
Retsevmo® 40 mg Hartkapseln
Retsevmo® 80 mg Hartkapseln
Retsevmo® 40 mg Hartkapseln
Jede Hartkapsel enthält 40 mg Selpercatinib.
Retsevmo® 80 mg Hartkapseln
Jede Hartkapsel enthält 80 mg Selpercatinib.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Hartkapseln.
Retsevmo® 40 mg Hartkapseln
Graue blickdichte Kapsel, 6 x 18 mm (Größe 2), auf der „Lilly“, „3977“ und „40 mg“ in schwarzer Farbe aufgedruckt ist.
Retsevmo® 80 mg Hartkapseln
Blaue blickdichte Kapsel, 8 x 22 mm (Größe 0), auf der „Lilly“, „2980“ und „80 mg“ in schwarzer Farbe aufgedruckt ist.
Retsevmo als Monotherapie wird angewendet zur Behandlung von Erwachsenen mit:
̶ |
fortgeschrittenem RET-Fusions-positiven nicht-kleinzelligen Lungenkarzinom (NSCLC), die zuvor nicht mit einem RET-Inhibitor behandelt wurden. Zur biomarkerbasierten Patientenauswahl siehe Abschnitt 4.2. |
Retsevmo als Monotherapie wird angewendet zur Behandlung von Erwachsenen und Kindern ab 2 Jahren mit:
̶ |
fortgeschrittenem RET-Fusions-positiven Schilddrüsenkarzinom, das refraktär für radioaktives Iod ist (wenn radioaktives Iod angemessen ist). Zur biomarkerbasierten Patientensauswahl siehe Abschnitt 4.2. |
̶ |
fortgeschrittenem RET-mutierten medullären Schilddrüsenkarzinom (MTC). Zur biomarkerbasierten Patientenauswahl siehe Abschnitt 4.2. |
̶ |
fortgeschrittenen RET-Fusions-positiven soliden Tumoren, wenn Behandlungsoptionen, die nicht auf RET abzielen, nur begrenzten klinischen Nutzen bieten oder ausgeschöpft sind (siehe Abschnitte 4.4 und 5.1). Zur biomarkerbasierten Patientenauswahl siehe Abschnitt 4.2. |
Die Behandlung mit Retsevmo sollte von onkologisch erfahrenen Ärzten eingeleitet und überwacht werden.
Patientenauswahl
Vor Beginn der Behandlung mit Retsevmo ist das Vorliegen einer RET-Genmutation (MTC) oder Fusion (alle anderen Tumorarten) mittels eines CE-gekennzeichneten In vitro-Diagnostikums (IVD) mit entsprechendem Verwendungszweck zu bestimmen. Ist kein CE-gekennzeichnetes IVD verfügbar, sollte ein alternativer validierter Test verwendet werden.
Dosierung
Die empfohlene, gewichtsabhängige Dosis von Retsevmo für Patienten ab 12 Jahren ist:
̶ |
weniger als 50 kg: 120 mg zweimal täglich. |
̶ |
50 kg oder mehr: 160 mg zweimal täglich. |
Die empfohlene Dosis von Retsevmo für pädiatrische Patienten im Alter von 2 bis unter 12 Jahren richtet sich nach den folgenden Kategorien der Körperoberfläche (BSA) (Tabelle 1):
Tabelle 1 Empfohlene Dosis für pädiatrische Patienten im Alter von 2 bis unter 12 Jahren:
Körperoberfläche (BSA) |
Empfohlene Dosis |
0,45 bis 0,65 m2 |
40 mg dreimal täglich |
0,66 bis 1,08 m2 |
80 mg zweimal täglich |
1,09 bis 1,52 m2 |
120 mg zweimal täglich |
≥1,53 m2 |
160 mg zweimal täglich |
Retsevmo wird für pädiatrische Patienten mit einer Körperoberfläche von weniger als 0,45 m² nicht empfohlen.
Für pädiatrische Patienten, die keine Kapseln schlucken können, sind Tabletten erhältlich (siehe Fachinformation für Tabletten).
Wenn der Patient sich erbricht oder eine Dosis auslässt, sollte er angewiesen werden, die nächste Dosis wie ursprünglich geplant einzunehmen; eine zusätzliche Dosis soll nicht eingenommen werden.
Die Behandlung sollte bis zum Krankheitsprogress oder inakzeptabler Toxizität fortgesetzt werden.
Die jeweils vorgesehene Selpercatinib Dosis sollte um 50 % reduziert werden, wenn sie parallel mit einem starken CYP3A-Inhibitor verabreicht wird. Wenn der CYP3A-Inhibitor abgesetzt wird, sollte Selpercatinib auf die Dosis erhöht werden, die vor Einnahme des Inhibitors verwendet wurde (nach 3‑5 Halbwertszeiten des CYP3A-Inhibitors).
Dosisanpassungen
Bestimmte Nebenwirkungen können eine Dosisunterbrechung und/oder Dosisreduktion erforderlich machen. Die Retsevmo Dosisanpassungen sind in Tabelle 2 und Tabelle 3 zusammengefasst.
Tabelle 2 Empfohlene Dosisanpassungen von Retsevmo bei Nebenwirkungen in Abhängigkeit von Körpergewicht und Körperoberfläche (BSA)
Dosis- |
Erwachsene und Jugendliche ≥ 50 kg |
Erwachsene und Jugendliche < 50 kg |
Kinder und Jugendliche von |
Kinder und Jugendliche von |
Kinder und Jugendliche von |
Kinder und Jugendliche von |
Startdosis |
160 mg zweimal täglich oral |
120 mg zweimal täglich oral |
160 mg zweimal täglich oral |
120 mg zweimal täglich oral |
80 mg zweimal täglich oral |
40 mg dreimal täglich oral |
Erste Dosis- |
120 mg zweimal täglich oral |
80 mg zweimal täglich oral |
120 mg zweimal täglich oral |
80 mg zweimal täglich oral |
40 mg zweimal täglich oral |
40 mg zweimal täglich oral |
Zweite Dosis- |
80 mg zweimal täglich oral |
40 mg zweimal täglich oral |
80 mg zweimal täglich oral |
40 mg zweimal täglich oral |
40 mg einmal täglich oral |
40 mg einmal täglich oral |
Dritte Dosis- |
40 mg zweimal täglich oral |
nicht |
40 mg zweimal täglich oral |
40 mg einmal täglich oral |
dauerhaft |
dauerhaft |
* |
Bei Patienten, die drei Dosisreduktionen nicht tolerieren, ist Selpercatinib dauerhaft abzusetzen. |
Dosisanpassungen bei pädiatrischen Patienten aufgrund von Nebenwirkungen sind auf Grundlage der aktuellen Dosis und gemäß dem schrittweisen Dosisstufen-Anpassungsschema vorzunehmen, wie es für Erwachsene bei den nachfolgend beschriebenen Nebenwirkungen dargestellt ist (Tabelle 3), sofern nicht anders angegeben.
Tabelle 3 Empfohlene Dosisanpassungen bei Nebenwirkungen
Unerwünschte Arzneimittelwirkung (ADR) |
Dosisanpassung |
||
Erhöhte Alanin-Aminotransferase (ALT) oder Aspartat-Aminotransferase (AST) |
Grad 3 oder Grad 4 |
• |
Behandlung unterbrechen bis zum Rückgang der Toxizität auf den Ausgangswert (siehe Abschnitt 4.4 und 4.8). Wiedereinnahme mit einer um 2 Stufen reduzierten Dosis. |
• |
Wenn Selpercatinib mindestens 2 Wochen ohne wiederkehrende Erhöhung von ALT oder AST vertragen wurde, Erhöhung der Dosis um 1 Stufe. |
||
• |
Wenn Selpercatinib mindestens 4 Wochen ohne wiederkehrende Erhöhung vertragen wurde, Erhöhung der Dosis auf die Dosis, die vor dem Auftreten der Grad 3 oder Grad 4 AST- oder ALT-Erhöhung eingenommen wurde. |
||
• |
Dauerhaftes Absetzen von Selpercatinib, wenn ALT oder AST Erhöhung Grad 3 oder 4 wiederholt auftritt trotz Dosisanpassungen. |
||
Überempfindlichkeit |
Alle Grade |
• |
Behandlung unterbrechen und Beginn einer Corticosteroid-Gabe von 1 mg/kg bis zum Rückgang der Toxizität (siehe Abschnitt 4.4 und 4.8). Neustart der Selpercatinib-Gabe mit 40 mg zweimal täglich unter Weiterführung der begleitenden Steroid-Behandlung. Abbruch der Selpercatinib-Einnahme bei wiederkehrender Überempfindlichkeit. |
• |
Wenn Selpercatinib nach mindestens 7 Tagen ohne wiederkehrende Überempfindlichkeit vertragen wird, wird die Selpercatinib Dosis jede Woche schrittweise um 1 Dosis-Level erhöht, bis die Dosis erreicht ist, die vor Auftreten der Überempfindlichkeit eingenommen wurde. Ausschleichen der Steroid-Dosis, nachdem die Selpercatinib Ziel-Dosis für mindestens 7 Tage vertragen wurde. |
||
QT-Intervall-Verlängerung |
Grad 3 |
• |
Bei einem QTcF-Intervall von > 500 ms wird die Behandlung unterbrochen, bis das QTcF-Intervall < 470 ms (oder ≤ 440 ms für Patienten im Alter von unter 12 Jahren) ist oder zum Ausgangwert zurückkehrt (siehe Abschnitt 4.4). |
• |
Fortsetzen der Selpercatinib Behandlung mit der nächstniedrigeren Dosis. |
||
Grad 4 |
• |
Dauerhaftes Absetzen von Selpercatinib, wenn die QT-Verlängerung nach zwei Dosisreduktionen inakzeptabel bleibt oder der Patient Anzeichen oder Symptome einer schweren Arrhythmie zeigt. |
|
Hypertonie |
Grad 3 |
• |
Vor Beginn der Behandlung sollte der Blutdruck des Patienten kontrolliert sein. |
• |
Selpercatinib sollte bei klinisch relevanter Hypertonie vorübergehend abgesetzt werden, bis diese mit einer antihypertensiven Therapie kontrolliert ist. Wenn klinisch indiziert, kann die Selpercatinib-Behandlung mit der nächstniedrigeren Dosis wiederaufgenommen werden. (siehe Abschnitt 4.4 und 4.8). |
||
Grad 4 |
• |
Selpercatinib sollte dauerhaft abgesetzt werden, wenn der klinisch signifikante Bluthochdruck nicht kontrolliert werden kann. |
|
Hämorrhagische Ereignisse |
Grad 3 |
• |
Bis zur Wiederherstellung sollte die Selpercatinib-Behandlung unterbrochen werden. Wiedereinnahme mit einer reduzierten Dosis. |
• |
Bei erneutem Auftreten von Ereignissen 3. Grades nach einer Dosisanpassung muss Selpercatinib dauerhaft abgesetzt werden. |
||
Grad 4 |
• |
Selpercatinib dauerhaft absetzen. |
|
Interstitielle Lungenerkrankung (ILD)/Pneumonitis |
Grad 2 |
• |
Selpercatinib bis zum Abklingen aussetzen. |
• |
Wiedereinnahme mit einer reduzierten Dosis. |
||
• |
Selpercatinib bei rezidivierender ILD/Pneumonitis absetzen. |
||
Grad 3 oder Grad 4 |
• |
Selpercatinib absetzen. |
|
Andere Nebenwirkungen |
Grad 3 oder Grad 4 |
• |
Bis zur Wiederherstellung sollte die Selpercatinib-Behandlung unterbrochen werden. Wiedereinnahme mit einer reduzierten Dosis. |
• |
Bei erneutem Auftreten von Ereignissen 4. Grades nach einer Dosisanpassung muss Selpercatinib dauerhaft abgesetzt werden. |
||
Besondere Patientengruppen
Ältere Patienten
Dosisanpassungen aufgrund des Alters sind nicht erforderlich (siehe Abschnitt 5.2).
Es wurden keine relevanten Unterschiede bei den unerwünschten Ereignissen oder der Wirksamkeit von Selpercatinib zwischen ≥ 65-jährigen und jüngeren Patienten beobachtet. Bei den ≥ 75-jährigen Patienten sind nur begrenzt Daten verfügbar.
Eingeschränkte Nierenfunktion
Bei Patienten mit leichter, mittelschwerer oder schwerer Nierenfunktionseinschränkung ist keine Dosisanpassung erforderlich. Für Patienten mit terminaler Niereninsuffizienz oder für dialysepflichtige Patienten liegen keine Daten vor (Abschnitt 5.2).
Eingeschränkte Leberfunktion
Die engmaschige Überwachung von Patienten mit eingeschränkter Leberfunktion ist wichtig. Es ist keine Dosisanpassung für Patienten mit leichter (Child-Pugh Klasse A) oder moderater (Child-Pugh Klasse B) Leberfunktionseinschränkung erforderlich. Erwachsene mit schwerer Leberfunktionseinschränkung (Child-Pugh Klasse C) sind mit 80 mg Selpercatinib zweimal täglich zu behandeln (Abschnitt 5.2). Die Anwendung von Selpercatinib bei Patienten unter 18 Jahren mit eingeschränkter Leberfunktion wurde nicht untersucht. Bei Patienten unter 18 Jahren mit schwerer Leberfunktionsstörung ist die Behandlung mit Vorsicht einzuleiten und Dosisanpassungen sollten auf Grundlage der klinischen Beurteilung und der individuellen Verträglichkeit erfolgen. Für pädiatrische Patienten unter 12 Jahren mit schwerer Leberfunktionsstörung ist die empfohlene Anfangsdosis in Tabelle 4 angegeben.
Tabelle 4 Empfohlene Anfangsdosis für pädiatrische Patienten unter 12 Jahren mit schwerer Leberfunktionsstörung
Körperoberfläche |
Empfohlene Anfangsdosis |
0,45 bis 0,65 m2 |
40 mg einmal täglich |
0,66 bis 1,08 m2 |
40 mg zweimal täglich |
1,09 bis 1,52 m2 |
40 mg zweimal täglich |
≥1,53 m2 |
80 mg zweimal täglich |
Kinder und Jugendliche
Retsevmo sollte bei Kindern unter 2 Jahren nicht angewendet werden, da keine Daten verfügbar sind.
Retsevmo ist zur Behandlung von RET-mutierten medullären Schilddrüsenkarzinomen (MTC), RET-Fusions-positiven Schilddrüsenkarzinomen sowie RET-Fusions-positiven soliden Tumoren bei Patienten ab einem Alter von 2 Jahren vorgesehen (siehe Abschnitt 4.4 und 5.1).
Patienten ab 12 Jahren sind gewichtsabhängig zu behandeln (siehe Abschnitt 4.2). Patienten im Alter von 2 bis unter 12 Jahren sind nach Körperoberfläche zu dosieren (siehe Abschnitt 4.2). Basierend auf den Ergebnissen einer präklinischen Studie (siehe Abschnitt 5.3) müssen offene Wachstumsfugen bei jugendlichen Patienten überwacht werden. Eine Unterbrechung oder ein Absetzen der Therapie sollte auf Grundlage des Schweregrads jeglicher Anomalien der Wachstumsfugen und einer individuellen Nutzen-Risiko-Bewertung abgewogen werden.
Art der Anwendung
Retsevmo ist zum Einnehmen bestimmt.
Die Kapseln sollen als Ganzes geschluckt werden (Patienten sollen die Kapsel vor dem Schlucken nicht öffnen, zerbrechen oder kauen) und können mit oder ohne Nahrung eingenommen werden. Für pädiatrische Patienten, die nicht in der Lage sind, Kapseln zu schlucken, stehen Tabletten zur Verfügung (siehe Fachinformation für Tabletten).
Die Patienten sollten die Kapseln jeden Tag ungefähr zur gleichen Uhrzeit einnehmen.
Im Fall einer gleichzeitigen Therapie mit einem Protonenpumpen-Inhibitor muss Retsevmo mit Nahrung eingenommen werden (siehe Abschnitt 4.5).
Retsevmo sollte 2 Stunden vor oder 10 Stunden nach der Einnahme von H2-Rezeptor-Antagonisten verabreicht werden (siehe Abschnitt 4.5).
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Tumorübergreifende Wirksamkeit
Der Nutzen von Selpercatinib wurde in einarmigen Studien an einer relativ kleinen Stichprobe von Patienten, deren Tumore RET-Genfusionen aufwiesen, gezeigt. Positive Wirkungen von Selpercatinib wurden auf der Grundlage der objektiven Ansprechrate und Ansprechdauer bei einer begrenzten Anzahl von Tumorarten gezeigt. Die Wirkung kann abhängig von Tumorart sowie begleitenden genomischen Veränderungen unterschiedlich stark ausgeprägt sein (siehe Abschnitt 5.1). Daher sollte Selpercatinib nur angewendet werden, wenn es keine Behandlungsoptionen gibt, für die ein klinischer Nutzen gezeigt wurde, oder wenn diese Behandlungsoptionen ausgeschöpft wurden (d. h. bei nicht zufriedenstellenden Behandlungsoptionen).
Interstitielle Lungenerkrankung (ILD)/Pneumonitis
Schwere, lebensbedrohliche oder tödliche Fälle von ILD/Pneumonitis wurden bei Patienten berichtet, die mit Selpercatinib behandelt wurden (siehe Abschnitt 4.8). Die Patienten sollten auf pulmonale Symptome überwacht werden, die auf eine ILD/Pneumonitis hindeuten. Die Behandlung mit Selpercatinib sollte unterbrochen und die Patienten sollten unverzüglich untersucht werden, wenn sie akute oder sich verschlechternde respiratorische Symptome aufweisen, die auf eine ILD hindeuten können (z. B. Dyspnoe, Husten und Fieber). Die medizinisch notwendige Behandlung sollte entsprechend eingeleitet werden. Abhängig vom Schweregrad der ILD/Pneumonitis sollte die Selpercatinib-Dosis ausgesetzt, reduziert oder dauerhaft abgesetzt werden (siehe Abschnitt 4.2).
Erhöhte Alaninaminotransferasen (ALT) / Aspartataminotransferasen (AST)
Bei Selpercatinib-Patienten wurden Anstiege der ALT- und/oder AST-Werte auf Grad ≥ 3 beobachtet (siehe Abschnitt 4.8). ALT und AST sollten vor Beginn der Selpercatinib-Therapie überprüft werden, alle 2 Wochen während der ersten 3 Monate der Behandlung, monatlich für die nächsten 3 Monate der Behandlung und ansonsten wenn klinisch indiziert. Basierend auf der Höhe des ALT- oder AST-Anstiegs kann eine Selpercatinib Dosisanpassung erforderlich sein (siehe Abschnitt 4.2).
Hypertonie
Es wurde über Hypertonie bei Selpercatinib-Patienten berichtet (siehe Abschnitt 4.8). Der Blutdruck des Patienten sollte vor und während der Selpercatinib-Behandlung überwacht und je nach Notwendigkeit mit einer antihypertensiven Standardtherapie behandelt werden. Basierend auf der Erhöhung des Blutdrucks kann eine Selpercatinib-Dosisanpassung erforderlich sein (siehe Abschnitt 4.2). Wenn eine klinisch relevante Hypertonie nicht mit einer antihypertensiven Therapie kontrolliert werden kann, sollte Selpercatinib dauerhaft abgesetzt werden.
QT-Intervall-Verlängerung
Es wurde über QT-Intervall-Verlängerung bei Selpercatinib-Patienten berichtet (siehe Abschnitt 5.1). Selpercatinib sollte bei Patienten mit angeborenem oder erworbenem Long-QT-Syndrom oder anderen klinischen Erkrankungen, die Arrhythmien prädisponieren, vorsichtig eingesetzt werden. Bevor eine Selpercatinib-Therapie begonnen wird, sollten Patienten ein QTcF-Intervall von ≤ 470 ms (≤ 440 ms bei Patienten unter 12 Jahren) und Serum-Elektrolyte im Normbereich aufweisen. Elektrokardiogramme und Serum-Elektrolyte sollten bei allen Patienten überwacht werden: nach 1 Woche Selpercatinib-Therapie, mindestens monatlich für die ersten 6 Monate und anderenfalls, wie klinisch indiziert, angepasst an die Häufigkeit von Risikofaktoren wie Durchfall, Erbrechen und/oder Übelkeit. Hypokaliämie, Hypomagnesiämie und Hypokalzämie sollten vor der Einleitung und während der Behandlung von Selpercatinib korrigiert werden. Überwachen Sie das QT-Intervall mit Hilfe von EKGs häufiger bei Patienten, die eine Behandlung mit begleitenden Arzneimitteln benötigen, von denen bekannt ist, dass sie das QT-Intervall verlängern.
Möglicherweise sind Dosisunterbrechungen oder Anpassungen von Selpercatinib erforderlich (siehe Abschnitt 4.2).
Hypothyreose
Es wurde über Hypothyreose bei Selpercatinib-Patienten berichtet (siehe Abschnitt 4.8). Bei allen Patienten wird vor Therapiebeginn empfohlen, eine Laborkontrolle der Schilddrüsenfunktion durchzuführen. Patienten mit bereits bestehender Hypothyreose sollten vor Beginn der Selpercatinib-Behandlung gemäß der Standardtherapie behandelt werden. Alle Patienten sollten während der Behandlung mit Selpercatinib engmaschig auf Anzeichen und Symptome einer Schilddrüsenfunktionsstörung beobachtet werden. Die Schilddrüsenfunktion sollte während der Behandlung mit Selpercatinib in regelmäßigen Abständen überprüft werden. Patienten, die eine Schilddrüsenfunktionsstörung entwickeln, sollten gemäß der Standardtherapie behandelt werden. Gegebenenfalls sprechen Patienten jedoch nicht ausreichend auf die Substitution mit Levothyroxin (T4) an, da Selpercatinib die Umwandlung von Levothyroxin in Triiodthyronin (T3) hemmen kann und eine Ergänzung mit Triiodthyronin erforderlich sein kann (siehe Abschnitt 4.5).
Starke CYP3A4-Induktoren
Die gleichzeitige Anwendung von starken CYP3A4-Induktoren sollte aufgrund des Risikos einer verminderten Wirksamkeit von Selpercatinib vermieden werden (siehe Abschnitt 4.5).
Frauen im gebärfähigen Alter / Kontrazeption bei Frauen und Männern
Frauen im gebärfähigen Alter müssen während der Behandung und für mindestens eine Woche nach der letzten Gabe von Selpercatinib eine sehr zuverlässige Methode zur Empfängnisverhütung anwenden. Männer mit Partnerinnen im gebärfähigen Alter sollten eine zuverlässige Methode zur Empfängnisverhütung während und für mindestens eine Woche nach der letzten Gabe von Selpercatinib anwenden (siehe Abschnitt 4.6).
Fertilität
Präklinische Sicherheitserkenntnisse weisen darauf hin, dass die männliche und weibliche Fertilität durch die Behandlung mit Retsevmo beeinträchtigt werden kann (siehe Abschnitt 4.6 und 5.3). Sowohl Männer als auch Frauen sollten sich vor der Behandlung Rat bezüglich des Erhalts der Fertilität einholen.
Überempfindlichkeit
Es wurde über das Auftreten von Überempfindlichkeitsreaktionen bei Selpercatinib-Patienten berichtet, wobei die Mehrheit der Fälle bei NSCLC-Patienten mit einer vorangegangenen Anti-PD-1/PD-L1-Immuntherapie beobachtet wurde (siehe Abschnitt 4.8). Anzeichen und Symptome einer Überempfindlichkeit beinhalteten Fieber, Ausschlag und Gelenk- oder Muskelschmerzen mit gleichzeitig verminderten Blutplättchen oder erhöhten Aminotransferasen.
Wenn Überempfindlichkeit auftritt, soll die Selpercatinib-Gabe unterbrochen und eine Steroid-Behandlung begonnen werden. Basierend auf der Schwere der Überempfindlichkeitsreaktion kann eine Dosisanpassung von Selpercatinib erforderlich sein (siehe Abschnitt 4.2). Die Steroid-Behandlung sollte weitergeführt werden, bis der Patient die Dosis erreicht hat, die vor Auftreten der Überempfindlichkeit eingenommen wurde, und dann ausgeschlichen werden. Selpercatinib soll bei wiederkehrender Überempfindlichkeit dauerhaft abgesetzt werden.
Hämorrhagien
Es wurde über schwere einschließlich tödlicher hämorrhagischer Ereignisse bei Selpercatinib-Patienten berichtet (siehe Abschnitt 4.8).
Selpercatinib muss bei Patienten mit lebensbedrohlichen oder wiederkehrenden schweren Hämorrhagien dauerhaft abgesetzt werden (siehe Abschnitt 4.2).
Tumorlysesyndrom (TLS)
Unter Selpercatinib-Behandlung wurden Fälle von TLS beobachtet. Risikofaktoren für TLS sind eine hohe Tumorlast, eine vorbestehende chronische Niereninsuffizienz, Oligurie, Dehydratation, Hypotonie und saurer Urin. Diese Patienten sollten engmaschig überwacht und wie klinisch indiziert behandelt werden, und eine geeignete Prophylaxe einschließlich Flüssigkeitszufuhr sollte in Betracht gezogen werden.
Epiphysenlösung des Femurkopfes bei pädiatrischen Patienten
Bei pädiatrischen Patienten (< 18 Jahre), die Selpercatinib erhielten, wurde über eine Epiphysenlösung des Femurkopfes berichtet (siehe Abschnitt 4.8). Die Patienten sollten auf Symptome überwacht werden, die auf eine Epiphysenlösung des Femurkopfes hinweisen, und entsprechend medizinisch sowie chirurgisch angemessen behandelt werden.
Schwere arzneimittelinduzierte Hautreaktionen (SCARs)
Das Stevens-Johnson-Syndrom (SJS), das lebensbedrohlich oder tödlich verlaufen kann, wurde in Zusammenhang mit der Behandlung mit Selpercatinib berichtet (siehe Abschnitt 4.8). Patienten sind über die Anzeichen schwerer arzneimittelinduzierter Hautreaktionen zu informieren und sollen beim Auftreten entsprechender Anzeichen oder Symptome unverzüglich ärztlichen Rat einholen. Treten Anzeichen auf, die auf eine solche Reaktion hindeuten, ist die Behandlung mit Selpercatinib sofort zu beenden und gegebenenfalls eine alternative Therapie in Erwägung zu ziehen. Entwickelt ein Patient unter Selpercatinib eine schwere Hautreaktion wie SJS, darf die Behandlung mit Selpercatinib zu keinem Zeitpunkt wieder aufgenommen werden.
Auswirkungen anderer Arzneimittel auf die Pharmakokinetik von Selpercatinib
Selpercatinib wird über CYP3A4 metabolisiert. Deshalb können Arzneimittel, die die CYP3A4-Enzymaktivität beeinflussen, die Pharmakokinetik von Selpercatinib verändern.
Selpercatinib ist in vitro ein Substrat des P-Glykoproteins (P-gp) und des Breast Cancer Resistance Proteins (BCRP), jedoch scheinen diese Transporter die orale Absorption von Selpercatinib nicht einzuschränken, da seine orale Bioverfügbarkeit 73 % beträgt und seine Exposition durch die parallele Verabreichung des P-gp-Inhibitors Rifampicin nur minimal erhöht wurde (Erhöhung der Selpercatinib AUC0 - 24 um etwa 6,5 % und des Cmax-Wertes um 19 %).
Wirkstoffe, die die Selpercatinib-Plasmakonzentrationen erhöhen können
Die parallele Verabreichung einer Einzeldosis 160 mg Selpercatinib mit Itraconazol, einem starken CYP3A-Inhibitor, erhöhte Cmax um 30 % und die AUC von Selpercatinib um 130 %, verglichen mit der alleinigen Selpercatinib-Gabe. Wenn starke CYP3A- und/oder P-gp-Inhibitoren parallel verabreicht werden müssen, einschließlich, aber nicht beschränkt auf, Ketoconazol, Itraconazol, Voriconazol, Ritonavir, Saquinavir, Telithromycin, Posaconazol und Nefazodon, sollte die Selpercatinib-Dosis reduziert werden (siehe Abschnitt 4.2).
Wirkstoffe, die die Selpercatinib-Plasmakonzentrationen vermindern können
Die parallele Verabreichung von Rifampicin, einem starken CYP3A4-Induktor, führte zu einem Rückgang der Selpercatinib-AUC um etwa 87 % und Cmax um etwa 70 % im Vergleich zur alleinigen Gabe von Selpercatinib. Deshalb sollte der gleichzeitige Einsatz von starken CYP3A4-Induktoren, einschließlich, aber nicht beschränkt auf, Carbamazepin, Phenobarbital, Phenytoin, Rifabutin, Rifampicin und Johanniskraut (Hypericum perforatum), vermieden werden.
Auswirkungen von Selpercatinib auf die Pharmakokinetik anderer Arzneimittel (Anstieg der Plasmakonzentration)
Empfindliche CYP2C8-Substrate
Selpercatinib erhöhte bei Repaglinid (einem CYP2C8-Substrat) die Cmax um etwa 91 % und die AUC um etwa 188 %. Daher sollte eine parallele Verabreichung mit empfindlichen CYP2C8-Substraten (z. B. Amodiaquin, Cerivastatin, Enzalutamid, Paclitaxel, Repaglinid, Torasemid, Sorafenib, Rosiglitazon, Buprenorphin, Selexipag, Dasabuvir and Montelukast) vermieden werden.
Empfindliche CYP3A4-Substrate
Selpercatinib erhöhte bei Midazolam (einem CYP3A4 Substrat) die Cmax um etwa 39 % und die AUC um etwa 54 %. Daher sollte eine gleichzeitige Anwendung mit empfindlichen CYP3A4-Substraten (z. B. Alfentanil, Avanafil, Buspiron, Conivaptan, Darifenacin, Darunavir, Ebastin, Lomitapid, Lovastatin, Midazolam, Naloxegol, Nisoldipin, Saquinavir, Simvastatin, Tipranavir, Triazolam, Vardenafil) vermieden werden.
Gleichzeitige Gabe von Arzneimitteln, die den Magen-pH beeinflussen
Selpercatinib weist eine pH-abhängige Löslichkeit auf mit geringerer Löslichkeit bei höheren pH-Werten. Bei Gabe mehrfacher täglicher Ranitidin-Dosen (H2-Rezeptor-Antagonist), die 2 Stunden nach der Selpercatinib-Dosis gegeben wurden, wurden keine klinisch signifikanten Unterschiede der Selpercatinib-Pharmakokinetik beobachtet.
Gleichzeitige Gabe von Protonenpumpen-Inhibitoren
Die gleichzeitige Gabe von mehrfachen täglichen Omeprazol-Dosen (einem Protonenpumpen-Inhibitor) verringerte die Selpercatinib AUC0 - INF und Cmax, wenn Selpercatinib auf nüchternen Magen eingenommen wurde. Die parallele Gabe von mehrfachen täglichen Omeprazol-Dosen hat die Selpercatinib AUC0 - INF und Cmax, bei Einnahme von Retsevmo mit Nahrung nicht wesentlich verändert.
Gleichzeitige Gabe von Arzneimitteln, die Transporter-Substrate sind
Selpercatinib inhibiert den renalen Transporter MATE1 (multidrug and toxin extrusion protein 1). In vivo können Selpercatinib-Interaktionen mit klinisch relevanten MATE1-Substraten, wie zum Beispiel Kreatinin, auftreten (siehe Abschnitt 5.2).
Selpercatinib ist ein in vitro Inhibitor von P-gp und BCRP. In vivo erhöhte Selpercatinib die Cmax und AUC von Dabigatran, einem P-gp-Substrat, um 43 % bzw. 38 %. Daher sollte bei Einnahme eines ausgeprägten P-gp-Substrates (z. B. Fexofenadin, Dabigatranetexilat, Colchicin, Saxagliptin), insbesondere bei solchen mit einer geringen therapeutischen Breite (z. B. Digoxin), Vorsicht geboten sein (siehe Abschnitt 5.2).
In vivo erhöhte Selpercatinib die Cmax und die AUC von Rosuvastatin, einem BCRP‑Substrat, um 71 % bzw. 80 %. Bei gleichzeitiger Anwendung eines BCRP‑Substrats (z. B. Rosuvastatin, Prazosin) ist Vorsicht geboten (siehe Abschnitt 5.2).
Arzneimittel, die bei gleichzeitiger Selpercatinib-Gabe weniger wirksam sein können
Selpercatinib könnte die D2-Deiodinase hemmen und dadurch die Umwandlung von Levothyroxin (T4) in Triiodthyronin (T3) verringern. Die Patienten könnten daher nicht ausreichend auf die Substitution mit Levothyroxin ansprechen und eine Ergänzung mit Triiodthyronin kann erforderlich sein (siehe Abschnitt 4.4).
Kinder und Jugendliche
Interaktionsstudien wurden nur bei Erwachsenen durchgeführt.
Frauen im gebärfähigen Alter/Kontrazeption bei Frauen und Männern
Frauen im gebärfähigen Alter müssen während der Behandlung und für mindestens eine Woche nach der letzten Selpercatinib-Dosis eine hochwirksame Verhütungsmethode anwenden. Männer mit Partnerinnen im gebärfähigen Alter sollen eine effektive Kontrazeption während der Behandlung und für mindestens eine Woche nach der letzten Selpercatinib-Dosis anwenden.
Schwangerschaft
Es liegen keine Daten zur Anwendung von Selpercatinib bei Schwangeren vor. Tierstudien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Der Einsatz von Retsevmo in der Schwangerschaft und bei gebärfähigen Frauen ohne Anwendung eines Verhütungsmittels wird nicht empfohlen. Es darf während der Schwangerschaft nur angewendet werden, wenn der potentielle Nutzen das potentielle Risiko für den Fötus rechtfertigt.
Stillzeit
Es ist nicht bekannt, ob Selpercatinib in die Muttermilch übergeht. Ein Risiko für gestillte Neugeborene/Kleinkinder kann nicht ausgeschlossen werden. Das Stillen sollte während der Behandlung mit Retsevmo und für mindestens eine Woche nach der letzten Dosis eingestellt werden.
Fertilität
Beim Menschen sind keine Daten zum Effekt von Selpercatinib auf die Fertilität verfügbar. Basierend auf Erkenntnissen von Tierstudien kann die männliche bzw. weibliche Fertilität durch die Behandlung mit Retsevmo beeinträchtigt werden (siehe Abschnitt 5.3). Sowohl Männer als auch Frauen sollten sich vor der Behandlung Rat über den Erhalt der Fertilität einholen.
Retsevmo kann geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen haben. Patienten sollten angewiesen werden, beim Steuern eines Fahrzeugs oder beim Bedienen von Maschinen vorsichtig zu sein, falls während der Behandlung mit Retsevmo Müdigkeit oder Schwindel auftreten (siehe Abschnitt 4.8).
Zusammenfassung des Sicherheitsprofils
Die integrierte Häufigkeit von Nebenwirkungen, die bei Patienten unter Selpercatinib Behandlung berichtet wurden, aus einer offenen, multizentrischen Phase 1/2-Studie mit Dosissteigerung (LIBRETTO-001) und aus zwei offenen, multizentrischen, randomisierten Phase-3-Vergleichsstudien (LIBRETTO-431 und LIBRETTO-531) sind zusammengefasst. Die häufigsten (≥ 1,0 %) schwerwiegenden Nebenwirkungen sind Pneumonie (5,3 %), Hämorrhagie (2,4 %), Bauchschmerzen (2,1 %), vermindertes Natrium im Blut (2,0 %), Diarrhö (1,5 %), Überempfindlichkeit (1,4 %), Erbrechen (1,3 %), erhöhtes Kreatinin im Blut (1,3 %), Fieber (1,3 %), Harnwegsinfektionen (1,3 %), erhöhte ALT (1,0 %) und erhöhte AST (1,0 %).
8,8 % der Patienten brachen die Retsevmo-Behandlung wegen während der Behandlung aufgetretener unerwünschter Ereignisse (unabhängig vom Kausalzusammenhang) während der Studie dauerhaft ab. Die häufigsten Nebenwirkungen, die zu einem dauerhaften Absetzen führten (3 oder mehr Patienten), waren erhöhte ALT-Werte (0,7 %), Fatigue (0,5 %), erhöhte AST-Werte (0,4 %), erhöhtes Bilirubin im Blut (0,3 %), Pneumonie (0,3 %), Thrombozytopenie (0,3 %), Hämorrhagie (0,3 %) und Überempfindlichkeit (0,3 %).
Tabellarische Auflistung der Nebenwirkungen
Die integrierte Häufigkeit und der Schweregrad der Nebenwirkungen, die bei Patienten berichtet wurden, die in den Studien LIBRETTO-001, LIBRETTO-431 und LIBRETTO-531 mit Selpercatinib behandelt wurden, sind in Tabelle 5 dargestellt. Die Nebenwirkungen sind gemäß der MedDRA-Systemorganklassen und Häufigkeit klassifiziert.
Häufigkeitsgruppen werden durch die folgende Konvention definiert: sehr häufig (≥ 1/10); häufig (≥ 1/100 bis < 1/10); gelegentlich (≥ 1/1 000 bis < 1/100); selten (≥ 1/10 000 bis < 1/1 000); sehr selten (< 1/10 000), und nicht bekannt (kann aus verfügbaren Daten nicht abgeschätzt werden).
Die mediane Behandlungszeit mit Selpercatinib betrug 30,09 Monate (Studie LIBRETTO-001), 16,7 Monate (Studie LIBRETTO-431) und 14,9 Monate (Studie LIBRETTO-531).
Tabelle 5 Nebenwirkungen bei Patienten mit Selpercatinib (N = 1188)
MedDRA Systemorganklasse |
MedDRA |
Häufigkeit aller Grade |
Häufigkeit von Grad ≥ 3 |
Infektionen und parasitäre Erkrankungen |
Harnwegsinfektionen a |
Sehr häufig |
Häufig |
Pneumonie b |
Sehr häufig |
Häufig |
|
Erkrankungen des Immunsystems c |
Überempfindlichkeit d |
Häufig |
Häufig |
Endokrine Erkrankungen |
Hypothyreose |
Sehr häufig |
- |
Stoffwechsel- und Ernährungsstörungen |
Verminderter Appetit |
Sehr häufig |
Gelegentlich |
Erkrankungen des Nervensystems |
Kopfschmerzen e |
Sehr häufig |
Häufig |
Schwindelgefühl f |
Sehr häufig |
Gelegentlich |
|
Herzerkrankungen |
EKG QT-Intervall-Verlängerung g |
Sehr häufig |
Häufig |
Gefäßerkrankungen |
Hypertonie h |
Sehr häufig |
Sehr häufig |
Hämorrhagie i |
Sehr häufig |
Häufig |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Interstitielle Lungenerkrankung/ |
Häufig |
Gelegentlich |
Chylothorax |
Häufig |
Gelegentlich |
|
Erkrankungen des Gastrointestinaltrakts |
Diarrhö k |
Sehr häufig |
Häufig |
Mundtrockenheit l |
Sehr häufig |
Gelegentlich |
|
Bauchschmerzen m |
Sehr häufig |
Häufig |
|
Obstipation |
Sehr häufig |
Gelegentlich |
|
Übelkeit |
Sehr häufig |
Häufig |
|
Erbrechen n |
Sehr häufig |
Häufig |
|
Stomatitis o |
Sehr häufig |
Gelegentlich |
|
Chylöser Aszites p |
Häufig |
Gelegentlich |
|
Erkrankungen der Haut und des Unterhautgewebes |
Ausschlag q |
Sehr häufig |
Häufig |
Stevens-Johnson-Syndrom r |
Nicht bekannt |
Nicht bekannt |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Epiphysenlösung des |
Häufig |
Häufig |
Erkrankungen des Fortpflanzungssystems und der Brust |
Erektile Dysfunktion t |
Sehr häufig |
Gelegentlich |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Ödeme u |
Sehr häufig |
Häufig |
Fatigue v |
Sehr häufig |
Häufig |
|
Fieber |
Sehr häufig |
Gelegentlich |
|
Untersuchungen w |
AST erhöht |
Sehr häufig |
Sehr häufig |
ALT erhöht |
Sehr häufig |
Sehr häufig |
|
Calcium erniedrigt |
Sehr häufig |
Häufig |
|
Lymphozytenzahl erniedrigt |
Sehr häufig |
Sehr häufig |
|
Anzahl weißer Blutkörperchen erniedrigt |
Sehr häufig |
Häufig |
|
Albumin erniedrigt |
Sehr häufig |
Häufig |
|
Kreatinin erhöht |
Sehr häufig |
Häufig |
|
Natrium erniedrigt |
Sehr häufig |
Sehr häufig |
|
Alkalische Phosphatase erhöht |
Sehr häufig |
Häufig |
|
Blutplättchen erniedrigt |
Sehr häufig |
Häufig |
|
Gesamtbilirubin erhöht |
Sehr häufig |
Häufig |
|
Neutrophilenzahl erniedrigt |
Sehr häufig |
Häufig |
|
Hämoglobin erniedrigt |
Sehr häufig |
Häufig |
|
Magnesium erniedrigt |
Sehr häufig |
Häufig |
|
Kalium erniedrigt |
Sehr häufig |
Häufig |
a |
Harnwegsinfektionen umfassen Harnwegsinfektionen, Blasenentzündungen, Urosepsis, Escherichia-Harnwegsinfektionen, Escherichia-Pyelonephritis, Niereninfektionen, Nitrit im Urin, Pyelonephritis, Urethritis, bakterielle Harnwegsinfektionen und urogenitale Pilzinfektionen. |
b |
Pneumonie umfasst Lungenentzündung, Lungeninfektionen, Aspirationspneumonie, Empyem, Lungenkonsolidierung, Pleurainfektion, bakterielle Lungenentzündung, Lungenentzündung durch Staphylokokken, atypische Lungenentzündung, Lungenabszess, Lungenentzündung durch Pneumocystis-jirovecii, Lungenentzündung durch Pneumokokken, respiratorische synzytiale virale Lungenentzündung, infektiöse Pleuritis mit Erguss und virale Pneumonie. |
c |
Überempfindlichkeitsreaktionen waren durch einen makulopapulösen Ausschlag gekennzeichnet, häufig mit vorangegangenem Fieber und assoziierten Gelenk- bzw. Muskelschmerzen während des ersten Behandlungszyklus des Patienten (typischerweise zwischen Tag 7 und 21). |
d |
Überempfindlichkeit umfasst Arzneimittel-Überempfindlichkeit und Überempfindlichkeit. |
e |
Kopfschmerzen umfasst Kopfschmerzen, Sinus-Kopfschmerzen und Spannungs-Kopfschmerzen. |
f |
Schwindelgefühl umfasst Schwindel, Vertigo, Präsynkope und posturaler Schwindel. |
g |
EKG QT-Intervall-Verlängerung umfasst EKG QT-Intervall-Verlängerung und anormales EKG QT-Intervall. |
h |
Hypertonie umfasst Hypertonie und erhöhten Blutdruck. |
i |
Hämorrhagie umfasst Epistaxis, Hämoptyse, Prellung, Hämaturie, rektale Blutung, Vaginalblutung, Hirnblutung, traumatisches Hämatom, Blut im Urin, konjunktivale Blutung, Ekchymose, Zahnfleischblutung, Hämatochezie, Petechien, Blutblase, spontane Hämatome, Abdominalwand-Hämatom, Analblutung, Angina bullosa haemorrhagica, disseminierte intravaskuläre Koagulation, Augenblutung, Magenblutung, gastrointestinale Blutung, intrakranielle Blutung, subkutane Blutung, haemorrhoidale Blutung, Leber-Hämatom, intraabdominale Blutung, Mundblutung, Speiseröhrenblutung, Beckenhämatom, periorbitales Hämatom, periorbitale Blutung, Rachenblutung, Lungenkontusion, Purpura, retroperitoneales Hämatom, Hautblutung, Subarachnoidalblutung, Divertikelblutung, Augenhämatom, Hämatemesis, Hämorrhagie, hämorrhagischer Schlaganfall, Leberblutung, Larynxblutung, Blutung im unteren Gastrointestinaltrakt, Meläna, Menorrhagie, okkultes Blut positiver Test, postprozedurale Blutung, postmenopausale Blutung, Netzhautblutung, Skleralblutung, subdurale Blutung, traumatischer Hämothorax, Tumorblutung, Blutung im oberen Gastrointestinaltrakt, Uterusblutung, Hämatom an der Gefäßpunktionsstelle, Hämarthrose und Hämatome. |
j |
Interstitielle Lungenerkrankung/Pneumonitis umfasst interstitielle Lungenerkrankung, Pneumonitis, Strahlenpneumonitis, restriktive Lungenerkrankung, akutes Atemnotsyndrom, Alveolitis, Bronchiolitis, Langerhans-Zell-Histiozytose, Strahlenschädigung der Lunge, zystische Lungenerkrankung, Lungeninfiltration und Opazität in der Lunge. |
k |
Diarrhö umfasst Diarrhö, Analinkontinenz, Stuhldrang, häufiger Stuhlgang und gastrointestinale Hypermotilität. |
l |
Mundtrockenheit umfasst Mundtrockenheit und trockene Mundschleimhaut. |
m |
Bauchschmerzen umfasst Bauchschmerzen, Oberbauchschmerzen, Magen-Darm-Beschwerden, Unterbauchschmerzen und Magen-Darm-Schmerzen. |
n |
Erbrechen umfasst erbrechen, Würgen und Aufstoßen. |
o |
Stomatitis umfasst Stomatitis, Geschwüre im Mund, Schleimhautentzündungen und Blasenbildung an der Mundschleimhaut. |
p |
Chylöser Aszites umfasst chylöser Aszites und Aszites chylös (MedDRA LLTs). |
q |
Ausschlag umfasst Ausschlag, makulopapulöser Ausschlag, Dermatitis, Exfoliation der Haut, makulöser Ausschlag, erythematöser Ausschlag, Urtikaria, allergische Dermatitis, exfoliativer Ausschlag, papulöser Ausschlag, morbilliformer Ausschlag, juckender Ausschlag, vesikulärer Ausschlag, Schmetterlingsausschlag, follikulärer Ausschlag, generalisierter Ausschlag, pustulöser Ausschlag und Hautreaktion. |
r |
Aus Daten nach der Markteinführung. |
s |
Eine Epiphysenlösung des Femurkopfes wurde bei pädiatrischen Patienten (< 18 Jahre), die mit Selpercatinib behandelt wurden (n = 47), häufig beobachtet (6,4 %). |
t |
Erektile Dysfunktion wurde in klinischen Studien bei männlichen Patienten, die mit Selpercatinib behandelt wurden (n = 986), sehr häufig (12,4 %) beobachtet. |
u |
Ödeme umfasst periphere Ödeme, Gesichtsödeme, periorbitale Ödeme, Gesichtsschwellungen, lokalisierte Ödeme, periphere Schwellungen, generalisierte Ödeme, Augenlidödeme, Augenschwellungen, Lymphödeme, Ödeme im Genitalbereich, Schwellung des Hodensacks, Angioödeme, Augenödeme, Ödeme, Ödeme im Hodensack, Hautödeme, Schwellungen, orbitale Ödeme, Hodenschwellung, vulvovaginale Schwellung, Orbitalschwellung, Penisödem, Schwellung um die Augenhöhle und Schwellung des Augenlids |
v |
Fatigue beinhaltet Fatigue, Asthenie und Unwohlsein (Malaise). |
w |
Basierend auf Labormessungen. Der Prozentsatz wird basierend auf der Anzahl der Patienten mit einem Ausgangswert und mindestens einem nachfolgenden Wert im Nenner berechnet. |
Beschreibung ausgewählter Nebenwirkungen bei Patienten, die Selpercatinib erhalten
Aminotransferase-Erhöhungen (AST/ALT erhöht)
Aufgrund von Labormessungen wurden ALT-Erhöhungen bei 59,4 % und AST-Erhöhungen bei 61 % der Patienten berichtet. ALT- oder AST-Erhöhungen Grad 3 oder 4 wurden bei 14,1 % bzw. 9,5 % der Patienten berichtet.
Die mediane Zeit bis zum ersten Auftreten betrug in LIBRETTO-001 für den AST-Anstieg 4,7 Wochen (Spanne: 0,7; 227,9) und für den ALT-Anstieg 4,4 Wochen (Spanne: 0,9; 186,1). In LIBRETTO-431 betrug sie für den AST-Anstieg 5,1 Wochen (Spanne: 0,7; 88,1) und für den ALT-Anstieg 5,1 Wochen (Spanne: 0,7; 110,9). In LIBRETTO-531 betrug sie für den AST-Anstieg 6,1 Wochen (Spanne: 0,1; 85,1) und für den ALT-Anstieg 6,1 Wochen (Spanne: 0,1; 85,1).
Eine Dosisanpassung wird für Patienten empfohlen, die eine ALT- oder AST-Erhöhung mit dem Schweregrad 3 oder 4 entwickeln (siehe Abschnitt 4.2).
QT-Intervall-Verlängerung
Bei den 837 Patienten in der Studie LIBRETTO-001, bei denen ein EKG durchgeführt wurde, zeigte die Überprüfung der Daten, dass bei 8,1 % der Patienten der maximale QTcF-Wert im Verlauf der Studie mehr als > 500 ms betrug, und 21,6 % der Patienten hatten eine maximale Verlängerung des QTcF-Intervalls um > 60 ms zum Ausgangswert. Von den 156 Patienten in LIBRETTO‑431, bei denen ein EKG durchgeführt wurde, betrug bei 5,1 % der Patienten der maximale QTcF-Wert im Verlauf der Studie mehr als > 500 ms, und 16,7 % der Patienten hatten eine maximale Verlängerung der QTcF-Intervalle um > 60 ms zum Ausgangswert. Von den 191 Patienten in LIBRETTO-531, bei denen ein EKG durchgeführt wurde, betrug bei 3,7 % der Patienten der maximale QTcF-Wert im Verlauf der Studie mehr als > 500 ms und 17,8 % der Patienten hatten eine maximale Verlängerung der QTcF-Intervalle um > 60 ms zum Ausgangswert.
In den Studien LIBRETTO-001, LIBRETTO-431 und LIBRETTO-531 gab es keine Berichte über Torsades de Pointes, Ereignisse des Grades ≥ 3 oder klinisch signifikante behandlungsbedingte Arrhythmien, ventrikuläre Tachykardie, Kammerflimmern oder Kammerflattern. Fatale Ereignisse wie plötzlicher Tod und Herzstillstand wurden bei Patienten mit signifikanter kardialer Vorgeschichte berichtet. In allen Studien brachen insgesamt zwei Patienten (0,2 %) die Behandlung mit Selpercatinib aufgrund einer QT-Verlängerung ab. Für Retsevmo ist möglicherweise eine Dosisunterbrechung oder -änderung erforderlich (siehe Abschnitte 4.2 und 4.4).
Hypertonie
Bei den 837 Patienten in der Studie LIBRETTO-001, bei denen Blutdruckmessungen durchgeführt wurden, betrug der mediane maximale Anstieg gegenüber dem systolischen Ausgangs-Blutdruck 32 mm Hg (Spanne: -15; + 100). Die Ergebnisse des diastolischen Blutdrucks waren ähnlich, aber die Anstiege waren von geringerem Ausmaß. In LIBRETTO-001 blieb der Ausgangs-Schweregrad während der Behandlung bei nur 10,3 % der Patienten unverändert, 40,7 % hatten eine Verschlechterung um einen Grad, 38,5 % um zwei Grade und 9,8 % um drei Grade. Ein während der Behandlung aufgetretenes unerwünschtes Ereignis einer Hypertonie wurde bei 44,8 % der Patienten mit hypertoner Vorgeschichte (28,2 % mit Grad 3 oder 4) und bei 41,7 % der Patienten ohne hypertone Vorgeschichte (14,1 % mit Grad 3 oder 4) berichtet.
Von den 154 mit Selpercatinib behandelten Patienten, bei denen der Blutdruck in LIBRETTO-431 gemessen wurde, blieb bei 23,4 % der Schweregrad gegenüber dem Ausgangswert unverändert, 49,4 % hatten eine Verschlechterung um einen Grad, 22,7 % hatten eine Verschlechterung um zwei Grade und 3,3 % hatten eine Verschlechterung um drei Grade.
Von den 192 mit Selpercatinib behandelten Patienten, bei denen der Blutdruck in LIBRETTO-531 gemessen wurde, blieb bei 20,8 % der Schweregrad gegenüber dem Ausgangswert unverändert, 43,8 % hatten eine Verschlechterung um einen Grad, 27,6 % hatten eine Verschlechterung um zwei Grade und 6,8 % hatten eine Verschlechterung um drei Grade.
Insgesamt wiesen in LIBRETTO-001 19,8 % der Patienten, in LIBRETTO-431 20,3 % der Patienten und in LIBRETTO-531 19,2 % der Patienten eine behandlungsbedingte Hypertonie Grad 3 (definiert als maximaler systolischer Blutdruck von mehr als 160 mm Hg) auf. Eine während der Behandlung aufgetretene Hypertonie vom Grad 4 wurde bei 0,1 % der Patienten in LIBRETTO-001 und bei keinem Patienten in LIBRETTO-431 und LIBRETTO-531 berichtet.
Zwei Patienten (0,2 %) brachen in LIBRETTO-001 die Behandlung aufgrund von Bluthochdruck dauerhaft ab, in LIBRETTO-431 und LIBRETTO-531 war es kein Patient. Bei Patienten, die eine Hypertonie entwickeln, wird eine Dosisanpassung empfohlen (siehe Abschnitt 4.2). Selpercatinib sollte dauerhaft abgesetzt werden, wenn eine klinisch signifikante Hypertonie mit einer antihypertensiven Therapie nicht kontrolliert werden kann (siehe Abschnitt 4.4).
Überempfindlichkeit
Anzeichen und Symptome einer Überempfindlichkeit beinhalteten Fieber, Ausschlag und Gelenk- oder Muskelschmerzen mit gleichzeitig verminderten Blutplättchen oder erhöhten Aminotransferasen.
In der Studie LIBRETTO-001 hatten 24,0 % (201/837) der Selpercatinib-Patienten zuvor eine Anti‑PD‑1/PD‑L1-Immuntherapie erhalten. Überempfindlichkeitsreaktionen traten bei insgesamt 5,7 % (48/837) der Selpercatinib-Patienten auf, wobei 1,9 % (16/837) einen Schweregrad 3 entwickelten.
Von den 48 Patienten mit einer Überempfindlichkeitsreaktion in LIBRETTO-001 hatten 54,2 % (26/48) ein NSCLC mit einer vorangegangenen Anti-PD‑1/PD‑L1-Immuntherapie.
Ein Schweregrad 3 trat bei 3,5 % (7/201) der Patienten auf, die zuvor eine Anti‑PD‑1/PD‑L1 Immunotherapie in LIBRETTO-001 erhalten hatten.
In LIBRETTO-001 betrug die mediane Zeit bis zum Auftreten 1,9 Wochen (Spanne: 0,7 bis 203,9 Wochen). Bei Patienten mit einer vorangegangenen Anti‑PD‑1/PD‑L1-Immuntherapie lag dieser Wert bei 1,7 Wochen und bei 4,4 Wochen bei Patienten, die Anti‑PD‑1/PD‑L1-Immuntherapie-naiv waren.
In der Studie LIBRETTO-431 wurden Patienten mit fortgeschrittenem oder metastasiertem NSCLC aufgenommen. Eine Überempfindlichkeitsreaktion trat bei insgesamt 1,9 % der Patienten (3/158) auf, die Selpercatinib erhielten, einschließlich einer Überempfindlichkeitsreaktion des Grades 3 bei 0,6 % der Patienten (1/158). In einer integrierten Analyse von Patienten mit NSCLC, die Selpercatinib erhielten und zuvor mit einer Anti-PD-1/PD-L1-Therapie behandelt wurden, trat gemäß den Studien LIBRETTO‑001 und LIBRETTO-431 (N = 205) bei 16,6 % der Patienten eine Überempfindlichkeit auf, einschließlich einer Überempfindlichkeit ≥ Grad 3 bei 5,9 % der Patienten.
In die Studie LIBRETTO-531 wurden Patienten mit fortgeschrittenem oder metastasiertem MTC aufgenommen. Eine Überempfindlichkeitsreaktion trat bei einem Patienten (0,5 % [1/193]) auf, der Selpercatinib erhielt. Bei diesem einen Patienten trat eine Grad 3 Überempfindlichkeitsreaktion auf.
Für Retsevmo ist möglicherweise eine Dosisunterbrechung oder -änderung erforderlich (siehe Abschnitt 4.2).
Hämorrhagien
Hämorrhagische Ereignisse ≥ Grad 3 sind bei 2,5 % der mit Selpercatinib behandelten Patienten in den Studien LIBRETTO-001,
LIBRETTO-431 und LIBRETTO-531 aufgetreten. Davon hatten 4 Patienten (0,5 %) in LIBRETTO-001 tödliche hämorrhagische Ereignisse, von denen zwei eine zerebrale Hämorrhagie erlitten und jeweils einer eine Tracheostomie-Blutung bzw. eine Hämoptyse erlitt. In LIBRETTO-431 und LIBRETTO-531 wurden keine tödlichen hämorrhagischen Ereignisse bei Patienten berichtet, die Selpercatinib erhielten. Die mediane Zeit bis zum Auftreten betrug 34,1 Wochen (Spanne: 0,1 Wochen bis 234,6 Wochen) in LIBRETTO‑001, 16,8 Wochen (Spanne 1,1 bis 94,1 Wochen) in LIBRETTO-431 und 10,7 Wochen (Spanne 1,0 and 124,1 Wochen) in LIBRETTO-531.
Selpercatinib sollte bei Patienten mit lebensbedrohlicher oder wiederkehrender schwerer Hämorrhagie dauerhaft abgesetzt werden (siehe Abschnitt 4.2).
Zusätzliche Informationen über spezielle Populationen
Kinder und Jugendliche
In LIBRETTO-001 wurden 3 Patienten < 18 Jahren (Spanne: 15 - 17) mit RET-mutiertem MTC eingeschlossen. In LIBRETTO-121 wurden 11 Patienten < 18 Jahren (Spanne: 2 - 17) mit RET-mutiertem MTC, 13 Patienten < 18 Jahren (Spanne: 6 - 17) mit RET-Fusions-positivem Schilddrüsenkarzinom und 6 Patienten < 18 Jahren (Spanne: 5 - 15 Jahre) mit RET‑veränderten soliden Tumoren, davon 3 Patienten mit RET‑Fusions-positiven soliden Tumoren eingeschlossen. Es gab einen Patienten im Alter von 12 Jahren mit RET-mutiertem MTC in LIBRETTO-531. Bei Patienten unter 18 Jahren, die mit Selpercatinib behandelt wurden, wurde von Fällen einer Epiphysenlösung des Femurkopfes berichtet (siehe Abschnitt 4.4). Es wurden keine sonstigen, für die Gruppe der Kinder und Jugendlichen unter 18 Jahren spezifischen Nebenwirkungen identifiziert.
Tabellarische Auflistung der Nebenwirkungen in LIBRETTO‑121
Die Häufigkeit und der Schweregrad der in der Studie LIBRETTO‑121 bei mit Selpercatinib behandelten Patienten ≤ 21 Jahren berichteten Nebenwirkungen, sind in Tabelle 6 dargestellt.
Die Nebenwirkungen sind gemäß der MedDRA‑Systemorganklasse und nach Häufigkeit klassifiziert.
Die Häufigkeitskategorien sind wie folgt definiert: sehr häufig (≥ 1/10); häufig (≥ 1/100 bis < 1/10); gelegentlich (≥ 1/1 000 bis < 1/100); selten (≥ 1/10 000 bis < 1/1 000); sehr selten (< 1/10 000) sowie nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Die mediane Behandlungsdauer mit Selpercatinib betrug 30,26 Monate.
Tabelle 6 Nebenwirkungen bei Patienten, die in der Studie LIBRETTO‑121 mit Selpercatinib behandelt wurden (n = 36)
MedDRA Systemorganklasse |
MedDRA |
Häufigkeit aller Grade # |
Häufigkeit von Grad ≥ 3 |
Infektionen und parasitäre Erkrankungen |
Harnwegsinfektionen a |
Sehr häufig |
Häufig |
Pneumonie b |
Häufig |
Häufig |
|
Erkrankungen des Immunsystems c |
Überempfindlichkeit d |
Sehr häufig |
Häufig |
Endokrine Erkrankungen |
Hypothyreose e |
Sehr häufig |
- |
Stoffwechsel- und Ernährungsstörungen |
Verminderter Appetit |
Häufig |
- |
Erkrankungen des Nervensystems |
Kopfschmerzen |
Sehr häufig |
- |
Schwindelgefühl |
Sehr häufig |
- |
|
Herzerkrankungen |
EKG QT-Intervall-Verlängerung f |
Sehr häufig |
Häufig |
Gefäßerkrankungen |
Hämorrhagie g |
Sehr häufig |
- |
Hypertonie |
Häufig |
Häufig |
|
Erkrankungen des Gastrointestinaltrakts |
Diarrhö h |
Sehr häufig |
Häufig |
Übelkeit |
Sehr häufig |
Häufig |
|
Bauchschmerzen i |
Sehr häufig |
- |
|
Erbrechen |
Sehr häufig |
Häufig |
|
Obstipation |
Sehr häufig |
Häufig |
|
Stomatitis |
Sehr häufig |
- |
|
Mundtrockenheit |
Häufig |
- |
|
Chylöser Aszites |
Häufig |
- |
|
Erkrankungen der Haut und des Unterhautgewebes |
Ausschlag j |
Sehr häufig |
- |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Schmerzen des Muskel- und Skelettsystems k |
Sehr häufig |
- |
Epiphysenlösung des Femurkopfes |
Häufig |
Häufig |
|
Erkrankungen des Fortpflanzungssystems und der Brust |
Erektile Dysfunktion l |
Sehr häufig |
Häufig |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber |
Sehr häufig |
- |
Fatigue m |
Sehr häufig |
- |
|
Ödeme n |
Sehr häufig |
- |
|
Gewicht erhöht |
Sehr häufig |
Sehr häufig |
|
Untersuchungeno |
Calcium erniedrigt |
Sehr häufig |
Sehr häufig |
ALT erhöht |
Sehr häufig |
Häufig |
|
Albumin erniedrigt |
Sehr häufig |
- |
|
AST erhöht |
Sehr häufig |
Häufig |
|
Alkalische Phosphatase erhöht |
Sehr häufig |
- |
|
Anzahl weißer Blutkörperchen erniedrigt |
Sehr häufig |
Häufig |
|
Neutrophilenzahl erniedrigt |
Sehr häufig |
Häufig |
|
Hämoglobin erniedrigt |
Sehr häufig |
Sehr häufig |
|
Gesamtbilirubin erhöht |
Sehr häufig |
Häufig |
|
Magnesium erniedrigt |
Sehr häufig |
Häufig |
|
Blutplättchen erniedrigt |
Sehr häufig |
Häufig |
|
Lymphozytenzahl erniedrigt |
Sehr häufig |
Sehr häufig |
|
Kalium erniedrigt |
Sehr häufig |
Häufig |
|
Kreatinin erhöht |
Sehr häufig |
Häufig |
|
Natrium erniedrigt |
Sehr häufig |
- |
# |
Von den 36 Patienten in LIBRETTO-121, waren 31 Patienten jünger als 18 Jahre und 5 Patienten waren zwischen 18 und 21 Jahre alt. |
a |
Harnwegsinfektionen umfassen Harnwegsinfektion und Zystitis. |
b |
Pneumonie umfasst Lungenentzündung sowie durch das respiratorische Synzytialvirus verursachte Pneumonie. |
c |
Überempfindlichkeitsreaktionen waren durch einen makulopapulösen Ausschlag gekennzeichnet, häufig mit vorangegangenem Fieber und assoziierten Gelenk- bzw. Muskelschmerzen während des ersten Behandlungszyklus des Patienten (typischerweise zwischen Tag 7 und 21). |
d |
Überempfindlichkeit umfasst Arzneimittel-Überempfindlichkeit und Überempfindlichkeit. |
e |
Hypothyreose umfasst Hypothyreose, erhöhtes Thyreoidea‑stimulierendes Hormon im Blut sowie erhöhtes Thyreoglobulin. |
f |
EKG‑QT‑Intervall‑Verlängerung umfasst verlängertes QT‑Intervall im Elektrokardiogramm sowie verlängerten QRS‑Komplex im Elektrokardiogramm. |
g |
Hämorrhagie umfasst Epistaxis, Hämaturie, verlängerte aktivierte partielle Thromboplastinzeit, anale Blutung, Nachweis von Blut im Urin, Hämoptyse, Menorrhagie und orale Blutung. |
h |
Diarrhö umfasst Diarrhö und Analinkontinenz. |
i |
Bauchschmerzen umfassen Bauchschmerzen, Oberbauchschmerzen und abdominelle Beschwerden. |
j |
Ausschlag umfasst makulopapulösen Ausschlag, Ausschlag, erythematösen Ausschlag und Urtikaria. |
k |
Muskuloskelettale Schmerzen umfassen Arthralgie, Schmerzen in den Extremitäten, Rückenschmerzen, nicht‑kardiale Brustschmerzen, Knochenschmerzen, muskuloskelettale Brustschmerzen, muskuloskelettale Schmerzen und Nackenschmerzen. |
l |
Erektile Dysfunktion bei männlichen Patienten, die mit Selpercatinib behandelt wurden. Der Nenner |
m |
Fatigue umfasst Fatigue, Asthenie und Unwohlsein (Malaise). |
n |
Ödeme umfassen Gesichtsödeme, periphere Ödeme, periorbitale Ödeme, generalisierte Ödeme, lokalisierte Ödeme sowie Schwellungen. |
o |
Basierend auf Laboruntersuchungen. Der Prozentsatz wird auf Grundlage der Anzahl der Patienten mit einem Ausgangswert und mindestens einem nachfolgenden Messwert als Nenner berechnet. |
Ältere Patienten
In der Studie LIBRETTO-001 waren Patienten, die Selpercatinib erhielten, zu 24,7 % zwischen 65 und 74 Jahre alt, zu 8,6 % zwischen 75 und 84 Jahren und zu 1,0 % 85 Jahre oder älter. In der Studie LIBRETTO-431 waren 26,6 % der Patienten, die Selpercatinib erhielten, zwischen 65 und 74 Jahre alt, 9,5 % zwischen 75 bis 84 Jahren und 1,3 % 85 Jahre oder älter. In der Studie LIBRETTO-531 waren 20,2 % der Patienten, die Selpercatinib erhielten, zwischen 65 und 74 Jahre alt, 5,2 % zwischen 75 bis 84 Jahren und keiner war 85 Jahre oder älter. Die Häufigkeit der gemeldeten schwerwiegenden, unerwünschten Ereignisse war in LIBRETTO-001 in den Altersgruppen von 65 bis 74 Jahren (58,0 %), 75 bis 84 Jahren (62,5 %) und ≥ 85 Jahren (100,0 %) größer als bei Patienten im Alter von < 65 Jahren (46,7 %); in LIBRETTO-431 war sie in den Altersgruppen 65 bis 74 Jahren (38,1 %), 75 bis 84 Jahren (46,7 %), ≥ 85 Jahren (50,0 %) größer als bei Patienten im Alter von < 65 Jahren (31,3 %). Die Häufigkeit der gemeldeten schwerwiegenden, unerwünschten Ereignisse war in LIBRETTO-531 in den Altersgruppen von 75 bis 84 Jahren (50 %) größer als bei Patienten im Alter von < 65 Jahren (20,8 %) und 65 bis 74 Jahren (17,9 %).
Die Häufigkeit der unerwünschten Ereignisse in der Studie LIBRETTO-001, die zum Abbruch von Selpercatinib führten, war in den Altersgruppen von 65 bis 74 Jahren (10,1 %), 75 bis 84 Jahren (19,4 %) und ≥ 85 Jahren (37,5 %) größer als bei Patienten im Alter von < 65 Jahren (7,6 %). Die Häufigkeit der unerwünschten Ereignisse in der Studie LIBRETTO-431, die zum Abbruch von Selpercatinib führten, war in den Altersgruppen von 65 bis 74 Jahren (14,3 %), 75 bis 84 Jahren (20,0 %) höher als bei Patienten im Alter von < 65 Jahren (7,1 %). Bei keinem Patienten im Alter von ≥ 85 Jahren wurde die Behandlung mit Selpercatinib aufgrund von unerwünschten Ereignissen abgebrochen. Die Häufigkeit der unerwünschten Ereignisse in der Studie LIBRETTO-531, die zum Abbruch von Selpercatinib führten, war in den Altersgruppen von 75 bis 84 Jahren (10 %) und 65 bis 74 Jahren (7,7 %) größer als bei Patienten im Alter von < 65 Jahren (3,5 %).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem anzuzeigen:
Bundesinstitut für Arzneimittel und
Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: https://www.bfarm.de
Symptome einer Überdosierung wurden nicht festgestellt. Im Falle einer vermuteten Überdosierung sollte eine symptomatische Behandlung durchgeführt werden.
Pharmakotherapeutische Gruppe: Antineoplastische und immunmodulierende Mittel, antineoplastische Mittel, Proteinkinase-Inhibitoren, ATC-Code: L01EX22
Wirkmechanismus
Selpercatinib ist ein Inhibitor der RET (rearranged during transfection) Rezeptor-Tyrosin-Kinase. Selpercatinib inhibierte den RET-Wildtyp, mehrere mutierte RET-Isoformen und VEGFR1 und VEGFR3 mit IC50-Werten im Bereich von 0,92 nM bis 67,8 nM. In anderen Enzym-Assays inhibierte Selpercatinib bei höheren Konzentrationen, die klinisch noch erreichbar waren, auch FGFR 1, 2 und 3. In einem Bindungs-Assay bei einer Konzentration von 1 μM Selpercatinib wurde eine signifikante Antagonist-Bindungsaktivität (> 50 %) für den 5-HT (Serotonin) Transporter (70,2 % Antagonist) und α2C Adrenorezeptor (51,7 % Antagonist) beobachtet. Die Konzentration von 1 μM ist etwa 7‑fach höher als die maximale freie Plasmakonzentration der wirksamen Selpercatinib-Dosis.
Bestimmte Punktmutationen in RET oder chromosomale Rearrangements, wie z. B. in-frame RET-Fusionen mit verschiedenen Partnern, können zur Bildung von konstitutiv aktivierten, chimären RET-Fusionsproteinen führen, die als onkogene Treiber durch Förderung der Zellproliferation von Tumorzelllinien wirken können. In in vitro und in vivo Tumormodellen zeigte Selpercatinib Antitumor-Aktivität in Zellen, die eine konstitutive Aktivierung des RET-Proteins infolge von Genfusionen und Mutationen aufweisen, wie z. B. CCDC6‑RET, KIF5B‑RET, RET V804M und RET M918T. Außerdem zeigte Selpercatinib bei Mäusen, denen intrakraniell ein vom Patienten stammender RET-Fusions-positiver Tumor implantiert wurde, eine Antitumor-Aktivität.
Pharmakodynamische Wirkungen
Herz-Elektrophysiologie
In einer ausführlichen QT-Studie mit positiver Kontrolle bei 32 gesunden Probanden wurde bei Selpercatinib-Konzentrationen, die denen ähnelten, die unter einem therapeutischen Dosierungsplan beobachtet wurden, keine große Änderung (das heißt > 20 ms) des QTcF-Intervalls festgestellt. Eine Expositions-Reaktions-Analyse zeigte, dass supra-therapeutische Konzentrationen zu einem Anstieg der QTc > 20 ms führen können.
Bei Patienten, die Selpercatinib erhielten, wurde über eine QT-Intervall-Verlängerung berichtet. Deshalb kann bei Patienten eine Dosisunterbrechung oder -änderung erforderlich sein (siehe Abschnitt 4.2 und 4.4).
Klinische Wirksamkeit und Sicherheit
Die Wirksamkeit von Retsevmo wurde bei erwachsenen Patienten mit fortgeschnittenem RET-Fusions-positiven NSCLC, RET-Fusions-positivem Schilddrüsen-Karzinom und anderen RET-Fusions-positiven soliden Tumoren ausgewertet und bei erwachsenen und jugendlichen Patienten mit RET-mutiertem MTC in einer multizentrischen, offenen, einarmigen klinischen Phase1/2 Studie, LIBRETTO-001, untersucht. Die Wirksamkeit von Retsevmo bei RET-Fusions-positivem NSCLC wurde in der Phase-3-Studie LIBRETTO-431 bestätigt (siehe Abschnitt Therapienaives RET-Fusions-positives NSCLC). Die Wirksamkeit von Retsevmo bei RET-mutiertem MTC wurde in der Phase-3-Studie LIBRETTO-531 bestätigt (siehe Abschnitt Vandetanib- und Cabozantinib-naives RET-mutiertes medulläres Schilddrüsenkarzinom (MTC)).
Die Studie LIBRETTO-001 umfasste zwei Teile: Phase 1 (Dosis-Eskalation) und Phase 2 (Dosis-Erweiterung). Das Hauptziel des Phase 1-Teils war es, die empfohlene Phase 2 Selpercatinib-Dosis zu ermitteln. Das Hauptziel des Phase 2-Teils war es, die Antitumor-Aktivität von Selpercatinib durch Gesamtansprechraten-Bestimmung zu beurteilen, wie durch eine unabhängige Kommission beurteilt. Es wurden Patienten mit nach RECIST 1.1 messbaren oder nicht-messbaren Erkrankungen, mit Nachweis einer RET-Genveränderung im Tumor eingeschlossen. Patienten mit stabilen Metastasen des zentralen Nervensystems (ZNS) konnten eingeschlossen werden, während Patienten mit symptomatischem, primärem ZNS-Befall bzw. ZNS-Metastasen, leptomeningealer Karzinomatose oder Rückenmarks-Kompression ausgeschlossen waren. Patienten mit bekannter primärer non-RET Treiberalteration wurden ausgeschlossen. Auch Patienten mit klinisch signifikanten, aktiven Herz-Kreislauf-Erkrankungen oder erlittenem Herzinfarkt, QTcF-Intervall > 470 ms waren ausgeschlossen.
Patienten im Phase 2-Teil der Studie erhielten Retsevmo 160 mg zweimal täglich bis zum Auftreten inakzeptabler Toxizität oder bis ein Fortschreiten der Erkrankung auftrat. Die Identifikation einer RET-Genveränderung wurde prospektiv in lokalen Laboratorien unter Verwendung von NGS (Next-Generation Sequencing), PCR (Polymerase-Kettenreaktion) oder FISH (Fluoreszenz-in-situ-Hybridisierung) ermittelt. Als primärer Parameter für die Wirksamkeit wurde die objektive Ansprechrate (ORR) gemäß RECIST 1.1 verwendet, die von einem verblindeten, unabhängigen Review Committee (IRC) bewertet wurde. Zu den sekundären Parametern für die Wirksamkeit gehörten die Dauer des Ansprechens (DOR), das progressionsfreie Überleben (PFS) und das Gesamtüberleben (OS).
Therapienaives RET-Fusions-positives NSCLC
LIBRETTO-431
Die Wirksamkeit von Retsevmo bei RET-Fusions-positivem NSCLC wurde in LIBRETTO-431 bestätigt, einer Phase 3 multizentrischen, randomisierten, offenen Vergleichsstudie, in der Selpercatinib mit einer platinbasierten und Pemetrexed-Therapie mit oder ohne Pembrolizumab bei Patienten mit fortgeschrittenem oder metastasiertem RET-Fusions-positivem NSCLC verglichen wurde. Erwachsene Patienten mit histologisch bestätigtem, inoperablem, lokal fortgeschrittenem oder metastasiertem NSCLC ohne vorherige systemische Therapie der metastasierenden Erkrankung waren geeignet. Patienten, die eine adjuvante oder neoadjuvante Therapie erhielten, und die letzte Dosis der systemischen Behandlung mindestens 6 Monate vor der Randomisierung abgeschlossen wurde, waren ebenfalls geeignet. Die Patienten erhielten zweimal täglich 160 mg Selpercatinib (Anfangsdosis) oder eine platinbasierte und Pemetrexed-Therapie mit oder ohne Pembrolizumab. Die Patienten wurden wie folgt stratifiziert: nach geografischer Region (Ostasien vs. anderenorts), Status in Bezug auf die vom Prüfarzt beurteilten Hirnmetastasen zu Studienbeginn (nicht vorhanden oder unbekannt vs. vorhanden) und ob der Prüfarzt (vor der Randomisierung) beabsichtigt hatte, den Patienten mit oder ohne Pembrolizumab zu behandeln. Der primäre Parameter für die Wirksamkeit war das PFS gemäß RECIST 1.1 bewertet durch BICR (Blinded Independent Central Review). Zu den sekundären Wirksamkeitsparametern gehörten OS, ORR/DOR/Krankheitskontrollrate (DCR) jeweils bewertet durch BICR. Ebenfalls eingeschlossen waren die intrakranielle ORR und DOR durch BICR sowie die Zeit bis zur Verschlechterung der Lungensymptome, erfasst mittels des Fragebogens zur Symptombewertung bei nicht-kleinzelligem Lungenkarzinom (NSCLC-SAQ).
Von den 261 Patienten, die in die Studie LIBRETTO-431 Intention to Treat (ITT)-Population aufgenommen und randomisiert wurden, wurden 212 danach stratifiziert, ob der Prüfarzt eine Pembrolizumab Therapie für den Patienten (vor der Randomisierung) vorgesehen hätte, um die ITT-Pembrolizumab-Population zu bilden. In der ITT-Pembrolizumab-Population erhielten 129 Patienten Selpercatinib, während 83 eine platinbasierte Pemetrexed-Chemotherapie mit Pembrolizumab erhielten. Das mediane Alter der Patienten in der ITT-Pembrolizumab-Population betrug 61,5 Jahre (Spanne 31 bis 84 Jahre). 53,3 % der Patienten waren weiblich. 41,3 % der Patienten waren Weiße, 56,3 % Asiaten, 1 % Schwarze. 67,9 % waren nie Raucher. In der ITT-Pembrolizumab-Population hatten 93 % eine metastasierende Erkrankung und 20,3 % der Patienten hatten zu Studienbeginn ZNS-Metastasen. Der ECOG-Performancestatus wurde als 0 - 1 (96,7 %) oder 2 (3,3 %) angegeben. Der häufigste Fusionspartner war KIF5B (44,8 %), gefolgt von CCDC6 (9,9 %). Die Studie erreichte ihren primären Endpunkt, nämlich die Verbesserung des PFS sowohl in der ITT-Pembrolizumab- als auch in der ITT-Population. Die primären Wirksamkeitsergebnisse für die ITT-Pembrolizumab-Population bei behandlungsnaiven Patienten mit RET-Fusions-positivem NSCLC sind in Tabelle 7 und Abbildung 1 zusammengefasst.
Tabelle 7 LIBRETTO-431: Zusammenfassung der Wirksamkeitsdaten (BICR-Bewertung, ITT-Pembrolizumab-Population)
Selpercatinib |
Kontrolle (platinbasierte Pemetrexed-Chemotherapie mit Pembrolizumab) |
|
Progressionsfreies Überleben |
N = 129 |
N = 83 |
Median [Monate] (95 % CI) |
24,84 (16,89; NE) |
11,17 (8,77; 16,76) |
Hazard Ratio (95 % CI) |
0,465 (0,309; 0,699) |
|
Stratifizierter log rank p-Wert |
0,0002 |
|
24 - Monats PFS Rate (%) (95 % CI) |
54,2 (43,6; 63,6) |
31,6 (20,1; 43,7) |
Objektives Ansprechen (CR + PR) |
||
% (95 % CI) |
83,7 (76,2; 89,6) |
65,1 (53,8; 75,2) |
komplette Remission (CR) n (%) |
9 (7,0) |
5 (6,0) |
partielle Remission (PR) n (%) |
99 (76,7) |
49 (59,0) |
Dauer des Ansprechens (Monate) * |
||
Median [Monate] (95 % CI) |
24,18 (17,94; NE) |
11,47 (9,66; 23,26) |
Anteil der Patienten (%) mit Ansprechdauer |
||
24 Monate (95 % CI) |
59,6 (47,5; 69,8) |
22,8 (6,3, 45,5) |
CI = Konfidenzintervall, CR = komplette Remission, NE = nicht auswertbar, PR = partielle Remission | ||
* |
Die mediane Dauer des Follow-ups betrug 17,97 Monate (25. Perzentil: 12,32; 75. Perzentil: 21,03) im Selpercatinib-Arm und 14,55 Monate (25. Perzentil: 9,69; 75. Perzentil: 20,73) im Kontrollarm. |
Cut-off Datum: 01. Mai 2023 | |
Abbildung 1. LIBRETTO-431: Kaplan-Meier-Diagramm des progressionsfreien Überlebens (BICR-Bewertung, ITT-Pembrolizumab-Population) |
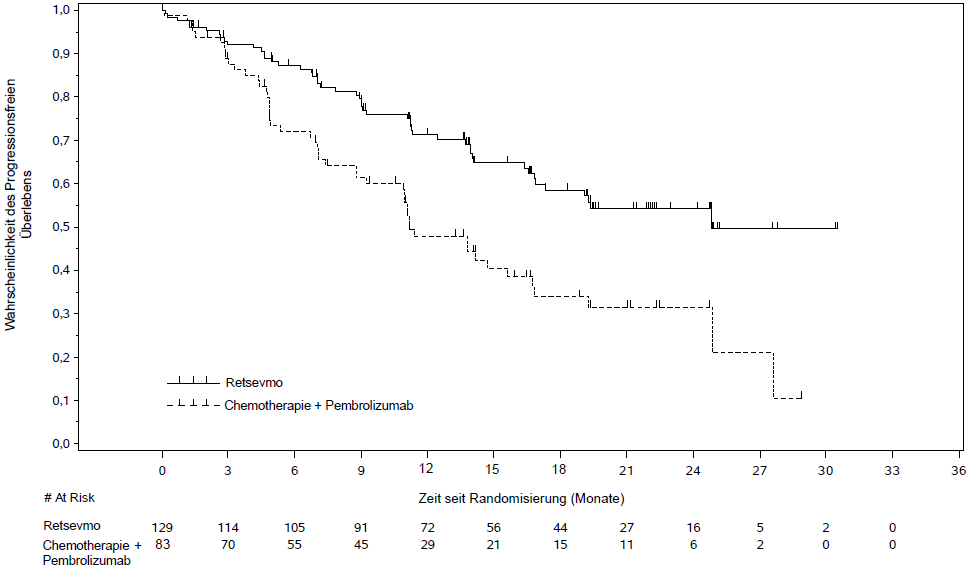 |
Cut-off Datum: 01. Mai 2023 |
Das OS war zum Zeitpunkt der primären PFS-Analyse nicht ausgereift. Zum Zeitpunkt einer aktualisierten Interim-Analyse des OS (43 % der im Voraus festgelegten OS-Ereignisse, die für die endgültige Analyse benötigt werden, mit einem Datenschnitt vom 1. Mai 2024) wurden in der ITT-Population 75 Ereignisse in beiden Therapiearmen beobachtet und die Hazard Ratio (HR) betrug 1,259 ([95 % CI: 0,777, 2,040]; p = 0,3496). Nach 30 Monaten betrug das OS 71 % (95 % CI: 63, 78) im Selpercatinib-Arm und 76 % (95 % CI: 66, 84) im Kontrollarm. Das OS kann durch das Ungleichgewicht in den Therapien, die nach einem Progress durchgeführt wurden, beeinflusst werden. Von 68 Patienten im Kontrollarm, mit fortgeschrittener Erkrankung, erhielten 50 Patienten (74 %) nach Progress Selpercatinib. Von 71 Patienten im Selpercatinib-Arm, mit fortgeschrittner Erkrankung, erhielten nach Progress 16 (23 %) eine Chemotherapie und/oder Immun-Checkpoint-Inhibitor-Therapie und 44 (62 %) setzten die Behandlung mit Selpercatinib fort.
In der ITT-Pembrolizumab-Population verzögerte Selpercatinib die Zeit bis zur Verschlechterung der von Patienten berichteten NSCLC-Symptome signifikant, gemessen anhand des Gesamtscores des NSCLC-SAQ (Symptom Assessment Questionnaire), definiert als ein Anstieg um ≥ 2 Punkte im Vergleich zur Kontrollgruppe (HR 0,34 [95 % CI: 0,20; 0,55]; die mediane Zeit wurde für den Selpercatinib-Arm nicht erreicht gegenüber 1,9 Monaten [95 % CI: 0,7; 6,6]) für den Kontrollarm. Darüber hinaus verzögerte Selpercatinib signifikant die Zeit bis zur bestätigten Verschlechterung der körperlichen Funktionsfähigkeit und hielt die allgemeine Lebensqualität im Laufe der Zeit aufrecht.
LIBRETTO-001
Von den 362 RET-Fusions-positiven NSCLC-Patienten, die in LIBRETTO-001 eingeschlossen wurden, waren 69 therapienaiv. Das mediane Alter betrug 63 Jahre (23 bis 92 Jahre). 62,3 % der Patienten waren weiblich. 69,6 % der Patienten waren Weiße, 18,8 % Asiaten, 5,8 % Schwarze und 69,6 % waren Nie-Raucher. Die meisten Patienten (98,6 %) hatten bei Studieneinschluss eine metastasierte Erkrankung; 23,2 % hatten nach Aussage des Prüfarztes zur Baseline ZNS-Metastasen. Der ECOG-Performancestatus wurde mit 0 ‑ 1 (94,2 %) oder 2 (5,8 %) angegeben. Der häufigste Fusionspartner war KIF5B (69,6 %), gefolgt von CCDC6 (14,5 %) und dann NCOA4 (1,4 %). Die Ergebnisse der Wirksamkeit von therapienaiven Patienten mit RET-Fusions-positiven NSCLC sind in Tabelle 8 zusammengefasst.
Tabelle 8 LIBRETTO-001: Objektives Ansprechen und Dauer des Ansprechens
Patienten für die Wirksamkeitsbeurteilung |
|
N |
69 |
Objektives Ansprechen (CR + PR) |
|
% (95 % CI) |
82,6 (71,6; 90,7) |
komplette Remission (CR) n (%) |
5 (7,2) |
partielle Remission (PR) n (%) |
52 (75,4) |
Dauer des Ansprechens (Monate) * |
|
Median, 95 % CI |
20,23 (15,4; 29,5) |
Anteil der Patienten (%) mit Ansprechdauer |
|
≥ 6 Monate (95 % CI) |
87,5 (75,5; 93,8) |
≥ 12 Monate (95 % CI) |
66,7 (52,4; 77,6) |
CI = Konfidenzintervall, CR = komplette Remission, PR = partielle Remission | |
* |
Die mediane Dauer des Follow-ups betrug 37,09 Monate (25. Perzentil: 24,0; 75. Perzentil: 45,1) |
Cut-off Datum: 13. Januar 2023 | |
Vorbehandeltes RET-Fusions-positives NSCLC
Insgesamt 247 Patienten hatten zuvor eine platinbasierte Chemotherapie in der Studie LIBRETTO-001 erhalten. Das mediane Alter betrug 61 Jahre (23 bis 81 Jahre). 56,7 % der Patienten waren weiblich. 43,7 % der Patienten waren Weiße, 47,8 % Asiaten, 4,9 % Schwarze und 66,8 % waren Nie‑Raucher. Die meisten Patienten (98,8 %) hatten bei Studieneinschluss eine metastasierte Erkrankung; 31,2 % hatten nach Aussage des Prüfarztes zur Baseline ZNS-Metastasen. Der ECOG-Performancestatus wurde mit 0 - 1 (97,1 %) oder 2 (2,8 %) angegeben. Der häufigste Fusionspartner war KIF5B (61,9 %), gefolgt von CCDC6 (21,5 %) und dann NCOA4 (2,0 %). Die mediane Anzahl systemischer Vortherapien betrug 2 (Spanne 1 – 15) und 43,3 % (n = 107/247) hatten 3 oder mehr systemische Vortherapien erhalten; dies waren Anti-PD1/PD-L1-Therapien (58,3 %), Multikinase-Inhibitoren (MKI) (31,6 %) und Taxane (34,8 %); 41,3 % hatten andere systemische Therapien. Die Ergebnisse der Wirksamkeit von vorbehandelten Patienten mit RET-Fusions-positivem NSCLC sind in Tabelle 9 zusammengefasst.
Tabelle 9 LIBRETTO-001: Objektives Ansprechen und Dauer des Ansprechens
Patienten für die Wirksamkeitsbeurteilung |
|
N |
247 |
Objektives Ansprechen (CR + PR) |
|
% (95 % CI) |
61,5 (55,2; 67,6) |
komplette Remission (CR) n (%) |
20 (8,1) |
partielle Remission (PR) n (%) |
132 (53,4) |
Dauer des Ansprechens (Monate) * |
|
Median (95 % CI) |
31,6 (20,4; 42,3) |
Anteil der Patienten (%) mit Ansprechdauer |
|
≥ 6 Monate (95 % CI) |
87,0 (80,4; 91,5) |
≥ 12 Monate (95 % CI) |
73,0 (65,0; 79,5) |
CI = Konfidenzintervall, CR = komplette Remission, PR = partielle Remission | |
* |
Die mediane Dauer des Follow-ups betrug 39,52 Monate (25. Perzentil: 24,6; 75. Perzentil: 45,0) |
Cut-off Datum: 13. Januar 2023 | |
ZNS Ansprechen in RET-Fusions-positivem NSCLC
In der Studie LIBRETTO-431 betrug die mit BICR beurteilte ZNS ORR bei den 17 mit Selpercatinib behandelten Patienten mit messbaren Hirnmetastasen zu Studienbeginn 82,4 % (14/17; 95 % CI: 56,6; 96,2) gegenüber 58,3 % (7/12; 95 % CI: 27,7 bis 84,4) bei den 12 Patienten im Kontrollarm der ITT-Pembrolizumab-Population. Komplette Remissionen wurden bei 6/17 der Patienten (35,3 %) im Selpercatinib-Arm gegenüber 2/12 der Patienten (16,7 %) im Kontrollarm beobachtet. Mit einer medianen Follow-up Dauer für DOR von 9,92 Monaten (95 % CI: 7,66; 18,10) im Selpercatinib-Arm und 12,68 Monaten (95 % CI: 2,79; NE) im Kontrollarm wurde die mediane DOR für Selpercatinib (95 % CI: 7,62; NE) im Vergleich zu 13,4 Monaten (95 % CI: 3,45; NE) mit der Kontrolle nicht erreicht. Bei 192 Patienten mit intrakraniellen Baseline-Scans betrug die ursachenspezifische Hazard Ratio für die Zeit bis zur ZNS-Progression, wie vom BICR bewertet, 0,28; 95 % CI: 0,12; 0,68 (HR von 0,17; 95 % CI: 0,04; 0,69 für 150 Patienten ohne intrakranielle Metastasen zu Studienbeginn und HR von 0,61; 95 % CI: 0,19; 1,92 für 42 Patienten mit intrakraniellen Metastasen zu Studienbeginn). 8 Patienten (6,7 %) im Selpercatinib-Arm hatten ein erstes Ereignis der ZNS-Progression im Vergleich zu 13 Patienten (18,1 %) im Kontrollarm.
In der Studie LIBRETTO-001 betrug die intrakranielle ORR nach Beurteilung des unabhängigen Review-Committees 84,6 % (22/26; 95 % CI: 65,1; 95,6) bei 26 Patienten mit messbarer Erkrankung. Eine komplette Remission wurde bei 7 (26,9 %) Patienten und partielle Remission bei 15 (57,5 %) Patienten beobachtet. Die mediane intrakranielle Ansprechdauer betrug 9,36 Monate (95 % CI: 7,4; 15,3).
Systemtherapienaives RET-Fusions-positives Schilddrüsenkarzinom
Von den RET-Fusions-positiven Schilddrüsenkarzinom Patienten, die keine andere systemische Therapie als radioaktives Iod erhielten und in LIBRETTO-001 aufgenommen wurden, wurden 24 Patienten mindestens 6 Monate lang beobachtet und waren Grundlage der Wirksamkeitsbeurteilung. Das mediane Alter betrug 60,5 Jahre (Spanne 20 bis 84 Jahre). 58,3 % der Patienten waren männlich. 75 % der Patienten waren Weiße. Der ECOG-Performancestatus wurde mit 0 - 1 (95,8 %) oder 2 (4,2 %) angegeben. 100 % der Patienten hatten eine Vorgeschichte mit einer metastasierten Erkrankung. 22 der 24 Patienten (91,7 %) erhielten vor der Aufnahme in die Studie radioaktives Iod und wurden daher als refraktär für radioaktives Iod eingestuft. Die verschiedenen Histologien waren bei den eingeschlossenen 24 Patienten papillär (n = 23) und gering differenziert (n = 1). Der häufigste Fusions-Partner war CCDC6 (62,5 %), gefolgt von NCOA4 (29,2 %). Die Ergebnisse der Wirksamkeit für systemtherapienaive Patienten mit RET-Fusions-positivem Schilddrüsenkarzinom sind in Tabelle 10 zusammengefasst.
Tabelle 10 LIBRETTO-001: Objektives Ansprechen und Dauer des Ansprechens
Patienten für die Wirksamkeitsbeurteilung |
|
N |
24 |
Objektives Ansprechen (CR + PR) |
|
% (95 % CI) |
95,8 (78,9; 99,9) |
komplette Remission (CR) n (%) |
5 (20,8) |
partielle Remission (PR) n (%) |
18 (75,0) |
Dauer des Ansprechens (Monate) * |
|
Median (95 % CI) |
NE (42,8; NE) |
Anteil der Patienten (%) mit Ansprechdauer |
|
≥ 12 Monate (95 % CI) |
100,0 (100,0; 100,0) |
≥ 24 Monate (95 % CI) |
94,4 (66,6; 99,2) |
≥ 36 Monate (95 % CI) |
88,9 (62,4; 97,1) |
CI = Konfidenzintervall, CR = komplette Remission, NE = nicht auswertbar, PR = partielle Remission | |
* |
Die mediane Dauer des Follow-ups betrug 54,8 Monate (25. Perzentil: 32,3; 75. Perzentil: 62,5) |
Cut-off Datum: 14. Februar 2025 | |
Vorbehandeltes RET-Fusions‑positives Schilddrüsen-Karzinom
Von den RET-Fusions-positiven Schilddrüsen-Karzinom Patienten, die mit einer anderen systemischen Therapie als radioaktivem Iod vorbehandelt und in LIBRETTO-001 aufgenommen wurden, wurden 41 Patienten mindestens 6 Monate lang beobachtet und waren Grundlage der Wirksamkeitsbeurteilung. Das mediane Alter betrug 58 Jahre (Spanne 25 bis 88 Jahre). 43,9 % der Patienten waren männlich. 58,5 % der Patienten waren Weiße, 29,3 % Asiaten und 7,3 % Schwarze. Der ECOG-Performancestatus wurde mit 0 oder 1 (92,7 %) oder 2 (7,3 %) angegeben. 100 % der Patienten hatten eine metastasierte Erkrankung. Die Patienten erhielten im Median 3 systemische Vortherapien (Spanne: 1 - 7). Die Häufigsten Vortherapien beinhalteten radioaktives Iod (73,2 %) und MKI (85,4 %). 9,8 % der Patienten hatten andere systemische Therapien erhalten. Die verschiedenen Histologien bei den eingeschlossenen 41 Patienten beinhalteten papilläre (n = 31), gering differenzierte (n = 5), anaplastische (n = 4) und Hürthle-Zellen (n = 1). Der häufigste Fusions-Partner war CCDC6 (61,0 %), gefolgt von NCOA4 (19,5 %).
Die Ergebnisse der Wirksamkeit für vorbehandelte RET-Fusions-positive Schilddrüsen-Karzinome sind in Tabelle 11 zusammengefasst.
Tabelle 11 LIBRETTO-001: Objektives Ansprechen und Dauer des Ansprechens
Patienten für die Wirksamkeitsbeurteilung |
|
N |
41 |
Objektives Ansprechen (CR + PR) |
|
% (95 % CI) |
85,4 (70,8; 94,4) |
komplette Remission (CR) n (%) |
5 (12,2) |
partielle Remission (PR) n (%) |
30 (73,2) |
Dauer des Ansprechens (Monate) * |
|
Median (95 % CI) |
26,7 (12,1; NE) |
Anteil der Patienten (%) mit Ansprechdauer |
|
≥ 12 Monate (95 % CI) |
71,7 (52,4; 84,2) |
≥ 24 Monate (95 % CI) |
50,7 (30,4; 67,8) |
CI = Konfidenzintervall, CR = komplette Remission, NE = nicht auswertbar, PR = partielle Remission | |
* |
Die mediane Dauer des Follow-ups betrug 33,87 Monate (25. Perzentil: 12,9; 75. Perzentil: 44,8) |
Cut-off Datum: 13. Januar 2023 | |
Vandetanib- und Cabozantinib-naives RET-mutiertes medulläres Schilddrüsen-Karzinom (MTC)
LIBRETTO-531
Die Wirksamkeit von Retsevmo bei RET-mutiertem MTC wurde in LIBRETTO-531 bestätigt, einer multizentrischen, randomisierten, offenen Phase-3-Vergleichsstudie, in der Selpercatinib bei Patienten mit progredientem, fortgeschrittenem, Kinase-Inhibitor-naivem, RET-mutiertem MTC mit Cabozantinib oder Vandetanib nach Wahl des Arztes verglichen wurde. Erwachsene oder jugendliche Patienten mit histologisch bestätigtem, inoperablem, lokal fortgeschrittenem oder metastasiertem MTC ohne vorherige Behandlung mit einem Kinase-Inhibitor waren geeignet. Die Patienten erhielten 160 mg Selpercatinib zweimal täglich (Anfangsdosis) oder nach Wahl des Arztes Cabozantinib (140 mg einmal täglich) oder Vandetanib (300 mg einmal täglich). Die Patienten wurden nach RET-Mutation (M918T vs. andere) stratifiziert. Ergänzend wurden die Patienten, die in den Kontrollarm randomisiert wurden, nach der vom Arzt beabsichtigten Behandlung (Cabozantinib vs. Vandetanib) stratifiziert. Als primärer Parameter für die Wirksamkeit war das PFS gemäß RECIST 1.1 nach BICR (Blinded Independent Central Review). Zu den wichtigsten sekundären Parametern für die Wirksamkeit gehörte das treatment failure-free survival (TFFS) und die Verträglichkeit. Andere sekundäre Parameter für die Wirksamkeit umfassten OS und ORR/DOR nach BICR.
Von den 291 Patienten, die in LIBRETTO-531 aufgenommen und randomisiert wurden, um die Intention to Treat (ITT)-Population zu bilden, wurden 193 in den Selpercatinib-Arm und 98 in den Kontrollarm randomisiert. Von den 98 Patienten, die in den Kontrollarm randomisiert wurden, wurden 73 auf Cabozantinib und 25 auf Vandetanib stratifiziert. Das mediane Alter der Patienten in der ITT-Population betrug 55 Jahre (Spanne: 12 bis 84 Jahre). 37,1 % der Patienten waren weiblich. 69,4 % der Patienten waren Weiße, 27,7 % Asiaten, 2,9 % Schwarze. Die meisten Patienten (77 %) hatten bei der Aufnahme eine metastasierte Erkrankung. Der ECOG-Performancesstatus wurde mit 0 - 1 (98,3 %) oder 2 (1 %) angegeben. Die häufigste Mutation war M918T (62,5 %). Die Studie erreichte ihren primären Endpunkt, nämlich die Verbesserung des PFS in der ITT-Population. Die Wirksamkeitsergebnisse für die ITT-Population sind in Tabelle 12 und Abbildung 2 zusammengefasst.
Tabelle 12 LIBRETTO-531: Zusammenfassung der Wirksamkeitsdaten (BICR-Bewertung, ITT-Population)
Selpercatinib |
Kontrolle (Cabozantinib oder Vandetanib) |
|
Progressionsfreies Überleben |
N = 193 |
N = 98 |
Median [Monate] (95 % CI) |
NE (NE; NE) |
16,76 (12,22; 25,10) |
Hazard Ratio (95 % CI) |
0,280 (0,165; 0,475) |
|
Stratifizierter log rank p-Wert |
< 0,0001 |
|
30-Monats PFS Rate (%) (95 % CI) |
76,4 (66,5; 83,8) |
24,8 (6,9; 48,3) |
Treatment failure-free survival * |
N = 193 |
N = 98 |
Median [Monate] (95 % CI) |
NE (NE; NE) |
13,93 (11,27; 25,10) |
Hazard Ratio (95 % CI) |
0,254 (0,153; 0,423) |
|
Stratifizierter log rank p-Wert |
< 0,0001 |
|
30-Monats TFFS Rate (%) 95 % CI |
75,8 (65,9; 83,2) |
25,3 (7,2; 48,8) |
Objektives Ansprechen (CR + PR) |
||
% (95 % CI) |
69,4 (62,4; 75,8) |
38,8 (29,1; 49,2) |
komplette Remission (CR) n (%) |
23 (11,9) |
4 (4,1) |
partielle Remission (PR) n (%) |
111 (57,5) |
34 (34,7) |
Dauer des Ansprechens (Monate) * |
||
Median (95 % CI) |
NE (NE; NE) |
16,56 (10,41; NE) |
Anteil der Patienten (%) mit Ansprechdauer |
||
≥ 24 Monate (95 % CI) |
79,1 (66,9; 87,2) |
NE (NE; NE) |
CI = Konfidenzintervall, CR = komplette Remission, NE = nicht auswertbar, PR = partielle Remission | ||
* |
Treatment failure-free survival ist definiert als die Zeit von der Randomisierung bis zum ersten Auftreten von: radiologisch dokumentiertem Fortschreiten der Erkrankung gemäß RECIST 1.1 oder inakzeptabler Toxizität, die nach ärztlicher Beurteilung zum Abbruch der Behandlung führt, oder Tod aufgrund einer beliebigen Ursache. |
# |
Die mediane Dauer des Follow-ups betrug im Selpercatinib-Arm 11,14 Monate (25. Perzentil: 5,62; 75. Perzentil: 16,62) und im Kontrollarm 12,81 Monate (25. Perzentil; 6,34 und 75. Perzentil: 15,51). |
Cut-off Datum: 22. Mai 2023 | |
Abbildung 2. LIBRETTO-531: Kaplan-Meier-Diagramm des progressionsfreien Überlebens (BICR-Bewertung, ITT-Population)
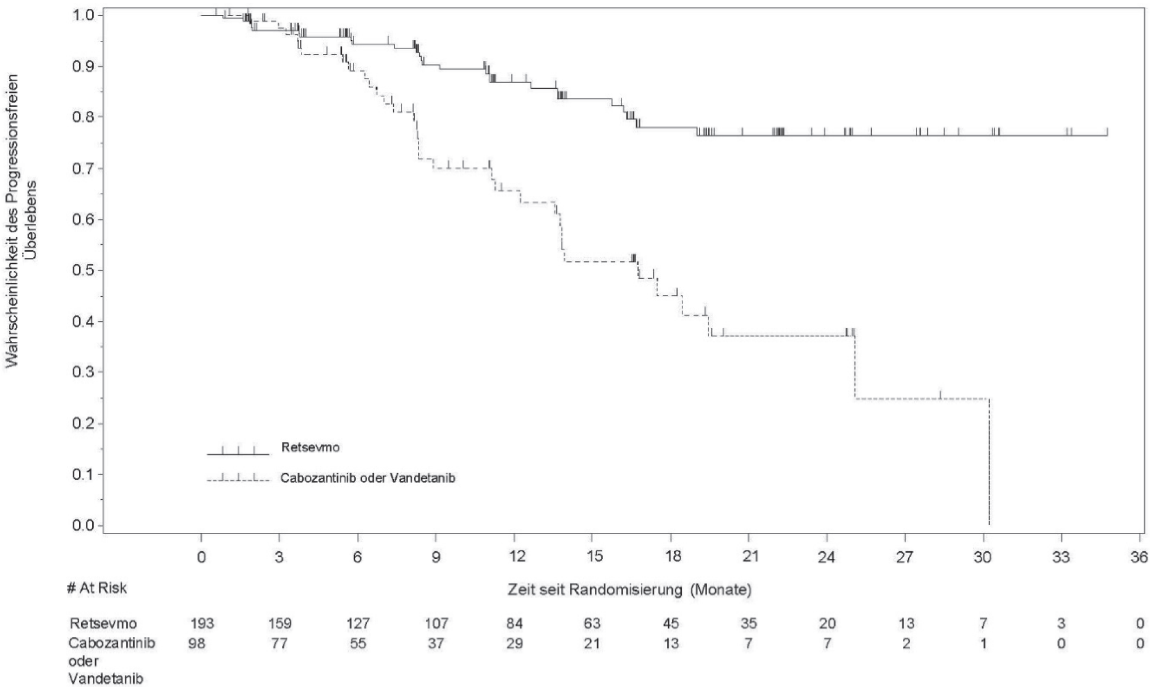
Cut-off Datum: 22. Mai 2023
Zum Zeitpunkt der primären PFS-Analyse wurden 18 OS Ereignisse in den beiden Armen beobachtet. In der ITT-Population betrug die OS HR 0,374 ([95 % CI: 0,147; 0,949]). Die Zensierungsrate betrug 95,9 % im Selpercatinib-Arm und 89,8 % im Kontrollarm.
Die Verträglichkeit wurde bei 242 Patienten (Selpercatinib-Arm, N = 161; Kontrollarm, N = 81) untersucht. Der Selpercatinib-Arm hatte einen statistisch signifikant geringeren Anteil an der Behandlungszeit, bei der die Patienten über „hohe Belastung durch Nebenwirkungen“ (8 %) berichteten, als der Kontrollarm (24 %) (95 % CI: -23 %, -10 %, p < 0,0001), wie durch Functional Assessment of Cancer Therapy Item GP5, Antwort 3 „Ziemlich viel“ oder 4 „Sehr viel“ beurteilt wurde.
Bei einer späteren OS Analyse mit einem Datenschnitt vom 11. März 2024 wurden 26 Ereignisse in den beiden Armen beobachtet und die HR betrug 0,275 (95 % CI: 0,124, 0,608). Die PFS HR für diese Analyse betrug 0,202 (95 % CI: 0,128; 0,320) und die ORR für Selpercatinib 82,4 % im Vergleich zu 43,9 % für den Kontrollarm.
LIBRETTO-001
Von den 324 RET-mutierten MTC-Patienten, die in LIBRETTO-001 eingeschlossen wurden, wurden 143 Patienten nicht mit Cabozantinib und/oder Vandetanib vorbehandelt. Von diesen Patienten waren 116 in Bezug auf andere systemische Vorbehandlungen therapienaiv und 27 haben eine andere systemische Vorbehandlung erhalten. Unter den Cabozantinib- und Vandetanib-naiven Patienten betrug das mediane Alter 57 Jahre (Spanne 15 bis 87 Jahre). 2 Patienten (1,4 %) waren < 18 Jahre alt. 58,0 % der Patienten waren männlich. 86,7 % der Patienten waren Weiße, 5,6 % Asiaten, 1,4 % Schwarze. Die meisten Patienten (97,9 %) hatten bei Studieneinschluss eine metastasierte Erkrankung. Der ECOG-Performancestatus wurde mit 0 oder 1 (95,9 %) oder 2 (4,2 %) angegeben. Die häufigste Mutation war M918T (60 %), gefolgt von extrazellulären Cystein-Mutationen (23,8 %). Die Ergebnisse der Wirksamkeit für nicht mit Cabozantinib und / oder Vandetanib vorbehandelten, RET-mutierten MTC‑Patienten sind in Tabelle 13 zusammengefasst.
Tabelle 13 LIBRETTO-001: Objektives Ansprechen und Dauer des Ansprechens
Patienten für die Wirksamkeitsbeurteilung |
|
N |
143 |
Objektives Ansprechen (CR + PR) |
|
% (95 % CI) |
82,5 (75,3; 88,4) |
komplette Remission (CR) n (%) |
34 (23,8) |
partielle Remission (PR) n (%) |
84 (58,7) |
Dauer des Ansprechens (Monate)* |
|
Median, 95 % CI |
NE (51,3; NE) |
Anteil der Patienten (%) mit Ansprechen nach |
|
12 Monaten (95 % CI) |
91,4 (84,6; 95,3) |
24 Monaten (95 % CI) |
84,1 (75,9; 89,7) |
CI = Konfidenzintervall, CR = komplette Remission, NE = nicht auswertbar, PR = partielle Remission | |
* |
Die mediane Dauer des Follow-ups betrug 39,4 Monate (25. Perzentil: 32,3; 75. Perzentil: 45,4). |
Cut-off Datum: 13. Januar 2023 | |
Vorbehandeltes RET-mutiertes medulläres Schilddrüsen-Karzinom
Von RET-mutierten MTC-Patienten, die in LIBRETTO-001 eingeschlossen wurden, wurden 152 Patienten zuvor mit Cabozantinib und/oder Vandetanib behandelt und waren Grundlage der Wirksamkeitsbeurteilung. Das mediane Alter betrug 58 Jahre (Spanne 17 bis 90 Jahre), 1 Patient (0,7 %) war < 18 Jahre alt. 63,8 % der Patienten waren männlich. 90,1 % der Patienten waren Weiße, 1,3 % Asiaten, 1,3 % Schwarze. Der ECOG-Performancestatus wurde mit 0 oder 1 (92,7 %) oder 2 (7,2 %) angegeben. 98,0 % der Patienten hatten eine metastasierte Erkrankung. Die häufigste Mutation war M918T (65,1 %), gefolgt von extrazellulären
Cystein-Mutationen (15,8 %). 100 % (n = 152) der Patienten mit systemischen Vortherapien erhielten im Median 2 systemische Therapien, 27,6 % (n = 42) erhielten 3 oder mehr.
Die Ergebnisse der Wirksamkeit für vorbehandeltes RET-mutiertes MTC sind in Tabelle 14 zusammengefasst.
Tabelle 14 LIBRETTO-001: Objektives Ansprechen und Dauer des Ansprechens
Patienten für die Wirksamkeitsbeurteilung |
|
N |
152 |
Objektives Ansprechen (CR + PR) |
|
% (95 % CI) |
77,6 (70,2; 84,0) |
komplette Remission (CR) n (%) |
19 (12,5) |
partielle Remission (PR) n (%) |
99 (65,1) |
Dauer des Ansprechens (Monate)* |
|
Median (95 % CI) |
45,3 (33,6; NE) |
Anteil der Patienten (%) mit Ansprechen nach |
|
12 Monaten (95 % CI) |
83,0 (74,6; 88,8) |
24 Monaten (95 % CI) |
66,4 (56,3; 74,7) |
CI = Konfidenzintervall, CR = komplette Remission, NE = nicht auswertbar, PR = partielle Remission | |
* |
Die mediane Dauer des Follow-ups betrug 38,3 Monate (25. Perzentil: 23,0; 75. Perzentil: 46,1). |
Cut-off Datum: 13. Januar 2023 | |
Andere RET-Fusions-positive solide Tumore
Die Wirksamkeit wurde bei 75 Patienten mit RET-Fusions-positiven Tumoren ausgenommen NSCLC und Schilddrüsenkarzinom ausgewertet, bei denen die Erkrankung während oder nach einer früheren systemischen Behandlung fortschritt oder bei denen keine zufriedenstellenden alternativen Behandlungsoptionen zur Verfügung standen. Das mediane Alter betrug 59 Jahre (Spanne 21 bis 92 Jahre); 50,7 % waren weiblich; 60,0 % waren Weiße, 34,7 % Asiaten, und 4,0 % Schwarze; der ECOG-Performancestatus betrug 0 - 1 (90,6 %) oder 2 (9,3 %) und 96,0 % der Patienten hatten eine metastasierte Erkrankung. 69 Patienten (92,0 %) hatten eine systemische Vortherapie mit einem Median von 2 systemischen Vortherapien (Spanne 0 bis 9) und 36,0 % hatten 3 oder mehr systemische Vortherapien erhalten. Keiner der Patienten war zuvor mit einem selektiven RET-Inhibitor behandelt worden. Die häufigsten Krebsarten waren Kolon- (29,3 %), Pankreas- (24,0 %) und Speicheldrüsenkrebs (6,7 %) gefolgt von Sarkomen (6,7 %) und Cholangiokarzinomen (6,7 %). Die häufigsten Fusionspartner waren NCOA4 (38,7 %), CCDC6 (20,0 %), und KIF5B (8,0 %). Die Ergebnisse der Wirksamkeit für RET-Fusions-positive solide Tumore ausgenommen NSCLC und Schilddrüsenkarzinom sind in Tabelle 15 und 16 zusammengefasst.
Tabelle 15 LIBRETTO-001: Objektives Ansprechen und Dauer des Ansprechens
Patienten für die Wirksamkeitsbeurteilung |
||
N |
75 |
|
Objektives Ansprechen (CR+PR) |
||
% (95 % CI) |
46,7 (35,1; 58,6) |
|
komplette Remission (CR) n (%) |
4 (5,3) |
|
partielle Remission (PR) n (%) |
31 (41,3) |
|
Dauer des Ansprechens (Monate) * | ||
Median (95 % CI) |
24,54 (11,2; 49,1) |
|
Anteil der Patienten (%) mit Ansprechen nach |
||
≥ 6 Monaten |
(95 % CI) |
82,0 (64,2; 91,5) |
≥ 12 Monaten |
(95 % CI) |
68,6 (49,3; 81,8) |
≥ 24 Monaten |
(95 % CI) |
52,5 (32,6; 69,0) |
≥ 36 Monaten |
(95 % CI) |
43,3 (24,0; 61,1) |
* |
Die mediane Dauer des Follow-ups betrug 32,23 Monate |
CI = Konfidenzintervall, CR = komplette Remission, NE = nicht auswertbar, PR = partielle Remission | |
Cut-off Datum: 14. Februar 2025 | |
Tabelle 16 LIBRETTO-001: Objektives Ansprechen und Dauer des Ansprechens nach Tumorgewebe
Tumorgewebe |
Patienten |
ORR (IRC Bewertung) |
DOR |
|
n (%) |
95 % CI |
|||
Kolorektal |
22 |
10 (45,5) |
24,4; 67,8 |
4,63; 36,14+ |
Pankreas |
18 |
9 (50,0) |
26,0; 74,0 |
2,50; 52,14 |
Speicheldrüse |
5 |
3 (60,0) |
14,7; 94,7 |
5,72; 37,19 |
Gallenwege |
5 |
2 (40,0) |
5,3; 85,3 |
7,36; 14,82 |
Sarkom |
5 |
2 (40,0) |
5,3; 85,3 |
3,71; 56,51+ |
Haut |
3 |
1 (33,3) |
0,8; 90,6 |
27,14 |
Unbekannter Primärtumor (CUP) |
3 |
1 (33,3) |
0,8; 90,6 |
9,23 |
Brust |
2 |
PR, CR |
15,8; 100,0 |
2,30+; 17,28 |
Xanthogranulom |
2 |
NE, NE a |
0,0; 84,2 |
NA |
Karzinoid |
1 |
PR |
2,5; 100,0 |
49,08 |
Ovar |
1 |
PR |
2,5; 100,0 |
28,55+ |
Pulmonales Karzinosarkom |
1 |
NE |
0,0; 97,5 |
NA |
Rektum neuroendokrin |
1 |
NE |
0,0; 97,5 |
NA |
Dünndarm |
1 |
CR |
2,5; 100,0 |
24,54 |
Neuroendokrin |
1 |
PR |
2,5; 100,0 |
23,13 |
Kleinzelliges Lungenkarzinom |
1 |
SD |
0,0; 97,5 |
NA |
Speiseröhre |
1 |
SD |
0,0; 97,5 |
NA |
Pankreas neuroendokrin |
1 |
PR |
2,5; 100,0 |
17,51+ |
Magen |
1 |
SD |
0,0; 97,5 |
NA |
+ |
kennzeichnet ein anhaltendes Ansprechen. |
a |
Ein Xanthogranulom Patient hatte eine Ausprägung, die nicht durch das IRC bewertet werden konnte, da die Haut der einzige Ort der Erkrankung war. Basierend auf der Einschätzung des Prüfers hatte dieser Patient eine komplette Remission. |
CI = Konfidenzintervall, CR = komplette Remission, DOR = Dauer des Ansprechens, NA = nicht zutreffend, NE = nicht auswertbar, ORR = objektive Ansprechrate, PR = partielle Remission, SD = stabile Erkrankung. | |
Cut-off Datum: 14. Februar 2025 | |
Aufgrund der Seltenheit von RET-Fusions-positivem Krebs wurden Patienten über eine Vielzahl von Tumorarten hinweg untersucht, wobei bei einigen Tumorarten aufgrund begrenzter Patientenzahlen Unsicherheiten in der Schätzung der ORR je Tumorart bestehen.
Die ORR in der Gesamtpopulation kann möglicherweise vom zu erwartenden Ansprechen bei einem bestimmten Tumortyp abweichen.
Kinder und Jugendliche
Bis zum 08. November 2024 wurde die Wirksamkeit bei 36 Patienten im Alter von 2 bis ≤ 20 Jahren untersucht, die im Rahmen von LIBRETTO‑121, einer Phase‑1/2‑Studie bei pädiatrischen Patienten, behandelt wurden.
Die Studie umfasst Patienten mit einem fortgeschrittenen soliden Tumor oder einem primären ZNS‑Tumor, der eine aktivierende RET‑Veränderung aufweist. Von diesen 36 Patienten waren 31 jünger als 18 Jahre. Von den 36 Patienten hatten 14 ein RET‑mutiertes medulläres Schilddrüsenkarzinom (MTC), 1 Patient ein RET‑Fusions‑positives MTC, 15 Patienten ein RET‑Fusions‑positives Schilddrüsenkarzinom, 3 Patienten RET‑Fusions‑positive andere solide Tumore sowie 3 Patienten RET‑mutierte andere solide Tumore.
Von den 14 Patienten mit RET‑mutiertem MTC waren 11 Patienten jünger als 18 Jahre, davon 5 Patienten jünger als 12 Jahre. Für alle 14 Patienten betrug gemäß Bewertung durch das unabhängige Review‑Komitee (IRC) die objektive Ansprechrate (ORR) 57,1 % (95 % CI: 28,9; 82,3). 5 Patienten zeigten eine bestätigte komplette Remission, während 3 Patienten eine bestätigte partielle Remission aufwiesen.
Für diese 14 Patienten war die mediane Ansprechdauer (in Monaten) nicht schätzbar (95 % CI: 24,9; NE). Der Anteil der Patienten mit einer Ansprechdauer von ≥ 24 Monaten betrug 100 % (95 % CI: 100; 100) und bei ≥ 30 Monaten 83,3 % (95 % CI: 27,3; 97,5).
Von den 15 Patienten mit RET‑Fusions positivem Schilddrüsenkarzinom waren 13 Patienten jünger als 18 Jahre, davon 3 Patienten jünger als 12 Jahre. Von diesen 15 Patienten waren 7 Patienten zuvor mit radioaktivem Iod behandelt worden. Für alle 15 Patienten betrug gemäß Bewertung durch das unabhängige Review‑Komitee (IRC) die objektive Ansprechrate (ORR) 53,3 % (95 % CI: 26,6; 78,7). 4 Patienten zeigten eine bestätigte komplette Remission, während 4 Patienten eine bestätigte partielle Remission aufwiesen. Für diese 15 Patienten war die mediane Ansprechdauer (in Monaten) nicht schätzbar (95 % CI: NE; NE). Der Anteil der Patienten mit einer Ansprechdauer von ≥ 42 Monaten betrug 100 % (95 % CI: 100; 100).
Von den 3 Patienten mit RET‑Fusions positiven anderen soliden Tumoren waren alle 3 Patienten jünger als 18 Jahre, davon 1 Patient jünger als 12 Jahre. Von diesen 3 Patienten zeigten 2 Patienten eine bestätigte partielle Remission.
Von den 3 Patienten mit RET‑mutierten anderen soliden Tumoren waren ebenfalls alle 3 Patienten jünger als 18 Jahre, davon 2 Patienten jünger als 12 Jahre. Bei diesen 3 Patienten mit RET‑mutierten anderen soliden Tumoren wurde kein Ansprechen auf die Behandlung mit Selpercatinib beobachtet.
Die Europäische Arzneimittel-Agentur hat für Selpercatinib eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in der pädiatrischen Altersklasse von 6 Monaten und jünger in soliden Tumoren gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Selpercatinib-Pharmakokinetik wurde bei Patienten mit lokal fortgeschrittenem oder metastasiertem soliden Tumor ausgewertet, denen, sofern nicht anders angegeben, zweimal täglich 160 mg verabreicht wurden. Der Selpercatinib AUC- und Cmax-Steady-state stieg linear bis überlinear von 20 mg einmal täglich bis 240 mg zweimal täglich proportional zur Dosis an.
Der Steady-state wurde nach ungefähr 7 Tagen erreicht, und das mediane Akkumulationsverhältnis nach Verabreichung von 160 mg zweimal täglich betrug das 3,4‑fache. Der mittlere Selpercatinib-Steady-state [Variationskoeffizient (CV %)] von Cmax betrug 2 980 (53 %) ng/ml und von AUC0 - 24h 51 600 (58 %) ng*h/ml.
In vivo Studien weisen darauf hin, dass Selpercatinib ein schwacher Inhibitor von P-gp ist.
In vitro Studien zeigen, dass Selpercatinib bei klinisch relevanten Konzentrationen CYP1A2, CYP2B6, CYP2C9, CYP2C19 oder CYP2D6 weder inhibiert noch induziert.
In vitro Studien zeigen, dass Selpercatinib MATE1 und BCRP inhibiert, während es OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, BSEP und MATE2‑K bei klinisch relevanten Konzentrationen nicht inhibiert. Selpercatinib kann das Serum-Kreatinin erhöhen, indem es die renale tubuläre Kreatinin-Sekretion über die Hemmung von MATE1 vermindert.
Die Hartkapsel- und Filmtablettendarreichungsformen von Selpercatinib sind bioäquivalent.
Resorption
Retsevmo wird nach einer oralen Gabe von 160 mg schnell aufgenommen - mit einem Tmax von ungefähr 2 Stunden. Die absolute orale Bioverfügbarkeit von Selpercatinib betrug im geometrischen Mittel 73,2 % (Spanne: 60,2 - 81,5 %).
Auswirkung von Nahrung
Bei gesunden Probanden war die Selpercatinib-AUC und Cmax nach oraler Verabreichung einer einzigen 160 mg Dosis auf nüchternen Magen im Vergleich zur Selpercatinib-AUC nach fettreichem Essen um 9 % erhöht und Cmax um 14 % erniedrigt. Diese Veränderungen wurden als nicht klinisch relevant erachtet. Deshalb kann Selpercatinib mit oder ohne Nahrung verabreicht werden.
Verteilung
Das durch die Populations-PK-Analyse geschätzte mittlere scheinbare Verteilungsvolumen (Vss/F) von Selpercatinib (CV %) beträgt nach oraler Verabreichung bei erwachsenen Patienten 203,1 Liter (69 %). Selpercatinib liegt in vitro zu 96 % gebunden an humanem Plasmaprotein vor und die Bindung ist unabhängig von der Konzentration. Das Verhältnis der Blut-/Plasma-Konzentration beträgt 0,7.
Biotransformation
Selpercatinib wird vorwiegend über CYP3A4 metabolisiert. Nach oraler Verabreichung einer einzelnen radioaktiv markierten [14C] 160 mg-Dosis von Selpercatinib an gesunde Probanden machte das unveränderte Selpercatinib 86 % der gemessenen radioaktiven Komponenten im Plasma aus.
Elimination
Die mittlere scheinbare Clearance (CL/F) von Selpercatinib (CV %) beträgt 5,5 l/h (45 %) und die Halbwertszeit nach oraler Verabreichung beträgt bei erwachsenen Patienten 26,5 Stunden. Nach oraler Verabreichung einer einzelnen radioaktiv markierten [14C] 160 mg-Dosis von Selpercatinib an gesunde Probanden wurden 69 % (14 % unverändert) der verabreichten Radioaktivität im Stuhl und 24 % im Urin (11,5 % unverändert) wiedergefunden.
Spezielle Populationen
Alter, Geschlecht und Körpergewicht
Alter (Spanne: 12 Jahre bis 92 Jahre) oder Geschlecht hatten keinen klinisch bedeutsamen Effekt auf die Pharmakokinetik von Retsevmo. Das scheinbare Verteilungsvolumen (Vss/F) und die scheinbare Clearance (CL/F) von Selpercatinib nehmen mit steigendem Körpergewicht (von 9,6 kg bis 179 kg) zu.
Patienten ab 12 Jahren mit einem Körpergewicht < 50 kg sollten die Retsevmo-Behandlung mit einer Dosis von 120 mg zweimal täglich starten, während Patienten mit ≥ 50 kg mit 160 mg zweimal täglich beginnen sollten.
Pädiatrische Patienten im Alter von 2 bis unter 12 Jahren sollten die Behandlung mit Retsevmo mit einer Dosis beginnen, die anhand ihrer Körperoberfläche bestimmt wird (siehe Tabelle 1).
Retsevmo wird für pädiatrische Patienten mit einer Körperoberfläche von weniger als 0,45 m² nicht empfohlen.
Eingeschränkte Leberfunktion
Die Selpercatinib-AUC0-∞ wurde bei Probanden mit einer Child-Pugh-Klassifikation von „leicht“ um 7 % und bei Probanden mit Child-Pugh-Klassifikation von „moderat“ um 32 % erhöht. Somit ist die Selpercatinib-Exposition (AUC) bei Probanden mit leichter und moderater Leberfunktionseinschränkung (Child-Pugh Klasse A und B) vergleichbar mit der Exposition bei gesunden Probanden, wenn eine Dosis von 160 mg verabreicht wird.
Die Selpercatinib-AUC0-∞ wurde bei Probanden mit schwerer Leberfunktionseinschränkung (Child-Pugh Klasse C) um 77 % erhöht. Klinische Daten über die Sicherheit von Selpercatinib bei Patienten mit schwerer Leberfunktionseinschränkung sind nur begrenzt verfügbar. Deshalb werden bei Patienten mit schwerer Leberfunktionseinschränkung Dosisanpassungen empfohlen (Abschnitt 4.2).
Eingeschränkte Nierenfunktion
In einer klinisch-pharmakologischen Studie, bei der eine Einzeldosis Selpercatinib 160 mg verabreicht wurde, war die Exposition (AUC) bei Patienten mit leichter, mittelschwerer oder schwerer Nierenfunktionseinschränkung unverändert. Patienten mit terminaler Niereninsuffizienz (eGFR < 15 ml/min) und dialysepflichtige Patienten wurden nicht untersucht.
Kinder und Jugendliche
Die Expositionen von Selpercatinib bei pädiatrischen Patienten ab einem Alter von 2 Jahren sind vergleichbar mit denen bei erwachsenen Patienten, die mit den empfohlenen Dosierungen behandelt wurden.
Zur Charakterisierung der Toxizität wurden Studien mit wiederholter Verabreichung an juvenilen und heranwachsenden/erwachsenen Ratten und heranwachsenden/erwachsenen Minischweinen durchgeführt. Zielorgane der Toxizität bei Ratten und Minischweinen waren das hämatopoetische System, das lymphatische Gewebe, die Zunge, der Pankreas, der Gastrointestinaltrakt, die epiphysäre Wachstumsplatte und das männliche Reproduktionsgewebe. Mit Ausnahme der testikulären Toxizität in heranwachsenden/erwachsenen und juvenilen Tieren und den Veränderungen der Wachstumsfugen bei juvenilen Ratten waren die Toxizitäten in diesen Organen reversibel. Eine reversible Toxizität in den Eierstöcken wurde nur bei Minischweinen beobachtet. Bei Gabe hoher Dosen führte die gastrointestinale Toxizität bei Minischweinen zur Morbidität, wobei die Exposition niedriger war als die beim Menschen bei empfohlener Dosierung. In einer Minischwein-Studie wiesen Weibchen eine leichte, reversible QTc-Verlängerung von etwa 12 % im Vergleich zur Kontrollgruppe und eine Verlängerung von 7 % im Vergleich zum Ausgangswert auf. Nur bei Ratten wurden folgende Zielorgane der Toxizität festgestellt: Schneidezahn, Leber, Vagina, Lunge, Brunner-Drüsen und Multi-Gewebe-Mineralisierung assoziiert mit Hyperphosphatämie. Diese Toxizitäten, die nur in den Organen von Ratten auftraten, waren reversibel.
Juvenile Toxizität
Eine Selpercatinib-Exposition, die etwa der 0,5‑ bis 2‑ fachen Exposition bei erwachsenen Patienten entsprach, führte zu einer erhöhten Sterblichkeit bei Ratten, die jünger als 21 Tage alt waren. Eine vergleichbare Exposition wurde von Ratten, die 21 Tage und älter waren, toleriert.
Juvenile und heranwachsende/erwachsene Ratten sowie heranwachsende/erwachsene Minischweine, alle mit offenen Wachstumsfugen, denen Selpercatinib verabreicht wurde, zeigten mikroskopische Veränderungen in Form von Hypertrophie, Hyperplasie und Dysplasie des Wachstumsfugenknorpels (Physe). Bei juvenilen Ratten war die Dysplasie an den Wachstumsfugen irreversibel und mit einer verringerten Femurlänge und einer Verringerung der Knochenmineraldichte verbunden. Skelettale Veränderungen wurden bei Expositionsleveln beobachtet, die denen von erwachsenen Patienten mit der empfohlenen Dosis von 160 mg BID entsprachen.
Juvenile männliche Ratten, denen Selpercatinib verabreicht wurde und die man nach Beendigung der Therapie das reproduktive Alter erreichen ließ, zeigten eine verringerte Reproduktionsleistung, wenn sie sich mit unbehandelten weiblichen Ratten paarten. Verringerte Fruchtbarkeits- und Kopulationsindizes, erhöhte Prä- und Postimplantationsverluste und eine verringerte Anzahl lebensfähiger Embryonen wurden bei einer Exposition beobachtet, die etwa dem 3,4‑fachen der wirksamen Exposition bei Erwachsenen entsprach.
Genotoxizität
Selpercatinib ist in therapeutischer Dosierung nicht genotoxisch. In einem in vivo Mikronukleus-Assay bei Ratten war Selpercatinib positiv bei Konzentrationen über das 7‑fache des Cmax-Wertes bei der menschlichen Dosis von 160 mg zweimal täglich. In einem in vitro Mikronukleus-Assay mit humanen peripheren Blutlymphozyten zeigte sich ein uneindeutiges Ergebnis bei einer Konzentration von etwa dem 485‑fachen Cmax bei der humanen Dosis.
Mutagenese
Selpercatinib hat in einem bakteriellen Mutagenitäts-Assay zu keinen Mutationen geführt.
Karzinogenität
In einer 2-jährigen Karzinogenitätsstudie mit Selpercatinib bei Ratten wurden bei einigen weiblichen Tieren Vaginaltumore bei Plasmakonzentrationen beobachtet, die denen bei erwachsenen Patienten ähnlich waren, die mit einer Dosis von 160 mg zweimal täglich behandelt wurden. Es wurden keine präneoplastischen Veränderungen im Fortpflanzungstrakt weiblicher Ratten beobachtet. Die klinische Relevanz dieser Befunde ist unbekannt. Selpercatinib war in dieser Studie bei männlichen Ratten nicht karzinogen.
Selpercatinib war in einer 6-monatigen Studie bei männlichen und weiblichen Mäusen nicht karzinogen.
Embryotoxizität / Teratogenität
Basierend auf Reproduktionsstudien an Tieren und dem Wirkmechanismus kann die Verabreichung von Selpercatinib bei schwangeren Frauen fetale Schädigungen auslösen. Bei schwangeren Ratten führte eine Behandlung mit Selpercatinib, die etwa der empfohlenen Humandosis von 160 mg zweimal täglich entsprach, während der Organentwicklung zur Zeit der mütterlichen Exposition zu Embryoletalität und Missbildungen.
Reproduktionstoxizität
Ergebnisse von den an Ratten und Minischweinen durchgeführten Studien deuten darauf hin, dass Selpercatinib die Fruchtbarkeit von Männern und Frauen beeinträchtigen könnte.
In einer Fertilitätsstudie an männlichen Ratten wurde eine dosisabhängige Depletion von Keimzellen und eine Retention von Spermatiden bei subklinischer AUC-definierter Exposition (das 0,2‑fache der klinischen Exposition bei der empfohlenen Humandosis) beobachtet. Diese Effekte waren mit vermindertem Gewicht der Organe, reduzierter Samen-Beweglichkeit und einer Erhöhung der Anzahl an abnormalen Spermien bei AUC-definierter Exposition, die etwa doppelt so hoch war wie die klinische Exposition bei empfohlener Humandosis, verbunden. Die mikroskopischen Befunde der Fertilitätsstudie an männlichen Ratten stimmten mit den Beobachtungen in Studien bei wiederholter Gabe an Ratten und Minischweinen überein, in denen eine dosisabhängige, nicht-reversible Hodendegeneration mit reduzierten Luminalspermien in den Nebenhoden bei subklinisch AUC-definierter Exposition assoziiert war (0,1- bis 0,4‑faches der klinischen Exposition bei empfohlener Humandosis).
In einer Fertilitäts- und frühen Embryonalstudie an weiblichen Ratten wurden eine Reduktion der Anzahl an Östruszyklen und Embryoletalität bei AUC-definierter Exposition, die etwa gleich der klinischen Exposition bei empfohlener Humandosis war, beobachtet. In Studien bei wiederholter Gabe an Ratten wurden reversible vaginale Muzifikationen mit individuellen Zellverhornungen und veränderten Östruszyklen bei klinisch relevanter AUC-definierter Exposition beobachtet. Bei Minischweinen wurden reduzierte Gelbkörper und/oder Gelbkörper-Zysten bei subklinisch AUC-definierter klinischer Exposition beobachtet (0,07- bis 0,3‑faches der klinischen Exposition bei empfohlener Humandosis).
Kapselinhalt
Mikrokristalline Cellulose
Hochdisperses Siliciumdioxid
Kapselhülle
Retsevmo® 40 mg Hartkapseln
Gelatine
Titandioxid (E 171)
Eisen(Ⅱ, Ⅲ)-oxid
Retsevmo® 80 mg Hartkapseln
Gelatine
Titandioxid (E 171)
Brillantblau FCF (E 133)
Kapseln: Zusammensetzung der schwarzen Farbe
Schellack
Ethanol 96 %,
2-Propanol (Ph. Eur.)
Butan-1-ol
Propylenglycol
Gereinigtes Wasser
Konzentrierte Ammoniak-Lösung
Kaliumhydroxid
Eisen(Ⅱ, Ⅲ)-oxid
Nicht zutreffend.
3 Jahre.
Dieses Arzneimittel bedarf keiner besonderen Vorsichtsmaßnahmen für die Aufbewahrung.
Plastikflasche
Jede Packung enthält 1 HDPE-Flasche mit einem Plastik-Schraubdeckel.
Retsevmo® 40 mg Hartkapseln
Retsevmo 40 mg Hartkapseln werden in HDPE-Flaschen mit 60 Kapseln bereitgestellt.
Retsevmo® 80 mg Hartkapseln
Retsevmo 80 mg Hartkapseln werden in HPDE-Flaschen mit 60 oder 120 Kapseln bereitgestellt.
Blisterpackung
Retsevmo® 40 mg Hartkapseln
werden in PCTFE/PVC-Blisterpackungen bereitgestellt. Diese sind mit einer Aluminiumfolie in einer Blisterkarte versiegelt und sind in Packungen mit 14, 42, 56 oder 168 Hartkapseln verfügbar.
Retsevmo® 80 mg Hartkapseln
werden in PCTFE/PVC-Blisterpackungen bereitgestellt. Diese sind mit einer Aluminiumfolie in einer Blisterkarte versiegelt und sind in Packungen mit 14, 28, 56 oder 112 Hartkapseln verfügbar.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Eli Lilly Nederland B.V.
Orteliuslaan 1000
3528 BD Utrecht
Niederlande
EU/1/20/1527/001
EU/1/20/1527/002
EU/1/20/1527/003
EU/1/20/1527/004
EU/1/20/1527/005
EU/1/20/1527/006
EU/1/20/1527/007
EU/1/20/1527/008
EU/1/20/1527/009
EU/1/20/1527/010
EU/1/20/1527/011
Datum der Erteilung der Zulassung: 11. Februar 2021
Datum der letzten Verlängerung der Zulassung: 12. Dezember 2025
Juni 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig
Retsevmo® 40 mg Hartkapseln
Inhalt |
PZN |
56 Hartkapseln |
17533568 |
168 Hartkapseln |
17533574 |
Retsevmo® 80 mg Hartkapseln
Inhalt |
PZN |
56 Hartkapseln |
17533580 |
168 Hartkapseln |
17533597 |
Lilly Deutschland GmbH
Werner-Reimers-Straße 2 - 4
D-61352 Bad Homburg
Tel. +49-(0) 6172 273 2222