
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Vyndaqel® 61 mg Weichkapseln
Jede Weichkapsel enthält 61 mg mikronisiertes Tafamidis.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Weichkapsel enthält nicht mehr als 44 mg Sorbitol (Ph.Eur.) (E 420).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Weichkapsel.
Rötlich-braune, opake, längliche (etwa 21 mm) Kapsel mit dem Aufdruck „VYN 61“ in Weiß.
Vyndaqel ist indiziert zur Behandlung der Wildtyp- oder hereditären Transthyretin-Amyloidose bei erwachsenen Patienten mit Kardiomyopathie (ATTR-CM).
Die Therapie sollte unter der Kontrolle eines in der Behandlung von Patienten mit Amyloidose oder Kardiomyopathie erfahrenen Arztes begonnen werden.
Wenn bei Patienten mit einer bestimmten Anamnese oder Anzeichen für Herzinsuffizienz oder Kardiomyopathie ein Verdacht besteht, muss ein mit der Behandlung von Amyloidose oder Kardiomyopathie erfahrener Arzt eine ätiologische Diagnose durchführen, um ATTR-CM zu bestätigen und eine AL-Amyloidose auszuschließen, bevor die Behandlung mit Tafamidis eingeleitet wird. Hierfür eignen sich die folgenden Untersuchungsverfahren: Knochenszintigrafie und Blut-/ Urin-Untersuchung und/ oder histologische Untersuchung einer Biopsie und Genotypisierung des Transthyretin (TTR), um es als Wildtyp oder hereditär zu charakterisieren.
Dosierung
Die empfohlene Dosierung beträgt eine Kapsel Vyndaqel 61 mg (Tafamidis) einmal täglich per os (siehe Abschnitt 5.1).
Vyndaqel 61 mg (Tafamidis) entspricht 80 mg Tafamidis-Meglumin. Tafamidis und Tafamidis-Meglumin sind auf Basis der mg-Angabe nicht gegeneinander austauschbar (siehe Abschnitt 5.2).
Die Behandlung mit Vyndaqel sollte so früh wie möglich im Verlauf der Erkrankung begonnen werden, wenn der klinische Nutzen in Bezug auf den Krankheitsfortschritt deutlicher ist. Im Gegensatz dazu liegt die Entscheidung über die Einleitung oder Fortsetzung einer Therapie bei einer weiter fortgeschrittenen Amyloid-bedingten Herzschädigung, z. B. der NYHA-Klasse III, im Ermessen eines in der Behandlung von Patienten mit Amyloidose oder Kardiomyopathie erfahrenen Arztes (siehe Abschnitt 5.1). Für Patienten mit NYHA-Klasse IV liegen begrenzte klinische Daten vor.
Wenn es nach der Einnahme zu Erbrechen kommt und die intakte Vyndaqel-Kapsel gefunden wird, sollte, sofern möglich, eine zusätzliche Dosis Vyndaqel eingenommen werden. Wenn keine Kapsel gefunden wird, ist keine zusätzliche Dosis notwendig, und die Einnahme von Vyndaqel kann am Folgetag wie gewohnt fortgesetzt werden.
Besondere Patientengruppen
Ältere Patienten
Bei älteren Patienten (≥ 65 Jahre) ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Eingeschränkte Leber- und Nierenfunktion
Bei Patienten mit eingeschränkter Nierenfunktion oder leichter bis mäßiger Einschränkung der Leberfunktion ist keine Dosisanpassung erforderlich. Es liegen begrenzte Daten zu Patienten mit starker Einschränkung der Nierenfunktion (Kreatinin-Clearance ≤ 30 ml/min) vor. Tafamidis wurde nicht an Patienten mit schwerer Beeinträchtigung der Leberfunktion untersucht, so dass bei diesen Patienten Vorsicht geboten ist (siehe Abschnitt 5.2).
Kinder und Jugendliche
Es gibt keinen relevanten Nutzen von Tafamidis bei Kindern und Jugendlichen.
Art der Anwendung
Zum Einnehmen.
Die Weichkapseln müssen im Ganzen geschluckt und dürfen nicht zerdrückt oder durchgeschnitten werden. Vyndaqel kann mit oder ohne Nahrung eingenommen werden.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Frauen im gebärfähigen Alter müssen während der Einnahme von Tafamidis und bis 1 Monat nach Ende der Behandlung mit Tafamidis eine adäquate Kontrazeption vornehmen (siehe Abschnitt 4.6).
Tafamidis sollte zu der Standardtherapie zur Behandlung von Patienten mit Transthyretin-Amyloidose hinzugefügt werden. Die Ärzte sollten die Patienten überwachen und die Notwendigkeit anderer Therapien im Rahmen dieser Standardtherapie fortwährend beurteilen, einschließlich der Notwendigkeit einer Organtransplantation. Da keine Daten zur Anwendung von Tafamidis bei Patienten nach Organtransplantation vorliegen, sollte Tafamidis bei Patienten, die eine Organtransplantation erhalten, abgesetzt werden.
Ein Anstieg in Leberfunktionstests und eine Verringerung von Thyroxin können auftreten (siehe Abschnitt 4.5 und 4.8).
Durch die gleichzeitige Anwendung von Tafamidis mit Substraten des Brustkrebs-Resistenz-Proteins (engl. breast cancer resistance protein, BCRP) kann sich die Exposition gegenüber dem BCRP-Substrat erhöhen. Im Fall einer gleichzeitigen Anwendung sollten die Patienten auf Nebenwirkungen im Zusammenhang mit dem BCRP-Substrat überwacht werden. Eine Dosisreduktion des BCRP-Substrats kann in Betracht gezogen werden (siehe Abschnitt 4.5).
Dieses Arzneimittel enthält nicht mehr als 44 mg Sorbitol (Ph. Eur.) (E 420) pro Kapsel. Sorbitol ist eine Quelle für Fructose.
Die additive Wirkung gleichzeitig angewendeter Sorbitol (oder Fructose) -haltiger Arzneimittel und die Einnahme von Sorbitol (oder Fructose) über die Nahrung ist zu berücksichtigen.
Der Sorbitolgehalt oral angewendeter Arzneimittel kann die Bioverfügbarkeit von anderen gleichzeitig oral angewendeten Arzneimitteln beeinflussen.
In einer klinischen Studie an gesunden Probanden bewirkten 20 mg Tafamidis-Meglumin keine Induktion oder Inhibition des Cytochrom-P450-Enzyms CYP3A4.
Tafamidis hemmt in vitro den Efflux-Transporter BCRP mit einer IC50 = 1,16 µm und könnte in klinischen relevanten Konzentrationen zu Arzneimittelinteraktionen mit Substraten dieses Transporters (z. B. Methotrexat, Rosuvastatin, Atorvastatin, Apixaban, Rivaroxaban, Imatinib) führen. In einer klinischen Studie an gesunden Teilnehmern erhöhte sich die Exposition gegenüber dem BCRP-Substrat Rosuvastatin nach mehreren täglichen Einnahmen von 61 mg Tafamidis um das etwa 2-Fache. Daher sollten die Patienten auf Nebenwirkungen im Zusammenhang mit dem BCRP-Substrat überwacht werden, wenn dieses gleichzeitig mit Tafamidis angewendet wird. Eine Dosisreduktion des BCRP-Substrats kann in Betracht gezogen werden (siehe Abschnitt 4.4).
In ähnlicher Weise hemmt Tafamidis die Aufnahmetransporter OAT1 und OAT3 (organische Anionen‑Transporter) mit einer IC50 = 2,9 µm bzw. IC50 = 2,36 µm und könnte in klinisch relevanten Konzentrationen zu Arzneimittelinteraktionen mit Substraten dieser Transporter (z. B. nicht‑steroidale Entzündungshemmer, Bumetanid, Furosemid, Lamivudin, Methotrexat, Oseltamivir, Tenofovir, Ganciclovir, Adefovir, Cidofovir, Zidovudin, Zalcitabin) führen. Basierend auf In-vitro-Daten wurde ermittelt, dass die prognostizierten maximalen Veränderungen der AUC von Substraten der OAT1 und OAT3 bei der Dosis von 61 mg Tafamidis unter 1,25 liegen. Daher wird nicht davon ausgegangen, dass eine Hemmung von OAT1- oder OAT3-Transportern durch Tafamidis zu klinisch signifikanten Wechselwirkungen führt.
Es wurden keine Wechselwirkungsstudien durchgeführt, die die Wirkung anderer Arzneimittel auf Tafamidis untersuchten.
Anomalien in Labortests
Tafamidis kann die Serumkonzentrationen des Gesamt-Thyroxins verringern, ohne gleichzeitige Veränderung des freien Thyroxins (T4) oder des Thyreotropins (Thyroid Stimulating Hormone, TSH). Diese Beobachtung hinsichtlich der Gesamt-Thyroxin-Werte ist wahrscheinlich das Ergebnis einer reduzierten Bindung von Thyroxin an oder dessen Verdrängung von TTR aufgrund der hohen Bindungsaffinität von Tafamidis an den TTR-Thyroxin-Rezeptor. Es wurden keine entsprechenden klinischen Befunde beobachtet, die mit einer Schilddrüsenfunktionsstörung im Einklang stehen.
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen wegen der langen Halbwertszeit während der Behandlung mit Tafamidis und nach Behandlungsende noch für 1 Monat kontrazeptive Maßnahmen durchführen.
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Tafamidis bei Schwangeren vor. Tierexperimentelle Studien haben eine Entwicklungstoxizität gezeigt (siehe Abschnitt 5.3). Die Anwendung von Tafamidis während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen.
Stillzeit
Die zur Verfügung stehenden Daten bei Tieren zeigten, dass Tafamidis in die Milch übergeht. Ein Risiko für das Neugeborene/ Kind kann nicht ausgeschlossen werden. Tafamidis soll während der Stillzeit nicht angewendet werden.
Fertilität
In nicht klinischen Studien wurde keine Beeinträchtigung der Fertilität beobachtet (siehe Abschnitt 5.3).
Auf der Grundlage des pharmakodynamischen und pharmakokinetischen Profils von Tafamidis wird kein oder ein zu vernachlässigender Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen angenommen.
Zusammenfassung des Sicherheitsprofils
Die Sicherheitsdaten spiegeln die Exposition von 176 Patienten mit ATTR-CM gegenüber 80 mg (Gabe als 4 x 20 mg) Tafamidis-Meglumin wider, das in einer 30-monatigen placebokontrollierten Studie täglich verabreicht wurde (siehe Abschnitt 5.1).
Die Häufigkeit von unerwünschten Ereignissen bei Patienten, die mit 80 mg Tafamidis-Meglumin behandelt wurden, war im Allgemeinen ähnlich und mit Placebo vergleichbar.
Die folgenden unerwünschten Ereignisse wurden im Vergleich zu Placebo häufiger bei Patienten, die mit 80 mg Tafamidis-Meglumin behandelt wurden, berichtet: Flatulenz (8 Patienten [4,5 %] gegenüber 3 Patienten [1,7 %]) und Anstieg im Leberfunktionstest (6 Patienten [3,4 %] gegenüber 2 Patienten [1,1 %]). Ein kausaler Zusammenhang wurde nicht festgestellt.
Es sind Sicherheitsdaten für 61 mg Tafamidis aus einer offenen Langzeitverlängerungsstudie verfügbar.
Tabellarische Auflistung der Nebenwirkungen
Die Nebenwirkungen sind im Folgenden nach MedDRA-Systemorganklassen (SOC) und Häufigkeitskategorien nach folgender Konvention aufgeführt: Sehr häufig (≥ 1/10), häufig (≥1/100 bis < 1/10) und gelegentlich (≥ 1/1.000 bis < 1/100). Innerhalb der Häufigkeitskategorie sind die Nebenwirkungen nach abnehmendem Schweregrad angegeben. In der nachstehenden Tabelle sind Nebenwirkungen aus kumulativen klinischen Daten von ATTR-CM Patienten aufgeführt.
Systemorganklasse |
Häufig |
Erkrankungen des Gastrointestinaltrakts |
Diarrhoe |
Erkrankungen der Haut und des Unterhautgewebes |
Hautausschlag |
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das aufgeführte nationale Meldesystem anzuzeigen.
Deutschland
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: https://www.bfarm.de
Österreich
Bundesamt für Sicherheit im Gesundheitswesen
Traisengasse 5
1200 WIEN
ÖSTERREICH
Fax: + 43 (0) 50 555 36207
Website: https://www.basg.gv.at/
Symptome
Es liegen nur minimale klinische Erfahrungen mit Überdosierungen vor. Während klinischer Prüfungen nahmen zwei Patienten mit der Diagnose ATTR-CM versehentlich eine Einzeldosis von 160 mg Tafamidis-Meglumin ein, ohne dass in diesem Zusammenhang unerwünschte Ereignisse auftraten. Die höchste an gesunde Probanden in einer klinischen Prüfung verabreichte Dosis betrug 480 mg Tafamidis-Meglumin als Einzeldosis. Bei dieser Dosis gab es ein gemeldetes behandlungsbedingtes unerwünschtes Ereignis, und zwar ein leichtes Hordeolum.
Behandlung
Im Fall einer Überdosierung sind je nach Bedarf unterstützende Standardmaßnahmen anzuwenden.
Pharmakotherapeutische Gruppe: Andere Mittel für das Nervensystem, ATC-Code: N07XX08
Wirkmechanismus
Tafamidis ist ein selektiver Stabilisator von TTR. Tafamidis bindet an den Thyroxin‑Bindungsstellen des TTR, stabilisiert so das Tetramer und verlangsamt die Spaltung in Monomere, den geschwindigkeitsbestimmenden Schritt im amyloidogenen Prozess.
Pharmakodynamische Wirkungen
Die Transthyretin-Amyloidose ist eine stark beeinträchtigende Erkrankung, die durch die Anreicherung verschiedener nicht löslicher fibrillärer Proteine, bzw. von Amyloid, in den Geweben verursacht wird, und zwar in Mengen, die ausreichen, um die normale Funktion zu beeinträchtigen. Die Spaltung des Transthyretin‑Tetramers in Monomere ist der geschwindigkeitsbestimmende Schritt in der Pathogenese der Transthyretin-Amyloidose. Die gefalteten Monomere durchlaufen eine partielle Denaturierung, wodurch alternativ gefaltete monomere amyloidogene Zwischenprodukte gebildet werden. Diese setzen sich dann fehlerhaft in lösliche Oligomere, Profilamente, Filamente und Amyloidfibrillen zusammen. Tafamidis bindet mit negativer Kooperativität an die beiden ThyroxinBindungsstellen der nativen tetrameren Form von Transthyretin und verhindert so die Aufspaltung in Monomere. Die Hemmung der Spaltung des TTR-Tetramers bildet die Grundlage für die Anwendung von Tafamidis bei Patienten mit ATTR-CM.
Ein TTR-Stabilisierungs-Assay wurde als pharmakodynamischer Marker verwendet und untersuchte die Stabilität des TTR-Tetramers.
Tafamidis stabilisierte sowohl das Wildtyp-TTR-Tetramer als auch die Tetramere von 14 TTR-Varianten, die nach einmal täglicher Dosisgabe von Tafamidis klinisch getestet wurden. Tafamidis stabilisierte außerdem das TTR-Tetramer für 25 ex vivo getestete Varianten, wodurch eine TTR-Stabilisierung von 40 amyloidogenen TTR-Genotypen gezeigt wurde.
In einer multizentrischen, internationalen, doppelblinden, placebokontrollierten, randomisierten Studie (siehe Abschnitt zur klinischen Wirksamkeit und Sicherheit) wurde in Monat 1 eine TTR-Stabilisierung festgestellt, die bis Monat 30 aufrechterhalten wurde.
Die Ergebnisse für mit Herzinsuffizienz assoziierte Biomarker (NT-proBNP und Troponin I) fielen zugunsten von Vyndaqel im Vergleich zu Placebo aus.
Klinische Wirksamkeit und Sicherheit
Die Wirksamkeit wurde in einer multizentrischen, internationalen, doppelblinden, placebokontrollierten, randomisierten, 3-armigen Studie bei 441 Patienten mit Wildtyp‑ oder hereditärer ATTR-CM nachgewiesen.
Die Patienten wurden entweder zu einmal täglich Tafamidis-Meglumin 20 mg (n = 88) oder 80 mg (verabreicht als vier 20-mg-Tafamidis-Meglumin-Kapseln, n = 176) oder zu einem identischen Placebo (n = 177) randomisiert, zusätzlich zur Standardtherapie (z. B. Diuretika), über einen Zeitraum von 30 Monaten. Die Behandlungszuweisung wurde nach Vorliegen oder Nichtvorliegen eines hereditären TTR-Genotyps sowie nach Schwere der Erkrankung bei Baseline (NYHA-Klasse) stratifiziert. Tabelle 1 beschreibt die demografischen Patientendaten und die Merkmale bei Baseline.
Tabelle 1: Demografische Patientendaten und Merkmale bei Baseline
Merkmal |
Gepooltes Tafamidis |
Placebo |
Alter — Jahr | ||
Mittelwert (Standardabweichung) |
74,5 (7,2) |
74,1 (6,7) |
Median (Minimum, Maximum) |
75 (46, 88) |
74 (51,89) |
Geschlecht — Anzahl (%) | ||
Männlich |
241 (91,3) |
157 (88,7) |
Weiblich |
23 (8,7) |
20 (11,3) |
TTR-Genotyp — Anzahl (%) | ||
ATTRm |
63 (23,9) |
43 (24,3) |
ATTRwt |
201 (76,1) |
134 (75,7) |
NYHA-Klasse — Anzahl (%) |
||
NYHA-Klasse I |
24 (9,1) |
13 (7,3) |
NYHA-Klasse II |
162 (61,4) |
101 (57,1) |
NYHA-Klasse III |
78 (29,5) |
63 (35,6) |
Abkürzungen: ATTRm = hereditäres Transthyretin-Amyloid, ATTRwt = Wildtyp-Transthyretin-Amyloid, NYHA = New York Heart Association.
Bei der primären Analyse wurde eine hierarchische Kombination unter Anwendung der Finkelstein-Schoenfeld-Methode (F-S) auf die Gesamtmortalität und die Häufigkeit der kardiovaskulär bedingten Hospitalisierungen, definiert als die Anzahl der Fälle, in denen ein Patient aufgrund kardiovaskulär bedingter Morbidität ins Krankenhaus eingewiesen wird, verwendet. Die Methode verglich jeden Patienten mit jedem anderen Patienten in jedem Stratum; dabei wurde paarweise auf hierarchische Weise vorgegangen, wobei zuerst die Gesamtmortalität gefolgt von der Häufigkeit der kardiovaskulär bedingten Hospitalisierungen, wenn eine Differenzierung der Patienten basierend auf der Mortalität nicht möglich war, herangezogen wurde.
Bei dieser Analyse wurde in der Gruppe mit der gepoolten Tafamidis-Dosis von 20 mg und 80 mg im Vergleich zu Placebo eine signifikante Reduzierung (p = 0,0006) der Gesamtmortalität und der Häufigkeit kardiovaskulär bedingter Hospitalisierungen gezeigt (Tabelle 2).
Tabelle 2: Primäranalyse der Gesamtmortalität und der Häufigkeit kardiovaskulär bedingter Hospitalisierungen mittels Finkelstein-Schoenfeld (F-S)-Methode
Primäranalyse |
Gepooltes Tafamidis |
Placebo |
Anzahl (%) der Patienten, die in Monat 30 noch lebten* |
186 (70,5) |
101 (57,1) |
Durchschnittliche kardiovaskulär bedingte Hospitalisierungen über einen Zeitraum von 30 Monaten (pro Patient pro Jahr) unter jenen Patienten, die in Monat 30 noch lebten† |
0,297 |
0,455 |
p-Wert basierend auf F-S-Methode |
0,0006 |
|
* Herztransplantationen und Implantationen von Systemen zur mechanischen Unterstützung der Herzfunktion werden als Anzeichen für ein nahendes Endstadium angesehen. Daher werden diese Patienten in der Analyse als mit Todesfällen gleichwertig behandelt. Aus diesem Grund sind diese Patienten in dem Wert „Anzahl der Patienten, die in Monat 30 noch lebten“ nicht enthalten, selbst wenn die Patienten basierend auf der Folgeuntersuchung des Vitalstatus in Monat 30 noch am Leben sind.
† Deskriptiver Mittelwert unter jenen, die den 30-Monats-Zeitraum überlebten.
Die Analyse der einzelnen Komponenten der Primäranalyse (Gesamtmortalität und kardiovaskulär bedingte Hospitalisierungen) zeigte außerdem signifikante Reduzierungen bei Tafamidis im Vergleich zu Placebo.
Die Hazard Ratio aus dem Cox-Proportional-Gefahrenmodell für die Gesamtmortalität für gepooltes Tafamidis betrug 0,698 (95 %-KI 0,508, 0,958), was eine 30,2%ige Reduzierung des Sterberisikos relativ zur Placebogruppe zeigt (p = 0,0259). Eine Kaplan-Meier-Kurve der Zeit bis zum Ereignis „Gesamtmortalität“ ist in Abbildung 1 gezeigt.
Abbildung 1: Gesamtmortalität*
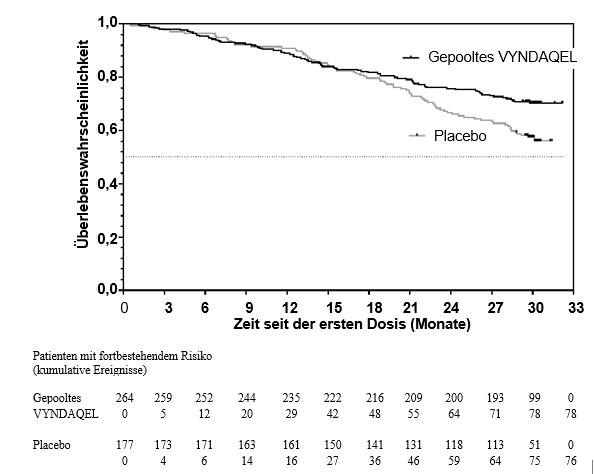
* Herztransplantationen und Systeme zur mechanischen Unterstützung der Herzfunktion wurden als Todesfälle behandelt. Hazard Ratio aus dem Cox‑Proportional-Gefahrenmodell mit Behandlung, TTR-Genotyp (hereditär und Wildtyp) und Baseline‑Klassifikation der New York Heart Association (NYHA) (NYHA-Klasse I und II kombiniert sowie NYHA-Klasse III) als Faktoren.
Unter Anwendung von Tafamidis gab es im Vergleich zu Placebo signifikant weniger kardiovaskulär bedingte Hospitalisierungen, mit einer Reduzierung des Risikos um 32,4 % (Tabelle 3).
Tabelle 3: Kardiovaskulär bedingte Hospitalisierungen
Gepooltes Tafamidis |
Placebo |
|
Gesamtanzahl (%) der Patienten mit kardiovaskulär bedingten Hospitalisierungen |
138 (52,3) |
107 (60,5) |
Kardiovaskulär bedingte Hospitalisierungen pro Jahr* |
0,4750 |
0,7025 |
Behandlungsunterschied gepooltes Tafamidis vs. Placebo (relatives Risiko)* |
0,6761 |
|
p-Wert* |
< 0,0001 |
|
Abkürzung: NYHA = New York Heart Association.
* Diese Analyse basierte auf einem Poisson-Regressionsmodell mit Behandlung, TTR-Genotyp (hereditär und Wildtyp), Baseline‑Klassifikation der New York Heart Association (NYHA) (NYHA-Klasse I und II kombiniert sowie NYHA-Klasse III), Interaktion zwischen Behandlung und TTR-Genotyp und Interaktion zwischen Behandlung und NYHA-Baseline-Klassifikation als Faktoren.
Der Behandlungseffekt von Tafamidis auf die Funktionsfähigkeit und den Gesundheitszustand wurde mithilfe des 6‑Minuten-Gehtests (6MGT) und des Kansas City Cardiomyopathy Questionnaire-Overall Summary (KCCQ-OS)-Score beurteilt (bestehend aus den Domänen Gesamtsymptome, körperliche Einschränkungen, Lebensqualität und soziale Einschränkungen). Ein signifikanter Behandlungseffekt zugunsten von Tafamidis wurde erstmals in Monat 6 beobachtet und blieb sowohl im Hinblick auf die Gehstrecke im 6MGT als auch im Hinblick auf den KCCQ-OS-Score bis Monat 30 konstant erhalten (Tabelle 4).
Tabelle 4: 6MGT und KCCQ-OS sowie Scores der Komponenten-Domänen
Endpunkte |
Baseline-Mittelwert (SD) |
Veränderung von Baseline bis Monat 30, Kleinste-Quadrate-Mittelwert (SE) |
Behandlungunterschied zu Kleinste-Quadrate-Mittelwert von Placebo (95 %-KI) |
p-Wert |
||
Gepooltes Tafamidis |
Placebo |
Gepooltes Tafamidis |
Placebo |
|||
6MGT* (Meter) |
350,55 |
353,26 |
-54,87 |
-130,55 |
75,68 |
p < 0,0001 |
KCCQ-OS* |
67,27 |
65,90 |
-7,16 |
-20,81 |
13,65 |
p < 0,0001 |
* Höhere Werte als Hinweis auf besseren Gesundheitszustand
Abkürzungen: 6MGT = 6-Minuten-Gehtest; KCCQ-OS = Kansas City Cardiomyopathy Questionnaire-Overall Summary; KI = Konfidenzintervall.
Die mit der F-S-Methode ermittelten Ergebnisse (dargestellt als Win‑Ratio) für den kombinierten Endpunkt und seine Komponenten (Gesamtmortalität und Häufigkeit kardiovaskulär bedingter Hospitalisierungen) fielen nach Dosis und in allen Subgruppen (Wildtyp, hereditär und NYHA‑Klasse I und II sowie NYHA-Klasse III) im Vergleich zu Placebo konstant zugunsten von Tafamidis aus, außer im Hinblick auf die Häufigkeit der kardiovaskulär bedingten Hospitalisierungen in NYHA‑Klasse III (Abbildung 2), die in der mit Tafamidis behandelten Gruppe höher ist als bei Placebo (siehe Abschnitt 4.2). Analysen des 6MGT und des KCCQ-OS fielen im Vergleich zu Placebo in jeder Subgruppe ebenfalls zugunsten von Tafamidis aus.
Abbildung 2: Mit der F-S-Methode ermittelte Ergebnisse und Komponenten nach Subgruppe und Dosis
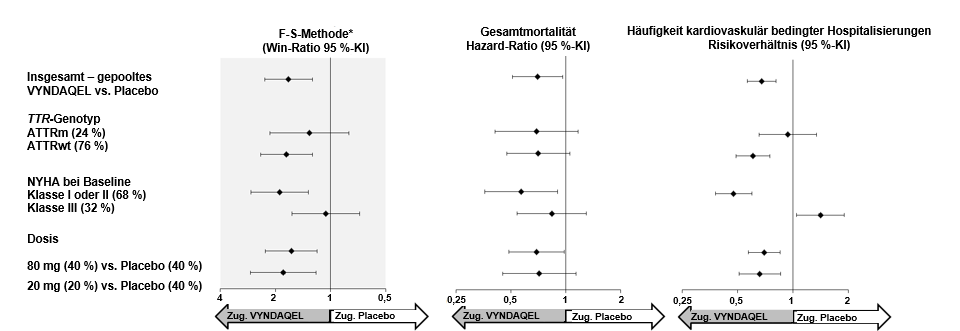
Abkürzungen: ATTRm = hereditäres Transthyretin-Amyloid, ATTRwt = Wildtyp-Transthyretin-Amyloid, F-S = Finkelstein‑Schoenfeld, KI = Konfidenzintervall.
* F-S-Ergebnisse dargestellt als Win-Ratio (basierend auf Gesamtmortalität und Häufigkeit der kardiovaskulär bedingten Hospitalisierungen). Die Win-Ratio ist die Anzahl der Paare mit „Gewinnen“ des behandelten Patienten dividiert durch die Anzahl der Paare mit „Gewinnen“ des Placebo-Patienten.
Herztransplantationen und Systeme zur mechanischen Unterstützung der Herzfunktion wurden als Todesfälle behandelt.
Bei der individuellen Anwendung der F-S-Methode auf jede Dosisgruppe reduzierte Tafamidis im Vergleich zu Placebo die Kombination aus Gesamtmortalität und Häufigkeit kardiovaskulär-bedingter Hospitalisierungen, und zwar sowohl für die 80-mg- als auch für die 20-mg-Dosis (p = 0,0030 bzw. p = 0,0048). Die Ergebnisse der Primäranalyse sowie der 6MWT und KCCQ-OS in Monat 30 waren im Vergleich zu Placebo bei der 80-mg- und 20-mg-Dosis von Tafamidis statistisch signifikant, mit ähnlichen Ergebnissen für beide Dosierungen.
Es sind keine Daten zur Wirksamkeit von 61 mg Tafamidis verfügbar, da diese Formulierung nicht in der doppelblinden, placebokontrollierten, randomisierten Phase-3-Studie untersucht wurde. Die relative Bioverfügbarkeit von 61 mg Tafamidis ist ähnlich wie bei 80 mg Tafamidis-Meglumin im Steady State (siehe Abschnitt 5.2).
Eine supra-therapeutische orale Einzeldosis von 400 mg Tafamidis-Meglumin-Lösung zeigte bei gesunden Probanden keine Verlängerung des QTc-Intervalls.
Die Europäische Arzneimittel-Agentur hat für Tafamidis eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen bei Transthyretin-Amyloidose gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Resorption
Bei einmal täglicher oraler Anwendung der Weichkapsel wird die maximale Spitzenkonzentration (Cmax) im Nüchternzustand im Median (tmax) innerhalb von 4 Stunden für 61 mg Tafamidis bzw. innerhalb von 2 Stunden für 80 mg Tafamidis-Meglumin (4 x 20 mg) nach der Einnahme erreicht. Die gleichzeitige Einnahme einer fett- und kalorienreichen Mahlzeit änderte die Geschwindigkeit, nicht aber das Ausmaß der Resorption. Diese Ergebnisse unterstützen die Einnahme von Tafamidis mit oder ohne Nahrung.
Verteilung
Tafamidis wird im Plasma in hohem Maße an Proteine gebunden (> 99 %). Das scheinbare Verteilungsvolumen im Steady State beträgt 18,5 Liter.
Das Ausmaß der Bindung von Tafamidis an Plasmaproteine wurde unter Verwendung von tierischem und menschlichem Plasma untersucht. Die Affinität von Tafamidis zu TTR ist höher als die zu Albumin. Daher bindet Tafamidis trotz der signifikant höheren Konzentrationen von Albumin (600 μM) im Vergleich zu TTR (3,6 μM) im Plasma bevorzugt an TTR.
Biotransformation und Elimination
Es gibt keine eindeutigen Belege für eine Exkretion von Tafamidis über die Galle beim Menschen. Präklinische Daten weisen darauf hin, dass Tafamidis über eine Glucuronidierung metabolisiert und über die Galle ausgeschieden wird. Diese Route der Biotransformation ist beim Menschen plausibel, da etwa 59 % der eingenommenen Gesamtdosis im Stuhl und etwa 22 % im Urin nachgewiesen werden. Basierend auf populationspharmakokinetischen Ergebnissen beträgt die scheinbare orale Clearance von Tafamidis 0,263 l/h und die populationsspezifische mittlere Halbwertszeit ca. 49 Stunden.
Dosis- und Zeitlinearität
Die Exposition durch eine einmal tägliche Einnahme von Tafamidis-Meglumin stieg mit einer Erhöhung der Dosis auf eine Einzeldosis von bis zu 480 mg und mehrere Dosen von bis zu 80 mg/Tag an. Im Allgemeinen war der Anstieg proportional oder fast proportional zur Dosis. Im Zeitverlauf stagnierte die Tafamidis-Clearance.
Die relative Bioverfügbarkeit von 61 mg Tafamidis ist ähnlich wie bei 80 mg Tafamidis-Meglumin im Steady State. Tafamidis und Tafamidis-Meglumin sind auf Basis der mg-Angaben nicht gegeneinander austauschbar.
Die pharmakokinetischen Parameter waren nach einmaliger und wiederholter Einnahme von 20 mg Tafamidis-Meglumin vergleichbar, was auf das Fehlen einer Induktion oder Inhibition des Tafamidis-Metabolismus hinweist.
Die Ergebnisse nach einmal täglicher Einnahme von 15 mg bis 60 mg Tafamidis‑Meglumin-Lösung zum Einnehmen über 14 Tage zeigten, dass der Steady State an Tag 14 erreicht wurde.
Besondere Patientengruppen
Eingeschränkte Leberfunktion
Die pharmakokinetischen Daten wiesen auf eine verminderte systemische Exposition (etwa 40 %) und erhöhte Gesamtclearance (0,52 l/h vs. 0,31 l/h) von Tafamidis-Meglumin bei Patienten mit mittelschwerer Beeinträchtigung der Leberfunktion (Child-Pugh-Score von 7 bis 9 einschließlich) im Vergleich zu gesunden Probanden hin, die auf einen höheren Anteil von nicht gebundenem Tafamidis zurückzuführen ist. Da Patienten mit mäßiger Beeinträchtigung der Leberfunktion niedrigere TTR‑Spiegel aufweisen als gesunde Probanden, ist eine Dosisanpassung nicht erforderlich, weil die Stöchiometrie von Tafamidis und seinem Zielprotein TTR für eine Stabilisierung des TTR-Tetramers ausreichend wäre. Bei Patienten mit schwerer Beeinträchtigung der Leberfunktion ist die Exposition gegenüber Tafamidis nicht bekannt.
Eingeschränkte Nierenfunktion
Tafamidis wurde nicht spezifisch in einer speziell angelegten Studie mit Patienten mit eingeschränkter Nierenfunktion untersucht. Der Einfluss der Kreatinin-Clearance auf die Pharmakokinetik von Tafamidis wurde in einer pharmakokinetischen Populationsanalyse bei Patienten mit einer Kreatinin-Clearance von mehr als 18 ml/min untersucht. Pharmakokinetische Schätzungen wiesen darauf hin, dass bei der scheinbaren oralen Clearance von Tafamidis zwischen Patienten mit einer Kreatinin-Clearance von weniger als 80 ml/min und Patienten mit einer Kreatinin-Clearance von mindestens 80 ml/min kein Unterscheid besteht. Eine Dosisanpassung wird bei Patienten mit eingeschränkter Nierenfunktion nicht für erforderlich gehalten.
Ältere Patienten
Auf der Grundlage von populationspharmakokinetischen Ergebnissen hatten Patienten im Alter von ≥ 65 Jahren einen um durchschnittlich 15 % niedrigeren Schätzwert für die scheinbare orale Clearance im Steady State als Patienten unter 65 Jahren. Allerdings führt der Unterscheid bei der Clearance im Vergleich zu jüngeren Patienten zu einer Erhöhung der mittleren Cmax und der AUC um < 20 % und ist nicht klinisch signifikant.
Pharmakokinetische/ pharmakodynamische Beziehungen
In-vitro-Daten legten nahe, dass Tafamidis die Cytochrom-P450-Enzyme CYP1A2, CYP3A4, CYP3A5, CYP2B6, CYP2C8, CYP2C9, CYP2C19 und CYP2D6 nicht signifikant hemmt. Es wird nicht davon ausgegangen, dass Tafamidis aufgrund der Induktion von CYP1A2, CYP2B6 oder CYP3A4 zu klinisch relevanten Arzneimittelwechselwirkungen führt.
In-vitro-Studien legen nahe, dass es unwahrscheinlich ist, dass Tafamidis in klinisch relevanten Konzentrationen systemische Wechselwirkungen mit Substraten der UDP-Glucuronosyltransferase (UGT) verursacht. Tafamidis kann die Aktivität von UGT1A1 im Darm hemmen.
Tafamidis zeigte ein geringes Potenzial zur systemischen und im Gastrointestinaltrakt stattfindenden Hemmung des Multidrug-Resistance-Proteins (MDR1) (auch als P-Glykoprotein bzw. P-gp bekannt) sowie zur Hemmung des organischen Kationentransporters 2 (OCT2), des Multidrug and Toxin Extrusion Transporter 1 (MATE1) und des MATE2K, des organische Anionen transportierenden Polypeptids 1B1 (OATP1B1) und des OATP1B3 in klinisch relevanten Konzentrationen.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Fertilität und frühen Embryonalentwicklung, Genotoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. In Studien zur Toxizität bei wiederholter Gabe und in Studien zur Karzinogenität erschien die Leber als ein Zielorgan der Toxizität bei den verschiedenen untersuchten Spezies. Lebereffekte wurden bei Expositionen von etwa dem Äquivalent der AUC im Steady State beim Menschen bei einer klinischen Dosis von 61 mg Tafamidis beobachtet.
In einer Studie zur Entwicklungstoxizität an Kaninchen wurden bei Expositionen von etwa dem ≥ 2,1‑fachen der AUC im Steady State beim Menschen bei einer klinischen Dosis von 61 mg Tafamidis eine geringe Zunahme von Skelettmissbildungen und -variationen, Totgeburten bei wenigen Weibchen, geringeres embryofetales Überleben und eine Reduktion des Fetalgewichts beobachtet.
In der Tafamidis-Studie zur prä- und postnatalen Entwicklung bei Ratten wurde nach Gabe von Dosen von 15 mg/kg/Tag und 30 mg/kg/Tag an die Muttertiere in der Gestation und Laktationszeit ein vermindertes Überleben und Gewicht der Jungtiere beobachtet. Ein vermindertes Gewicht der Jungtiere war bei 15 mg/kg/Tag bei Männchen mit einer verzögerten sexuellen Reifung (Separation des Präputiums) verbunden. Bei 15 mg/kg/Tag wurde eine beeinträchtigte Leistung in einem Water-Maze-Test für Lernen und Gedächtnis beobachtet. Der NOAEL für Lebensfähigkeit und Wachstum der Nachkommen der F1-Generation betrug nach Gabe einer Dosis an die Muttertiere in der Gestation und Laktationszeit mit Tafamidis 5 mg/kg/Tag (humanäquivalente Tafamidis-Dosis = 0,8 mg/kg/Tag), was etwa der klinischen Dosis von 61 mg Tafamidis entspricht.
Kapselhülle
Gelatine (E 441)
Glycerol (E 422)
Eisen(III)-oxid (E 172)
Sorbitan
Sorbitol (Ph. Eur.) (E 420)
Mannitol (Ph. Eur.) (E 421)
Gereinigtes Wasser
Kapselinhalt
Macrogol 400 (E 1521)
Polysorbat 20 (E 432)
Povidon (K 90)
Butylhydroxytoluol (Ph. Eur.) (E 321)
Drucktinte (Opacode weiß)
Ethanol
2-Propanol (Ph. Eur.)
Gereinigtes Wasser
Macrogol 400 (E 1521)
Polyvinylacetatphthalat
Propylenglycol (E 1520)
Titandioxid (E 171)
Ammoniumhydroxid 28 % (E 527)
Nicht zutreffend.
2 Jahre
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Perforierte Blisterpackung zur Abgabe von Einzeldosen (PVC/ PA/ Al/ PVC//Al).
Packungsgrößen: eine Packung mit 30 x 1 Weichkapsel und eine Mehrfachpackung mit 90 (3 Packungen mit 30 x 1) Weichkapseln.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Brüssel
Belgien
EU/1/11/717/003
EU/1/11/717/004
Datum der Erteilung der Zulassung: 16. November 2011
Datum der letzten Verlängerung der Zulassung: 22. Juli 2016
März 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
VERKAUFSABGRENZUNG IN DEUTSCHLAND
Verschreibungspflichtig
REZEPTPFLICHT/APOTHEKENPFLICHT IN ÖSTERREICH
Rezept- und apothekenpflichtig, wiederholte Abgabe verboten
PACKUNGSGRÖSSEN IN DEUTSCHLAND
Vyndaqel® 61 mg Blister mit 30 Weichkapseln (N1)
PACKUNGSGRÖSSEN IN ÖSTERREICH
Vyndaqel® 61 mg Blister mit 30 Weichkapseln
REPRÄSENTANT IN DEUTSCHLAND
PFIZER PHARMA GmbH
Friedrichstr. 110
10117 Berlin
Tel.: 030 550055-51000
Fax: 030 550054-10000
REPRÄSENTANT IN ÖSTERREICH
Pfizer Corporation Austria Ges.m.b.H.
Floridsdorfer Hauptstraße 1
A-1210 Wien
Tel.: +43 (0)1 521 15-0