuvor einer bilateralen Orchiektomie unterzogen. Etwa 11 % der Patienten erhielten zuvor eine Behandlung mit Docetaxel (maximal 6 Zyklen, letzte Dosis ≤ 2 Monate vor Randomisierung und anhaltendes Ansprechen). Die co-primären Wirksamkeitsendpunkte der Studie waren das Gesamtüberleben (overall survival, OS) und das radiografisch progressionsfreie Überleben (radiographic progression-free survival, rPFS). Die Ergebnisse für die Wirksamkeit in der TITAN-Studie sind in Tabelle 3 zusammengefasst.
Tabelle 3. Zusammenfassung der Ergebnisse zur Wirksamkeit für die Intent-to-treat mHSPC-Population (TITAN-Studie)
Endpunkt |
Apalutamid |
Placebo |
Primäres Gesamtüberlebena |
||
Todesfälle (%) |
83 (16 %) |
117 (22 %) |
Median, Monate (95% KI) |
NE (NE, NE) |
NE (NE, NE) |
Hazard Ratio (95% KI)b |
0,671 (0,507; 0,890) |
|
p-Wertc |
0,0053 |
|
Aktualisiertes Gesamtüberlebend |
||
Todesfälle (%) |
170 (32 %) |
235 (45 %) |
Median, Monate (95% KI) |
NE (NE, NE) |
52 (42, NE) |
Hazard Ratio (95% KI)b |
0,651 (0,534; 0,793) |
|
p-Wertc,e |
<0,0001 |
|
Radiografisches progressionsfreies Überleben |
||
Radiografische Progression der Erkrankung oder Tod (%) |
134 (26 %) |
231 (44 %) |
Median, Monate (95% KI) |
NE (NE, NE) |
22,08 (18,46; 32,92) |
Hazard Ratio (95% KI)b |
0,484 (0,391; 0,600) |
|
p-Wertc |
< 0,0001 |
|
a Dies basiert auf der vorab festgelegten Zwischenanalyse mit einer medianen Nachbeobachtungszeit von 22 Monaten. | ||
Eine konsistente Verbesserung des rPFS wurde über alle Subgruppen hinweg beobachtet, einschließlich bei Patienten mit hoher oder niedriger Tumorlast (high- or low-volume disease), Metastasierungsstadium bei der Diagnose (M0 oder M1), vorheriger Anwendung von Docetaxel (Ja vs Nein), Alter (< 65, ≥ 65 oder ≥ 75 Jahre), PSA-Wert bei Baseline oberhalb des Medians (Ja oder Nein) und Anzahl der Knochenmetastasen (≤ 10 oder > 10).
Eine konsistente Verbesserung des OS wurde über alle Subgruppen hinweg beobachtet, einschließlich bei Patienten mit hoher oder niedriger Tumorlast (high- or low-volume disease), Metastasierungs-stadium bei der Diagnose (M0 oder M1) und Gleason-Score bei Diagnose (≤ 7 vs. > 7).
Kombination mit Darolutamid und Docetaxel (ARASENS-Studie)
Die Wirksamkeit und Sicherheit von Darolutamid in Kombination mit Docetaxel wurden in einer multizentrischen, doppelblinden, placebokontrollierten Phase-III-Studie (ARASENS) bei Patienten mit mHSPC beurteilt. Insgesamt wurden 1306 Patienten im Verhältnis 1:1 randomisiert und erhielten oral zweimal täglich entweder 600 mg Darolutamid (n = 651) oder passendes Placebo (n = 655), zusammen mit 75 mg/m2 Docetaxel für 6 Zyklen. Die Behandlung mit Darolutamid oder Placebo wurde fortgeführt bis eines der folgenden Ereignisse eintrat: symptomatische progrediente Erkrankung, Änderung der anti-neoplastischen Therapie, nicht akzeptable toxische Wirkung, Tod oder Absetzen der Behandlung.
Primärer Wirksamkeitsendpunkt war das Gesamtüberleben (OS).
Tabelle 4. Wirksamkeitsergebnisse aus der ARASENS-Studie
Wirksamkeitsparameter |
Anzahl (%) der Patienten mit Ereignissen |
Median (Monate) (95 % KI) |
Hazard Ratiob |
||
Darolutamid + Docetaxel |
Placebo+ Docetaxel |
Darolutamid |
Placebo+ |
||
Gesamtüberlebend |
229 (35,2 %) |
304 (46,5 %) |
NE |
48,9 |
0,675 |
a Ein Patient im Placeboarm wurde von allen Analysen ausgeschlossen | |||||
Die folgenden sekundären Wirksamkeitsendpunkte zeigten einen statistisch signifikanten Vorteil zugunsten der Patienten im Darolutamid+Docetaxel-Arm im Vergleich zu Patienten im Placebo+Docetaxel-Arm: Zeit bis zum Auftreten eines kastrationsresistenten Prostatakarzinoms (medianes NE vs. 19,1 Monate; HR = 0,357, p < 0,0001); Zeit bis zum ersten symptomatischen Skelettereignis (medianes NE vs. NE Monate; HR = 0,712, p = 0,0081); Zeit bis zum Beginn einer nachfolgenden antineoplastischen Chemotherapie (medianes NE vs. 25,3 Monate; HR = 0,388, p < 0,0001); Zeit bis zur Schmerzprogression (medianes NE vs. 27,5 Monate; HR = 0,792, p = 0,0058); symptomatische skelettereignisfreie Überlebenszeit (Median 51,2 vs. 39,7 Monate; HR = 0,609, p < 0,0001).
Kombination mit Abirateronacetat und Prednison oder Prednisolon für Patienten mit neu diagnostiziertem Hochrisiko-Prostatakarzinom (Studie 3011)
In die Studie 3011 wurden Patienten eingeschlossen, die maximal 3 Monate vor Randomisierung neu mit mHSPC diagnostiziert wurden und Hochrisiko-Prognosefaktoren aufwiesen. Hochrisiko-Prognose war definiert als Vorliegen von mindestens 2 der folgenden 3 Risikofaktoren: (1) Gleason-Score von ≥8; (2) Vorliegen von mindestens 3 Läsionen in der Knochenszintigraphie; (3) Vorliegen von messbaren viszeralen Metastasen (ausgeschlossen Lymphknotenbefall). Im aktiven Arm wurde Abirateronacetat in einer Dosierung von 1000 mg täglich in Kombination mit niedrig dosiertem Prednison 5 mg einmal täglich zusätzlich zur ADT (LHRH-Agonist oder Orchiektomie) gegeben. Letztere entsprach der Standardbehandlung. Patienten im Kontroll-Arm erhielten ADT und Placebo für sowohl Abirateronacetat als auch Prednison. Das mediane Alter der in Studie 3011 (n = 1199) eingeschlossenen Patienten betrug 67 Jahre.
Co-primare Wirksamkeitsendpunkte waren Gesamtüberleben (overall survival, OS) und radiographisch progressionsfreies Überleben (radiographic progression free survival, rPFS). Neben den co-primären Endpunkten wurde auch der Nutzen anhand folgender Faktoren beurteilt: Zeit bis zum nächsten skelettalen Ereignis (skeletal-related event, SRE), Zeit bis zur Folgetherapie des Prostatakarzinoms, Zeit bis zum Beginn einer Chemotherapie, Zeit bis zur Schmerzprogression und Zeit bis zur PSA-Progression.
Radiographisch progressionsfreies Überleben war definiert als Zeit von der Randomisierung bis zum Auftreten einer radiographischen Progression oder zum Tod aufgrund jeglicher Ursache.
Es wurde ein signifikanter Unterschied im rPFS zwischen den Behandlungsgruppen beobachtet (siehe Tabelle 5).
Tabelle 5. Radiographisch progressionsfreies Überleben – Stratifizierte Analyse, Intent-To-Treat-Population (Studie PCR3011)
AA-P |
Placebo |
|
Randomisierte Patienten |
597 |
602 |
Zeit bis Ereignis (in Monaten) |
33,02 (29,57, NE) |
14,78 (14,69, 18,27) |
p-Werta |
< 0,0001 |
|
Hinweis: + = zensierte Beobachtung, NE (not estimable) = nicht auswertbar. Die radiographische Progression und der Tod sind in der Definition des rPFS-Ereignisses berücksichtigt. AA-P = Patienten, die Abirateronacetat und Prednison erhielten. | ||
Im Vergleich zu Placebo plus ADT war eine statistisch signifikante Verbesserung des Gesamtüberlebens (OS) zugunsten von AA-P plus ADT mit einer 34%igen Reduktion des Risikos zu versterben zu beobachten (HR = 0,66; 95 % KI: 0,56; 0,78; p < 0,0001), (siehe Tabelle 6).
Tabelle 6. Gesamtüberleben der Patienten, die entweder mit Abirateron oder mit Placebos in Studie PCR3011 behandelt wurden (Intent-to-Treat Analyse)
Gesamtüberleben |
Abirateron mit Prednison |
Placebos |
Todesfälle (%) |
275 (46 %) |
343 (57 %) |
Hazard-Ratio (95 % KI)1 |
0,66 (0,56; 0,78) |
|
NE = nicht auswertbar | ||
Kombination mit Docetaxel (STAMPEDE- und CHAARTED-Studie)
STAMPEDE-Studie
Die Sicherheit und Wirksamkeit von Docetaxel mit gleichzeitiger Anwendung des Behandlungsstandards (Androgendeprivationstherapie) bei Patienten mit lokal fortgeschrittenem hormonsensitiven Hochrisiko-Prostatakarzinom oder mit metastasiertem hormonsensitivem Prostatakarzinom wurden in einer randomisierten, multizentrischen, multi-Arm multi-Stage (MAMS) Studie mit einem Phase-II/III-Studiendesign (STAMPEDE – MRC PR08) untersucht. Insgesamt wurden 1776 männliche Patienten in die folgenden Behandlungsarme randomisiert:
— Behandlungsstandard + Docetaxel 75 mg/m2 alle 3 Wochen über 6 Zyklen,
— Behandlungsstandard allein.
Zur Anwendung von Docetaxel wurden kontinuierlich 5 mg Prednison oder Prednisolon zweimal täglich als Begleitmedikation verabreicht. Von den 1776 randomisierten Patienten hatten 1086 (61 %) eine metastasierte Erkrankung, von denen 362 Patienten auf Docetaxel in Kombination
mit Behandlungsstandard randomisiert wurden und 724 Patienten den Behandlungsstandard allein erhielten.
Bei Patienten mit metastasiertem Prostatakarzinom war das mediane Gesamtüberleben in der Docetaxel-Gruppe signifikant länger im Vergleich zu der Gruppe, die nur den Behandlungsstandard erhielt. Durch das Hinzufügen von Docetaxel zum Behandlungsstandard verlängerte sich das mediane Gesamtüberleben um 19 Monate (HR = 0,76; 95 % KI = 0,62 – 0,92; p = 0,005).
Die Wirksamkeitsergebnisse aus dem Docetaxel-Behandlungsarm gegenüber dem Kontrollarm für Patienten mit metastasiertem Prostatakarzinom sind in der nachfolgenden Tabelle zusammengefasst:
Tabelle 7. Wirksamkeit von Docetaxel in Kombination mit Prednison oder Prednisolon und dem Behandlungsstandard bei der Behandlung von Patienten mit metastasiertem hormonsensitivem Prostatakarzinom (STAMPEDE)
Endpunkt |
Docetaxel + Behandlungsstandard |
Behandlungsstandard allein |
Anzahl der Patienten mit metastasiertem Prostatakarzinom |
362 |
724 |
Medianes Gesamtüberleben (Monate) |
62 |
43 |
95 % KI |
51-73 |
40-48 |
Adjustierte Hazard Ratio |
0,76 |
|
Überleben ohne Therapieversagenb |
0,4 |
12 |
95 % KI |
16,8-25,2 |
9,6- 12 |
Adjustierte Hazard Ratio |
0,66 |
|
a p-Wert berechnet aus dem Likelihood-Quotienten-Test und adjustiert für alle Stratifikationsfaktoren (außer Zentrum und geplante Hormontherapie) und stratifiziert nach Studienphase | ||
CHAARTED-Studie
Die Sicherheit und Wirksamkeit der Anwendung von Docetaxel zu Beginn einer Androgendeprivationstherapie (ADT) bei Patienten mit metastasiertem hormonsensitivem Prostatakarzinom wurden in einer randomisierten, multizentrischen Phase-III-Studie (CHAARTED) untersucht.
Insgesamt wurden 790 männliche Patienten in die beiden Behandlungsgruppen randomisiert.
— ADT + Docetaxel 75 mg/m2 alle 3 Wochen über 6 Zyklen mit Beginn der ADT
— ADT allein
Das mediane Gesamtüberleben in der Docetaxel-Gruppe war signifikant länger als in der Gruppe mit ADT allein. Durch das Hinzufügen von Docetaxel zur Androgendeprivationstherapie verlängerte sich das mediane Gesamtüberleben um 13,6 Monate (HR = 0,61, 95 % KI = 0,47 – 0,80, p = 0,0003).
Die Wirksamkeitsergebnisse aus dem Docetaxel-Behandlungsarm gegenüber dem Kontrollarm sind in der nachfolgenden Tabelle zusammengefasst.
Tabelle 8. Wirksamkeit von Docetaxel und ADT bei der Behandlung von Patienten mit metastasiertem hormonsensitivem Prostatakarzinom (CHAARTED)
Endpunkt |
Docetaxel + ADT |
ADT allein |
Anzahl an Patienten |
397 |
393 |
Medianes Gesamtüberleben (Monate) aller Patienten |
57,6 |
44,0 |
95 % KI |
49,1-72,8 |
34,4-49,1 |
Progressionsfreies Überleben |
19,8 |
11,6 |
95 % KI |
16,7-22,8 |
11,8-14,3 |
Adjustierte Hazard Ratio |
0,60 |
- |
95 % KI |
0,51-0,72 |
- |
p-Wert* |
< 0,0001 |
- |
PSA-Ansprechen** nach 6 Monaten – n (%) |
127 (32,0) |
77 (19,6) |
PSA-Ansprechen** nach 12 Monaten – n (%) |
110 (27,7) |
66 (16,8) |
Zeit bis zum kastrationsresistenten Prostatakarzinomb |
||
Median (Monate) |
20,2 |
11,7 |
Zeit bis zur klinischen Progressionc |
33,0 |
19,8 |
a Zeit bis Event-Variablen: Stratifizierter Log-Rank-Test | ||
Nicht metastasiertes kastrationsresistentes Prostatakarzinom (nmCRPC)
Kombination mit Enzalutamid (PROSPER-Studie)
Die PROSPER-Studie schloss 1401 Patienten mit asymptomatischem, nicht metastasiertem Hochrisiko-CRPC ein, die ihre Androgenentzugstherapie (definiert als LHRH Analogon oder vorangegangene bilaterale Orchiektomie) fortsetzten. Die Patienten mussten eine Verdopplungszeit des prostataspezifischen Antigens (Prostate Specific Antigen Doubling Time, PSADT) von ≤ 10 Monate, einen PSA-Wert von ≥ 2 ng/ml und die Bestätigung einer nicht metastasierten Erkrankung mittels einer verblindeten unabhängigen zentralen Bewertung aufweisen.
Die Patienten erhielten randomisiert im Verhältnis 2:1 entweder Enzalutamid in einer Dosis von 160 mg einmal täglich (N = 933) oder Placebo (N = 468). Die Patienten wurden nach der PSADT (< 6 Monate oder ≥ 6 Monate) und der Anwendung von gezielt in den Knochen wirkenden Arzneimitteln (ja oder nein) stratifiziert.
Das metastasenfreie Überleben (MFS) war der primäre Endpunkt, definiert als die Zeit ab der Randomisierung bis zu radiologischer Progression oder Tod innerhalb von 112 Tagen nach Absetzen der Behandlung ohne Nachweis einer radiologischen Progression, je nachdem, was zuerst eintrat. Wichtigste in der Studie beurteilte sekundäre Endpunkte waren Zeit bis zur PSA-Progression, Zeit bis zur ersten Anwendung einer neuen antineoplastischen Therapie (TTA) und Gesamtüberleben (OS). Weitere sekundäre Endpunkte waren Zeit bis zur Anwendung einer zytotoxischen Chemotherapie und Chemotherapie-freies Überleben.
Enzalutamid zeigte im Vergleich zu Placebo eine statistisch signifikante Reduktion des relativen Risikos von radiologischer Progression oder Tod um 71 % [HR = 0,29 (95 %-KI: 0,24; 0,35), p < 0,0001]. Das mediane MFS betrug 36,6 Monate (95 %-KI: 33,1; nicht erreicht) im Enzalutamid-Arm versus 14,7 Monate (95 %-KI: 14,2; 15,0) im Placebo-Arm (siehe Tabelle 9).
Enzalutamid zeigte im Vergleich zu Placebo eine statistisch signifikante Reduktion des relativen Risikos einer PSA-Progression um 93 % [HR = 0,07 (95 %-KI: 0,05; 0,08), p < 0,0001]. Die mediane Zeit bis zur PSA-Progression betrug 37,2 Monate (95 %-KI: 33,1; nicht erreicht) im Enzalutamid-Arm versus 3,9 Monate (95 %-KI: 3,8; 4,0) im Placebo-Arm.
Enzalutamid zeigte im Vergleich zu Placebo eine statistisch signifikante Verzögerung der Zeit bis zur ersten Anwendung einer neuen antineoplastischen Therapie [HR = 0,21 (95 %-KI: 0,17; 0,26), p < 0,0001]. Die mediane Zeit bis zur ersten Anwendung einer neuen antineoplastischen Therapie betrug 39,6 Monate (95 %-KI: 37,7; nicht erreicht) im Enzalutamid-Arm versus 17,7 Monate (95 %-KI: 16,2; 19,7) im Placebo-Arm.
Tabelle 9. Zusammenfassung der Wirksamkeitsergebnisse in der PROSPER-Studie (Intent-to-Treat-Analyse)
Enzalutamid |
Placebo |
||
Primärer Endpunkt | |||
Metastasenfreies Überleben | |||
Anzahl Ereignisse (%) |
219 (23,5) |
228 (48,7) |
|
Median, Monate (95 %-KI)1 |
36,6 (33,1; nicht erreicht) |
14,7 (14,2; 15,0) |
|
Hazard Ratio (95 %-KI)2 |
0,29 (0,24; 0,35) |
||
p-Wert3 |
p < 0,0001 |
||
Wichtigste sekundäre Wirksamkeitsendpunkte | |||
Gesamtüberleben4 |
|||
Anzahl Ereignisse (%) |
288 (30,9) |
178 (38,0) |
|
Median, Monate (95 %-KI)1 |
67,0 (64,0; nicht erreicht) |
56,3 (54,4; 63,0) |
|
Hazard Ratio (95 %-KI)2 |
0,734 (0,608; 0,885) |
||
p-Wert3 |
p = 0,0011 |
||
Zeit bis zur PSA-Progression |
|||
Anzahl Ereignisse (%) |
208 (22,3) |
324 (69,2) |
|
Median, Monate (95 %-KI)1 |
37,2 (33,1; nicht erreicht) |
3,9 (3,8; 4,0) |
|
Hazard Ratio (95 %-KI)2 |
0,07 (0,05; 0,08) |
||
p-Wert3 |
p < 0,0001 |
||
Zeit bis zur ersten Anwendung einer neuen antineoplastischen Therapie |
|||
Anzahl Ereignisse (%) |
142 (15,2) |
226 (48,3) |
|
Median, Monate (95 %-KI)1 |
39,6 (37,7; nicht erreicht) |
17,7 (16,2; 19,7) |
|
Hazard Ratio (95 %-KI)2 |
0,21 (0,17; 0,26) |
||
p-Wert3 |
p < 0,0001 |
||
1 Auf Basis von Kaplan-Meier-Schätzungen. | |||
Kombination mit Apalutamid (SPARTAN-Studie)
Insgesamt 1207 Studienteilnehmer mit nmCRPC wurden 2:1 randomisiert und erhielten in einer multizentrischen, doppelblinden klinischen Studie (Studie ARN-509-003) entweder Apalutamid oral in einer Dosis von 240 mg einmal täglich in Kombination mit einer Androgendeprivationstherapie (ADT) (medikamentöse Kastration oder vorherige chirurgische Kastration) oder Placebo mit ADT. Die eingeschlossenen Patienten hatten eine Verdopplungszeit des Prostataspezifischen Antigens (PSADT) von ≤ 10 Monaten. Daher wurde bei ihnen ein hohes Risiko für eine unmittelbar drohende Metastasierung und Tod aufgrund von Prostatakrebs angenommen. Alle nicht chirurgisch kastrierten Studienteilnehmer erhielten während der Studie durchgängig eine fortlaufende ADT. Die PSA-Ergebnisse waren verblindet und wurden nicht als Grund für einen Behandlungsabbruch verwendet.
Primärer Endpunkt war metastasenfreies Überleben (metastasis-free survival, MFS), definiert als Zeit von der Randomisierung bis zum Zeitpunkt des ersten Nachweises von BICR-bestätigten Fernmetastasen in Knochen oder Weichteilen oder bis zum Tod jeglicher Ursache, je nachdem, was zuerst eintrat. Die Behandlung mit Apalutamid verbesserte das metastasenfreie Überleben signifikant. Apalutamid verminderte das relative Risiko für Fernmetastasen oder Tod im Vergleich zu Placebo um 70 % (HR = 0,30; 95 %-KI: 0,24; 0,36; p < 0,0001). Das mediane MFS betrug 41 Monate unter Apalutamid und 16 Monate unter Placebo.
Unter Apalutamid wurde eine konsistente Verbesserung des MFS für alle vordefinierten Subgruppen beobachtet, wie Alter, ethnische Zugehörigkeit, Region, Lymphknotenstatus, Anzahl der früheren Hormontherapien, PSA-Wert bei Baseline, PSA-Verdopplungszeit, ECOG-Status bei Baseline und Anwendung von osteoprotektiven Wirkstoffen.
Unter Berücksichtigung aller Daten zeigten die mit Apalutamid und ADT behandelten Studienteilnehmer gegenüber den allein mit ADT Behandelten eine signifikante Verbesserung bei den folgenden sekundären Endpunkten: Zeit bis zur Metastasierung (HR = 0,28; 95 %-KI: 0,23; 0,34; p < 0,0001); progressionsfreies Überleben (progression-free survival, PFS) (HR = 0,30; 95 %-KI: 0,25; 0,36; p < 0,0001); Zeit bis zur symptomatischen Progression (HR = 0,57; 95 %-KI: 0, 44; 0,73; p < 0,0001); Gesamtüberleben (overall survival, OS) (HR = 0,78; 95 %-KI: 0,64; 0,96; p = 0,0161) und Zeit bis zur Einleitung einer zytotoxischen Chemotherapie (HR = 0,63; 95 %-KI: 0,49, 0,81; p = 0,0002).
Bei einem medianen Nachbeobachtungszeitraum von 52,0 Monaten zeigten die Ergebnisse, dass die Behandlung mit Apalutamid das Risiko zu versterben im Vergleich zu Placebo signifikant um 22 % reduzierte (HR = 0,784; 95 %-KI: 0,643, 0,956; 2-seitiges p = 0,0161). Das mediane Gesamtüberleben (OS) betrug 73,9 Monate im Apalutamid-Arm und 59,9 Monate im Placebo-Arm. Das vorab festgelegte Signifikanzniveau (p ≤ 0,046) wurde überschritten und statistische Signifikanz erreicht. Diese Verbesserung zeigte sich, obwohl 19 % der Patienten im Placebo-Arm Apalutamid als Folgetherapie erhielten.
Die Behandlung mit Apalutamid reduzierte das Risiko für die Einleitung einer zytotoxischen Chemotherapie signifikant um 37 % im Vergleich zu Placebo (HR = 0,629; 95 %-KI: 0,489, 0,808; p = 0,0002) und zeigte damit eine statistisch signifikante Verbesserung für Apalutamid im Vergleich zu Placebo. Die mediane Zeit bis zur Einleitung einer zytotoxischen Chemotherapie wurde für beide Behandlungsarme nicht erreicht.
Das PFS-2, definiert als die Zeit bis zum Tod oder Fortschreiten der Erkrankung durch PSA, radiographische oder symptomatische Progression unter oder nach der ersten Folgetherapie war bei mit Apalutamid behandelten Studienteilnehmern im Vergleich zu den mit Placebo Behandelten länger. Die Ergebnisse zeigten eine 44%ige Reduktion des PFS-2-Risikos unter Apalutamid im Vergleich zu Placebo (HR = 0,565, 95 %-KI: 0,471, 0,677; p < 0,0001).
Kombination mit Darolutamid (ARAMIS-Studie)
Die Wirksamkeit und Sicherheit von Darolutamid wurden in einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase-III-Studie (ARAMIS) bei Patienten mit nicht-metastasiertem (beurteilt mittels konventioneller CT-, Knochenscan- oder MRT-Aufnahmen) kastrationsresistentem Prostatakarzinom mit einer Verdopplungszeit des prostataspezifischen Antigens (PSADT) von ≤ 10 Monaten beurteilt. Alle Patienten erhielten begleitend ein Luteinisierendes-Hormon-Releasing-Hormon (LHRH)-Analogon oder hatten sich zuvor einer bilateralen Orchiektomie unterzogen.
Patienten wurden in die Studie aufgenommen, wenn während der Androgendeprivationstherapie nach dem Nadir in mindestens 1-wöchigen Abständen drei ansteigende Werte für das Prostataspezifische Antigen (PSA) festgestellt wurden, der PSA-Spiegel beim Screening ≥ 2 ng/ml und der Kastrationsspiegel für Testosteron im Serum < 1,7 nmol/l betrug.
Insgesamt erhielten 1509 Patienten, randomisiert im Verhältnis 2:1, entweder zweimal täglich 600 mg Darolutamid oral (n = 955) oder Placebo (n = 554). Patienten mit Beckenlymphknoten von < 2 cm Größe in der kurzen Achse unterhalb der Aortenbifurkation konnten in die Studie aufgenommen werden. Das Vorliegen oder Fehlen von Metastasen wurde durch eine unabhängige zentrale radiologische Beurteilung festgestellt. Eingeschlossen in diese Analysen waren 89 Patienten, bei denen retrospektiv Metastasen zu Studienbeginn identifiziert wurden. Die Randomisierung war nach PSADT (≤ 6 Monate oder > 6 Monate) und Anwendung einer zielgerichteten Therapie gegen Osteoklasten bei Studieneintritt (ja oder nein) stratifiziert.
Die Behandlung mit Darolutamid führte im Vergleich zu Placebo zu einer Verbesserung des MFS (siehe Tabelle 10). Die Ergebnisse zu MFS stimmten über alle Subgruppen der Patienten hinweg überein, unabhängig von PSADT, vorheriger Anwendung von Osteoprotektiva oder lokoregionärer Erkrankung. Weitere Subgruppen mit einheitlichen MFS-Ergebnissen waren PSA-Ausgangswert, Gleason-Score bei Diagnose, Alter, geographische Region, ECOG-PS-Ausgangswert, ethnische Zugehörigkeit und Anzahl vorheriger Hormontherapien.
Tabelle 10. Wirksamkeitsergebnisse aus der ARAMIS-Studie
Wirksamkeitsparameter |
Anzahl (%) der Patienten mit Ereignissen |
Median (Monate) (95 % KI) |
Hazard Ratiob |
||
Darolutamid |
Placeboa |
Darolutamid |
Placeboa |
||
Metastasenfreies Überlebenc |
221 (23,1 %) |
216 (39,0 %) |
40,4 |
18,4 |
0,413 |
Gesamtüberleben |
148 (15,5 %) |
106 (19,1 %) |
NE |
NE |
0,685 |
Zeit bis zur Schmerzprogressionc,d |
251 (26,3 %) |
178 (32,1 %) |
40,3 |
25,4 |
0,647 |
Zeit bis zum Einleiten einer ersten zytotoxischen Chemotherapie |
127 (13,3 %) |
98 (17,7 %) |
NE |
NE |
0,579 |
Zeit bis zum ersten symptomatischen Skelettereignis |
29 (3,0 %) |
28 (5,1 %) |
NE |
NE |
0,484 |
ª Einschließlich 170 Patienten, die zu open-label Darolutamid gewechselt sind | |||||
Die Behandlung mit Darolutamid führte zu einem längeren progressionsfreien Überleben (PFS; Median 36,8 vs. 14,8 Monate; HR = 0,380; nominaler p-Wert < 0,000001) und einer längeren Zeit bis zur PSA-Progression (Median 29,5 Monate vs. 7,2 Monate; HR = 0,164; nominaler p-Wert < 0,000001). Über alle Überlebensparameter (MFS, OS und PFS) hinweg wurde eine konsistente Wirkung beobachtet. Patienten, die in der ARAMIS-Studie (Doppelblindzeit) Darolutamid erhielten, zeigten eine signifikant höhere bestätigte PSA-Ansprechrate (definiert als Reduktion um ≥ 50 % gegenüber dem Ausgangswert) als Patienten, die Placebo erhielten: 84,0 % vs. 7,9 % (Differenz = 76,1 %; p < 0,000001) (nominaler p-Wert, nur zur Information).
Metastasiertes kastrationsresistentes Prostatakarzinom (mCRPC)
In klinischen Studien konnte bei Patienten mit metastasiertem, kastrationsresistentem Prostatakarzinom der Nutzen einer zusätzlichen Wirkstoffgabe, wie etwa Inhibitoren der Androgensynthese (z. B. Abirateronacetat), Androgen-Rezeptor-Inhibitoren (z. B. Enzalutamid), Taxane (z. B. Docetaxel oder Cabazitaxel), PARP-Inhibitoren (z.B. Olaparib), oder Strahlentherapeutika (z. B. Radium-223, 177Lutetium-PSMA-617) zusätzlich zu GnRH-Agonisten, wie Leuprorelinacetat gezeigt werden.
Freisetzung
Der Wirkstoff Leuprorelinacetat wird nach Injektion der Depotsuspension Sixantone kontinuierlich aus dem Milchsäurepolymer über den Zeitraum von 6 Monaten freigesetzt. Das Polymer wird dabei wie chirurgisches Nahtmaterial resorbiert.
Resorption
Die Leuprorelinspiegel im Serum steigen nach einmaliger s.c. Applikation von Sixantone bei Prostatakrebspatienten rasch an und fallen dann nach einigen Tagen auf ein Plateau ab. Innerhalb von 1,8 Stunden werden im Mittel maximale Serumspiegel von 102 ng/ml gemessen. In der Plateauphase befinden sich nachweisbare Spiegel im Serum bis > 26 Wochen nach der Applikation. Bei einigen Patienten waren Leuprorelinspiegel bis zu 30 Wochen nachweisbar. Abb. 1 zeigt den Verlauf des Leuprorelinspiegels nach einmaliger Gabe von Sixantone.
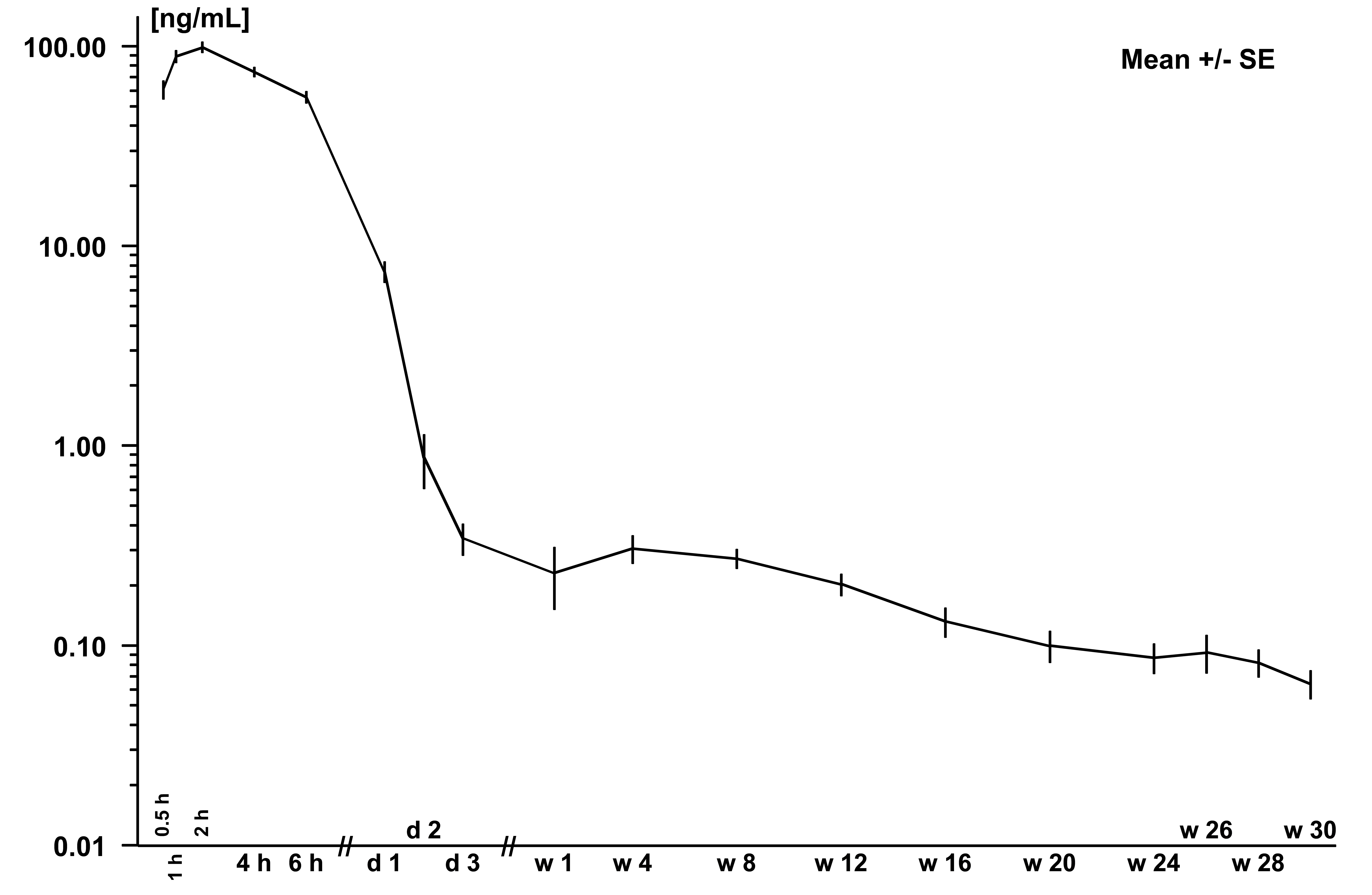
Abb. 1. Leuprorelinspiegel im Serum nach einmaliger s.c. Applikation von Sixantone bei Prostatakrebspatienten
Ein erster Wiederanstieg des Testosteronspiegels wird im Mittel nach 200 Tagen beobachtet, wenn keine weitere Injektion von Sixantone erfolgt.
Verteilung und Elimination
Das Verteilungsvolumen von Leuprorelin beträgt bei Männern 36 l, die totale Clearance liegt bei 139,6 ml/min (bestimmt unter Anwendung von Enantone Monats-Depot).
Bei wiederholter Gabe kommt es zu einer anhaltenden Senkung des Testosteronspiegels in den Kastrationsbereich, ohne dass der Testosteronspiegel wie nach erstmaliger Injektion einen vorübergehenden Anstieg zeigt.
Besondere Patientengruppen
Patienten mit eingeschränkter Nierenfunktion/Leberfunktion
Bei Patienten mit einer eingeschränkten Nierenfunktion (Kreatininwert 1,4 ml bis 2 mg/dl) ist der Leuprorelinserumspiegel genauso wie bei Patienten mit normaler Nierenfunktionen.
Bei Patienten mit schwerer Nierenfunktion (Kreatininwert > 2 mg/dl) wurden nach Gabe von Leuprorelin (als Enantone Monats-Depot) teilweise höhere Leuprorelinserumspiegel gemessen. Klinisch scheint diese Beobachtung jedoch ohne Relevanz.
Bei Patienten mit eingeschränkter Leberfunktion zeigten sich keine Auswirkungen auf den Leuprorelinserumspiegel.
Präklinische Studien mit Leuprorelinacetat zeigten bei beiden Geschlechtern Auswirkungen auf das Fortpflanzungssytem, die aufgrund der bekannten pharmakologischen Wirkungen zu erwarten sind. Diese Wirkungen sind im Prinzip nach einer Erholungsphase reversibel (siehe Abschnitt 5.1).
Leuprorelinacetat zeigte keine teratogene Wirkung. Aufgrund der pharmakologischen Wirkungen auf das Reproduktionssystem zeigte sich Embryotoxizität und -letalität bei Kaninchen.
Studien zur Karzinogenität wurden an Ratten und Mäusen über 24 Monate durchgeführt. Bei Ratten wurde nach subkutaner Injektion eine dosisabhängige Zunahme von gutartigen Hypophysenhyperplasien und gutartigen Hypophysenadenomen bei Dosierungen von 0,6 bis 4 mg/kg/Tag beobachtet. Kein derartiger Effekt wurde bei Mäusen beobachtet, so dass der Effekt an Ratten als Spezies-spezifisch angesehen werden kann.
Leuprorelinacetat wirkte nicht mutagen in einer Reihe von in vitro und in vivo Untersuchungen.
Retardmikrokapseln:
270,0 mg Polymilchsäure
Mannitol (Ph.Eur.)
Suspensionsmittel:
Mannitol (Ph.Eur.)
Carmellose-Natrium
Polysorbat 80
Essigsäure 99 % (zur Einstellung des pH-Wertes)
Wasser für Injektionszwecke
Nicht zutreffend. Sixantone soll nicht gleichzeitig mit anderen Arzneimitteln injiziert werden.
Die Retardmikrokapseln und das Suspensionsmittel sind 36 Monate haltbar.
Bei Auftreten einer Verfärbung der Retardmikrokapseln und/oder Trübung des klaren Suspensionsmittels vor der Suspendierung darf die Zweikammerspritze nicht mehr verwendet werden. Nach der Zubereitung entsteht eine milchig-trübe Suspension.
Die chemische und physikalische Stabilität der gebrauchsfertigen Zubereitung wurde für 24 Stunden bei 25 °C nachgewiesen. Aus mikrobiologischer Sicht sollte die gebrauchsfertige Zubereitung sofort verwendet werden. Wenn die gebrauchsfertige Zubereitung nicht sofort verwendet wird, ist der Anwender für die Dauer und die Bedingungen der Aufbewahrung verantwortlich.
Vor der Injektion ist die Zweikammerspritze erneut vorsichtig gegen die Handfläche zu klopfen, um eine gleichmäßige Wirkstoffverteilung zu gewährleisten.
Nicht über 25 °C lagern. Zweikammerspritze im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
1 Zweikammerspritze mit 352,9 mg Retardmikrokapseln und 1 ml Suspensionsmittel.
Die Zweikammerspritze (Glas EP Typ I) mit Luer-Lock-Verschluss (Polypropylen) und Stopfensystem (Chlorbutyl-Gummi) ist versiegelt in einer Blisterpackung.
Reste von Arzneimitteln und deren Kontaktmaterialen sollten entsprechend der gesetzlichen Bestimmungen entsorgt werden. Entsorgen Sie Arzneimittel auf keinen Fall im Abwasser.
Anleitung zur Herstellung der Sixantone Suspension
Spritzenstempel bis zum Anschlag eindrehen.
Spritze während der Zubereitung mit der Kanüle (Nadelseite) senkrecht nach oben halten (um ein Austreten von Suspensionsmittel zu vermeiden).
Das Stopfensystem langsam bis zur blauen Markierung vorschieben.
Dabei gelangt das Suspensionsmittel über den Bypass in die innere Wirkstoffkammer.
Der Spritzenstempel darf jetzt nicht mehr zurückgezogen werden.
Spritze zur Herstellung einer milchigen Suspension mit der Kanüle senkrecht nach oben halten und diese vorsichtig gegen die Handfläche klopfen, um das Pulver und das Suspensionsmittel zu mischen, bis eine homogene Suspension entsteht (nicht waagerecht oder nach unten halten, da sonst Suspensionsmittel austreten kann). Die fertige Suspension sollte milchig und ohne sichtbare Klümpchen sein. Starkes Klopfen oder Schütteln vermeiden, um Blasenbildung zu limitieren.
Vor der Injektion die Schutzkappe von der Injektionsnadel gerade nach oben abziehen, nicht abdrehen! Dann die Luft über der Suspension vorsichtig herausdrücken.
Die Spritze ist jetzt injektionsbereit.
Eine Aspiration ist bei subkutan liegender Injektionsnadel möglich.
Handhabung nach der Injektion
Sicherheitsvorrichtung an der Kanüle bis zum fühlbaren/hörbaren Einrasten entsprechend der Pfeilmarkierung ganz nach vorne schieben.
Spritze bitte gemäß den örtlichen Vorschriften entsorgen. Spritzen und Nadeln nicht über den Hausmüll entsorgen.
Ein Video mit der Anleitung zur Herstellung der Suspension erhalten Sie, indem Sie mit einem Smartphone/Tablet den QR-Code in der Fach- oder der Gebrauchsinformation scannen.
Dieselbe Information finden Sie auch unter folgendem Link: https://www.prostata.de/handhabung/leuprorelin

Takeda GmbH
Byk-Gulden-Str. 2
78467 Konstanz
Tel.: +49 (0)800 825 332 5
Fax: +49 (0)800 825 332 9
E-Mail: medinfoEMEA@takeda.com
63660.00.00
Datum der Erteilung der Zulassung: 08. Juli 2008
Datum der letzten Verlängerung der Zulassung: 03. Februar 2014
Mai 2026
Verschreibungspflichtig.
Mitvertrieb
APOGEPHA Arzneimittel GmbH
Kyffhäuserstr. 27
01309 Dresden
Telefon: 0351 3363-3
Telefax: 0351 3363-440
E-Mail: info@apogepha.de
Art.-Nr. 1107103936