
NUBEQA® 300 mg Filmtabletten
Jede Filmtablette enthält 300 mg Darolutamid.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 186 mg Lactose‑Monohydrat (siehe Abschnitt 4.4).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette (Tablette).
Weiße bis weißgraue, ovale Tabletten mit einer Länge von 16 mm und einer Breite von 8 mm, mit der Prägung „300“ auf der einen und „BAYER“ auf der anderen Seite.
NUBEQA wird angewendet zur Behandlung erwachsener Männer mit
nicht-metastasiertem kastrationsresistentem Prostatakarzinom (nmCRPC), die ein hohes Risiko für die Entwicklung von Metastasen aufweisen (siehe Abschnitt 5.1).
metastasiertem hormonsensitivem Prostatakarzinom (mHSPC) in Kombination mit einer Androgendeprivationstherapie (siehe Abschnitt 5.1).
metastasiertem hormonsensitivem Prostatakarzinom (mHSPC) in Kombination mit Docetaxel und einer Androgendeprivationstherapie (siehe Abschnitt 5.1).
Die Behandlung sollte von einem Facharzt mit Erfahrung in der Behandlung des Prostatakarzinoms begonnen und überwacht werden.
Dosierung
Die empfohlene Dosis beträgt 600 mg Darolutamid (zwei 300‑mg‑Tabletten) zweimal täglich, entsprechend einer Tagesgesamtdosis von 1200 mg (siehe Abschnitt 5.2).
Die Behandlung mit Darolutamid ist bis zur Krankheitsprogression oder bis zum Auftreten einer inakzeptablen Toxizität fortzusetzen.
Eine medikamentöse Kastration mit einem Luteinisierenden-Hormon-Releasing-Hormon-(LHRH) –Agonisten oder -Antagonisten (GnRH-Analogon) soll während der Behandlung von Patienten, die nicht chirurgisch kastriert sind, fortgeführt werden.
Metastasiertes hormonsensitives Prostatakarzinom (mHSPC) – Behandlung mit Darolutamid in Kombination mit Docetaxel
Patienten mit mHSPC sollten die Behandlung mit Darolutamid in Kombination mit Docetaxel beginnen (siehe Abschnitt 5.1). Der erste von 6 Behandlungszyklen mit Docetaxel sollte innerhalb von 6 Wochen nach Beginn der Darolutamid-Behandlung verabreicht werden. Die Empfehlungen in der Produktinformation von Docetaxel sollten eingehalten werden. Die Behandlung mit Darolutamid ist bis zur Krankheitsprogression oder bis zum Auftreten einer inakzeptablen Toxizität fortzusetzen, selbst wenn ein Docetaxel-Zyklus aufgeschoben, unterbrochen oder abgebrochen wird.
Versäumte Dosis
Wenn eine Dosis versäumt wurde, ist diese nachzuholen, sobald sich der Patient vor dem nächsten vorgesehenen Einnahmezeitpunkt daran erinnert. Der Patient sollte nicht zwei Dosen zusammen einnehmen, um die versäumte Einnahme zu ersetzen.
Dosisanpassung
Falls bei einem Patienten eine toxische Wirkung vom Schweregrad ≥ 3 oder eine nicht tolerierbare Nebenwirkung in Zusammenhang mit Darolutamid auftritt (siehe Abschnitte 4.4 und 4.8), sollte die Behandlung unterbrochen oder die Dosierung auf 300 mg zweimal täglich reduziert werden bis sich die Symptome verbessern. Die Behandlung kann danach mit einer Dosis von 600 mg zweimal täglich fortgesetzt werden.
Eine Dosisreduktion auf weniger als 300 mg zweimal täglich wird nicht empfohlen, weil die Wirksamkeit nicht nachgewiesen ist.
Besondere Patientengruppen
Ältere Patienten
Bei älteren Patienten ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Nierenfunktionsstörung
Bei Patienten mit leichter bis mäßiger Nierenfunktionsstörung ist keine Dosisanpassung erforderlich.
Bei Patienten mit schwerer Nierenfunktionsstörung (eGFR 15‑29 ml/min/1,73 m2), die keine Hämodialyse erhalten, beträgt die empfohlene Anfangsdosis 300 mg zweimal täglich (siehe Abschnitte 4.4 und 5.2).
Leberfunktionsstörung
Bei Patienten mit leichter Leberfunktionsstörung ist keine Dosisanpassung erforderlich.
Die verfügbaren Daten zur Pharmakokinetik von Darolutamid bei mäßiger Leberfunktionsstörung sind begrenzt.
Darolutamid wurde bei Patienten mit schwerer Leberfunktionsstörung nicht untersucht.
Bei Patienten mit mäßiger bis schwerer Leberfunktionsstörung (Child‑Pugh-Klassen B und C) beträgt die empfohlene Anfangsdosis 300 mg zweimal täglich (siehe Abschnitte 4.4 und 5.2).
Kinder und Jugendliche
Es gibt keinen relevanten Nutzen von Darolutamid bei Kindern und Jugendlichen.
Art der Anwendung
NUBEQA ist zum Einnehmen.
Die Tabletten müssen als Ganzes zu einer Mahlzeit geschluckt werden (siehe Abschnitt 5.2).
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Frauen, die schwanger sind oder schwanger werden können (siehe Abschnitt 4.6).
Nierenfunktionsstörung
Die verfügbaren Daten bei Patienten mit schwerer Nierenfunktionsstörung sind begrenzt.
Da die Exposition erhöht sein kann, sollten diese Patienten engmaschig auf Nebenwirkungen überwacht werden (siehe Abschnitte 4.2 und 5.2).
Leberfunktionsstörung
Die verfügbaren Daten von Patienten mit mäßiger Leberfunktionsstörung sind begrenzt. Bei Patienten mit schwerer Leberfunktionsstörung wurde Darolutamid nicht untersucht.
Da die Exposition erhöht sein kann, sollten diese Patienten engmaschig auf Nebenwirkungen überwacht werden (siehe Abschnitte 4.2 und 5.2).
Kürzlich aufgetretene kardiovaskuläre Erkrankung
Patienten mit einer klinisch relevanten kardiovaskulären Erkrankung in den vergangenen 6 Monaten, einschließlich Schlaganfall, Myokardinfarkt, schwerer/ instabiler Angina pectoris, koronarer oder peripher-arterieller Bypass-Operation und symptomatischer Herzinsuffizienz, waren von den klinischen Studien ausgeschlossen. Daher ist die Sicherheit von Darolutamid bei diesen Patienten nicht erwiesen.
Wenn NUBEQA verordnet wird, sollten Patienten mit einer klinisch relevanten kardiovaskulären Erkrankung hinsichtlich dieser Erkrankungen entsprechend den geltenden Leitlinien behandelt werden.
Hepatotoxizität
Im Falle von erhöhten Leberwerten, die auf eine idiosynkratische arzneimittelinduzierte Leberschädigung hinweisen, setzen Sie die Behandlung mit Darolutamid dauerhaft ab (siehe Abschnitt 4.8).
Gleichzeitige Anwendung mit anderen Arzneimitteln
Die Anwendung von starken CYP3A4- und P‑gp-Induktoren während der Behandlung mit Darolutamid kann zu einer verminderten Plasmakonzentration von Darolutamid führen und wird nicht empfohlen, es sei denn, es ist keine alternative Therapieoption verfügbar. Bei gleichzeitiger Anwendung sollte ein alternatives Arzneimittel mit geringerem Potential, CYP3A4 oder P‑gp zu induzieren, in Erwägung gezogen werden (siehe Abschnitt 4.5).
Die Patienten sind bzgl. Nebenwirkungen von BCRP-, OATP1B1- und OATP1B3-Substraten zu überwachen, da die gleichzeitige Verabreichung von Darolutamid einen Anstieg der Plasmakonzentrationen dieser Substrate bewirken kann.
Die gleichzeitige Verabreichung von Rosuvastatin sollte vermieden werden, es sei denn, es ist keine alternative Therapieoption verfügbar (siehe Abschnitt 4.5).
Androgendeprivationstherapie kann das QT-Intervall verlängern
Bei Patienten mit Risikofaktoren für eine QT-Verlängerung in der Anamnese und bei Patienten, die als Begleitmedikation Arzneimittel erhalten, die das QT-Intervall verlängern können (siehe Abschnitt 4.5), sollten Ärzte vor Beginn einer Behandlung mit NUBEQA das Nutzen-Risiko-Verhältnis, einschließlich des Potentials für Torsade-de-Pointes, abschätzen.
Angaben zu sonstigen Bestandteilen
NUBEQA enthält Lactose. Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactase‑Mangel oder Glucose‑Galactose‑Malabsorption sollten dieses Arzneimittel nicht anwenden.
Wirkungen anderer Arzneimittel auf Darolutamid
CYP3A4- und P‑gp‑Induktoren
Darolutamid ist ein Substrat von CYP3A4 und P‑Glycoprotein (P‑gp).
Die Anwendung von starken und moderaten CYP3A4‑Induktoren und P‑gp‑Induktoren (z. B. Carbamazepin, Phenobarbital, Johanniskraut, Phenytoin und Rifampicin) während der Behandlung mit Darolutamid wird nicht empfohlen, es sei denn, es ist keine alternative Therapieoption verfügbar. Bei gleichzeitiger Anwendung sollte ein alternatives Arzneimittel ohne oder mit nur schwacher Induktion von CYP3A4 oder P‑gp in Erwägung gezogen werden.
Die wiederholte Verabreichung von Rifampicin (600 mg), einem starken CYP3A4- und P‑gp‑Induktor, mit einer Einzeldosis Darolutamid (600 mg) zusammen mit einer Mahlzeit führte zu einer Abnahme der mittleren Exposition (AUC0‑72) um 72 % und zu einer Abnahme der Cmax von Darolutamid um 52 %.
CYP3A4-, P‑gp- und BCRP‑Inhibitoren
Darolutamid ist ein Substrat von CYP3A4, P‑gp und dem Breast Cancer Resistance Protein (BCRP).
Bei Verabreichung von CYP3A4‑, P‑gp‑ oder BCRP‑Inhibitoren ist keine klinisch bedeutsame Arzneimittelwechselwirkung zu erwarten. Darolutamid kann zusammen mit CYP3A4‑, P‑gp‑ oder BCRP‑Inhibitoren gegeben werden. Die gleichzeitige Anwendung von Darolutamid mit einem kombinierten P‑gp‑ und starkem CYP3A4‑Inhibitor erhöht die Darolutamid-Exposition, dies könnte das Risiko von Darolutamid-Nebenwirkungen erhöhen. Es wird empfohlen, Patienten häufiger auf Darolutamid-Nebenwirkungen hin zu überwachen und die Darolutamid-Dosis nach Bedarf zu ändern.
Die Verabreichung von Itraconazol (200 mg zweimal täglich an Tag 1 und einmal täglich an den 7 folgenden Tagen), einem starken CYP3A4‑, P‑gp‑ und BCRP‑Inhibitor, mit einer Einzeldosis Darolutamid (600 mg an Tag 5 zu einer Mahlzeit) führte zu einer 1,7‑fachen Zunahme der mittleren Exposition (AUC0‑72) und einem 1,4‑fachen Anstieg der Cmax von Darolutamid.
UGT1A9‑Inhibitoren
Darolutamid ist ein Substrat von UGT1A9.
Bei Verabreichung von UGT1A9-Inhibitoren ist keine klinisch relevante Arzneimittelwechselwirkung zu erwarten.
Darolutamid kann zusammen mit UGT1A9-Inhibitoren verabreicht werden.
Eine pharmakokinetische Populationsanalyse ergab, dass die gleichzeitige Verabreichung von UGT1A9-Inhibitoren mit Darolutamid zu einer 1,2‑fachen Expositionszunahme (AUC0‑72) gegenüber Darolutamid führte.
Docetaxel
Bei Patienten mit mHSPC führte die Anwendung von Darolutamid in Kombination mit Docetaxel zu keinen klinisch relevanten Veränderungen der Pharmakokinetik von Darolutamid (siehe Abschnitt 5.1).
Wirkungen von Darolutamid auf andere Arzneimittel
Substrate von BCRP, OATP1B1 und OATP1B3
Darolutamid hemmt das Breast Cancer Resistance Protein (BCRP) und die organischen Anion‑Transporterpolypeptide (OATP) 1B1 und 1B3.
Die gleichzeitige Verabreichung von Rosuvastatin sollte vermieden werden, es sei denn, es ist keine therapeutische Alternative verfügbar. Es sollte eine andere Begleitmedikation mit schwächerer Hemmung von BCRP, OATP1B1 und OATP1B3 in Erwägung gezogen werden.
Die Verabreichung von Darolutamid (600 mg zweimal täglich über 5 Tage) vor der zusätzlichen Gabe einer Einzeldosis Rosuvastatin (5 mg) zusammen mit einer Mahlzeit führte zu einer ca. 5‑fachen Zunahme der mittleren Exposition (AUC) und der Cmax von Rosuvastatin.
Die gleichzeitige Anwendung von Darolutamid mit anderen BCRP-Substraten sollte nach Möglichkeit vermieden werden.
Die gleichzeitige Gabe von Darolutamid kann einen Anstieg der Plasmakonzentrationen anderer begleitend verabreichter Substrate von BCRP, OATP1B1 und OATP1B3 (z. B. Methotrexat, Sulfasalazin, Fluvastatin, Atorvastatin, Pitavastatin) bewirken. Es wird daher empfohlen, die Patienten bzgl. Nebenwirkungen der Substrate von BCRP, OATP1B1 und OATP1B3 zu überwachen. Darüber hinaus sind bei der gleichzeitigen Verabreichung mit Darolutamid die entsprechenden Empfehlungen in den Produktinformationen dieser Substrate zu beachten.
P‑gp‑Substrate
Bei Verabreichung von P‑gp‑Substraten ist keine klinisch relevante Arzneimittelwechselwirkung zu erwarten. Darolutamid kann zusammen mit P‑gp‑Substraten (z. B. Digoxin, Verapamil oder Nifedipin) gegeben werden. Die gleichzeitige Verabreichung von Darolutamid und dem sensitiven P‑gp‑Substrat Dabigatranetexilat ergab keinen Anstieg der Exposition (AUC und Cmax) gegenüber Dabigatran.
CYP3A4‑Substrate
Darolutamid ist ein schwacher Induktor von CYP3A4.
Bei Verabreichung eines CYP-Substrates ist keine klinisch relevante Arzneimittelwechselwirkung zu erwarten. Darolutamid kann zusammen mit CYP‑Substraten (z. B. Warfarin, L‑Thyroxin, Omeprazol) gegeben werden.
Die Verabreichung von Darolutamid (600 mg zweimal täglich über 9 Tage) vor der zusätzlichen Gabe einer Einzeldosis des sensitiven CYP3A4‑Substrats Midazolam (1 mg) zusammen mit einer Mahlzeit führte zu einer Abnahme der mittleren Exposition (AUC) und der Cmax von Midazolam um 29 % bzw. 32 %.
Bei klinisch relevanten Konzentrationen bewirkte Darolutamid in vitro keine Hemmung des Stoffwechsels ausgewählter CYP‑Substrate.
Docetaxel
Bei Patienten mit mHSPC führte die Anwendung von Darolutamid in Kombination mit Docetaxel zu keinen klinisch relevanten Veränderungen der Pharmakokinetik von Docetaxel (siehe Abschnitt 5.1).
Arzneimittel, die das QT‑Intervall verlängern
Da eine Androgendeprivationstherapie das QT‑Intervall verlängern kann, sollte die gleichzeitige Verabreichung von Arzneimitteln, die bekanntermaßen das QT‑Intervall verlängern, oder von Arzneimitteln, die Torsade‑de‑Pointes verursachen können, sorgfältig abgewogen werden. Hierzu zählen Arzneimittel wie Antiarrhythmika der Klasse IA (z. B. Chinidin, Disopyramid) oder der Klasse III (z. B. Amiodaron, Sotalol, Dofetilid, Ibutilid), Methadon, Moxifloxacin und Antipsychotika (z. B. Haloperidol).
Dieses Arzneimittel ist nicht zur Anwendung bei Frauen im gebärfähigen Alter bestimmt. Es darf nicht bei Frauen angewendet werden, die schwanger sind oder sein könnten bzw. stillen (siehe Abschnitte 4.1 und 4.3).
Frauen im gebärfähigen Alter/ Kontrazeption bei Männern und Frauen
Es ist nicht bekannt, ob Darolutamid oder dessen Metabolite in das Sperma übergehen. Hat der Patient Geschlechtsverkehr mit einer Frau im gebärfähigen Alter, muss während der Behandlung und für eine Woche nach Beenden der Behandlung mit NUBEQA eine hochwirksame Empfängnisverhütungsmethode (jährliche Versagerquote < 1 %) angewendet werden, um eine Schwangerschaft zu vermeiden.
Schwangerschaft
Aufgrund seines Wirkmechanismus‘ kann Darolutamid das ungeborene Kind schädigen. Es wurden keine präklinischen Studien zur Reproduktionstoxizität durchgeführt (siehe Abschnitt 5.3.).
Es ist nicht bekannt, ob Darolutamid oder seine Metabolite in das Sperma übergehen. Hat der Patient Geschlechtsverkehr mit einer schwangeren Frau, müssen während der Behandlung und für eine Woche nach Beenden der Behandlung mit NUBEQA Kondome verwendet werden. Die Exposition des Fetus gegenüber einem Androgenrezeptorhemmer mittels Übertragung von Samen an die schwangere Frau muss vermieden werden, da so die Entwicklung des Fetus beeinträchtigt werden könnte.
Stillzeit
Es ist nicht bekannt, ob Darolutamid oder dessen Metabolite in die Muttermilch übergehen. Es wurden keine tierexperimentellen Studien durchgeführt, um die Ausscheidung von Darolutamid oder dessen Metabolite in die Muttermilch zu bewerten (siehe Abschnitt 5.3). Ein Risiko für das gestillte Kind kann nicht ausgeschlossen werden.
Fertilität
Es liegen keine Daten zu Auswirkungen von Darolutamid auf die Fertilität beim Menschen vor. Basierend auf tierexperimentellen Studien kann NUBEQA die Fruchtbarkeit bei Männern mit Fortpflanzungspotential beeinträchtigen (siehe Abschnitt 5.3).
NUBEQA hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die am häufigsten beobachteten Nebenwirkungen bei Patienten mit
nmCRPC oder mHSPC, die Darolutamid erhalten, sind Fatigue/ Erschöpfungszustände (13,7 %).
mHSPC, die Darolutamid in Kombination mit Docetaxel erhalten, sind Ausschlag (16,6 %) und Hypertonie (13,8 %).
Weitere Informationen zur Sicherheit bei der Anwendung von Darolutamid in Kombination mit anderen Arzneimitteln sind der Produktinformation der einzelnen Arzneimittel zu entnehmen.
Tabellarische Auflistung der Nebenwirkungen
Nebenwirkungen, die bei Patienten mit nmCRPC oder mHSPC unter der Behandlung mit Darolutamid beobachtet wurden, sind in Tabelle 1 aufgeführt. Nebenwirkungen, die bei Patienten mit mHSPC unter der Behandlung mit Darolutamid in Kombination mit Docetaxel beobachtet wurden, sind Tabelle 2 zu entnehmen.
Nebenwirkungen werden nach Systemorganklasse angegeben. Sie werden nach ihrer Häufigkeit geordnet aufgelistet. Bei den Häufigkeitsangaben werden folgende Kategorien zugrunde gelegt: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1.000, < 1/100); selten (≥ 1/10.000, < 1/1.000); sehr selten (< 1/10.000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Innerhalb der einzelnen Häufigkeitskategorien sind die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 1: Nebenwirkungen, die bei nmCRPC- und mHSPC-Patienten unter Behandlung mit Darolutamid in der ARAMIS‑ und der ARANOTE-Studie berichtet wurdena
Systemorganklasse | Sehr häufig | Häufig |
Herzerkrankungen | Ischämische Herzerkrankungb | |
Erkrankungen der Haut und des Unterhautzellgewebes | Ausschlagd | |
Skelettmuskulatur‑, Bindegewebs‑ und Knochenerkrankungen | Schmerzen in einer Extremität | |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | Fatigue/ Erschöpfungszuständee | |
Untersuchungenf | Neutrophilenzahl verringert |
a Die mediane Expositionsdauer in der ARAMIS- und der ARANOTE-Studie betrug bei Patienten, die mit Darolutamid behandelt wurden, 18,2 Monate (Spanne: 0,0 bis 44,3 Monate) und 11,6 Monate (Spanne: 0,0 bis 40,5 Monate) bei Patienten, die Placebo erhielten.
b Umfasst Arteriosklerose der Koronararterien, koronare Herzerkrankung, Koronararterienverschluss, Stenose der Koronararterien, akutes Koronarsyndrom, akuter Myokardinfarkt, Angina pectoris, instabile Angina pectoris, Myokardinfarkt, myokardiale Ischämie.
c Umfasst Herzinsuffizienz, akute Herzinsuffizienz, chronische Herzinsuffizienz, Stauungsinsuffizienz, kardiogenen Schock, Herzinsuffizienz mit erhaltener Auswurffraktion.
d Umfasst Ausschlag, makulösen Ausschlag, makulo-papulösen Ausschlag, papulösen Ausschlag, pustulösen Ausschlag, Erythem, Dermatitis.
e Umfasst Fatigue sowie Asthenie, Lethargie und Unwohlsein.
f Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. Die Inzidenz beruht auf Werten, die als auffällige Laborwerte berichtet wurden.
Tabelle 2: Nebenwirkungen, die bei Patienten mit mHSPC unter der Behandlung mit Darolutamid in Kombination mit Docetaxel in der ARASENS‑Studie berichtet wurdena, b
Systemorganklasse | Sehr häufig | Häufig |
Gefäßerkrankungen | Hypertoniec | |
Erkrankungen der Haut und des Unterhautzellgewebes | Ausschlagd, e | |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | Frakturen | |
Erkrankungen der Geschlechtsorgane und der Brustdrüse | Gynäkomastie | |
Untersuchungenf | Neutrophilenzahl verringert |
a Die mediane Expositionsdauer betrug bei Patienten, die mit Darolutamid+Docetaxel behandelt wurden, 41,0 Monate (Spanne: 0,1 bis 56,5 Monate) und 16,7 Monate (Spanne: 0,3 bis 55,8 Monate) bei Patienten, die Placebo+Docetaxel erhielten.
b Die Inzidenzen der Nebenwirkungen sind möglicherweise nicht nur Darolutamid allein zuzuschreiben, sondern könnten teilweise auch auf die in Kombination angewendeten Arzneimittel zurückzuführen sein.
c Umfasst Hypertonie, erhöhten Blutdruck, hypertensiven Notfall.
d Umfasst Ausschlag, Medikamentenausschlag, erythematösen Ausschlag, follikulären Ausschlag, makulösen Ausschlag, makulo-papulösen Ausschlag, papulösen Ausschlag, Ausschlag mit Juckreiz, pustulösen Ausschlag, blasigen Hautausschlag, Erythem, Dermatitis.
e Die Inzidenz war in den ersten 6 Behandlungsmonaten am höchsten.
f Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03. Die Inzidenz beruht auf Werten, die als auffällige Laborwerte berichtet wurden.
Beschreibung ausgewählter Nebenwirkungen
Leberfunktionstests
Unter der Behandlung mit Darolutamid wurden Fälle von idiosynkratischen arzneimittelinduzierten Leberschäden mit Erhöhungen vom Schweregrad 3 und 4 der Alanin-Aminotransferase (ALT) und / oder Aspartat-Aminotransferase (AST) auf das ≥ 5-Fache bzw. das ≥ 20-Fache der Obergrenze des Normwerts (upper limit of normal [ULN]) berichtet, einschließlich erhöhter Transaminasen bei gleichzeitigem Anstieg des Gesamtbilirubins auf das ≥ 2-Fache ULN. Die Zeit bis zum Auftreten lag zwischen 1 Monat und 12 Monaten nach Beginn der Behandlung mit Darolutamid. Nach Absetzen von Darolutamid waren die ALT- und AST-Erhöhungen in vielen Fällen reversibel. Für spezifische Empfehlungen siehe Abschnitt 4.4.
Nicht-metastasiertes kastrationsresistentes Prostatakarzinom (nmCRPC) und metastasiertes hormonsensitives Prostatakarzinom (mHSPC) – Behandlung mit Darolutamid
Fatigue
Fatigue/ Erschöpfungszustände wurden von 13,7 % der mit Darolutamid behandelten Patienten und von 11,7 % der mit Placebo behandelten Patienten berichtet. Ereignisse bis zum 3. Schweregrad wurden von 0,4 % der mit Darolutamid behandelten Patienten und von 0,9 % der mit Placebo behandelten Patienten berichtet. Bei den meisten der Patienten trat Fatigue (ohne Asthenie, Lethargie oder Unwohlsein) auf (10,0 % der mit Darolutamid behandelten Patienten und 8,5 % der mit Placebo behandelten Patienten).
Frakturen
Frakturen traten bei 4,1 % der mit Darolutamid behandelten Patienten und bei 3,2 % der mit Placebo behandelten Patienten auf.
Ischämische Herzkrankheit und Herzinsuffizienz
Eine ischämische Herzkrankheit trat bei 3,4 % der mit Darolutamid behandelten Patienten und bei 2,2 % der mit Placebo behandelten Patienten auf. Ereignisse vom Schweregrad 5 traten bei 0,4 % der mit Darolutamid behandelten Patienten und bei 0,4 % der mit Placebo behandelten Patienten auf. Herzinsuffizienz trat bei 1,6 % der mit Darolutamid behandelten Patienten und bei 0,9 % der mit Placebo behandelten Patienten auf.
Neutrophilenzahl verringert
Bei 17,3 % der mit Darolutamid behandelten Patienten und bei 7,4 % der mit Placebo behandelten Patienten wurde eine verringerte Neutrophilenzahl als auffälliger Laborwert berichtet. Die mediane Zeit bis zum Nadir betrug 225 Tage. Die auffälligen Laborwerte waren überwiegend vom Schweregrad 1 oder 2. Eine verringerte Neutrophilenzahl vom Grad 3 und 4 wurde bei 2,6 % bzw. 0,3 % der Patienten berichtet. Die Behandlung mit Darolutamid wurde bei lediglich einem Patienten aufgrund von Neutropenie dauerhaft abgesetzt. Die Neutropenie war bei 83 % der Patienten entweder vorübergehend oder reversibel und nicht mit klinisch bedeutsamen Anzeichen oder Symptomen verbunden.
Bilirubin im Blut erhöht
Bei 16,1 % der mit Darolutamid behandelten Patienten und bei 6,1 % der mit Placebo behandelten Patienten wurde ein erhöhter Bilirubinspiegel als auffälliger Laborwert berichtet. Die Episoden waren überwiegend vom Schweregrad 1 oder 2, nicht mit klinisch bedeutsamen Anzeichen oder Symptomen verbunden und nach dem Absetzen von Darolutamid reversibel. Ein erhöhter Bilirubinspiegel 3. und 4. Grades wurde bei 0,2 % der mit Darolutamid behandelten Patienten und bei 0 % der mit Placebo behandelten Patienten berichtet. Im Darolutamid‑Arm betrug die mittlere Zeit bis zum ersten Auftreten erhöhten Bilirubins 187 Tage, und die mittlere Dauer der ersten Episode betrug 172 Tage. Bei einem der Patienten wurde die Behandlung mit Darolutamid aufgrund eines erhöhten Bilirubinspiegels abgesetzt.
ALT und AST erhöht
Bei 13,3 % der mit Darolutamid behandelten Patienten und bei 9,7 % der mit Placebo behandelten Patienten wurde ein erhöhter ALT-Wert als auffälliger Laborwert berichtet. Bei 22,0 % der mit Darolutamid behandelten Patienten und bei 13,4 % der mit Placebo behandelten Patienten wurde ein erhöhter AST‑Wert als auffälliger Laborwert berichtet. Die Episoden waren überwiegend vom Schweregrad 1 oder 2, nicht mit klinisch bedeutsamen Anzeichen oder Symptomen verbunden und nach dem Absetzen von Darolutamid reversibel. Ein ALT-Anstieg 3. und 4. Grades wurde bei 0,9 % der mit Darolutamid behandelten Patienten und bei 0,3 % der mit Placebo behandelten Patienten berichtet. Ein AST‑Anstieg 3. und 4. Grades wurde bei 1,2 % der mit Darolutamid behandelten Patienten und bei 0,3 % der mit Placebo behandelten Patienten berichtet. Im Darolutamid-Arm betrug die mittlere Zeit bis zum ersten Auftreten eines erhöhten ALT-Wertes 253 Tage und eines erhöhten AST‑Wertes 257 Tage. Die mittlere Dauer der ersten Episode betrug 122 Tage (erhöhter ALT-Wert) bzw. 121 Tage (erhöhter AST-Wert). Bei 2 bzw. 3 Patienten wurde die Behandlung mit Darolutamid aufgrund eines ALT- bzw. AST‑Anstiegs abgesetzt.
Metastasiertes hormonsensitives Prostatakarzinom (mHSPC) – Behandlung mit Darolutamid in Kombination mit Docetaxel
Hypertonie
In der ARASENS-Studie wurde bei 13,8 % der mit Darolutamid+Docetaxel behandelten Patienten und bei 9,4 % der Patienten, die Placebo+Docetaxel erhielten, über Hypertonie berichtet.
Den Berichten zufolge trat eine Hypertonie vom Schweregrad 3 bei 6,4 % der Patienten, die mit Darolutamid+Docetaxel behandelt wurden, auf im Vergleich zu 3,5 % der Patienten unter der Behandlung mit Placebo+Docetaxel. Ein Patient in jedem Behandlungsarm hatte eine Hypertonie vom Schweregrad 4.
Im Arm mit Darolutamid+Docetaxel wurde über einen Fall einer Hypertonie vom Schweregrad 5 mit Arteriosklerose vom Schweregrad 5 berichtet. Dieser Patient wies eine langjährige Anamnese mit Hypertonie und Rauchen auf, und der Fall trat mehr als 3 Jahre nach Beginn der Behandlung mit Darolutamid auf. In beiden Behandlungsarmen wurden Hypertonieereignisse häufiger bei Patienten gemeldet, bei denen anamnestisch keine Hypertonie bekannt war.
Frakturen
Frakturen traten bei 7,5 % der mit Darolutamid+Docetaxel behandelten Patienten und bei 5,1 % der mit Placebo+Docetaxel behandelten Patienten auf.
Neutrophilenzahl verringert
Bei 50,6 % der mit Darolutamid+Docetaxel behandelten Patienten und bei 45,5 % der mit Placebo+Docetaxel behandelten Patienten wurde eine verringerte Neutrophilenzahl als auffälliger Laborwert berichtet. Eine Verringerung der Neutrophilenzahl vom Schweregrad 3 und 4 wurde bei 34,4 % der mit Darolutamid+Docetaxel behandelten Patienten und bei 31,4 % der mit Placebo+Docetaxel behandelten Patienten berichtet. In beiden Behandlungsarmen war das Auftreten einer verringerten Neutrophilenzahl und einer Neutropenie in den ersten Monaten der Behandlung am höchsten, danach nahmen Häufigkeit und Schweregrad der Ereignisse ab.
Bilirubin im Blut erhöht
Bei 19,6 % der mit Darolutamid+Docetaxel behandelten Patienten und bei 10,0 % der mit Placebo+Docetaxel behandelten Patienten wurde ein erhöhter Bilirubinspiegel als auffälliger Laborwert berichtet. Die Ereignisse waren überwiegend vom Schweregrad 1 oder 2. Ein erhöhter Bilirubinspiegel 3. oder 4. Grades wurde bei 0,5 % der mit Darolutamid+Docetaxel behandelten Patienten und bei 0,3 % der mit Placebo+Docetaxel behandelten Patienten berichtet.
ALT und AST erhöht
Bei 42,3 % der mit Darolutamid+Docetaxel behandelten Patienten und bei 38,0 % der mit Placebo+Docetaxel behandelten Patienten wurde ein erhöhter Wert für Alanin-Aminotransferase (ALT) als auffälliger Laborwert berichtet. Bei 43,9 % der mit Darolutamid+Docetaxel behandelten Patienten und bei 39,3 % der mit Placebo+Docetaxel behandelten Patienten wurde ein erhöhter Wert für Aspartat-Aminotransferase (AST) als auffälliger Laborwert berichtet. ALT- und AST-Anstiege waren überwiegend vom Schweregrad 1. ALT-Anstiege 3. und 4. Grades wurden bei 3,7 % der mit Darolutamid+Docetaxel behandelten Patienten berichtet sowie bei 3,0 % der Patienten, die mit Placebo+Docetaxel behandelt wurden. AST-Anstiege 3. und 4. Grades wurden bei 3,6 % der mit Darolutamid+Docetaxel behandelten Patienten berichtet sowie bei 2,3 % der Patienten, die mit Placebo+Docetaxel behandelt wurden.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
Die höchste klinisch untersuchte Dosis von Darolutamid betrug 900 mg zweimal täglich, entsprechend einer Tagesgesamtdosis von 1800 mg. Bei dieser Dosis wurden keine dosislimitierenden Toxizitäten beobachtet.
Unter Berücksichtigung der sättigbaren Resorption (siehe Abschnitt 5.2) und fehlenden Hinweisen auf eine akute Toxizität ist nicht zu erwarten, dass die Einnahme einer höheren als der empfohlenen Dosis von Darolutamid toxische Wirkungen nach sich zieht.
Bei Einnahme einer höheren als der empfohlenen Dosis kann die Behandlung mit Darolutamid wie geplant mit der nächsten Dosis fortgesetzt werden.
Es gibt kein spezifisches Antidot für Darolutamid und die Symptome einer Überdosierung sind nicht bekannt.
Pharmakotherapeutische Gruppe: Endokrine Therapie, Antiandrogene; ATC‑Code: L02BB06
Wirkmechanismus
Darolutamid ist ein Androgenrezeptor (AR)-Inhibitor mit einer flexiblen, polar substituierten Pyrazolstruktur, der mit hoher Affinität direkt an die Liganden-bindende Domäne des Rezeptors bindet.
Darolutamid hemmt kompetitiv die Androgenbindung, die Translokation des AR in den Zellkern sowie die AR‑vermittelte Transkription. Ein Hauptmetabolit, Keto-Darolutamid, zeigte ähnliche In‑vitro‑Aktivität wie Darolutamid. Die Behandlung mit Darolutamid vermindert die Proliferation der Prostatakarzinomzellen, wodurch es zu einer ausgeprägten antitumoralen Aktivität kommt.
Pharmakodynamische Wirkungen
Nach zweimal täglicher oraler Gabe von 600 mg Darolutamid wurde im Vergleich zu Placebo keine Verlängerung (d. h. mehr als 10 ms) des mittleren QTcF‑Intervalls beobachtet.
Klinische Wirksamkeit und Sicherheit
Die Wirksamkeit und Sicherheit wurden in drei randomisierten, placebokontrollierten, multizentrischen Phase‑III-Studien bei Patienten mit nmCRPC (ARAMIS) und mHSPC (ARANOTE und ARASENS) nachgewiesen. Alle Patienten erhielten begleitend einen Luteinisierendes-Hormon-Releasing-Hormon-(LHRH-)Agonisten oder -Antagonisten oder hatten sich zuvor einer bilateralen Orchiektomie unterzogen.
Nicht-metastasiertes kastrationsresistentes Prostatakarzinom (nmCRPC) – Behandlung mit Darolutamid
Die Wirksamkeit und Sicherheit von Darolutamid wurden in einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase-III-Studie (ARAMIS) bei Patienten mit nicht-metastasiertem (beurteilt mittels konventioneller CT‑, Knochenscan‑ oder MRT‑Aufnahmen) kastrationsresistentem Prostatakarzinom mit einer Verdopplungszeit des prostataspezifischen Antigens (PSADT) von ≤ 10 Monaten beurteilt.
Patienten wurden in die Studie aufgenommen, wenn während der Androgendeprivationstherapie nach dem Nadir in mindestens 1‑wöchigen Abständen drei ansteigende Werte für das Prostataspezifische Antigen (PSA) festgestellt wurden, der PSA‑Spiegel beim Screening ≥ 2 ng/ml und der Kastrationsspiegel für Testosteron im Serum < 1,7 nmol/l betrug.
Patienten mit einer Vorgeschichte von Krampfanfällen konnten in die Studie aufgenommen werden. In der Darolutamid-Behandlungsgruppe waren 12 Patienten (0,21 %) mit einer Vorgeschichte von Krampfanfällen.
Patienten mit unkontrollierter Hypertonie oder Patienten mit kürzlich (in den letzten 6 Monaten) erlittenem Schlaganfall, Myokardinfarkt, schwerer/ instabiler Angina pectoris, koronarer oder peripher-arterieller Bypass-Operation, Herzinsuffizienz der NYHA-Klasse III oder IV (NYHA = New York Heart Association) waren von der Studie ausgeschlossen.
Patienten, die zuvor bereits mit AR‑Inhibitoren der zweiten Generation wie Enzalutamid, Apalutamid und Darolutamid oder CYP17‑Enzyminhibitoren wie Abirateronacetat behandelt wurden, sowie Patienten, die innerhalb von 28 Tagen vor der Randomisierung eine systemische Corticosteroid‑Therapie mit einem Dosisäquivalent von mehr als 10 mg Prednison/Tag erhalten hatten, waren von der Studie ausgeschlossen.
Insgesamt erhielten 1509 Patienten, randomisiert im Verhältnis 2:1, entweder zweimal täglich 600 mg Darolutamid oral (n = 955) oder Placebo (n = 554).
Patienten mit Beckenlymphknoten von < 2 cm Größe in der kurzen Achse unterhalb der Aortenbifurkation konnten in die Studie aufgenommen werden. Das Vorliegen oder Fehlen von Metastasen wurde durch eine unabhängige zentrale radiologische Beurteilung festgestellt. Eingeschlossen in diese Analysen waren 89 Patienten, bei denen retrospektiv Metastasen zu Studienbeginn identifiziert wurden. Die Randomisierung war nach PSADT (≤ 6 Monate oder > 6 Monate) und Anwendung einer zielgerichteten Therapie gegen Osteoklasten bei Studieneintritt (ja oder nein) stratifiziert.
Die folgenden demografischen Merkmale der Patienten und ihre Krankheitscharakteristika waren in den Behandlungsgruppen ausgewogen. Das mediane Alter betrug 74 Jahre (Spanne: 48‑95) und 9 % der Patienten waren 85 Jahre oder älter. Die Verteilung nach ethnischer Zugehörigkeit war wie folgt: 79 % kaukasisch, 13 % asiatisch und 3 % schwarz. Die meisten Patienten (73 %) wiesen bei der Diagnose einen Gleason‑Score von 7 oder höher auf. Die mediane PSADT betrug 4,5 Monate. Neun Prozent (9 %) der Patienten hatten sich zuvor einer Orchiektomie, 25 % der Patienten einer Prostatektomie und 50 % der Patienten mindestens einer Strahlentherapie unterzogen. Sechsundsiebzig Prozent (76 %) der Patienten hatten zuvor mehr als eine hormonablative Therapie erhalten. Bei Studieneintritt wiesen die Patienten einen Performance‑Status nach der Eastern Cooperative Oncology Group (ECOG PS) von 0 (69 %) oder 1 (31 %) auf.
Die Behandlung mit Darolutamid wurde fortgeführt, bis eine verblindete zentrale Beurteilung mittels konventioneller Bildgebungsverfahren (CT, Knochenscan, MRT) eine radiologische Krankheitsprogression feststellte, eine unzumutbare Toxizität auftrat oder die Einwilligung zurückgezogen wurde.
Primärer Wirksamkeitsendpunkt war das metastasenfreie Überleben (MFS). Sekundäre Endpunkte umfassten Gesamtüberleben (OS), Zeit bis zur Schmerzprogression, Zeit bis zur Einleitung einer ersten zytotoxischen Chemotherapie gegen das Prostatakarzinom und Zeit bis zu den ersten symptomatischen Knochenereignissen (definiert als Auftreten eines der folgenden Ereignisse: externe Bestrahlung zur Linderung der skelettbezogenen Symptome, neue symptomatische pathologische Knochenfraktur, Rückenmarkskompression oder tumorbedingte orthopädische Operation).
Die Behandlung mit Darolutamid führte im Vergleich zu Placebo zu einer Verbesserung des MFS (siehe Tabelle 3 und Abbildung 1).
Die Ergebnisse zu MFS stimmten über alle Subgruppen der Patienten hinweg überein, unabhängig von PSADT, vorheriger Anwendung von Osteoprotektiva oder lokoregionärer Erkrankung. Weitere Subgruppen mit einheitlichen MFS‑Ergebnissen waren PSA‑Ausgangswert, Gleason‑Score bei Diagnose, Alter, geographische Region, ECOG‑PS‑Ausgangswert, ethnische Zugehörigkeit und Anzahl vorheriger Hormontherapien.
Nach der Primäranalyse des MFS wurde nach der Entblindung der Studie jenen Patienten, die Placebo erhalten hatten, eine Behandlung mit open-label Darolutamid angeboten (Crossover-Option). Von den 554 Patienten, die in den Placeboarm randomisiert worden waren, wechselten 170 (31 %) zu einer Behandlung mit Darolutamid. Die Analyse des Gesamtüberlebens wurde nicht um die Störeffekte des Crossovers bereinigt.
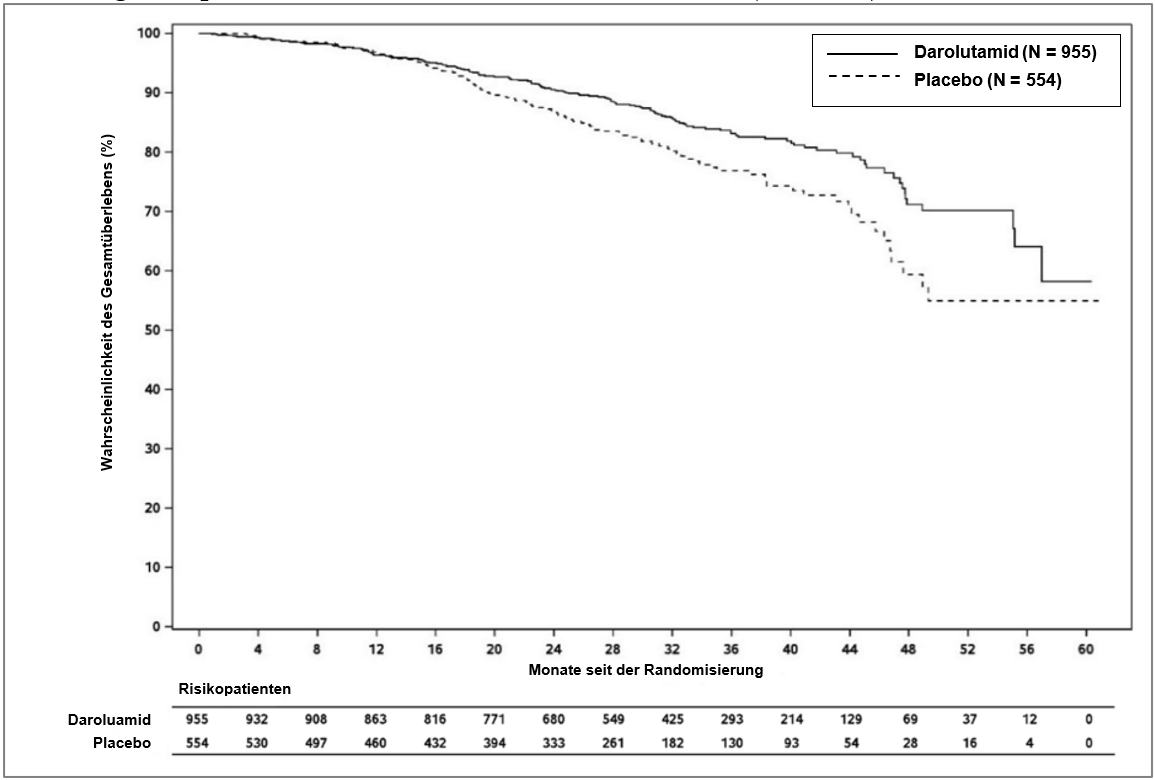
Zum Zeitpunkt der finalen Analyse, führte die Behandlung mit Darolutamid im Vergleich zu Placebo zu einer signifikanten Verbesserung hinsichtlich des Gesamtüberlebens (Median wurde in beiden Gruppen nicht erreicht, siehe Tabelle 3 und Abbildung 2).
Außerdem führte die Behandlung mit Darolutamid im Vergleich zu Placebo zu statistisch signifikanten Verlängerungen der Zeit bis zur Schmerzprogression, der Zeit bis zur Einleitung einer ersten zytotoxischen Chemotherapie und der Zeit bis zum ersten symptomatischen Skelettereignis (siehe Tabelle 3).
Zum Zeitpunkt der finalen Analyse betrug die mediane Behandlungsdauer bei Patienten, die mit Darolutamid behandelt wurden, 33,3 Monate (Spanne: 0,0 bis 74,0 Monate) während der kombinierten doppelblinden und unverblindeten Phase.
Alle Analysen wurden im vollständigen Analysesatz durchgeführt.
Tabelle 3: Wirksamkeitsergebnisse aus der ARAMIS‑Studie
Wirksamkeitsparameter | Anzahl (%) der Patienten mit Ereignissen | Median (Monate) (95% KI) | Hazard Ratiob | ||
Darolutamid | Placeboa | Darolutamid | Placeboa | ||
Metastasenfreies Überlebenc | 221 (23,1 %) | 216 (39,0 %) | 40,4 | 18,4 | 0,413 |
Gesamtüberleben | 148 (15,5 %) | 106 (19,1 %) | NE | NE | 0,685 |
Zeit bis zur Schmerzprogressionc,d | 251 (26,3 %) | 178 (32,1 %) | 40,3 | 25,4 | 0,647 |
Zeit bis zum Einleiten einer ersten zytotoxischen Chemotherapie | 127 (13,3 %) | 98 (17,7 %) | NE | NE | 0,579 |
Zeit bis zum ersten symptomatischen Skelettereignis | 29 (3,0 %) | 28 (5,1 %) | NE | NE | 0,484 |
a Einschließlich 170 Patienten, die zu open-label Darolutamid gewechselt sind
b Hazard Ratio < 1 zugunsten Darolutamid
c Für MFS und Zeit bis zur Schmerzprogression wird die zum Zeitpunkt des primären Abschlusses durchgeführte Analyse als die finale Analyse betrachtet.
d Von den Patienten berichtete Ergebnisse gemäß dem Fragebogen Brief Pain Inventory‑Short Form.
NE: Nicht erreicht
Die Behandlung mit Darolutamid führte zu einem längeren progressionsfreien Überleben (PFS; Median 36,8 vs. 14,8 Monate; HR = 0,380; nominaler p‑Wert < 0,000001) und einer längeren Zeit bis zur PSA‑Progression (Median 29,5 Monate vs. 7,2 Monate; HR = 0,164; nominaler p‑Wert < 0,000001). Über alle Überlebensparameter (MFS, OS und PFS) hinweg wurde eine konsistente Wirkung beobachtet.
Abbildung 1: Kaplan‑Meier‑Kurven für das metastasenfreie Überleben (ARAMIS)
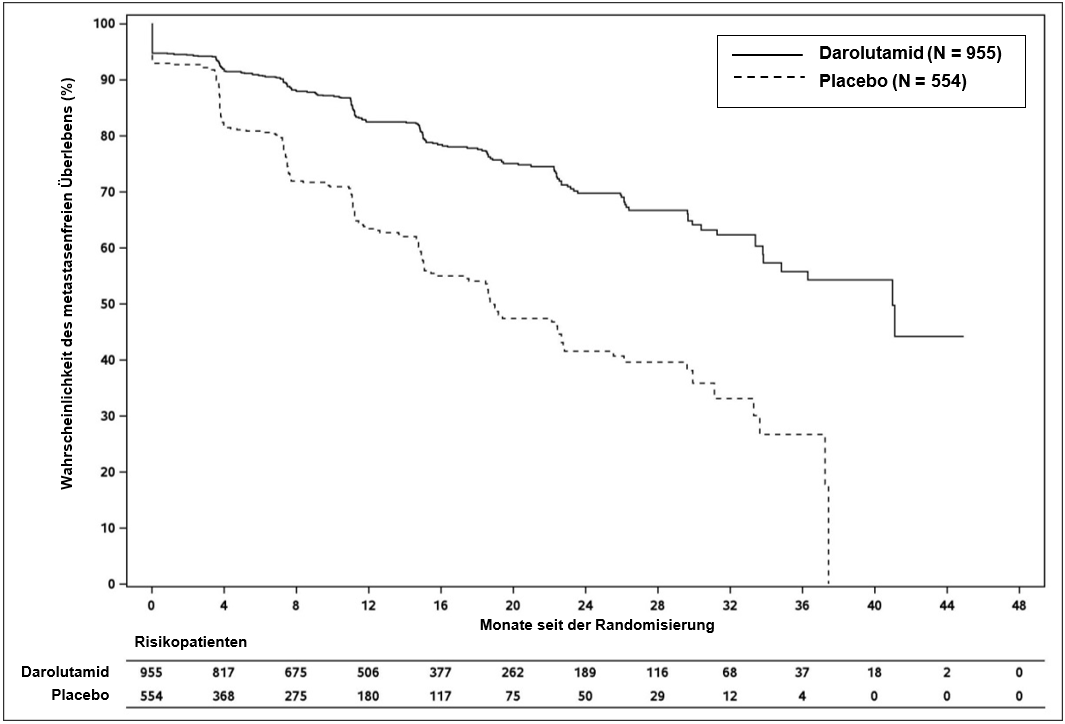
Abbildung 2: Kaplan‑Meier‑Kurven für das Gesamtüberleben (ARAMIS)
Patienten, die in der ARAMIS‑Studie (Doppelblindzeit) Darolutamid erhielten, zeigten eine signifikant höhere bestätigte PSA‑Ansprechrate (definiert als Reduktion um ≥ 50 % gegenüber dem Ausgangswert) als Patienten, die Placebo erhielten: 84,0 % vs. 7,9 % (Differenz = 76,1 %; p < 0,000001) (nominaler p-Wert, nur zur Information).
Metastasiertes hormonsensitives Prostatakarzinom (mHSPC) – Behandlung mit Darolutamid
Die Wirksamkeit und Sicherheit von Darolutamid wurden in einer multizentrischen, doppelblinden, placebokontrollierten Phase-III-Studie (ARANOTE) bei Patienten mit mHSPC untersucht. Insgesamt wurden 669 Patienten im Verhältnis 2:1 randomisiert und erhielten 600 mg Darolutamid oral zweimal täglich (n = 446) oder ein entsprechendes Placebo (n = 223). Die Behandlung mit Darolutamid bzw. Placebo wurde fortgeführt bis eine Krankheitsprogression eintrat, die antineoplastische Therapie geändert wurde, eine unzumutbare Toxizität auftrat, der Patient verstarb oder die Einwilligung zurückgezogen wurde.
Das Vorliegen von Metastasen wurde durch eine unabhängige zentrale radiologische Beurteilung festgestellt. Patienten, die nur eine regionale Lymphknotenbeteiligung (M0) aufwiesen, wurden aus der Studie ausgeschlossen. Die Randomisierung erfolgte stratifiziert nach Vorliegen viszeraler Metastasen und Anwendung einer vorherigen lokalen Therapie.
Patienten, die zuvor bereits mit AR‑Inhibitoren der zweiten Generation wie Enzalutamid, Apalutamid und Darolutamid oder mit CYP17‑Enzyminhibitoren wie Abirateronacetat oder mit einer Chemotherapie einschließlich Docetaxel behandelt worden waren, waren von der Studie ausgeschlossen.
Die folgenden demografischen Merkmale der Patienten und Krankheitscharakteristika waren in den Behandlungsgruppen ausgewogen. Das mediane Alter betrug 70 Jahre (Spanne: 43‑93) und 4,2 % der Patienten waren 85 Jahre oder älter. Die Verteilung nach ethnischer Zugehörigkeit war wie folgt: 56,2 % kaukasisch, 31,2 % asiatisch und 9,7 % schwarz oder afroamerikanisch und 2,8 % andere. Die meisten Patienten (68,3 %) wiesen bei der Diagnose einen Gleason‑Score von 8 oder höher auf. Zu Studienbeginn hatten 49,8 %, 47,2 % bzw. 3,0 % einen ECOG-PS von 0, 1 und 2. Bei 72,5 % der Patienten handelte es sich um eine neu diagnostizierte Erkrankung und bei 21,7 % um eine rezidivierte Erkrankung. Zu Studienbeginn befanden sich 3,8 % der Patienten im Krankheitsstadium M1a (nur nicht-regionale Lymphknotenmetastasen), 77,1 % im Krankheitsstadium M1b (Knochenmetastasen mit oder ohne Lymphknotenmetastasen) und 19,1 % im Krankheitsstadium M1c (viszerale Metastasen mit oder ohne Lymphknotenmetastasen oder mit oder ohne Knochenmetastasen). Der mediane PSA-Ausgangswert lag bei 21,3 µg/l. 70,6 % der Patienten wiesen eine hochvolumige Erkrankung auf und 29,4 % eine geringvolumige Erkrankung. Eine hochvolumige Erkrankung wurde definiert als Vorhandensein von viszeralen Metastasen oder 4 oder mehr Knochenläsionen, mit mindestens einer Metastase außerhalb der Wirbelsäule und der Beckenknochen. Patienten mit Krampfanfällen in der Anamnese wurden in die Studie aufgenommen, und 1 Patient (0,2 %) wurde in den Darolutamid-Arm aufgenommen.
Primärer Wirksamkeitsendpunkt war das radiologische progressionsfreie Überleben (rPFS). Sekundäre Endpunkte waren das Gesamtüberleben (OS), die Zeit bis zum Beginn einer nachfolgenden Krebstherapie, die Zeit bis zum Auftreten eines kastrationsresistenten Prostatakarzinoms, die Zeit bis zur PSA-Progression, die Rate von PSA-Werten unter der Nachweisgrenze und die Zeit bis zur Schmerzprogression. Die Schmerzprogression wurde anhand der von den Patienten gemäß dem (Patient Related Outcome, PRO) Brief-Pain-Inventory‑Short-Form(BPI-SF)-Fragebogen berichteten Ereignisse ausgewertet. Sie war definiert als Verschlechterung um mindestens 2 Punkte im Vergleich zum Nadir oder Beginn der Anwendung kurz- oder langwirksamer Opioide bei malignen Erkrankungen über ≥ 7 aufeinanderfolgende Tage. Der Daten-Cut-off für die rPFS-Analyse war der 7. Juni 2024. Nach der primären rPFS-Analyse und der Entblindung der Studie wurde den Placebo-Patienten eine offene Behandlung mit Darolutamid angeboten (Crossover-Option). Von den 63 Patienten, die zum Zeitpunkt des Daten-Cut-offs der primären Analyse noch Placebo erhielten, wechselten 60 (95 %) zur Darolutamid-Behandlung. Der Daten-Cut-off für die finale OS-Analyse war der 10. Januar 2025. Die OS-Analyse wurde nicht um Störeffekte des Crossovers adjustiert.
Tabelle 4: Wirksamkeitsergebnisse aus der ARANOTE-Studie
Wirksamkeitsparameter | Anzahl (%) der Patienten mit Ereignissen | Median (Monate) (95% KI) | Hazard Ratioa | ||
Darolutamid | Placebo | Darolutamid | Placebo | ||
Radiologisches progressionsfreies Überlebenc | 128 (28,7 %) | 94 (42,2 %) | A | 25,0 | 0,541 |
Gesamtüberlebend | 115 (25,8 %) | 70 (31,4 %) | A | A | 0,776 |
a Hazard Ratio < 1 zugunsten Darolutamid
b Basierend auf stratifiziertem Log-Rank-Test
c Der Daten-Cut-off für die rPFS-Analyse war der 7. Juni 2024.
d Der p-Wert für das OS erreichte zum Zeitpunkt der finalen OS-Analyse nicht den vorab festgelegten Grenzwert für statistische Signifikanz. Der Daten-Cut-off war der 10. Januar 2025.
A: Wert aufgrund zensierter Daten nicht schätzbar.
Abbildung 3: Kaplan‑Meier‑Kurven für das radiologische progressionsfreie Überleben; mHSPC-Population (ARANOTE)a
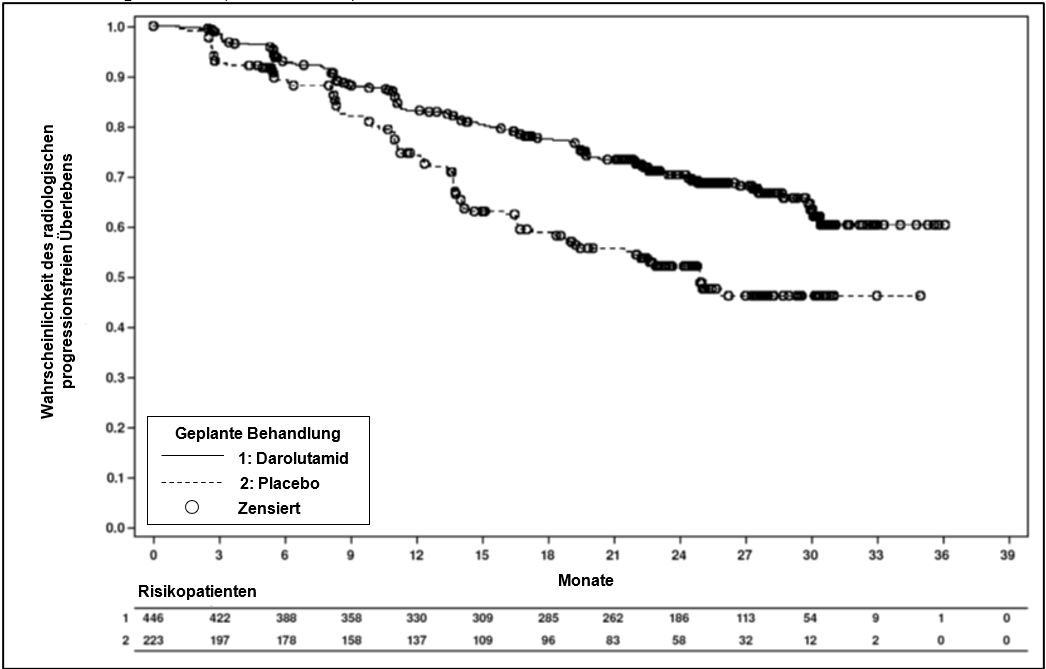
a Die rPFS-Rate nach 12 Monaten betrug 83,1 % (95% KI, 79,5 % bis 86,7 %) im Darolutamid-Arm vs. 74,1 % (95% KI, 68,0 % bis 80,2 %) im Placebo. Die rPFS-Rate nach 24 Monaten betrug 70,3 % (95% KI, 65,7 % bis 74,9 %) im Darolutamid-Arm vs. 52,1 % (95% KI, 44,7 % bis 59,5 %) im Placeboarm. Der Daten-Cut-off war der 7. Juni 2024.
Abbildung 4: Kaplan‑Meier‑Kurven für das Gesamtüberleben; mHSPC-Population (ARANOTE)a
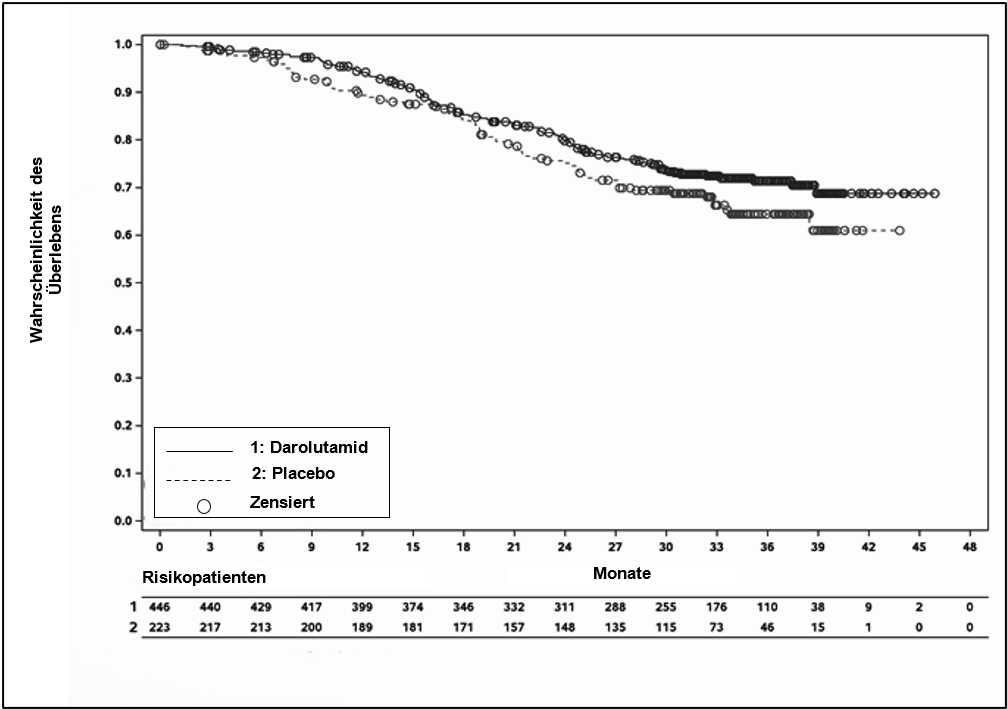
a Die OS-Rate nach 36 Monaten betrug 71,4 % (95% KI, 66,8 % bis 76,0 %) im Darolutamid-Arm vs. 64,4 % (95% KI, 57,3 % bis 71,4 %) im Placeboarm. Der Daten-Cut-off war der 10. Januar 2025.
Metastasiertes hormonsensitives Prostatakarzinom (mHSPC) – Behandlung mit Darolutamid in Kombination mit Docetaxel
Die Wirksamkeit und Sicherheit von Darolutamid in Kombination mit Docetaxel wurden in einer multizentrischen, doppelblinden, placebokontrollierten Phase-III-Studie (ARASENS) bei Patienten mit mHSPC beurteilt. Insgesamt wurden 1.306 Patienten im Verhältnis 1:1 randomisiert und erhielten oral zweimal täglich entweder 600 mg Darolutamid (n = 651) oder passendes Placebo (n = 655), zusammen mit 75 mg/m2 Docetaxel für 6 Zyklen. Die Behandlung mit Darolutamid oder Placebo wurde fortgeführt bis eines der folgenden Ereignisse eintrat: symptomatische progrediente Erkrankung, Änderung der anti-neoplastischen Therapie, nicht akzeptable toxische Wirkung, Tod oder Absetzen der Behandlung.
Das Vorliegen von Metastasen wurde durch eine unabhängige zentrale radiologische Beurteilung festgestellt. Patienten, die nur eine regionale Lymphknotenbeteiligung (M0) aufwiesen, wurden aus der Studie ausgeschlossen. Die Randomisierung erfolgte stratifiziert nach Ausmaß der Erkrankung (nur nicht-regionale Lymphknotenmetastasen [M1a], Knochenmetastasen mit oder ohne Lymphknotenmetastasen [M1b] oder viszerale Metastasen mit oder ohne Lymphknotenmetastasen oder mit oder ohne Knochenmetastasen [M1c]) sowie nach der Konzentration der alkalischen Phosphatase (< oder ≥ Obergrenze des Normwerts) bei Studieneintritt. Patienten mit Hirnmetastasen durften an der Studie teilnehmen, es wurden jedoch keine Patienten mit Hirnmetastasen aufgenommen.
Die folgenden demografischen Merkmale und Krankheitscharakteristika der Patienten waren in den Behandlungsgruppen ausgewogen. Das mediane Alter betrug 67 Jahre (Spanne: 41‑89) und 0,5 % der Patienten waren 85 Jahre oder älter. Die Verteilung nach ethnischer Zugehörigkeit war wie folgt: 52 % kaukasisch, 36 % asiatisch und 4 % schwarz. Die meisten Patienten (78 %) wiesen bei der Diagnose einen Gleason‑Score von 8 oder höher auf. Bei 71 % der Patienten betrug der ECOG-Performance-Status 0 und bei 29 % lag dieser Wert bei 1. Bei 86,1 % der Patienten handelte es sich um eine neu diagnostizierte Erkrankung und bei 12,9 % um eine rezidivierte Erkrankung. Bei Studieneintritt befanden sich 3 % der Patienten im Krankheitsstadium M1a, 79,5 % in M1b und 17,5 % in M1c. Die Konzentration der alkalischen Phosphatase lag bei 44,5 % der Patienten < ULN und bei 55,5 % der Patienten ≥ ULN. Der mediane PSA-Ausgangswert lag in der Gruppe mit Darolutamid bei 30,3 µg/l und in der Placebogruppe bei 24,2 µg/l. Patienten mit Krampfanfällen in der Anamnese wurden in die Studie aufgenommen, und 4 dieser Patienten (0,6 %) wurden in den Arm mit Darolutamid+Docetaxel aufgenommen.
77,0 % der Patienten hatten eine hochvolumige Erkrankung und 23,0 % eine geringvolumige Erkrankung. Eine hochvolumige Erkrankung wurde definiert als Vorhandensein von viszeralen Metastasen oder 4 oder mehr Knochenläsionen, mit mindestens einer Metastase außerhalb der Wirbelsäule und der Beckenknochen. Etwa 25 % der Patienten erhielten eine Begleittherapie mit Bisphosphonaten oder Denosumab.
Primärer Wirksamkeitsendpunkt war das Gesamtüberleben (OS). Sekundäre Endpunkte waren die Zeit bis zum Auftreten eines kastrationsresistenten Prostatakarzinoms, Zeit bis zur Schmerzprogression, Überleben ohne symptomatisches Skelettereignis (symptomatic skeletal event free survival, SSE‑FS), Zeit bis zum ersten symptomatischen Skelettereignis (SSE), Zeit bis zum Einleiten einer anschließenden antineoplastischen Therapie, Zeit bis zur Verschlechterung der krankheitsbezogenen körperlichen Symptome und Zeit bis zum Beginn der Anwendung von Opioiden über ≥ 7 aufeinanderfolgende Tage. Die Schmerzprogression wurde anhand der von den Patienten gemäß dem Brief Pain Inventory‑Short Form (BPI-SF) Fragebogen berichteten Ereignisse ausgewertet. Sie war definiert als Verschlechterung um mindestens 2 Punkte im Vergleich zum Nadir oder Beginn der Anwendung kurz- oder langwirksamer Opioide gegen Schmerzen über ≥ 7 aufeinanderfolgende Tage.
Die mediane Behandlungsdauer betrug bei Patienten, die mit Darolutamid+Docetaxel behandelt wurden, 41,0 Monate (Spanne: 0,1 bis 56,5 Monate) und bei Patienten, die Placebo+Docetaxel erhielten, 16,7 Monate (Spanne: 0,3 bis 55,8 Monate). 87,6 % der Patienten im Arm mit Darolutamid+Docetaxel und 85,5 % der Patienten im Arm mit Placebo+Docetaxel erhielten sechs vollständige Docetaxel-Zyklen, und 1,5 % bzw. 2,0 % der Patienten wurde kein Docetaxel verabreicht.
Tabelle 5: Wirksamkeitsergebnisse aus der ARASENS-Studie
Wirksamkeitsparameter | Anzahl (%) der Patienten mit Ereignissen | Median (Monate) (95% KI) | Hazard Ratiob | ||
Darolutamid + Docetaxel | Placebo + Docetaxel | Darolutamid + Docetaxel | Placebo + Docetaxel | ||
Gesamtüberlebend | 229 (35,2 %) | 304 (46,5 %) | NE | 48,9 | 0,675 |
a Ein Patient im Placeboarm wurde von allen Analysen ausgeschlossen
b Hazard Ratio < 1 zugunsten Darolutamid
c Basierend auf stratifiziertem Log-Rank-Test
d Die Ergebnisse zum Gesamtüberleben stimmten über alle Patientensubgruppen hinweg überein, einschließlich Ausmaß der Erkrankung und Konzentration der alkalischen Phosphatase
NE: nicht erreicht
Die folgenden sekundären Wirksamkeitsendpunkte zeigten einen statistisch signifikanten Vorteil zugunsten der Patienten im Darolutamid+Docetaxel-Arm im Vergleich zu Patienten im Placebo+Docetaxel-Arm: Zeit bis zum Auftreten eines kastrationsresistenten Prostatakarzinoms (medianes NE vs. 19,1 Monate; HR = 0,357, p < 0,0001); Zeit bis zum ersten symptomatischen Skelettereignis (medianes NE vs. NE Monate; HR = 0,712, p = 0,0081); Zeit bis zum Beginn einer nachfolgenden antineoplastischen Chemotherapie (medianes NE vs. 25,3 Monate; HR = 0,388, p < 0,0001); Zeit bis zur Schmerzprogression (medianes NE vs. 27,5 Monate; HR = 0,792, p = 0,0058); symptomatische skelettereignisfreie Überlebenszeit (Median 51,2 vs. 39,7 Monate; HR = 0,609, p < 0,0001).
Abbildung 5: Kaplan‑Meier‑Kurven für das Gesamtüberleben (ARASENS)a
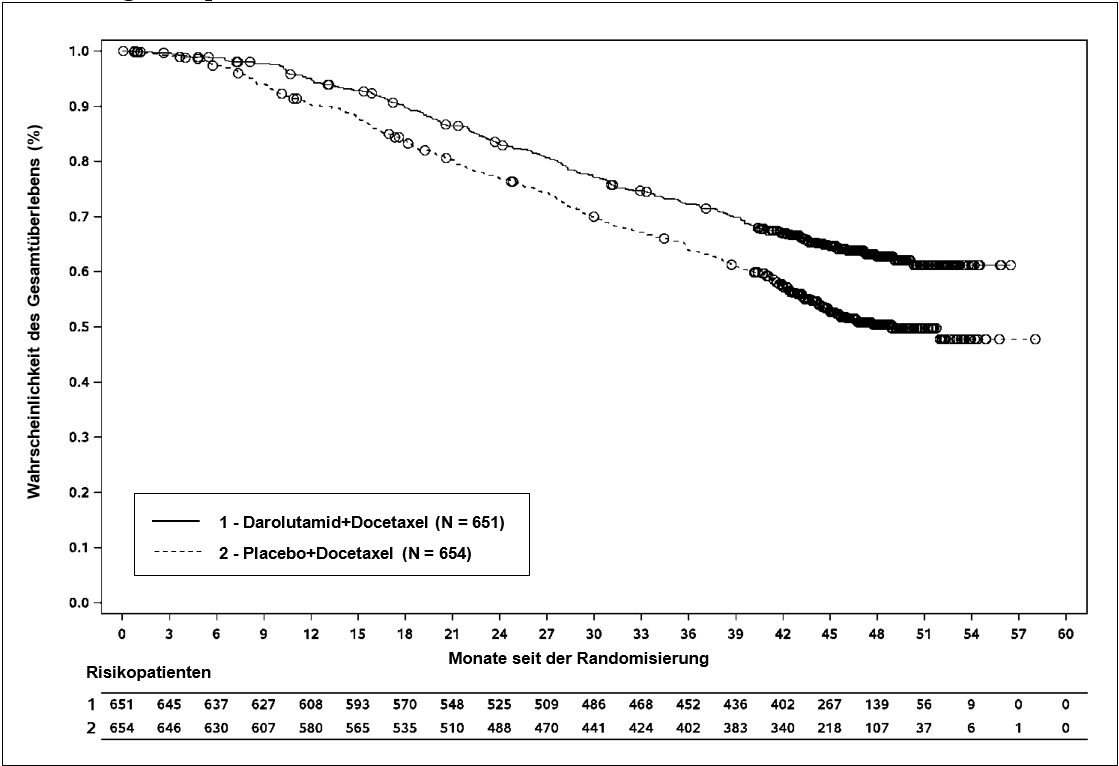
a Die Gesamtüberlebensrate nach 36 Monaten betrug 72,3 % (95% KI, 68,8 % bis 75,8 %) im Arm mit Darolutamid+Docetaxel versus 63,8 % (95% KI, 60,1 % bis 67,6 %) im Arm mit Placebo+Docetaxel.
Die Gesamtüberlebensrate nach 48 Monaten betrug 62,7 % (95% KI, 58,7 % bis 66,7 %) im Arm mit Darolutamid+Docetaxel versus 50,4 % (95% KI, 46,3 % bis 54,6 %) im Arm mit Placebo+Docetaxel.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Darolutamid eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen zu malignen Neoplasmen der Prostata gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Allgemeine Einführung
Darolutamid besteht aus zwei Diastereomeren [(S,R)‑Darolutamid und (S,S)‑Darolutamid], die über den im Blutkreislauf vorkommenden Hauptmetaboliten Keto‑Darolutamid ineinander umgewandelt werden. In vitro zeigen alle drei Substanzen eine ähnliche pharmakologische Aktivität. Darolutamid ist in wässrigen Lösungsmitteln über einen großen pH‑Bereich nur schwer löslich und in organischen Lösungsmitteln im Allgemeinen besser löslich.
Resorption
Nach oraler Gabe von 600 mg (2 Tabletten zu 300 mg) zweimal täglich wurden maximale Darolutamid-Plasmakonzentrationen im Steady State von 4,79 mg/l (Variationskoeffizient: 30,9 %) bei nmCRPC-Patienten in der ARAMIS-Studie und von 3,84 mg/l (Variationskoeffizient: 35,6 %) bei mHSPC-Patienten in der ARASENS-Studie erreicht. Die mediane Zeit bis zum Erreichen maximaler Plasmakonzentrationen betrug 3 – 4 Stunden. Auf Basis von AUC0‑12-Daten im Steady‑State veränderte sich das Verhältnis der beiden Diastereomere, (S,R)‑Darolutamid und (S,S)‑Darolutamid, von 1:1 in der Tablette zu einem ungefähren Verhältnis von 1:9 im Plasma. Nach oraler Gabe zusammen mit einer Mahlzeit wird der Steady‑State nach 2‑5 Tagen wiederholter zweimal täglicher Dosisgabe erreicht.
Die absolute Bioverfügbarkeit nach oraler Gabe einer Tablette NUBEQA mit 300 mg Darolutamid im Nüchternzustand beträgt im Vergleich zu einer intravenösen Injektion ca. 30 %. Bei Gabe zusammen mit einer Mahlzeit war die Bioverfügbarkeit von Darolutamid um den Faktor 2,0 bis 2,5 erhöht. Für den Hauptmetaboliten Keto‑Darolutamid wurde ein ähnlicher Anstieg der Exposition beobachtet.
Verteilung
Das scheinbare Verteilungsvolumen von Darolutamid nach intravenöser Verabreichung beträgt 119 l, was darauf hindeutet, dass Darolutamid umfassend über den gesamten Körper sowohl in intrazelluläre als auch extrazelluläre Flüssigkeitsräume verteilt wird.
Darolutamid ist mäßig stark (zu 92 %) an humane Plasmaproteine gebunden, wobei es zwischen den beiden Diastereomeren keine Unterschiede gibt. Der Hauptmetabolit von Darolutamid, Keto‑Darolutamid, ist stark (zu 99,8 %) an Plasmaproteine gebunden.
Ob Darolutamid die Blut‑Hirn‑Schranke passiert, wurde klinisch nicht untersucht. Expositionen gegenüber Darolutamid im Gehirn sind jedoch hinsichtlich der AUC0‑24 sehr niedrig: 4,5 % der Plasmaexposition nach einer Einzeldosis bei Ratten und 1,9‑3,9 % nach wiederholter Gabe an Mäusen. Dies weist auf eine geringe Passage von Darolutamid über die intakte Blut‑Hirn‑Schranke bei Ratten und Mäusen hin und auf eine geringe Wahrscheinlichkeit, dass Darolutamid die intakte Blut‑Hirn‑Schranke beim Menschen in einem klinisch bedeutsamen Umfang passiert.
Biotransformation
Die Diastereomere (S,R)‑Darolutamid und (S,S)‑Darolutamid können über den Metaboliten Keto‑Darolutamid ineinander umgewandelt werden, wobei bevorzugt (S,S)‑Darolutamid entsteht.
Nach oraler Einzelgabe von 300 mg 14 C‑markiertem Darolutamid als Lösung zum Einnehmen ist Keto‑Darolutamid der einzige Hauptmetabolit mit einer ca. zweifach höheren Gesamtexposition im Plasma im Vergleich zu Darolutamid. Darolutamid und Keto‑Darolutamid sind zusammen für 87,4 % der 14 C‑Radioaktivität im Plasma verantwortlich, was darauf hindeutet, dass allen anderen Metaboliten nur eine untergeordnete Bedeutung zukommt.
Darolutamid wird hauptsächlich über einen oxidativen Stoffwechselweg abgebaut, der im Wesentlichen von CYP3A4 vermittelt wird, wie auch durch direkte Glucuronidierung, die vor allem durch UGT1A9 und UGT1A1 erfolgt. Darüber hinaus wurde gezeigt, dass hauptsächlich die AKR1C‑Isoformen die Reduktion von Keto‑Darolutamid zu den Wirkstoffdiastereomeren katalysieren.
Elimination
Die effektive Halbwertszeit von Darolutamid und Keto‑Darolutamid im Plasma von Patienten beträgt ca. 18 bis 20 Stunden. Von den beiden Diastereomeren, aus denen Darolutamid besteht, hat (S,R)‑Darolutamid mit 9 Stunden eine kürzere effektive Halbwertszeit als (S,S)‑Darolutamid mit 22 Stunden. Die Clearance von Darolutamid nach intravenöser Verabreichung betrug 116 ml/min (VK: 39,7 %). Insgesamt 63,4 % der wirkstoffbezogenen Stoffe werden mit dem Urin (ungefähr 7 % in unveränderter Form) und 32,4 % mit dem Stuhl ausgeschieden. Mehr als 95 % der Dosis wurden innerhalb von 7 Tagen nach der Verabreichung wiedergefunden.
Linearität/ Nicht‑Linearität
Im Dosisbereich von 100 bis 700 mg (nach Einzeldosis und im Steady‑State) steigt die Exposition gegenüber den beiden Diastereomeren und dem Hauptmetaboliten Keto-Darolutamid nahezu dosisproportional linear an. Aufgrund der gesättigten Resorption wurde bei 900 mg zweimal täglich kein weiterer Anstieg der Exposition gegenüber Darolutamid beobachtet.
Besondere Patientengruppen
Ältere Patienten
Bei Patienten zwischen 65 und 95 Jahren wurden keine klinisch relevanten Unterschiede in der Pharmakokinetik von Darolutamid beobachtet.
Nierenfunktionsstörung
In einer klinischen Studie zur Pharmakokinetik waren AUC und Cmax von Darolutamid bei Patienten mit schwerer Nierenfunktionsstörung (geschätzte glomeruläre Filtrationsrate [eGFR] 15 ‑ 29 ml/min/1,73 m2) im Vergleich zu gesunden Probanden um das 2,5‑ bzw. 1,6‑Fache erhöht.
Eine pharmakokinetische Populationsanalyse deutet darauf hin, dass die Exposition (AUC) gegenüber Darolutamid bei Patienten mit leichter, mäßiger und schwerer Nierenfunktionsstörung (eGFR 15 ‑ 89 ml/min/1,73 m2) im Vergleich zu Patienten mit normaler Nierenfunktion um das 1,1-, 1,3- bzw. ca. 1,5‑Fache erhöht ist.
Die Pharmakokinetik von Darolutamid wurde bei Patienten mit dialysepflichtiger terminaler Niereninsuffizienz (eGFR < 15 ml/min/1,73 m2) nicht untersucht.
Leberfunktionsstörung
In einer klinischen Studie zur Pharmakokinetik waren Cmax und AUC von Darolutamid bei Patienten mit mäßiger Leberfunktionsstörung (Child‑Pugh‑Klasse B) im Vergleich zu gesunden Probanden um das 1,5‑ bzw. 1,9‑Fache erhöht. Für Patienten mit schwerer Leberfunktionsstörung (Child‑Pugh‑Klasse C) liegen keine Daten vor.
Unterschiede in Bezug auf die ethnische Zugehörigkeit
Es wurden keine klinisch relevanten Unterschiede in der Pharmakokinetik von Darolutamid in Bezug auf die ethnische Zugehörigkeit (kaukasisch, japanisch, asiatisch außer japanisch, schwarz oder afroamerikanisch) beobachtet. Eine pharmakokinetische Populationsanalyse ergab bei japanischen Patienten in beiden Studien, ARAMIS und ARASENS, im Vergleich zu Patienten aus allen anderen Regionen einen geometrischen mittleren Anstieg der Exposition (AUC) bis auf das 1,56‑Fache (90 % KI: 1,43 bis 1,70).
Systemische Toxizität
In Studien zur Toxizität bei wiederholter Gabe an Ratten und Hunden waren die wichtigsten Ergebnisse Veränderungen der männlichen Reproduktionsorgane (Abnahme des Organgewichts mit Atrophie der Prostata und der Nebenhoden). Diese Effekte traten bei systemischen Expositionen auf, die im Bereich oder unterhalb der erwarteten humantherapeutischen Exposition liegen (auf der Basis von AUC‑Vergleichen). Zusätzliche Veränderungen im Reproduktionsgewebe umfassten einen minimalen Anstieg der Vakuolisierung in der Hypophyse, Atrophie und reduzierte Sekretion der Samenbläschen und Brustdrüsen bei Ratten sowie testikuläre Hypospermie, Dilatation und Degeneration der Hodenkanälchen bei Hunden. Die in den männlichen Reproduktionsorganen beider Tierspezies festgestellten Veränderungen standen im Einklang mit der pharmakologischen Aktivität von Darolutamid und hatten sich nach 4‑ bis 8‑wöchigen Erholungsphasen ganz oder teilweise zurückgebildet.
Embryotoxizität/ Teratogenität
Es wurden keine Studien zur Entwicklungstoxizität durchgeführt.
Reproduktionstoxizität
Es wurden keine Studien zur Reproduktionstoxizität durchgeführt. Basierend auf Studienergebnissen zur Toxizität bei wiederholter Gabe an Ratten und Hunden wird die männliche Fertilität wahrscheinlich beeinträchtigt. Dies steht im Einklang mit der pharmakologischen Aktivität von Darolutamid.
Genotoxizität und Karzinogenität
Darolutamid induzierte im mikrobiologischen Mutagenesetest (Ames‑Test) keine Mutationen. Bei hohen Konzentrationen induzierte Darolutamid in vitro strukturelle Chromosomenaberrationen bei kultivierten Humanlymphozyten. In vivo wurde jedoch im kombinierten Knochenmark-Mikronukleus-Test und Comet‑Assay in Leber und Duodenum der Ratte keine Genotoxizität bei Expositionen höher als die maximale Exposition beim Menschen beobachtet.
Bei oraler Gabe von Darolutamid über 6 Monate an männliche transgene rasH2-Mäuse zeigte sich bei Dosen von bis zu 1.000 mg/kg/Tag kein karzinogenes Potenzial. Diese Dosis entspricht für Darolutamid dem 0,9‑ bis 1,3‑Fachen bzw. für Keto-Darolutamid dem 2,1‑ bis 2,3‑Fachen der klinischen Exposition (AUC) unter der empfohlenen klinischen Tagesdosis von 1.200 mg/Tag. Ausgehend von dieser Studie kann ein karzinogenes Risiko von Darolutamid nicht vollständig ausgeschlossen werden.
Sicherheitspharmakologie
In vitro führte Darolutamid zu einer schwachen Hemmung des hERG-vermittelten Kaliumstroms und des L‑Typ-Calciumkanals. In vivo bewirkte Darolutamid bei narkotisierten Hunden eine geringfügige Verkürzung der QT‑Intervalldauer, dieser Effekt wurde jedoch bei wachen Hunden nicht festgestellt.
Tablettenkern
Calciumhydrogenphosphat (E 341)
Croscarmellose‑Natrium
Lactose‑Monohydrat
Magnesiumstearat (E 470b)
Povidon (E 1201)
Filmüberzug
Hypromellose
Lactose‑Monohydrat
Macrogol (E 1521)
Titandioxid (E 171)
Nicht zutreffend
3 Jahre
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
PVC/ Aluminiumfolie‑Blisterpackungen mit 16 Filmtabletten.
Jede Packung enthält 112 Filmtabletten.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Bayer AG
51368 Leverkusen
Deutschland
EU/1/20/1432/001 112 Filmtabletten
Datum der Erteilung der Zulassung: 27. März 2020
Datum der letzten Verlängerung der Zulassung: 24. Oktober 2024
Juli 2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
--------------------------------------------------------------------------------------------------------------------------
Verschreibungspflichtig
Bayer Vital GmbH
51368 Leverkusen
Tel.: +49 (0)214-30 513 48
Fax: +49 (0)214-2605 516 03
E-Mail: medical-information@bayer.com
DE/7