innohep® 20.000 Anti-Xa I.E./ml Durchstechflaschen Injektionslösung
Tinzaparin-Natrium 20.000 Anti-Xa I.E./ml
Sonstige Bestandteile mit bekannter Wirkung:
Benzylalkohol (10 mg/ml), Natriummetabisulfit (1,83 mg/ml) und Natrium (bis zu 40 mg/ml ).
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
Injektionslösung
Die farblose 2 ml-Durchstechflasche enthält eine klare, farblose oder leicht gelbliche Flüssigkeit. Sie ist nicht getrübt und es entsteht kein Bodensatz bei Lagerung.
Zur Behandlung von Venenthrombosen und thromboembolischen Erkrankungen einschließlich tiefer Venenthrombosen und Lungenembolien bei Erwachsenen.
Langzeitbehandlung venöser Thromboembolien und Rezidivprophylaxe bei erwachsenen Patienten mit aktiver Tumorerkrankung.
Bei bestimmten Patienten mit Lungenembolien (z. B. Patienten mit schwerer hämodynamischer Instabilität) kann eine alternative Behandlung wie z. B. eine Operation oder Thrombolyse angezeigt sein.
Dosierung
Behandlung von Erwachsenen
175 Anti-Xa I.E./kg Körpergewicht sollen einmal täglich für mindestens 6 Tage gegeben werden, bis eine gleichwertige orale Antikoagulation erreicht ist.
Längerfristige Behandlung bei erwachsenen Patienten mit aktiver Tumorerkrankung
175 Anti-Xa I.E./kg Körpergewicht sollen einmal täglich subkutan über eine empfohlene Behandlungsdauer von 6 Monaten gegeben werden. Der Nutzen einer weitergeführten Behandlung mit Antikoagulanzien, die über die Dauer von 6 Monaten hinausgeht, soll evaluiert werden.
Neuroaxiale Anästhesie
Bei Patienten, die eine neuroaxiale Anästhesie erhalten, sind therapeutische Dosen von innohep (175 I.E./kg) kontraindiziert (siehe Abschnitt 4.3). Wenn eine neuroaxiale Anästhesie geplant ist, sollte innohep spätestens 24 Stunden vor dem Eingriff abgesetzt werden. Die Gabe von innohep soll frühestens 4-6 Stunden nach der spinalen Anästhesie oder nach Entfernen des Katheters fortgesetzt werden.
Austauschbarkeit
Für die Austauschbarkeit mit anderen niedermolekularen Heparinen (NMH) siehe Abschnitt 4.4.
Kinder und Jugendliche
Zu Sicherheit und Wirksamkeit von innohep® bei Kindern und Jugendlichen unter 18 Jahren liegen keine ausreichenden Erfahrungen vor. Derzeit verfügbare Daten sind in Abschnitt 5.2 beschrieben. Es kann jedoch keine Dosierungsempfehlung gegeben werden.
Eingeschränkte Nierenfunktion
Wird eine Beeinträchtigung der Nieren vermutet, soll die Nierenfunktion anhand der Kreatinin-Clearance abgeschätzt werden (unter Verwendung einer Formel auf Basis des Serum-Kreatinins).
Die Anwendung von innohep bei Patienten mit einer Kreatinin-Clearance unter 30 ml/min wird nicht empfohlen, da für diese Patientengruppe keine Dosis ermittelt wurde. Die vorhandene Datenlage belegt, dass bei Patienten mit einer Kreatinin-Clearance bis ≥ 20 ml/min keine Akkumulation stattfindet.
Bei Bedarf kann bei diesen Patienten, wenn der Nutzen die Risiken überwiegt, die Behandlung mit innohep mit einer Anti-Xa-Überwachung begonnen werden (siehe Abschnitt 4.4, Eingeschränkte Nierenfunktion). In diesem Fall ist die Dosis von innohep, falls erforderlich, auf Basis der Anti-Faktor-Xa-Aktivität anzupassen. Liegt diese unter- oder oberhalb des gewünschten Bereichs, sollte die Dosis von innohep entsprechend erhöht bzw. reduziert und die Messung der Anti-Faktor-Xa-Aktivität nach 3-4 neuen Dosen wiederholt werden. Dosisanpassungen sollten solange erfolgen, bis die gewünschte Anti-Faktor-Xa-Aktivität erreicht ist. Als Orientierung dienen hier mittlere Werte zwischen 0,5 und 1,5 I.E. Anti-Faktor-Xa/ml, die im Zeitraum von 4 bis 6 Stunden nach Gabe an gesunde Probanden und an Patienten ohne stark eingeschränkte Nierenfunktion gemessen wurden. Die Bestimmung der Anti-Faktor-Xa-Aktivität erfolgte mithilfe eines Chromogen-Assays.
Ältere
innohep® sollte bei älteren Patienten mit der Standard-Dosis angewendet werden. Bei der Behandlung von älteren Patienten, deren Nierenfunktion eingeschränkt ist, ist Vorsicht geboten. Bei Verdacht einer eingeschränkten Nierenfunktion, siehe Abschnitt 4.2: Eingeschränkte Nierenfunktion und Abschnitt 4.4: Eingeschränkte Nierenfunktion.
Art der Anwendung
Parenterale Produkte sollten vor der Anwendung visuell inspiziert werden. Das Arzneimittel darf nicht angewendet werden, wenn Eintrübungen oder Niederschlag zu beobachten sind. Die Flüssigkeit kann sich während der Lagerung gelb verfärben, ist aber dennoch verwendbar.
Die Gabe erfolgt mittels subkutaner Injektion wie folgt: innohep® kann in die Bauchhaut, in die Außenseite des Oberschenkels, den unteren Rückenbereich, den Oberschenkel oder den Oberarm injiziert werden. Die Injektion darf nicht im Bereich des Bauchnabels oder von Narben oder in Wunden erfolgen. Bei Injektionen in die Bauchhaut sollte der Patient auf dem Rücken liegen und die Injektion alternierend auf der rechten und auf der linken Seite vorgenommen werden. Die Luftblase in der Spritze darf nicht herausgedrückt werden. Zur Injektion sollte an der Injektionsstelle eine Hautfalte gebildet werden.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Aktuelle oder anamnestisch bekannte immunvermittelte Heparin-induzierte Thrombozytopenie (Typ II) (siehe Abschnitt 4.4).
Akute schwere Blutungen oder Zustände, die schwere Blutungen begünstigen. Eine schwere Blutung ist dadurch definiert, dass eines der drei nachfolgenden Kriterien erfüllt ist:
Auftreten in einem kritischen Bereich oder Organ (z. B. intrakranial, intraspinal, intraokular, retroperitoneal, intraartikulär oder perikardial, intrauterin oder intramuskulär mit Kompartmentsyndrom),
Abfall des Hämoglobin-Wertes um 20 g/l (1,24 mmol/l) oder mehr, oder
Transfusion von zwei oder mehr Einheiten von Vollblut oder roten Blutkörperchen erforderlich.
Septische Endocarditis.
innohep® Durchstechflaschen zur Mehrfachentnahme enthalten 10 mg/ml Benzylalkohol als Konservierungsmittel. Diese Formulierung darf aufgrund des Risikos der Schnappatmung nicht bei Frühgeborenen oder Neugeborenen angewendet werden.
Bei Patienten, die eine neuroaxiale Anästhesie erhalten, sind therapeutische Dosen von innohep® (175 I.E./kg) kontraindiziert. Wenn eine neuroaxiale Anästhesie geplant ist, soll innohep® spätestens 24 Stunden vor dem Eingriff abgesetzt werden. Die Gabe von innohep® sollte frühestens 4 - 6 Stunden nach der spinalen Anästhesie oder nach Entfernen des Katheters fortgesetzt werden. Patienten sollen engmaschig auf Anzeichen und Symptome einer neurologischen Verletzung hin beobachtet werden.
Blutungsneigung
innohep® darf Patienten mit Blutungsneigung nur mit Vorsicht verabreicht werden. Zu Patienten mit einem Risiko für schwere Blutungen siehe Abschnitt 4.3. Die Kombination mit Arzneimitteln, die die Funktion der Blutplättchen oder die Gerinnung beeinflussen, soll vermieden oder aber engmaschig überwacht werden (siehe Abschnitt 4.5).
Intramuskuläre Injektionen
Aufgrund des Risikos von Hämatomen sollte innohep nicht intramuskulär verabreicht werden. Darüber hinaus sollten gleichzeitige intramuskuläre Injektionen vermieden werden.
Heparin-induzierte Thrombozytopenie
Die Thrombozytenzahl soll aufgrund des Risikos immunvermittelter, Heparin-induzierter Thrombozytopenie (Typ II) vor Beginn der Behandlung und in regelmäßigen Abständen danach gemessen werden. Bei Patienten, die eine immunvermittelte, Heparin-induzierte Thrombozytopenie (Typ II) entwickeln, muss innohep® abgesetzt werden (siehe Abschnitt 4.3 und 4.8). Die Thrombozytenzahl normalisiert sich für gewöhnlich innerhalb von 2 - 4 Wochen nach Absetzen.
Regelmäßige Kontrollen der Thrombozytenzahl sind auch bei der Langzeitbehandlung von tumorassoziierten Thrombosen erforderlich, insbesondere im ersten Behandlungsmonat, vor dem Hintergrund, dass Tumorerkrankungen und deren Behandlungen, wie z. B. Chemotherapie, ebenfalls eine Thrombozytopenie verursachen können.
Hyperkaliämie
Heparin-Produkte können die adrenale Sekretion von Aldosteron unterdrücken, was zu einer Hyperkaliämie führt. Risikofaktoren schließen Diabetes mellitus, chronisches Nierenversagen, vorbestehende metabolische Azidose, erhöhte Plasma-Kaliumspiegel vor Behandlungsbeginn, die gleichzeitige Therapie mit Arzneimitteln, die den Plasma-Kaliumspiegel erhöhen können, sowie die langfristige Anwendung von innohep® ein. Bei Risikopatienten sollte der Kaliumspiegel vor Beginn der Behandlung mit innohep® gemessen und dann in regelmäßigen Abständen kontrolliert werden. Eine Heparin-bedingte Hyperkaliämie ist normalerweise nach Beenden der Behandlung reversibel, allerdings sollten andere Therapieansätze in Betracht gezogen werden, wenn die Behandlung mit innohep® als lebensrettend eingestuft wird (z. B. Verringerung der Kalium-Einnahme, Absetzen anderer Arzneimittel, die das Kalium-Gleichgewicht beeinflussen können).
Herzklappenprothesen
Über therapeutische Misserfolge wurde bei Patienten mit Herzklappenprothesen berichtet, die eine volle gerinnungshemmende Dosis von innohep® oder anderen niedermolekularen Heparinen erhalten haben. Die Anwendung von innohep® wird für diese Patientengruppe nicht empfohlen.
Eingeschränkte Nierenfunktion
Die Anwendung von innohep bei Patienten mit einer Kreatinin-Clearance unter 30 ml/min wird nicht empfohlen, da für diese Patientengruppe keine Dosis ermittelt wurde. Die vorhandene Datenlage belegt, dass bei Patienten mit einer Kreatinin-Clearance bis ≥ 20 ml/min keine Akkumulation stattfindet.
Bei Bedarf kann bei diesen Patienten, wenn der Nutzen die Risiken überwiegt, die Behandlung mit innohep mit Vorsicht unter einer Anti-Xa-Überwachung erfolgen (siehe Abschnitt 4.2). Obwohl eine Anti-Xa-Überwachung zur Vorhersage eines Blutungsrisikos nur bedingt geeignet ist, ist sie die am besten geeignete Methode zur Messung der pharmakodynamischen Wirkung von innohep.
Ältere Patienten
Bei älteren Menschen ist die Wahrscheinlichkeit einer eingeschränkten Nierenfunktion erhöht (siehe Abschnitt 4.4: Eingeschränkte Nierenfunktion). Deshalb sollte innohep® bei älteren Patienten nur mit Vorsicht angewendet werden.
Austauschbarkeit
Niedermolekulare Heparine sind aufgrund ihrer unterschiedlichen pharmakokinetischen Eigenschaften und biologischen Aktivität nicht austauschbar. Die Umstellung auf ein anderes niedermolekulares Heparin muss, insbesondere während der Langzeitbehandlung, mit besonderer Vorsicht erfolgen. Hierbei sind die spezifischen Dosierungsanleitungen des jeweiligen Produkts zu befolgen.
Hinweise zu sonstigen Bestandteilen
Die Durchstechflasche zur Mehrfachentnahme von innohep® enthält 10 mg/ml des Konservierungsmittels Benzylalkohol. Benzylalkohol kann allergische Reaktionen hervorrufen.
Benzylalkohol kann bei Säuglingen und Kindern bis zu 3 Jahren toxische und anaphylaktoide Reaktionen hervorrufen. Große Mengen Benzylalkohol sollten wegen des Risikos der Akkumulation und Toxizität (metabolische Azidose) nur mit Vorsicht und wenn absolut nötig angewendet werden,
insbesondere bei Personen mit eingeschränkter Leber- und Nierenfunktion und in der Schwangerschaft und Stillzeit.
innohep® 20.000 Anti-Xa I.E./ml enthält Natriummetabisulfit. Metabisulfite können in seltenen Fällen schwere Überempfindlichkeitsreaktionen und Bronchospasmen hervorrufen. Darreichungsformen von innohep®, die Natriummetabisulfit enthalten, müssen bei Patienten mit Asthma mit Vorsicht angewendet werden.
Dieses Arzneimittel enthält bis zu 40 mg Natrium pro ml, entsprechend 2 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
Der gerinnungshemmende Effekt von innohep® kann durch andere, das Gerinnungssystem beeinflussende Arzneimittel verstärkt werden, z.B. solche, die die Thrombozytenfunktion inhibieren (z. B. Acetylsalicylsäure und andere nicht-steroidale Entzündungshemmer), thrombolytisch wirksame Mittel, Vitamin K-Antagonisten, aktiviertes C-Protein, direkte Faktor Xa- und IIa-Inhibitoren. Solche Kombinationen sollen vermieden oder nur unter engmaschiger Überwachung gegeben werden (siehe Abschnitt 4.4).
Schwangerschaft
Die Behandlung schwangerer Frauen mit Antikoagulanzien sollte durch einen Spezialisten erfolgen.
Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Abschnitt 5.3).
Weitergehende Erfahrungen an schwangeren Frauen (mehr als 2.200 Schwangerschaftsverläufe) deuten nicht auf ein Fehlbildungsrisiko oder eine fetale/neonatale Toxizität von Tinzaparin hin. Tinzaparin ist nicht plazentagängig. innohep® kann während aller Trimester der Schwangerschaft angewendet werden, wenn dies aus klinischer Sicht notwendig ist.
Epidurale Anästhesie
Bei Patienten, die eine neuroaxiale Anästhesie erhalten, sind therapeutische Dosen von innohep® (175 I.E./kg) aufgrund des Risikos von Spinalhämatomen kontraindiziert. Deshalb sollte eine epidurale Anästhesie bei schwangeren Frauen immer bis mindestens 24 Stunden nach Gabe der letzten therapeutischen Dosis von innohep® verschoben werden. Prophylaktische Dosen können angewendet werden, solange ein Abstand von mindestens 12 Stunden zwischen der letzten innohep®-Gabe und dem Setzen der Kanüle bzw. des Katheters eingehalten wird.
Schwangere Frauen mit Herzklappenprothese
Bei Frauen mit Herzklappenprothese wurde über Therapieversagen bei vollen gerinnungshemmenden Dosen von innohep® oder anderen niedermolekularen Heparinen berichtet. innohep® kann für die Anwendung bei dieser Patientenpopulation nicht empfohlen werden.
Sonstige Bestandteile
innohep® Durchstechflaschen enthalten Benzylalkohol. Da dieses Konservierungsmittel die Plazentaschranke passieren und zur Akkumulation und Toxizität (metabolische Azidose) führen kann, sollten während der Schwangerschaft innohep®-Formulierungen ohne Benzylalkohol (Fertigspritzen) verwendet werden.
Stillzeit
Tierexperimentelle Studien belegen, dass Tinzaparin minimal über die Muttermilch ausgeschieden wird.
Es ist nicht bekannt, ob Tinzaparin beim Menschen in die Muttermilch übergeht. Obwohl die orale Aufnahme von niedermolekularen Heparinen unwahrscheinlich ist, kann ein Risiko für Neugeborene/Kleinkinder nicht ausgeschlossen werden. Bei Risikopatientinnen ist die Gefahr von venösen Thromboembolien während der ersten 6 Wochen nach der Geburt des Kindes besonders hoch. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen oder die Behandlung mit innohep® zu unterbrechen ist. Dabei sind sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau abzuwägen.
Sonstige Bestandteile:
innohep® Durchstechflaschen enthalten Benzylalkohol. Aufgrund des Risikos der Akkumulation und Toxizität
(metabolische Azidose), sollte in der Stillzeit die innohep® Darreichungsform ohne Benzylalkohol (Fertigspritzen) verabreicht werden.
Fertilität
Hinsichtlich der Fertilität liegen keine klinischen Studien mit innohep® vor.
innohep® hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Die am häufigsten berichteten Nebenwirkungen sind Blutungsereignisse, Anämie aufgrund von Blutungen und Reaktionen an der Einstichstelle.
Blutungen können in verschiedenen Schweregraden in allen Organen auftreten. Komplikationen können vor allem bei Verabreichung hoher Dosen auftreten. Obwohl schwere Blutungen nur gelegentlich auftreten, wurde in einigen Fällen über Todesfälle oder anhaltende Behinderung berichtet.
Eine immunvermittelte, Heparin-induzierte Thrombozytopenie (Typ II) manifestiert sich weitgehend innerhalb von 5 bis 14 Tagen nach Erhalt der ersten Dosis. Darüber hinaus wurde bei Patienten, die im Vorfeld mit Heparin behandelt wurden, eine rasch einsetzende Form beschrieben. Eine immunvermittelte, Heparin-induzierte Thrombozytopenie (Typ II) kann mit einer arteriellen und venösen Thrombose assoziiert sein. Die Gabe von innohep® muss in allen Fällen von immunvermittelter, Heparin-induzierter Thrombozytopenie abgebrochen werden (siehe Abschnitt 4.4).
In seltenen Fällen kann innohep® eine Hyperkaliämie aufgrund von Hypoaldosteronismus hervorrufen. Patienten mit Diabetes mellitus oder eingeschränkter Nierenfunktion haben ein erhöhtes Risiko (siehe Abschnitt 4.4).
In manchen Fällen können schwere allergische Reaktionen auftreten. Diese beinhalten seltene Fälle von Hautnekrosen, toxische Hautausschläge (z. B. Stevens-Johnson-Syndrom), Angioödeme und Anaphylaxien. Die Behandlung sollte beim geringsten Verdacht auf solch schwere Reaktionen sofort abgebrochen werden.
Die Abschätzung der Häufigkeit von Nebenwirkungen basiert auf einer zusammengefassten Analyse von Daten aus klinischen Studien und Spontanberichten.
Die unerwünschten Wirkungen sind anhand des MedDRA System Organklassen (SOC) aufgelistet, wobei die einzelnen unerwünschten Wirkungen nach ihrer Häufigkeit, beginnend mit der am häufigsten berichteten unerwünschten Wirkung, aufgelistet werden. Innerhalb einer Häufigkeitsgruppierung sind die unerwünschten Wirkungen nach abnehmendem Schweregrad gelistet.
Sehr häufig (≥1/10)
Häufig (≥1/100, <1/10)
Gelegentlich (≥1/1.000, <1/100)
Selten (≥1/10.000, <1/1.000)
Sehr selten (<1/10.000)
Erkrankungen des Blutes und des Lymphsystems | |
Häufig (≥1/100, <1/10) |
Anämie (inkl. erniedrigtem Hämoglobinwert) |
Gelegentlich (≥1/1.000, <1/100) |
Thrombozytopenie (Typ I) (inkl. reduzierter Thrombozytenzahl) |
Selten (≥1/10.000, <1/1.000) |
Heparin-induzierte Thrombozytopenie (Typ II), Thrombozytose |
Erkrankungen des Immunsystems | |
Gelegentlich (≥1/1.000, <1/100) |
Überempfindlichkeit |
Selten (≥1/10.000, <1/1.000) |
Anaphylaktische Reaktionen |
Stoffwechsel- und Ernährungsstörungen | |
Selten (≥1/10.000, <1/1.000) |
Hyperkaliämie |
Gefäßerkrankungen | |
Häufig (≥1/100, <1/10) |
Blutung |
Gelegentlich (≥1/1.000, <1/100) |
Blutergüsse, Ecchymose und Purpura |
Leber- und Gallenerkrankungen | |
Gelegentlich (≥1/1.000, <1/100) |
erhöhte Leberenzymwerte (inkl. erhöhte Transaminasen, ALT, AST und GGT) |
Erkankungen der Haut und des Unterhautzellgewebes | |
Gelegentlich (≥1/1.000, <1/100) |
Dermatitis (inkl. allergischer und bullöser Dermatitis) |
Selten (≥1/10.000, <1/1.000) |
Toxische Hautausschläge (inkl. Stevens-Johnson- Syndrom) |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |
Selten (≥1/10.000, <1/1.000) |
Osteoporose (in Verbindung mit Langzeitanwendung) |
Erkrankungen der Geschlechtsorgane und der Brustdrüse | |
Selten (≥1/10.000, <1/1.000) |
Priapismus |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Häufig (≥1/100, <1/10) |
Reaktionen an der Einstichstelle (inkl. Hämatombildung an der Einstichstelle, Blutung, Schmerzen, Juckreiz, Knötchenbildung, Erythem und Extravasation) |
Tumorpatienten unter Langzeitbehandlung
In einer Studie mit Tumorpatienten unter Langzeitbehandlung (6 Monate) mit innohep war die Gesamtrate unerwünschter Arzneimittelreaktionen vergleichbar mit der bei anderen innohep behandelten Patienten. Tumorpatienten haben im Allgemeinen ein erhöhtes Blutungsrisiko, das durch höheres Alter, Begleiterkrankungen, chirurgische Interventionen und Begleitbehandlungen weiter beeinflusst wird. Die Inzidenz hämorrhagischer Ereignisse war demnach erwartungsgemäß höher als unter Kurzzeitanwendung und ähnlich der bei Tumorpatienten unter Langzeitbehandlung mit Antikoagulanzien beobachteten Inzidenz.
Kinder und Jugendliche
Begrenzte Informationen aus einer Studie und Markterfahrungen weisen darauf hin, dass das Nebenwirkungs-Profil bei Kindern und Jugendlichen dem von Erwachsenen gleicht.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
Blutungen stellen die Hauptkomplikation einer Überdosierung dar. Aufgrund der relativ kurzen Halbwertszeit von innohep® (siehe Abschnitt 5.2) können kleinere Blutungen durch Abbruch der Behandlung konservativ behandelt werden. Schwere Blutungen können die Gabe des Antidots Protaminsulfat notwendig machen. Patienten sollten sorgfältig überwacht werden.
Pharmakotherapeutische Gruppe: Antithrombotische Mittel, Heparingruppe, ATC-Code: B01AB10
Wirkmechanismus
Tinzaparin-Natrium ist ein niedermolekulares Heparin vom Schwein mit einem Anti-Xa-/Anti-IIa-Verhältnis zwischen 1,5 und 2,5. Tinzaparin-Natrium wird durch enzymatische Depolymerisation von konventionellem unfraktionierten Heparin hergestellt. Wie konventionelles Heparin, wirkt Tinzaparin-Natrium als Antikoagulans durch Verstärkung der Antithrombin-III-Inhibition von aktivierten Koagulationsfaktoren, in erster Linie Faktor Xa.
Die biologische Aktivität von Tinzaparin-Natrium ist gegen den aktuellen „Internationalen Standard für Heparin niedriger Molekülmasse“ standardisiert, ausgedrückt in Anti-Xa Internationalen Einheiten (I.E.).
Die Anti-Xa-Aktivität von Tinzaparin-Natrium liegt zwischen 70 und 120 I.E./mg. Die Anti-IIa-Aktivität von Tinzaparin-Natrium ist etwa 55 I.E./mg. Der charakteristische Wert für die mittlere Molekülmasse von Tinzaparin-Natrium liegt bei 6.500 Dalton.
Pharmakodynamische Wirkungen
Tinzaparin verfügt über eine hohe Antithrombin-Aktivität (Anti-IIa), ein niedriges Anti-Xa/Anti-IIa-Verhältnis und hemmt die Thrombinbildung mit fast der gleichen Wirksamkeit wie unfraktioniertes Heparin. Zusätzlich zur Anti-Xa/IIa-Aktivität wurde bei Patienten eine Induktion von TFPI (Tissue Factor Pathway Inhibitor) festgestellt.
Klinische Wirksamkeit und Sicherheit
Erstbehandlung von akuter tiefer Venenthrombose und Lungenembolie
In einer klinischen, doppelblinden Studie wurde Tinzaparin (175 I.E./kg einmal täglich subkutan) mit dosisangepasstem Heparin verglichen, das durch kontinuierliche intravenöse Infusion zur Erstbehandlung von Patienten mit proximaler Venenthrombose verabreicht wurde. Alle Patienten begannen an Tag 2 eine orale Antikoagulationstherapie mit Warfarin und wurden mindestens 6 Tage lang mit Tinzaparin oder Heparin behandelt. Bei 6 der 213 Patienten, die Tinzaparin erhielten (2,8 %), und bei 15 der 219 Patienten, die Heparin erhielten (6,9 %), kam es während des 3-monatigen Nachbeobachtungszeitraums der Studie erneut zu einer venösen Thromboembolie (VTE) (p = 0,07). Schwerwiegende Blutungen, bei denen ein Zusammenhang mit der Erstbehandlung festgestellt wurde, traten bei einem Patienten auf, der Tinzaparin erhielt (0,5 %), und bei 11 Patienten, die Heparin erhielten (5,0 %), was einer Risikoreduktion von 91 % entspricht (p = 0,006). In der Tinzaparin-Gruppe kam es zu 10 Todesfällen (4,7 %), in der Heparin-Gruppe zu 21 Todesfällen (9,6 %), was einer Risikoreduktion von 51 % entspricht (p = 0,049).
In einer unverblindeten Studie (THESEE) wurden 612 Patienten mit symptomatischer Lungenembolie randomisiert und wurden während der ersten 8 Tage der Behandlung mit Tinzaparin (175 I.E./kg einmal täglich subkutan) oder dosisangepasstem intravenösem Heparin behandelt. Die orale Antikoagulationstherapie wurde an den Tagen 1 – 3 eingeleitet und mindestens 3 Monate lang durchgeführt. Basierend auf einem kombinierten Endpunkt (rezidivierende VTE, schwerwiegende Blutungen und Tod) hatten 9 der 308 Patienten in der Heparin-Gruppe (2,9 %) und 9 der 304 Patienten in der Tinzaparin-Gruppe (3,0 %) mindestens einen Endpunkt an Tag 8 erreicht (absolute Differenz: -0,1 %; 95-%-Kl: -2,7 bis 2,6).
Verlängerte Behandlung von einer akuter tiefer Venenthrombose und Lungenembolie
In einer Subanalyse („Main-LITE cancer“) einer randomisierten, unverblindeten klinischen Studie wurde Tinzaparin (175 I.E./kg subkutan einmal täglich) im Rahmen einer 3-monatigen Behandlung bei Patienten mit proximaler Venenthrombose mit Warfarin verglichen. Unter den 200 Patienten mit Krebs (100 Patienten in jeder Gruppe) traten in der Warfarin-Gruppe mehr Fälle von rezidivierenden VTE nach 12 Monaten auf (16 %) als in der Tinzaparin-Gruppe (7 %) (absolute Differenz: -9,0; 95-%-KI: -21,7 bis -0,7). Innerhalb von 3 Monaten traten bei 7 % der Patienten in beiden Gruppen schwerwiegende Blutungen auf. Nach einem Jahr betrug die Mortalität in beiden Gruppen 47 %
In einer unverblindeten, randomisierten Studie mit 241 Patienten mit akuter proximaler tiefer Venenthrombose (TVT), davon 69 Patienten mit Krebs, wurde Tinzaparin (175 I.E./kg subkutan einmal täglich) während einer 6-monatigen TVT-Behandlung mit einem oralen Vitamin-K-Antagonisten (VKA) verglichen. Unter den Patienten mit Krebs war die Inzidenz rezidivierender VTE in der Tinzaparin-Gruppe geringer (2/36 [5,5 %]) im Vergleich zu 7/33 [21,2 %]). In der Tinzaparin-Gruppe trat eine schwerwiegende Blutung auf, verglichen mit 3 in der VKA-Gruppe.
In einer kontrollierten, unverblindeten, randomisierten klinischen Studie (CATCH) wurden die Wirksamkeit und die Sicherheit von Tinzaparin nach einer 6-monatigen Behandlung einer akuten, symptomatischen TVT oder Lungenembolie bei Patienten mit aktiver Krebserkrankung mit Warfarin verglichen. An der Studie nahmen 900 Patienten mit einer Nierenfunktion entsprechend einer Kreatinin-Clearance (KrCl) von bis zu 20 ml/min teil. Patienten mit einer Thrombozytenzahl unter 50 × 109/l wurden nicht in die Studie eingeschlossen. Die Patienten in der Tinzaparin-Gruppe erhielten während des gesamten Behandlungszeitraums (6 Monate) einmal täglich eine therapeutische Dosis Tinzaparin (175 I.E./kg subkutan) und wurden mit Patienten verglichen, die 5 – 10 Tage lang einmal täglich Tinzaparin erhielten, gefolgt von dosisangepasstem Warfarin (INR: 2,0 – 3,0) für 6 Monate. Die Ergebnisse zur Wirksamkeit (TVT in den unteren Extremitäten und Lungenembolie) und Sicherheit (Blutungen, Heparin-induzierte Thrombozytopenie und Tod) wurden von einem verblindeten Ausschuss bewertet. Rezidivierende VTE traten bei 31 von 449 Patienten in der Tinzaparin-Gruppe und bei 45 von 451 Patienten in der Warfarin-Gruppe auf (kumulative 6-Monats-Inzidenz: 7,2 % unter Tinzaparin im Vergleich zu 10,5 % unter Warfarin; Hazard Ratio [HR]: 0,65; 95-%-KI: 0,41 – 1,03; p = 0,07). Eine symptomatische TVT trat bei 12 Patienten in der Tinzaparin-Gruppe und bei 24 Patienten in der Warfarin-Gruppe auf (HR: 0,48; 95-%-Kl: 0,24 – 0,96; p = 0,04). Es gab keinen signifikanten Unterschied in Bezug auf schwerwiegende Blutungsereignisse (HR: 0,89; 95-%-KI: 0,40 – 1,99; p = 0,77) oder die Mortalität beliebiger Ursache (1,08; 95-%-KI: 0,85 – 1,36; p = 0,54), andererseits gab es jedoch in der Tinzaparin-Gruppe ein statistisch signifikant verringertes Risiko für klinisch relevante, nicht schwerwiegende Blutungen im Vergleich zur Warfarin-Gruppe (HR: 0,58; 95-%-KI: 0,40 – 0,84; p = 0,004).
In einer vorspezifizierten Sekundäranalyse der CATCH-Studie, bei der konkurrierende Ergebnisse für eine Regressionsanalyse der Zeit bis zur ersten klinisch relevanten Blutung (KRB; schwerwiegende und klinisch relevante, nicht schwerwiegende Ereignisse) verwendet wurden, war das Risiko für das Auftreten mindestens eines KRB-Ereignisses während der 6-monatigen Studie in der Tinzaparin-Gruppe (n = 60/449) signifikant geringer als in der Warfarin-Gruppe (n = 78/451), HR: 0,64; 95-%-Kl: 0,45 – 0,89; p = 0,009. Die kumulativen Inzidenzraten von KRB in den beiden Gruppen unterschieden sich fast von Anfang an und zeigten während des 6-monatigen Behandlungszeitraums weiterhin einen Nutzen für die Tinzaparin-Patienten (siehe Abbildung 1). In einer multivariaten Analyse für alle Behandlungsgruppen wurde festgestellt, dass das Risiko für KRB mit einem Alter von > 75 Jahren (HR: 1,83) und einer intrakraniellen malignen Erkrankung (HR: 1,97) anstieg.
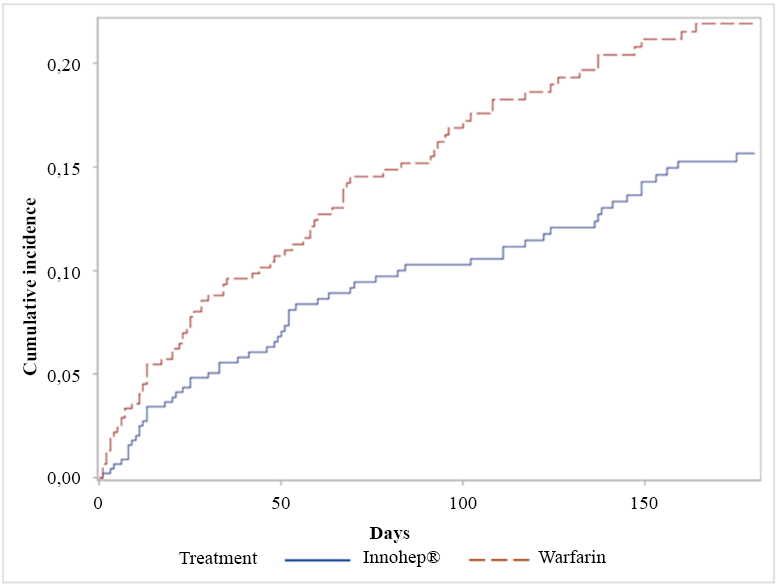
In einer Sekundäranalyse der CATCH-Studie wurde die Auswirkung einer eingeschränkten Nierenfunktion (definiert als glomeruläre Filtrationsrate [GFR] < 60 ml/min/1,73 m2) auf die Wirksamkeit und Sicherheit der Antikoagulationstherapie bei Patienten mit krebsassoziierter Thrombose bewertet. Die Studienpopulation für diese Analyse umfasste 864 Patienten (96 %), für die zum Zeitpunkt der Randomisierung ein GFR-Wert aus einem Zentrallabor vorlag. Davon wiesen 131 Patienten (15 %) eine eingeschränkte Nierenfunktion auf (69 Patienten in der Tinzaparin-Gruppe und 62 Patienten in der Warfarin-Gruppe). Eine eingeschränkte Nierenfunktion war bei Patienten mit krebsassoziierter Thrombose, die eine Antikoagulationstherapie erhielten, mit einem statistisch signifikanten Anstieg rezidivierender VTE und schwerwiegender Blutungen assoziiert, aber es wurde kein signifikanter Anstieg klinisch relevanter Blutungen (KRB) oder der Mortalität beobachtet. Die Langzeitbehandlung mit Tinzaparin in therapeutischer Dosis ohne Dosisanpassung bei Patienten mit eingeschränkter Nierenfunktion führte im Vergleich zu Warfarin nicht zu einer erhöhten Inzidenz rezidivierender VTE, KRB, schwerwiegender Blutungen oder einer erhöhten Mortalität.
In eine prospektive, unverblindete klinische Studie („TICAT“) wurden insgesamt 247 Patienten mit aktiver Krebserkrankung und neu diagnostizierter TVT und/oder Lungenembolie eingeschlossen. Die durchschnittliche Behandlungsdauer mit Tinzaparin (175 I.E./kg subkutan einmal täglich) betrug 15,6 (SD: 13,2) Monate. Die Inzidenz rezidivierender VTE sank während der Studie von 4,5 % in den ersten 6 Monaten (95-%-KI: 2,2 % – 7,8 %) auf 1,1 % (95-%-KI: 0,1 % – 3,9 %) in den Monaten 7 – 12 (p = 0,08). Die Inzidenz klinisch relevanter Blutungen betrug 0,9 % pro Patientenmonat (95-%-KI: 0,5 % – 1,6 %) in den ersten 6 Monaten und 0,6 % pro Patientenmonat (95-%-KI: 0,2 % – 1,4 %) in den Monaten 7 – 12. Ein Patient (0,4 %) starb aufgrund einer rezidivierenden Lungenembolie und 2 Patienten (0,8 %) starben aufgrund von Blutungen.
Besondere Patientenpopulationen
Population mit eingeschränkter Nierenfunktion
Das Sicherheitsprofil von Tinzaparin (175 I.E./kg einmal täglich) über einen Zeitraum von bis zu 30 Tagen wurde in einer Studie mit 200 stationären älteren Patienten mit einer Kreatinin-Clearance (KrCl) von > 20 ml/min untersucht. Die Anti-Xa-Aktivität im Plasma wurde regelmäßig gemessen. Das Durchschnittsalter betrug 85,2 Jahre (Bereich: 70 bis 102) und die durchschnittliche KrCl betrug 51,2 ± 22,9 ml/min. Bei einem Todesfall bestand der Verdacht, dass er mit der Antikoagulationstherapie zusammenhing. Es wurden drei schwerwiegende Blutungen (1,5 %) gemeldet. Eine Heparin-induzierte Thrombozytopenie wurde bei 2 Patienten (1 %) bestätigt. Es wurde keine Korrelation zwischen der Anti-Xa-Aktivität und der KrCl oder dem Alter festgestellt.
Die absolute Bioverfügbarkeit, die auf der Anti-Xa Aktivität nach subkutaner Verabreichung beruht, beträgt ungefähr 90 % und die Dauer bis zum Erreichen der maximalen Aktivität liegt bei 4 - 6 Stunden. Die terminale Halbwertszeit liegt bei circa 3,7 Stunden.
Tinzaparin-Natrium wird in der Leber durch Depolymerisation geringfügig metabolisiert und über die Nieren in unveränderter oder nahezu unveränderter Form ausgeschieden.
Besondere Patientenpopulationen
Schwangere Frauen
Die pharmakokinetische Aktivität von Tinzaparin wurde an schwangeren Frauen untersucht. Daten sequentieller pharmakokinetischer Überwachungen an 55 schwangeren Frauen legen den Schluss nahe, dass sich die pharmakokinetischen Eigenschaften bei Schwangeren im Vergleich zu den pharmakokinetischen Eigenschaften von nicht‑schwangeren Frauen nicht unterscheiden.
Eingeschränkte Nierenfunktion
Tinzaparin hat ein hohes durchschnittliches Molekulargewicht, und es gibt klinische und präklinische Hinweise auf eine signifikante nicht-renale Elimination von Tinzaparin.
Nach Verabreichung einer intravenösen Bolusinjektion bei Dialysepatienten ist die beobachtete Halbwertszeit kürzer als nach subkutaner Verabreichung bei gesunden Freiwilligen (etwa 2,5 Stunden gegenüber etwa 3,7 Stunden).
In einer prospektiven Studie wurde untersucht, ob sich Tinzaparin (175 I.E. Anti-Xa/kg einmal täglich subkutan) während einer 10-tägigen Behandlung bei 30 stationären Patienten im Alter von über 70 Jahren, die eine therapeutische Dosis zur Behandlung einer akuten thromboembolischen Erkrankung erhielten, akkumuliert. Die Plasmaspiegel von Anti-Xa und Anti-IIa sowie die aktivierte partielle Thromboplastinzeit (aPTT) wurden vor der ersten Injektion bei den Spitzenwerten, d. h. 5 Stunden nach der zweiten Injektion (Tag 2) sowie an den Tagen 5, 7 und 10, bestimmt. Die Patienten waren im Durchschnitt 87 Jahre alt (Bereich: 71 – 96 Jahre), hatten ein mittleres Körpergewicht von 62,7 kg (Bereich: 38 – 90 kg) und eine mittlere KrCl von 40,6 ± 15,3 ml/min (Bereich: 20 – 72 ml/min). Da keiner der Patienten eine Anti-Xa-Aktivität von mehr als 1,5 I.E./ml aufwies, wurde keine Dosisanpassung vorgenommen. Der durchschnittliche maximale Anti-Xa-Spiegel betrug 0,66 ± 0,20 I.E./ml (Bereich: 0,26 – 1,04) an Tag 2. Nach wiederholter täglicher Behandlung mit Tinzaparin über 10 Tage war kein progredienter Anstieg der Anti-Xa- oder Anti-IIa-Aktivität festzustellen. Es wurde keine Korrelation zwischen der Anti-Xa- und Anti-IIa-Aktivität und dem Alter, dem Gewicht oder der KrCl festgestellt. Es traten keine schwerwiegenden Blutungen auf, und es gab keine thromboembolischen Komplikationen oder Todesfälle.
Kinder und Jugendliche
Vorläufige Daten vom Gebrauch von Tinzaparin weisen darauf hin, dass jüngere Kinder einschließlich Neugeborenen und Kleinkindern Tinzaparin schneller abbauen und deshalb höhere Dosen benötigen als ältere Kinder. Dennoch reichen die Daten nicht aus, um Dosierungsempfehlungen zu geben, siehe Abschnitt 4.2.
Heparine und niedermolekulare Heparine (NMH) sind normalerweise nur wenig toxisch; dies trifft auch auf Tinzaparin-Natrium zu. Der wesentliche Effekt, der in Studien zur akuten, subakuten und chronischen Toxizität, Reproduktionstoxizität und Mutagenität beobachtet wurde, sind Blutungen, die durch die Verabreichung sehr hoher Dosen verursacht wurden.
Nach intramuskulärer Applikation von niedermolekularem Heparin am Tier wurden nekrotisierende Hämatome beobachtet. Osteoporotische Effekte traten in einer Studie über 12 Monate an Ratten auf. Untersuchungen an Ratten und Kaninchen ergaben keinen Hinweis auf ein teratogenes Potenzial von niedermolekularem Heparin in Dosierungen bis 25 mg/kg Körpergewicht. Gewichtsabnahme gegenüber Kontrollgruppen wurde bei mit 10 mg/kg Körpergewicht pränatal exponierten Feten beobachtet.
Benzylalkohol
Natriummetabisulfit (Ph. Eur.) (E 223)
Natriumhydroxid
Wasser für Injektionszwecke.
Dieses Arzneimittel darf nicht mit anderen Injektionslösungen gemischt werden.
2 Jahre
Nach Anbruch konnte die chemische und physikalische Stabilität über 28 Tage bei 30° C gezeigt werden. Aus mikrobiologischer Sicht kann das Arzneimittel nach Anbruch maximal 28 Tage bei 30° C aufbewahrt werden.
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Zu den Aufbewahrungsbedingungen nach Anbruch des Arzneimittels siehe Abschnitt 6.3.
2 ml Durchstechflasche zur Mehrfachentnahme.
Packungsgrößen: 1 und 10 Durchstechflaschen
Klinikpackung: 10 Durchstechflaschen
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
LEO Pharma A/S
Industriparken 55
2750 Ballerup
Dänemark
Örtlicher Vertreter:
LEO Pharma GmbH
Siemensstraße 5b
63263 Neu-Isenburg
Telefon: 06102 / 201 - 0
Telefax: 06102 / 201 - 200
www.leo-pharma.de
37710.00.00
Datum der Erteilung der Zulassung: 17. April 1997
Datum der letzten Verlängerung der Zulassung: 09. März 2007
September 2025
Verschreibungspflichtig