
Polivy® 30 mg Pulver zur Herstellung eines Infusionslösungskonzentrats.
Polivy® 140 mg Pulver zur Herstellung eines Infusionslösungskonzentrats.
Polivy 30 mg Pulver zur Herstellung eines Infusionslösungskonzentrats
Jede Durchstechflasche mit Pulver zur Herstellung eines Infusionslösungskonzentrats enthält 30 mg Polatuzumab Vedotin.
Nach Rekonstitution enthält jeder ml 20 mg Polatuzumab Vedotin.
Polivy 140 mg Pulver zur Herstellung eines Infusionslösungskonzentrats
Jede Durchstechflasche mit Pulver zur Herstellung eines Infusionslösungskonzentrats enthält 140 mg Polatuzumab Vedotin.
Nach Rekonstitution enthält jeder ml 20 mg Polatuzumab Vedotin.
Polatuzumab Vedotin ist ein Antikörper-Wirkstoff-Konjugat, bestehend aus dem Mitosehemmstoff Monomethyl-Auristatin-E (MMAE), der kovalent an einen gegen CD79b gerichteten monoklonalen Antikörper gebunden ist (rekombinantes humanisiertes Immunglobulin G1 [IgG1]), hergestellt mittels rekombinanter DNA-Technologie in Ovarialzellen des chinesischen Hamsters.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Polivy 30-mg-Durchstechflasche enthält 1,8 mg Polysorbat 20.
Jede Polivy 140-mg-Durchstechflasche enthält 8,4 mg Polysorbat 20.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Pulver zur Herstellung eines Infusionslösungskonzentrats (Pulver zur Herstellung eines Konzentrats).
Weißer bis gräulich-weißer Lyophilisat-Kuchen.
Polivy in Kombination mit Rituximab, Cyclophosphamid, Doxorubicin und Prednison (R-CHP) wird angewendet zur Behandlung erwachsener Patienten mit bisher unbehandeltem diffusem großzelligem B-Zell-Lymphom (DLBCL).
Polivy in Kombination mit Bendamustin und Rituximab wird angewendet zur Behandlung erwachsener Patienten mit rezidivierendem oder refraktärem diffusem großzelligem B-Zell-Lymphom (DLBCL), die nicht für eine hämatopoetische Stammzelltransplantation in Frage kommen.
Polivy darf nur unter Aufsicht von in der Diagnose und Behandlung von Krebspatienten erfahrenem medizinischem Fachpersonal angewendet werden.
Dosierung
Diffuses großzelliges B-Zell-Lymphom
Bisher unbehandelte Patienten
Die empfohlene Dosis von Polivy beträgt 1,8 mg/kg, angewendet als intravenöse Infusion alle 21 Tage, in Kombination mit Rituximab, Cyclophosphamid, Doxorubicin und Prednison (R-CHP) über 6 Zyklen. Polivy, Rituximab, Cyclophosphamid und Doxorubicin können in beliebiger Reihenfolge an Tag 1 nach der Gabe von Prednison angewendet werden. Prednison wird an den Tagen 1 – 5 eines jeden Zyklus gegeben. Die Zyklen 7 und 8 bestehen aus Rituximab als Monotherapie.
Die Fachinformationen der Chemotherapeutika, die in Kombination mit Polivy bei Patienten mit bisher unbehandeltem DLBCL angewendet werden, sind zu beachten.
Patienten mit rezidivierender oder refraktärer Erkrankung
Die empfohlene Dosis von Polivy beträgt 1,8 mg/kg, angewendet als intravenöse Infusion alle 21 Tage, in Kombination mit Bendamustin und Rituximab über 6 Zyklen. Polivy, Bendamustin und Rituximab können in beliebiger Reihenfolge an Tag 1 eines jeden Zyklus angewendet werden. Wird Bendamustin mit Polivy angewendet, beträgt die empfohlene Dosis von Bendamustin 90 mg/m2/Tag an Tag 1 und Tag 2 eines jeden Zyklus und die empfohlene Dosis von Rituximab 375 mg/m2 an Tag 1 eines jeden Zyklus. Aufgrund begrenzter klinischer Erfahrungen bei Patienten, die mit 1,8 mg/kg Polivy bei einer Gesamtdosis von > 240 mg behandelt werden, wird empfohlen, die Dosis von 240 mg/Zyklus nicht zu überschreiten.
Bisher unbehandelte Patienten und Patienten mit rezidivierender oder refraktärer Erkrankung
Wenn nicht bereits eine Prämedikation besteht, ist bei Patienten vor Anwendung von Polivy eine Prämedikation mit einem Antihistaminikum und einem Antipyretikum zu verabreichen.
Verspätete oder versäumte Dosen
Wenn eine geplante Dosis von Polivy versäumt wurde, ist sie so bald wie möglich nachzuholen und das Anwendungsschema so anzupassen, dass ein 21-tägiges Intervall zwischen den Dosen beibehalten wird.
Dosisanpassungen
Wenn bei einem Patienten eine Reaktion im Zusammenhang mit einer Infusion auftritt, ist die Infusionsrate von Polivy zu verlangsamen oder die Anwendung zu unterbrechen. Die Anwendung von Polivy ist umgehend und dauerhaft abzubrechen, wenn bei einem Patienten eine lebensbedrohliche Reaktion auftritt.
Es gibt verschiedene mögliche Dosisanpassungen für Polivy bei Patienten mit bisher unbehandeltem DLBCL und bei Patienten mit rezidivierender oder refraktärer Erkrankung.
Für Dosisanpassungen bei peripherer Neuropathie (Abschnitt 4.4) siehe Tabelle 1.
Tabelle 1: Dosisanpassungen von Polivy bei peripherer Neuropathie (PN)
Anwendungs-gebiet |
Schweregrad der PN an Tag 1 eines jeden Zyklus |
Dosisanpassungen |
Bisher unbehandeltes DLBCL |
Grad 2a |
Sensorische Neuropathie:
Motorische Neuropathie:
Bei gleichzeitiger sensorischer und motorischer Neuropathie ist die strengste oben genannte Dosisanpassung zu befolgen. |
Grad 3a |
Sensorische Neuropathie:
Motorische Neuropathie:
Bei gleichzeitiger sensorischer und motorischer Neuropathie ist die strengste oben genannte Dosisanpassung zu befolgen. |
|
Grad 4 |
Behandlung mit Polivy abbrechen. |
|
R/R DLBCL |
Grad 2 - 3 |
Behandlung mit Polivy unterbrechen bis Verbesserung auf Grad ≤ 1. |
Grad 4 |
Behandlung mit Polivy abbrechen. |
a R-CHP kann weiterhin angewendet werden.
Für Dosisanpassungen bei Myelosuppression (Abschnitt 4.4), siehe Tabelle 2.
Tabelle 2: Dosisanpassungen von Polivy, Chemotherapie und Rituximab bei Myelosuppression
Anwendungs- |
Schweregrad der Myelosuppression an Tag 1 eines jeden Zyklus |
Dosisanpassung |
Bisher unbehandeltes DLBCL |
Grad 3 ‑ 4 Neutropenie |
Jegliche Behandlung unterbrechen, bis Wiederanstieg der ANC* auf > 1.000/µl.
|
Grad 3 – 4 Thrombozytopenie |
Jegliche Behandlung unterbrechen, bis Wiederanstieg der Thrombozyten auf > 75.000/µl.
|
|
R/R DLBCL |
Grad 3 ‑ 4 Neutropenie1 |
Jegliche Behandlung unterbrechen, bis Wiederanstieg der ANC auf > 1.000/µl.
|
Grad 3 – 4 Thrombozytopenie1 |
Jegliche Behandlung unterbrechen, bis Wiederanstieg der Thrombozyten auf > 75.000/µl.
|
1 Wenn die Primärursache lymphombedingt ist, muss die Dosis von Bendamustin möglicherweise nicht reduziert werden.
*ANC: absolute Neutrophilenzahl (absolute neutrophil count)
Für Dosisanpassungen bei Reaktionen im Zusammenhang mit einer Infusion (Abschnitt 4.4), siehe Tabelle 3.
Tabelle 3: Dosisanpassungen von Polivy bei Reaktionen im Zusammenhang mit einer Infusion (infusion-related reaction, IRR)
Anwendungs- |
Schweregrad der IRR an Tag 1 eines jeden Zyklus |
Dosisanpassung |
Bisher unbehandeltes und R/R DLBCL |
Grad 1 – 3 |
Unterbrechung der Infusion von Polivy und Einleitung einer unterstützenden Behandlung. |
Grad 4 |
Infusion von Polivy sofort abbrechen. |
Besondere Patientengruppen
Ältere Patienten
Bei Patienten im Alter von ≥ 65 Jahre ist keine Dosisanpassung von Polivy erforderlich (siehe Abschnitt 5.2).
Nierenfunktionsstörung
Bei Patienten mit einer Kreatinin-Clearance (CrCl) ≥ 30 ml/min ist keine Dosisanpassung von Polivy erforderlich. Eine Dosisempfehlung für Patienten mit CrCl < 30 ml/min wurde aufgrund begrenzter Daten nicht bestimmt.
Leberfunktionsstörung
Die Anwendung von Polivy bei Patienten mit mäßiger oder schwerer Leberfunktionsstörung (Bilirubin größer als 1,5 × obere Normalgrenze [upper limit of normal, ULN]) ist zu vermeiden.
Eine Anpassung der Anfangsdosis ist nicht erforderlich, wenn Polivy bei Patienten mit leichter Leberfunktionsstörung (Bilirubin größer als ULN bis kleiner oder gleich 1,5 × ULN oder Aspartat-Aminotransferase [AST] größer als ULN) angewendet wird.
In der Studienpopulation mit leichter Leberfunktionsstörung [definiert als AST oder ALT > 1,0 bis 2,5 × ULN oder Gesamtbilirubin > 1,0 bis 1,5 × ULN] gab es einen Anstieg der unkonjugierten MMAE-Exposition um weniger als 40 %, der nicht als klinisch signifikant eingestuft wurde.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Polivy bei Kindern und Jugendlichen unter 18 Jahren ist nicht untersucht. Es liegen keine Daten vor.
Art der Anwendung
Polivy wird intravenös angewendet.
Die Initialdosis von Polivy ist als 90-minütige intravenöse Infusion zu verabreichen. Die Patienten sind während der Infusion und für mindestens 90 Minuten nach Beendigung der Initialdosis auf IRR/Überempfindlichkeitsreaktionen zu überwachen.
Wenn die vorherige Infusion gut vertragen wurde, kann die nachfolgende Dosis von Polivy als 30‑minütige Infusion verabreicht werden und die Patienten sind während der Infusion und für mindestens 30 Minuten nach Beendigung der Infusion zu überwachen.
Polivy muss unter aseptischen Bedingungen und unter Aufsicht von Angehörigen von Gesundheitsberufen rekonstituiert und verdünnt werden. Polivy ist als intravenöse Infusion durch ein eigenes Infusionssystem, das mit einem sterilen, pyrogenfreien, Inline- oder Add-on-Filter (0,2 oder 0,22 Mikrometer Porengröße) mit niedriger Proteinbindungskapazität und einem Katheter ausgestattet ist, anzuwenden. Polivy darf nicht als intravenöse Druck- oder Bolusinjektion verabreicht werden.
Hinweise zur Rekonstitution und Verdünnung des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
Vorsichtsmaßnahmen vor Handhabung oder Anwendung des Arzneimittels
Polivy enthält eine zytotoxische Komponente, die kovalent an den monoklonalen Antikörper gebunden ist. Angemessene Verfahren für die korrekte Handhabung und Beseitigung sind zu befolgen (siehe Abschnitt 6.6).
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Aktive schwere Infektionen (siehe Abschnitt 4.4).
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Myelosuppression
Schwerwiegende und schwere Neutropenie und febrile Neutropenie wurden bereits im ersten Zyklus bei Patienten berichtet, die mit Polivy behandelt wurden. Die prophylaktische Gabe von Granulozyten-Kolonie-stimulierendem Faktor (G-CSF) war in der klinischen Entwicklung vorgeschrieben und ist in Erwägung zu ziehen. Thrombozytopenie oder Anämie von Grad 3 oder 4 können ebenfalls unter Anwendung von Polivy auftreten. Das große Blutbild ist vor jeder Anwendung von Polivy zu überprüfen. Eine häufigere Überwachung der Laborwerte und/oder ein Aufschub oder ein Abbruch der Behandlung mit Polivy ist bei Patienten mit Neutropenie und/oder Thrombozytopenie von Grad 3 oder 4 in Betracht zu ziehen (siehe Abschnitt 4.2).
Periphere Neuropathie (PN)
Eine PN wurde bei mit Polivy behandelten Patienten bereits im ersten Zyklus berichtet und das Risiko steigt mit aufeinanderfolgenden Dosen an. Bei Patienten mit vorbestehender PN kann es zu einer Verschlechterung dieser Nebenwirkung kommen. Bei der PN, die unter Behandlung mit Polivy berichtet wurde, handelt es sich überwiegend um sensorische PN. Dennoch wurde auch motorische und sensomotorische PN berichtet. Patienten sind auf Symptome einer PN, wie Hypästhesie, Hyperästhesie, Parästhesie, Dysästhesie, neuropathische Schmerzen, Gefühl des Brennens, Muskelschwäche und Gangstörungen zu überwachen. Bei Patienten, bei denen eine neue PN auftritt oder sich eine PN verschlechtert, kann ein Aufschub, eine Dosisreduktion oder ein Abbruch der Behandlung mit Polivy erforderlich sein (siehe Abschnitt 4.2).
Infektionen
Schwerwiegende, lebensbedrohliche oder tödlich verlaufende Infektionen, einschließlich opportunistische Infektionen, wie Pneumonie (einschließlich Pneumocystis jirovecii und andere durch Pilze verursachte Pneumonien), Bakteriämie, Sepsis, Herpesinfektion und Zytomegalievirus-Infektion, wurden bei Patienten unter Behandlung mit Polivy berichtet (siehe Abschnitt 4.8). Die Reaktivierung latenter Infektionen wurde berichtet. Patienten sind während der Behandlung engmaschig auf Anzeichen bakterieller, mykotischer oder viraler Infektionen zu überwachen und müssen medizinischen Rat einholen, wenn bei ihnen Anzeichen und Symptome auftreten. Eine antiinfektive Prophylaxe ist über den gesamten Zeitraum der Behandlung mit Polivy in Betracht zu ziehen. Bei Vorliegen einer aktiven schweren Infektion ist Polivy nicht zu verabreichen. Polivy und jegliche gleichzeitige Chemotherapie ist bei Patienten, bei denen schwerwiegende Infektionen auftreten, abzubrechen.
Humaner Immundefizienz-Virus (HIV)
Polivy wurde bei Patienten mit HIV nicht untersucht. Hinsichtlich der gleichzeitigen Anwendung von CYP3A-Inhibitoren, siehe Abschnitt 4.5.
Immunisierung
Lebend- oder attenuierte Lebendimpfstoffe sind nicht gleichzeitig mit der Behandlung anzuwenden. Es wurden keine Studien bei Patienten durchgeführt, die kurz vor Behandlungsbeginn Lebendimpfstoffe erhalten haben.
Progressive multifokale Leukoenzephalopathie (PML)
PML wurde unter Behandlung mit Polivy berichtet (siehe Abschnitt 4.8). Die Patienten sind engmaschig auf neue oder sich verschlechternde neurologische, kognitive oder Verhaltensveränderungen, die auf eine PML hinweisen, zu überwachen. Wenn Verdacht auf eine PML besteht, ist die Behandlung mit Polivy und jegliche gleichzeitige Chemotherapie zu unterbrechen und wenn die Diagnose bestätigt wird, dauerhaft abzusetzen.
Tumorlysesyndrom (TLS)
Für Patienten mit hoher Tumorlast und schnell proliferierendem Tumor kann ein erhöhtes Risiko für die Entwicklung eines TLS bestehen. Geeignete Maßnahmen/eine geeignete Prophylaxe in Übereinstimmung mit lokalen Leitlinien sind vor Beginn der Behandlung mit Polivy zu ergreifen. Während einer Behandlung mit Polivy sind Patienten engmaschig auf das Auftreten eines TLS zu überwachen.
Reaktion im Zusammenhang mit einer Infusion
Polivy kann IRR verursachen, einschließlich schwere Fälle. Verzögerte IRR bis zu 24 Stunden nach der Behandlung mit Polivy sind aufgetreten. Vor der Behandlung mit Polivy sind ein Antihistaminikum und ein Antipyretikum zu verabreichen und die Patienten sind während der Infusion engmaschig zu überwachen. Tritt eine IRR auf, ist die Infusion zu unterbrechen und entsprechende medizinische Maßnahmen sind einzuleiten (siehe Abschnitt 4.2).
Embryo-fetale Toxizität
Basierend auf dem Wirkmechanismus und präklinischen Studien kann Polivy bei Anwendung an Schwangeren den Fötus schädigen (siehe Abschnitt 5.3). Schwangere sind über das Risiko für den Fötus aufzuklären.
Frauen im gebärfähigen Alter sind anzuweisen, während der Behandlung mit Polivy und für mindestens 9 Monate nach der letzten Dosis, eine zuverlässige Verhütungsmethode anzuwenden (siehe Abschnitt 4.6). Männliche Patienten mit Partnerinnen im gebärfähigen Alter sind anzuweisen, während der Behandlung mit Polivy und für mindestens 6 Monate nach der letzten Dosis, eine zuverlässige Verhütungsmethode anzuwenden (siehe Abschnitt 4.6).
Fertilität
In präklinischen Studien führte Polatuzumab Vedotin zu testikulärer Toxizität und kann die männliche Reproduktionsfähigkeit und Fertilität beeinträchtigen (siehe Abschnitt 5.3).
Deswegen wird Männern, die mit Polivy behandelt werden, geraten, vor Behandlungsbeginn Spermaproben einfrieren und aufbewahren zu lassen (siehe Abschnitt 4.6).
Ältere Patienten
Von den 435 bisher unbehandelten DLBCL-Patienten, die in der Studie GO39942 mit Polivy in Kombination mit R-CHP behandelt wurden, waren 227 (52,2 %) ≥ 65 Jahre. Patienten ≥ 65 Jahre hatten eine Inzidenz schwerwiegender Nebenwirkungen von 39,2 % und Patienten < 65 Jahre von 28,4 %. Eine vergleichbare Inzidenz schwerwiegender Nebenwirkungen wurde bei älteren Patienten im R-CHOP-Behandlungsarm festgestellt.
Von den 151 vorbehandelten DLBCL-Patienten, die in der Studie GO29365 mit Polivy in Kombination mit Bendamustin und Rituximab (BR) behandelt wurden, waren 103 (68 %) ≥ 65 Jahre. Patienten ≥ 65 Jahre hatten eine vergleichbare Inzidenz schwerwiegender Nebenwirkungen (55 %) zu Patienten < 65 Jahre (56 %). Klinische Studien mit Polivy schlossen keine ausreichende Anzahl an Patienten ≥ 65 Jahre ein, um zu ermitteln, ob sie anders auf Polivy ansprechen als jüngere Patienten.
Hepatotoxizität
Schwerwiegende Fälle von Hepatotoxizität, die vergleichbar mit einem hepatozellulären Schaden waren, einschließlich Erhöhungen der Transaminasen und/oder des Bilirubins, sind bei Patienten unter Behandlung mit Polivy aufgetreten (siehe Abschnitt 4.8). Eine vorbestehende Lebererkrankung, erhöhte Ausgangs-Leberenzymwerte und gleichzeitig angewendete Arzneimittel können das Risiko erhöhen. Die Leberenzym- und Bilirubinwerte sind zu überwachen (siehe Abschnitt 4.2).
Läsionen an der Infusionsstelle durch Extravasation
Im Falle einer Extravasation nach Gabe von Polatuzumab Vedotin wurden Schädigungen der Haut und des Weichteilgewebes innerhalb von Stunden bis Wochen beobachtet (siehe Abschnitt 4.8). Vergewissern Sie sich vor Beginn der Behandlung mit Polivy, dass ein guter venöser Zugang besteht, und überwachen Sie die Infusionsstelle während der Verabreichung auf eine mögliche Extravasation. Falls eine Extravasation auftritt, ist die Infusion abzubrechen, das Gewebe auf Schäden zu überwachen und die Behandlung gemäß den lokalen klinischen Leitlinien durchzuführen.
Nach klinischem Ermessen kann die verbleibende Dosis in einer anderen Extremität verabreicht werden.
Sonstige Bestandteile mit bekannter Wirkung
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro Dosis, d. h. es ist nahezu „natriumfrei“.
Dieses Arzneimittel enthält Polysorbat 20. Jede Durchstechflasche mit Polivy 30 mg Pulver zur Herstellung eines Infusionslösungskonzentrats enthält 1,8 mg Polysorbat 20. Jede Durchstechflasche mit Polivy 140 mg Pulver zur Herstellung eines Infusionslösungskonzentrats enthält 8,4 mg Polysorbat 20, entsprechend 1,2 mg/ml. Polysorbat 20 kann allergische Reaktionen hervorrufen.
Es wurden keine klinischen Studien beim Menschen mit Polatuzumab Vedotin zu Wechselwirkungen mit anderen Arzneimitteln durchgeführt.
Wechselwirkungen mit gleichzeitig angewendeten Arzneimitteln, die CYP3A4-Inhibitoren, -Substrate oder -Induktoren sind und mit gleichzeitig angewendeten Arzneimitteln, die P-gp-Inhibitoren sind
Basierend auf physiologiebasierten pharmakokinetischen Modellen (PBPK) von MMAE, welches aus Polatuzumab Vedotin freigesetzt wird, können starke CYP3A4- und P-gp-Inhibitoren (z. B. Ketoconazol) die Fläche unter der Konzentrations-Zeitkurve (area under the concentration-time curve, AUC) von unkonjugiertem MMAE um 48 % erhöhen. Bei gleichzeitiger Behandlung mit CYP3A4-Inhibitoren ist Vorsicht geboten. Patienten, die gleichzeitig starke CYP3A4-Inhibitoren erhalten (z. B. Boceprevir, Clarithromycin, Cobicistat, Indinavir, Itraconazol, Nefazodon, Nelfinavir, Posaconazol, Ritonavir, Saquinavir, Telaprevir, Telithromycin, Voriconazol), sind engmaschiger auf Anzeichen für Toxizitäten zu überwachen.
Unkonjugiertes MMAE verändert voraussichtlich die AUC gleichzeitig angewendeter Arzneimittel, die CYP3A4-Substrate sind (z. B. Midazolam), nicht.
Starke CYP3A4-Induktoren (z. B. Rifampicin, Carbamazepin, Phenobarbital, Phenytoin, Johanniskraut [Hypericum perforatum]) können die Exposition von unkonjugiertem MMAE verringern.
Arzneimittelwechselwirkungen von Rituximab, Bendamustin, Cyclophosphamid und Doxorubicin in Kombination mit Polatuzumab Vedotin
Die Pharmakokinetik (PK) von Rituximab, Bendamustin, Cyclophosphamid und Doxorubicin wird durch die gleichzeitige Anwendung von Polatuzumab Vedotin nicht beeinflusst. Basierend auf PK-Populationsanalysen wird gleichzeitig angewendetes Rituximab mit um 24 % erhöhter Plasma-AUC von antikörperkonjugiertem MMAE (acMMAE) und um 37 % verringerter unkonjugierter Plasma-AUC von MMAE in Verbindung gebracht. Die Plasma-AUC von acMMAE und unkonjugierter MMAE für Polivy plus R-CHP entsprechen denen in anderen Studien mit Polivy. Es ist keine Dosisanpassung erforderlich.
Bendamustin hat keine Wirkung auf acMMAE- und unkonjugierte MMAE-Plasma-AUC.
Frauen im gebärfähigen Alter/Kontrazeption bei Männern und Frauen
Frauen
Frauen im gebärfähigen Alter sind anzuweisen, während der Behandlung mit Polatuzumab Vedotin und für mindestens 9 Monate nach der letzten Dosis, eine zuverlässige Verhütungsmethode anzuwenden.
Männer
Männliche Patienten von Partnerinnen im gebärfähigen Alter sind anzuweisen, während der Behandlung mit Polatuzumab Vedotin und für mindestens 6 Monate nach der letzten Dosis, eine zuverlässige Verhütungsmethode anzuwenden.
Schwangerschaft
Es gibt keine Daten zu Schwangeren unter Behandlung mit Polivy. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Basierend auf dem Wirkmechanismus und präklinischen Studien kann Polatuzumab Vedotin den Fötus schädigen, wenn es bei Schwangeren angewendet wird. Bei Frauen im gebärfähigen Alter ist vor der Behandlung ein Schwangerschaftstest durchzuführen. Die Anwendung von Polivy während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die keine Verhütungsmethode anwenden, wird nicht empfohlen, es sei denn der potenzielle Nutzen für die Mutter überwiegt das potenzielle Risiko für den Fötus.
Stillzeit
Es ist nicht bekannt, ob Polatuzumab Vedotin oder dessen Metabolite in die Muttermilch übergehen. Ein Risiko für den gestillten Säugling kann nicht ausgeschlossen werden. Frauen sollten während einer Behandlung mit Polivy und mindestens 3 Monate nach der letzten Dosis nicht stillen.
Fertilität
In präklinischen Studien führte Polatuzumab Vedotin zu testikulärer Toxizität und kann die männliche Reproduktionsfähigkeit und Fertilität beeinträchtigen (siehe Abschnitt 5.3).
Deswegen wird Männern, die mit diesem Arzneimittel behandelt werden, geraten, vor Behandlungsbeginn Spermaproben einfrieren und aufbewahren zu lassen. Männern, die mit Polivy behandelt werden, wird empfohlen während der Behandlung und bis zu 6 Monate nach der letzten Dosis kein Kind zu zeugen.
Polivy hat geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. IRR, PN, Ermüdung (Fatigue) und Schwindelgefühl können während der Behandlung mit Polivy auftreten (siehe Abschnitte 4.4 und 4.8).
Zusammenfassung des Sicherheitsprofils
Die Sicherheit von Polivy wurde bei 435 Patienten in der Studie GO39942 (POLARIX) bewertet. Die in Abschnitt 4.8 beschriebenen Nebenwirkungen wurden identifiziert:
während der Behandlung und der Nachbeobachtung von bisher unbehandelten DLBCL-Patienten, die in der pivotalen klinischen Prüfung GO39942 (POLARIX) Polivy plus R-CHP (n = 435) oder R-CHOP (n = 438) erhielten. In der Gruppe Polivy plus R-CHP erhielten 91,7 % der Patienten 6 Zyklen Polivy versus 88,5 % der Patienten, die in der R-CHOP-Gruppe 6 Zyklen Vincristin erhielten.
Bei bisher unbehandelten DLBCL-Patienten, die mit Polivy plus R-CHP behandelt wurden:
Die am häufigsten berichteten Nebenwirkungen (≥ 30 %) bei mit Polivy plus R-CHP behandelten bisher unbehandelten DLBCL-Patienten, waren periphere Neuropathie (52,9 %), Übelkeit (41,6 %), Neutropenie (38,4 %) und Diarrhö (30,8 %).
Schwerwiegende Nebenwirkungen wurden bei 24,1 % der mit Polivy plus R-CHP behandelten Patienten berichtet.
Die häufigsten schwerwiegenden Nebenwirkungen, die bei ≥ 5 % der Patienten berichtet wurden, waren febrile Neutropenie (10,6 %) und Pneumonie (5,3 %).
Die Nebenwirkung, die bei > 1 % der mit Polivy plus R-CHP behandelten Patienten zu einem Abbruch der Therapie führte, war Pneumonie (1,1 %).
Die Sicherheit von Polivy wurde bei 151 Patienten in der Studie GO29365 bewertet. Die in Abschnitt 4.8 beschriebenen Nebenwirkungen wurden identifiziert:
während der Behandlung und der Nachbeobachtung von vorbehandelten DLBCL-Patienten (n = 151) in der pivotalen klinischen Prüfung GO29365. Das schließt Patienten aus der Run-in-Phase (n = 6), randomisierte Patienten (n = 39) und Patienten aus der erweiterten Kohorte (n = 106), die Polivy plus BR, verglichen zu randomisierten Patienten (n = 39), die nur BR erhielten, ein. Die Patienten erhielten in den Behandlungsarmen im Median 5 Behandlungszyklen, wohingegen Patienten im Vergleichsarm im Median 3 Behandlungszyklen erhielten.
Bei vorbehandelten DLBCL-Patienten, die mit Polivy plus BR behandelt wurden:
Die am häufigsten berichteten Nebenwirkungen (≥ 30 %) (alle Grade) bei vorbehandelten DLBCL-Patienten, die mit Polivy plus BR behandelt wurden, waren Neutropenie (45,7 %), Diarrhö (35,8 %), Übelkeit (33,1 %), Thrombozytopenie (32,5 %), Anämie (31,8 %) und periphere Neuropathie (30,5 %).
Schwerwiegende Nebenwirkungen wurden bei 41,7 % der mit Polivy plus BR behandelten Patienten berichtet.
Die häufigsten schwerwiegenden Nebenwirkungen, die bei ≥ 5 % der Patienten berichtet wurden, waren febrile Neutropenie (10,6 %), Sepsis (9,9 %), Pneumonie (8,6 %) und Fieber (7,9 %).
Die Nebenwirkung, die bei > 5 % der mit Polivy plus BR behandelten Patienten zu einem Abbruch der Therapie führte, war Thrombozytopenie (7,9 %).
Tabellarische Auflistung der Nebenwirkungen aus klinischen Prüfungen
Die Nebenwirkungen, die bei 586 mit Polivy behandelten Patienten aufgetreten sind, sind in Tabelle 4 dargestellt. Die Nebenwirkungen sind nachfolgend gemäß MedDRA nach Systemorganklasse (system organ class = SOC) und Häufigkeitskategorien aufgelistet. Die folgenden Häufigkeitskategorien wurden verwendet: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 4: Tabellarische Auflistung der Nebenwirkungen, die in klinischen Prüfungen bei mit Polivy behandelten Patienten auftraten
Infektionen und parasitäre Erkrankungen | |
Sehr häufig |
Pneumoniea, Infektion der oberen Atemwege |
Häufig |
Sepsisa, Herpesvirus-Infektiona, Zytomegalievirus-Infektion, Harnwegsinfektionc |
Erkrankungen des Blutes und des Lymphsystems | |
Sehr häufig |
Febrile Neutropenie, Neutropenie, Thrombozytopenie, Anämie, Leukopenie |
Häufig |
Lymphopenie, Panzytopenie |
Stoffwechsel- und Ernährungsstörungen | |
Sehr häufig |
Hypokaliämie, Appetit vermindert |
Häufig |
Hypokalzämie, Hypalbuminämie |
Erkrankungen des Nervensystems | |
Sehr häufig |
Periphere Neuropathie |
Häufig |
Schwindelgefühl |
Augenerkrankungen | |
Gelegentlich |
Sehen verschwommenb |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
Sehr häufig |
Husten |
Häufig |
Pneumonitis, Dyspnoec |
Erkrankungen des Gastrointestinaltrakts | |
Sehr häufig |
Diarrhö, Übelkeit, Obstipation, Erbrechen, Mukositisc, Abdominalschmerz |
Erkrankungen der Haut und des Unterhautgewebes | |
Sehr häufig |
Alopeziec |
Häufig |
Pruritus, Hautinfektionc, Ausschlagc, trockene Hautc |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |
Häufig |
Arthralgie, Myalgiec |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Sehr häufig |
Fieber, Ermüdung (Fatigue), Asthenie |
Häufig |
Peripheres Ödemc, Schüttelfrost |
Gelegentlich |
Extravasation an der Infusionsstelle |
Untersuchungen | |
Sehr häufig |
Gewicht erniedrigt |
Häufig |
Lipase erhöhtb, Hypophosphatämie |
Leber- und Gallenerkrankungen | |
Häufig |
Transaminasen erhöht |
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen | |
Sehr häufig |
Reaktion im Zusammenhang mit einer Infusion |
a Nebenwirkungen, die mit tödlichem Ausgang assoziiert sind
b Nebenwirkungen, die nur bei rezidivierendem oder refraktärem DLBCL beobachtet wurden.
c Nebenwirkungen, die nur bei bisher unbehandeltem DLBCL beobachtet wurden.
Die aufgelisteten Nebenwirkungen wurden sowohl bei bisher unbehandeltem DLBCL als auch bei rezidivierendem oder refraktärem DLBCL beobachtet, sofern nicht mit Fußnote versehen
Seltene und sehr seltene Nebenwirkungen: keine
Beschreibung ausgewählter Nebenwirkungen
Myelosuppression
In der placebokontrollierten Studie GO39942 (POLARIX) brachen 0,5 % der Patienten im Behandlungsarm mit Polivy plus R-CHP die Studienbehandlung aufgrund einer Neutropenie ab. Im Behandlungsarm mit R-CHOP brach kein Patient die Studienbehandlung aufgrund einer Neutropenie ab. Thrombozytopenische Ereignisse führten bei 0,2 % der Patienten im Behandlungsarm mit Polivy plus R-CHP zu einem Abbruch der Studienbehandlung, während es im Behandlungsarm mit R-CHOP bei keinem Patienten zu einem Abbruch kam. Weder im Behandlungsarm mit Polivy plus R-CHP noch im Behandlungsarm mit R-CHOP brach ein Patient die Behandlung aufgrund von Anämie ab.
In der unverblindeten Studie GO29365 brachen 4 % der Patienten in den Behandlungsarmen mit Polivy plus BR die Anwendung von Polivy aufgrund von Neutropenie ab, während es im Behandlungsarm mit BR bei 2,6 % der Patienten zu einem Abbruch kam. Thrombozytopenische Ereignisse führten bei 7,9 % der Patienten in den Behandlungsarmen mit Polivy plus BR zu einem Behandlungsabbruch und bei 5,1 % der Patienten im Behandlungsarm mit BR. Kein Patient brach die Behandlung aufgrund einer Anämie ab, weder in den Behandlungsarmen mit Polivy plus BR noch im Behandlungsarm mit BR. In den Behandlungsarmen mit Polivy plus BR wurden Neutropenien, Thrombozytopenien bzw. Anämien von jeweils Grad 3 oder höher bei 40,4 % der Patienten, bei 25,8 % der Patienten bzw. bei 12,6 % der Patienten festgestellt.
Periphere Neuropathie (PN)
In der placebokontrollierten Studie GO39942 (POLARIX) wurde im Behandlungsarm mit Polivy plus R-CHP bei 39,1 % der Patienten eine PN Grad 1, bei 12,2 % der Patienten eine PN Grad 2 und bei 1,6 % der Patienten eine PN Grad 3 berichtet. Im Behandlungsarm mit R-CHOP wurde bei 37,2 % der Patienten eine PN Grad 1, bei 15,5 % der Patienten eine PN Grad 2 und bei 1,1 % der Patienten eine PN Grad 3 berichtet. Es wurde keine PN Grad 4 - 5 im Behandlungsarm mit Polivy plus R-CHP oder im Behandlungsarm mit R-CHOP berichtet. Aufgrund einer PN brachen 0,7 % der Patienten im Behandlungsarm mit Polivy plus R-CHP die Behandlung ab, im Vergleich zu 2,3 % der Patienten im Behandlungsarm mit R-CHOP. 4,6 % der Patienten im Behandlungsarm mit Polivy plus R-CHP benötigten eine Dosisreduktion der Studienbehandlung aufgrund von PN, im Vergleich zu 8,2 % im Behandlungsarm mit R-CHOP. Im Behandlungsarm mit Polivy plus R-CHP betrug die mediane Zeit bis zum Auftreten des ersten PN-Ereignisses 2,27 Monate im Vergleich zu 1,87 Monaten im Behandlungsarm mit R-CHOP. PN-Ereignisse klangen bei 57,8 % der Patienten im Behandlungsarm mit Polivy plus R-CHP zum klinischen Stichtag ab, verglichen mit 66,9 % in der R-CHOP-Gruppe. Die mediane Zeit bis zum Verschwinden der peripheren Neuropathie betrug 4,04 Monate im Behandlungsarm mit Polivy plus R-CHP im Vergleich zu 4,6 Monaten im Behandlungsarm mit R-CHOP.
In der unverblindeten Studie GO29365 wurde in den Behandlungsarmen mit Polivy plus BR bei 15,9 % der Patienten eine PN Grad 1 und bei 12,6 % der Patienten eine PN Grad 2 berichtet. Im Behandlungsarm mit BR wurde bei 2,6 % der Patienten eine PN Grad 1 und bei 5,1 % der Patienten eine PN Grad 2 berichtet. Eine PN Grad 3 wurde in den Behandlungsarmen mit Polivy plus BR berichtet und im Behandlungsarm mit BR berichtete kein Patient PN-Ereignisse. Es wurde keine PN Grad 4 - 5 in den Behandlungsarmen mit Polivy plus BR oder im Behandlungsarm mit BR berichtet. Aufgrund einer PN brachen 2,6 % der Patienten die Behandlung mit Polivy ab und 2,0 % der Patienten benötigten eine Dosisreduktion. Kein Patient brach im Behandlungsarm mit BR die Behandlung ab oder benötigte aufgrund einer PN eine Dosisreduktion. In den Behandlungsarmen mit Polivy plus BR betrug die mediane Zeit bis zum ersten Auftreten einer PN 1,6 Monate und 39,1 % der Patienten mit PN-Ereignissen berichteten im Verlauf über ein Abklingen der Ereignisse.
Infektionen
In der placebokontrollierten Studie GO39942 (POLARIX) wurden bei 49,7 % der Patienten im Behandlungsarm mit Polivy plus R-CHP und bei 42,7 % der Patienten im Behandlungsarm mit R-CHOP Infektionen, einschließlich Pneumonie und andere Arten von Infektionen, berichtet. Infektionen von Grad 3 - 4 traten bei 14,0 % der Patienten im Behandlungsarm mit Polivy plus R‑CHP und bei 11,2 % der Patienten im Behandlungsarm mit R-CHOP auf. Im Behandlungsarm mit Polivy plus R-CHP wurden bei 14,0 % der Patienten schwerwiegende Infektionen und bei 1,1 % der Patienten tödliche Infektionen berichtet. Im Behandlungsarm mit R-CHOP wurden bei 10,3 % der Patienten schwerwiegende Infektionen und bei 1,4 % der Patienten tödliche Infektionen berichtet. 7 Patienten (1,6 %) im Behandlungsarm mit Polivy plus R-CHP brachen die Behandlung aufgrund einer Infektion ab, verglichen mit 10 Patienten (2,3 %) im Behandlungsarm mit R-CHOP.
In der unverblindeten Studie GO29365 wurden Infektionen, einschließlich Pneumonie und anderer Arten von Infektionen, bei 48,3 % der Patienten in den Behandlungsarmen mit Polivy plus BR und bei 51,3 % der Patienten im Behandlungsarm mit BR berichtet. In den Behandlungsarmen mit Polivy plus BR wurden bei 27,2 % der Patienten schwerwiegende Infektionen und bei 6,6 % der Patienten tödlich verlaufende Infektionen berichtet. Im Behandlungsarm mit BR wurden bei 30,8 % der Patienten schwerwiegende Infektionen und bei 10,3 % der Patienten tödlich verlaufende Infektionen berichtet. In den Behandlungsarmen mit Polivy plus BR brachen vier Patienten (2,6 %) die Behandlung aufgrund von einer Infektion ab, im Vergleich zu zwei Patienten (5,1 %) im Behandlungsarm mit BR.
Progressive multifokale Leukoenzephalopathie (PML)
In der placebokontrollierten Studie GO39942 (POLARIX) wurden keine Fälle einer PML berichtet.
In der unverblindeten Studie GO29365 kam es bei einem mit Polivy plus Bendamustin und Obinutuzumab behandelten Patienten zu einer tödlich verlaufenen PML. Dieser Patient hatte drei vorangegangene Therapielinien erhalten, die Anti-CD20-Antikörper enthielten.
Lebertoxizität
In der placebokontrollierten Studie GO39942 (POLARIX) wurde bei 10,6 % der Patienten im Behandlungsarm mit Polivy plus R-CHP und bei 7,3 % der Patienten im Behandlungsarm mit R‑CHOP eine Lebertoxizität berichtet. Im Behandlungsarm mit Polivy plus R-CHP waren die meisten Ereignisse von Grad 1 - 2 (8,7 %); Ereignisse von Grad 3 wurden bei 1,8 % der Patienten berichtet. Es traten keine Ereignisse von Grad 4 oder 5 auf. Schwerwiegende Ereignisse einer Lebertoxizität wurden bei 1 Patienten (0,2 %) berichtet und waren reversibel.
In einer anderen Studie wurden zwei Fälle schwerwiegender Lebertoxizität (hepatozellulärer Schaden und Steatosis hepatis) berichtet, die reversibel waren.
Gastrointestinale Toxizität
In der placebokontrollierten Studie GO39942 (POLARIX) wurde gastrointestinale Toxizität bei 76,1 % der Patienten im Behandlungsarm mit Polivy plus R-CHP berichtet, im Vergleich zu 71,9 % der Patienten im Behandlungsarm mit R-CHOP. Die meisten Ereignisse waren von Grad 1 - 2 und Ereignisse von Grad ≥ 3 wurden bei 9,7 % der Patienten im Behandlungsarm mit Polivy plus R-CHP gegenüber 8,2 % der Patienten im Behandlungsarm mit R-CHOP berichtet. Die häufigsten Ereignisse gastrointestinaler Toxizität waren Übelkeit und Diarrhö.
In der unverblindeten Studie GO29365 wurde gastrointestinale Toxizität bei 72,8 % der Patienten in den Behandlungsarmen mit Polivy plus BR berichtet, im Vergleich zu 66,7 % der Patienten im Behandlungsarm mit BR. Die meisten Ereignisse waren Grad 1 ‑ 2 und Grad 3 – 4 Ereignisse wurden bei 16,5 % der Patienten in den Behandlungsarmen mit Polivy plus BR berichtet, im Vergleich zu 12,9 % der Patienten im Behandlungsarm mit BR. Die häufigsten gastrointestinalen Ereignisse waren Diarrhö und Übelkeit.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51‑59
63225 Langen
Tel: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
anzuzeigen.
Aus klinischen Prüfungen liegen keine Erfahrungen zur Überdosierung beim Menschen vor. Die bisher höchste getestete Dosis beträgt 2,4 mg/kg, die als intravenöse Infusion angewendet wurde. In dieser Dosis traten PN-Ereignisse mit höherer Häufigkeit und höherem Schweregrad auf. Bei Patienten, bei denen es zu einer Überdosierung gekommen ist, ist die Infusion sofort abzubrechen und eine engmaschige Überwachung ist angezeigt.
Pharmakotherapeutische Gruppe: antineoplastische Arzneimittel; monoklonale Antikörper und Antikörper-Wirkstoff-Konjugate, ATC-Code: L01FX14
Wirkmechanismus
Polatuzumab Vedotin ist ein gegen CD79b-gerichtetes Antikörper-Wirkstoff-Konjugat, das bevorzugt an B-Zellen einen Mitosehemmstoff abgibt (Monomethylauristatin E oder MMAE), was zur Abtötung maligner B-Zellen führt. Das Polatuzumab-Vedotin-Molekül besteht aus MMAE, welches über einen spaltbaren Linker kovalent an einen humanisierten monoklonalen Immunglobulin-G1-Antikörper gebunden ist. Der monoklonale Antikörper bindet mit hoher Affinität und Selektivität an CD79b, eine Zelloberflächenkomponente des B-Zell-Rezeptors. Die CD79b-Expression ist auf normale Zellen innerhalb der B-Zelllinien (mit Ausnahme von Plasmazellen) und auf maligne B-Zellen beschränkt. CD79b wird bei > 95 % der diffusen großzelligen B-Zell-Lymphome exprimiert. Nach Bindung an CD79b wird Polatuzumab Vedotin schnell internalisiert und der Linker durch lysosomale Proteasen abgespalten, um die intrazelluläre Freisetzung von MMAE zu ermöglichen. MMAE bindet an Mikrotubuli und tötet sich teilende Zellen durch Mitosehemmung und Induktion der Apoptose.
Pharmakodynamische Wirkungen
Kardiale Elektrophysiologie
Basierend auf EKG-Daten aus zwei unverblindeten Studien bei Patienten mit vorbehandelten malignen B-Zell-Erkrankungen verlängerte Polatuzumab Vedotin in der empfohlenen Dosierung nicht das mittlere QTc-Intervall in klinisch relevantem Ausmaß.
Klinische Wirksamkeit und Sicherheit
Bisher unbehandeltes DLBCL
Die Wirksamkeit von Polivy wurde in einer internationalen, multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie (POLARIX, GO39942) an 879 Patienten mit bisher unbehandeltem DLBCL untersucht.
Geeignete Patienten waren zwischen 18 – 80 Jahre alt, hatten einen Internationalen Prognostischen Index(IPI)-Score von 2 – 5 und einen Eastern-Cooperative-Oncology-Group-Performance-Status (ECOG-PS) von 0 – 2. Zu den Histologien gehörten DLBCL [nicht anderweitig spezifiziert (not otherwise specified, NOS), ABC-(activated B-cell-)DLBCL, GCB-(germinal center B-cell)DLBCL], HGBCL (NOS, Double-Hit, Triple-Hit) und andere Subtypen des großzelligen B-Zell-Lymphoms (EBV-positiv, T-Zell-/Histiozyten-reich). Die Patienten hatten kein bekanntes ZNS-Lymphom und keine periphere Neuropathie > Grad 1.
Die Patienten wurden im Verhältnis 1:1 randomisiert und erhielten entweder Polivy plus R-CHP oder R-CHOP für sechs 21-tägige Zyklen, gefolgt von zwei zusätzlichen Zyklen mit Rituximab allein in beiden Armen. Die Patienten wurden nach IPI-Score (2 vs. 3 - 5), Vorhandensein oder Nichtvorhandensein einer Bulky-Disease (Läsion ≥ 7,5 cm) und nach geografischer Region stratifiziert.
Polivy wurde intravenös in einer Dosis von 1,8 mg/kg an Tag 1 der Zyklen 1 - 6 verabreicht. R-CHP oder R-CHOP wurden ab Tag 1 der Zyklen 1 - 6 verabreicht, gefolgt von Rituximab allein an Tag 1 der Zyklen 7 - 8. Die Dosierung in jedem Behandlungsarm war wie folgt:
Polivy + R-CHP-Arm: Polivy 1,8 mg/kg, Rituximab 375 mg/m², Cyclophosphamid 750 mg/m², Doxorubicin 50 mg/m² sowie Prednison oral 100 mg/Tag, an den Tagen 1 - 5 jedes Zyklus.
R-CHOP-Arm: Rituximab 375 mg/m², Cyclophosphamid 750 mg/m², Doxorubicin 50 mg/m², Vincristin 1,4 mg/m² sowie Prednison oral 100 mg/Tag, an den Tagen 1 - 5 jedes Zyklus.
Die beiden Behandlungsgruppen waren hinsichtlich der demografischen Merkmale und der Krankheitsmerkmale zu Behandlungsbeginn insgesamt ausgewogen. Das mediane Alter betrug 65 Jahre (Spanne 19 bis 80 Jahre), 53,6 % der Patienten waren hellhäutig und 53,8 % waren männlich. 43,8 % hatten eine Bulky-Disease, 38,0 % einen IPI-Wert von 2, 62,0 % einen IPI-Wert von 3 bis 5 und 88,7 % eine Erkrankung von Grad 3 oder 4. Die Mehrheit der Patienten (84,2 %) hatte DLBCL (einschließlich NOS, ABC und GCB).
Bei 211 Patienten wurde kein Ergebnis für die Ursprungszelle (cell of origin, COO) gemeldet. Von der COO-auswertbaren Population (n = 668) wiesen 33,1 % der Patienten ein ABC-artiges DLBCL und 52,7 % der Patienten ein GCB-artiges DLBCL auf, das mittels Genexpressionsanalysen bestimmt wurde.
Der primäre Endpunkt der Studie war das vom Prüfarzt bewertete progressionsfreie Überleben. Die mediane Dauer der Nachbeobachtung betrug 28,2 Monate. Die Wirksamkeitsergebnisse sind in Tabelle 5 und in Abbildung 1 zusammengefasst.
Tabelle 5: Zusammenfassung der Wirksamkeit bei Patienten mit bisher unbehandeltem DLBCL aus der Studie GO39942 (POLARIX)
Polivy + R-CHP |
R-CHOP |
|
Primärer Endpunkt |
||
Progressionsfreies Überleben1,* |
||
Anzahl (%) der Patienten mit Ereignis |
107 (24,3 %) |
134 (30,5 %) |
HR (95‑%‑KI) |
0,73 [0,57; 0,95] |
|
p-Wert3,** |
0,0177 |
|
2-Jahres-Schätzung für PFS (%) |
76,7 |
70,2 |
[95‑%‑KI] |
[72,65; 80,76] |
[65,80; 74,61] |
Wesentliche sekundäre Endpunkte |
||
Ereignisfreies Überleben (EFSeff)1 |
||
Anzahl (%) der Patienten mit Ereignis |
112 (25,5 %) |
138 (31,4%) |
HR [95‑%‑KI] |
0,75 [0,58; 0,96] |
|
p-Wert3,** |
0,0244 |
|
Objektive Ansprechrate (objective response rate, ORR) zu Behandlungsende2 |
||
Patienten mit Ansprechen (%) (CR, PR) |
376 (85,5 %) |
368 (83,8 %) |
Unterschied der Ansprechrate (%) [95‑%‑KI] |
1,63 [-3,32; 6,57] |
|
Vollständiges Ansprechen (%) (CR)2,* |
||
Patienten mit Ansprechen (%) |
343 (78,0 %) |
325 (74,0 %) |
Unterschied der Ansprechrate (%) [95‑%‑KI] |
3,92 [-1,89; 9,70] |
|
Partielles Ansprechen (%) (PR) |
33 (7,5 %) |
43 (9,8 %) |
95‑%‑KI Clopper-Pearson |
[5,22; 10,37] |
[7,18; 12,97] |
INV: Prüfarzt (investigator); BICR: Verblindetes unabhängiges Prüfkomitee (Blinded independent central review); KI: Konfidenzintervall; HR: Hazard Ratio; PFS: Progressionsfreies Überleben (Progression free survival); EFSeff: Ereignisfreies Überleben bezogen auf Wirksamkeit (Event free survival efficacy): wird verwendet, um EFS-Ereignisse widerzuspiegeln, die die Wirksamkeit adressieren, und ist definiert als Zeit vom Datum der Randomisierung bis zum frühesten Auftreten eines der folgenden Ereignisse: Fortschreiten der Erkrankung/Rezidiv, Tod jedweder Ursache, vom Prüfarzt bewertete primäre wirksamkeitsbezogene Notwendigkeit (außer Fortschreiten der Erkrankung/Rezidiv), die zur Einleitung einer nicht protokollgemäßen Anti-Lymphom-Behandlung (non-protocol specified anti-lymphoma treatment, NALT) geführt hat oder wenn nach Abschluss der Behandlung eine Biopsie entnommen wurde, die unabhängig davon, ob eine NALT eingeleitet wurde oder nicht, eine positive Resterkrankung ergab; CMH: Cochran-Mantel-Haenszel
1) INV-bewertet
2) BICR-bewertet
3) stratifizierter Log-Rank-Test
* gemäß Lugano-2014-Kriterien
** stratifiziert nach IPI (2 vs. 3 - 5), Vorhandensein oder Abwesenheit von Bulky-Disease, geografische Region
Bei der Zwischenanalyse war der wesentliche sekundäre Endpunkt OS unreif und unterschied sich statistisch nicht [stratifizierte Hazard Ratio von 0,94 (95-%-KI: 0,65; 1,37); p = 0,7524].
Abbildung 1: Kaplan-Meier-Kurve des Prüfarzt-bewerteten progressionsfreien Überlebens (PFS) in der Studie GO39942 (POLARIX)
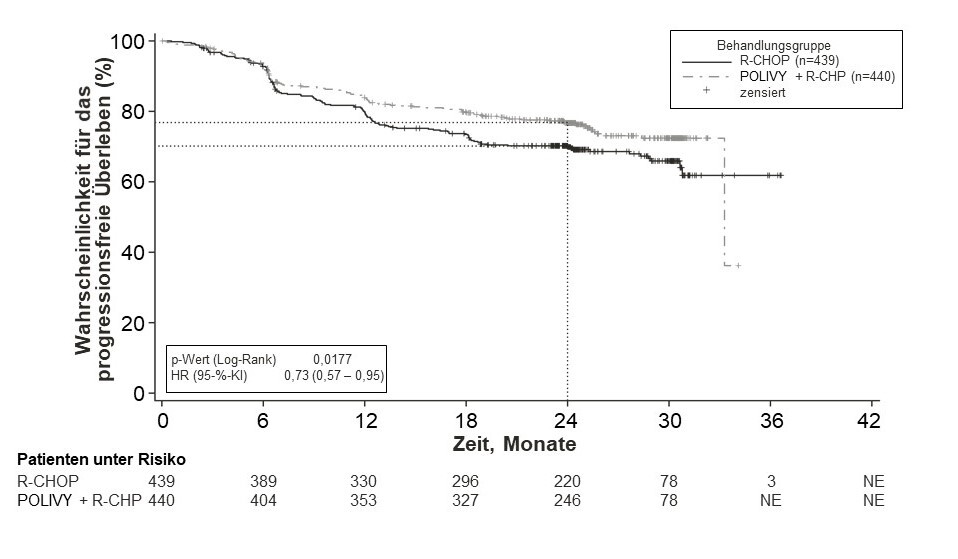
Patienten mit rezidivierender oder refraktärer Erkrankung
Die Wirksamkeit von Polivy wurde in einer internationalen, multizentrischen, unverblindeten Studie (GO29365), die eine randomisierte Kohorte von 80 Patienten mit vorbehandeltem DLBCL einschloss, bewertet. Die Patienten wurden 1:1 randomisiert, um entweder Polivy plus BR oder BR allein über sechs 21‑Tage-Zyklen zu erhalten. Patienten wurden nach Ansprechdauer auf die letzte vorangegangene Behandlung von ≤ 12 Monaten oder > 12 Monaten stratifiziert.
Geeignete Patienten kamen nicht für eine autologe hämatopoetische Stammzelltransplantation (HSCT) in Frage und hatten nach Erhalt von mindestens einem vorangegangenen systemischen Chemotherapieregime eine rezidivierende oder refraktäre Erkrankung. Die Studie schloss Patienten mit vorangegangener allogener HSCT, Lymphom des zentralen Nervensystems, transformiertem indolenten Lymphom, follikulärem Lymphom von Grad 3b, signifikanter kardiovaskulärer oder pulmonaler Erkrankung, aktiven Infektionen, AST oder Alanin-Aminotransferase (ALT) > 2,5 × ULN oder Gesamtbilirubin ≥ 1,5 × ULN, Kreatinin > 1,5 × ULN (oder CrCl < 40 ml/min) aus, es sei denn, die veränderten Werte waren auf das zugrundeliegende Lymphom zurückzuführen.
Polivy wurde an Tag 2 des 1. Zyklus und an Tag 1 der Zyklen 2 - 6 intravenös in einer Dosierung von 1,8 mg/kg verabreicht. Bendamustin wurde an den Tagen 2 und 3 des 1. Zyklus und den Tagen 1 und 2 der Zyklen 2 ‑ 6 intravenös in einer Dosierung von 90 mg/m2/Tag verabreicht. Rituximab wurde an Tag 1 der Zyklen 1 ‑ 6 in einer Dosierung von 375 mg/m2 verabreicht.
Unter den 80 Patienten, die randomisiert einer Behandlung mit Polivy plus BR (n = 40) oder BR allein (n = 40) zugeteilt wurden, war die Mehrheit hellhäutig (71 %) und männlich (66 %) und das mediane Alter betrug 69 Jahre (Spanne: 30 ‑ 86 Jahre). 64 der 80 Patienten (80 %) hatten einen ECOG-PS von 0 – 1 und 14 der 80 Patienten (18 %) hatten einen ECOG-PS von 2. Die Mehrheit der Patienten (98 %) hatten ein nicht anderweitig spezifiziertes (not otherwise specified, NOS) DLBCL. Insgesamt hatten 48 % der Patienten eine ABC-(activated B-cell-)DLBCL und 40 % hatten eine GCB-(germinal center B-cell like-)DLBCL. Die primären Gründe, warum Patienten nicht für eine HSCT in Frage kamen, waren Alter (40 %), nicht ausreichendes Ansprechen auf eine Behandlung (26 %) und Therapieversagen einer vorangegangenen Transplantation (20 %). Die mediane Anzahl vorangegangener Therapien betrug 2 (Spanne: 1 ‑ 7). 29 % (n = 23) der Patienten hatten eine vorangegangene Therapie, 25 % (n = 20) hatten 2 vorangegangene Therapien und 46 % (n = 37) hatten 3 oder mehr vorangegangene Therapien erhalten. Alle Patienten, mit Ausnahme eines Patienten im Arm Polatuzumab Vedotin + BR der randomisierten Phase–II-Studie, hatten noch keine Bendamustin-Behandlung erhalten. 80 % der Patienten hatten eine refraktäre Erkrankung. Bei Patienten, die Polatuzumab Vedotin plus BR erhielten und deren CD3+-Lymphozytenzahl ausgewertet wurde, betrug die absolute CD3+-Lymphozytenzahl > 200 Zellen/µl bei 95 % der Patienten, die vor der Therapie (n = 134) analysiert wurden, bei 79 % der Patienten, die am Ende der Behandlung (n = 72) bzw. bei 83 % der Patienten, die 6 Monate nach Ende der Behandlung (n = 18) analysiert wurden.
Der primäre Endpunkt der Studie war die Gesamtansprechrate (complete response, CR) am Ende der Behandlung (6 ‑ 8 Wochen nach Tag 1 des 6. Behandlungszyklus oder der letzten Studienbehandlung), bewertet mittels PET-CT durch ein unabhängiges Prüfkomitee (Independent Review Committee, IRC).
Tabelle 6: Zusammenfassung der Wirksamkeit bei Patienten mit vorbehandeltem DLBCL aus Studie GO29365
Polivy + Bendamustin + Rituximab |
Bendamustin + Rituximab |
|
Mediane Beobachtungszeit 22 Monate |
||
Primärer Endpunkt |
||
Gesamtansprechrate* (IRC-bewertet) zum Behandlungsende** |
||
Patienten mit Ansprechen (%) |
16 (40,0) |
7 (17,5) |
Unterschied der Ansprechrate (%) [95‑%‑KI] |
22,5 [2,6; 40,2] |
|
p‑Wert (CMH Chi-Quadrat-Test***) |
0,0261 |
|
Wesentliche sekundäre und exploratorische Endpunkte |
||
Dauer des Ansprechens (INV-bewertet) |
||
Anzahl der in die Analyse eingeschlossenen Patienten |
28 |
13 |
Mediane Dauer des Ansprechens (95‑%‑KI), Monate |
10,3 (5,6; nb) |
4,1 (2,6; 12,7) |
0,44 (0,20; 0,95) | ||
p‑Wert (Log-Rank-Test, stratifiziert***) |
0,0321 |
|
Gesamtansprechrate* (INV-bewertet) zu Behandlungsende** |
||
Patienten mit Ansprechen (%) (CR, PR) |
19 (47,5) |
7 (17,5) |
Unterschied der Ansprechrate (%) [95‑%‑KI] |
30,0 [9,5; 47,4] |
|
p‑Wert (CMH Chi-Quadrat-Test***) |
0,0036 |
|
Vollständiges Ansprechen (%) (CR) |
17 (42,5) |
6 (15,0) |
Unterschied der Ansprechrate (%) [95‑%‑KI] |
27,5 [7,7; 44,7] |
|
p‑Wert (CMH Chi-Quadrat-Test***) |
0,0061 |
|
Partielles Ansprechen (%) (PR) |
2 (5,0) |
1 (2,5) |
Beste Gesamtansprechrate* (INV-bewertet) |
||
Patienten mit Ansprechen (%) (CR, PR) |
28 (70,0) |
13 (32,5) |
Unterschied der Ansprechrate (%) [95‑%‑KI] |
37,5 [15,6; 54,7] |
|
Vollständiges Ansprechen (%) (CR) |
23 (57,5) |
8 (20,0) |
95‑%‑KI Clopper-Pearson |
[40,9; 73,0] |
[9,1; 35,7] |
Partielles Ansprechen (%) (PR) |
5 (12,5) |
5 (12,5) |
KI: Konfidenzintervall; CMH: Cochran-Mantel-Haenszel; CR: vollständiges Ansprechen (complete response); DOR: Dauer des Ansprechens (duration of response); HR: Hazard Ratio; INV: Prüfarzt (investigator); IRC: Unabhängiges Prüfkomitee (Independent Review Committee); nb: nicht bewertbar; PR: partielles Ansprechen (partial response)
* gemäß modifizierten Lugano-2014-Kriterien: Knochenmarksbestätigung des PET-CT CR erforderlich. PET-CT PR erforderte das Erreichen sowohl der PET-CT- als auch der CT-Kriterien.
** 6 - 8 Wochen nach Tag 1 des 6. Zyklus oder letzte Studienbehandlung
*** Stratifizierung durch Ansprechdauer auf die vorangegangene Therapie (≤ 12 Monate vs. > 12 Monate)
Das Gesamtüberleben (overall survival, OS) war ein explorativer Endpunkt mit nicht-kontrolliertem Fehler 1. Art. Das mediane OS im Polivy plus BR-Arm betrug 12,4 Monate (95‑%‑KI: 9,0; nb) gegenüber 4,7 Monaten (95‑%‑KI: 3,7; 8,3) im Kontrollarm. Der nicht-adjustierte Schätzer für OS-HR betrug 0,42. Unter Berücksichtigung des Einflusses von Baseline-Kovariablen wurde das OS-HR auf 0,59 angepasst. Zu den Kovariablen gehörten der primär refraktäre Status, die Anzahl der vorherigen Therapielinien, der IPI und eine vorherige Stammzelltransplantation.
Das vom Prüfer bewertete progressionsfreie Überleben (progression free survival, PFS) war ein explorativer Endpunkt mit nicht-kontrolliertem Fehler 1. Art. Das mediane PFS im Polivy plus BR-Arm betrug 7,6 Monate (95‑%‑KI: 6,0; 17,0) gegenüber 2,0 Monaten (95‑%‑KI: 1,5; 3,7) im Kontrollarm. Der nicht-adjustierte Schätzer für PFS HR betrug 0,34.
Immunogenität
Wie bei allen therapeutischen Proteinen besteht die Möglichkeit einer Immunantwort bei Patienten, die mit Polatuzumab Vedotin behandelt wurden. In den Studien GO39442 (POLARIX) und GO29365 wurden bei 1,4 % (6/427) bzw. 5,2 % (12/233) der Patienten Anti-Polatuzumab-Vedotin-Antikörper nachgewiesen, von denen keiner ein neutralisierender Antikörper war. Aufgrund der limitierten Anzahl der Anti-Polatuzumab-Vedotin-Antikörper-positiven Patienten, können im Hinblick auf eine potenzielle Auswirkung der Immunogenität auf die Wirksamkeit oder Sicherheit keine Schlussfolgerungen gezogen werden.
Immunogenitäts-Testergebnisse hängen stark von mehreren Faktoren ab, darunter Testempfindlichkeit und -spezifizität, Testmethoden, Handhabung der Proben, Zeitpunkt der Probennahme, gleichzeitig angewendete Arzneimittel und Grunderkrankungen. Aus diesen Gründen ist ein Vergleich der Inzidenz von Antikörpern gegen Polatuzumab Vedotin mit der Inzidenz von Antikörpern gegen andere Substanzen eventuell irreführend.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Polivy eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen zur Behandlung reifer B-Zell-Neoplasien gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Plasmaverfügbarkeit von antikörperkonjugiertem MMAE (acMMAE) stieg gegenüber dem Dosisbereich von Polatuzumab Vedotin von 0,1 bis 2,4 mg/kg dosisproportional an. Nach der ersten Dosis von 1,8 mg/kg Polatuzumab Vedotin betrug die mittlere maximale Konzentration (Cmax) von acMMAE 803 (± 233) ng/ml und die Fläche unter der Konzentrations-Zeit-Kurve ab Zeitpunkt null bis unendlich (AUCinf) betrug 1.860 (± 966) Tag • ng/ml. Basierend auf der PK-Populationsanalyse stieg die AUC von acMMAE in Zyklus 3 um etwa 30 % über die AUC aus Zyklus 1 und erreichte mehr als 90 % der AUC von Zyklus 6. Die terminale Halbwertszeit für acMMAE in Zyklus 6 betrug etwa 12 Tage (95‑%‑KI von 8,1 - 19,5 Tage). Basierend auf einer PK-Populationsanalyse beträgt die vorhergesagte acMMAE-Konzentration am Ende von Zyklus 6 ca. 80 % des theoretischen Steady-State-Wertes. Expositionen von unkonjugiertem MMAE, der zytotoxischen Komponente von Polatuzumab Vedotin, stiegen über den Dosisbereich von Polatuzumab Vedotin von 0,1 bis 2,4 mg/kg dosisproportional an. Die Plasmakonzentrationen von MMAE folgten einer durch die Bildungsrate begrenzten Kinetik. Nach der ersten Dosis von 1,8 mg/kg Polatuzumab Vedotin betrug die Cmax 6,82 (± 4,73) ng/ml, die Zeit bis zur maximalen Plasmakonzentration etwa 2,5 Tage und die terminale Halbwertszeit etwa 4 Tage. Die Plasmaverfügbarkeiten von unkonjugiertem MMAE betragen < 3 % der acMMAE-Expositionen. Basierend auf der PK-Populationsanalyse kommt es nach wiederholter dreiwöchentlicher Dosierung zu einer Verringerung der unkonjugierten MMAE-Exposition im Plasma (AUC).
Basierend auf einer Post-hoc-Analyse von populationspharmakokinetischen Simulationen wird eine um nicht mehr als 55 % erhöhte Exposition gegenüber unkonjugiertem MMAE bei Patienten mit einem Körpergewicht von mehr als 100 kg angenommen.
Resorption
Polivy wird als intravenöse Infusion verabreicht. Es wurden keine Studien mit anderen Darreichungsformen durchgeführt.
Verteilung
Der Populationsschätzer des zentralen Verteilungsvolumens von acMMAE betrug 3,15 l, was annähernd dem Plasmavolumen entsprach. In vitro bindet MMAE mäßig an menschliche Plasmaproteine (71 - 77 %). In vitro geht MMAE nicht signifikant in menschliche rote Blutzellen über; die Blut-Plasma-Ratio beträgt 0,79 zu 0,98.
In-vitro-Daten zeigen, dass MMAE ein P-gp-Substrat ist, aber in klinisch relevanten Konzentrationen kein Inhibitor von P-gp ist.
Biotransformation
Man nimmt an, dass Polatuzumab Vedotin bei Patienten einen Katabolismus durchläuft, der zur Produktion kleiner Peptide, Aminosäuren, unkonjugiertem MMAE und zugehörigen Kataboliten von unkonjugiertem MMAE führt. Die Spiegel der MMAE-Metabolite wurden in menschlichem Plasma nicht gemessen.
In-vitro-Studien zeigen, dass MMAE ein Substrat für CYP3A4/5 ist, aber keine wichtigen CYP-Enzyme induziert. MMAE ist ein schwacher, zeitabhängiger Inhibitor von CYP3A4/5, der aber in klinisch relevanten Konzentrationen CYP3A4/5 nicht kompetitiv inhibiert.
MMAE ist kein Inhibitor von CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 oder CYP2D6.
Elimination
Basierend auf einer PK-Populationsanalyse wird das Konjugat (acMMAE) primär durch nicht-spezifische lineare Clearance mit einem Wert von 0,9 l/Tag eliminiert. In-vivo-Studien mit Ratten, die Polatuzumab Vedotin erhielten (mit radioaktiv markiertem MMAE), zeigten, dass der Großteil der Radioaktivität mit den Faeces ausgeschieden wird und der kleinere Teil der Radioaktivität mit dem Urin.
Kinder und Jugendliche
Es wurden keine Studien zur Untersuchung der Pharmakokinetik von Polatuzumab Vedotin bei Kindern und Jugendlichen (< 18 Jahre) durchgeführt.
Ältere Patienten
Basierend auf PK-Populationsanalysen mit Patienten zwischen 19 ‑ 89 Jahren hatte das Alter keine Auswirkung auf die Pharmakokinetik von acMMAE und unkonjugiertem MMAE. Bei Patienten < 65 Jahre (n = 394) und Patienten ≥ 65 Jahre (n = 495) wurden basierend auf PK-Populationsanalysen keine signifikanten Unterschiede in der Pharmakokinetik von acMMAE und unkonjugiertem MMAE beobachtet.
Nierenfunktionsstörung
Basierend auf PK-Populationsanalysen mit Patienten mit leichter (CrCl 60 ‑ 89 ml/min, n = 361) oder mäßiger (CrCl 30 - 59 ml/min, n = 163) Nierenfunktionsstörung, waren Expositionen von acMMAE und unkonjugiertem MMAE vergleichbar zu den Expositionen bei Patienten mit normaler Nierenfunktion (CrCl ≥ 90 ml/min, n = 356). Zur Bewertung der Auswirkungen einer schweren Nierenfunktionsstörung (CrCl 15 ‑ 29 ml/min, n = 4) auf die PK liegen unzureichende Daten vor. Zu Patienten mit Nierenerkrankung im Endstadium oder Dialysepatienten liegen keine Daten vor.
Leberfunktionsstörung
Basierend auf PK-Populationsanalysen sind bei Patienten mit leichter Leberfunktionsstörung [AST oder ALT > 1,0 bis 2,5 × ULN oder Gesamtbilirubin > 1,0 bis 1,5 × ULN, n = 133] die acMMAE-Expositionen gegenüber Patienten mit normaler Leberfunktion (n = 737) vergleichbar, wohingegen die AUC von unkonjugiertem MMAE um nicht mehr als 40 % höher liegt.
Zur Bewertung der Auswirkungen einer mäßigen Leberfunktionsstörung (Gesamtbilirubin > 1,5 ‑ 3 × ULN, n = 11) auf die PK liegen unzureichende Daten vor. Zu Patienten mit schwerer Leberfunktionsstörung oder Lebertransplantation liegen begrenzte Daten vor.
Systemische Toxizität
Sowohl bei Ratten als auch bei Cynomolgus-Affen schlossen die vorherrschenden systemischen Toxizitäten, die mit der Anwendung von MMAE und Polatuzumab Vedotin in Verbindung standen, reversible Knochenmarkstoxizität und damit verbundene Auswirkungen auf periphere Blutzellen ein.
Genotoxizität
Es wurden keine speziellen Mutagenitätsstudien mit Polatuzumab Vedotin durchgeführt. Im bakteriellen Rückmutationstest (Ames-Test) oder dem L5178Y-Maus-Lymphom-Mutationstest war MMAE nicht mutagen.
MMAE zeigte in einer Knochenmarks-Mikronukleusstudie an Ratten genotoxische Eigenschaften, vermutlich über einen aneugenischen Mechanismus. Dieser Mechanismus stimmt mit der pharmakologischen Wirkung von MMAE als Mikrotubuli zerstörende Substanz überein.
Kanzerogenität
Es wurden keine speziellen Kanzerogenitätsstudien mit Polatuzumab Vedotin und/oder MMAE durchgeführt.
Beeinträchtigung der Fertilität
Es wurden keine speziellen Fertilitätsstudien an Tieren mit Polatuzumab Vedotin durchgeführt. Die Ergebnisse der 4-wöchigen Studie zur Toxizität bei Ratten zeigen jedoch das Potenzial von Polatuzumab Vedotin, die männliche Zeugungsfähigkeit und Fertilität zu beeinträchtigen. Die Degeneration der testikulären seminiferen Tubuli war nach einer 6-wöchigen behandlungsfreien Zeitspanne nicht reversibel und korrelierte mit verringertem Hodengewicht und dem Gesamtbefund von kleinen und/oder weichen Hoden nach Nekropsie von Männchen, die Dosen von ≥ 2 mg/kg erhalten hatten und sich davon erholten.
Reproduktionstoxizität
Es wurden keine speziellen Teratogenitätsstudien mit Polatuzumab Vedotin bei Tieren durchgeführt. Die Behandlung trächtiger Ratten mit MMAE in einer Dosierung von 0,2 mg/kg führte zu Embryoletalität und fetalen Missbildungen (einschließlich vorstehender Zunge, verdrehten Gliedmaßen, Gastroschisis und Agnathie).
Die systemische Exposition (AUC) bei Ratten mit Dosen von 0,2 mg/kg MMAE liegt bei etwa 50 % der AUC von Patienten, die die empfohlene Dosis von 1,8 mg/kg Polivy alle 21 Tage erhielten.
Bernsteinsäure
Natriumhydroxid (zur pH-Einstellung)
Saccharose
Polysorbat 20 (E 432)
Das Arzneimittel darf, außer mit den unter Abschnitt 6.6 aufgeführten, nicht mit anderen Arzneimitteln gemischt oder verdünnt werden.
Ungeöffnete Durchstechflasche
30 Monate
Rekonstituierte Lösung
Aus mikrobiologischer Sicht soll die rekonstituierte Lösung sofort verwendet werden. Falls die Lösung nicht unmittelbar verwendet wird, liegen die Aufbewahrungszeiten und -bedingungen bis zur Anwendung in der Verantwortung des Anwenders und sollten normalerweise 24 Stunden gekühlt (2 °C – 8 °C) nicht überschreiten, es sei denn, die Rekonstitution hat unter kontrollierten und validierten aseptischen Bedingungen stattgefunden. Die chemische und physikalische Haltbarkeit der rekonstituierten Lösung wurde über 72 Stunden gekühlt (2 °C – 8 °C) bzw. 24-stündiger Lagerung bei Raumtemperatur (9 °C – 25 °C) nachgewiesen.
Verdünnte Lösung
Aus mikrobiologischer Sicht soll die zubereitete Infusionslösung sofort verwendet werden. Falls die Lösung nicht unmittelbar verwendet wird, liegen die Aufbewahrungszeiten und -bedingungen bis zur Anwendung in der Verantwortung des Anwenders und sollten normalerweise 24 Stunden gekühlt (2 °C – 8 °C) nicht überschreiten, es sei denn, die Verdünnung hat unter kontrollierten und validierten aseptischen Bedingungen stattgefunden. Eine chemische und physikalische Haltbarkeit der zubereiteten Infusionslösung wurde für die in Tabelle 7 aufgeführten Zeitspannen nachgewiesen. Verdünnte Lösung ist zu verwerfen, wenn die Aufbewahrungszeit die in Tabelle 7 aufgeführten Zeitspannen überschritten hat.
Tabelle 7: Zeitspannen, für die eine chemische und physikalische Haltbarkeit der zubereiteten Infusionslösung gezeigt wurde
Verdünnungsmittel zur Zubereitung der Infusionslösung |
Aufbewahrungszeit der Infusionslösung1 |
|
Natriumchlorid 9 mg/ml (0,9 %) |
Bis zu 72 Stunden gekühlt (2 °C – 8 °C) oder |
|
Natriumchlorid 4,5 mg/ml (0,45 %) |
Bis zu 72 Stunden gekühlt (2 °C – 8 °C) oder |
|
5 % Glucose |
Bis zu 72 Stunden gekühlt (2 °C – 8 °C) oder |
|
1 Zur Sicherstellung der Produktstabilität, angegebene Aufbewahrungszeiten nicht überschreiten. | ||
Im Kühlschrank lagern (2 °C – 8 °C).
Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Rekonstitution und Verdünnung des Arzneimittels, siehe Abschnitt 6.3.
Polivy 30 mg Pulver zur Herstellung eines Infusionslösungskonzentrats:
6-ml-Durchstechflasche (Glasart I, farblos) mit Stopfen (Fluorharzlaminat) und Aluminiumsiegel mit Flip-off-Deckel aus Kunststoff verschlossen. Enthält 30 mg Polatuzumab Vedotin. Packung mit einer Durchstechflasche.
Polivy 140 mg Pulver zur Herstellung eines Infusionslösungskonzentrats:
20-ml-Durchstechflasche (Glasart I, farblos) mit Stopfen (Fluorharzlaminat) und Aluminiumsiegel mit Flip-off-Deckel aus Kunststoff verschlossen. Enthält 140 mg Polatuzumab Vedotin. Packung mit einer Durchstechflasche.
Allgemeine Vorsichtsmaßnahmen
Polivy enthält eine zytotoxische Komponente und muss unter Aufsicht eines in der Anwendung von zytotoxischen Substanzen erfahrenen Arztes verabreicht werden. Verfahren für die korrekte Handhabung und Beseitigung von antineoplastischen und zytotoxischen Arzneimitteln sind zu befolgen.
Das rekonstituierte Arzneimittel enthält keine Konservierungsstoffe und ist nur zum einmaligen Gebrauch bestimmt. Während der gesamten Handhabung dieses Arzneimittels ist eine geeignete aseptische Technik anzuwenden.
Polivy muss mit sterilem Wasser für Injektionszwecke rekonstituiert werden und vor der Anwendung in einem intravenösen Infusionsbeutel verdünnt werden, der eine 9-mg/ml-Natriumchlorid- Injektionslösung (0,9 %) oder 4,5-mg/ml-Natriumchlorid-Injektionslösung (0,45 %) oder 5-%-Glucoselösung enthält.
Die rekonstituierte Lösung sowie die Infusionslösung dürfen nicht eingefroren oder direktem Sonnenlicht ausgesetzt werden.
Anweisungen zur Rekonstitution
Polivy 30 mg: Injizieren Sie mit einer sterilen Spritze langsam 1,8 ml steriles Wasser für Injektionszwecke in die Polivy 30-mg-Durchstechflasche, um eine Einmaldosis der Lösung mit 20 mg/ml Polatuzumab Vedotin zu erhalten. Richten Sie den Strahl auf die Wand der Durchstechflasche und nicht direkt auf den Lyophilisat-Kuchen.
Polivy 140 mg: Injizieren Sie mit einer sterilen Spritze langsam 7,2 ml steriles Wasser für Injektionszwecke in die Polivy 140-mg-Durchstechflasche, um eine Einmaldosis der Lösung mit 20 mg/ml Polatuzumab Vedotin zu erhalten. Richten Sie den Strahl auf die Wand der Durchstechflasche und nicht direkt auf den Lyophilisat-Kuchen.
Schwenken Sie die Durchstechflasche vorsichtig, bis der Inhalt vollständig aufgelöst ist. Nicht schütteln.
Prüfen Sie die rekonstituierte Lösung auf Verfärbungen und Partikel. Die zubereitete Lösung muss farblos bis leicht bräunlich, klar bis leicht opaleszent und frei von sichtbaren Partikeln sein. Nicht verwenden, wenn die rekonstituierte Lösung eine Verfärbung oder eine Trübung aufweist oder sichtbare Partikel enthält.
Anweisungen zur Verdünnung
Polivy muss in einem intravenösen Infusionsbeutel mit einem minimalen Volumen von 50 ml, der eine 9-mg/ml-Natriumchlorid-Injektionslösung oder 4,5-mg/ml Natriumchlorid-Injektionslösung oder 5-%-Glucoselösung enthält, auf eine finale Konzentration von 0,72 ‑ 2,7 mg/ml verdünnt werden.
Bestimmen Sie das Volumen der benötigten rekonstituierten 20 mg/ml Lösung anhand der erforderlichen Dosis (siehe unten):
Gesamtdosis von Polivy (ml), |
= |
Dosis Polivy (mg/kg) x Körpergewicht des Patienten (kg) |
Konzentration der rekonstituierten Lösung in der Durchstechflasche (20 mg/ml) |
Entnehmen Sie das erforderliche Volumen der rekonstituierten Lösung aus der Polivy Durchstechflasche unter Verwendung einer sterilen Spritze und verdünnen Sie es im intravenösen Infusionsbeutel. Verwerfen Sie in der Durchstechflasche übrig gebliebene rekonstituierte Lösung.
Mischen Sie vorsichtig den intravenösen Infusionsbeutel indem Sie ihn langsam umdrehen. Nicht schütteln.
Prüfen Sie den Inhalt des intravenösen Infusionsbeutels auf Partikel und verwerfen Sie die Lösung, wenn Partikel vorhanden sind.
Vermeiden Sie den Transport der zubereiteten Infusionslösung, da Bewegung zu Aggregation führen kann. Wenn die zubereitete Infusion transportiert werden soll, entnehmen Sie die Luft aus dem Infusionsbeutel und begrenzen Sie den Transport auf 30 Minuten bei Raumtemperatur (9 °C ‑ 25 °C) oder 24 Stunden gekühlt (bei 2 °C ‑ 8 °C). Wenn die Luft entnommen wurde, ist ein Infusionsset mit einem Einstechdorn und Belüftung erforderlich, um eine korrekte Dosierung während der Infusion sicherzustellen. Die gesamte Aufbewahrungs- und Transportzeit der verdünnten Lösung darf die in Tabelle 7 definierte Aufbewahrungszeit nicht überschreiten (siehe Abschnitt 6.3).
Polivy muss über ein eigenes Infusionssystem verabreicht werden, das mit einem sterilen, pyrogenfreien, In-line- oder Add-on-Filter (0,2 oder 0,22 Mikrometer Porengröße) mit niedriger Proteinbindungskapazität und einem Katheter ausgestattet ist.
Polivy ist kompatibel mit intravenösen Infusionsbeuteln mit Produktkontaktflächen aus Polyvinylchlorid (PVC) oder Polyolefinen, wie Polyethylen (PE) und Polypropylen. Zusätzlich wurden keine Inkompatibilitäten mit Infusionssets oder Infusionshilfen mit Produktkontaktflächen aus PVC, PE, Polyurethan, Polybutadien, Acrylnitril-Butadien-Styrol, Polycarbonat, Polyetherurethan, Fluorethylenpropylen oder Polytetrafluorethylen oder mit Filtermembranen aus Polyethersulfon oder Polysulfon beobachtet.
Hinweise zur Beseitigung
Polivy ist nur zur Einmalanwendung.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Roche Registration GmbH
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Deutschland
EU/1/19/1388/001
EU/1/19/1388/002
Datum der Erteilung der Zulassung: 16. Januar 2020
Datum der letzten Verlängerung der Zulassung: 03. Dezember 2021
März 2026
Verschreibungspflichtig
Roche Pharma AG
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Telefon (07624) 14-0
Telefax (07624) 1019
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.