
YERVOY 5 mg/ml Konzentrat zur Herstellung einer Infusionslösung
Jeder ml des Konzentrats enthält 5 mg Ipilimumab.
Eine 10‑ml‑Durchstechflasche enthält 50 mg Ipilimumab.
Eine 40‑ml‑Durchstechflasche enthält 200 mg Ipilimumab.
Ipilimumab ist ein vollständig humaner Anti‑CTLA‑4‑Antikörper (IgG1κ), der mittels rekombinanter DNA‑Technologie aus Ovarialzellen des Chinesischen Hamsters gewonnen wird.
Sonstige Bestandteile mit bekannter Wirkung:
Jeder ml Konzentrat enthält 0,1 mmol Natrium, was 2,30 mg Natrium entspricht.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
Konzentrat zur Herstellung einer Infusionslösung (steriles Konzentrat).
Klare bis leicht opaleszierende, farblose bis blass gelbe Flüssigkeit, die helle (wenige) Schwebstoffe enthalten kann und einen pH‑Wert von 7,0 und eine Osmolarität von 260‑300 mOsm/kg hat.
Melanom
YERVOY ist als Monotherapie oder in Kombination mit Nivolumab zur Behandlung des fortgeschrittenen (nicht resezierbaren oder metastasierten) Melanoms bei Erwachsenen und Jugendlichen ab einem Alter von 12 Jahren indiziert (für weitere Informationen siehe Abschnitt 4.4).
Im Vergleich zur Nivolumab‑Monotherapie wurde in der Kombination Nivolumab mit Ipilimumab nur bei Patienten mit niedriger Tumor‑PD‑L1‑Expression ein Anstieg des progressionsfreien Überlebens (progression‑free survival, PFS) und des Gesamtüberlebens (overall survival, OS) gezeigt (siehe Abschnitte 4.4 und 5.1).
Nierenzellkarzinom (renal cell carcinoma, RCC)
YERVOY ist in Kombination mit Nivolumab für die Erstlinientherapie des fortgeschrittenen Nierenzellkarzinoms bei Erwachsenen mit intermediärem/ungünstigem Risikoprofil indiziert (siehe Abschnitt 5.1).
Nicht‑kleinzelliges Lungenkarzinom (non‑small cell lung cancer, NSCLC)
YERVOY ist in Kombination mit Nivolumab und 2 Zyklen platinbasierter Chemotherapie für die Erstlinientherapie des metastasierten nicht‑kleinzelligen Lungenkarzinoms (NSCLC) bei Erwachsenen, deren Tumoren keine sensitivierende EGFR‑Mutation oder ALK‑Translokation aufweisen, indiziert.
Malignes Pleuramesotheliom (MPM)
YERVOY ist in Kombination mit Nivolumab für die Erstlinientherapie des nicht‑resezierbaren malignen Pleuramesothelioms bei Erwachsenen indiziert.
Kolorektalkarzinom (colorectal cancer, CRC) mit Mismatch‑Reparatur‑Defizienz (Mismatch repair deficient, dMMR) oder hoher Mikrosatelliteninstabilität (microsatellite instability high, MSI‑H)
YERVOY ist in Kombination mit Nivolumab zur Behandlung des Kolorektalkarzinoms mit Mismatch‑Reparatur‑Defizienz oder hoher Mikrosatelliteninstabilität bei Erwachsenen in den folgenden Fällen indiziert:
Erstlinientherapie des nicht resezierbaren oder metastasierten Kolorektalkarzinoms
Behandlung des metastasierten Kolorektalkarzinoms nach vorheriger fluoropyrimidinbasierter Kombinationschemotherapie (siehe Abschnitt 5.1).
Plattenepithelkarzinom des Ösophagus (esophageal squamous cell carcinoma, ESCC)
YERVOY ist in Kombination mit Nivolumab für die Erstlinienbehandlung des nicht resezierbaren fortgeschrittenen, rezidivierten oder metastasierten Plattenepithelkarzinoms des Ösophagus mit Tumorzell‑PD‑L1‑Expression ≥ 1 % bei Erwachsenen indiziert.
Hepatozelluläres Karzinom (hepatocellular carcinoma, HCC)
YERVOY ist in Kombination mit Nivolumab für die Erstlinientherapie des nicht resezierbaren oder fortgeschrittenen hepatozellulären Karzinoms bei Erwachsenen indiziert.
Die Behandlung muss von einem auf dem Gebiet der Krebsbehandlung erfahrenen Facharzt eingeleitet und überwacht werden.
PD‑L1‑Testung
Falls im Anwendungsgebiet angegeben, sollten die Patienten für eine Behandlung mit YERVOY basierend auf einer mittels validierten Tests bestätigten Tumor‑PD‑L1‑Expression selektiert werden (siehe Abschnitte 4.1, 4.4 und 5.1).
MSI‑/MMR‑Testung
Falls im Anwendungsgebiet angegeben, sollen die Patienten für eine Behandlung mit YERVOY mittels eines IVDs (In-vitro-Diagnostikum) mit CE-Kennzeichnung und entsprechendem Verwendungszweck beurteilten MSI‑H‑/dMMR‑Tumorstatus selektiert werden. Wenn das IVD mit CE-Kennzeichnung nicht verfügbar ist, sollte ein alternativer validierter Test verwendet werden (siehe Abschnitte 4.1, 4.4 und 5.1).
Dosierung
YERVOY als Monotherapie
Melanom
Erwachsene und Jugendliche ab einem Alter von 12 Jahren
Das empfohlene Induktionsregime für YERVOY liegt bei 3 mg/kg, intravenös über einen Zeitraum von 30 Minuten verabreicht, alle 3 Wochen für insgesamt 4 Dosen. Die Patienten sollten, sofern es die Verträglichkeit erlaubt, das gesamte Induktionsregime (4 Dosen) erhalten, unabhängig davon, ob neue Läsionen auftreten oder bestehende Läsionen weiter wachsen. Die Beurteilung des Tumoransprechens sollte erst nach Abschluss der Induktionstherapie durchgeführt werden.
YERVOY in Kombination mit Nivolumab
Melanom
Bei Erwachsenen und Jugendlichen ab einem Alter von 12 Jahren und einem Gewicht von mindestens 50 kg beträgt die empfohlene Dosis 3 mg/kg Ipilimumab in Kombination mit 1 mg/kg Nivolumab, die alle 3 Wochen für die ersten 4 Dosen intravenös infundiert wird. Anschließend folgt eine zweite Phase, in welcher Nivolumab als Monotherapie in einer Dosierung von entweder 240 mg alle 2 Wochen oder 480 mg alle 4 Wochen (siehe Abschnitte 5.1 und 5.2) intravenös infundiert wird wie in Tabelle 1 dargestellt. In der Monotherapie‑Phase sollte die erste Nivolumab‑Dosis wie folgt verabreicht werden:
3 Wochen nach der letzten Dosis der Kombination von Nivolumab und Ipilimumab, wenn 240 mg alle 2 Wochen gegeben werden, oder
6 Wochen nach der letzten Dosis der Kombination von Nivolumab und Ipilimumab, wenn 480 mg alle 4 Wochen gegeben werden.
Bei Jugendlichen ab einem Alter von 12 Jahren und einem Gewicht von weniger als 50 kg beträgt die empfohlene Dosis 3 mg/kg Ipilimumab in Kombination mit 1 mg/kg Nivolumab, die alle 3 Wochen für die ersten 4 Dosen intravenös infundiert wird. Anschließend folgt eine zweite Phase, in welcher Nivolumab als Monotherapie in einer Dosierung von entweder 3 mg/kg alle 2 Wochen oder 6 mg/kg alle 4 Wochen (siehe Abschnitte 5.1 und 5.2) intravenös infundiert wird wie in Tabelle 1 dargestellt. In der Monotherapie‑Phase sollte die erste Nivolumab‑Dosis wie folgt verabreicht werden:
3 Wochen nach der letzten Dosis der Kombination von Nivolumab und Ipilimumab, wenn 3 mg/kg alle 2 Wochen gegeben werden, oder
6 Wochen nach der letzten Dosis der Kombination von Nivolumab und Ipilimumab, wenn 6 mg/kg alle 4 Wochen gegeben werden.
Tabelle 1: Empfohlene Dosierungen und Infusionszeiten zur intravenösen Verabreichung von Ipilimumab in Kombination mit Nivolumab – Melanom
Kombinationsphase, alle 3 Wochen für 4 Dosierungszyklen |
Monotherapiephase |
|
Nivolumab |
Erwachsene und Jugendliche ab einem Alter von 12 Jahren: |
Erwachsene und Jugendliche (ab einem Alter von 12 Jahren und einem Gewicht von mindestens 50 kg): |
Ipilimumab |
Erwachsene und Jugendliche ab einem Alter von 12 Jahren: |
- |
Nierenzellkarzinom
Die empfohlene Dosis beträgt 1 mg/kg Ipilimumab in Kombination mit 3 mg/kg Nivolumab, die alle 3 Wochen für die ersten 4 Dosen intravenös infundiert wird. Anschließend folgt eine zweite Phase, in welcher Nivolumab als Monotherapie in einer Dosierung von entweder 240 mg alle 2 Wochen oder 480 mg alle 4 Wochen intravenös infundiert wird, wie in Tabelle 2 dargestellt. In der Monotherapie‑Phase sollte die erste Nivolumab‑Dosis wie folgt verabreicht werden:
3 Wochen nach der letzten Dosis der Kombination von Nivolumab und Ipilimumab, wenn 240 mg alle 2 Wochen gegeben werden, oder
6 Wochen nach der letzten Dosis der Kombination von Nivolumab und Ipilimumab, wenn 480 mg alle 4 Wochen gegeben werden.
Tabelle 2: Empfohlene Dosierungen und Infusionszeiten zur intravenösen Verabreichung von Ipilimumab in Kombination mit Nivolumab für RCC
Kombinationsphase, alle 3 Wochen für 4 Dosierungszyklen |
Monotherapiephase |
|
Nivolumab |
3 mg/kg über 30 Minuten |
240 mg alle 2 Wochen über 30 Minuten oder |
Ipilimumab |
1 mg/kg über 30 Minuten |
- |
dMMR‑ oder MSI‑H‑Kolorektalkarzinom
Die empfohlene Dosis für die Erstlinientherapie des dMMR‑ oder MSI‑H‑Kolorektalkarzinoms beträgt 1 mg/kg Ipilimumab in Kombination mit 240 mg Nivolumab, die alle 3 Wochen für maximal 4 Dosen intravenös verabreicht wird, gefolgt von einer Monotherapie mit Nivolumab intravenös in einer Dosierung von entweder 240 mg alle 2 Wochen oder 480 mg alle 4 Wochen, wie in Tabelle 3 dargestellt. In der Monotherapiephase soll die erste Nivolumab-Dosis 3 Wochen nach der letzten Dosis der Kombination von Nivolumab und Ipilimumab verabreicht werden. Die Behandlung mit Nivolumab soll bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate bei Patienten ohne Progression der Erkrankung fortgesetzt werden.
Die empfohlene Dosis für Patienten, die zuvor eine fluoropyrimidinbasierte Kombinationschemotherapie für die Behandlung des dMMR‑ oder MSI‑H‑Kolorektalkarzinoms erhalten haben, beträgt 1 mg/kg Ipilimumab in Kombination mit 3 mg/kg Nivolumab, die alle 3 Wochen für die ersten 4 Dosen intravenös verabreicht wird, gefolgt von einer Monotherapie mit Nivolumab intravenös in einer Dosierung von 240 mg alle 2 Wochen, wie in Tabelle 3 dargestellt. In der Monotherapiephase sollte die erste Nivolumab-Dosis 3 Wochen nach der letzten Dosis der Kombination von Ipilimumab und Nivolumab verabreicht werden.
Tabelle 3: Empfohlene Dosierungen und Infusionszeiten zur intravenösen Verabreichung von Ipilimumab in Kombination mit Nivolumab für dMMR‑ oder MSI‑H‑Kolorektalkarzinom
Kombinationsphase, alle 3 Wochen für 4 Dosierungszyklen |
Monotherapiephase |
||
Nivolumab |
Erstlinientherapie |
240 mg über 30 Minuten |
240 mg alle 2 Wochen über 30 Minuten oder |
Nach vorheriger fluoropyrimidinbasierter Kombinationschemotherapie |
3 mg/kg über 30 Minuten |
240 mg alle 2 Wochen über 30 Minuten |
|
Ipilimumab |
1 mg/kg über 30 Minuten |
- |
|
Malignes Pleuramesotheliom
Die empfohlene Dosis beträgt 1 mg/kg Ipilimumab intravenös über 30 Minuten alle 6 Wochen in Kombination mit 360 mg Nivolumab intravenös über 30 Minuten alle 3 Wochen. Die Behandlung wird bei Patienten ohne Progression der Erkrankung bis zu 24 Monate fortgesetzt.
Plattenepithelkarzinom des Ösophagus
Die empfohlene Dosis beträgt 1 mg/kg Ipilimumab intravenös über 30 Minuten alle 6 Wochen in Kombination mit entweder 3 mg/kg Nivolumab alle 2 Wochen oder 360 mg Nivolumab alle 3 Wochen intravenös über 30 Minuten. Die Behandlung sollte bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate bei Patienten ohne Progression der Erkrankung fortgesetzt werden.
Hepatozelluläres Karzinom
Die empfohlene Dosis beträgt 3 mg/kg Ipilimumab in Kombination mit 1 mg/kg Nivolumab, die alle 3 Wochen für bis zu 4 Dosen intravenös verabreicht wird. Anschließend folgt eine zweite Phase, in welcher die Nivolumab‑Monotherapie entweder mit 240 mg alle 2 Wochen oder 480 mg alle 4 Wochen (siehe Abschnitte 5.1 und 5.2) intravenös verabreicht wird, wie in Tabelle 4 dargestellt. Es wird empfohlen, die Behandlung bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate fortzusetzen. In der Monotherapiephase soll die erste Nivolumab‑Dosis wie folgt verabreicht werden:
3 Wochen nach der letzten Dosis der Kombination von Nivolumab und Ipilimumab, wenn 240 mg alle 2 Wochen oder 480 mg alle 4 Wochen gegeben werden.
Tabelle 4: Empfohlene Dosierungen und Infusionszeiten zur intravenösen Verabreichung von Ipilimumab in Kombination mit Nivolumab für HCC
Kombinationsphase, alle 3 Wochen für 4 Dosierungszyklen |
Monotherapiephase |
|
Nivolumab |
1 mg/kg über 30 Minuten |
240 mg alle 2 Wochen über 30 Minuten oder |
Ipilimumab |
3 mg/kg über 30 Minuten |
- |
YERVOY in Kombination mit Nivolumab und Chemotherapie
Nicht‑kleinzelliges Lungenkarzinom
Die empfohlene Dosis beträgt 1 mg/kg Ipilimumab intravenös über 30 Minuten alle 6 Wochen in Kombination mit 360 mg Nivolumab intravenös über 30 Minuten alle 3 Wochen und platinbasierter Chemotherapie alle 3 Wochen. Nach 2 Zyklen Chemotherapie wird die Behandlung mit 1 mg/kg Ipilimumab intravenös alle 6 Wochen in Kombination mit 360 mg Nivolumab intravenös alle 3 Wochen fortgesetzt. Die Behandlung sollte bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate bei Patienten ohne Progression der Erkrankung fortgesetzt werden.
Dauer der Behandlung
Die Behandlung mit YERVOY in Kombination mit Nivolumab sollte fortgesetzt werden, solange der klinische Nutzen besteht oder bis die Behandlung vom Patienten nicht mehr vertragen wird (oder bis zur maximalen Therapiedauer, soweit diese für eine Indikation festgelegt ist).
Untypisches Ansprechen (z. B. eine initiale vorübergehende Zunahme der Tumorgröße oder kleine, neue Läsionen innerhalb der ersten Monate gefolgt von einer Schrumpfung des Tumors) wurde beobachtet. Bei klinisch stabilen Patienten mit initialen Anzeichen einer Krankheitsprogression wird empfohlen, die Behandlung mit YERVOY in Kombination mit Nivolumab fortzusetzen bis eine Krankheitsprogression bestätigt ist.
Vor Einleitung und vor jeder erneuten Gabe von YERVOY müssen sowohl die Leberwerte mittels Leberfunktionstests (LFTs) als auch die Schilddrüsenwerte analysiert werden. Zusätzlich müssen die Patienten während der Behandlung mit YERVOY auf Anzeichen oder Symptome von immunvermittelten Nebenwirkungen einschließlich Diarrhö und Kolitis untersucht werden (siehe Tabellen 5A, 5B und Abschnitt 4.4).
Kinder unter 12 Jahren
Die Sicherheit und Wirksamkeit von YERVOY bei Kindern unter 12 Jahren ist nicht nachgewiesen.
Dauerhafter Behandlungsabbruch oder das Aufschieben von Dosen
Die Behandlung immunvermittelter Nebenwirkungen kann das Aufschieben einer Dosis oder einen dauerhaften Abbruch der Behandlung mit YERVOY und die Einleitung einer Therapie mit systemischen hochdosierten Corticosteroiden erfordern. In einigen Fällen kann eine zusätzliche Therapie mit anderen Immunsuppressiva in Betracht gezogen werden (siehe Abschnitt 4.4).
Eine Dosissteigerung oder ‑reduktion wird nicht empfohlen. Je nach individueller Sicherheit und Verträglichkeit ist möglicherweise ein Aufschieben einer Dosis oder ein dauerhafter Abbruch der Behandlung erforderlich.
Die Richtlinien für einen permanenten Abbruch oder das Aufschieben von Dosen sind in den Tabellen 5A und 5B für YERVOY als Monotherapie und in Tabelle 5C für YERVOY in Kombination mit Nivolumab oder Verabreichung der zweiten Behandlungsphase (Nivolumab‑Monotherapie), welche der Kombinationsphase folgt, aufgeführt. Detaillierte Empfehlungen für die Behandlung von immunvermittelten Nebenwirkungen sind im Abschnitt 4.4 beschrieben.
Tabelle 5A: Wann muss YERVOY als Monotherapie dauerhaft abgesetzt werden?
Setzen Sie YERVOY dauerhaft ab, wenn Patienten folgende Nebenwirkungen aufweisen. Die Behandlung dieser Nebenwirkungen kann auch eine systemische hochdosierte Corticosteroidtherapie erfordern, wenn es sich dabei nachweislich oder mutmaßlich um immunvermittelte Nebenwirkungen handelt (für detaillierte Behandlungsrichtlinien siehe Abschnitt 4.4). | |
Nebenwirkungen |
NCI‑CTCAE v4 Grada |
Gastrointestinal: |
|
Hepatisch: |
|
Haut: |
|
Neurologisch: |
|
Sonstige Organsystemeb: |
|
a Toxizitätsgrade entsprechen den Kriterien des National Cancer Institute (National Cancer Institute Common Terminology Criteria for Adverse Events), Version 4.0 (NCI‑CTCAE v4). | |
Tabelle 5B: Wann sollte eine Dosis von YERVOY als Monotherapie aufgeschoben werden?
Schieben Sie eine YERVOY‑Dosisa bei Patienten mit folgenden immunvermittelten Nebenwirkungen auf. Für detaillierte Behandlungsrichtlinien siehe Abschnitt 4.4. | |
Nebenwirkungen |
Maßnahme |
Gastrointestinal: |
|
Hepatisch: | |
Haut: | |
Endokrin: | |
Neurologisch: | |
Sonstige mäßige Nebenwirkungenc | |
a Es wird keine Dosisreduktion von YERVOY empfohlen. | |
Tabelle 5C: Empfohlene Behandlungsmodifikationen für YERVOY in Kombination mit Nivolumab oder Verabreichung der zweiten Behandlungsphase (Nivolumab‑Monotherapie), welche der Kombinationsbehandlung folgt
Immunvermittelte Nebenwirkungen |
Schweregrad |
Behandlungsmodifikation |
|
Immunvermittelte |
Pneumonitis Grad 2 |
Dosis(en) aufschieben bis sich die Symptome zurückgebildet haben, radiologisch erkennbare Veränderungen sich gebessert haben und die Behandlung mit Corticosteroiden beendet ist |
|
Pneumonitis Grad 3 oder 4 |
Setzen Sie die Behandlung dauerhaft ab |
||
Immunvermittelte Kolitis |
Diarrhö oder Kolitis Grad 2 |
Dosis(en) aufschieben bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden, falls erforderlich, beendet ist |
|
Grad 3 oder 4 Diarrhö oder Kolitis |
Setzen Sie die Behandlung dauerhaft ab |
||
Immunvermittelte Hepatitis ohne HCC |
Erhöhung der Aspartat-Aminotransferase (AST), Alanin-Aminotransferase (ALT) oder Gesamtbilirubin Grad 2 |
Dosis(en) aufschieben bis die Laborwerte auf den Ausgangswert zurückgegangen sind und die Behandlung mit Corticosteroiden, falls erforderlich, beendet ist |
|
Erhöhung von AST, ALT, oder Gesamtbilirubin Grad 3 oder 4 |
Setzen Sie die Behandlung dauerhaft ab |
||
Immunvermittelte Hepatitis mit HCC |
Falls der AST/ALT‑Ausgangswert im Normalbereich liegt und auf > 3- und ≤ 10-mal Obergrenze des Normbereichs (upper limit of normal, ULN) ansteigt |
Dosis(en) aufschieben, bis die Laborwerte auf den Ausgangswert zurückgegangen sind und die Behandlung mit Corticosteroiden, falls erforderlich, beendet ist |
|
|
Setzen Sie die Behandlung dauerhaft ab |
||
Immunvermittelte Nephritis und Nierenfunktionsstörung |
Kreatinin‑Erhöhung Grad 2 oder 3 |
Dosis(en) aufschieben bis das Kreatinin auf den Ausgangswert zurückgegangen ist und die Behandlung mit Corticosteroiden beendet ist |
|
Kreatinin‑Erhöhung Grad 4 |
Setzen Sie die Behandlung dauerhaft ab |
||
Immunvermittelte |
Symptomatische Grad 2 oder 3 Hypothyreose, Hyperthyreose, Hypophysitis, |
Dosis(en) aufschieben bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden (falls nötig bei Symptomen akuter Entzündung) beendet ist. Die Behandlung sollte begleitend zur Hormonersatztherapiea fortgeführt werden, sofern keine Symptome auftreten |
|
Grad 4 Hypothyreose |
Setzen Sie die Behandlung dauerhaft ab |
||
Immunvermittelte |
Hautausschlag Grad 3 |
Dosis(en) aufschieben bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden beendet ist |
|
Hautausschlag Grad 4 |
Setzen Sie die Behandlung dauerhaft ab |
||
Stevens‑Johnson Syndrom (SJS) oder toxische epidermale Nekrolyse (TEN) |
Setzen Sie die Behandlung dauerhaft ab (siehe Abschnitt 4.4) |
||
Immunvermittelte |
Grad 2 Myokarditis |
Dosis(en) aufschieben bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden beendet istb |
|
Grad 3 oder 4 Myokarditis |
Setzen Sie die Behandlung dauerhaft ab |
||
Andere immunvermittelte Nebenwirkungen |
Grad 3 (erstes Auftreten) |
Dosis(en) aufschieben |
|
Grad 4 oder wiederauftretender Grad 3; trotz Behandlungsmodifikation persistierender Grad 2 oder 3; Fälle, in denen die Corticosteroiddosis nicht auf 10 mg Prednison oder das entsprechende Äquivalent pro Tag reduziert werden kann |
Setzen Sie die Behandlung dauerhaft ab |
||
Myokarditis-Myositis- |
Grad 2 Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom |
Dosis(en) aufschieben, bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden beendet ist |
|
Grad 3 oder 4 Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom |
Setzen Sie die Behandlung dauerhaft ab |
||
Hinweis: Toxizitätsgrade entsprechen den Kriterien des National Cancer Institute (National Cancer Institute Common Terminology Criteria for Adverse Events), Version 4.0 (NCI CTCAE v4). | |||
YERVOY in Kombination mit Nivolumab muss dauerhaft abgesetzt werden bei:
Grad 4 oder wiederauftretenden Grad 3 Nebenwirkungen,
Grad 2 oder 3 Nebenwirkungen, die trotz Behandlung persistieren.
Wenn YERVOY in Kombination mit Nivolumab angewendet wird, soll bei Aufschiebung des einen Wirkstoffes auch die Gabe des anderen Wirkstoffs aufgeschoben werden. Wenn die Behandlung nach einer Pause wieder aufgenommen wird, sollte aufgrund individueller Beurteilung des Patienten entweder die Kombinationsbehandlung oder Nivolumab‑Monotherapie wieder aufgenommen werden.
Spezielle Patientenpopulationen
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von YERVOY als Monotherapie bei Kindern unter 12 Jahren ist nicht nachgewiesen. Es stehen nur sehr begrenzt Daten zur Verfügung. YERVOY sollte bei Kindern unter 12 Jahren nicht verwendet werden.
Die Sicherheit und Wirksamkeit von YERVOY in Kombination mit Nivolumab bei Kindern unter 18 Jahren ist nicht erwiesen, außer bei Jugendlichen ab einem Alter von 12 Jahren mit Melanom. Zurzeit vorliegende Daten werden in den Abschnitten 4.2, 4.8, 5.1 und 5.2 beschrieben.
Ältere Menschen
Zwischen älteren (≥ 65 Jahre) und jüngeren Patienten (< 65 Jahre) wurden keine allgemeinen Unterschiede im Hinblick auf Sicherheit und Wirksamkeit festgestellt. Daten von Erstlinien‑RCC‑Patienten ab 75 Jahren sind zu begrenzt, um Rückschlüsse auf diese Population zu ziehen (siehe Abschnitt 5.1). In dieser Patientengruppe ist keine spezifische Dosisanpassung erforderlich (siehe Abschnitt 5.1).
Eingeschränkte Nierenfunktion
Sicherheit und Wirksamkeit von YERVOY wurden bei Patienten mit eingeschränkter Nierenfunktion nicht untersucht. Auf der Grundlage von Daten zur Populationspharmakokinetik ist bei Patienten mit leichter bis mäßiger Niereninsuffizienz keine spezielle Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Eingeschränkte Leberfunktion
Sicherheit und Wirksamkeit von YERVOY wurden bei Patienten mit eingeschränkter Leberfunktion nicht untersucht. Basierend auf populationspharmakokinetischen Daten ist bei Patienten mit leicht eingeschränkter Leberfunktion keine spezielle Dosisanpassung erforderlich (siehe Abschnitt 5.2). Bei Patienten mit Transaminasewerten ≥ 5 x ULN oder Bilirubinwerten > 3 x ULN zu Therapiebeginn muss YERVOY mit Vorsicht eingesetzt werden (siehe Abschnitt 5.1).
Art der Anwendung
YERVOY ist zur intravenösen Anwendung. Die empfohlene Infusionsdauer beträgt 30 Minuten.
YERVOY kann unverdünnt oder mittels einer Natriumchloridlösung 9 mg/ml (0,9 %) für Injektionszwecke oder Glucoselösung 50 mg/ml (5 %) für Injektionszwecke in einer Konzentration zwischen 1 und 4 mg/ml verdünnt intravenös verabreicht werden.
YERVOY darf nicht durch intravenöse Druck‑ oder Bolusinjektion verabreicht werden.
Wenn YERVOY in Kombination mit Nivolumab oder in Kombination mit Nivolumab und Chemotherapie angewendet wird, soll Nivolumab zuerst gegeben werden, gefolgt von YERVOY, gefolgt von Chemotherapie (wenn zutreffend) am gleichen Tag. Für jede Infusion sind separate Infusionsbeutel und Filter zu verwenden.
Anweisungen zur Herstellung und Handhabung des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Beurteilung des PD‑L1‑Status
Es ist wichtig, für die Beurteilung des PD‑L1‑Status eine gut validierte und robuste Methode zu verwenden.
Beurteilung des MSI‑/MMR‑Status
Es ist wichtig, für die Beurteilung des MSI‑H‑ und dMMR‑Tumorstatus eine gut validierte und robuste Methode zu verwenden.
Ipilimumab in Kombination mit Nivolumab
Wenn Ipilimumab in Kombination angewendet wird, lesen Sie die Fachinformationen der anderen Kombinationstherapie‑Komponenten bevor Sie mit der Behandlung beginnen. Für weitere Informationen zu Warnhinweisen und Vorsichtsmaßnahmen im Zusammenhang mit der Nivolumab‑Behandlung, lesen Sie in der Nivolumab‑Fachinformation nach. Die meisten immunvermittelten Nebenwirkungen verbesserten sich oder verschwanden bei geeignetem Nebenwirkungsmanagement, einschließlich Einleitung einer Corticosteroidbehandlung und Behandlungsmodifikationen (siehe Abschnitt 4.2). Bei Nivolumab in Kombination mit Ipilimumab traten im Vergleich zu Nivolumab als Monotherapie bei höheren Häufigkeiten immunbedingte Nebenwirkungen auf.
Bei der Kombinationstherapie wurden auch kardiale und pulmonale Nebenwirkungen einschließlich Lungenembolie berichtet. Patienten sollten fortlaufend auf kardiale und pulmonale Nebenwirkungen, sowie vor und regelmäßig während der Behandlung auf klinische Anzeichen und Symptome und Laborwertabweichungen, die Störungen des Elektrolythaushalts und Deydratation erkennen lassen, überwacht werden. Ipilimumab in Kombination mit Nivolumab muss bei lebensbedrohlichen oder schweren wiederauftretenden kardialen und pulmonalen Nebenwirkungen abgesetzt werden (siehe Abschnitt 4.2).
Patienten sollten engmaschig überwacht werden (mindestens bis zu 5 Monate nach der letzten Dosis), da Nebenwirkungen unter Ipilimumab in Kombination mit Nivolumab jederzeit während oder nach der Behandlung auftreten können.
Immunvermittelte Reaktionen
Ipilimumab wird mit entzündlichen Nebenwirkungen aufgrund einer erhöhten oder übermäßigen Immunaktivität (immunvermittelte Nebenwirkungen) in Verbindung gebracht, die wahrscheinlich auf den Wirkungsmechanismus des Präparats zurückzuführen sind. Immunvermittelte Nebenwirkungen, die schwerwiegend bis lebensbedrohlich sein können, können Verdauungstrakt, Leber, Haut, Nervensystem, endokrines System oder andere Organsysteme betreffen. Obwohl die meisten immunvermittelten Nebenwirkungen während der Induktionsphase auftraten, wurde auch Monate nach der letzten Ipilimumab‑Dosis über deren Auftreten berichtet. Solange keine andere Ursache ermittelt wurde, müssen Diarrhö, erhöhte Stuhlfrequenz, blutiger Stuhl, LFT‑Erhöhungen, Hautausschlag und Endokrinopathie als immunvermittelt und als im Zusammenhang mit Ipilimumab stehend betrachtet werden. Eine frühzeitige Diagnose und adäquate Behandlung sind von entscheidender Bedeutung, um das Auftreten lebensbedrohlicher Komplikationen zu minimieren.
Eine systemische hochdosierte Therapie mit Corticosteroiden zusammen mit oder ohne andere Immunsuppressiva kann für die Behandlung schwerer immunvermittelter Nebenwirkungen erforderlich sein.
Spezifische Richtlinien für Ipilimumab zur Behandlung von immunvermittelten Nebenwirkungen sind im Folgenden für die Monotherapie und die Kombinationstherapie mit Nivolumab aufgeführt.
Bei vermuteten immunvermittelten Nebenwirkungen sollte zur Bestätigung der Ätiologie oder zum Ausschluss anderer Ursachen eine angemessene Abklärung durchgeführt werden. In Abhängigkeit vom Schweregrad der Nebenwirkung sollte die Behandlung mit Ipilimumab oder Ipilimumab in Kombination mit Nivolumab aufgeschoben und die Patienten mit Corticosteroiden behandelt werden. Wenn eine Immunsuppression mit Corticosteroiden zur Behandlung von Nebenwirkungen eingesetzt wird, welche infolge der Kombinationstherapie aufgetreten sind, sollte die Corticosteroidtherapie nach Besserung der Nebenwirkungen über mindestens einen Monat ausgeschlichen werden. Ein zu schnelles Ausschleichen kann zur Verschlechterung oder Wiederauftreten der Nebenwirkung führen. Wenn es trotz Corticosteroidanwendung zu einer Verschlechterung oder keiner Besserung kommt, sollten zusätzlich nicht‑steroidale Immunsuppressiva gegeben werden.
Die Behandlung mit Ipilimumab in Kombination mit Nivolumab sollte nicht fortgesetzt werden, solange der Patient immunsuppressive Dosen von Corticosteroiden oder andere Immunsuppressiva erhält. Prophylaktisch sollten Antibiotika gegeben werden, um opportunistische Infektionen bei Patienten zu verhindern, die immunsuppressiv behandelt werden.
Ipilimumab in Kombination mit Nivolumab muss bei jeder schweren wiederauftretenden immunvermittelten Nebenwirkung und bei jeder lebensbedrohlichen immunvermittelten Nebenwirkung dauerhaft abgesetzt werden.
Immunvermittelte gastrointestinale Nebenwirkungen
Ipilimumab als Monotherapie
Ipilimumab ist mit schwerwiegenden immunvermittelten gastrointestinalen Nebenwirkungen assoziiert. In klinischen Studien wurde über Todesfälle aufgrund gastrointestinaler Perforation berichtet (siehe Abschnitt 4.8).
Bei Patienten, die in einer Phase‑III‑Studie beim fortgeschrittenen (nicht resezierbaren oder metastasierten) Melanom (MDX010‑20, siehe Abschnitt 5.1) Ipilimumab 3 mg/kg als Monotherapie erhielten, betrug die mediane Zeit bis zum Auftreten schwerer oder tödlicher (Grad 3‑5) immunvermittelter gastrointestinaler Nebenwirkungen im Median 8 Wochen (Bereich 5 bis 13 Wochen) ab Therapiebeginn. Unter protokolldefinierten Behandlungsrichtlinien gingen die Symptome in den meisten Fällen (90 %) innerhalb eines medianen Zeitraums von 4 Wochen (Bereich 0,6 bis 22 Wochen) nach Beginn der Behandlung zurück (definiert als Verbesserung zu leichten Symptomen [Grad 1] oder weniger oder zum Schweregrad bei Behandlungsbeginn).
Patienten müssen sorgfältig auf gastrointestinale Symptome überwacht werden, die auf eine immunvermittelte Kolitis oder gastrointestinale Perforation hinweisen können. Dazu können Diarrhö, eine verstärkte Darmtätigkeit, Abdominalschmerz oder Hämatochezie mit oder ohne Fieber zählen. In klinischen Studien war die immunvermittelte Kolitis mit nachweisbaren entzündlichen Veränderungen der Darmschleimhaut mit oder ohne Ulzeration und einer lymphozytären und neutrophilen Infiltration assoziiert. Nach Markteinführung wurden Fälle von Zytomegalievirus (cytomegalovirus, CMV)‑Infektionen bzw. Reaktivierung des Virus bei Patienten mit einer gegenüber Corticosteroiden refraktären Kolitis gemeldet. Bei Auftreten von Diarrhö oder Kolitis sollten Stuhlproben auf Infektionen untersucht werden, um Infektionen oder andere Ursachen auszuschließen.
Behandlungsempfehlungen für Diarrhö oder Kolitis basieren auf dem Schweregrad der Symptome (gemäß der NCI‑CTCAE v4 Klassifizierung des Schweregrads). Patienten mit leichter bis mäßiger (Grad 1 oder 2) Diarrhö (Steigerung um bis zu 6 Stuhlgänge pro Tag) oder Verdacht auf leichte bis mäßige Kolitis (z. B. Abdominalschmerz oder Blut im Stuhl) können weiter mit Ipilimumab behandelt werden. Es empfiehlt sich eine Behandlung der Symptome (z. B. Loperamid, Flüssigkeitsersatz) und eine sorgfältige Beobachtung. Wenn leichte bis mäßige Symptome erneut auftreten oder über 5‑7 Tage andauern, sollte die nächste geplante Dosis Ipilimumab aufgeschoben und eine Corticosteroidtherapie (z. B. Prednison 1 mg/kg oral einmal täglich oder ein entsprechender Wirkstoff) eingeleitet werden. Wenn eine Rückbildung auf Grad 0 bis 1 oder bis zum Ausgangswert erreicht wird, kann die Therapie mit Ipilimumab wieder aufgenommen werden (siehe Abschnitt 4.2).
Bei Patienten mit schwerer (Grad 3 oder 4) Diarrhö oder Kolitis muss Ipilimumab dauerhaft abgesetzt werden (siehe Abschnitt 4.2) und es sollte unverzüglich eine systemische, hochdosierte intravenöse Corticosteroidtherapie eingeleitet werden (in klinischen Studien wurde Methylprednisolon 2 mg/kg/Tag eingesetzt). Sobald die Diarrhö und die anderen Symptome unter Kontrolle sind, sollte das Ausschleichen des Corticosteroids nach klinischem Ermessen des behandelnden Arztes eingeleitet werden. In klinischen Studien führte ein rasches Ausschleichen (über einen Zeitraum von < 1 Monat) bei einigen Patienten zu einem erneuten Auftreten der Diarrhö bzw. Kolitis. Patienten müssen auf Anzeichen einer gastrointestinalen Perforation oder Peritonitis untersucht werden.
Die Erfahrungen aus klinischen Studien zur Behandlung einer gegenüber Corticosteroiden refraktären Diarrhö oder Kolitis sind begrenzt. Die Zugabe eines alternativen Immunsuppressivums zum Corticosteroidregime sollte bei der gegenüber Corticosteroiden refraktären Kolitis erwogen werden, wenn andere Ursachen ausgeschlossen sind (einschließlich Zytomegalievirus‑Infektion/‑Reaktivierung, abgeklärt mit viraler PCR in der Biopsie, und andere virale, bakterielle und parasitäre Ursachen). In klinischen Studien wurde, sofern nicht kontraindiziert, eine Einzeldosis Infliximab 5 mg/kg zugegeben. Infliximab darf jedoch nicht bei Verdacht auf gastrointestinale Perforation oder Sepsis eingesetzt werden (siehe in der Fachinformation von Infliximab).
Immunvermittelte Kolitis
Ipilimumab in Kombination mit Nivolumab
Unter Ipilimumab in Kombination mit Nivolumab wurden schwere Diarrhö oder Kolitis beobachtet (siehe Abschnitt 4.8). Patienten sollten auf Diarrhö und weitere Symptome einer Kolitis wie Abdominalschmerz und Schleim oder Blut im Stuhl überwacht werden. Infektionen und krankheitsbedingte Ätiologien sind auszuschließen.
Bei Diarrhö oder Kolitis von Grad 4 muss Ipilimumab in Kombination mit Nivolumab dauerhaft abgesetzt werden und es sollte eine Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden.
Eine Diarrhö oder Kolitis von Grad 3, die bei Ipilimumab in Kombination mit Nivolumab auftritt, erfordert ein dauerhaftes Absetzen der Behandlung und die Initiierung von Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent.
Bei Diarrhö oder Kolitis von Grad 2 sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben werden. Bei anhaltender Diarrhö oder Kolitis sollte mit Corticosteroiden in einer Dosierung von 0,5 bis 1 mg/kg/Tag Methylprednisolon‑Äquivalent behandelt werden. Bei einer Besserung kann die Behandlung mit Ipilimumab in Kombination mit Nivolumab nach dem Ausschleichen der Corticosteroide (sofern erforderlich) fortgesetzt werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, sollte die Dosis auf 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent erhöht werden und Ipilimumab in Kombination mit Nivolumab muss dauerhaft abgesetzt werden.
Immunvermittelte Pneumonitis
Ipilimumab in Kombination mit Nivolumab
Unter Ipilimumab in Kombination mit Nivolumab wurden schwere Pneumonitis oder interstitielle Lungenerkrankung, auch mit tödlichem Verlauf, beobachtet (siehe Abschnitt 4.8). Die Patienten sollten auf Anzeichen und Symptome einer Pneumonitis wie beispielsweise radiologische Veränderungen (z. B. fokale milchglasartige Dichteanhebung, fleckige Infiltrate), Dyspnoe und Hypoxie überwacht werden. Infektionen und krankheitsbedingte Ätiologien sind auszuschließen.
Bei Pneumonitis von Grad 3 oder 4 muss Ipilimumab in Kombination mit Nivolumab dauerhaft abgesetzt werden und es sollte eine Behandlung mit Corticosteroiden in einer Dosierung von 2 bis 4 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden.
Bei (symptomatischer) Pneumonitis von Grad 2 muss Ipilimumab in Kombination mit Nivolumab aufgeschoben und mit einer Behandlung mit Corticosteroiden in einer Dosierung von 1 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden. Bei einer Besserung kann die Behandlung mit Ipilimumab in Kombination mit Nivolumab nach dem Ausschleichen der Corticosteroide fortgesetzt werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, sollte die Dosis auf 2 bis 4 mg/kg/Tag Methylprednisolon‑Äquivalent erhöht werden und Ipilimumab in Kombination mit Nivolumab muss dauerhaft abgesetzt werden.
Immunvermittelte Hepatotoxizität
Ipilimumab als Monotherapie
Ipilimumab wird mit schwerwiegenden immunvermittelten Hepatotoxizitäten in Zusammenhang gebracht. In klinischen Studien wurde über Todesfälle infolge von Leberversagen berichtet (siehe Abschnitt 4.8).
Bei Patienten, die in der Studie MDX010‑20 Ipilimumab 3 mg/kg als Monotherapie erhielten, lag der Zeitraum bis zum Ausbruch mäßiger bis schwerer oder tödlicher (Grad 2‑5) immunvermittelter Hepatotoxizitäten zwischen 3 und 9 Wochen ab Behandlungsbeginn. Unter protokolldefinierten Behandlungsrichtlinien gingen die Symptome innerhalb eines Zeitraums von 0,7 bis 2 Wochen zurück.
Die Lebertransaminase‑ und Bilirubinwerte müssen vor Verabreichung jeder Ipilimumab‑Dosis bewertet werden, da frühzeitige Veränderungen der Laborwerte auf eine beginnende immunvermittelte Hepatitis hinweisen können (siehe Abschnitt 4.2). LFT‑Erhöhungen können auch ohne klinische Symptome auftreten. Eine Erhöhung der AST und ALT oder des Gesamtbilirubins sollte untersucht werden, um andere Ursachen eines Leberschadens wie Infektionen, Tumorprogression oder Begleitmedikation auszuschließen, und bis zum Rückgang der Symptome beobachtet werden. Leberbiopsien von Patienten mit immunvermittelten Hepatotoxizitäten zeigten Hinweise auf eine akute Entzündungsreaktion (Neutrophile, Lymphozyten und Makrophagen).
Bei Patienten mit einer Erhöhung der Transaminasen oder des Gesamtbilirubins von Grad 2, sollte die nächste geplante Ipilimumab‑Dosis aufgeschoben werden; LFTs müssen bis zur Normalisierung überwacht werden. Bei Verbesserung kann die Ipilimumab‑Therapie wieder aufgenommen werden (siehe Abschnitt 4.2).
Bei Patienten mit einer Transaminase‑ oder Gesamtbilirubinerhöhung von Grad 3 oder 4, muss die Therapie dauerhaft abgebrochen werden (siehe Abschnitt 4.2), und es sollte unverzüglich eine systemische, hochdosierte intravenöse Corticosteroidtherapie (z. B. mit Methylprednisolon 2 mg/kg täglich oder einem entsprechenden Wirkstoff) eingeleitet werden. In diesem Fall müssen die LFTs bis zur Normalisierung kontrolliert werden. Sobald die Symptome abgeklungen sind und die LFTs eine anhaltende Verbesserung aufweisen oder auf Normalwerte zurückgegangen sind, sollte das Ausschleichen des Corticosteroids nach klinischem Ermessen eingeleitet werden. Das Ausschleichen sollte über einen Zeitraum von mindestens 1 Monat erfolgen. LFT‑Erhöhungen während der Ausschleichphase können durch Erhöhung der Corticosteroiddosis und langsameres Ausschleichen behandelt werden.
Bei Patienten mit signifikanten LFT‑Erhöhungen, die sich als refraktär gegenüber einer Corticosteroidtherapie erweisen, kann die Zugabe von anderen Immunsuppressiva zum Corticosteroidregime in Betracht gezogen werden. Bei Patienten, die auf die Corticosteroidtherapie nicht ansprachen oder bei denen es während des Ausschleichens des Corticosteroids zu einer LFT‑Erhöhung kam, die nicht auf eine Erhöhung der Corticosteroiddosis ansprach, wurde in klinischen Studien Mycophenolat‑Mofetil eingesetzt (siehe Fachinformation von Mycophenolat‑Mofetil).
Ipilimumab in Kombination mit Nivolumab
Unter Ipilimumab in Kombination mit Nivolumab wurden Fälle von schwerer Hepatitis beobachtet (siehe Abschnitt 4.8). Patienten sollten auf Anzeichen und Symptome einer Hepatitis wie Anstieg der Transaminasen und des Gesamtbilirubins überwacht werden. Infektionen und krankheitsbedingte Ätiologien sind auszuschließen.
Bei Erhöhung der Transaminasen oder des Gesamtbilirubins von Grad 3 oder 4 muss Ipilimumab in Kombination mit Nivolumab dauerhaft abgesetzt werden und es sollte eine Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden.
Bei Erhöhung der Transaminasen oder des Gesamtbilirubins von Grad 2 sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben werden. Bei anhaltenden Erhöhungen dieser Laborwerte sollte mit Corticosteroiden in einer Dosierung von 0,5 bis 1 mg/kg/Tag Methylprednisolon‑Äquivalent behandelt werden. Bei einer Besserung kann die Behandlung mit Ipilimumab in Kombination mit Nivolumab nach dem Ausschleichen der Corticosteroide (sofern erforderlich) fortgesetzt werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, sollte die Dosis auf 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent erhöht werden und Ipilimumab in Kombination mit Nivolumab muss dauerhaft abgesetzt werden.
Immunvermittelte Nebenwirkungen der Haut
Vorsicht ist geboten, wenn Ipilimumab als Monotherapie oder Ipilimumab in Kombination mit Nivolumab bei Patienten angewendet werden soll, bei denen zuvor während einer früheren immunstimulierenden Krebsbehandlung schwere oder lebensbedrohliche Nebenwirkungen der Haut aufgetreten sind.
Ipilimumab als Monotherapie
Ipilimumab wird mit schwerwiegenden Nebenwirkungen der Haut in Verbindung gebracht, die immunvermittelt sein könnten. Es wurden seltene Fälle von toxischer epidermaler Nekrolyse (TEN) (einschließlich Stevens‑Johnson‑Syndrom (SJS)) beobachtet, einige mit tödlichem Ausgang. Außerdem wurden auch seltene Fälle von Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen (Drug Reaction with Eosinophilia and Systemic Symptoms = DRESS) in klinischen Studien und nach Markteinführung berichtet (siehe Abschnitt 4.8).
DRESS tritt in Form eines Hautausschlags mit Eosinophilie in Verbindung mit einem oder mehreren der folgenden Symptome auf: Fieber, Lymphadenopathie, Gesichtsödem und Beteiligung der inneren Organe (Leber, Niere, Lunge). DRESS kann eine lange Latenzzeit (2 bis 8 Wochen) zwischen Arzneimittelexposition und Auftreten der Krankheit haben.
Durch Ipilimumab induzierter Hautausschlag und Pruritus waren überwiegend leicht bis mäßig (Grad 1 oder 2) und sprachen auf eine symptomatische Behandlung an. Bei Patienten, die in der Studie MDX010‑20 Ipilimumab 3 mg/kg als Monotherapie erhielten, betrug der Zeitraum bis zum Auftreten mäßiger bis schwerer oder tödlicher (Grad 2‑5) Nebenwirkungen der Haut im Median 3 Wochen (Bereich 0,9‑16 Wochen) ab Behandlungsbeginn. Unter protokolldefinierten Behandlungsrichtlinien gingen die Symptome in den meisten Fällen (87 %) innerhalb eines medianen Zeitraums von 5 Wochen nach Therapiebeginn zurück (Bereich 0,6 bis 29 Wochen).
Durch Ipilimumab induzierter Hautausschlag und Pruritus sollten je nach Schweregrad behandelt werden. Patienten mit einem leichten bis mäßigen (Grad 1 oder 2) Hautausschlag können weiter mit Ipilimumab behandelt werden. Zusätzlich sollte eine symptomatische Behandlung erfolgen (z. B. mit Antihistaminika). Bei leichtem bis mäßigem Ausschlag oder leichtem Pruritus, der 1 bis 2 Wochen anhält und auf topische Corticosteroide nicht anspricht, sollte eine orale Corticosteroidtherapie eingeleitet werden (z. B. mit Prednison 1 mg/kg einmal täglich oder einem gleichwertigen Wirkstoff).
Bei Patienten mit einem schweren (Grad 3) Hautausschlag sollte die nächste geplante Ipilimumab‑Dosis aufgeschoben werden. Wenn sich die anfänglichen Symptome bis zu einem leichten Stadium verbessert haben (Grad 1) oder abgeklungen sind, kann die Ipilimumab‑Therapie wieder aufgenommen werden (siehe Abschnitt 4.2).
Bei Patienten mit einem sehr schweren (Grad 4) Hautausschlag oder schwerem (Grad 3) Pruritus muss Ipilimumab dauerhaft abgesetzt werden (siehe Abschnitt 4.2), und unverzüglich eine systemische, hochdosierte intravenöse Corticosteroidtherapie (z. B. mit Methylprednisolon 2 mg/kg/Tag) eingeleitet werden. Sobald der Ausschlag oder Pruritus unter Kontrolle ist, sollte nach klinischem Ermessen das Ausschleichen des Corticosteroids eingeleitet werden. Das Ausschleichen sollte über einen Zeitraum von mindestens 1 Monat erfolgen.
Ipilimumab in Kombination mit Nivolumab
Unter Ipilimumab in Kombination mit Nivolumab wurden schwere Hautausschläge beobachtet (siehe Abschnitt 4.8). Die Behandlung mit Ipilimumab in Kombination mit Nivolumab sollte bei Hautausschlag von Grad 3 aufgeschoben und bei Hautausschlag von Grad 4 abgesetzt werden. Schwerer Hautausschlag sollte mit hochdosierten Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent behandelt werden.
In seltenen Fällen wurden SJS und TEN berichtet, darunter waren auch einige Todesfälle. Wenn Symptome oder Anzeichen für SJS oder TEN auftreten, sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab abgesetzt und der Patient in eine spezialisierte Abteilung zur Beurteilung und Behandlung überwiesen werden. Wenn sich beim Patienten unter der Anwendung von Ipilimumab in Kombination mit Nivolumab SJS oder TEN entwickelt haben, wird die dauerhafte Absetzung der Behandlung empfohlen (siehe Abschnitt 4.2).
Immunvermittelte neurologische Nebenwirkungen
Ipilimumab als Monotherapie
Ipilimumab ist mit schwerwiegenden immunvermittelten neurologischen Nebenwirkungen assoziiert. In klinischen Studien wurde über Todesfälle durch das Guillain‑Barré‑Syndrom berichtet. Myasthenia gravis‑ähnliche Symptome wurden ebenfalls berichtet (siehe Abschnitt 4.8). Patienten können Muskelschwäche aufweisen. Zudem können sensorische Neuropathien auftreten.
Eine ungeklärte motorische Neuropathie, Muskelschwäche oder sensorische Neuropathie von > 4 Tagen muss abgeklärt und nichtentzündliche Ursachen wie Krankheitsprogression, Infektionen, metabolisches Syndrom und Begleitmedikation sollten ausgeschlossen werden. Bei Patienten mit mäßiger (Grad 2) Neuropathie (motorisch, mit oder ohne sensorischer Störung), die vermutlich auf Ipilimumab zurückzuführen ist, sollte die nächste geplante Dosis aufgeschoben werden. Wenn sich die neurologischen Symptome wieder zum ursprünglichen Zustand zurückgebildet haben, kann der Patient die Ipilimumab‑Therapie wieder aufnehmen (siehe Abschnitt 4.2).
Bei Patienten mit schwerer (Grad 3 oder 4) sensorischer Neuropathie, die vermutlich auf Ipilimumab zurückzuführen ist, muss Ipilimumab dauerhaft abgesetzt werden (siehe Abschnitt 4.2). Die Patienten müssen gemäß den geltenden Richtlinien zur Behandlung der sensorischen Neuropathie behandelt werden und eine Behandlung mit intravenösen Corticosteroiden (z. B. Methylprednisolon 2 mg/kg/Tag) sollte unverzüglich eingeleitet werden.
Fortschreitende Anzeichen einer motorischen Neuropathie müssen als immunvermittelt betrachtet und entsprechend behandelt werden. Bei Patienten mit schwerer (Grad 3 oder 4) motorischer Neuropathie muss Ipilimumab, unabhängig von der Ursache, dauerhaft abgesetzt werden (siehe Abschnitt 4.2).
Immunvermittelte Nephritis und Nierenfunktionsstörung
Ipilimumab in Kombination mit Nivolumab
Unter der Behandlung mit Ipilimumab in Kombination mit Nivolumab wurden schwere Nephritis und Nierenfunktionsstörungen beobachtet (siehe Abschnitt 4.8). Patienten sind auf Anzeichen und Symptome einer Nephritis oder Nierenfunktionsstörung zu überwachen. Bei den meisten Patienten tritt eine asymptomatische Kreatininerhöhung im Serum auf. Infektionen und krankheitsbedingte Ätiologien sind auszuschließen.
Bei einer Kreatininerhöhung im Serum von Grad 4 muss Ipilimumab in Kombination mit Nivolumab dauerhaft abgesetzt werden und es sollte mit einer Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden.
Bei einer Kreatininerhöhung im Serum von Grad 2 oder 3 sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben und eine Behandlung mit Corticosteroiden in einer Dosierung von 0,5 bis 1 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden. Bei einer Besserung kann die Behandlung mit Ipilimumab in Kombination mit Nivolumab nach dem Ausschleichen der Corticosteroide fortgesetzt werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, sollte die Dosis auf 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent erhöht werden und Ipilimumab in Kombination mit Nivolumab muss dauerhaft abgesetzt werden.
Immunvermittelte Endokrinopathie
Ipilimumab als Monotherapie
Ipilimumab kann eine Entzündung der Organe des endokrinen Systems verursachen, was sich als Hypophysitis, Hypopituitarismus, Nebenniereninsuffizienz, Hypothyreose, Typ‑1‑Diabetes‑mellitus und diabetische Ketoazidose manifestiert (siehe Abschnitte 4.2 und 4.8). Patienten können unspezifische Symptome aufweisen, die anderen Ursachen wie Hirnmetastasen oder der zugrundeliegenden Erkrankung ähneln können. Zu den häufigsten Beschwerden gehören Kopfschmerzen und Ermüdung/Fatigue. Die Symptome können aber auch Gesichtsfeldausfälle, Verhaltensänderungen, Elektrolytstörungen und Hypotonie umfassen. Eine Nebennierenkrise muss als Ursache der Symptome des Patienten ausgeschlossen werden. Die klinischen Erfahrungen mit einer Ipilimumab‑assoziierten Endokrinopathie sind begrenzt.
Bei Patienten, die in der Studie MDX010‑20 Ipilimumab 3 mg/kg als Monotherapie erhielten, lag der Zeitraum bis zum Auftreten mäßiger bis sehr schwerer (Grad 2‑4) immunvermittelter Endokrinopathien zwischen 7 und fast 20 Wochen ab Behandlungsbeginn. Die in klinischen Studien beobachteten Fälle immunvermittelter Endokrinopathien konnten in der Regel durch eine immunsuppressive Therapie und Hormonersatztherapie kontrolliert werden.
Sollten Anzeichen einer akuten Nebennierenkrise auftreten, wie z. B. schwere Dehydrierung, Hypotonie oder Schock, empfiehlt sich die sofortige intravenöse Verabreichung von Corticosteroiden mit mineralcorticoider Wirkung. Zudem muss das Vorliegen einer Sepsis oder Infektionen abgeklärt werden. Wenn Anzeichen für eine Nebenniereninsuffizienz, jedoch nicht für eine akute Nebennierenkrise, bestehen, sollten weitere Untersuchungen einschließlich Labortests und bildgebender Verfahren in Betracht gezogen werden. Bevor eine Corticosteroidtherapie eingeleitet wird, kann eine Auswertung der Laborergebnisse zur Einstufung der endokrinen Funktion durchgeführt werden. Wenn die bildgebende Hypophysendiagnostik oder Labortests der endokrinen Funktion Auffälligkeiten ergeben, empfiehlt sich eine kurze hochdosierte Corticosteroidtherapie (z. B. mit Dexamethason 4 mg alle 6 Stunden oder einem entsprechenden Wirkstoff), um die Entzündung der betroffenen Drüse zu behandeln. Die nächste geplante Ipilimumab‑Dosis sollte zudem aufgeschoben werden (siehe Abschnitt 4.2). Derzeit ist noch unbekannt, ob die Corticosteroidtherapie die Drüsenfunktion wiederherstellt. Zusätzlich sollte eine geeignete Hormonersatztherapie eingeleitet werden. Diese kann langfristig erforderlich sein.
Bei symptomatischem Diabetes sollte Ipilimumab nicht weiter angewendet und bei Bedarf mit einer Insulinersatztherapie begonnen werden. Die Überwachung des Blutzuckerspiegels sollte fortgeführt werden um die Anwendung eines angemessenen Insulinersatzes sicherzustellen. Die Anwendung von Ipilimumab muss bei lebensbedrohlichem Diabetes dauerhaft eingestellt werden.
Sobald die Symptome und Veränderungen der Laborwerte unter Kontrolle sind und sich eine sichtbare Verbesserung des Allgemeinzustands des Patienten zeigt, kann die Behandlung mit Ipilimumab wieder aufgenommen und das Ausschleichen des Corticosteroids nach klinischem Ermessen eingeleitet werden. Das Ausschleichen sollte über einen Zeitraum von mindestens 1 Monat erfolgen.
Ipilimumab in Kombination mit Nivolumab
Unter Ipilimumab in Kombination mit Nivolumab wurden schwere Endokrinopathien, einschließlich Hypothyreose, Hyperthyreose, Nebenniereninsuffizienz (einschließlich sekundäre Nebenniereninsuffizienz), Hypophysitis (einschließlich Hypophyseninsuffizienz), Diabetes mellitus und diabetische Ketoazidose beobachtet (siehe Abschnitt 4.8).
Patienten sollten hinsichtlich klinischer Anzeichen und Symptome von Endokrinopathien, Hyperglykämie und Veränderungen der Schilddrüsenfunktion überwacht werden (zu Beginn der Behandlung, regelmäßig während der Behandlung und wenn es nach klinischer Beurteilung angezeigt ist). Patienten können mit Ermüdung/Fatigue, Kopfschmerzen, psychischen Veränderungen, Abdominalschmerz, Veränderung der Stuhlgewohnheiten und Hypotonie oder unspezifischen Symptomen vorstellig werden, die anderen Ursachen, wie etwa Gehirnmetastasen oder der zugrundeliegenden Erkrankung, ähneln können. Bis eine andere Ätiologie identifiziert worden ist, sollten Anzeichen oder Symptome von Endokrinopathien als immunvermittelt betrachtet werden.
Bei symptomatischer Hypothyreose sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben und bei Bedarf mit einer Hormonersatztherapie begonnen werden. Bei symptomatischer Hyperthyreose sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben und bei Bedarf mit einer Behandlung mit Thyreostatika begonnen werden. Bei Verdacht auf eine akute Entzündung der Schilddrüse sollte auch eine Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent in Betracht gezogen werden. Bei einer Besserung kann die Behandlung mit Ipilimumab in Kombination mit Nivolumab nach dem Ausschleichen der Corticosteroide (sofern erforderlich) fortgesetzt werden. Die Schilddrüsenfunktion sollte weiterhin überwacht werden, um sicherzustellen, dass die passende Hormonersatztherapie angewandt wird. Bei lebensbedrohlicher Hyperthyreose oder Hypothyreose muss Ipilimumab in Kombination mit Nivolumab dauerhaft abgesetzt werden.
Bei symptomatischer Nebenniereninsuffizienz von Grad 2 sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben und bei Bedarf mit einer physiologischen Corticosteroid‑Ersatztherapie begonnen werden. Bei schwerwiegender (Grad 3) oder lebensbedrohlicher (Grad 4) Nebenniereninsuffizienz muss Ipilimumab in Kombination mit Nivolumab dauerhaft abgesetzt werden. Die Nebennierenfunktion und Hormonspiegel sollten weiterhin überwacht werden um sicherzustellen, dass die passende Corticosteroid‑Ersatztherapie angewandt wird.
Bei symptomatischer Hypophysitis von Grad 2 oder 3 sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben und bei Bedarf mit einer Hormonersatztherapie begonnen werden. Bei Verdacht auf akute Entzündung der Hypophyse sollte auch eine Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent in Betracht gezogen werden. Bei einer Besserung kann die Behandlung mit Ipilimumab in Kombination mit Nivolumab nach dem Ausschleichen der Corticosteroide (sofern erforderlich) fortgesetzt werden. Bei lebensbedrohlicher (Grad 4) Hypophysitis muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt werden. Die Hypophysenfunktion und Hormonspiegel sollten weiterhin überwacht werden, um sicherzustellen, dass die passende Hormonersatztherapie angewandt wird.
Bei symptomatischem Diabetes sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben und bei Bedarf mit einer Insulinersatztherapie begonnen werden. Der Blutzuckerspiegel sollte weiterhin überwacht werden, um sicherzustellen, dass die passende Insulinersatztherapie angewandt wird. Bei lebensbedrohlichem Diabetes muss Ipilimumab in Kombination mit Nivolumab dauerhaft abgesetzt werden.
Infusionsreaktionen
Ipilimumab als Monotherapie und in Kombination mit Nivolumab
In klinischen Studien mit Ipilimumab oder Ipilimumab in Kombination mit Nivolumab wurden schwere Infusionsreaktionen berichtet (siehe Abschnitt 4.8). Falls eine schwere oder lebensbedrohliche Infusionsreaktion auftritt, muss die Ipilimumab‑Infusion bzw. die Infusion von Ipilimumab in Kombination mit Nivolumab abgesetzt und eine geeignete medizinische Behandlung eingeleitet werden. Patienten mit leichter oder mäßiger Infusionsreaktion können Ipilimumab oder Ipilimumab in Kombination mit Nivolumab unter engmaschiger Überwachung und dem Einsatz von Prämedikation gemäß lokalen Behandlungsrichtlinien zur Prophylaxe von infusionsbedingten Reaktionen erhalten.
Andere immunvermittelte Nebenwirkungen
Ipilimumab als Monotherapie
Folgende, vermutlich immunvermittelte Nebenwirkungen wurden bei Patienten beobachtet, die in der Studie MDX010‑20 mit Ipilimumab 3 mg/kg als Monotherapie behandelt wurden: Uveitis, Eosinophilie, Lipaseerhöhung und Glomerulonephritis. Weiter wurden bei Patienten, die in der Studie MDX010‑20 mit Ipilimumab 3 mg/kg + gp100‑Peptid‑Vakzine behandelt wurden, Iritis, hämolytische Anämie, Amylaseerhöhungen, multiples Organversagen und Pneumonitis beobachtet. Nach Markteinführung wurden Fälle von Vogt‑Koyanagi‑Harada‑Syndrom, seröser Netzhautablösung und nicht‑infektiöser Zystitis berichtet (siehe Abschnitte 4.2 und 4.8).
Sollten diese Ereignisse schwerwiegend (Grad 3 oder 4) sein, kann eine unverzügliche systemische hochdosierte Corticosteroidtherapie und ein Abbruch der Behandlung mit Ipilimumab erforderlich sein (siehe Abschnitt 4.2). Bei Uveitis, Iritis, seröser Netzhautablösung oder Episkleritis im Zusammenhang mit Ipilimumab ist eine topische Behandlung mit corticosteroidhaltigen Augentropfen angezeigt. Bei Patienten mit Ipilimumab‑bedingten Augenentzündungen wurde vorübergehender Sehverlust berichtet.
Nach dem Inverkehrbringen wurde bei Patienten, die mit Ipilimumab behandelt wurden, über eine Abstoßung solider Organtransplantate berichtet. Die Behandlung mit Ipilimumab kann das Abstoßungsrisiko bei Empfängern solider Organtransplantate erhöhen. Bei diesen Patienten sollte der Nutzen der Behandlung mit Ipilimumab gegen das Risiko einer möglichen Organabstoßung abgewogen werden.
Ipilimumab als Monotherapie oder in Kombination mit einem PD‑1‑ oder PD‑L1‑Inhibitor
Eine hämophagozytische Lymphohistiozytose (HLH) wurde mit Ipilimumab als Monotherapie und Ipilimumab in Kombination mit einem PD‑1‑ oder PD‑L1‑Inhibitor (einschließlich Nivolumab) beobachtet. Vorsicht ist geboten, wenn Ipilimumab als Monotherapie oder in Kombination mit einem PD‑1‑ oder PD‑L1‑Inhibitor gegeben wird. Wenn HLH bestätigt wird, sollte die Gabe von Ipilimumab oder Ipilimumab in Kombination mit einem PD‑1‑ oder PD‑L1‑Inhibitor abgebrochen und die Behandlung von HLH eingeleitet werden.
Ipilimumab in Kombination mit Nivolumab
Die folgenden immunvermittelten Nebenwirkungen wurden bei weniger als 1 % der in klinischen Studien mit Ipilimumab in Kombination mit Nivolumab behandelten Patienten dosis‑ und tumorartenübergreifend berichtet: Pankreatitis, Uveitis, Demyelinisierung, autoimmune Neuropathie (einschließlich Gesichtsnerv‑ und Abduzensparese), Guillain Barré‑Syndrom, Myasthenia gravis, Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom, myasthenes Syndrom, aseptische Meningitis, Enzephalitis, Gastritis, Sarkoidose, Duodenitis, Myositis, Myokarditis, Rhabdomyolyse und Myelitis. Nach Markteinführung wurden Fälle von Vogt‑Koyanagi‑Harada‑Syndrom, seröser Netzhautablösung und nicht‑infektiöser Zystitis berichtet (siehe Abschnitte 4.2 und 4.8). Bei Patienten mit Ipilimumab‑bedingten Augenentzündungen wurde vorübergehender Sehverlust berichtet.
Bei vermuteten immunvermittelten Nebenwirkungen sollte zur Bestätigung der Ätiologie oder zum Ausschluss anderer Ursachen eine angemessene Abklärung durchgeführt werden. In Abhängigkeit vom Schweregrad der Nebenwirkung sollte die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben und Corticosteroide gegeben werden. Bei einer Besserung kann die Behandlung mit Ipilimumab in Kombination mit Nivolumab nach dem Ausschleichen der Corticosteroide fortgesetzt werden. Ipilimumab in Kombination mit Nivolumab muss bei jeder schweren wiederauftretenden immunvermittelten Nebenwirkung und bei jeder lebensbedrohlichen immunvermittelten Nebenwirkung dauerhaft abgesetzt werden.
Es wurden Fälle von Myotoxizität (Myositis, Myokarditis und Rhabdomyolyse) mit Ipilimumab in Kombination mit Nivolumab berichtet, manche davon mit tödlichem Ausgang. Wenn ein Patient Anzeichen und Symptome einer Myotoxizität entwickelt, sollte er engmaschig überwacht und unverzüglich an einen Spezialisten zur Beurteilung und Behandlung überwiesen werden. Je nach Schweregrad der Myotoxizität sollte Ipilimumab in Kombination mit Nivolumab aufgeschoben oder abgesetzt werden (siehe Abschnitt 4.2) und eine geeignete Behandlung eingeleitet werden.
Die Diagnose einer Myokarditis erfordert ein hohes Maß an Aufmerksamkeit. Patienten mit kardialen oder kardiopulmonalen Symptomen sollten auf eine mögliche Myokarditis untersucht werden. Falls eine Myokarditis vermutet wird, sollte unverzüglich eine Hochdosistherapie mit Steroiden (Prednison 1 ‑ 2 mg/kg/Tag oder Methylprednisolon 1 ‑ 2 mg/kg/Tag) eingeleitet werden und unverzüglich eine kardiologische Untersuchung mit umfassender Diagnostik nach aktuellen klinischen Leitlinien veranlasst werden. Sobald die Diagnose einer Myokarditis bestätigt wurde, sollte Ipilimumab in Kombination mit Nivolumab aufgeschoben oder dauerhaft abgesetzt werden (siehe Abschnitt 4.2).
Fälle von Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom (das sich als Überlappung von entweder zwei oder allen drei Erkrankungen äußert), einige davon mit tödlichem Ausgang, wurden unter Ipilimumab in Kombination mit Nivolumab berichtet. Eine frühzeitige Erkennung und aggressive Behandlung sind unerlässlich, um der damit verbundenen Morbidität und dem Mortalitätsrisiko entgegenzuwirken.
Bei Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom Grad 3 oder 4 muss Ipilimumab in Kombination mit Nivolumab dauerhaft abgesetzt werden (siehe Abschnitt 4.2). Eine Behandlung mit Corticosteroiden soll gemäß klinischer Indikation eingeleitet werden.
Bei Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom Grad 2 soll die Behandlung mit Ipilimumab in Kombination mit Nivolumab aufgeschoben werden und eine Behandlung mit Corticosteroiden gemäß klinischer Indikation eingeleitet werden (siehe Abschnitt 4.2). Bei einer Besserung kann die Fortsetzung der Behandlung mit Ipilimumab in Kombination mit Nivolumab nach dem Ausschleichen der Corticosteroide in Betracht gezogen werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, soll die Corticosteroid‑Dosis gemäß klinischer Indikation angepasst werden und Ipilimumab in Kombination mit Nivolumab muss dauerhaft abgesetzt werden.
Krankheitsspezifische Vorsichtsmaßnahmen
Melanom
Patienten mit okulärem Melanom, primärem ZNS‑Melanom und aktiven Gehirnmetastasen waren nicht in die Studie MDX010‑20 eingeschlossen (siehe Abschnitt 5.1).
Patienten mit okulärem Melanom waren nicht in die klinische Studie CA184‑169 eingeschlossen. Allerdings waren Patienten mit Gehirnmetastasen in diese Studie eingeschlossen, sofern sie frei von neurologischen Symptomen waren, die mit metastatischen Gehirnläsionen zusammenhängen und wenn sie in den letzten 10 Tagen vor Beginn der Ipilimumab‑Therapie keine systemische Corticosteroidtherapie benötigten oder erhalten haben (siehe Abschnitt 5.1).
Patienten mit okulärem Melanom, aktiven Gehirnmetastasen und vorheriger Therapie mit Ipilimumab waren nicht in die pädiatrische Studie CA184‑070 eingeschlossen (siehe Abschnitt 5.1).
Patienten mit okulärem Melanom, aktiven Gehirnmetastasen und vorheriger Therapie mit CTLA‑4‑, PD‑1‑, PD‑L1‑ oder CD137‑zielorientierten Wirkstoffen waren nicht in die pädiatrische Studie CA184‑178 eingeschlossen (siehe Abschnitt 5.1).
Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, aktiven Hirnmetastasen oder Autoimmunerkrankung und Patienten, die vor Studienbeginn systemische Immunsuppressiva erhalten hatten, waren von den klinischen Studien mit Ipilimumab in Kombination mit Nivolumab ausgeschlossen. Patienten mit okulärem/uvealem Melanom waren von den klinischen Studien zum Melanom ausgeschlossen. Ohne weitere Daten sollte Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Im Vergleich zur Nivolumab‑Monotherapie wurde in der Kombination Ipilimumab mit Nivolumab nur bei Patienten mit niedriger Tumor‑PD‑L1‑Expression ein Anstieg des progressionsfreien Überlebens (PFS) gezeigt. Die Verbesserung des Gesamtüberlebens war bei Patienten mit hoher Tumor‑PD‑L1‑Expression (PD‑L1≥1 %) bei der Behandlung mit Ipilimumab in Kombination mit Nivolumab und der Behandlung mit Nivolumab als Monotherapie ähnlich. Bevor eine Behandlung mit der Kombination eingeleitet wird, wird den Ärzten empfohlen, die individuellen Patienten‑ und Tumorcharakteristika sorgfältig unter Berücksichtigung des beobachteten Nutzens und der Toxizität der Kombination relativ zur Nivolumab‑Monotherapie zu bewerten (siehe Abschnitte 4.8 und 5.1).
Anwendung von Ipilimumab in Kombination mit Nivolumab bei Melanom‑Patienten mit schnell fortschreitender Krankheit
Ärzte sollten das verzögerte Einsetzen der Wirkung von Ipilimumab in Kombination mit Nivolumab berücksichtigen, bevor sie eine Behandlung bei Patienten mit schnell fortschreitender Krankheit beginnen (siehe Abschnitt 5.1).
Nierenzellkarzinom
Patienten mit einer Vorgeschichte gleichzeitig aufgetretener Hirnmetastasen, Patienten mit einer aktiven Autoimmunerkrankung oder bei denen medizinische Bedingungen vorliegen, die eine systemische Immunsuppression erfordern, wurden von den klinischen Studien mit Ipilimumab in Kombination mit Nivolumab ausgeschlossen (siehe Abschnitt 4.5 und 5.1). Da keine weiteren Daten vorliegen, sollte Ipilimumab in Kombination mit Nivolumab bei diesen Patientenpopulationen nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall mit Vorsicht angewendet werden.
Nicht‑kleinzelliges Lungenkarzinom
Patienten mit einer aktiven Autoimmunerkrankung, einer symptomatischen interstitiellen Lungenerkrankung, mit Erkrankungen, die eine systemische immunsuppressive Therapie erforderlich machen, mit aktiven (unbehandelten) Hirnmetastasen sowie Patienten, die bereits zuvor eine systemische Behandlung für die fortgeschrittene Erkrankung erhalten haben oder die sensitivierende EGFR‑Mutationen oder ALK‑Translokationen aufwiesen, waren von der pivotalen klinischen Studie zur Erstlinientherapie des NSCLC ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Die Daten von älteren Patienten (≥ 75 Jahre) sind begrenzt (siehe Abschnitt 5.1). Bei diesen Patienten sollte Ipilimumab in Kombination mit Nivolumab und Chemotherapie mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Malignes Pleuramesotheliom
Patienten mit primärem Mesotheliom des Peritoneums, Perikards oder der Tunica vaginalis testis sowie Patienten mit interstitieller Lungenerkrankung, aktiver Autoimmunerkrankung, mit Erkrankungen, die eine systemische Immunsuppression erfordern, und Patienten mit Hirnmetastasen (soweit nicht chirurgisch reseziert oder mit stereotaktischer Radiotherapie behandelt, und ohne Weiterentwicklung innerhalb von 3 Monaten vor Einschluss in die Studie) waren von der pivotalen Studie zur Erstlinientherapie des MPM ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten sollte Ipilimumab in Kombination mit Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
dMMR‑ oder MSI‑H‑Kolorektalkarzinom
Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, aktiven Hirnmetastasen oder leptomeningealen Metastasen, aktiver Autoimmunerkrankung oder Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von den klinischen Studien beim metastasierten dMMR‑ oder MSI‑H‑Kolorektalkarzinom ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten sollte Ipilimumab in Kombination mit Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Plattenepithelkarzinom des Ösophagus
Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, mit einer Vorgeschichte von gleichzeitig aufgetretenen Hirnmetastasen, mit aktiver Autoimmunerkrankung, mit Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, oder mit erhöhtem Risiko für Blutungen oder Fisteln aufgrund von offensichtlicher Tumorinvasion in angrenzende Organe des ösophagealen Tumors waren von der klinischen Studie im ESCC ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten sollte Ipilimumab in Kombination mit Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
In der ESCC‑Erstlinienstudie wurde eine höhere Anzahl an Todesfällen innerhalb der ersten 4 Monate bei Patienten beobachtet, die mit Ipilimumab in Kombination mit Nivolumab behandelt wurden, verglichen mit den mit Chemotherapie behandelten Patienten. Ärzte sollten das verzögerte Einsetzen der Wirkung von Ipilimumab in Kombination mit Nivolumab berücksichtigen, bevor sie eine Behandlung bei Patienten mit prognostisch ungünstigeren Faktoren und/oder einem aggressiven Krankheitsverlauf beginnen (siehe Abschnitt 5.1).
Hepatozelluläres Karzinom
Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, vorheriger Lebertransplantation, Child‑Pugh‑C‑Lebererkrankung, einer Vorgeschichte gleichzeitig aufgetretener Hirnmetastasen, einer Vorgeschichte von hepatischer Enzephalopathie (innerhalb von 12 Monaten vor der Randomisierung), klinisch signifikantem Aszites, einer HIV-Infektion oder aktiver Koinfektion mit Hepatitis-B-Virus (HBV) und Hepatitis-C-Virus (HCV) oder HBV und Hepatitis-D-Virus (HDV), aktiver Autoimmunerkrankung oder Erkrankungen, die eine systemische Immunsuppression erfordern, wurden von der klinischen Studie im HCC ausgeschlossen (siehe Abschnitt 4.5 und 5.1). Die Daten zu HCC‑Patienten mit Child‑Pugh B sind begrenzt. Ohne weitere Daten muss Ipilimumab in Kombination mit Nivolumab, gefolgt von Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Beim HCC wurde eine höhere Anzahl an Todesfällen innerhalb der ersten 6 Monate bei Patienten beobachtet, die mit Ipilimumab in Kombination mit Nivolumab behandelt wurden, verglichen mit den mit Lenvatinib oder Sorafenib behandelten Patienten. Mit prognostisch ungünstigen Faktoren kann ein höheres Sterberisiko assoziiert sein. Ärzte sollten dieses Risiko bei Patienten mit prognostisch ungünstigeren Faktoren vor Beginn der Therapie mit Ipilimumab in Kombination mit Nivolumab berücksichtigen.
Patienten mit einer Autoimmunerkrankung
Patienten mit Autoimmunerkrankungen in der Vorgeschichte (außer Vitiligo und angemessen kontrollierten endokrinen Fehlfunktionen wie Hypothyreose) sowie Patienten, die eine systemische Immunsuppression wegen einer bestehenden Autoimmunerkrankung oder zum Erhalt eines transplantierten Organs benötigen, wurden in klinischen Studien nicht untersucht. Ipilimumab ist ein Verstärker der T‑Zellfunktion, der die Immunantwort aktiviert (siehe Abschnitt 5.1) und sich störend auf die immunsupprimierende Therapie auswirken kann, was zur Exazerbation der zugrundeliegenden Erkrankung oder einem erhöhten Risiko einer Transplantatabstoßung führen kann. Ipilimumab sollte bei Patienten mit schweren aktiven Autoimmunerkrankungen, bei denen eine weitere Immunaktivierung möglicherweise lebensgefährlich sein kann, vermieden werden. Bei anderen Patienten mit Autoimmunerkrankungen in der Vorgeschichte sollte Ipilimumab mit Vorsicht nach sorgfältiger Abwägung des individuellen klinischen Nutzen‑Risiko‑Verhältnisses angewendet werden.
Patienten mit kontrollierter Natriumdiät
Dieses Arzneimittel enthält 23 mg Natrium pro 10‑ml‑Durchstechflasche bzw. 92 mg Natrium pro 40‑ml‑Durchstechflasche, entsprechend 1,15 % bzw. 4,60 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g. Dies sollte bei der Behandlung von Patienten mit kontrollierter Natriumdiät berücksichtigt werden.
Gleichzeitige Anwendung von Vemurafenib
In einer Phase‑I‑Studie wurden bei gleichzeitiger Anwendung von Ipilimumab (3 mg/kg) und Vemurafenib (960 mg zweimal täglich oder 720 mg zweimal täglich) asymptomatische Grad 3 Erhöhungen von Transaminasen (ALT/AST mehr als 5‑fach über dem Normwert) und Bilirubin (Gesamtbilirubin mehr als 3‑fach über dem Normwert) berichtet. Aufgrund dieser vorläufigen Daten wird die gleichzeitige Anwendung von Ipilimumab und Vemurafenib nicht empfohlen.
Sequenzielle Anwendung von Vemurafenib
In einer Phase‑II‑Studie zeigten Patienten mit BRAF‑mutiertem metastasiertem Melanom bei sequenzieller Anwendung von Vemurafenib gefolgt von 10 mg/kg Ipilimumab eine höhere Inzidenz von Grad 3+ Nebenwirkungen der Haut als bei einer alleinigen Ipilimumab‑Behandlung. Vorsicht ist angezeigt, wenn Ipilimumab nach einer vorherigen Vemurafenib‑Behandlung angewendet wird.
Kinder und Jugendliche
Es liegen begrenzte Daten zur Sicherheit der Anwendung von Ipilimumab bei Jugendlichen ab einem Alter von 12 Jahren vor, jedoch keine Langzeitdaten.
Es liegen nur sehr begrenzte Daten bei Kindern unter 12 Jahren vor. Deswegen sollte Ipilimumab bei Kindern unter 12 Jahren nicht angewendet werden.
Vor Beginn der Behandlung mit Ipilimumab‑Monotherapie bei Jugendlichen im Alter von 12 Jahren oder älter, sind die Ärzte dazu angehalten, in Anbetracht der begrenzten verfügbaren Daten, des beobachteten Nutzens und der Toxizität der Ipilimumab‑Monotherapie bei Kindern und Jugendlichen, jeden Patienten sorgfältig individuell einzuschätzen (siehe Abschnitte 4.8 und 5.1).
Ipilimumab ist ein humaner monoklonaler Antikörper, der nicht mithilfe von Cytochrom‑P450‑Enzymen (CYPs) oder anderen Enzymen des Arzneimittelmetabolismus abgebaut wird.
Eine Wechselwirkungsstudie mit Ipilimumab, angewendet bei Erwachsenen als Monotherapie und in Kombination mit Chemotherapie (Dacarbazin oder Paclitaxel/Carboplatin) wurde bei Patienten mit behandlungsnaivem fortgeschrittenem Melanom zur Ermittlung der Wechselwirkung mit CYP‑Isoenzymen (insbesondere CYP1A2, CYP2E1, CYP2C8 und CYP3A4) durchgeführt. Zwischen Ipilimumab und Paclitaxel/Carboplatin, Dacarbazin oder seinem Metaboliten, 5‑Aminoimidazol‑4‑carboxamid (AIC) wurde keine klinisch relevante pharmakokinetische Arzneimittelwechselwirkung beobachtet.
Sonstige Wechselwirkungen
Corticosteroide
Die Verwendung systemischer Corticosteroide vor dem Behandlungsbeginn mit Ipilimumab sollte vermieden werden, da sie die pharmakodynamische Aktivität und Wirksamkeit von Ipilimumab beeinträchtigen könnten. Dennoch können systemische Corticosteroide oder andere Immunsuppressiva nach dem Beginn der Ipilimumab‑Therapie eingesetzt werden, um immunvermittelte Nebenwirkungen zu behandeln. Die Verwendung von systemischen Corticosteroiden nach dem Behandlungsbeginn mit Ipilimumab scheint die Wirksamkeit von Ipilimumab nicht zu beeinträchtigen.
Antikoagulantien
Die Verwendung von Antikoagulantien erhöht bekannterweise das Risiko einer Gastrointestinalblutung. Da diese zu den Nebenwirkungen von Ipilimumab zählt (siehe Abschnitt 4.8), sollten Patienten, die einer gleichzeitigen antikoagulativen Behandlung bedürfen, engmaschig überwacht werden.
Schwangerschaft
Bisher liegen keine Erfahrungen zur Anwendung von Ipilimumab bei Schwangeren vor. Bei tierexperimentellen Reproduktionsstudien wurde Reproduktionstoxizität festgestellt (siehe Abschnitt 5.3). Humanes IgG1 passiert die Plazentaschranke. Das potenzielle Risiko der Behandlung für den sich entwickelnden Fetus ist nicht bekannt. Die Anwendung von YERVOY während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen, es sei denn, der klinische Nutzen überwiegt das potenzielle Risiko.
Stillzeit
Ipilimumab wurde in sehr geringen Mengen in der Milch von Cynomolgus‑Affen nachgewiesen, die während der Trächtigkeit behandelt wurden. Es ist nicht bekannt, ob Ipilimumab in die Muttermilch übergeht. Die Ausscheidung von IgGs in die humane Muttermilch ist im Allgemeinen begrenzt und IgGs weisen eine niedrige orale Bioverfügbarkeit auf. Eine signifikante systemische Exposition des Säuglings ist nicht zu erwarten und es werden keine Auswirkungen auf gestillte Neugeborene/Kinder erwartet. Da jedoch Nebenwirkungen beim gestillten Kind nicht ausgeschlossen werden können, muss unter Abwägung des Nutzens des Stillens für das Kind und des Nutzens der Behandlung für die Mutter eine Entscheidung darüber getroffen werden, ob das Stillen oder die Behandlung mit YERVOY unterbrochen werden soll.
Fertilität
Es wurden keine Studien durchgeführt, um die Auswirkung von Ipilimumab auf die Fertilität zu untersuchen. Daher ist die Auswirkung von Ipilimumab auf die männliche oder weibliche Fertilität unbekannt.
YERVOY hat einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Aufgrund potenzieller Nebenwirkungen, wie Ermüdung/Fatigue (siehe Abschnitt 4.8), sollten Patienten angewiesen werden, beim Autofahren oder beim Bedienen von Maschinen vorsichtig zu sein, bis sie sicher sind, nicht durch Ipilimumab beeinträchtigt zu werden.
Ipilimumab als Monotherapie (siehe Abschnitt 4.2)
a. Zusammenfassung des Sicherheitsprofils
Ipilimumab wurde in einem klinischen Programm zur Untersuchung der Wirkung bei unterschiedlichen Dosierungen und Tumorarten bei ungefähr 10 000 Patienten angewendet. Sofern nicht anders angegeben, beziehen sich die unten beschriebenen Daten auf die Exposition gegenüber Ipilimumab in einer Dosis von 3 mg/kg in klinischen Studien bei Melanomen. In der Phase‑III‑Studie MDX010‑20 (siehe Abschnitt 5.1) erhielten die Patienten im Median 4 Dosen (Bereich 1‑4).
Ipilimumab ist am häufigsten mit Nebenwirkungen assoziiert, die aus der erhöhten oder übermäßigen Immunaktivität resultieren. Die meisten dieser Nebenwirkungen, einschließlich schwerwiegender Ereignisse, klangen nach Einleitung einer geeigneten Therapie oder Abbruch der Behandlung mit Ipilimumab wieder ab (siehe Abschnitt 4.4 zur Behandlung von immunvermittelten Nebenwirkungen).
Bei Patienten, die in der Studie MDX010‑20 3 mg/kg Ipilimumab als Monotherapie erhielten, waren die am häufigsten beobachteten Nebenwirkungen (≥ 10 % der Patienten) Diarrhö, Ausschlag, Pruritus, Ermüdung/Fatigue, Übelkeit, Erbrechen, verminderter Appetit und Abdominalschmerz. Die Mehrzahl dieser Nebenwirkungen war leicht bis mäßig (Grad 1 oder 2). Die Behandlung mit Ipilimumab wurde bei 10 % der Patienten wegen Nebenwirkungen abgebrochen.
b. Tabellarische Aufstellung der Nebenwirkungen
In Tabelle 6 sind die Nebenwirkungen von Patienten mit fortgeschrittenem Melanom aufgeführt, die in klinischen Studien (n = 767) mit Ipilimumab 3 mg/kg behandelt wurden, und die Nebenwirkungen, die im Rahmen der Überwachung nach der Markteinführung berichtet wurden.
Diese Nebenwirkungen sind nach Systemorganklassen und Häufigkeit geordnet. Häufigkeiten sind wie folgt definiert: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1 000, < 1/100); selten (≥ 1/10 000, < 1/1 000); sehr selten (< 1/10 000), nicht bekannt (kann aus den verfügbaren Daten nach Markteinführung nicht abgeschätzt werden). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt. Die Häufigkeit immunvermittelter Nebenwirkungen bei HLA‑A2*0201‑positiven Patienten, die in der Studie MDX010‑20 mit Ipilimumab behandelt wurden, war vergleichbar mit jenen im gesamten klinischen Programm.
Das Sicherheitsprofil von Ipilimumab 3 mg/kg bei chemotherapienaiven Patienten aus klinischen Studien der Phasen‑II und III (N = 75; behandelt), bei behandlungsnaiven Patienten in zwei retrospektiven Beobachtungsstudien (N = 273 und N = 157) und in der Studie CA184‑169 (N = 362) war ähnlich dem bei vorbehandeltem fortgeschrittenem Melanom.
Die Sicherheitsdaten für Patienten mit inoperablem oder metastasierendem Melanom, die mit Ipilimumab (3 mg/kg, mit mindestens 3 Jahren Nachbeobachtung) behandelt und in die multinationale, prospektive, Beobachtungsstudie CA184‑143 (N = 1151) aufgenommen wurden, waren ähnlich wie in den klinischen Studien mit Ipilimumab beim fortgeschrittenen Melanom.
Tabelle 6: Nebenwirkungen bei Patienten mit fortgeschrittenem Melanom unter Behandlung mit Ipilimumab 3 mg/kga
Infektionen und parasitäre Erkrankungen | |
Häufig |
Sepsisb, Harnweginfektion, Infektion der Atemwege |
Gelegentlich |
Septischer Schockb, Pneumonie |
Gutartige, bösartige und nicht spezifizierte Neubildungen (einschl. Zysten und Polypen) | |
Häufig |
Tumorschmerzen |
Gelegentlich |
Paraneoplastisches Syndrom |
Erkrankungen des Blutes und des Lymphsystems | |
Häufig |
Anämie, Lymphopenie, Thrombozytopenie, Neutropenie |
Gelegentlich |
Hämolytische Anämieb, Eosinophilie |
Nicht bekannt |
Hämophagozytische Lymphohistiozytosee |
Erkrankungen des Immunsystems | |
Gelegentlich |
Hypersensitivität |
Sehr selten |
Anaphylaktische Reaktion |
Nicht bekannt |
Abstoßung eines soliden Organtransplantatse |
Endokrine Erkrankungen | |
Häufig |
Hypopituitarismus (einschließlich Hypophysitis)c, Hypothyreosec |
Gelegentlich |
Nebenniereninsuffizienzc, Sekundäre Nebenniereninsuffizienzd, Hyperthyreosec, Hypogonadismus |
Selten |
Autoimmune Thyroiditisd, Thyroiditisd |
Stoffwechsel‑ und Ernährungsstörungen | |
Sehr häufig |
Verminderter Appetit |
Häufig |
Dehydrierung, Hypokaliämie, Gewichtsabnahme, Hyponatriämie |
Gelegentlich |
Alkalose, Hypophosphatämie, Tumorlysesyndrom, Hypokalziämied |
Selten |
Typ‑1‑Diabetes‑mellitus (einschließlich diabetischer Ketoazidose)h |
Psychiatrische Erkrankungen | |
Häufig |
Verwirrtheit, Depression |
Gelegentlich |
Veränderung der psychischen Verfassung, verminderte Libido |
Erkrankungen des Nervensystems | |
Häufig |
Periphere sensorische Neuropathie, Schwindel, Kopfschmerzen, Lethargie, kraniale Neuropathie, Gehirnödeme, periphere Neuropathie |
Gelegentlich |
Guillain‑Barré‑Syndromb,c, Meningitis (aseptisch), zentrale autoimmune Neuropathie (Enzephalitis)d, Synkope, Ataxie, Tremor, Myoklonie, Dysarthrie |
Selten |
Myasthenia gravisd |
Nicht bekannt |
Myelitis |
Augenerkrankungen | |
Häufig |
Verschwommenes Sehen, Augenschmerzen |
Gelegentlich |
Uveitisc, Glaskörperblutung, Iritisc, Augenödemd, Blepharitisd, verminderte Sehschärfe, Fremdkörpergefühl in den Augen, Konjunktivitis |
Selten |
Vogt‑Koyanagi‑Harada‑Syndrome, seröse Netzhautablösung |
Herzerkrankungen | |
Häufig |
Arrhythmie, Vorhofflimmern |
Gefäßerkrankungen | |
Häufig |
Hypotonie, Hautrötungen, Hitzewallungen |
Gelegentlich |
Vaskulitis, Angiopathieb, periphere Ischämie, orthostatische Hypotonie |
Selten |
Arteriitis temporalisd |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
Häufig |
Dyspnoe, Husten, allergische Rhinitis |
Gelegentlich |
Respiratorische Insuffizienz, akutes respiratorisches Distress-Syndromb, Lungeninfiltration, Lungenödeme, Pneumonitis |
Erkrankungen des Gastrointestinaltrakts | |
Sehr häufig |
Diarrhöc, Erbrechen, Übelkeit, Verstopfung, Abdominalschmerz |
Häufig |
Gastrointestinale Hämorrhagie, Kolitisb, c, gastroösophageale Refluxkrankheit, Schleimhautentzündungd, Gastroenteritis, Stomatitis |
Gelegentlich |
Gastrointestinale Perforationb, c, Dickdarmperforationb, c, intestinale Perforationb, c, Peritonitisb Divertikulitis, Pankreatitis, Enterokolitis, Magengeschwür, Dickdarmgeschwür, Ösophagitis, Ileusd, Proktitisd |
Selten |
Exokrine Pankreasinsuffizienz; Zöliakie |
Leber‑ und Gallenerkrankungen | |
Häufig |
Leberfunktionsstörungen |
Gelegentlich |
Leberversagenb, c, Hepatitis, Hepatomegalie, Gelbsucht |
Erkrankungen der Haut und des Unterhautgewebes | |
Sehr häufig |
Ausschlagc, Pruritusc |
Häufig |
Dermatitis, Erythem, Vitiligo, Urtikaria, Ekzemd, Alopezie, Nachtschweiß, trockene Haut |
Gelegentlich |
Toxische epidermale Nekrolyseb, c, leukozytoklastische Vaskulitis, Hautabschälung, Veränderung der Haarfarbed |
Selten |
Erythema multiformed, Psoriasisd, Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen (DRESS)d |
Nicht bekannt |
Pemphigoid |
Skelettmuskulatur‑, Bindegewebs‑ und Knochenerkrankungen | |
Sehr häufig |
Muskel‑ und Skelettschmerzenf |
Häufig |
Arthralgie, Myalgie, Muskelspasmus, Arthritis |
Gelegentlich |
Rheumatische Polymyalgie, Myositisd Muskelschwäched |
Selten |
Polymyositisd |
Erkrankungen der Nieren und Harnwege | |
Häufig |
Nierenversagenb |
Gelegentlich |
Glomerulonephritisc, autoimmune Nephritisd, Nierentubulusazidose, Hämaturied, nicht infektiöse Zystitisg, Proteinuried |
Erkrankungen der Geschlechtsorgane und der Brustdrüse | |
Gelegentlich |
Amenorrhoe |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Sehr häufig |
Ermüdung/Fatigue, Reaktionen an der Injektionsstelle, Fieber, Ödeme, Schmerzen |
Häufig |
Schüttelfrost, Asthenie, grippeähnliche Erkrankungd |
Gelegentlich |
Multiples Organversagenb,c, systemisches inflammatorisches Response-Syndromd, infusionsbedingte Reaktionen |
Untersuchungen | |
Häufig |
Erhöhte Alanin‑Aminotransferasec, erhöhte Aspartat‑Aminotransferasec, erhöhte alkalische Phosphatase im Blutd, erhöhte Bilirubinwerte, erhöhte Lipasec |
Gelegentlich |
Erhöhte Gamma‑Glutamyltransferased, erhöhte Kreatininwerte, Anstieg des thyreotropen Hormons im Blut, Verminderung des Cortisolspiegels, Verminderung des Corticotropinspiegels, erhöhte Amylasec, positive antinukleäre Antikörperd, Verminderung des Testosteronspiegels |
Selten |
Verminderung des thyreotropen Hormons im Blutd, Verminderung des Thyroxinspiegelsd, anomaler Prolaktinspiegel im Blutd |
Die in Tabelle 6 angegebenen Häufigkeiten der Nebenwirkungen sind möglicherweise nicht vollständig auf Ipilimumab zurückzuführen, da auch die Grunderkrankung dazu beitragen kann. | |
Weitere Nebenwirkungen, die nicht in Tabelle 6 aufgeführt sind, wurden bei Patienten in klinischen Studien beim Melanom, die andere Dosen von Ipilimumab (entweder < oder > 3 mg/kg) erhielten, berichtet. Diese zusätzlichen Nebenwirkungen traten, sofern nicht anders vermerkt, mit einer Häufigkeit von < 1 % auf: Meningismus, Myokarditis, Perikarderguss, Kardiomyopathie, autoimmune Hepatitis, Erythema nodosum, autoimmune Pankreatitis, Hyperpituitarismus, Hypoparathyroidismus, infektiöse Peritonitis, Episkleritis, Skleritis, Raynaud-Syndrom, palmar‑plantares Erythrodysästhesie‑Syndrom, Zytokin‑Freisetzungs‑Syndrom, Sarkoidose, Verminderung des Gonadotropinspiegels, Leukopenie, Polyzythämie, Lymphozytose, okulare Myositis, neurosensorische Hypakusis.
Insgesamt war das Sicherheitsprofil von Ipilimumab 3 mg/kg in der klinischen Studie CA184‑169 (N = 362) übereinstimmend mit dem, welches für Ipilimumab bei Patienten mit fortgeschrittenem Melanom bekannt ist.
Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) (siehe Abschnitt 4.2)
a. Zusammenfassung des Sicherheitsprofils
Wenn Ipilimumab in Kombination angewendet wird, lesen Sie die Fachinformation des bzw. der anderen Arzneimittel(s), bevor Sie mit der Behandlung beginnen. Weitere Informationen zum Sicherheitsprofil der anderen Arzneimittel, die in Kombination mit Ipilimumab eingesetzt werden, finden Sie in den jeweiligen Fachinformationen.
Im zusammengefassten Datensatz von Ipilimumab, verabreicht in Kombination mit Nivolumab (mit oder ohne Chemotherapie) über die genannten Tumorarten (n = 2626) mit einer Mindest‑Nachbeobachtungszeit von 6 bis 47 Monaten, waren die häufigsten Nebenwirkungen (≥ 10 %) Ermüdung/Fatigue (47 %), Diarrhö (35 %), Hautausschlag (37 %), Übelkeit (27 %), Pruritus (29 %), Muskel‑ und Skelettschmerzen (26 %), Fieber (23 %), verminderter Appetit (22 %), Husten (21 %), Abdominalschmerz (18 %), Erbrechen (18 %), Obstipation (18 %), Arthralgie (18 %), Dyspnoe (17 %), Hypothyreose (16 %), Kopfschmerzen (15 %), Infektionen der oberen Atemwege (13 %), Ödeme (13 %) und Schwindelgefühl (10 %). Die Häufigkeit von Grad‑3‑5‑Nebenwirkungen war 66 % für Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie), mit 1,0 % tödlichen Nebenwirkungen, welche auf die Studienmedikation zurückzuführen sind. Bei Patienten, die mit Ipilimumab 3 mg/kg in Kombination mit Nivolumab 1 mg/kg für Melanom behandelt wurden, wurden die folgenden Nebenwirkungen, im Vergleich zu der Häufigkeitsrate, die im zusammengefassten Datensatz von Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) berichtet wurde, mit einer ≥ 10 % höheren Häufigkeitsrate berichtet: Ermüdung/Fatigue (62 %), Hautausschlag (57 %), Diarrhö (52 %), Übelkeit (42 %), Pruritus (40 %), Fieber (36 %) und Kopfschmerzen (26 %). Bei Patienten, die mit Ipilimumab 1 mg/kg in Kombination mit Nivolumab 360 mg und Chemotherapie für NSCLC behandelt wurden, wurden die folgenden Nebenwirkungen, im Vergleich zu der Häufigkeitsrate, die im zusammengefassten Datensatz von Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) berichtet wurde, mit einer ≥ 10 % höheren Häufigkeitsrate berichtet: Anämie (32 %) und Neutropenie (15 %).
b. Tabellarische Aufstellung der Nebenwirkungen
In Tabelle 7 sind die Nebenwirkungen aufgeführt, die aus dem zusammengefassten Datensatz für die mit Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) (n = 2626) behandelten Patienten und nach der Markteinführung stammen. Diese Nebenwirkungen sind nach Systemorganklassen und Häufigkeit geordnet. Häufigkeiten sind wie folgt definiert: sehr häufig (≥ 1/10); häufig (≥ 1/100 bis < 1/10); gelegentlich (≥ 1/1 000 bis < 1/100); selten (≥ 1/10 000 bis < 1/1 000); sehr selten (< 1/10 000), nicht bekannt (kann aus den verfügbaren Daten nach Markteinführung nicht abgeschätzt werden). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 7: Nebenwirkungen unter Ipilimumab in Kombination mit anderen Arzneimitteln
Kombination mit Nivolumab (mit oder ohne Chemotherapie) |
|
Infektionen und parasitäre Erkrankungen | |
Sehr häufig |
Infektionen der oberen Atemwege |
Häufig |
Pneumonie, Bronchitis, Konjunktivitis |
Selten |
Aseptische Meningitis |
Erkrankungen des Blutes und des Lymphsystems | |
Sehr häufig |
Anämieb,i, Thrombozytopenieb, Leukopenieb, Lymphopenieb, Neutropenieb |
Häufig |
Eosinophilie |
Gelegentlich |
Febrile Neutropenie |
Nicht bekannt |
Hämophagozytische Lymphohistiozytoseg |
Erkrankungen des Immunsystems | |
Häufig |
Infusionsbedingte Reaktion (einschließlich Zytokin-Freisetzungssyndrom), Hypersensibilität |
Selten |
Sarkoidose |
Nicht bekannt |
Abstoßung eines soliden Organtransplantatsf |
Endokrine Erkrankungen | |
Sehr häufig |
Hypothyreose |
Häufig |
Hyperthyreose, Thyroiditis, Nebenniereninsuffizienz, Hypophysitis, Hypophyseninsuffizienz, Diabetes mellitus |
Gelegentlich |
Diabetische Ketoazidose |
Selten |
Hypoparathyreoidismus |
Stoffwechsel‑ und Ernährungsstörungen | |
Sehr häufig |
Verminderter Appetit, Hyperglykämieb, Hypoglykämieb |
Häufig |
Dehydrierung, Hypoalbuminämie, Hypophosphatämie, Gewichtsabnahme |
Gelegentlich |
Metabolische Azidose |
Nicht bekannt |
Tumorlyse‑Syndromg |
Erkrankungen des Nervensystems | |
Sehr häufig |
Kopfschmerzen |
Häufig |
Schwindelgefühl, periphere Neuropathie |
Gelegentlich |
Polyneuropathie, Peroneuslähmung, autoimmune Neuropathie (einschließlich Gesichtsnerv‑ und Abduzensparese), Enzephalitis, Myasthenia gravis, Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndromk |
Selten |
Guillain‑Barré‑Syndrom, Neuritis, Myelitis (einschließlich transverse Myelitis) |
Augenerkrankungen | |
Häufig |
Verschwommenes Sehen, trockene Augen |
Gelegentlich |
Uveitis, Episkleritis |
Selten |
Vogt‑Koyanagi‑Harada‑Syndrom, seröse Netzhautablösung |
Herzerkrankungen | |
Häufig |
Tachykardie, Vorhofflimmern |
Gelegentlich |
Myokarditisa, Arrhythmie (einschließlich ventrikulärer Arrhythmie)a, Bradykardie |
Nicht bekannt |
Perikardiale Erkrankungenh |
Gefäßerkrankungen | |
Häufig |
Hypertonie |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
Sehr häufig |
Husten, Dyspnoe |
Häufig |
Pneumonitisa, Lungenemboliea, Pleuraerguss |
Erkrankungen des Gastrointestinaltrakts | |
Sehr häufig |
Diarrhö, Erbrechen, Übelkeit, Abdominalschmerz, Obstipation |
Häufig |
Kolitisa, Pankreatitis, Stomatitis, Gastritis, trockener Mund |
Gelegentlich |
Duodenitis |
Selten |
Darmperforationa, exokrine Pankreasinsuffizienz, Zöliakie |
Leber‑ und Gallenerkrankungen | |
Häufig |
Hepatitis |
Erkrankungen der Haut und des Unterhautgewebes | |
Sehr häufig |
Hautausschlagc, Pruritus |
Häufig |
Alopezie, Vitiligo, Urtikaria, trockene Haut, Erythem |
Gelegentlich |
Stevens‑Johnson‑Syndrom, Erythema multiforme, Psoriasis, andere Lichenerkrankungenj |
Selten |
Toxische epidermale Nekrolysea,d, Lichen sclerosus |
Skelettmuskulatur‑, Bindegewebs‑ und Knochenerkrankungen | |
Sehr häufig |
Muskel‑ und Skelettschmerzene, Arthralgie |
Häufig |
Muskelkrämpfe, muskuläre Schwäche, Arthritis |
Gelegentlich |
Rheumatische Polymyalgie, Myopathie, Myositis (einschließlich Polymyositis)a |
Selten |
Spondyloarthropathie, Sjögren‑Syndrom, Rhabdomyolysea |
Erkrankungen der Nieren und Harnwege | |
Häufig |
Nierenversagen (einschließlich akutes Nierenversagen)a |
Gelegentlich |
Tubulointerstitielle Nephritis, Nephritis |
Selten |
Nicht‑infektiöse Zystitis |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Sehr häufig |
Ermüdung/Fatigue, Fieber, Ödeme (einschließlich peripheres Ödem) |
Häufig |
Schmerzen in der Brust, Schmerzen, Schüttelfrost |
Untersuchungen | |
Sehr häufig |
Anstieg der alkalischen Phosphataseb, AST-Anstiegb, ALT‑Anstiegb, Anstieg des Gesamt‑Bilirubinsb, Kreatinin‑Anstiegb, Amylase‑Anstiegb, Lipase‑Anstiegb, Hyponatriämieb, Hyperkaliämieb, Hypokaliämieb, Hyperkalziämieb, Hypokalziämieb |
Häufig |
Hypernatriämieb, Hypermagnesiämieb, TSH-Anstieg, Gamma‑Glutamyltransferase erhöht |
Die in Tabelle 7 angegebenen Häufigkeiten der Nebenwirkungen sind möglicherweise nicht vollständig auf Ipilimumab allein oder in Kombination mit anderen Arzneimitteln zurückzuführen, da auch die Grunderkrankung oder die in Kombination verwendeten Arzneimittel dazu beitragen können. | |
Beschreibung einzelner Nebenwirkungen
Wenn nicht anders gekennzeichnet, beruhen die Daten bezüglich der Ipilimumab‑Monotherapie auf Patienten, die in der Phase‑III‑Studie beim fortgeschrittenen (nicht resezierbaren oder metastasierten) Melanom (MDX010‑20, siehe Abschnitt 5.1) entweder Ipilimumab 3 mg/kg als Monotherapie (n = 131) oder Ipilimumab 3 mg/kg in Kombination mit der Peptid‑Vakzine gp100 (n = 380) erhalten haben.
Ipilimumab in Kombination ist mit immunvermittelten Nebenwirkungen assoziiert. Diese immunvermittelten Nebenwirkungen sind mit einer adäquaten medizinischen Behandlung meist reversibel. Bei einem größeren Anteil der Patienten, die Ipilimumab in Kombination mit Nivolumab erhielten, war im Allgemeinen dauerhaftes Absetzen der Behandlung erforderlich, als bei Patienten, die eine Nivolumab‑Monotherapie erhielten. Tabelle 8 zeigt den prozentualen Anteil der Patienten mit immunvermittelten Nebenwirkungen, bei welchen die Therapie dauerhaft abgesetzt werden musste. Außerdem zeigt Tabelle 8 für die Patienten, bei welchen eine Nebenwirkung auftrat, den Prozentsatz an Patienten, der mit hochdosierten Corticosteroiden (mindestens 40 mg Prednison‑Äquivalent täglich) behandelt werden musste. Die Behandlungsrichtlinien für diese Nebenwirkungen sind in Abschnitt 4.4 beschrieben.
Tabelle 8: Immunvermittelte Nebenwirkungen, welche zum dauerhaften Absetzen der Therapie führen oder welche eine Behandlung mit hochdosierten Corticosteroiden erfordern
Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) |
|
Immunvermittelte Nebenwirkung, welche zum dauerhaften Absetzen der Therapie führt | |
Pneumonitis |
2,1 |
Kolitis |
6 |
Hepatitis |
5 |
Nephritis und Nierenfunktionsstörung |
1,1 |
Endokrinopathien |
2,2 |
Haut |
1,0 |
Hypersensibilität/Infusionsbedingte Reaktion |
0,3 |
Immunvermittelte Nebenwirkung, welche eine Behandlung mit hochdosierten Corticosteroiden erforderta,b | |
Pneumonitis |
59 |
Kolitis |
32 |
Hepatitis |
39 |
Nephritis und Nierenfunktionsstörung |
27 |
Endokrinopathien |
18 |
Haut |
8 |
Hypersensibilität/Infusionsbedingte Reaktion |
18 |
a mindestens 40 mg Prednison‑Äquivalent täglich | |
Immunvermittelte gastrointestinale Nebenwirkungen
Ipilimumab ist mit schwerwiegenden immunvermittelten gastrointestinalen Nebenwirkungen assoziiert. Bei < 1 % der Patienten, die Ipilimumab 3 mg/kg in Kombination mit gp100 erhalten haben, wurden Todesfälle aufgrund von gastrointestinalen Perforationen berichtet.
In der Gruppe, die Ipilimumab 3 mg/kg als Monotherapie erhielt, wurden Diarrhö und Kolitis jeden Schweregrads in 27 % bzw. 8 % der Fälle berichtet. Die Häufigkeit einer schweren (Grad 3 oder 4) Diarrhö und einer schweren (Grad 3 oder 4) Kolitis lag jeweils bei 5 %. Die mediane Zeit bis zum Einsetzen einer schweren oder tödlichen immunvermittelten gastrointestinalen Nebenwirkung (Grad 3‑5) lag bei 8 Wochen (Bereich 5 bis 13 Wochen) ab Behandlungsbeginn. Unter protokolldefinierten Behandlungsrichtlinien war in den meisten Fällen (90 %) ein Rückgang der Symptome (definiert als Verbesserung zu leichten [Grad 1] Symptomen oder weniger oder zum Schweregrad bei Behandlungsbeginn) innerhalb eines medianen Zeitraums von 4 Wochen (Bereich 0,6 bis 22 Wochen) vom Beginn der Symptome bis zur Besserung zu verzeichnen. In klinischen Studien war die immunvermittelte Kolitis mit Anzeichen einer Schleimhautentzündung mit oder ohne Ulzerationen und einer lymphozytischen und neutrophilen Infiltration assoziiert.
Immunvermittelte Kolitis
Bei Patienten, die mit Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Diarrhö oder Kolitis 26,0 % (682/2626). Fälle von Grad 2, Grad 3 bzw. Grad 4 wurden bei 8,1 % (212/2626), 6,4 % (167/2626) bzw. 0,2 % (4/2626) der Patienten berichtet. Bei zwei Patienten (< 0,1 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 1,4 Monate (Spanne: 0,0 ‑ 48,9). Bei 618 Patienten (91 %) kam es zu einer Rückbildung nach einer medianen Zeit von 2,9 Wochen (Spanne: 0,1 ‑ 170,0+). Bei Patienten, die mit Ipilimumab 3 mg/kg in Kombination mit Nivolumab 1 mg/kg für Melanom behandelt wurden, war die Häufigkeit von Diarrhö oder Kolitis 46,7 %, einschließlich Grad 2 (13,6 %), Grad 3 (15,8 %) und Grad 4 (0,4 %).
Immunvermittelte Pneumonitis
Bei Patienten, die mit Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Pneumonitis, einschließlich einer interstitiellen Lungenerkrankung 6,0 % (157/2626). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 3,0 % (78/2626), 1,0 % (27/2626) bzw. 0,3 % (8/2626) der Patienten berichtet. Bei vier Patienten (0,2 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 2,7 Monate (Spanne: 0,1 ‑ 56,8). Bei 129 Patienten (82,2 %) kam es zu einer Rückbildung nach einer medianen Zeit von 6,1 Wochen (Spanne: 0,1+ ‑ 149,3+).
Immunvermittelte Hepatotoxizität
Ipilimumab ist mit einer schwerwiegenden immunvermittelten Hepatotoxizität assoziiert. Ein tödliches Leberversagen wurde bei < 1 % der Patienten unter einer Ipilimumab 3 mg/kg Monotherapie berichtet.
Ein Anstieg der AST und der ALT jeden Schweregrads wurde bei 1 % bzw. 2 % der Patienten berichtet. Es gab keine Berichte eines schweren (Grad 3 oder 4) Anstiegs der AST oder ALT. Der Zeitraum bis zum Ausbruch mäßiger bis schwerer oder tödlicher (Grad 2‑5) immunvermittelter Hepatotoxizitäten lag zwischen 3 und 9 Wochen ab Behandlungsbeginn. Unter protokolldefinierten Behandlungsrichtlinien gingen die Symptome innerhalb eines Zeitraums von 0,7 bis 2 Wochen zurück. In klinischen Studien zeigten Leberbiopsien von Patienten, die eine immunvermittelte Hepatotoxizität hatten, Anzeichen einer akuten Entzündung (Neutrophile, Lymphozyten und Makrophagen).
Bei Patienten, die Ipilimumab in einer höheren als der empfohlenen Dosierung zusammen mit Dacarbazin erhielten, traten immunvermittelte Hepatotoxizitäten häufiger auf, als bei Patienten, die Ipilimumab 3 mg/kg als Monotherapie erhielten.
Bei Patienten, die mit Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Anomalien bei Leberfunktionstests 21,2 % (556/2626). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 5,0 % (132/2626), 8,3 % (218/2626) bzw. 1,3 % (34/2626) der Patienten berichtet. Bei sieben Patienten (0,3 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 1,5 Monate (Spanne: 0,0 – 36,6). Bei 482 Patienten (87,0 %) kam es zu einer Rückbildung nach einer medianen Zeit von 5,9 Wochen (Spanne: 0,1 ‑ 175,9+). Bei Patienten, die mit Ipilimumab 3 mg/kg in Kombination mit Nivolumab 1 mg/kg für Melanom behandelt wurden, war die Häufigkeit von Anomalien bei Leberfunktionstests 30,1 %, einschließlich Grad 2 (6,9 %), Grad 3 (15,8 %) und Grad 4 (1,8 %). Bei Patienten, die mit Ipilimumab 3 mg/kg in Kombination mit Nivolumab 1 mg/kg für HCC behandelt wurden, war die Häufigkeit von Anomalien bei Leberfunktionstests 34,3 %, einschließlich Grad 2 (8,4 %), Grad 3 (14,2 %) und Grad 4 (2,7 %).
Immunvermittelte Nebenwirkungen der Haut
Ipilimumab wird mit schwerwiegenden Nebenwirkungen der Haut in Verbindung gebracht, die immunvermittelt sein könnten. Bei < 1 % der Patienten, die Ipilimumab in Kombination mit gp100 erhielten, wurde über Todesfälle aufgrund toxischer epidermaler Nekrolyse (einschließlich SJS) berichtet (siehe Abschnitt 5.1). Bei Anwendung von Ipilimumab in klinischen Studien und nach Markteinführung wurde in seltenen Fällen Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen (DRESS) berichtet. Nach Markteinführung wurden gelegentliche Fälle von Pemphigoid berichtet.
In der Gruppe, die Ipilimumab 3 mg/kg als Monotherapie erhielt, wurden Hautausschlag und Pruritus jeden Schweregrads bei jeweils 26 % der Patienten berichtet. Durch Ipilimumab induzierter Hautausschlag und Pruritus waren überwiegend leicht bis mäßig (Grad 1 oder 2) und sprachen auf eine symptomatische Behandlung an. Der Zeitraum bis zum Auftreten mäßiger bis schwerer oder tödlicher Nebenwirkungen (Grad 2‑5) der Haut betrug im Median 3 Wochen (Bereich 0,9‑16 Wochen) ab Behandlungsbeginn. Unter protokolldefinierten Behandlungsrichtlinien gingen die Symptome in den meisten Fällen (87 %) innerhalb eines medianen Zeitraums von 5 Wochen vom Behandlungsbeginn zurück (Bereich 0,6 bis 29 Wochen).
Bei Patienten, die mit Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Hautausschlag 46,1 % (1210/2626). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 14,3 % (375/2626), 4,6 % (120/2626) bzw. 0,1 % (3/2626) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 0,7 Monate (Spanne: 0,0 ‑ 33,8). Bei 843 Patienten (70 %) kam es zu einer Rückbildung nach einer medianen Zeit von 12,1 Wochen (Spanne: 0,1 ‑ 268,7+). Bei Patienten, die mit Ipilimumab 3 mg/kg in Kombination mit Nivolumab 1 mg/kg für Melanom behandelt wurden, war die Häufigkeit von Hautausschlag 65,2 %, einschließlich Grad 2 (20,3 %) und Grad 3 (7,8 %).
Immunvermittelte neurologische Nebenwirkungen
Ipilimumab ist mit schwerwiegenden immunvermittelten neurologischen Nebenwirkungen assoziiert. Bei < 1 % der Patienten, die Ipilimumab 3 mg/kg in Kombination mit gp100 erhielten, wurde über Todesfälle durch das Guillain‑Barré‑Syndrom berichtet. Myasthenia gravis‑ähnliche Symptome wurden ebenfalls bei < 1 % der Patienten, die Ipilimumab in klinischen Studien in höheren Dosen erhielten, berichtet.
Immunvermittelte Nephritis und Nierenfunktionsstörung
Bei Patienten, die mit Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Nephritis oder Nierenfunktionsstörungen 5,4 % (141/2626). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 2,0 % (52/2626). 0,8 % (21/2626) bzw. 0,4 % (11/2626) der Patienten berichtet. Bei zwei Patienten (< 0,1 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 2,6 Monate (Spanne; 0,0 ‑ 34,8). Bei 110 Patienten (78,0 %) kam es zu einer Rückbildung nach einer medianen Zeit von 5,9 Wochen (Spanne: 0,1 ‑ 172,1+).
Immunvermittelte Endokrinopathie
In der Gruppe, die Ipilimumab 3 mg/kg als Monotherapie erhielt, wurden bei 4 % der Patienten Hypopituitarismus jeden Schweregrads berichtet. Nebenniereninsuffizienz, Hyperthyreose und Hypothyreose jeden Schweregrads wurden bei jeweils 2 % der Patienten berichtet. Die Häufigkeit eines schweren (Grad 3 oder 4) Hypopituitarismus betrug 3 % der Patienten. Der Zeitraum bis zum Auftreten mäßiger bis sehr schwerer (Grad 2‑4) immunvermittelter Endokrinopathie betrug zwischen 7 und fast 20 Wochen ab Behandlungsbeginn. Die in klinischen Studien beobachteten Fälle immunvermittelter Endokrinopathie konnten in der Regel durch Hormonersatztherapie kontrolliert werden.
Bei Patienten, die mit Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Schilddrüsenerkrankungen 23,2 % (608/2626). Schilddrüsenerkrankungen mit dem Schweregrad 2 bzw. 3 wurden bei 12,7 % (333/2626) bzw. 1,0 % (27/2626) der Patienten berichtet. Hypophysitis (einschließlich lymphozytische Hypophysitis) von Grad 2 bzw. Grad 3 trat bei 1,9 % (49/2626) bzw. 1,5 % (40/2626) der Patienten auf. Hypophyseninsuffizienz von Grad 2 bzw. Grad 3 trat bei 0,6 % (16/2626) bzw. 0,5 % (13/2626) der Patienten auf. Nebenniereninsuffizienz (einschließlich sekundärer Nebennierenrindeninsuffizienz, akuter Nebennierenrindeninsuffizienz, Verminderung des Corticotropinspiegels und immunvermittelter Nebenniereninsuffizienz) von Grad 2, Grad 3 bzw. Grad 4 trat bei 2,7 % (72/2626) bzw. 1,6 % (43/2626) bzw. 0,2 % (4/2626) der Patienten auf. Diabetes mellitus (einschließlich Typ‑1‑Diabetes mellitus und diabetischer Ketoazidose) vom Schweregrad 1, 2, 3 bzw. 4 trat bei < 0,1 % (1/2626), 0,3 % (8/2626), 0,3 % (7/2626) bzw. 0,2 % (6/2626) der Patienten auf. Die mediane Zeit bis zum Auftreten dieser Endokrinopathien betrug 2,1 Monate (Spanne: 0,0 ‑ 28,1). Bei 297 Patienten (40,0 %) kam es zu einer Rückbildung. Die Zeit bis zum Auftreten der Rückbildung betrug 0,3 bis 257,1+ Wochen.
Infusionsreaktionen
Bei Patienten, die mit Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Hypersensibilität/Infusionsreaktionen 4,5 % (118/2626). Fälle mit Grad 1, Grad 2, Grad 3 bzw. Grad 4 wurden bei 1,9 % (49/2626), 2,4 % (62/2626), 0,2 % (6/2626) bzw. < 0,1 % (1/2626) der Patienten berichtet. Bei Patienten mit MPM, die mit Ipilimumab 1 mg/kg in Kombination mit Nivolumab 3 mg/kg behandelt wurden, war die Häufigkeit von Hypersensibilität/Infusionsreaktionen 12 %.
Immunogenität
Weniger als 2 % der Patienten mit fortgeschrittenem Melanom, die in klinischen Studien der Phasen II und III Ipilimumab erhielten, entwickelten Antikörper gegen Ipilimumab. Keiner zeigte infusionsbedingte Überempfindlichkeiten, Reaktionen an der Applikationsstelle oder anaphylaktische Reaktionen. Es wurden keine neutralisierenden Antikörper gegen Ipilimumab entdeckt. Insgesamt wurde kein offensichtlicher Zusammenhang zwischen Antikörperentwicklung und Nebenwirkungen beobachtet.
Von denjenigen Patienten, die mit Ipilimumab in Kombination mit Nivolumab behandelt wurden und bei welchen das Vorhandensein von Anti‑Ipilimumab‑Antikörpern auswertbar war, lag die Inzidenz von Anti‑Ipilimumab‑Antikörpern zwischen 6,3 und 13,7 %. Neutralisierende Antikörper gegen Ipilimumab lagen im Bereich zwischen 0 und 0,4 %. Von den Patienten, die mit Ipilimumab in Kombination mit Nivolumab und Chemotherapie behandelt wurden und deren Daten hinsichtlich des Auftretens von gegen Ipilimumab gerichteten Antikörpern oder neutralisierenden Antikörpern gegen Ipilimumab auswertbar waren, betrug die Inzidenz von gegen Ipilimumab gerichteten Antikörpern 7,5 %, und die Inzidenz von neutralisierenden Antikörpern gegen Ipilimumab 1,6 %. Bei Patienten, die hinsichtlich des Vorhandenseins von Anti‑Nivolumab‑Antikörpern auswertbar waren, betrug die Inzidenz von Anti‑Nivolumab‑Antikörpern 26 % bei Nivolumab 3 mg/kg und Ipilimumab 1 mg/kg alle 3 Wochen, 24,9 % bei Nivolumab 3 mg/kg alle 2 Wochen und Ipilimumab 1 mg/kg alle 6 Wochen, 37,8 % bei Nivolumab 1 mg/kg und Ipilimumab 3 mg/kg alle 3 Wochen, und 33,8 % bei Nivolumab 360 mg alle 3 Wochen in Kombination mit Ipilimumab 1 mg/kg alle 6 Wochen und Chemotherapie. Die Inzidenz von neutralisierenden Antikörpern gegen Nivolumab betrug 0,8 % bei Nivolumab 3 mg/kg und Ipilimumab 1 mg/kg alle 3 Wochen, 1,5 % bei Nivolumab 3 mg/kg alle 2 Wochen und Ipilimumab 1 mg/kg alle 6 Wochen, 4,6 % bei Nivolumab 1 mg/kg und Ipilimumab 3 mg/kg alle 3 Wochen, und 2,6 % bei Nivolumab 360 mg alle 3 Wochen in Kombination mit Ipilimumab 1 mg/kg alle 6 Wochen und Chemotherapie.
Bei Verabreichung in Kombination mit Nivolumab war die CL von Ipilimumab in Gegenwart von Anti‑Ipilimumab‑Antikörpern unverändert und es gab keinen Hinweis auf ein verändertes Toxizitätsprofil.
Laborwertanomalien
Der Anteil der Patienten, bei denen es unter der Therapie mit Ipilimumab in Kombination mit Nivolumab (mit oder ohne Chemotherapie) zu einer Verschlechterung der Laborwerte gegenüber dem Ausgangswert auf Grad 3 oder 4 kam, war 4,8 % für Anämie, 1,8 % für Thrombozytopenie, 2,2 % für Leukopenie, 6,9 % für Lymphozytopenie, 3,3 % für Neutropenie, 2,7 % für Anstieg der alkalischen Phosphatase, 9,8 % für AST‑Anstieg, 9,3 % für ALT‑Anstieg, 2,3 % für Anstieg des Gesamtbilirubins, 1,8 % für Kreatininanstieg, 1,4 % für Hypoalbuminämie, 7,1 % für Hyperglykämie, 0,7 % für Hypoglykämie, 7,8 % für Amylase‑Anstieg, 16,3 % für Lipase‑Anstieg, 0,8 % für Hypokalziämie, 0,2 % für Hypernatriämie, 0,8 % für Hyperkalziämie, 2,0 % für Hyperkaliämie, 0,8 % für Hypermagnesiämie, 0,4 % für Hypomagnesiämie, 3,0 % für Hypokaliämie und 8,7 % für Hyponatriämie.
Bei Patienten, die mit Ipilimumab 3 mg/kg in Kombination mit Nivolumab 1 mg/kg für Melanom behandelt wurden, kam es bei einem höheren Anteil von Patienten zu einer Verschlechterung der Laborwerte auf Grad 3 oder 4 ALT‑Anstieg (15,3 %) gegenüber dem Ausgangswert.
Kinder und Jugendliche
Ipilimumab als Monotherapie
Es wurden keine neuen Nebenwirkungen bei Jugendlichen ab 12 Jahren berichtet.
In der Studie CA184‑070 wurden keine immunvermittelten Nebenwirkungen (irAR) von ≥ Grad 3 für den einen Patienten im Alter von 12 Jahren oder älter berichtet, der mit Ipilimumab 3 mg/kg behandelt wurde. Bei 2 (25,0 %) der 8 Patienten, die mit 5 mg/kg behandelt wurden und bei 1 (11,1 %) der 9 Patienten, die mit 10 mg/kg behandelt wurden, wurden Grad 3–4 Ereignisse berichtet. Keines der Ereignisse verlief tödlich. Die Art der immunvermittelten Nebenwirkungen (irARs) waren konsistent mit denen, die bei Erwachsenen beobachtet wurden. Am häufigsten wurden Ereignisse von irARs über alle Gruppen verteilt in den Kategorien gastrointestinal (0 [3 mg/kg], 62,5 % [5 mg/kg] und 44,4 % [10 mg/kg]), Leberfunktion (0 [3 mg/kg], 75,0 % [5 mg/kg], 33,3 % [10 mg/kg]) und Haut (0 [3 mg/kg], 25,0 % [5 mg/kg], 33,3 % [10 mg/kg]) berichtet. Es wurden keine neuen oder unerwarteten irARs in dieser Studie beobachtet. Es gab offenkundig keine Unterschiede im Spektrum der irARs bei Erwachsenen und Kindern und Jugendlichen.
In der Studie CA184‑178 wurden keine neuen oder unerwarteten irARs beobachtet und die berichteten irARs waren in der Häufigkeit, Intensität und dem betroffenen Organ dem ähnlich, was in Studien mit erwachsenen Patienten berichtet wurde. 2 Patienten in der 10 mg/kg Gruppe erlitten Hyperglykämie, eine endokrine irAR, von Grad 1 und Grad 3, während der Studie. Weitere endokrine Abweichungen wurden nicht berichtet.
Eine Zusammenfassung der Nebenwirkungen bei Jugendlichen ab 12 Jahren sowie bei erwachsenen Patienten ist in Tabelle 9 dargestellt.
Tabelle 9: Zusammenfassung der Nebenwirkungen nach bis zu 4 Dosen von 3, 5 und 10 mg/kg, alle behandelten Patienten
Anzahl der Patienten (%) |
|||||||
Alter ≥ 12 bis 21 Jahre |
Alter 12 bis < 18 Jahre |
Erwachsene |
|||||
Fortgeschrittenes Melanom und Nicht‑Melanom solide Tumore |
Fortgeschrittenes Melanom |
Fortgeschrittenes Melanom |
|||||
CA184‑070 |
CA184‑178 |
CA184‑004/ |
CA184‑004/ |
||||
3 mg/kg |
5 mg/kg |
10 mg/kg |
3 mg/kg |
10 mg/kg |
3 mg/kg |
10 mg/kg |
|
Alle Todesfälle, n (%) |
1 (100,0) |
4 (50,0) |
2 (22,2) |
2 (50,0) |
3 (37,5) |
26 (23,4) |
71 (21,8) |
Behandlungsbezogene |
0 |
0 |
0 |
0 |
0 |
2 (1,8) |
6 (1,8) |
Schwere Nebenwirkungen |
1 (100,0) |
7 (87,5) |
4 (44,4) |
1 (25,0) |
6 (75,0) |
50 (45,0) |
168 (51,7) |
Schwere Arzneimittelbezogene |
1 (100,0) |
5 (62,5) |
4 (44,4) |
1 (25,0) |
5 (62,5) |
19 (17,1) |
95 (29,2) |
Nebenwirkungen, |
0 |
3 (37,5) |
2 (22,2) |
1 (25,0) |
5 (62,5) |
12 (10,8) |
88 (27,1) |
Arzneimittelbezogene |
0 |
3 (37,5) |
2 (22,2) |
1 (25,0) |
5 (62,5) |
9 (8,1) |
61 (18,8) |
Immunvermittelte |
1 (100,0) |
7 (87,5) |
7 (77,8) |
2 (50,0) |
4 (50,0) |
68 (61,3) |
234 (72,0) |
Nebenwirkung, n (%) |
1 (100,0) |
8 (100,0) |
9 (100,0) |
4 (100,0) |
8 (100,0) |
108 (97,3) |
315 (96,9) |
Arzneimittelbezogene |
1 (100,0) |
7 (87,5) |
9 (100,0) |
2 (50,0) |
7 (87,5) |
88 (79,3) |
274 (84,3) |
MedDRA v.17.0 für CA184070, v.19.0 für CA184178 und V.12.1 für den Sicherheitspool für Erwachsene (Adult Safety Pool). NA = nicht bewertet (not assessed) | |||||||
Ipilimumab in Kombination mit Nivolumab
Die Sicherheit von Ipilimumab (1 mg/kg alle 3 Wochen) in Kombination mit Nivolumab (1 mg/kg oder 3 mg/kg für die ersten 4 Dosen, gefolgt von Nivolumab 3 mg/kg als Monotherapie alle 2 Wochen) wurde bei 33 pädiatrischen Patienten im Alter von ≥ 1 bis < 18 Jahren (einschließlich 20 Patienten im Alter von 12 bis < 18 Jahren) mit rezidivierenden oder refraktären soliden oder hämatologischen Tumoren, einschließlich fortgeschrittenen Melanomen, in der klinischen Studie CA209070 untersucht. Das Sicherheitsprofil bei pädiatrischen Patienten war allgemein dem Sicherheitsprofil ähnlich, das bei Erwachsenen beobachtet wurde, die mit Ipilimumab in Kombination mit Nivolumab behandelt wurden. Es wurden keine neuen Sicherheitssignale beobachtet.
Die häufigsten Nebenwirkungen (berichtet bei mindestens 20 % der pädiatrischen Patienten) bei Ipilimumab in Kombination mit Nivolumab waren Ermüdung/Fatigue (33,3 %) und makulopapulöser Ausschlag (21,2 %). Die Mehrzahl der für Ipilimumab in Kombination mit Nivolumab berichteten Nebenwirkungen war vom Schweregrad 1 oder 2. Bei zehn Patienten (30 %) trat(en) eine oder mehrere Nebenwirkung(en) vom Grad 3 bis 4 auf.
In der klinischen Studie CA209908 mit 74 pädiatrischen Patienten mit hochgradig malignen primären Tumoren des Zentralnervensystems (ZNS) (siehe Abschnitt 5.1) wurden keine neuen Sicherheitssignale im Vergleich zu den Daten aus indikationsübergreifenden Studien mit Erwachsenen beobachtet.
Ältere Menschen
Bei Patienten ab 75 Jahren mit MPM, die Ipilimumab in Kombination mit Nivolumab erhalten hatten, trat eine höhere Rate an schwerwiegenden Nebenwirkungen und an Therapieabbrüchen aufgrund von Nebenwirkungen auf (68 % bzw. 35 %) im Vergleich zu allen Patienten (54 % bzw. 28 %). Daten von dMMR‑ oder MSI‑H‑CRC‑Patienten, die 75 Jahre oder älter waren, sind limitiert (siehe Abschnitt 5.1). Bei Patienten ab 75 Jahren mit HCC traten höhere Raten an schwerwiegenden Nebenwirkungen und an Therapieabbrüchen aufgrund von Nebenwirkungen auf (67 % bzw. 35 %) im Vergleich zu allen Patienten, die Ipilimumab mit Nivolumab erhalten hatten, (53 % bzw. 27 %).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen‑Risiko‑Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung anzuzeigen am:
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51-59
63225 Langen
Tel: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
Die maximal verträgliche Dosis von Ipilimumab wurde nicht bestimmt. In klinischen Studien erhielten Patienten bis zu 20 mg/kg ohne erkennbare toxische Wirkung.
Bei Überdosierung müssen die Patienten sorgfältig auf Anzeichen oder Symptome von Nebenwirkungen beobachtet und es muss eine adäquate symptomatische Behandlung eingeleitet werden.
Pharmakotherapeutische Gruppe: Antineoplastische Mittel, monoklonale Antikörper und Antikörper‑Wirkstoff‑Konjugate, Andere monoklonale Antikörper und Antikörper‑Wirkstoff‑Konjugate, ATC‑Code: L01FX04.
Wirkmechanismus
CTLA‑4 (Cytotoxic T‑Lymphocyte Antigen‑4) spielt eine entscheidende Rolle bei der Regulation der T‑Zell‑Aktivität. Ipilimumab ist ein CTLA‑4‑Immun‑Checkpoint‑Inhibitor, der die vom CTLA‑4‑Signalweg induzierten inhibitorischen Signale auf die T‑Zellen blockiert. Dadurch erhöht sich die Anzahl der Tumor‑reaktiven T‑Effektorzellen, welche dann den Tumor direkt angreifen können. Eine CTLA‑4‑Blockade kann auch zu einer Reduzierung der regulatorischen T‑Zellfunktion führen, was wiederum eine Erhöhung der Anti‑Tumor‑Immunantwort bewirken kann. Ipilimumab kann durch selektive Depletion von regulatorischen T‑Zellen in der Tumorumgebung das Verhältnis von intratumoralen T‑Effektorzellen zu regulatorischen T‑Zellen erhöhen, was das Absterben von Tumorzellen begünstigt.
Pharmakodynamische Wirkungen
Bei Patienten mit Melanomen, die Ipilimumab erhielten, stieg die mittlere absolute periphere Lymphozytenzahl (ALC) im peripheren Blut während der gesamten Induktionsphase an. In Phase‑II‑Studien erfolgte dieser Anstieg dosisabhängig. In der Studie MDX010‑20 (siehe Abschnitt 5.1) ließ Ipilimumab 3 mg/kg mit oder ohne gp100 die ALC während der gesamten Induktionsphase ansteigen, während in der Kontrollgruppe mit Patienten, die nur gp100‑Peptid‑Vakzine allein erhielten, keine signifikante Veränderung der ALC zu beobachten war.
Im peripheren Blut der Patienten mit Melanom wurde nach der Behandlung mit Ipilimumab, gemäß dem Wirkungsmechanismus, ein mittlerer Anstieg im Prozentsatz der aktivierten HLA‑DR+, CD4+ und CD8+ T‑Zellen beobachtet. Außerdem wurde nach der Behandlung mit Ipilimumab ein mittlerer Anstieg des Prozentsatzes der zentralen Gedächtnis‑ (CCR7+ CD45RA‑) CD4+ und CD8+ T‑Zellen und ein geringerer, aber dennoch signifikanter mittlerer Anstieg im Prozentsatz der Effektor‑Gedächtnis‑ (CCR7‑ CD45RA‑) CD8+ T‑Zellen beobachtet.
Klinische Wirksamkeit und Sicherheit
Ipilimumab in Kombination mit Nivolumab
Weitere Informationen zur klinischen Wirksamkeit und Sicherheit im Zusammenhang mit den Dosierungsempfehlungen von Nivolumab als Monotherapie nach der Kombinationstherapie mit Ipilimumab, finden Sie in der Fachinformation zu Nivolumab.
Basierend auf einer Modellierung der Dosis/Expositionswirksamkeit und der Sicherheitsbeziehungen gibt es keine klinisch signifikanten Unterschiede in der Wirksamkeit und Sicherheit zwischen einer Nivolumab Dosis von 240 mg alle 2 Wochen oder 3 mg/kg alle 2 Wochen. Basierend auf diesen Zusammenhängen gab es außerdem keine klinisch signifikanten Unterschiede zwischen einer Nivolumab Dosis von 480 mg alle 4 Wochen oder 3 mg/kg alle 2 Wochen beim fortgeschrittenen Melanom und Nierenzellkarzinom.
Klinische Studien mit Ipilimumab als Monotherapie
Melanom
Der Gesamtüberlebensvorteil von Ipilimumab in der empfohlenen Dosis von 3 mg/kg bei vorbehandelten Patienten mit fortgeschrittenem (nicht resezierbarem oder metastasiertem) Melanom wurde in einer Phase‑III‑Studie (MDX010‑20) gezeigt. Patienten mit okulärem Melanom, primärem ZNS‑Melanom, aktiven Gehirnmetastasen, humanem Immundefizienzvirus (HIV), Hepatitis B und Hepatitis C waren nicht in die klinische Studie MDX010‑20 eingeschlossen. Von klinischen Studien ausgeschlossen waren auch Patienten mit einem ECOG Performance‑Status > 1 und Mukosamelanom. Ausgeschlossen aus den klinischen Studien waren auch Patienten ohne Lebermetastasen, die eine Ausgangs‑AST > 2,5 x ULN aufwiesen, Patienten mit Lebermetastasen, die eine Ausgangs‑AST > 5 x ULN aufwiesen und Patienten mit einem Gesamtbilirubin ≥ 3 x ULN als Ausgangswert.
Patienten mit Autoimmunerkrankungen in der Vorgeschichte: siehe auch Abschnitt 4.4.
MDX010‑20
Für diese doppelblinde Phase‑III‑Studie wurden Patienten mit fortgeschrittenem (nicht resezierbarem oder metastasiertem) Melanom rekrutiert, die zuvor bereits nach Schemata behandelt worden waren, die einen oder mehrere der folgenden Wirkstoffe enthielten: IL‑2, Dacarbazin, Temozolomid, Fotemustin oder Carboplatin. Die Patienten wurden im Verhältnis 3:1:1 randomisiert und mit Ipilimumab 3 mg/kg in Kombination mit einer experimentellen gp100‑Peptid‑Vakzine (gp100), mit Ipilimumab 3 mg/kg als Monotherapie oder mit gp100 allein behandelt. Alle Patienten in dieser Studie waren vom Typ HLA‑A2*0201; dieser HLA‑Typ unterstützt die Immunpräsentation von gp100. Patienten wurden unabhängig von ihrem B‑RAF‑Mutationsstatus zu Therapiebeginn eingeschlossen. Die Patienten erhielten Ipilimumab je nach Verträglichkeit alle 3 Wochen für 4 Dosen (Induktionstherapie). Patienten mit erkennbarem Anstieg der Tumorlast vor Abschluss der Induktionsphase blieben bei gegebener Verträglichkeit weiter in der Induktionstherapie, sofern ihr Performance‑Status gut war. Die Untersuchung des Tumoransprechens auf Ipilimumab wurde ungefähr in Woche 12, nach Abschluss der Induktionstherapie, durchgeführt.
Eine weitere zusätzliche Behandlung mit Ipilimumab (erneute Behandlung) wurde Patienten angeboten, bei denen es nach anfänglichem klinischem Ansprechen (partial response, PR oder complete response, CR) oder nach einer Stabilisierung der Erkrankung (stable disease, SD, gemäß modifizierten WHO‑Kriterien) zu einem Fortschreiten der Erkrankung mehr als 3 Monate nach der ersten Untersuchung des Tumors kam. Der primäre Endpunkt war das Gesamtüberleben (Overall Survival, OS) in der Ipilimumab+ gp100‑Gruppe im Vergleich zur gp100‑Gruppe. Wichtige sekundäre Endpunkte waren OS in der Ipilimumab+ gp100‑Gruppe im Vergleich zur Ipilimumab Monotherapie‑Gruppe und in der Ipilimumab Monotherapie‑Gruppe im Vergleich zur gp100‑Gruppe.
Insgesamt wurden 676 Patienten randomisiert: 137 für die Ipilimumab‑Monotherapie, 403 für die Behandlung mit Ipilimumab + gp100 und 136 für die Behandlung mit gp100 allein. Die Mehrzahl der Patienten erhielt während der Induktionsphase alle 4 Dosen. 32 Patienten wurden erneut behandelt: 8 Patienten in der Ipilimumab‑Monotherapie‑Gruppe, 23 Patienten in der Ipilimumab + gp100‑Gruppe und 1 Patient in der gp100‑Gruppe. Die Beobachtungszeit betrug bis zu 55 Monate. Die klinischen Ausgangsmerkmale waren in allen Behandlungsgruppen gut ausgewogen. Das Medianalter lag bei 57 Jahren. Die Mehrzahl (71‑73 %) der Patienten befand sich im Krankheitsstadium M1c, und 37‑40 % der Patienten wiesen zu Beginn einen erhöhten Laktatdehydrogenase (LDH)‑Wert auf. Insgesamt waren 77 Patienten früher bereits wegen Gehirnmetastasen behandelt worden.
Die Ipilimumab‑Studiengruppen zeigten gegenüber der gp100‑Kontrollgruppe in Bezug auf das Gesamtüberleben einen statistisch signifikanten Vorteil. Die Hazard Ratio (HR) für den Vergleich des Gesamtüberlebens zwischen der Ipilimumab Monotherapie‑Gruppe und der gp100‑Gruppe lag bei 0,66 (95 % CI: 0,51‑0,87; p = 0,0026).
In einer Subgruppenanalyse war der Gesamtüberlebensvorteil in den meisten Subgruppen (Status der Metastasierung, Interleukin‑2‑Vortherapie, LDH zu Therapiebeginn, Alter, Geschlecht und Art und Häufigkeit der Vorbehandlung) konsistent. Jedoch sind die Daten für Frauen über 50 Jahre, die einen Vorteil von Ipilimumab für das Gesamtüberleben zeigen, limitiert. Da Subgruppenanalysen immer nur eine sehr kleine Anzahl von Patienten einschließen, lassen sich keine endgültigen Schlüsse aus diesen Daten ziehen.
Die medianen und geschätzten Überlebensraten nach 1 und 2 Jahren sind in Tabelle 10 aufgeführt.
Tabelle 10: Gesamtüberleben in MDX010‑20
Ipilimumab 3 mg/kg |
gp100a |
|
Median Monate (95 % CI) |
10 Monate |
6 Monate |
Überlebensrate nach 1 Jahr % (95 % CI) |
46 % (37,0‑54,1) |
25 % (18,1‑32,9) |
Überlebensrate nach 2 Jahren % (95 % CI) |
24 % (16,0‑31,5) |
14 % (8,0‑20,0) |
a gp100‑Peptid‑Vakzine ist eine experimentelle Kontrolle. | ||
In der Ipilimumab 3 mg/kg Monotherapie‑Gruppe lag das Gesamtüberleben für Patienten mit SD (stable disease) bzw. PD (progressive disease) im Median bei 22 Monaten bzw. 8 Monaten. Zum Zeitpunkt der Analyse waren die Mediane für die Patienten mit CR (complete response) bzw. PR (partial response) noch nicht erreicht.
Bei Patienten mit erneuter Behandlung lag die Best Overall Response Rate (BORR) bei 38 % (3/8 Patienten) in der Ipilimumab Monotherapie‑Gruppe und 0 % in der gp100‑Gruppe. Die Disease Control Rate (DCR, definiert als CR + PR + SD) lag bei 75 % (6/8 Patienten) und 0 %. Da diese Analysen auf der Basis einer sehr kleinen Patientenzahl gemacht wurden, lassen sich hieraus keine endgültigen Schlüsse für die Wirksamkeit einer erneuten Behandlung mit Ipilimumab ziehen.
Das Einsetzen oder die Aufrechterhaltung der klinischen Aktivität nach Behandlung mit Ipilimumab war mit oder ohne Einsatz systemischer Corticosteroide ähnlich.
CA184‑169
Eine doppelblinde Phase‑III‑Studie, in welcher Patienten mit vorbehandeltem oder nicht‑vorbehandeltem Stadium‑III‑ oder Stadium‑IV‑Melanom eingeschlossen waren. Insgesamt wurden 727 Patienten randomisiert, 362 um Ipilimumab 3 mg/kg zu erhalten und 365 um Ipilimumab 10 mg/kg alle 3 Wochen für 4 Dosen zu erhalten. In der Ipilimumab 10 mg/kg Gruppe betrug das mediane OS (95 % CI) 16 Monate (11,63; 17,84) und in der Ipilimumab 3 mg/kg Gruppe betrug das mediane OS (95 % CI) 12 Monate (9,86; 13,27). Das Gesamtüberleben verglichen zwischen der Ipilimumab 10 mg/kg und 3 mg/kg Gruppe zeigte ein HR = 0,84 (95 % CI: 0,70; 0,99; P‑Wert = 0,04). Es wurde kein statistisch signifikanter Unterschied in Bezug auf das progressionsfreie Überleben (PFS) zwischen der 10 mg/kg und der 3 mg/kg Gruppe gesehen. (HR 0,89 mit einem 95 % CI von 0,76; 1,04 und Log‑Rank‑Test P‑Wert = 0,1548). BORR war ähnlich in der 10 mg/kg und 3 mg/kg Gruppe. BORR betrug in der 10 mg/kg Gruppe 15,3 % (95 % CI: 11,8; 19,5) und in der 3 mg/kg Gruppe 12,2 % (95 % CI: 9,0; 16,0). Ipilimumab 10 mg/kg war mit höheren Nebenwirkungsraten verbunden als die 3‑mg/kg‑Dosis. Die Häufigkeiten von schwerwiegenden Nebenwirkungen betrugen in der 10 mg/kg Gruppe 37 % und in der 3 mg/kg Gruppe 18 %. Die 3 häufigsten schwerwiegenden Nebenwirkungen waren Diarrhö (10,7 % vs. 5,5 %), Kolitis (8,0 % vs 3,0 %) und Hypophysitis (4,4 % vs 1,9 %). Nebenwirkungen, die zum Therapieabbruch geführt haben, traten mit 31 % in der 10 mg/kg und mit 19 % in der 3 mg/kg Gruppe auf. Nebenwirkungen, die zum Tod führten, traten bei 4 beziehungsweise 2 Patienten auf.
In der empfohlenen Dosierung von 3 mg/kg war das mediane OS in der Subgruppe von Frauen ≥ 50 Jahre ähnlich verglichen mit dem der Gesamtpopulation: (11,40 vs. 11,53 Monate). Das mediane OS in der Subgruppe mit Gehirnmetastasen betrug zu Beginn 5,67 Monate bei der empfohlenen Dosis von 3 mg/kg.
Andere Studien mit Ipilimumab als Monotherapie
Melanom
CA184‑332 und CA184‑338
Das Gesamtüberleben bei Ipilimumab 3 mg/kg Monotherapie bei chemotherapienaiven Patienten aus klinischen Studien der Phasen‑II und III (N = 78; randomisiert), und bei behandlungsnaiven Patienten in zwei retrospektiven Beobachtungsstudien (N = 273 und N = 157) war weitgehend konsistent. 12,1 % und 33,1 % der Patienten, die in die beiden Beobachtungsstudien eingeschlossen wurden, hatten bei der Diagnose des fortgeschrittenen Melanoms Hirnmetastasen. Medianes OS und geschätzte 1‑Jahres‑, 2‑Jahres‑, 3‑Jahres‑ und 4‑Jahres‑Überlebensraten sind in Tabelle 11 dargestellt. Die bezifferten 1‑Jahres‑, 2‑Jahres‑ und 3‑Jahres‑Überlebensraten bei chemotherapienaiven Patienten aus klinischen Studien der Phasen‑II und III waren 54,1 % (95 % CI: 42,5‑65,6), 31,6 % (95 % CI: 20,7‑42,9) und 23,7 % (95 % CI: 14,3‑34,4).
Tabelle 11: Gesamtüberleben in Beobachtungsstudien
CA184‑338 |
CA184‑332 |
|
Medianes OS (95 % CI) |
14 Monate |
10 Monate |
Überlebensrate nach 1 Jahr % (95 % CI) |
59 % (52,5‑64,3) |
44 % (35,5‑51,4) |
Überlebensrate nach 2 Jahren % (95 % CI) |
39 % (33,1‑44,8) |
26 % (18,9‑33,3) |
Überlebensrate nach 3 Jahren % (95 % CI) |
31 % (25,5‑36,7) |
22 % (15,5‑29,2) |
Überlebensrate nach 4 Jahren % (95 % CI) |
26 % (20,4‑31,3) |
22 % (15,5‑29,2) |
Patienten mit Hirnmetastasen in der Studie CA184‑332 hatten ein medianes Gesamtüberleben (OS) von 7 Monaten (95 % CI: 5,06 ‑ 12,81) und Patienten ohne Hirnmetastasen hatten ein medianes OS von 14,1 Monaten (95 % CI: 9,96‑Nicht geschätzt).
Patienten mit Hirnmetastasen in der Studie CA184‑338 hatten ein medianes Gesamtüberleben (OS) von 6,3 Monaten (95 % CI: 3,2 ‑ 12,0) und Patienten ohne Hirnmetastasen hatten ein medianes OS von 17,7 Monaten (95 % CI: 13,6 – 12,1).
Der Langzeitüberlebensvorteil der Behandlung mit Ipilimumab (bei 3 mg/kg) wurde in einer gepoolten Analyse von Gesamtüberlebensdaten aus klinischen Studien bei behandlungsnaiven und vorbehandelten Patienten mit fortgeschrittenem Melanom nachgewiesen (N = 965). Die Kaplan‑Meier‑Kurve des Gesamtüberlebens zeigte ein Plateau ab etwa dem Jahr 3 (OS‑Rate = 21 % [95 % CI: 17‑24]), das sich bei einigen Patienten bis in das Jahr 10 erstreckte (siehe Abbildung 1).
Abbildung 1: Gesamtüberleben unter Ipilimumab 3 mg/kg ‑ gepoolte Analyse
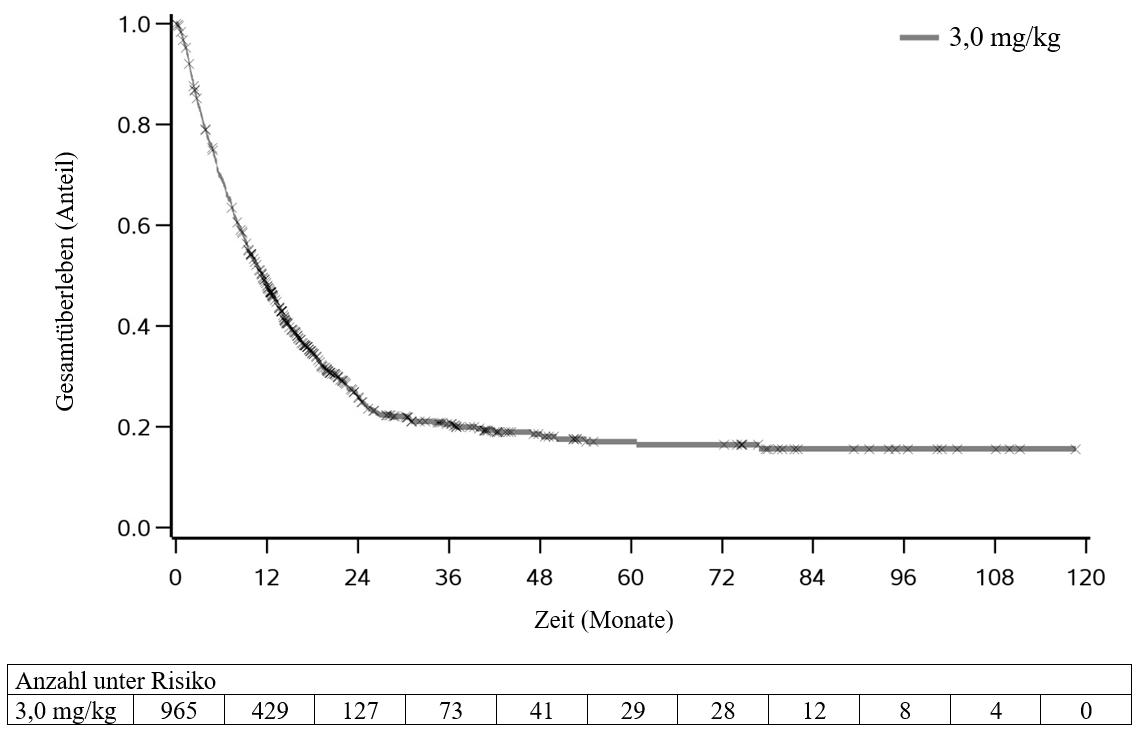
Klinische Studien mit Ipilimumab in Kombination mit Nivolumab
Melanom
Randomisierte Phase‑III‑Studie mit Ipilimumab in Kombination mit Nivolumab oder Nivolumab als Monotherapie im Vergleich gegen Ipilimumab als Monotherapie (CA209‑067)
Die Sicherheit und Wirksamkeit von Ipilimumab 3 mg/kg in Kombination mit Nivolumab 1 mg/kg, Nivolumab 3 mg/kg vs. Ipilimumab‑Monotherapie 3 mg/kg zur Behandlung des fortgeschrittenen (nicht resezierbaren oder metastasierten) Melanoms wurden in einer randomisierten, doppelblinden Phase‑III‑Studie (CA209‑067) untersucht. Die Unterschiede zwischen den beiden Nivolumab‑enthaltenden Gruppen wurden deskriptiv untersucht. In die Studie wurden erwachsene Patienten mit bestätigtem nicht resezierbaren Melanom im Stadium III oder IV eingeschlossen. Die Patienten mussten einen ECOG‑Performance‑Status 0 oder 1 haben. Es wurden Patienten eingeschlossen, die bisher keine systemischen Vortherapien zur Behandlung des nicht resezierbaren oder metastasierten Melanoms erhalten haben. Adjuvante oder neoadjuvante Vortherapie war erlaubt, wenn diese mindestens 6 Wochen vor Einschluss in die Studie abgeschlossen worden war. Patienten mit akuter Autoimmunerkrankung, okulärem/uvealem Melanom, aktiven Hirnmetastasen oder leptomeningealen Metastasen waren von der Studie ausgeschlossen.
Insgesamt wurden 945 Patienten entweder für Ipilimumab in Kombination mit Nivolumab (n = 314), Nivolumab als Monotherapie (n = 316) oder Ipilimumab als Monotherapie (n = 315) randomisiert. Patienten im Kombinationsarm erhielten Nivolumab in einer Dosierung von 1 mg/kg Körpergewicht über 60 Minuten und Ipilimumab in einer Dosierung von 3 mg/kg Körpergewicht über 90 Minuten jeweils als intravenös verabreichte Infusion alle 3 Wochen für die ersten vier Gaben, gefolgt von Nivolumab in einer Dosierung von 3 mg/kg Körpergewicht als Monotherapie alle 2 Wochen. Patienten im Nivolumab‑Monotherapie‑Arm erhielten Nivolumab in einer Dosierung von 3 mg/kg Körpergewicht alle 2 Wochen. Patienten im Vergleichsarm erhielten Ipilimumab in einer Dosierung von 3 mg/kg Körpergewicht und Placebo für Nivolumab als intravenöse Infusion alle 3 Wochen für 4 Gaben und anschließend Placebo alle 2 Wochen. Die Randomisierung wurde mittels PD‑L1‑Expressionsstatus stratifiziert (≥ 5 % gegenüber < 5 % Expression auf der Tumorzellmembran), BRAF‑Status und M‑Stadium gemäß der Einstufung des American Joint Committee on Cancer (AJCC). Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen bestand oder bis die Behandlung nicht mehr vertragen wurde. Tumorbeurteilungen wurden zum ersten Mal 12 Wochen nach Randomisierung und dann im ersten Jahr alle 6 Wochen und anschließend alle 12 Wochen durchgeführt. Die primären Wirksamkeitskriterien waren das progressionsfreie Überleben (progression free survival = PFS) sowie Gesamtüberleben (Overall Survival = OS). Sekundäre Wirksamkeitskriterien waren die objektive Ansprechrate (objective response rate = ORR) und die Dauer des Ansprechens.
Die Ausgangsmerkmale der Gruppen waren ähnlich verteilt. Das mittlere Alter betrug 61 Jahre (Spanne: 18 ‑ 90), 65 % waren Männer und 97 % waren weiß. Der ECOG‑Performance‑Status war 0 bei 73 % der Patienten und 1 bei 27 % der Patienten. 93 % der Patienten hatten einen Krankheitsstatus von IV gemäß AJCC, 58 % hatten einen Krankheitsstatus von M1c bei Studienbeginn. 22% der Patienten hatten eine adjuvante Vortherapie erhalten. 32 % der Patienten hatten ein BRAF‑Mutations‑positives Melanom und 26,5 % der Patienten hatten eine PD‑L1‑Expression in der Tumorzellmembran von ≥ 5 %. 4 % der Patienten hatten Hirnmetastasen in der Vorgeschichte und 36 % der Patienten hatten zu Studienbeginn einen LDH‑Ausgangsspiegel über dem oberen Normbereich (upper limit of normal, ULN). Bei den Patienten mit quantifizierbarer Tumor‑PD‑L1‑Expression war die Verteilung der Patienten zwischen den 3 Behandlungsgruppen ausgeglichen. Die Tumor‑PD‑L1‑Expression wurde mittels PD‑L1‑IHC‑28‑8‑PharmDx‑Assay ermittelt.
In der primären Analyse (minimale Nachbeobachtungszeit 9 Monate) betrug das mediane PFS 6,9 Monate in der Nivolumab‑Gruppe verglichen mit 2,9 Monaten in der Ipilimumab‑Gruppe (HR = 0,57; 99,5 %; CI: 0,43; 0,76; p < 0,0001). Das mediane PFS betrug 11,5 Monate in der Ipilimumab‑in‑Kombination‑mit‑Nivolumab‑Gruppe verglichen mit 2,9 Monaten in der Ipilimumab‑Gruppe (HR = 0,42; 99,5 %; CI: 0,31; 0,57; p < 0,0001).
Die PFS‑Ergebnisse aus einer deskriptiven Analyse (mit einer minimalen Nachbeobachtungszeit von 90 Monaten) sind in Abbildung 2 dargestellt (gesamte randomisierte Population), Abbildung 3 zeigt einen Tumor‑PD‑L1‑5‑%‑Cut‑off und Abbildung 4 einen Tumor‑PD‑L1‑1‑%‑Cut‑off.
Abbildung 2: Progressionsfreies Überleben (CA209067)
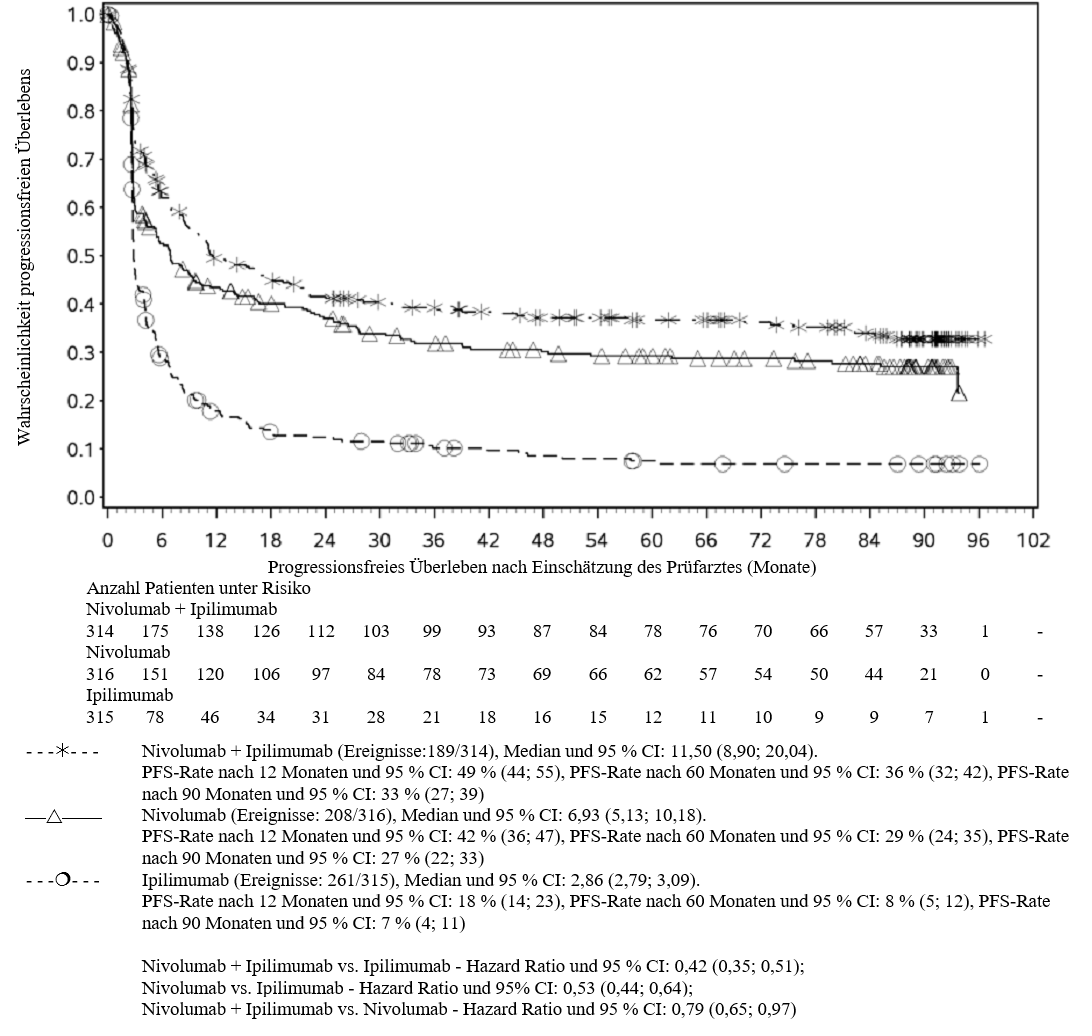
Abbildung 3: Progressionsfreies Überleben bei PD‑L1‑Expression: 5‑%‑Cut‑off (CA209067)
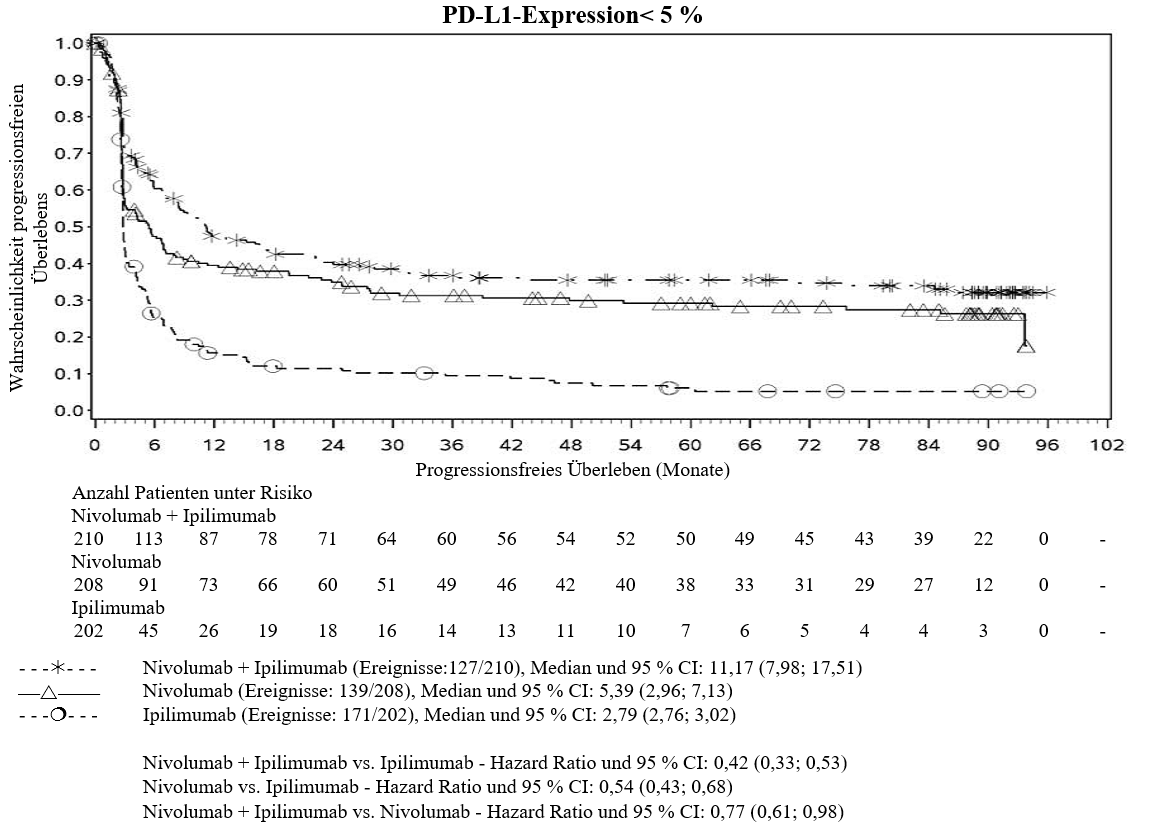
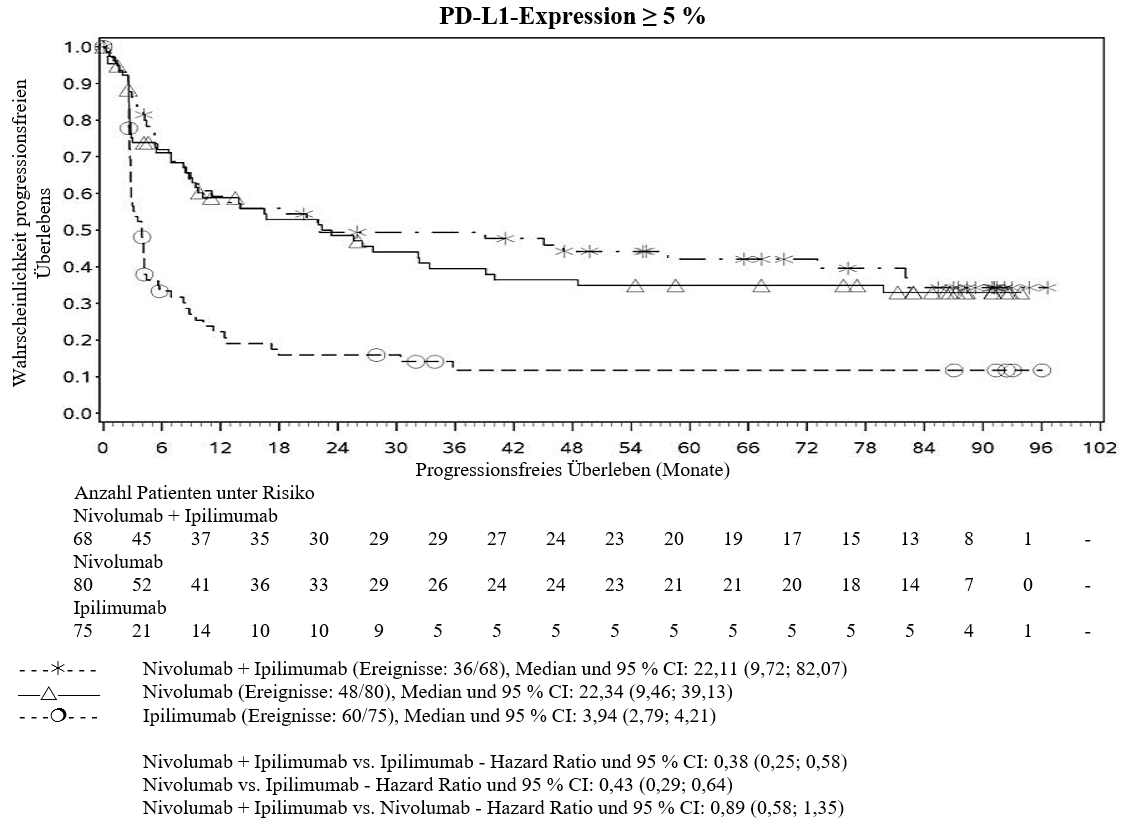
Abbildung 4: Progressionsfreies Überleben bei PD‑L1‑Expression: 1‑%‑Cut‑off (CA209067)
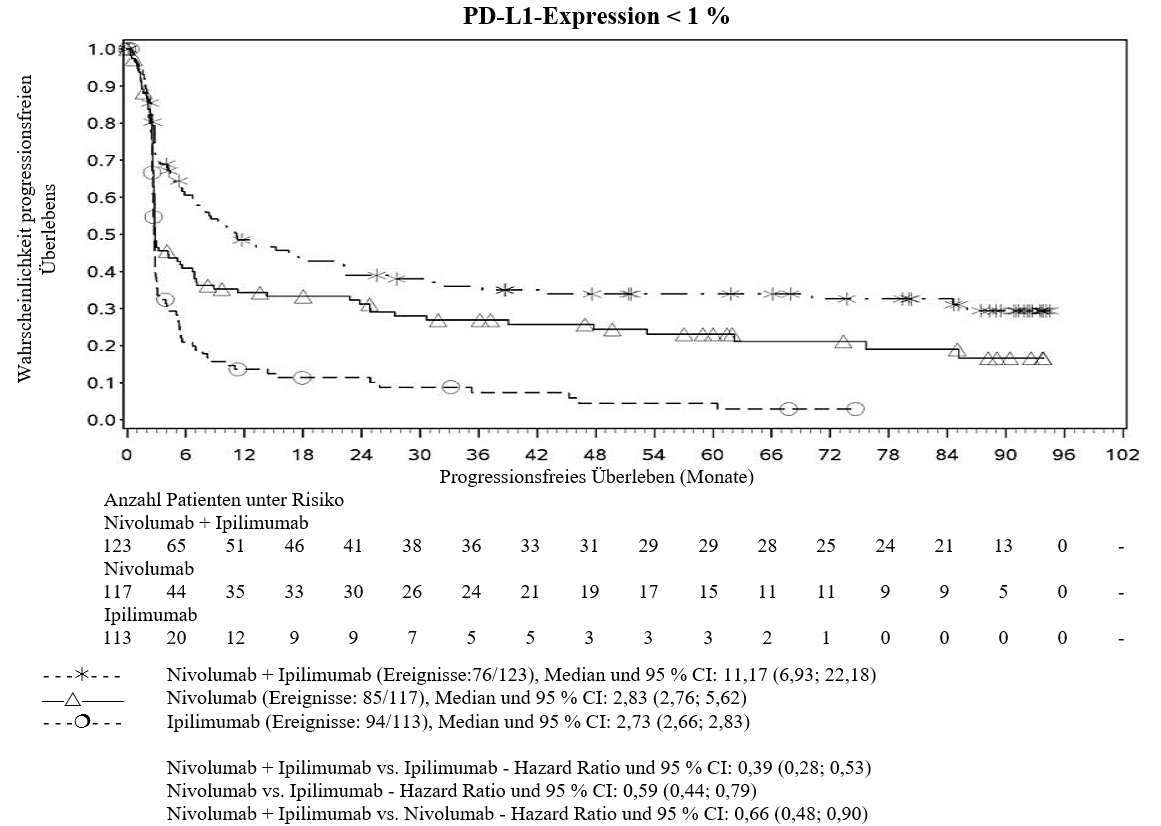
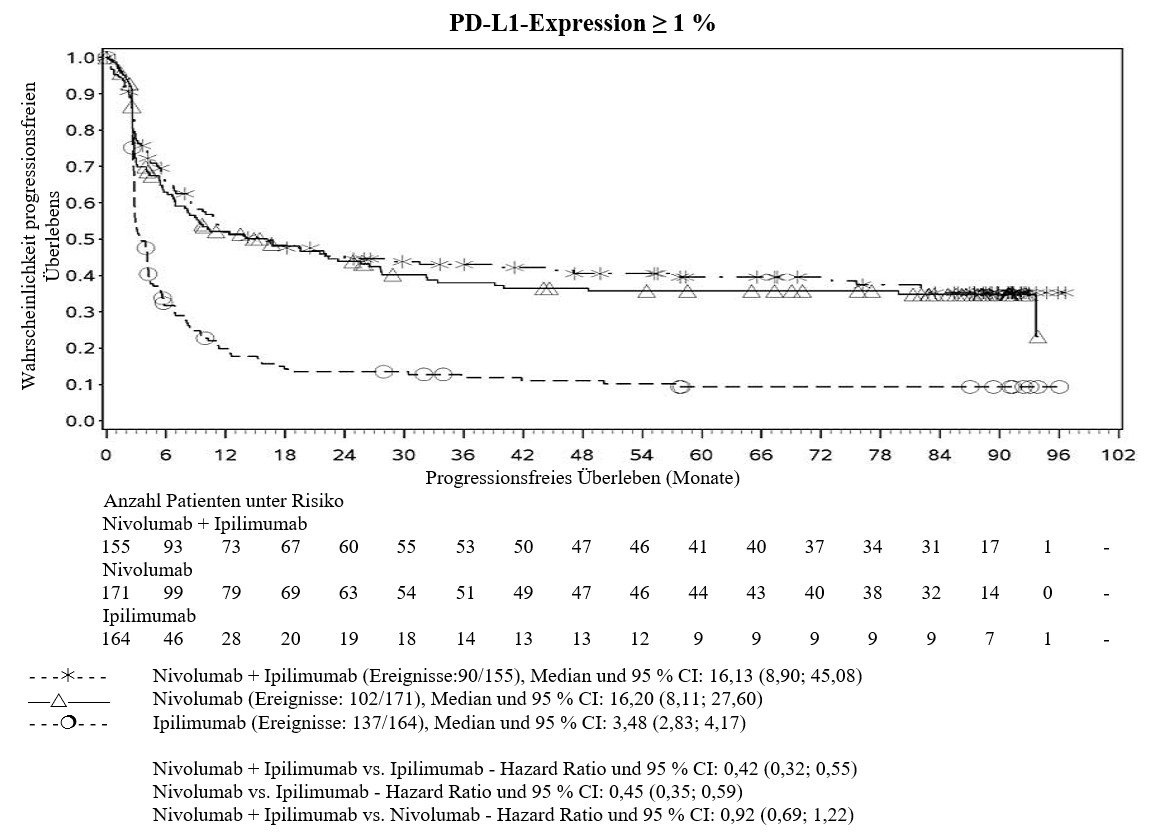
Die finale (primäre) OS‑Analyse wurde durchgeführt, nachdem alle Patienten eine minimale Nachbeobachtungszeit von 28 Monaten erreicht hatten. Nach 28 Monaten wurde das mediane OS in der Nivolumab‑Gruppe nicht erreicht verglichen mit 19,98 Monaten in der Ipilimumab‑Gruppe (HR = 0,63; 98 % CI: 0,48; 0,81; p‑Wert: < 0,0001). Das mediane OS wurde in der Patientengruppe, die mit der Kombination aus Nivolumab und Ipilimumab behandelt wurde, nicht erreicht. Im Vergleich zur Ipilimumab‑Gruppe beträgt die HR = 0,55 (98 %CI: 0,42; 0,72; p‑Wert: < 0,0001).
Die OS‑Ergebnisse einer zusätzlichen deskriptiven Analyse nach einer minimalen Nachbeobachtungszeit von 90 Monaten waren konsistent mit den Ergebnissen der ursprünglichen primären Analyse. Die OS‑Ergebnisse dieser Nachbeobachtungsanalyse sind in Abbildung 5 (alle randomisierten Populationen), Abbildung 6 und 7 (Tumor‑PD‑L1‑5‑%‑und‑1‑%‑Cut‑off) dargestellt.
Die OS‑Analyse war nicht darauf ausgerichtet, die nachfolgend erhaltenen Therapien zu erfassen. Anschließende systemische Therapien erhielten 36,0 % der Patienten, die die Kombination erhalten hatten, 49,1 % der Nivolumab‑Monotherapie‑Patienten und 66,3 % der Ipilimumab‑Patienten. Anschließende Immuntherapien (einschließlich Anti‑PD1‑Therapie, Anti‑CTLA‑4‑Antikörper oder andere Immuntherapien) erhielten 19,1 % der Patienten, die die Kombination erhalten hatten, 34,2 % der Nivolumab‑Monotherapie‑Patienten und 48,3 % der Ipilimumab‑Patienten.
Abbildung 5: Gesamtüberleben (CA209067) - Minimale Nachbeobachtungszeit 90 Monate
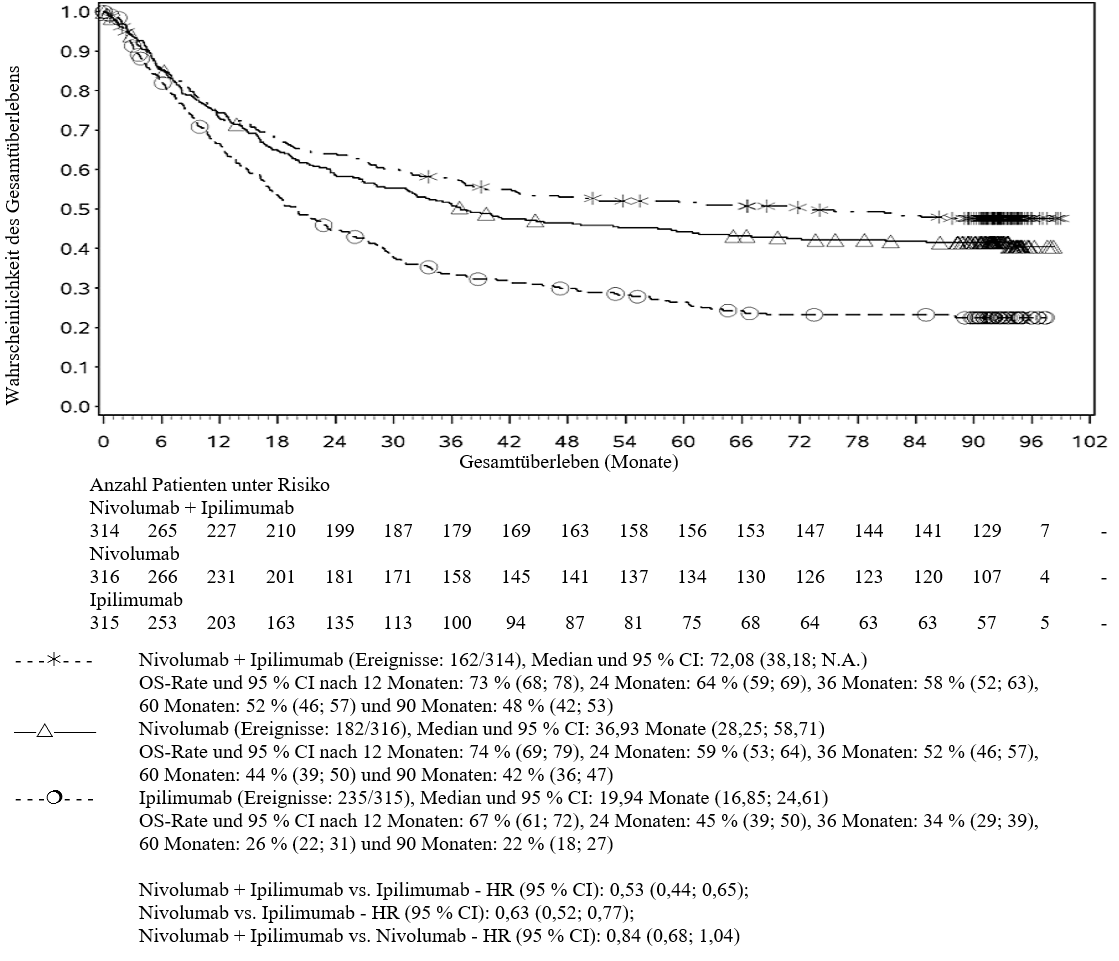
Abbildung 6: Gesamtüberleben nach PD‑L1‑Expression: 5‑%‑Cut‑off (CA209067) ‑ Minimale Nachbeobachtungszeit 90 Monate
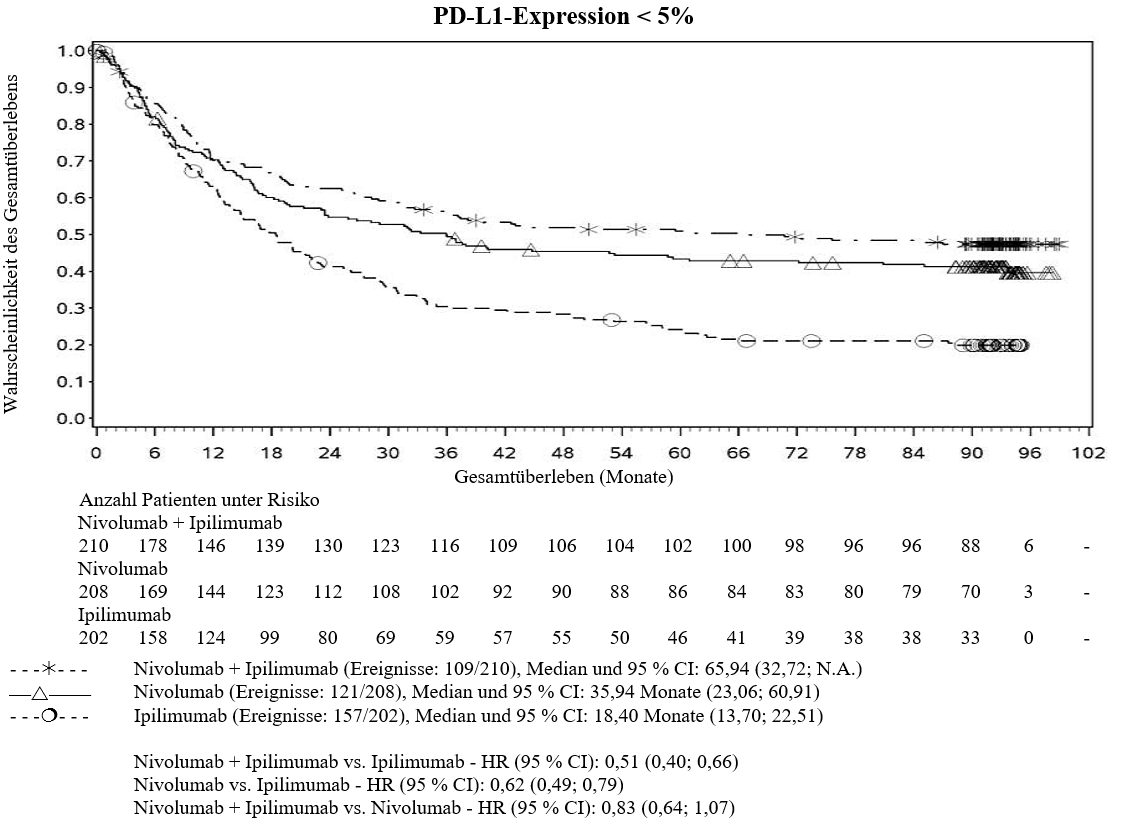
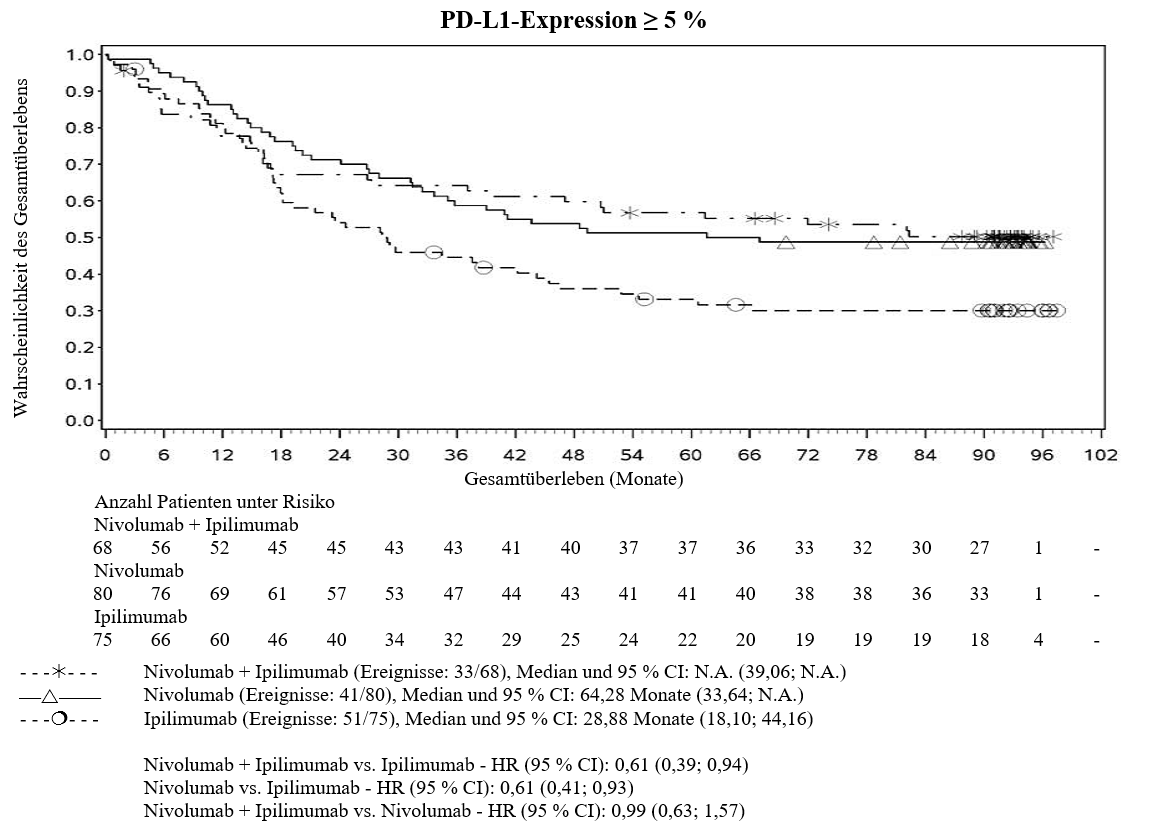
Abbildung 7: Gesamtüberleben nach PD‑L1‑Expression: 1‑%‑Cut‑off (CA209067) ‑ Minimale Nachbeobachtungszeit 90 Monate
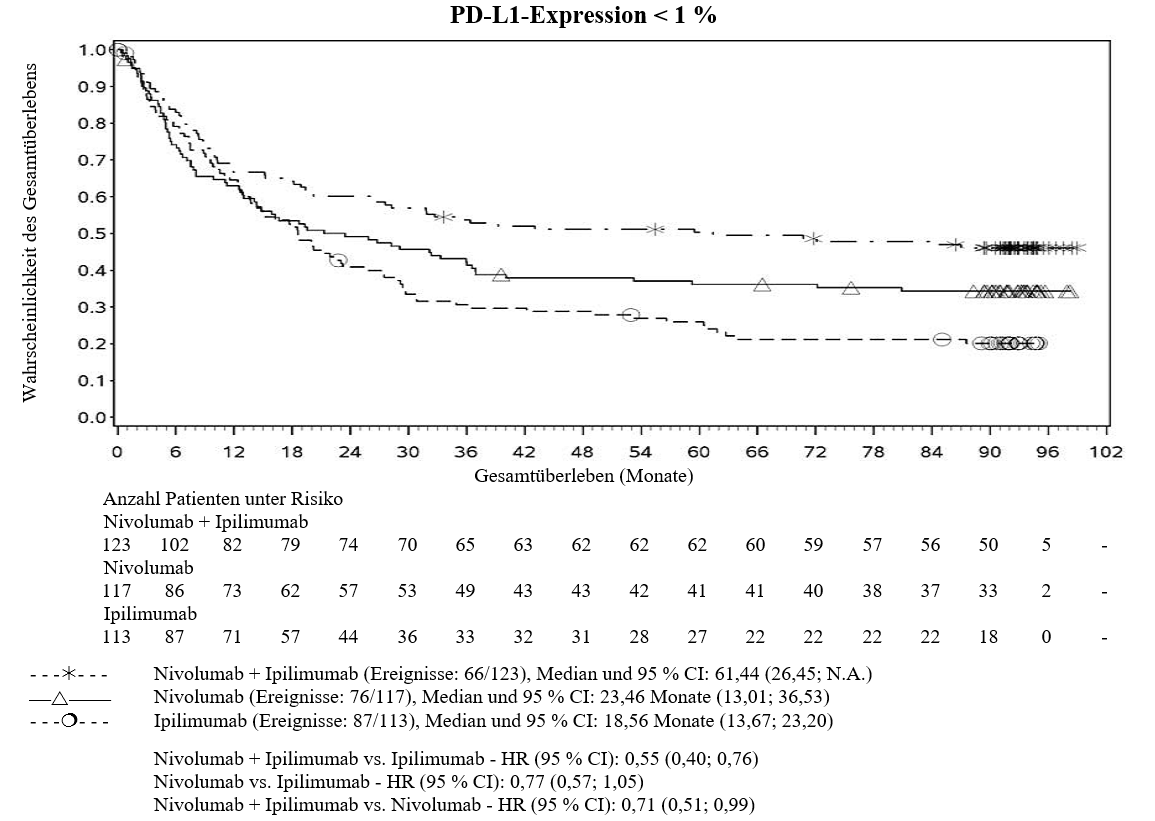
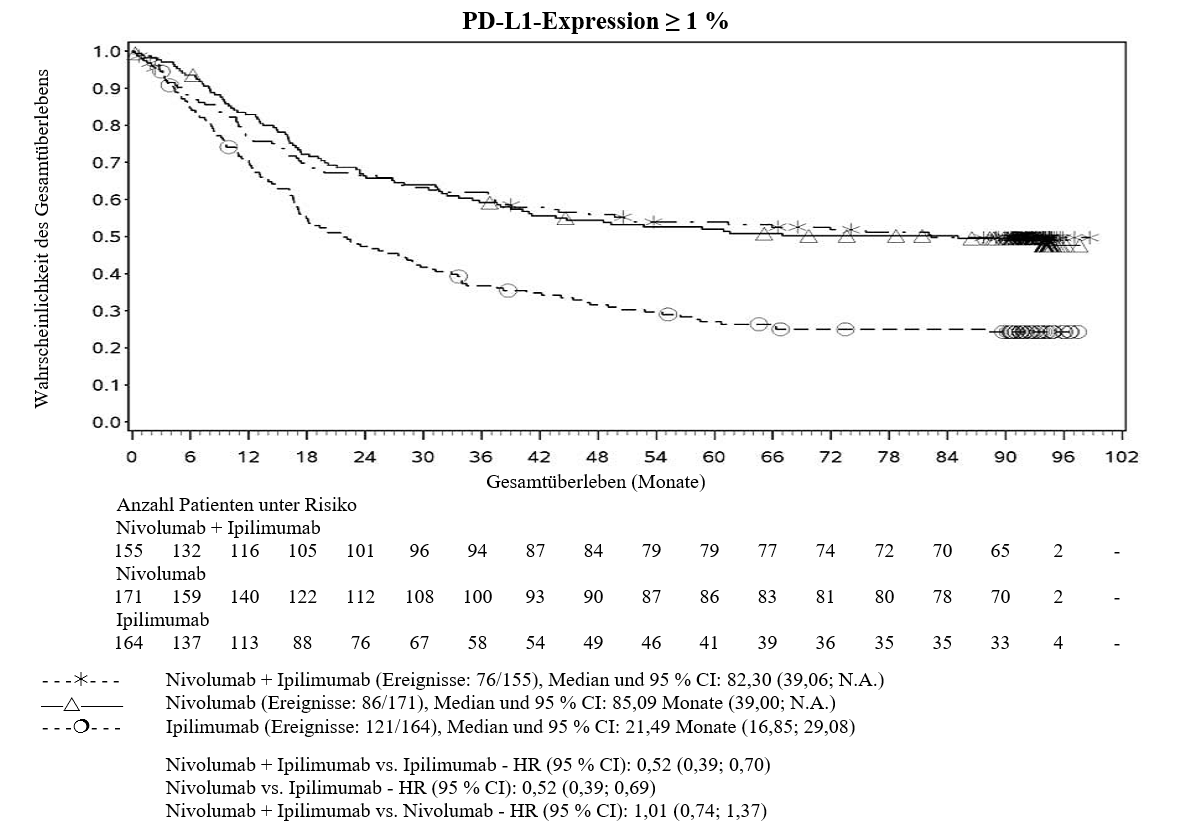
Minimale Nachbeobachtungszeit für die ORR‑Analyse waren 90 Monate. Das Ansprechen ist in Tabelle 12 zusammengefasst.
Tabelle 12: Objektives Ansprechen (CA209067)
Nivolumab + Ipilimumab |
Nivolumab |
Ipilimumab |
|
Objektives Ansprechen |
183 (58 %) |
142 (45 %) |
60 (19 %) |
(95 % CI) |
(52,6; 63,8) |
(39,4; 50,6) |
(14,9; 23,8) |
Odds Ratio (vs. Ipilimumab) |
6,35 |
3,5 |
|
(95 % CI) |
(4,38; 9,22) |
(2,49; 5,16) |
|
Vollständiges Ansprechen |
71 (23 %) |
59 (19 %) |
19 (6 %) |
Teilweises Ansprechen |
112 (36 %) |
83 (26 %) |
41 (13 %) |
Stabile Erkrankung (Stable Disease = SD) |
38 (12 %) |
29 (9 %) |
69 (22 %) |
Ansprechdauer |
|||
Median (Zeitspanne), Monate |
N.A. |
90,8 |
19,3 |
Anteil ≥ 12 Monate Ansprechdauer |
68 % |
73 % |
44 % |
Anteil ≥ 24 Monate Ansprechdauer |
58 % |
63 % |
30 % |
ORR (95 % CI) bei Tumor‑PD‑L1‑Expression | |||
< 5 % |
56 % (48,7; 62,5) |
43 % (36; 49,8) |
18 % (12,8; 23,8) |
≥ 5 % |
72 % (59,9; 82,3) |
59 % (47,2; 69,6) |
21 % (12,7; 32,3) |
< 1 % |
54 % (44,4; 62,7) |
36 % (27,2; 45,3) |
18 % (11,2; 26,0) |
≥ 1 % |
65 % (56,4; 72) |
55 % (47,2; 62,6) |
20 % (13,7; 26,4) |
Patienten in beiden Nivolumab‑enthaltenden‑Armen zeigten einen signifikanten Nutzen bzgl. PFS und OS und ein größeres ORR verglichen mit Ipilimumab Monotherapie. Die Resultate bezüglich des progressionsfreien Überlebens nach 18 Monaten, Nachbeobachtung und ORR‑ und OS‑Ergebnisse nach 28 Monaten Nachbeobachtungszeit waren in den verschiedenen Patienten‑Subgruppen konstant, so bei Patienten mit unterschiedlichem ECOG‑Status, BRAF‑Mutationsstatus, M‑Stadium, Alter, Hirnmetastasen in der Anamnese und LDH‑Ausgangsspiegel. Diese Beobachtungen wurden auch mit den OS‑Ergebnissen nach einer minimalen Nachbeobachtungszeit von 90 Monaten beibehalten.
Bei den 131 Patienten, die die Kombinationstherapie aufgrund von Nebenwirkungen nach 28 Monaten Nachbeobachtungszeit abgebrochen haben, war die Ansprechrate 71 % (93/131). Von diesen 71 % erreichten 20 % (26/131) der Patienten ein vollständiges Ansprechen. Das mediane Gesamtüberleben war noch nicht erreicht.
Patienten in beiden Nivolumab‑enthaltenden‑Armen hatten ein größeres objektives Ansprechen als Patienten im Ipilimumab‑Arm, unabhängig vom PD‑L1‑Expressionsstatus. Nach 90 Monaten Nachbeobachtungszeit war das objektive Ansprechen für die Kombination aus Nivolumab und Ipilimumab über alle Tumor‑PD‑L1‑Expressionsgruppen größer als für die Nivolumab‑Monotherapie (siehe Tabelle 12), wobei die beste Gesamtansprechrate für das vollständige Ansprechen mit einer verbesserten Überlebensrate korrelierte.
Nach 90 Monaten Nachbeobachtungszeit betrug die mediane Ansprechdauer bei Patienten mit Tumor‑PD‑L1‑Expressionsstatus ≥ 5 % 78,19 Monate im Kombinations‑Arm (Spanne: 18,07 ‑ N.A.) und 77,21 Monate im Nivolumab‑Monotherapie‑Arm (Spanne: 26,25 ‑ N.A.). Sie betrug 31,28 Monate (Spanne: 6,08 ‑ N.A.) im Ipilimumab‑Arm. Bei einer Tumor‑PD‑L1‑Expression < 5 % wurde die mediane Ansprechdauer im Kombinations‑Arm (Spanne: 61,93 ‑ N.A.) nicht erreicht und betrug 90,84 Monate im Nivolumab‑Monotherapie‑Arm (Spanne: 50,43 ‑ N.A.). Sie betrug 19,25 Monate im Ipilimumab‑Monotherapie‑Arm (Spanne: 5,32 ‑ 47,44).
Bezüglich der relevanten Endpunkte Tumoransprechen, PFS und OS konnte kein klarer Grenzwert zur PD‑L1‑Expression verlässlich definiert werden. Ergebnisse von exploratorischen multivarianten Analysen zeigten, dass auch andere Patienten‑ und Tumorcharakteristika (z. B. ECOG‑Status, M‑Stadium, Ausgangs‑LDH, BRAF‑Mutationsstatus, PD‑L1‑Status und Geschlecht) zum Überlebensresultat beitragen könnten.
Wirksamkeit bei BRAF-Status:
BRAF[V600]‑Mutation‑positive und BRAF‑Wildtyp‑Patienten, welche zu Ipilimumab in Kombination mit Nivolumab randomisiert wurden, hatten nach 90 Monaten Nachbeobachtungszeit ein medianes PFS von 16,76 Monaten (95 % CI: 8,28; 32,0) bzw. 11,17 Monaten (95 % CI: 7,0; 19,32) während die in den Nivolumab‑Monotherapie‑Arm randomisierten Patienten ein medianes PFS von 5,62 Monaten (95 % CI: 2,79; 9,46) bzw. 8,18 Monaten (95 % CI: 5,13; 19,55) hatten. BRAF[V600]‑Mutations‑positive bzw. BRAF‑Wildtyp‑Patienten, welche zur Ipilimumab‑Monotherapie randomisiert wurden, hatten ein medianes PFS von 3,09 Monaten (95 % CI: 2,79; 5,19) bzw. 2,83 Monaten (95 % CI: 2,76; 3,06).
Nach 90 Monaten Nachbeobachtungszeit hatten BRAF[V600]‑Mutations‑positive bzw. BRAF‑Wildtyp‑Patienten, welche zu Ipilimumab in Kombination mit Nivolumab randomisiert wurden, ein ORR von 67,0 % (95 % CI: 57,0; 75,9; n = 103) bzw. 54,0 % (95 % CI: 47,1; 60,9; n = 211) während die in den Nivolumab‑Monotherapie‑Arm randomisierten Patienten ein ORR von 37,87 % (95 % CI: 28,2; 48,1; n = 98) bzw. 48,2 % (95 % CI: 41,4; 55,0; n = 218) hatten. BRAF[V600]‑Mutations‑positive bzw. BRAF‑Wildtyp‑Patienten, welche zu Ipilimumab‑Monotherapie randomisiert wurden, hatten ein ORR von 23,0 % (95 % CI: 15,2; 32,5; n = 100) bzw. 17,2 % (95 % CI: 12,4; 22,9; n = 215),
Nach 90 Monaten Nachbeobachtungszeit wurde für BRAF[V600]‑Mutations‑positive Patienten das mediane OS im Kombinations‑Arm nicht erreicht und betrug 45,5 Monate im Nivolumab‑Monotherapie‑Arm. Das mediane OS für BRAF[V600]‑Mutations‑positive Patienten im Ipilimumab‑Monotherapie‑Arm betrug 24,6 Monate. Für BRAF‑Wildtyp‑Patienten betrug das mediane OS 39,06 Monate im Kombinations‑Arm, 34,37 Monate im Nivolumab‑Monotherapie‑Arm und 18,5 Monate im Ipilimumab‑Monotherapie‑Arm. Die Hazard Ratios des Gesamtüberlebens waren im Ipilimumab‑in‑Kombination‑mit‑Nivolumab‑Arm versus Nivolumab‑Monotherapie‑Arm für BRAF[V600]‑Mutations‑positive Patienten 0,66 (95 % CI: 0,44; 0,98) und für BRAF‑Wildtyp‑Patienten 0,95 (95 % CI: 0,74; 1,22).
Randomisierte Phase‑II‑Studie mit Ipilimumab in Kombination mit Nivolumab und Ipilimumab (CA209‑069)
Die Studie CA209‑069 war eine randomisierte, doppelblinde Phase‑II‑Studie, in der Nivolumab mit Ipilimumab im Vergleich zu Ipilimumab allein bei 142 Patienten mit fortgeschrittenem (nicht‑resezierbarem oder metastasiertem) Melanom evaluiert wurde. Die Einschlusskriterien dieser Studie waren denen der Studie CA209067 ähnlich und der primäre Wirksamkeitsendpunkt war die vom Prüfarzt bewertete Ansprechrate bei Patienten mit BRAF‑Wildtyp‑Melanom (77 % der Patienten). Die vom Prüfarzt bewertete Ansprechrate betrug 61 % (95 % CI: 48,9; 72,4) im Kombinations‑Arm (n = 72) versus 11 % (95 % CI: 3,0; 25,4) im Ipilimumab‑Monotherapie‑Arm (n = 37). Die geschätzten OS‑Raten nach 2 bzw. 3 Jahren betrugen 68 % (95 % CI: 56; 78) bzw. 61 % (95 % CI: 49; 71) für die Kombination (n = 73) und 53 % (95 % CI: 36; 68) bzw. 44 % (95 % CI: 28; 60) für Ipilimumab‑Monotherapie (n = 37).
Nierenzellkarzinom (renal cell carcinoma, RCC)
Randomisierte Phase‑III‑Studie mit Ipilimumab in Kombination mit Nivolumab vs. Sunitinib (CA209‑214)
Die Sicherheit und Wirksamkeit von Ipilimumab 1 mg/kg in Kombination mit Nivolumab 3 mg/kg zur Behandlung des fortgeschrittenen/metastasierten Nierenzellkarzinoms (RCC) wurden in einer randomisierten, offenen Phase‑III‑Studie (CA209‑214) untersucht. In die Studie wurden Patienten eingeschlossen (ab 18 Jahren) mit nicht vorbehandeltem, fortgeschrittenem oder metastasiertem Nierenzellkarzinom mit klarzelliger Komponente. Die primäre Population zur Untersuchung der Wirksamkeit, bestand aus Patienten mit intermediärem/ungünstigem Risikoprofil mit mindestens einem von sechs prognostischen Risikofaktoren nach den International Metastatic RCC Database Consortium (IMDC)‑Kriterien (weniger als ein Jahr seit dem Zeitpunkt der initialen Nierenzellkarzinom‑Diagnose bis zur Randomisierung, Karnofsky‑Performance‑Status < 80 %, Hämoglobin geringer als die untere Normgrenze, korrigiertes Calcium größer als 10 mg/dl, Anzahl der Blutplättchen größer als die obere Normgrenze und absolute Anzahl an Neutrophilen größer als die obere Normgrenze). Diese Studie schloss Patienten unabhängig von ihrem Tumor‑PD‑L1‑Status ein. Patienten mit einem Karnofsky‑Performance‑Status < 70 % und Patienten mit Gehirnmetastasen oder Gehirnmetastasen in der Vorgeschichte, Patienten mit aktiver Autoimmunerkrankung oder Patienten mit einer Erkrankung, die eine Behandlung mit einer systemischen Immunsuppression erfordert, waren von der Studie ausgeschlossen. Die Patienten wurden nach IMDC‑Prognostic‑Score und Region stratifiziert.
Insgesamt wurden 1096 Patienten in die Studie randomisiert, von denen 847 Patienten ein RCC mit intermediärem/ungünstigem Risikoprofil aufwiesen und entweder für 4 Dosiszyklen alle 3 Wochen 1 mg/kg Ipilimumab (n = 425) intravenös über 30 Minuten in Kombination mit 3 mg/kg Nivolumab intravenös über 60 Minuten verabreicht bekamen, gefolgt von einer Nivolumab‑Monotherapie mit 3 mg/kg alle 2 Wochen oder über 4 Wochen mit Sunitinib (n = 422) 50 mg täglich peroral behandelt wurden, gefolgt von einer 2‑wöchigen Einnahmepause in jedem Behandlungszyklus. Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen bestand oder bis die Behandlung nicht mehr vertragen wurde. Die erste Tumorbewertung fand 12 Wochen nach Randomisierung statt und wurde im ersten Jahr alle 6 Wochen und danach alle 12 Wochen bis zum Fortschreiten der Erkrankung oder dem Behandlungsende, je nachdem was später eintrat, wiederholt. Eine Weiterbehandlung nach einer durch den Prüfarzt festgestellten Progession gemäß RECIST, Version 1.1 war erlaubt, wenn der Patient nach Einschätzung des Prüfarztes einen klinischen Nutzen hatte und die Studienmedikation tolerierte. Die primären Wirksamkeitsendpunkte waren OS, ORR und PFS bei Patienten mit intermediärem/ungünstigem Risikoprofil, welche durch ein Blinded Independent Central Review (BICR) bestimmt wurden.
Die Ausgangsmerkmale waren in beiden Gruppen etwa gleich verteilt. Das mediane Alter war 61 Jahre (Spanne: 21 ‑ 85) mit 38 % ≥ 65 Jahre und 8 % ≥ 75 Jahre. Die Mehrheit der Patienten war männlich (73 %) und kaukasisch (87 %) und 31 % bzw. 69 % der Patienten hatten einen Ausgangs‑KPS von 70 bis 80 % bzw. 90 bis 100 %. Die mediane Zeit von der initialen Diagnose bis zur Randomisierung betrug 0,4 Jahre sowohl in der Ipilimumab 1 mg/kg in Kombination mit Nivolumab 3 mg/kg als auch in der Sunitinib Gruppe. Die mediane Behandlungszeit betrug 7,9 Monate (Spanne: 1 Tag ‑ 21,4+ Monate) bei Ipilimumab mit Nivolumab behandelten Patienten und 7,8 Monate (Spanne: 1 Tag ‑ 20,2+ Monate) bei mit Sunitinib behandelten Patienten. 29 % der Patienten wurden mit Ipilimumab mit Nivolumab über eine Progression hinaus weiterbehandelt.
Die Wirksamkeitsergebnisse für Patienten mit intermediärem/ungünstigem Risikoprofil sind in Tabelle 13 dargestellt (primäre Analyse mit minimaler Nachbeobachtungszeit von 17,5 Monaten und mit minimaler Nachbeobachtungszeit von 60 Monaten) und in Abbildung 8 (minimale Nachbeobachtungszeit von 60 Monaten).
Die OS‑Ergebnisse einer zusätzlichen deskriptiven Analyse, mit einer minimalen Nachbeobachtungszeit von 60 Monaten, zeigen Resultate, die mit der ursprünglichen primären Analyse übereinstimmen.
Tabelle 13: Wirksamkeitsergebnisse für Patienten mit intermediärem/ungünstigem Risikoprofil (CA209214)
Nivolumab + Ipilimumab |
Sunitinib |
|
Primäre Analyse |
||
Gesamtüberleben | ||
Ereignisse |
140 (33 %) |
188 (45 %) |
Hazard‑Ratioa |
0,63 |
|
99,8 % CI |
(0,44; 0,89) |
|
p‑Wertb, c |
< 0,0001 |
|
Median (95 % CI) |
NE (28,2; NE) |
25,9 (22,1; NE) |
Rate (95 % CI) |
||
Nach 6 Monaten |
89,5 (86,1; 92,1) |
86,2 (82,4; 89,1) |
Nach 12 Monaten |
80,1 (75,9; 83,6) |
72,1 (67,4; 76,2) |
Progressionsfreies Überleben | ||
Ereignisse |
228 (53,6 %) |
228 (54,0 %) |
Hazard‑Ratioa |
0,82 |
|
99,1 % CI |
(0,64; 1,05) |
|
p‑Wertb,h |
0,0331 |
|
Median (95 % CI) |
11,6 (8,71; 15,51) |
8,4 (7,03; 10,81) |
Bestätigtes objektives Ansprechen (BICR) |
177 (41,6 %) |
112 (26,5 %) |
(95 % CI) |
36,9 (46,5) |
(22,4; 31,0) |
Differenz des ORR (95 % CI)d |
16,0 (9,8; 22,2) |
|
p‑Werte,f |
< 0,0001 |
|
Vollständiges Ansprechen |
40 (9,4 %) |
5 (1,2 %) |
Teilweises Ansprechen |
137 (32,2 %) |
107 (25,4 %) |
Stabile Erkrankung (Stable Disease = SD) |
133 (31,3 %) |
188 (44,5 %) |
Mediane Ansprechdauerg | ||
Monate (Spanne) |
NE (1,4+‑25,5+) |
18,17 (1,3+‑23,6+) |
Mediane Zeit zum Ansprechen | ||
Monate (Spanne) |
2,8 (0,9 ‑ 11,3) |
3,0 (0,6 ‑ 15,0) |
Aktualisierte Analyse* |
||
Gesamtüberleben | ||
Ereignisse |
242 (57 %) |
282 (67 %) |
Hazard Ratioa |
0,68 |
|
95 % CI |
(0,58; 0,81) |
|
Median (95 % CI) |
46,95 (35,35; 57,43) |
26,64 (22,08; 33,54) |
Rate (95 % CI) |
||
Nach 24 Monaten |
66,3 (61,5; 70,6) |
52,4 (47,4; 57,1) |
Nach 36 Monaten |
54,6 (49,7; 59,3) |
43,7 (38,7; 48,5) |
Nach 48 Monaten |
49,9 (44,9; 54,6) |
35,8 (31,1; 40,5) |
Nach 60 Monaten |
43,0 (38,1; 47,7) |
31,3 (26,8; 35,9) |
Progressionsfreies Überleben | ||
Ereignisse |
245 (57,6 %) |
253 (60,0 %) |
Hazard Ratioa |
0,73 |
|
95 % CI) |
(0,61; 0,87) |
|
Median (95 % CI)) |
11,6 (8,44; 16,63) |
8,3 (7,03; 10,41) |
Bestätigtes objektives Ansprechen (BICR) |
179 (42,1 %) |
113 (26,8 %) |
(95 % CI) |
(37,4; 47,0) |
(22,6; 31,3) |
Differenz des ORR (95 % CI)d,e |
16,2 (10,0; 22,5) |
|
Vollständiges Ansprechen |
48 (11,3 %) |
9 (2,1 %) |
Teilweises Ansprechen |
131 (30,8 %) |
104 (24,6 %) |
Stabile Erkrankung (Stable Disease = SD) |
131 (30,8 %) |
187 (44,3 %) |
Mediane Ansprechdauerg | ||
Monate (Spanne) |
NE (50,89 ‑ NE) |
19,38 (15,38 ‑ 25,10) |
Mediane Zeit bis zum Ansprechen | ||
Monate (Spanne) |
2,8 (0,9 ‑ 35,0) |
3,1 (0,6 ‑ 23,6) |
a Basierend auf einem stratifizierten proportionalen Hazard‑Modell. | ||
Abbildung 8: Kaplan‑Meier‑Kurven des Gesamtüberlebens bei Patienten mit intermediärem/ungünstigem Risikoprofil (CA209214) Minimale Nachbeobachtungszeit 60 Monate
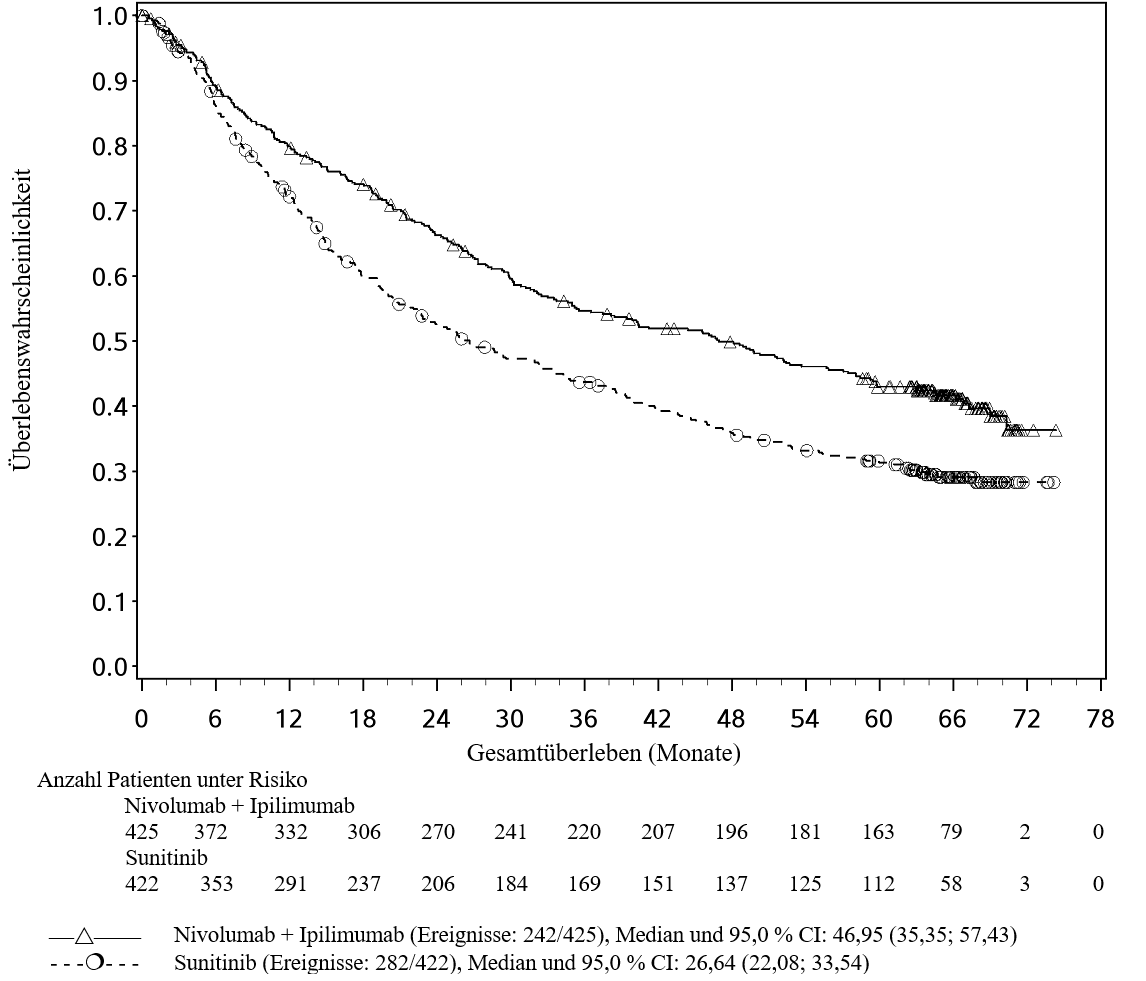
Eine aktualisierte deskriptive OS‑Analyse wurde durchgeführt als alle Patienten ein minimales Follow‑up von 24 Monaten erreicht hatten. Zum Zeitpunkt der Analyse betrug das Hazard Ratio 0,66 (99,8 % CI 0,48‑0,91) mit 166/425 Ereignissen im Kombinations‑Arm und 209/422 Ereignissen im Sunitinib‑Arm. Bei Patienten mit intermediärem/ungünstigem Risikoprofil wurde ein Überlebensvorteil für den Ipilimumab‑in‑Kombination‑mit‑Nivolumab‑Arm im Vergleich zum Sunitinib‑Arm unabhängig von der Tumor‑PD‑L1‑Expression beobachtet. Das mediane OS bei Tumor‑PD‑L1‑Expression ≥ 1 % wurde für Ipilimumab in Kombination mit Nivolumab nicht erreicht und betrug im Sunitinib‑Arm 19,61 Monate (HR = 0,52; 95 % CI: 0,34; 0,78). Das mediane OS bei Tumor‑PD‑L1‑Expression < 1 % betrug 34,7 Monate für Ipilimumab in Kombination mit Nivolumab und betrug 32,2 Monate im Sunitinib‑Arm (HR = 0,70; 95 % CI: 0,54; 0,92).
In CA209214 wurden auch 249 Patienten mit günstigem Risikoprofil nach IMDC‑Kriterien im Ipilimumab‑plus‑Nivolumab‑Arm (n = 125) oder Sunitinib‑Arm (n = 124) eingeschlossen. Diese Patienten waren nicht Teil der ausgewerteten primären Population zur Untersuchung der Wirksamkeit. Patienten mit günstigem Risikoprofil zeigten bei minimaler Nachbeobachtungszeit von 24 Monaten unter Ipilimumab plus Nivolumab im Vergleich zu Sunitinib eine OS‑Hazard‑Ratio von 1,13 (95 % CI: 0,64; 1,99, p = 0,6710). Bei einer minimalen Nachbeobachtungszeit von 60 Monaten betrug die OS‑Hazard‑Ratio 0,94 (95 % CI: 0,65; 1,37).
Es liegen keine Daten zur Erstlinientherapie beim RCC mit Ipilimumab in Kombination mit Nivolumab bei Patienten mit ausschließlich nicht‑klarzelliger Histologie vor.
Der Anteil von Patienten ≥ 75 Jahre stellte 8 % der Patienten mit intermediärem/ungünstigem Risikoprofil in CA209214 dar. Die Kombination von Ipilimumab und Nivolumab zeigte in dieser Subgruppe gegenüber der Gesamtpopulation numerisch einen geringeren Effekt auf OS (HR 0,97; 95 % CI: 0,48; 1,95) bei einer minimalen Nachbeobachtungszeit von 17,5 Monaten. Aufgrund der geringen Größe dieser Subgruppe, lassen sich daraus keine eindeutigen Schlussfolgerungen ziehen.
Erstlinientherapie des Nicht‑kleinzelligen Lungenkarzinoms
Randomisierte Phase‑III‑Studie mit Ipilimumab in Kombination mit Nivolumab und 2 Zyklen platinbasierter Chemotherapie vs. 4 Zyklen platinbasierter Chemotherapie (CA2099LA)
Die Sicherheit und Wirksamkeit von Ipilimumab 1 mg/kg alle 6 Wochen in Kombination mit Nivolumab 360 mg alle 3 Wochen und 2 Zyklen platinbasierter Chemotherapie wurden in einer randomisierten, offenen Phase‑III‑Studie (CA2099LA) untersucht. In die Studie wurden Patienten (18 Jahre oder älter) mit histologisch bestätigtem nicht‑plattenepithelialem oder plattenepithelialem, Stadium‑IV‑ oder rezidiviertem NSCLC (gemäß 7. Klassifikation der International Association for the Study of Lung Cancer), ECOG‑Performance‑Status 0 oder 1, und ohne vorherige Therapie zur Behandlung des NSCLC (einschließlich EGFR‑ und ALK‑Inhibitoren) eingeschlossen. Patienten wurden unabhängig vom PD‑L1‑Status des Tumors eingeschlossen.
Patienten mit sensitivierenden EGFR‑Mutationen oder ALK‑Translokationen, aktiven (unbehandelten) Hirnmetastasen, karzinomatöser Meningitis, aktiver Autoimmunerkrankung oder mit einer Erkrankung, die eine Behandlung mit einer systemischen Immunsuppression erfordert, waren von der Studie ausgeschlossen. Patienten mit behandelten Hirnmetastasen konnten in die Studie eingeschlossen werden, wenn sich die neurologische Symptomatik mindestens 2 Wochen vor Einschluss in die Studie auf den Ausgangsbefund zurückgebildet hatte und die Patienten entweder keine Corticosteroide erhielten oder eine stabile oder abnehmende Dosierung von < 10 mg Prednison‑Äquivalent pro Tag. Die Randomisierung wurde nach Histologie (plattenepithelial vs. nicht‑plattenepithelial), Tumor‑PD‑L1‑Expression (≥ 1 % vs. < 1 %) und Geschlecht (männlich vs. weiblich) stratifiziert.
Insgesamt wurden 719 Patienten entweder für Ipilimumab in Kombination mit Nivolumab und platinbasierter Chemotherapie (n = 361) oder für platinbasierte Chemotherapie (n = 358) randomisiert. Patienten im Behandlungsarm Ipilimumab in Kombination mit Nivolumab und platinbasierter Chemotherapie erhielten Ipilimumab 1 mg/kg intravenös über 30 Minuten alle 6 Wochen in Kombination mit Nivolumab 360 mg intravenös über 30 Minuten alle 3 Wochen und platinbasierter Chemotherapie alle 3 Wochen für 2 Zyklen. Patienten im Chemotherapie‑Arm erhielten platinbasierte Chemotherapie alle 3 Wochen über 4 Zyklen; Patienten mit nicht plattenepithelialer Histologie konnten eine Erhaltungstherapie mit Pemetrexed bekommen.
Platinbasierte Chemotherapie bestand aus Carboplatin (AUC 5 oder 6) und Pemetrexed 500 mg/m2, oder Cisplatin 75 mg/m2 und Pemetrexed 500 mg/m2 bei nicht‑plattenepithelialem NSCLC, oder aus Carboplatin (AUC 6) und Paclitaxel 200 mg/m2 bei plattenepithelialem NSCLC.
Die Behandlung wurde bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate fortgesetzt. Die Behandlung konnte über die Progression hinaus fortgeführt werden, wenn der Patient klinisch stabil war und nach Einschätzung des Prüfarztes einen klinischen Nutzen von der Behandlung hatte. Patienten, die die Kombinationstherapie aufgrund einer Nebenwirkung, die Ipilimumab zugeordnet wurde, abbrechen mussten, konnten mit Nivolumab‑Monotherapie weiter behandelt werden. Tumorbewertungen wurden in den ersten 12 Monaten alle 6 Wochen nach der ersten Dosis der Studienmedikation und danach alle 12 Wochen bis zur Progression der Erkrankung oder bis zur Beendigung der Studienmedikation durchgeführt.
In der Studie CA2099LA waren die Ausgangsmerkmale in allen Gruppen etwa gleich. Das mediane Alter war 65 Jahre (Spanne: 26 ‑ 86) mit 51 % ≥ 65 Jahre und 10 % ≥ 75 Jahre. Die Mehrheit der Patienten war weiß (89 %) und männlich (70 %). Der Ausgangs‑ECOG‑Performance‑Status war 0 (31 %) oder 1 (68 %), 57 % der Patienten hatten eine PD‑L1‑Expression ≥ 1 % und 37 % eine PD‑L1‑Expression < 1 %, 31 % hatten eine plattenepitheliale und 69 % eine nicht‑plattenepitheliale Histologie, 17 % hatten Hirnmetastasen und 86 % waren früher oder derzeit Raucher. Kein Patient erhielt zuvor eine Immuntherapie.
Das primäre Wirksamkeitskriterium der Studie CA2099LA war das Gesamtüberleben (Overall Survival = OS). Weitere Wirksamkeitsendpunkte waren das progressionsfreie Überleben (progression free survival = PFS), die objektive Ansprechrate (objective response rate = ORR) und die Dauer des Ansprechens, bestimmt von einem unabhängigen zentralen Komitee (Blinded Independent Central Review, BICR).
Die Studie zeigte zum Zeitpunkt der präspezifizierten Interimsanalyse nach 351 Ereignissen (87 % der für die finale Analyse geplanten Anzahl Ereignisse) eine statistisch signifikante Verbesserung des OS, PFS und ORR für Patienten, die in den Behandlungsarm Ipilimumab in Kombination mit Nivolumab und platinbasierter Chemotherapie gegenüber platinbasierter Chemotherapie alleine randomisiert worden waren. Die minimale Nachbeobachtungszeit für OS betrug 8,1 Monate.
Die Wirksamkeitsergebnisse sind in Abbildung 9 (aktualisierte OS‑Analyse mit einer minimalen Nachbeobachtungszeit von 12,7 Monaten) und Tabelle 14 (primäre Analyse mit einer minimalen Nachbeobachtungszeit von 8,1 Monaten) dargestellt.
Eine aktualisierte Wirksamkeitsanalyse wurde durchgeführt, nachdem alle Patienten eine minimale Nachbeobachtungszeit von 12,7 Monaten erreicht hatten (siehe Abbildung 9). Zum Zeitpunkt dieser Analyse betrug die Hazard‑Ratio für OS 0,66 (95 % CI: 0,55; 0,80) und die Hazard‑Ratio für PFS 0,68 (95 % CI: 0,57; 0,82).
Abbildung 9: Kaplan‑Meier‑Kurven des Gesamtüberlebens (CA2099LA)
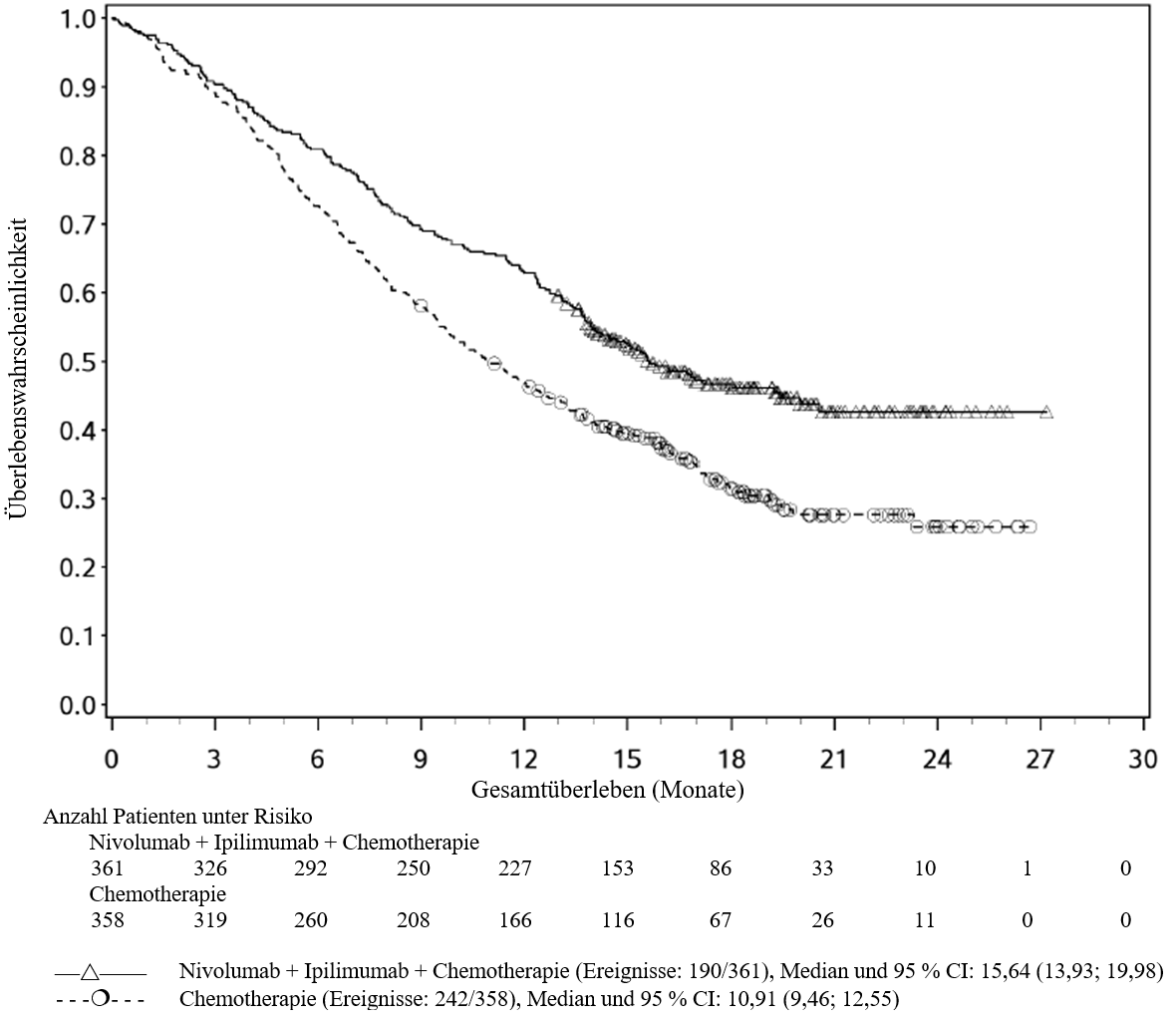
Tabelle 14: Wirksamkeitsergebnisse (CA2099LA)
Ipilimumab + Nivolumab + Chemotherapie |
Chemotherapie |
|
Gesamtüberleben | ||
Ereignisse |
156 (43,2 %) |
195 (54,5 %) |
Hazard‑Ratio |
0,69 |
|
Log‑Rank‑stratifizierter p‑Wertb |
0,0006 |
|
Median (Monate) |
14,1 |
10,7 |
Rate (95 % CI) nach 6 Monaten |
80,9 (76,4; 84,6) |
72,3 (67,4; 76,7) |
Progressionsfreies Überleben | ||
Ereignisse |
232 (64,3 %) |
249 (69,6 %) |
Hazard‑Ratio |
0,70 |
|
Log‑Rank‑stratifizierter p‑Wert c |
0,0001 |
|
Median (Monate)d |
6,83 |
4,96 |
Rate (95 % CI) nach 6 Monaten |
51,7 (46,2; 56,8) |
35,9 (30,5; 41,3) |
Gesamtansprechene |
136 (37,7 %) |
90 (25,1 %) |
(95 % CI) |
(32,7; 42,9) |
(20,7; 30,0) |
CMH‑Test‑stratifizierter p‑Wertf |
0,0003 |
|
Vollständiges Ansprechen |
7 (1,9 %) |
3 (0,8 %) |
Teilweises Ansprechen |
129 (35,7 %) |
87 (24,3 %) |
Ansprechdauer | ||
Median (Monate) |
10,02 |
5,09 |
% mit einer Dauer ≥ 6 Monateg |
74 |
41 |
a Mit einem stratifizierten Cox‑Modell für proportionale Hazards berechnet. | ||
Systemische Folgetherapien erhielten jeweils 28,8 % und 41,1 % der Patienten im Kombinations‑ bzw. im Chemotherapie‑Arm. Eine nachfolgende Immuntherapie (einschließlich Anti‑PD‑L1, Anti‑PD‑1 und Anti‑CTLA‑4) erhielten jeweils 3,9 % und 27,9 % der Patienten im Kombinations‑ bzw. im Chemotherapie‑Arm.
Deskriptive Subgruppen‑Analysen der Studie CA2099LA zeigten einen OS‑Vorteil für die Kombination Ipilimumab mit Nivolumab und Chemotherapie gegenüber der Chemotherapie sowohl bei Patienten mit plattenepithelialer Histologie (HR (95 % CI) 0,65 (0,46; 0,93), n = 227) als auch bei Patienten mit nicht‑plattenepithelialer Histologie (HR (95 % CI) 0,72 (0,55; 0,93), n = 492).
Tabelle 15 fasst die Wirksamkeitsergebnisse von OS, PFS und ORR nach Tumor PD‑L1‑Expression in prädefinierten Subgruppen‑Analysen zusammen.
Tabelle 15: Wirksamkeitsergebnisse nach Tumor‑PD‑L1‑Expression (CA2099LA)
Ipilimumab |
Chemotherapie |
Ipilimumab |
Chemotherapie |
Ipilimumab |
Chemotherapie |
Ipilimumab |
Chemotherapie |
|
PD‑L1 < 1 % |
PD‑L1 ≥ 1% |
PD‑L1 ≥ 1 % bis 49 % |
PD‑L1 ≥ 50 % |
|||||
OS |
0,65 |
0,67 |
0,69 |
0,64 |
||||
PFS |
0,77 |
0,67 |
0,71 |
0,59 |
||||
ORR % |
31,1 |
20,9 |
41,9 |
27,6 |
37,8 |
24,5 |
48,7 |
30,9 |
a Hazard‑Ratio basierend auf einem nicht‑stratifizierten proportionalen Cox‑Hazard‑Modell. | ||||||||
Es wurden insgesamt 70 NSCLC Patienten im Alter ≥ 75 Jahre in die Studie CA2099LA eingeschlossen (37 Patienten in den Arm Ipilimumab in Kombination mit Nivolumab und Chemotherapie und 33 Patienten in den Chemotherapie‑Arm). Eine Hazard‑Ratio von 1,36 (95 % CI: 0,74; 2,52) für OS und eine Hazard‑Ratio von 1,12 (95 % CI: 0,64; 1,96) für PFS wurden in dieser Subgruppe für Ipilimumab in Kombination mit Nivolumab und Chemotherapie vs. Chemotherapie beobachtet. Die ORR betrug 27,0 % im Arm Ipilimumab in Kombination mit Nivolumab und Chemotherapie und 15,2 % im Chemotherapie‑Arm. Dreiundvierzig Prozent der Patienten im Alter ≥ 75 Jahre brachen die Behandlung mit Ipilimumab in Kombination mit Nivolumab und Chemotherapie ab. Die Daten zur Wirksamkeit und Sicherheit von Ipilimumab in Kombination mit Nivolumab und Chemotherapie sind in dieser Patientenpopulation limitiert.
In einer Subgruppen‑Analyse bei Patienten, die nie geraucht hatten, wurde ein geringerer Vorteil im Gesamtüberleben für Ipilimumab in Kombination mit Nivolumab und Chemotherapie verglichen mit Chemotherapie beobachtet. Aufgrund der kleinen Anzahl an Patienten können aus diesen Daten jedoch keine definitiven Schlussfolgerungen gezogen werden.
Malignes Pleuramesotheliom
Randomisierte Phase‑III‑Studie von Ipilimumab in Kombination mit Nivolumab vs. Chemotherapie (CA209743)
Die Sicherheit und Wirksamkeit von Ipilimumab 1 mg/kg alle 6 Wochen in Kombination mit Nivolumab 3 mg/kg alle 2 Wochen wurde in einer randomisierten, offenen Phase‑III‑Studie (CA209743) untersucht. Die Studie schloss Patienten (18 Jahre oder älter) mit histologisch bestätigtem und zuvor unbehandeltem malignen Pleuramesotheliom mit epitheloider oder nicht‑epitheloider Histologie, einem ECOG‑Performance‑Status von 0 oder 1, und ohne palliative Radiotherapie innerhalb von 14 Tagen vor der ersten Studientherapie ein. Patienten wurden unabhängig vom PD‑L1‑Status ihres Tumors eingeschlossen.
Patienten mit primärem Mesotheliom des Peritoneums, Perikards oder der Tunica vaginalis testis, Patienten mit interstitieller Lungenerkrankung, aktiver Autoimmunerkrankung, Erkrankungen, die eine systemische Immunsuppression erfordern, und Patienten mit Hirnmetastasen (soweit nicht operativ reseziert oder mit stereotaktischer Radiotherapie behandelt und ohne Weiterentwicklung innerhalb von 3 Monaten vor Einschluss in die Studie) waren von der Studie ausgeschlossen. Die Randomisierung erfolgte stratifiziert nach Histologie (epitheloid vs. sarkomatoid oder gemischte Histologie) und Geschlecht (männlich vs. weiblich).
Insgesamt wurden 605 Patienten entweder für Ipilimumab in Kombination mit Nivolumab (n = 303) oder Chemotherapie (n = 302) randomisiert. Patienten im Behandlungsarm Ipilimumab in Kombination mit Nivolumab erhielten Ipilimumab 1 mg/kg intravenös über 30 Minuten alle 6 Wochen in Kombination mit Nivolumab 3 mg/kg intravenös über 30 Minuten alle 2 Wochen für bis zu 2 Jahre. Patienten im Chemotherapie‑Arm erhielten Chemotherapie für bis zu 6 Zyklen (ein Zyklus war 21 Tage). Die Chemotherapie bestand aus Cisplatin 75 mg/m2 und Pemetrexed 500 mg/m2 oder Carboplatin 5 AUC und Pemetrexed 500 mg/m2.
Die Behandlung wurde bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate fortgesetzt. Die Behandlung konnte über die Progression hinaus fortgeführt werden, wenn der Patient klinisch stabil war und nach Einschätzung des Prüfarztes einen klinischen Nutzen von der Behandlung hatte. Patienten, die die Kombinationstherapie aufgrund einer Nebenwirkung, die Ipilimumab zugeordnet wurde, abbrechen mussten, konnten mit Nivolumab‑Monotherapie weiter behandelt werden. Tumorbewertungen wurden in den ersten 12 Monaten alle 6 Wochen nach der ersten Dosis der Studienmedikation und danach alle 12 Wochen bis zur Progression der Erkrankung oder bis zur Beendigung der Studienmedikation durchgeführt.
In der Studie CA209743 waren die Ausgangsmerkmale in allen Gruppen etwa gleich. Das mediane Alter war 69 Jahre (Spanne: 25 ‑ 89) mit 72 % ≥ 65 Jahre und 26 % ≥ 75 Jahre. Die Mehrheit der Patienten war weiß (85 %) und männlich (77 %). Der Ausgangs ECOG‑Performance‑Status war 0 (40 %) oder 1 (60 %), 80 % der Patienten hatten eine PD‑L1‑Expression ≥ 1% und 20 % eine PD‑L1‑Expression < 1 %, 75 % hatten eine epitheloide und 25 % eine nicht‑epitheloide Histologie.
Das primäre Wirksamkeitskriterium der Studie CA209743 war das Gesamtüberleben (Overall Survival = OS). Wichtige sekundäre Wirksamkeitsendpunkte waren das progressionsfreie Überleben (Progression‑Free Survival = PFS), die objektive Ansprechrate (Objective Response Rate = ORR) und die Dauer des Ansprechens, bestimmt von einem unabhängigen zentralen Komitee (Blinded Independent Central Review, BICR) anhand der modifizierten RECIST‑Kriterien für das Pleuramesotheliom. Deskriptive Analysen für diese sekundären Endpunkte sind in Tabelle 16 dargestellt.
Die Studie zeigte zum Zeitpunkt der präspezifizierten Interimsanalyse nach 419 Ereignissen (89 % der für die finale Analyse geplanten Anzahl Ereignisse) eine statistisch signifikante Verbesserung des OS für Patienten, die in den Behandlungsarm Ipilimumab in Kombination mit Nivolumab randomisiert worden waren, gegenüber der Chemotherapie. Die minimale Nachbeobachtungszeit für OS betrug 22 Monate.
Die Wirksamkeitsergebnisse sind in Abbildung 10 und Tabelle 16 dargestellt.
Abbildung 10: Kaplan‑Meier‑Kurven des Gesamtüberlebens (CA209743)
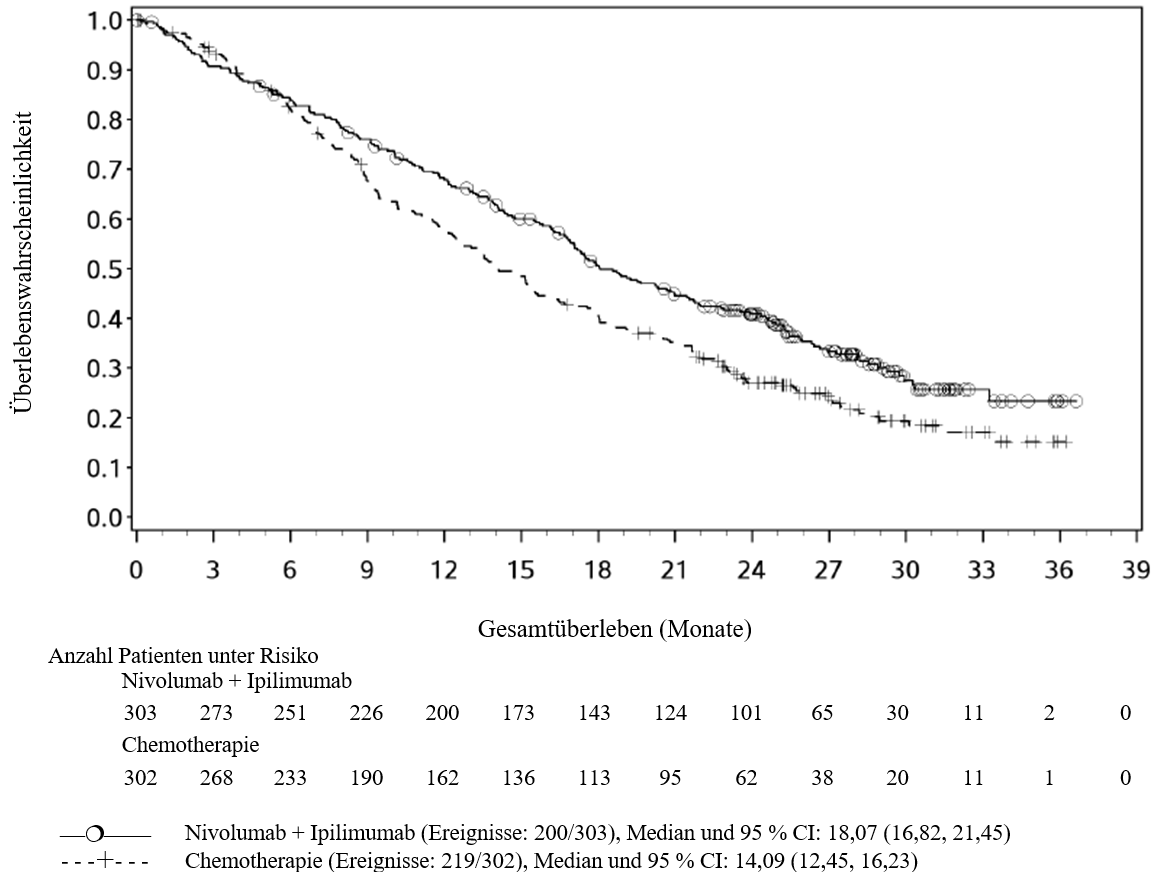
Tabelle 16: Wirksamkeitsergebnisse (CA209743)
Ipilimumab + Nivolumab |
Chemotherapie |
|
Gesamtüberleben | ||
Ereignisse |
200 (66 %) |
219 (73 %) |
Hazard‑Ratio |
0,74 |
|
Log‑Rank‑stratifizierter p‑Wertb |
0,002 |
|
Median (Monate)c |
18,1 |
14,1 |
Rate (95 % CI) nach 24 Monatenc |
41 % (35,1; 46,5) |
27 % (21,9; 32,4) |
Progressionsfreies Überleben | ||
Ereignisse |
218 (72 %) |
209 (69 %) |
Hazard‑Ratio |
1,0 |
|
Median (Monate)c |
6,8 |
7,2 |
Gesamtansprechen |
40 % |
43 % |
(95 % CI) |
(34,1; 45,4) |
(37,1; 48,5) |
Vollständiges Ansprechen |
1,7 % |
0 |
Teilweises Ansprechen |
38 % |
43 % |
Ansprechdauer | ||
Median (Monate)c |
11,0 |
6,7 |
a Mit einem stratifizierten Cox‑Modell für proportionale Hazards berechnet. | ||
Systemische Folgetherapien erhielten jeweils 44,2 % bzw. 40,7 % der Patienten im Kombinations‑ bzw. im Chemotherapie‑Arm. Eine nachfolgende Immuntherapie (einschließlich Anti‑PD‑1, Anti‑PD‑L1 und Anti‑CTLA‑4) erhielten jeweils 3,3 % bzw. 20,2 % der Patienten im Kombinations‑ bzw. im Chemotherapie‑Arm.
Tabelle 17 fasst die Wirksamkeitsergebnisse von OS, PFS und ORR nach Histologie in präspezifizierten Subgruppen‑Analysen zusammen.
Tabelle 17: Wirksamkeitsergebnisse nach Histologie (CA209743)
Epitheloid |
Nicht‑epitheloid |
|||
Ipilimumab |
Chemotherapie |
Ipilimumab |
Chemotherapie |
|
Gesamtüberleben | ||||
Ereignisse |
157 |
164 |
43 |
55 |
Hazard‑Ratio |
0,85 |
0,46 |
||
Median (Monate) |
18,73 |
16,23 |
16,89 |
8,80 |
Rate (95 % CI) nach 24 Monaten |
41,2 |
31,8 |
39,5 |
9,7 |
Progressionsfreies Überleben | ||||
Hazard‑Ratio |
1,14 |
0,58 |
||
Median (Monate) |
6,18 |
7,66 |
8,31 |
5,59 |
Gesamtansprechen |
38,6 % |
47,2 % |
43,3 % |
26,9 % |
(95 % CI)b |
(32,3; 45,1) |
(40,7; 53,8) |
(31,2; 56,0) |
(16,8; 39,1) |
Ansprechdauer |
8,44 |
6,83 |
24,02 |
4,21 |
Median (Monate) |
(7,16; 14,59) |
(5,59; 7,13) |
(8,31; N.A.) |
(2,79; 7,03) |
a Hazard‑Ratio mit einem unstratifizierten Cox‑Modell für proportionale Hazards berechnet. | ||||
Tabelle 18 fasst die Wirksamkeitsergebnisse von OS, PFS und ORR nach Tumor‑PD‑L1‑Expression zu Studienbeginn in präspezifizierten Subgruppen‑Analysen zusammen.
Tabelle 18: Wirksamkeitsergebnisse nach Tumor‑PD‑L1‑Expression (CA209743)
PD‑L1 < 1 % |
PD‑L1 ≥ 1 % |
|||
Ipilimumab |
Chemotherapie |
Ipilimumab |
Chemotherapie |
|
Gesamtüberleben | ||||
Ereignisse |
40 |
58 |
150 |
157 |
Hazard‑Ratio |
0,94 |
0,69 |
||
Median (Monate) |
17,3 |
16,5 |
18,0 |
13,3 |
Rate (95 % CI) nach 24 Monaten |
38,7 |
24,6 |
40,8 |
28,3 |
Progressionsfreies Überleben | ||||
Hazard Ratio |
1,79 |
0,81 |
||
Median (Monate) |
4,1 |
8,3 |
7,0 |
7,1 |
Gesamtansprechen |
21,1 % |
38,5 % |
43,5 % |
44,3 % |
(95 % CI)c |
(11,4; 33,9) |
(27,7; 50,2) |
(37,1; 50,2) |
(37,6; 51,1) |
a Hazard‑Ratio mit einem unstratifizierten Cox‑Modell für proportionale Hazards berechnet. | ||||
Es wurden insgesamt 157 MPM‑Patienten im Alter ≥ 75 Jahre in die Studie CA209743 eingeschlossen (78 Patienten in den Arm Ipilimumab in Kombination mit Nivolumab und 79 Patienten in den Chemotherapie‑Arm). Eine Hazard‑Ratio von 1,02 (95 % CI: 0,70; 1,48) für OS wurde in dieser Subgruppe für Ipilimumab in Kombination mit Nivolumab vs. Chemotherapie beobachtet. Im Verhältnis zu allen Patienten, die Ipilimumab in Kombination mit Nivolumab erhielten, zeigten sich für Patienten im Alter ≥ 75 Jahre höhere Raten an schwerwiegenden Nebenwirkungen und an Therapieabbrüchen aufgrund von Nebenwirkungen (siehe Abschnitt 4.8). Aufgrund des explorativen Charakters dieser Subgruppenanalyse können jedoch keine definitiven Schlussfolgerungen gezogen werden.
dMMR‑ oder MSI‑H‑Kolorektalkarzinom
Offene Studie zu Nivolumab in Kombination mit Ipilimumab vs. Chemotherapie bei Patienten mit dMMR‑ oder MSI‑H‑Kolorektalkarzinom, die im metastasierten Setting nicht vorbehandelt worden sind
Die Sicherheit und Wirksamkeit von 1 mg/kg Ipilimumab in Kombination mit 240 mg Nivolumab alle 3 Wochen für maximal 4 Dosen, gefolgt von einer Nivolumab‑Monotherapie mit 480 mg alle 4 Wochen für die Erstlinienbehandlung des nicht resezierbaren oder metastasierten Kolorektalkarzinoms mit bekanntem MSI‑H‑ oder dMMR‑Status wurden in einer randomisierten, mehrarmigen, offenen Phase‑III‑Studie (CA2098HW) untersucht. Die Studienbehandlungsarme umfassten eine Nivolumab‑Monotherapie, Nivolumab in Kombination mit Ipilimumab oder eine Chemotherapie nach Wahl des Prüfarztes. Der MSI‑H‑ bzw. dMMR‑Tumorstatus wurde gemäß lokalem Behandlungsstandard mittels PCR, NGS bzw. IHC‑Assays bestimmt. Die zentrale Beurteilung des MSI‑H‑Status mittels PCR (Idylla MSI)‑Test und des dMMR‑Status mittels IHC (Omnis MMR)‑Test wurde retrospektiv an Tumorproben der Patienten durchgeführt, die für die Bestimmung des lokalen MSI‑H‑/dMMR‑Status verwendet worden waren. Die primäre Population zur Untersuchung der Wirksamkeit bestand aus Patienten mit durch zentrale Testung bestätigtem MSI‑H‑/dMMR‑Status. Patienten mit symptomatischen Hirnmetastasen, mit aktiver Autoimmunerkrankung, Patienten, die systemische Corticosteroide oder Immunsuppressiva einnahmen oder mit Checkpoint‑Inhibitoren vorbehandelt worden waren, waren von der Studie ausgeschlossen. Die Randomisierung wurde nach Lage des Tumors (rechts vs. links) stratifiziert. Die in den Chemotherapie‑Arm randomisierten Patienten konnten bei Krankheitsprogression nach BICR eine Kombinationstherapie mit Ipilimumab plus Nivolumab erhalten.
Insgesamt wurden 303 nicht vorbehandelte Patienten im metastasierten Setting in der Studie randomisiert, wobei 202 Patienten der Kombinationstherapie mit Nivolumab plus Ipilimumab und 101 Patienten der Chemotherapie‑Gruppe zugewiesen wurden. Hiervon hatten 255 Patienten einen durch zentrale Beurteilung bestätigten MSI‑H‑/dMMR‑Status, 171 im Ipilimumab-plus-Nivolumab-Kombinations-Arm und 84 im Chemotherapie-Arm. Die Patienten im Ipilimumab-plus-Nivolumab-Arm erhielten 1 mg/kg Ipilimumab alle 3 Wochen in Kombination mit 240 mg Nivolumab alle 3 Wochen für maximal 4 Dosen, gefolgt von einer Nivolumab-Monotherapie mit 480 mg alle 4 Wochen. Die Patienten im Chemotherapie‑Arm erhielten: mFOLFOX6 (Oxaliplatin, Leucovorin und Fluorouracil) mit oder ohne entweder Bevacizumab oder Cetuximab: Bolus-Regime mit Oxaliplatin 85 mg/m2, Leucovorin 400 mg/m2 und Fluorouracil 400 mg/m2, gefolgt von Fluorouracil 2 400 mg/m2 über 46 Stunden alle 2 Wochen. Bevacizumab 5 mg/kg oder Cetuximab 500 mg/m2, verabreicht vor mFOLFOX6 alle 2 Wochen; oder FOLFIRI (Irinotecan, Leucovorin und Fluorouracil) mit oder ohne entweder Bevacizumab oder Cetuximab: Bolus-Regime mit Irinotecan 180 mg/m2, Leucovorin 400 mg/m2 und Fluorouracil 400 mg/m2 und Fluorouracil 2 400 mg/m2 über 46 Stunden alle 2 Wochen. Bevacizumab 5 mg/kg oder Cetuximab 500 mg/m2, verabreicht vor FOLFIRI alle 2 Wochen. Die Behandlung wurde bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bei der Kombinationstherapie mit Ipilimumab plus Nivolumab bis zu 24 Monate fortgesetzt. Patienten, die die Kombinationstherapie aufgrund einer Nebenwirkung, die Ipilimumab zugeordnet wurde, abbrechen mussten, konnten mit Nivolumab als Monotherapie weiterbehandelt werden. Tumorbeurteilungen wurden gemäß RECIST Version 1.1 während der ersten 24 Wochen alle 6 Wochen, dann alle 8 Wochen bis Woche 96, anschließend alle 16 Wochen bis Woche 146 und danach alle 24 Wochen durchgeführt.
Die Ausgangsmerkmale aller randomisierten, zuvor nicht aufgrund einer metastasierten Erkrankung behandelten Patienten waren wie folgt: Das mediane Alter betrug 63 Jahre (Spanne: 21 bis 87) mit 46 % ≥ 65 Jahre und 18 % ≥ 75 Jahre; 46 % der Patienten waren männlich und 86 % waren Kaukasier. Der ECOG‑Performance‑Status zu Studienbeginn war 0 (54 %) oder ≥ 1 (46 %); die Lage des Tumors war bei 68 % der Patienten rechts- und bei 32 % linksseitig; und 39 Patienten der 223 Patienten mit bekanntem Status hatten ein bestätigtes Lynch‑Syndrom. Die Ausgangsmerkmale der zuvor nicht aufgrund einer metastasierten Erkrankung behandelten Patienten mit zentral bestätigtem MSI‑H‑/dMMR‑Status waren konsistent mit denen aller randomisierten, nicht vorbehandelten Patienten. Von den 101 in den Chemotherapie‑Arm randomisierten Patienten erhielten 88 eine Chemotherapie gemäß Protokoll, einschließlich Oxaliplatin‑basierter (58 %) und Irinotecan‑basierter (42 %) Behandlungsschemata. Darüber hinaus erhielten 66 Patienten entweder Bevacizumab (64 %) oder Cetuximab (11 %) als zielgerichtete Therapie.
Ein primärer Wirksamkeitsendpunkt der Studie war das durch ein BICR gemäß RECIST 1.1‑Kriterien bestätigte progressionsfreie Überleben (PFS). Weitere Wirksamkeitsendpunkte umfassten objektive Ansprechrate (ORR) nach BICR, Gesamtüberleben (OS) und Dauer des Ansprechens.
Die Studie erreichte den primären Endpunkt bei der geplanten Zwischenanalyse und zeigte eine statistisch signifikante Verbesserung des PFS nach BICR bei Patienten mit zentral bestätigtem MSI‑H‑/dMMR‑Status im Ipilimumab-plus-Nivolumab-Arm im Vergleich zum Chemotherapie-Arm. Die Ergebnisse bezüglich des progressionsfreien Überlebens basierend auf der BICR-Beurteilung sind in Tabelle 19 und Abbildung 11 dargestellt. Zum Zeitpunkt der Zwischenanalyse waren die anderen Endpunkte, einschließlich der Daten aus dem Nivolumab‑Monotherapie‑Arm, aufgrund der Testhierarchie noch nicht getestet.
Tabelle 19: Wirksamkeitsergebnisse der Erstlinienbehandlung bei zentral bestätigtem MSI‑H‑/dMMR‑Kolorektalkarzinom (CA2098HW)a
Ipilimumab + Nivolumab |
Chemotherapie |
|
Progressionsfreies Überleben |
||
Ereignisse |
48 (28 %) |
52 (62 %) |
Hazard Ratio |
0,21 |
|
95 % CI |
(0,14; 0,32) |
|
p‑Wertb |
< 0,0001 |
|
Median (95 % CI) (Monate) |
NR (38,4; NR) |
5,9 (4,4; 7,8) |
a Mediane Nachbeobachtungsdauer 31,5 Monate (Spanne: 6,1 bis 48,4 Monate). | ||
Abbildung 11: Kaplan‑Meier‑Kurven des PFS in der Erstlinie bei Patienten mit zentral bestätigtem MSI‑H‑/dMMR‑Kolorektalkarzinom (CA2098HW)
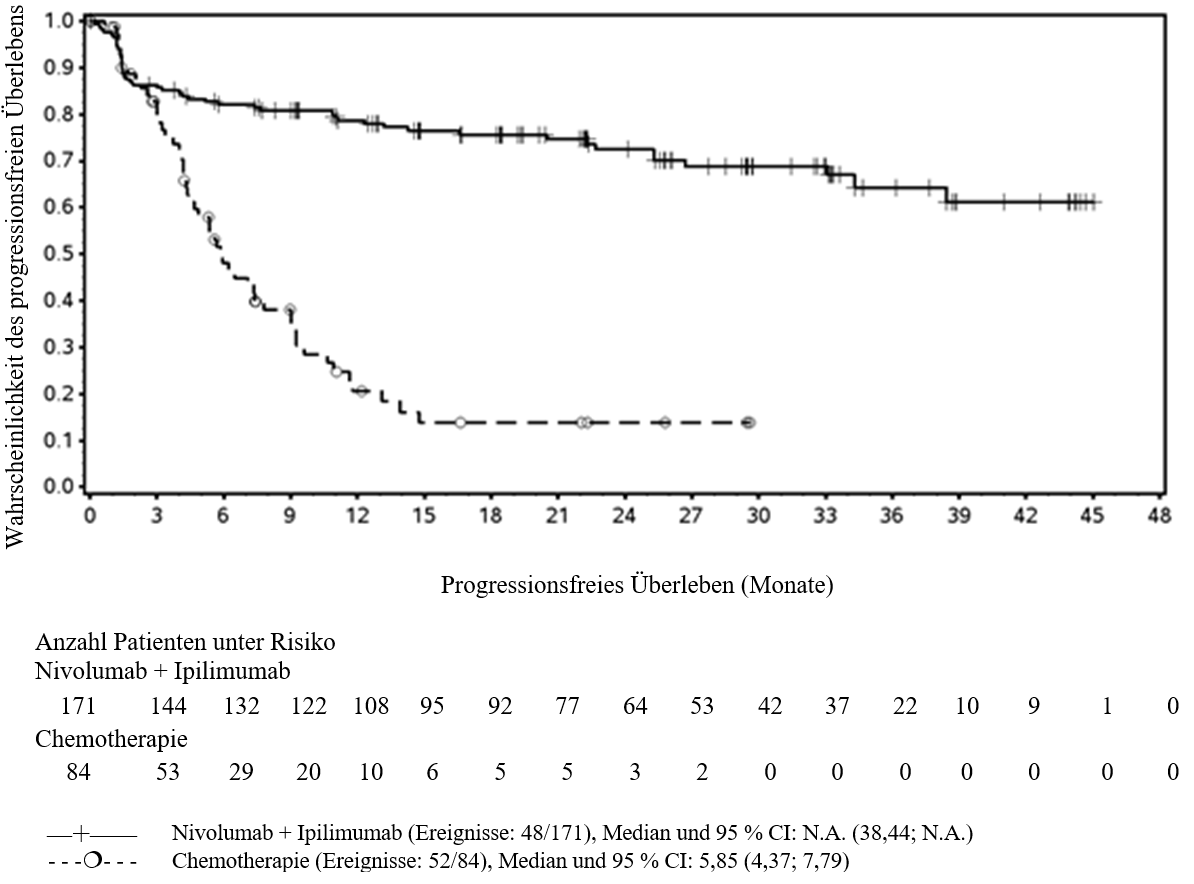
Offene Studie zu Nivolumab in Kombination mit Ipilimumab bei Patienten mit dMMR‑ oder MSI‑H‑Kolorektalkarzinom, die zuvor eine fluoropyrimidinbasierte Kombinationschemotherapie erhalten hatten
Die Sicherheit und Wirksamkeit von Ipilimumab 1 mg/kg in Kombination mit Nivolumab 3 mg/kg zur Behandlung des metastasierten Kolorektalkarzinoms mit Mismatch‑Reparatur‑Defizienz (dMMR) oder hoher Mikrosatelliteninstabilität (MSI‑H) wurde in einer multizentrischen, offenen, einarmigen Phase‑II‑Studie untersucht (CA209142).
In die Studie wurden Patienten (18 Jahre und älter) mit lokal bestimmtem dMMR‑ oder MSI‑H‑Status eingeschlossen, die einen Progress während oder nach einer Therapie mit Fluoropyrimidin und Oxaliplatin oder Irinotecan hatten, oder die vorher gegen eine dieser Therapien intolerant waren. Patienten, deren letzte Vortherapie im adjuvanten Setting durchgeführt wurde, sollten mit oder innerhalb von 6 Monaten nach Abschluss der adjuvanten Chemotherapie progredient geworden sein. Die Patienten wiesen einen ECOG‑Performance‑Status von 0 oder 1 auf und wurden unabhängig vom Tumor‑PD‑L1‑Status eingeschlossen. Patienten mit aktiven Hirnmetastasen, mit aktiver Autoimmunerkrankung oder mit Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von der Studie ausgeschlossen.
Insgesamt wurden 119 Patienten behandelt und erhielten Ipilimumab 1 mg/kg intravenös über 90 Minuten in Kombination mit Nivolumab 3 mg/kg intravenös über 60 Minuten alle 3 Wochen für 4 Dosen gefolgt von einer Nivolumab‑Monotherapie mit 3 mg/kg alle 2 Wochen. Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen bestand oder bis die Behandlung nicht mehr vertragen wurde. Untersuchungen des Tumors nach RECIST Version 1.1 wurden alle 6 Wochen für die ersten 24 Wochen und dann alle 12 Wochen durchgeführt. Das primäre Wirksamkeitskriterium war das von den Prüfärzten bewertete ORR. Sekundäre Wirksamkeitskriterien waren ORR, bestätigt durch BICR, und die Krankheitskontrollrate. Die ORR‑Analyse beinhaltete die Ansprechdauer und die Zeit bis zum Ansprechen. Explorative Wirksamkeitskriterien schlossen PFS und OS mit ein.
Das mediane Alter war 58 Jahre (Spanne: 21‑88) wobei 32 % ≥ 65 und 9 % ≥ 75 Jahre alt waren, 59 % waren männlich und 92 % waren weiß. Der Ausgangs‑ECOG‑Performance‑Status betrug 0 (45 %) oder 1 (55 %), 25 % der Patienten hatten BRAF‑Mutationen, 37 % hatten KRAS‑Mutationen und bei 12 % war der Mutationsstatus unbekannt. Von den 119 behandelten Patienten, hatten 109 vorher eine fluoropyrimidinbasierte Chemotherapie im metastasierten und 9 im adjuvanten Setting erhalten. Vor Einschluss in die Studie hatten von den 119 behandelten Patienten bereits 118 (99 %) Fluorouracil, 111 (93 %) Oxaliplatin und 87 (73 %) Irinotecan als Bestandteil von vorherigen Therapieregimen erhalten; 82 Patienten (69 %) hatten eine Vortherapie mit Fluoropyrimidin, Oxaliplatin und Irinotecan erhalten. 23 %, 36 %, 24 % und 16 % haben 1, 2, 3 bzw. 4 oder mehr Vortherapien erhalten und 29 % der Patienten hatten einen EGFR‑Inhibitor erhalten.
Die Wirksamkeitsergebnisse (minimale Nachbeobachtungszeit 46,9 Monate; mediane Nachbeobachtungszeit 51,1 Monate) sind in Tabelle 20 dargestellt.
Tabelle 20: Wirksamkeitsergebnisse (CA209142) bei Patienten mit dMMR‑ oder MSI‑H‑CRC *
Ipilimumab + Nivolumab |
|
(n = 119) | |
Bestätigtes objektives Ansprechen, n (%) |
77 (64,7) |
(95 % CI) |
(55,4; 73,2) |
Vollständiges Ansprechen |
15 (12,6) |
Teilweises Ansprechen (Partial Response = PR), n (%) |
62 (52,1) |
Stabile Erkrankung (Stable Disease = SD), n (%) |
25 (21,0) |
Ansprechdauer |
|
Median (Spanne) Monate |
NR (1,4; 58,0+) |
Mediane Zeit bis zum Ansprechen |
|
Monate (Spanne) |
2,8 (1,1; 37,1) |
* nach Beurteilung des Prüfarztes | |
Das ORR, bestätigt durch BICR, war 61,3 % (95 % CI: 52,0; 70,1), einschließlich einer CR‑Rate von 20,2 % (95 % CI: 13,4; 28,5), PR‑Rate von 41,2 % (95 % CI: 32,2; 50,6) und mit einer stabilen Krankheit, die bei 22,7 % berichtet wurde. Die Beurteilung nach BICR war im Allgemeinen konsistent mit der Beurteilung durch die Prüfärzte. Ein bestätigtes Ansprechen wurde unabhängig vom BRAF‑ oder KRAS‑Mutationsstatus und des Tumor‑PD L1‑Expressionslevels beobachtet.
Von den 119 Patienten waren 11 (9,2 %) Patienten ≥ 75 Jahre. Das durch die Prüfärzte beurteilte ORR betrug bei den Patienten ≥ 75 Jahren 45,5 % (95 % CI: 16,7; 76,6).
Plattenepithelkarzinom des Ösophagus
Randomisierte Phase‑III‑Studie mit Ipilimumab in Kombination mit Nivolumab vs. Chemotherapie als Erstlinientherapie (CA209648)
Sicherheit und Wirksamkeit von Ipilimumab in Kombination mit Nivolumab wurden in einer randomisierten, aktiv‑kontrollierten, offenen Phase‑III‑Studie untersucht (CA209648). In die Studie wurden erwachsene Patienten (18 Jahre und älter) mit nicht vorbehandeltem, nicht resezierbarem fortgeschrittenen, rezidivierten oder metastasierten Plattenepithelkarzinom des Ösophagus (ESCC) eingeschlossen. Patienten wurden unabhängig vom Tumor‑PD‑L1‑Status eingeschlossen und die Tumorzell‑PD‑L1‑Expression wurde mithilfe des PD‑L1‑IHC‑28‑8‑pharmDx‑Assays bestimmt. Die Patienten mussten ein Plattenepithelkarzinom oder ein adenosquamöses Karzinom des Ösophagus aufweisen und nicht geeignet sein für eine Chemo‑Strahlentherapie und/oder Operation. Vorangegangene adjuvante, neoadjuvante oder definitive Chemo‑, Strahlen‑ oder Chemoradiotherapie war zulässig, wenn sie als Teil einer kurativen Behandlung vor Beginn der Studie gegeben wurde. Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, mit symptomatischen Hirnmetastasen, mit aktiver Autoimmunerkrankung, Patienten, die systemische Corticosteroide oder Immunsuppressiva einnahmen, oder Patienten mit einem erhöhten Risiko für Blutungen und Fisteln aufgrund von offensichtlicher Tumorinvasion in angrenzende Organe des ösophagealen Tumors waren von der klinischen Studie ausgeschlossen. Die Randomisierung wurde nach Tumorzell‑PD‑L1‑Status (≥ 1 % vs. < 1 % oder unbestimmt), Region (Ostasien vs. übriges Asien vs. Rest der Welt), ECOG‑Performance‑Status (0 vs. 1) und Anzahl der Organe mit Metastasen (≤ 1 vs. ≥ 2) stratifiziert.
Insgesamt wurden 649 Patienten randomisiert und erhielten entweder Ipilimumab in Kombination mit Nivolumab (n = 325) oder Chemotherapie (n = 324). Davon hatten 315 Patienten eine Tumorzell‑PD‑L1‑Expression ≥ 1 %, 158 im Ipilimumab‑plus‑Nivolumab‑Arm und 157 im Chemotherapie‑Arm. Patienten im Ipilimumab‑plus‑Nivolumab‑Arm erhielten 1 mg/kg Ipilimumab alle 6 Wochen in Kombination mit 3 mg/kg Nivolumab alle 2 Wochen. Patienten im Chemotherapie‑Arm erhielten Fluorouracil 800 mg/m2/Tag intravenös von Tag 1 bis Tag 5 (für 5 Tage) und Cisplatin 80 mg/m2 intravenös an Tag 1 (eines vierwöchigen Zyklus). Die Behandlung wurde bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate fortgesetzt. Patienten, welche die Kombinationstherapie aufgrund einer Nebenwirkung, die Ipilimumab zugeordnet wurde, abbrechen mussten, konnten mit der Nivolumab‑Monotherapie weiter behandelt werden.
Die Basischarakteristika der Behandlungsgruppen waren im Allgemeinen ausgewogen. Bei Patienten mit einer Tumorzell‑PD‑L1‑Expression ≥ 1 % betrug das mediane Alter 63 Jahre (Spanne: 26 ‑ 85), 8,2 % waren ≥ 75 Jahre, 81,8 % waren männlich, 73,1 % waren Asiaten und 23,3 % waren weiß. Patienten hatten histologisch bestätigte Plattenepithelkarzinome (98,9 %) oder adenosquamöse Karzinome (1,1 %) des Ösophagus. Der Ausgangs‑ECOG‑Performance‑Status war 0 (45,2 %) oder 1 (54,8 %).
Die primäre Erfassung der Wirksamkeit erfolgte durch die Erhebung der Endpunkte PFS (durch BICR) und OS beurteilt bei Patienten mit Tumorzell‑PD‑L1‑Expression ≥ 1 %. Sekundäre Endpunkte gemäß der vordefinierten hierarchischen Testung enthielten OS, PFS (durch BICR) und ORR (durch BICR) bei allen randomisierten Patienten. Tumorbeurteilungen wurden gemäß RECIST Version 1.1 alle 6 Wochen bis zu und einschließlich Woche 48, danach alle 12 Wochen durchgeführt.
Bei der primären, präspezifizierten Analyse mit einer minimalen Nachbeobachtungszeit von 13,1 Monaten zeigte die Studie eine statistisch signifikante Verbesserung des OS für Patienten mit einer Tumorzell‑PD‑L1‑Expression ≥ 1 %. Die Wirksamkeitsergebnisse sind in Tabelle 21 dargestellt.
Tabelle 21: Wirksamkeitsergebnisse bei Patienten mit Tumorzell‑PD‑L1 ≥ 1 % (CA209648)
Ipilimumab + Nivolumab |
Chemotherapiea |
|
Gesamtüberleben | ||
Ereignisse |
106 (67,1 %) |
121 (77,1 %) |
Hazard‑Ratio (98,6 % CI)b |
0,64 (0,46; 0,90) |
|
p‑Wertc |
0,0010 |
|
Median (95 % CI) (Monate)d |
13,70 (11,24; 17,02) |
9,07 (7,69; 9,95) |
Rate (95 % CI) nach 12 Monatend |
57,1 (49,0; 64,4) |
37,1 (29,2; 44,9) |
Progressionsfreies Überlebene | ||
Ereignisse |
123 (77,8 %) |
100 (63,7 %) |
Hazard‑Ratio (98,5 % CI)b |
1,02 (0,73; 1,43) |
|
p‑Wertc |
0,8958 |
|
Median (95 % CI) (Monate)d |
4,04 (2,40; 4,93) |
4,44 (2,89; 5,82) |
Rate (95 % CI) nach 12 Monatend |
26,4 (19,5; 33,9) |
10,5 (4,7; 18,8) |
Gesamtansprechen, n (%)e |
56 (35,4) |
31 (19,7) |
(95 % CI) |
(28,0; 43,4) |
(13,8; 26,8) |
Vollständiges Ansprechen |
28 (17,7) |
8 (5,1) |
Teilweises Ansprechen |
28 (17,7) |
23 (14,6) |
Ansprechdauere | ||
Median (95 % CI) (Monate)d |
11,83 (7,10; 27,43) |
5,68 (4,40; 8,67) |
Spanne |
1,4+; 34,5+ |
1,4+; 31,8+ |
a Fluorouracil und Cisplatin. | ||
Bei einer aktualisierten deskriptiven Auswertung mit einer minimalen Nachbeobachtungszeit von 20 Monaten, waren die Verbesserungen des OS konsistent mit der primären Analyse. Das mediane OS betrug 13,70 Monate (95 % CI: 11,24; 17,41) für Ipilimumab plus Nivolumab vs. 9,07 Monate (95% CI: 7,69; 10,02) für Chemotherapie (HR = 0,63; 95 % CI: 0,49; 0,82). Das mediane PFS betrug 4,04 Monate (95 % CI: 2,40; 4,93) für Ipilimumab plus Nivolumab vs. 4,44 Monate (95 % CI: 2,89; 5,82) für Chemotherapie (HR = 1,02; 95 % CI: 0,77; 1,34). Die ORR betrug 35,4 % (95 % CI: 28,0; 43,4) für Ipilimumab plus Nivolumab vs. 19,7 % (95 % CI: 13,8; 26,8) für Chemotherapie.
Die Kaplan‑Meier‑Kurven für das Gesamtüberleben mit einer minimalen Nachbeobachtungszeit von 20 Monaten sind in Abbildung 12 dargestellt.
Abbildung 12: Kaplan‑Meier‑Kurven des Gesamtüberlebens bei Patienten mit Tumorzell‑PD‑L1 ≥ 1 % (CA209648)
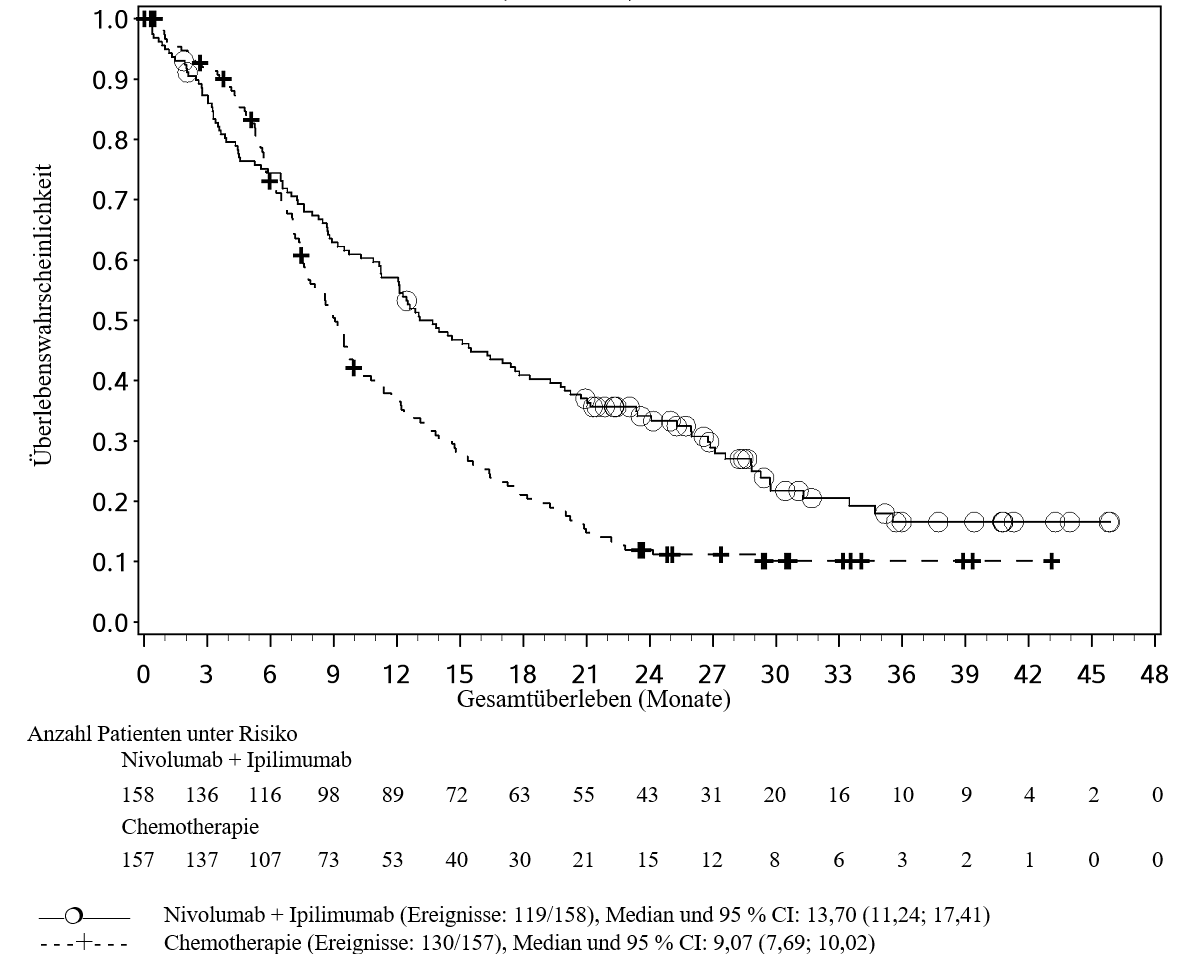
Basierend auf Datenschnitt: 23‑Aug‑2021, minimale Nachbeobachtungszeit von 20 Monaten
Hepatozelluläres Karzinom
Die Sicherheit und Wirksamkeit von 3 mg/kg Ipilimumab in Kombination mit 1 mg/kg Nivolumab alle 3 Wochen für maximal 4 Dosen, gefolgt von einer Nivolumab‑Monotherapie mit 480 mg alle 4 Wochen für die Erstlinienbehandlung des nicht resezierbaren oder fortgeschrittenen hepatozellulären Karzinoms (HCC) wurden in einer randomisierten, aktiv kontrollierten, offenen Phase‑III‑Studie (CA2099DW) untersucht. In die Studie eingeschlossen wurden erwachsene Patienten (ab 18 Jahren) mit histologisch bestätigtem HCC, Child‑Pugh‑Klassifikation A, ECOG‑Performance‑Status 0 oder 1, die keine vorherige systemische Therapie für die fortgeschrittene Erkrankung erhalten hatten. Eine Ösophagogastroduodenoskopie war vor der Aufnahme in die Studie nicht vorgeschrieben. In die Studie wurden Erwachsene eingeschlossen, deren Erkrankung nicht durch einen operativen Eingriff und/oder lokoregionäre Therapien behandelt werden konnte oder danach fortgeschritten ist. Eine vorausgegangene neoadjuvante oder adjuvante systemische Therapie war zulässig. Patienten mit aktiver Autoimmunerkrankung, Hirnmetastasen oder leptomeningealen Metastasen, vorheriger Lebertransplantation, einer Vorgeschichte von hepatischer Enzephalopathie (innerhalb von 12 Monaten vor der Randomisierung), klinisch signifikantem Aszites, Erkrankungen, die eine systemische Immunsuppression erfordern, einer HIV-Infektion oder aktiver Koinfektion mit Hepatitis-B-Virus (HBV) und Hepatitis-C-Virus (HCV) oder HBV und Hepatitis-D-Virus (HDV), waren von der Studie ausgeschlossen. Die Randomisierung wurde nach Ätiologie (HBV vs. HCV vs. nicht‑viral), makrovaskulärer Invasion und/oder extrahepatischer Manifestation (vorhanden oder nicht vorhanden) und Alpha-Fetoprotein-Werten (≥ 400 oder < 400 ng/ml) stratifiziert.
Insgesamt wurden 668 Patienten randomisiert und erhielten Ipilimumab in Kombination mit Nivolumab (n = 335) oder eine Behandlung mit Lenvatinib oder Sorafenib nach Wahl des Prüfarztes (n = 333). In dem Arm, der eine durch den Prüfarzt ausgewählte Behandlung vorsah, erhielten 85 % der behandelten Patienten Lenvatinib und 15 % Sorafenib. Die Patienten im Ipilimumab-plus-Nivolumab-Arm erhielten 3 mg/kg Ipilimumab alle 3 Wochen in Kombination mit 1 mg/kg Nivolumab alle 3 Wochen für bis zu maximal 4 Dosen, gefolgt von einer Nivolumab-Monotherapie mit 480 mg alle 4 Wochen. Patienten in dem Arm, der eine durch den Prüfarzt ausgewählte Behandlung vorsah, erhielten entweder 8 mg Lenvatinib täglich peroral (bei einem Körpergewicht von < 60 kg) oder 12 mg täglich peroral (bei einem Körpergewicht von ≥ 60 kg) oder Sorafenib 400 mg zweimal täglich peroral. Die Behandlung wurde bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate fortgesetzt. Patienten, die die Kombinationstherapie aufgrund einer Nebenwirkung, die Ipilimumab zugeordnet wurde, abbrechen mussten, konnten mit Nivolumab als Monotherapie weiterbehandelt werden. Tumorbeurteilungen wurden bei Studienbeginn, nach Randomisierung in Woche 9 und Woche 16, dann alle 8 Wochen bis zu 48 Wochen und anschließend alle 12 Wochen bis zum Progress der Erkrankung, dem Abbruch der Behandlung oder Einleitung der nachfolgenden Therapie durchgeführt.
Die bei Studieneintritt vorliegenden Merkmale der Patienten waren im Allgemeinen über die Behandlungsgruppen hinweg gleichmäßig verteilt. Das mediane Alter betrug 66 Jahre (Spanne: 20 bis 89); 53 % der Patienten waren ≥ 65 Jahre und 16 % waren ≥ 75 Jahre, 53 % der Patienten waren Kaukasier, 44 % waren Asiaten, 2,2 % waren Schwarze und 82 % waren männlich. Der ‑ECOG‑Performance‑Status bei Einschluss in die Studie war 0 (71 %) oder 1 (29 %). Vierunddreißig Prozent (34 %) der Patienten hatten eine HBV‑Infektion, 28 % eine HCV‑Infektion und bei 36 % gab es keine Nachweise für eine HBV- oder HCV‑Infektion. Neunzehn Prozent (19 %) der Patienten hatten eine alkoholbedingte Lebererkrankung und 11 % eine nichtalkoholische Fettlebererkrankung. Die Mehrheit der Patienten wies bei Studieneintritt BCLC‑Stadium C (73 %) auf, 19 % Stadium B und 6 % Stadium A. Der Anteil der Patienten mit Child‑Pugh‑Scores von 5, 6 und ≥ 7 betrug 77 %, 20 % bzw. 3 %. Bei insgesamt 54 % der Patienten lag eine extrahepatische Ausbreitung vor, 25 % hatten eine makrovaskuläre Invasion und 33 % wiesen AFP‑Werte ≥ 400 µg/l auf.
Die Studie zeigte eine statistisch signifikante Verbesserung des OS und der ORR für Patienten, die in den Behandlungsarm Ipilimumab in Kombination mit Nivolumab randomisiert worden waren, im Vergleich zu Lenvatinib oder Sorafenib nach Wahl des Prüfarztes. Die Wirksamkeitsergebnisse sind in Tabelle 22 und Abbildung 13 dargestellt.
Tabelle 22: Wirksamkeitsergebnisse bei HCC in der Erstlinie (CA2099DW)a
Ipilimumab + Nivolumab |
Lenvatinib oder Sorafenib |
|
Gesamtüberleben |
||
Ereignisse |
194 (58 %) |
228 (68 %) |
Median (Monate) |
23,7 |
20,6 |
Hazard Ratio (95 % CI)b |
0,79 (0,65; 0,96) |
|
p‑Wertc |
0,0180 |
|
Gesamtansprechen, n (%)d |
121 (36,1) |
44 (13,2) |
(95 % CI) |
(31,0; 41,5) |
(9,8; 17,3) |
p‑Werte |
< 0,0001 |
|
Vollständiges Ansprechen (%) |
23 (6,9) |
6 (1,8) |
Teilweises Ansprechen (%) |
98 (29,3) |
38 (11,4) |
Ansprechdauer (Monate)d |
||
Median |
30,4 |
12,9 |
a Mindest‑Nachbeobachtungszeit: 26,8 Monate. Mediane Nachbeobachtungszeit: 35,2 Monate. | ||
Abbildung 13: Kaplan‑Meier‑Kurven des OS in der Erstlinie bei Patienten mit HCC (CA2099DW)
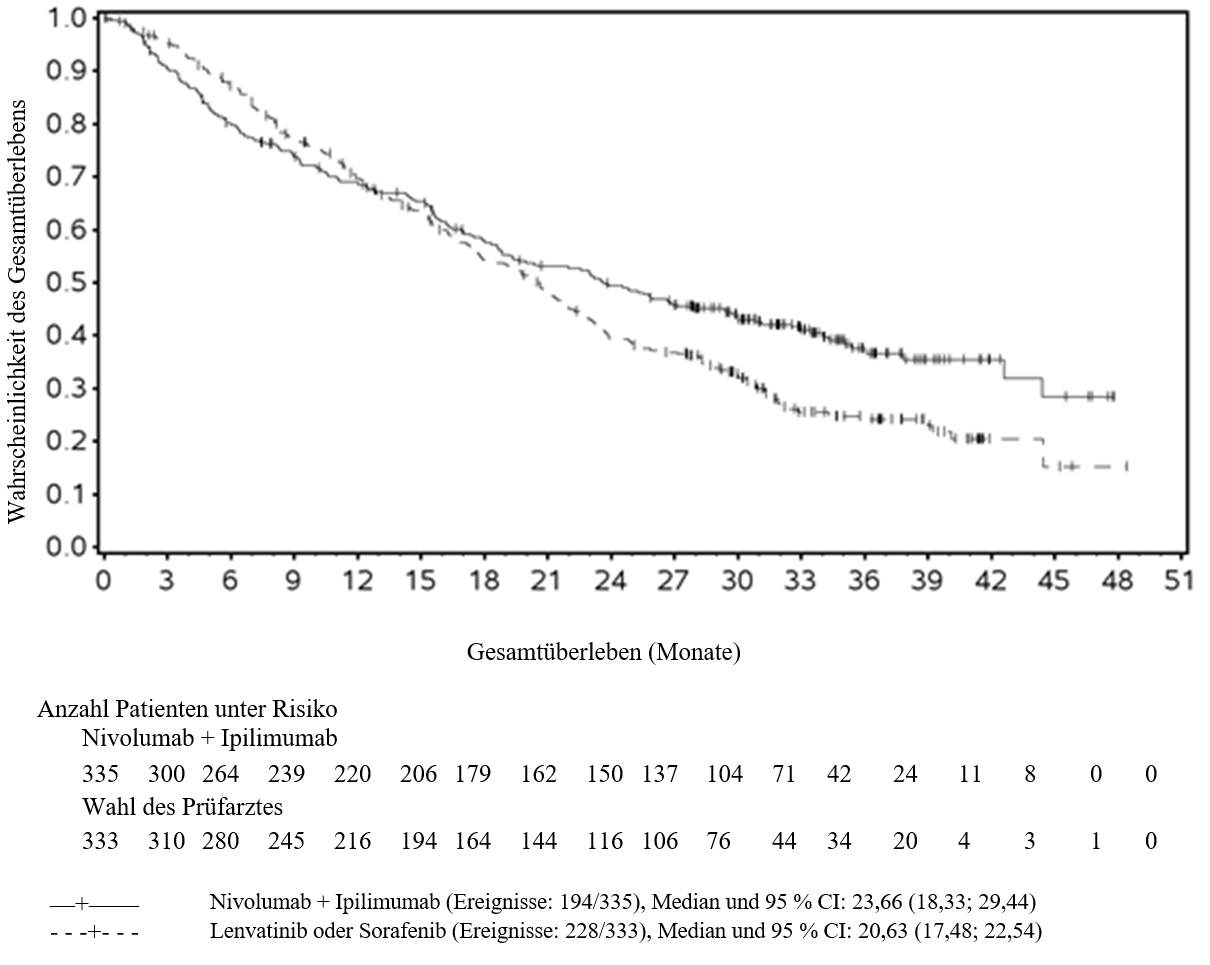
Kinder und Jugendliche
Ipilimumab als Monotherapie
Die Studie CA184‑070 war eine multizentrische, open‑label Phase‑I‑Dosiseskalierungsstudie, bei pädiatrischen Patienten im Alter von ≥ 1 bis ≤ 21 Jahren mit messbaren/evaluierbaren, nicht behandelbaren, rezidivierenden oder refraktären festen malignen Tumoren ohne Aussicht auf kurative Behandlung durch eine Standardtherapie. In die Studie waren 13 Patienten < 12 Jahre und 20 Patienten ≥ 12 Jahre eingeschlossen. Ipilimumab wurde alle 3 Wochen für 4 Dosen verabreicht und danach alle 12 Wochen sofern keine dosislimitierende Toxizität (DLT) oder Progression vorlagen. Die primären Endpunkte waren Sicherheit und Pharmakokinetik (PK). Von den Patienten, die 12 Jahre oder älter waren mit fortgeschrittenem Melanom wurde Ipilimumab 5 mg/kg bei 3 Patienten und Ipilimumab 10 mg/kg bei 2 Patienten angewendet. Eine stabile Erkrankung wurde bei 2 Patienten mit der Ipilimumab‑Dosis von 5 mg/kg erreicht, eine mit einer Dauer von > 22 Monaten.
Die Studie CA184‑178 war eine nicht‑randomisierte, multizentrische, offene Phase‑II‑Studie bei jugendlichen Patienten im Alter von 12 bis < 18 Jahren mit zuvor behandeltem oder unbehandeltem, nicht resezierbarem malignem Melanom im Stadium III oder IV. Ipilimumab wurde alle 3 Wochen für 4 Dosen verabreicht. Der primäre Wirksamkeitsendpunkt war die 1‑Jahres‑Überlebensrate. Die sekundären Wirksamkeitsendpunkte der besten Gesamtansprechrate (BORR), der stabilen Krankheit (SD), der Krankheitskontrollrate (DCR) und des progressionsfreien Überlebens (PFS) basierten auf mWHO‑Kriterien und wurden durch die Bewertung des Prüfarztes bestimmt. Das Gesamtüberleben (OS) wurde ebenfalls bewertet. Die Tumorbeurteilung wurde in Woche 12 durchgeführt. Alle Patienten wurden mindestens 1 Jahr lang beobachtet. Ipilimumab 3 mg/kg wurde 4 Patienten und Ipilimumab 10 mg/kg wurde 8 Patienten verabreicht. Die meisten Patienten waren männlich (58 %) und kaukasisch (92 %). Das mediane Alter lag bei 15 Jahren. Eine stabile Erkrankung wurde unter Ipilimumab 3 mg/kg bei einem Patienten für 260 Tage und unter Ipilimumab 10 mg/kg bei einem Patienten für etwa 14 Monate erreicht. 2 Patienten, die mit 10 mg/kg Ipilimumab behandelt wurden, zeigten eine partielle Remission, von denen eine länger als 1 Jahr eine dauerhafte Remission zeigte. Zusätzliche Wirksamkeitsergebnisse sind in Tabelle 23 dargestellt.
Tabelle 23: Wirksamkeitsergebnisse aus CA184178
Ipilimumab 3 mg/kg |
Ipilimumab 10 mg/kg |
|
1‑Jahres‑OS (%) (95 % CI) |
75 % (12,8; 96,1) |
62,5 % (22,9; 86,1) |
BORR (%) (95 % CI) |
0 % (0; 60,2) |
25 % (3,2; 65,1) |
SD (n/N)a |
1/4 |
1/8 |
DCR (%) (95 % CI) |
25 % (0,6; 80,6) |
37,5 % (8,5; 75,5) |
Medianes PFS (Monate) (95 % CI) |
2,6 (2,3; 8,5) |
2,9 (0,7;NEa) |
Medianes OS (Monate) (95 % CI) |
18,2 (8,9; 18,2) |
Nicht erreicht (5,2; NE) |
a NE = nicht abschätzbar (not estimable) | ||
Ipilimumab in Kombination mit Nivolumab
Bei der Studie CA209070 handelte es sich um eine offene, einarmige Dosisbestätigungs‑ und Dosiserweiterungsstudie der Phase I/II zu Nivolumab als Einzelwirkstoff und in Kombination mit Ipilimumab bei pädiatrischen und jungen erwachsenen Patienten mit rezidivierenden oder refraktären soliden oder hämatologischen Tumoren, einschließlich Neuroblastomen, Osteosarkomen, Rhabdomyosarkomen, Ewing‑Sarkomen, fortgeschrittenen Melanomen, cHL und Non‑Hodgkin‑Lymphomen (NHL). Unter den 126 behandelten Patienten befanden sich 97 pädiatrische Patienten im Alter von 12 Monaten bis < 18 Jahren. Von den 97 pädiatrischen Patienten wurden 64 mit der Nivolumab‑Monotherapie (3 mg/kg intravenös verabreicht über 60 Minuten alle 2 Wochen) und 33 mit Ipilimumab in Kombination mit Nivolumab (Nivolumab 1 mg/kg oder 3 mg/kg intravenös verabreicht über 60 Minuten in Kombination mit Ipilimumab 1 mg/kg intravenös verabreicht über 90 Minuten alle 3 Wochen für die ersten 4 Dosen, gefolgt von Nivolumab 3 mg/kg als Monotherapie alle 2 Wochen) behandelt. Die Patienten erhielten entweder Nivolumab als Monotherapie für im Median 2 Dosen (Spanne: 1, 89) oder Ipilimumab in Kombination mit Nivolumab für im Median 2 Dosen (Spanne: 1, 24). Die wichtigsten primären Wirksamkeitskriterien waren Sicherheit, Verträglichkeit und Antitumoraktivität, beurteilt nach deskriptiver ORR und deskriptivem OS.
Unter den 64 mit der Nivolumab‑Monotherapie behandelten pädiatrischen Patienten befanden sich 60 Patienten mit auswertbarem Ansprechen (Melanom n = 1, solide Tumoren n = 47 und hämatologische Tumoren n = 12). Bei den 48 pädiatrischen Patienten mit auswertbarem Ansprechen mit Melanom oder soliden Tumoren wurde kein objektives Ansprechen beobachtet. Bei den 12 pädiatrischen Patienten mit auswertbarem Ansprechen mit hämatologischen Tumoren lag die ORR bei 25,0 % (95 % CI: 5,5; 57,2), darunter 1‑mal vollständiges Ansprechen bei cHL und 2‑mal teilweises Ansprechen, eines bei cHL und ein anderes bei NHL. In den deskriptiven Analysen für die 64 mit der Nivolumab‑Monotherapie behandelten pädiatrischen Patienten betrug das mediane OS 6,67 Monate (95 % CI: 5,98; NA); 6,14 Monate (95 % CI: 5,39; 24,67) für Patienten mit Melanom oder soliden Tumoren; für Patienten mit hämatologischen Tumoren wurde es nicht erreicht.
Bei den 30 pädiatrischen Patienten mit auswertbarem Ansprechen, welche mit Ipilimumab in Kombination mit Nivolumab behandelt wurden (nur solide Tumoren mit Ausnahme des Melanoms), wurde kein objektives Ansprechen beobachtet. Bei den 33 pädiatrischen Patienten, die mit Ipilimumab in Kombination mit Nivolumab behandelt wurden, betrug in einer deskriptiven Analyse das mediane OS 8,25 Monate (95 % CI: 5,45; 16,95).
Bei der Studie CA209908 handelt es sich um eine offene, sequenzielle klinische Studie der Phase Ib/II mit Nivolumab‑Monotherapie und Ipilimumab in Kombination mit Nivolumab bei pädiatrischen und heranwachsenden Patienten mit hochgradig malignen primären Tumoren des ZNS, einschließlich diffusem intrinsischen Ponsgliom (DIPG), hochmalignem Gliom, Medulloblastom, Ependymom und anderen rezidivierenden Subtypen von hochgradig malignen ZNS‑Erkrankungen (z. B. Pineoblastom, atypische teratoide/rhabdoide Tumoren und embryonalen ZNS‑Tumoren). Von den 151 pädiatrischen Patienten (im Alter von ≥ 6 Monaten bis < 18 Jahren), die in die Studie eingeschlossen wurden, wurden 77 mit Nivolumab‑Monotherapie (3 mg/kg alle 2 Wochen) und 74 mit Ipilimumab 1 mg/kg in Kombination mit Nivolumab 3 mg/kg, alle 3 Wochen für 4 Dosen, gefolgt von einer Nivolumab‑Monotherapie 3 mg/kg alle 2 Wochen behandelt. Das primäre Wirksamkeitskriterium in der DIPG‑Kohorte war das Gesamtüberleben (Overall Survival = OS). Für alle anderen Tumorarten war das primäre Wirksamkeitskriterium das vom Prüfarzt bewertete progressionsfreie Überleben (Progression‑Free Survival = PFS), basierend auf den RANO‑Kriterien. Das mediane OS in der DIPG‑Kohorte betrug 10,97 Monate (80 % CI: 9,92; 12,16) bei Patienten, die mit Nivolumab‑Monotherapie behandelt wurden, und 10,50 Monate (80 % CI: 9,10; 12,32) bei Patienten, die mit Ipilimumab in Kombination mit Nivolumab behandelt wurden. Bei allen anderen untersuchten pädiatrischen ZNS‑Tumorarten lag das mediane PFS zwischen 1,23 und 2,35 Monaten bei Patienten, die mit Nivolumab‑Monotherapie behandelt wurden und zwischen 1,45 und 3,09 Monaten bei Patienten, die mit Ipilimumab in Kombination mit Nivolumab behandelt wurden. In der Studie wurde kein objektives Ansprechen beobachtet, mit Ausnahme eines Ependymom‑Patienten, der mit Nivolumab‑Monotherapie behandelt wurde und ein teilweises Ansprechen zeigte. Die in der Studie CA209908 beobachteten Ergebnisse bezüglich OS, PFS und Objektiver Ansprechrate (Objective Response Rate = ORR) deuten nicht auf einen klinisch bedeutsamen Nutzen hin, der über die zu erwartenden Beobachtungen in dieser Patientenpopulation hinausgeht.
Die Pharmakokinetik von Ipilimumab wurde bei 785 Patienten mit fortgeschrittenem Melanom untersucht, die Induktionsdosen zwischen 0,3 und 10 mg/kg erhielten (alle 3 Wochen für 4 Dosen). Cmax, Cmin und AUC verhielten sich bei Ipilimumab innerhalb des untersuchten Dosisbereichs proportional zur Dosis. Bei wiederholter Ipilimumab‑Dosis alle 3 Wochen war die Clearance (CL) zeitinvariant, und es wurde nur eine minimale systemische Akkumulation beobachtet, ersichtlich aus einem höchstens 1,5‑fachen Akkumulationsindex. Der Steady‑State‑Level von Ipilimumab wurde mit der dritten Dosis erreicht. Bei einer Populations‑Pharmakokinetik‑Analyse wurden folgende mittlere Parameter (Variationskoeffizient in Prozent) von Ipilimumab ermittelt: Terminale Halbwertszeit von 15,4 Tagen (34,4 %); systemische CL von 16,8 ml/h (38,1 %) und Verteilungsvolumen im Steady‑State von 7,47 l (10,1 %). Die mit dem Ipilimumab‑Induktionsregime in der Dosierung 3 mg/kg im Steady‑State erreichte mittlere Ipilimumab Cmin (Variationskoeffizient in Prozent) war 19,4 µg/ml (74,6 %).
Die CL von Ipilimumab stieg mit höherem Körpergewicht und höherer LDH vor Therapiebeginn an; bei einer Verabreichung auf mg/kg‑Basis ist jedoch bei erhöhten LDH‑Werten oder höherem Körpergewicht keine Anpassung der Dosis erforderlich. Die CL wurde nicht beeinflusst von Alter (Bereich 23‑88 Jahre), Geschlecht, dem gleichzeitigen Einsatz von Budesonid oder Dacarbazin, Performance‑Status, HLA‑A2*0201‑Status, leicht eingeschränkter Leberfunktion, eingeschränkter Nierenfunktion, Immunogenität und einer früheren Tumortherapie. Die Auswirkung der ethnischen Zugehörigkeit wurde nicht untersucht, da nicht genug Daten von Patientengruppen nicht‑kaukasischer Herkunft vorlagen. Es wurden keine kontrollierten Studien zur Pharmakokinetik von Ipilimumab bei Kindern und Jugendlichen oder bei Patienten mit Leber‑ oder Nierenfunktionsstörung durchgeführt.
Basierend auf einer Exposure‑Response‑Analyse von 497 Patienten mit fortgeschrittenem Melanom, war das Gesamtüberleben unabhängig von einer früheren systemischen Tumortherapie und stieg mit höherer Ipilimumab Cmin (Steady State)‑Plasmakonzentration.
YERVOY in Kombination mit Nivolumab: Als Ipilimumab 1 mg/kg in Kombination mit Nivolumab 3 mg/kg angewendet wurde, sank die CL von Ipilimumab um 1,5 % und stieg die CL von Nivolumab um 1 %. Dies wurde als nicht klinisch relevant erachtet. Als Ipilimumab 3 mg/kg in Kombination mit Nivolumab 1 mg/kg angewendet wurde, stieg die CL von Ipilimumab um 9 % und die CL von Nivolumab um 29 % an. Dies wurde als nicht klinisch relevant erachtet.
Die CL von Ipilimumab stieg bei der Gabe in Kombination mit Nivolumab um 5,7 % an, wenn Anti‑Ipilimumab‑Antikörper präsent waren. Die CL von Nivolumab stieg bei der Gabe in Kombination mit Ipilimumab um 20 %, wenn Anti‑Nivolumab‑Antikörper präsent waren. Diese Veränderungen wurden als nicht klinisch relevant erachtet.
YERVOY in Kombination mit Nivolumab und Chemotherapie: Wenn Ipilimumab 1 mg/kg alle 6 Wochen in Kombination mit Nivolumab 360 mg alle 3 Wochen und mit 2 Zyklen Chemotherapie verabreicht wurde, stieg die CL von Ipilimumab um circa 22 % an und die CL von Nivolumab sank um circa 10 %, was als nicht klinisch relevant betrachtet wurde.
Eingeschränkte Nierenfunktion
Bei der Analyse der Populations‑Pharmakokinetik der Daten aus klinischen Studien von Patienten mit metastasiertem Melanom hatten vorbestehende leicht und mäßig eingeschränkte Nierenfunktion keinen Einfluss auf die Ipilimumab‑CL. Es sind nur begrenzt klinische und pharmakokinetische Daten bei vorbestehender schwer eingeschränkter Nierenfunktion verfügbar; es kann daher nicht bestimmt werden, inwieweit eine Dosisanpassung erforderlich ist.
Eingeschränkte Leberfunktion
Bei der Analyse der Populations‑Pharmakokinetik der Daten aus klinischen Studien von Patienten mit metastasiertem Melanom hatte eine vorbestehende leicht eingeschränkte Leberfunktion keinen Einfluss auf die Ipilimumab‑CL. Es sind nur begrenzt klinische und pharmakokinetische Daten bei vorbestehender mäßig eingeschränkter Leberfunktion verfügbar; es kann daher nicht bestimmt werden, inwieweit eine Dosisanpassung erforderlich ist. Es gab keine Patienten mit vorbestehender schwer eingeschränkter Leberfunktion in klinischen Studien.
Kinder und Jugendliche
Bei der Ipilimumab‑Monotherapie nahm basierend auf einer Populations‑PK‑Analyse unter Verwendung von verfügbaren gepoolten Daten von 565 Patienten aus 4 Phase‑II‑Erwachsenenstudien (N = 521) und 2 pädiatrischen Studien (N = 44) die CL von Ipilimumab mit steigendem Ausgangskörpergewicht zu. Das Alter (2‑87 Jahre) hatte keine klinisch relevante Auswirkung auf die CL von Ipilimumab. Die geschätzte geometrische mittlere CL beträgt bei jugendlichen Patienten im Alter von ≥ 12 bis < 18 Jahren 8,72 ml/h. Expositionen bei Jugendlichen sind vergleichbar mit denen bei Erwachsenen, die die gleiche mg/kg‑Dosis erhalten. Basierend auf der Simulation in Erwachsenen und pädiatrischen Patienten wird eine vergleichbare Exposition bei Erwachsenen und Kindern und Jugendlichen in der empfohlenen Dosis von 3 mg/kg alle 3 Wochen erreicht.
Für Ipilimumab in Kombination mit Nivolumab wird erwartet, dass die Expositionen von Ipilimumab und Nivolumab bei pädiatrischen Patienten ab 12 Jahren bei der empfohlenen Dosis mit denen bei erwachsenen Patienten vergleichbar sind.
In Toxizitätsstudien an Affen mit wiederholter intravenöser Verabreichung wurde Ipilimumab im Allgemeinen gut vertragen. Immunvermittelte Nebenwirkungen wurden selten (ca. 3 %) beobachtet und schlossen Kolitis (welche in einem Fall tödlich war), Dermatitis und infusionsbedingte Reaktionen (möglicherweise aufgrund einer akuten Zytokin‑Freisetzung wegen einer schnellen Injektion) ein. In einer Studie wurde ein verringertes Gewicht von Schilddrüse und Hoden festgestellt ohne histologische Befunde. Die klinische Relevanz dieser Beobachtung ist nicht bekannt.
Die Wirkungen von Ipilimumab auf die prä‑ und postnatale Entwicklung wurden in einer Studie an Cynomolgus‑Affen untersucht. Trächtige Affen erhielten nach Einsetzen der Organogenese im ersten Trimester bis zur Geburt alle 3 Wochen Ipilimumab mit entweder gleichen oder höheren Expositionen (AUC) als diejenigen, die mit der klinischen Dosierung von 3 mg/kg Ipilimumab assoziiert werden. In den ersten zwei Trimestern der Trächtigkeit wurden keine mit der Behandlung in Zusammenhang stehenden Nebenwirkungen gefunden. Mit Beginn des dritten Trimesters traten im Vergleich zur Kontrollgruppe in beiden Ipilimumab‑Gruppen höhere Abortraten, mehr Totgeburten und Frühgeburten (mit entsprechendem geringerem Geburtsgewicht) und Jungensterblichkeit auf; diese Ergebnisse waren dosisabhängig. Zusätzlich wurden externe oder viszerale Anomalien im Urogenitaltrakt zweier Jungtiere festgestellt, die Ipilimumab in utero ausgesetzt waren. Ein weibliches Jungtier wies eine einseitige Agenesie der linken Niere und des Harnleiters auf und ein männliches Jungtier hatte eine unperforierte Harnröhre mit damit verbundener Harnstauung und ein subkutanes Skrotumödem. Der Bezug dieser Missbildungen zur Behandlung ist nicht geklärt.
Studien zum mutagenen und karzinogenen Potenzial von Ipilimumab wurden nicht durchgeführt. Auch Fertilitätsstudien wurden für Ipilimumab nicht durchgeführt.
Trometamolhydrochlorid (2‑Amino‑2‑hydroxymethyl‑1,3‑propandiol‑hydrochlorid)
Natriumchlorid
Mannitol (E 421)
Pentetsäure (Diethylen‑triaminpentaessigsäure)
Polysorbat 80
Natriumhydroxid (zum Einstellen des pH‑Werts)
Salzsäure (zum Einstellen des pH‑Werts)
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Ungeöffnete Durchstechflasche
3 Jahre
Nach Öffnen
Aus mikrobiologischer Sicht sollte das Arzneimittel nach Anbruch der Durchstechflasche sofort infundiert oder verdünnt und infundiert werden. Für die unverdünnte bzw. verdünnte Infusionslösung (zwischen 1 und 4 mg/ml) konnte bei Lagertemperaturen von 25 °C und 2 °C bis 8 °C eine chemische und physikalische Stabilität von 24 Stunden nach Anbruch nachgewiesen werden. Wenn die Infusionslösung (unverdünnt oder verdünnt) nicht sofort verwendet wird, kann sie bis zu 24 Stunden im Kühlschrank (2 °C bis 8 °C) oder bei Raumtemperatur (20 °C bis 25 °C) aufbewahrt werden.
Im Kühlschrank lagern (2 °C ‑ 8 °C).
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Anbruch oder Verdünnung des Arzneimittels, siehe Abschnitt 6.3.
10 ml Konzentrat zur Herstellung einer Infusionslösung in einer Durchstechflasche (Glas Typ 1) mit einem Stopfen (beschichtetes Butylgummi) und Flip‑Off‑Verschluss (Aluminium). Packungsgröße 1.
40 ml Konzentrat zur Herstellung einer Infusionslösung in einer Durchstechflasche (Glas Typ 1) mit einem Stopfen (beschichtetes Butylgummi) und Flip‑Off‑Verschluss (Aluminium). Packungsgröße 1.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Die Zubereitung sollte, besonders im Hinblick auf die Asepsis, durch geschultes Personal im Einklang mit den Richtlinien zur guten klinischen Praxis durchgeführt werden.
Berechnung der Dosis:
Ipilimumab‑Monotherapie oder Ipilimumab in Kombination mit Nivolumab:
Die verordnete Dosis für den Patienten wird in mg/kg angegeben. Berechnen Sie die notwendige Gesamtdosis ausgehend von dieser verschriebenen Dosis. Möglicherweise wird mehr als eine Durchstechflasche YERVOY‑Konzentrat benötigt, um die Gesamtdosis für den Patienten zu erhalten.
Jede 10‑ml‑Durchstechflasche YERVOY‑Konzentrat enthält 50 mg Ipilimumab; jede 40‑ml‑Durchstechflasche YERVOY‑Konzentrat enthält 200 mg Ipilimumab.
Die Gesamtdosis Ipilimumab in mg = das Körpergewicht des Patienten x die verordnete Dosis in mg/kg
Das benötigte Volumen des Ipilimumab‑Konzentrats, um die Dosis zuzubereiten, (ml) = die Gesamtdosis in mg, dividiert durch 5 (die Stärke des YERVOY‑Konzentrats beträgt 5 mg/ml).
Zubereitung der Infusion:
Achten Sie bei der Zubereitung der Infusion auf eine aseptische Durchführung.
YERVOY kann für die intravenöse Verabreichung verwendet werden, entweder
ohne Verdünnung nach der Überführung in ein Infusionsbehältnis mittels einer geeigneten sterilen Spritze;
oder
nach der bis zu 5‑fachen Verdünnung der Ausgangsmenge des Konzentrats (bis zu 4 Anteilen Verdünnungsmittel zu 1 Anteil Konzentrat). Die Endkonzentration sollte bei 1 bis 4 mg/ml liegen. Um das YERVOY‑Konzentrat zu verdünnen, verwenden Sie entweder:
Natriumchloridlösung 9 mg/ml (0,9 %) für Injektionszwecke; oder
Glucoselösung 50 mg/ml (5 %) für Injektionszwecke
SCHRITT 1
Lassen Sie die entsprechende Anzahl an Durchstechflaschen YERVOY für etwa 5 Minuten bei Raumtemperatur stehen.
Untersuchen Sie das YERVOY‑Konzentrat auf Schwebstoffteilchen oder Verfärbung. Das YERVOY‑Konzentrat ist eine klare bis leicht opaleszierende, farblose bis blass gelbe Flüssigkeit, die helle (wenige) Schwebstoffe enthalten kann. Verwenden Sie das Konzentrat nicht bei einer hohen Menge an Schwebstoffen oder Anzeichen einer Verfärbung.
Entnehmen Sie die benötigte Menge YERVOY‑Konzentrat mit einer geeigneten sterilen Spritze.
SCHRITT 2
Überführen Sie das Konzentrat in eine sterile entlüftete Glasflasche oder einen Infusionsbeutel (PVC oder nicht‑PVC).
Verdünnen Sie das Konzentrat gegebenenfalls mit der benötigten Menge Natriumchloridlösung 9 mg/ml (0,9 %) für Injektionszwecke oder Glucoselösung 50 mg/ml (5 %) für Injektionszwecke. Um das Zubereiten zu erleichtern, kann das Konzentrat auch direkt in einen vorgefüllten Infusionsbeutel, der die entsprechende Menge Natriumchloridlösung 9 mg/ml (0,9 %) für Injektionszwecke oder Glucoselösung 50 mg/ml (5 %) für Injektionszwecke enthält, gegeben werden. Infusion vorsichtig durch manuelle Drehung mischen.
Anwendung:
Die YERVOY‑Infusion darf nicht als intravenöse Druck‑ oder Bolus‑Injektion verabreicht werden.
Verabreichen Sie die YERVOY‑Infusion intravenös über einen Zeitraum von 30 Minuten.
Die YERVOY‑Infusion sollte nicht gleichzeitig mit anderen Arzneimitteln über dieselbe intravenöse Infusionsleitung infundiert werden. Verwenden Sie eine gesonderte Infusionsleitung.
Verwenden Sie ein Infusionsset und einen sterilen, pyrogenfreien In‑Line‑Filter mit geringer Proteinbindung (Porengröße: 0,2 bis 1,2 μm).
Die YERVOY‑Infusion ist kompatibel mit:
PVC‑Infusionssets
In‑Line‑Filtern aus Polyethersulfon (0,2 bis 1,2 μm) und Nylon (0,2 μm)
Spülen Sie die Infusionsleitung am Ende der Infusion mit Natriumchloridlösung 9 mg/ml (0,9 %) für Injektionszwecke oder Glucoselösung 50 mg/ml (5 %) für Injektionszwecke.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Bristol‑Myers Squibb Pharma EEIG
Plaza 254
Blanchardstown Corporate Park 2
Dublin 15, D15 T867
Irland
EU/1/11/698/001‑002
Datum der Erteilung der Zulassung: 13. Juli 2011
Datum der letzten Verlängerung der Zulassung: 21. April 2016
Mai 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel‑Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig
Bristol-Myers Squibb GmbH & Co. KGaA
Arnulfstraße 29
80636 München
Medizinische Information
Telefon: 0800 0752002
E-Mail: medwiss.info@bms.com
www.bmsmedinfo.de