
XELJANZ® 1 mg/ml Lösung zum Einnehmen
Jeder ml Lösung zum Einnehmen enthält Tofacitinibcitrat, entsprechend 1 mg Tofacitinib.
Sonstige Bestandteile mit bekannter Wirkung
Jeder ml Lösung zum Einnehmen enthält 2,39 mg Propylenglycol.
Jeder ml Lösung zum Einnehmen enthält 0,9 mg Natriumbenzoat.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Lösung zum Einnehmen
Klare, farblose Lösung.
Tofacitinib ist indiziert zur Behandlung der aktiven polyartikulären juvenilen idiopathischen Arthritis (Rheumafaktor-positive [RF+] oder -negative [RF-] Polyarthritis und erweiterte Oligoarthritis) und der juvenilen Psoriasis-Arthritis (PsA) bei Patienten ab einem Alter von 2 Jahren, die auf eine vorangegangene krankheitsmodifizierende antirheumatische (DMARD-)Therapie unzureichend angesprochen haben.
Tofacitinib kann in Kombination mit Methotrexat (MTX) angewendet werden oder als Monotherapie, wenn MTX nicht vertragen wird oder eine Fortsetzung der Behandlung mit MTX ungeeignet ist.
Die Behandlung sollte von Fachärzten eingeleitet und überwacht werden, die über Erfahrung in der Diagnose und Behandlung von Krankheiten verfügen, für die Tofacitinib indiziert ist.
Dosierung
Tofacitinib kann als Monotherapie oder in Kombination mit Methotrexat (MTX) angewendet werden.
Die empfohlene Dosis bei Patienten ab einem Alter von 2 Jahren basiert auf den folgenden Gewichtskategorien:
Tabelle 1: Tofacitinib-Dosis bei Patienten mit polyartikulärer juveniler idiopathischer Arthritis und juveniler PsA ab einem Alter von zwei Jahren
Körpergewicht (kg) |
Dosisschema |
10 ‑ < 20 |
3,2 mg (3,2 ml Lösung zum Einnehmen) zweimal täglich |
20 ‑ < 40 |
4 mg (4 ml Lösung zum Einnehmen) zweimal täglich |
≥ 40 |
5 mg (5 ml Lösung zum Einnehmen oder eine 5 mg Filmtablette) zweimal täglich |
Patienten mit einem Körpergewicht ≥ 40 kg, die mit Tofacitinib 5 ml Lösung zum Einnehmen zweimal täglich behandelt werden, können auf Tofacitinib 5 mg Filmtabletten zweimal täglich umgestellt werden. Patienten mit einem Körpergewicht < 40 kg können nicht von der Tofacitinib Lösung zum Einnehmen umgestellt werden.
Dosisanpassung
Eine Dosisanpassung ist bei gleichzeitiger Anwendung mit MTX nicht erforderlich.
Unterbrechen und Absetzen der Behandlung
Verfügbare Daten lassen darauf schließen, dass eine klinische Verbesserung innerhalb von 18 Wochen nach der Einleitung der Behandlung mit Tofacitinib eintritt. Eine Fortsetzung der Behandlung bei Patienten, bei denen es innerhalb dieses Zeitraums zu keiner klinischen Verbesserung kommt, sollte sorgfältig überdacht werden.
Falls es bei einem Patienten zu einer schwerwiegenden Infektion kommt, ist die Behandlung mit Tofacitinib zu unterbrechen, bis die Infektion unter Kontrolle ist.
Bei dosisbezogenen anormalen Laborbefunden wie Lymphopenie, Neutropenie und Anämie kann eine Dosisunterbrechung erforderlich sein. Entsprechend den Angaben in den nachfolgenden Tabellen 2, 3 und 4 richten sich die Empfehlungen für eine Unterbrechung oder einen Abbruch der Behandlung nach dem Schweregrad der Laborwertveränderungen (siehe Abschnitt 4.4).
Bei Kindern und Jugendlichen mit einer absoluten Lymphozytenzahl (absolute lymphocyte count, ALC) unter 750 Zellen/mm3 sollte keine Behandlung eingeleitet werden.
Tabelle 2: Niedrige absolute Lymphozytenzahl
Niedrige absolute Lymphozytenzahl (ALC) (siehe Abschnitt 4.4) | |
Laborwert |
Empfehlung |
ALC größer oder gleich 750 |
Dosis sollte beibehalten werden. |
ALC 500-750 |
Bei einer anhaltenden Abnahme (2 Werte in Folge in diesem Bereich bei Routineuntersuchungen) sollte die Dosis reduziert oder die Behandlung unterbrochen werden, bis die ALC größer als 750 ist. Bei Patienten, die zweimal täglich 5 mg Tofacitinib erhalten, sollte die Behandlung unterbrochen werden. Bei einer ALC über 750 sollte die Behandlung im klinisch angemessenen Rahmen wieder aufgenommen werden. |
ALC unter 500 |
Wenn der Laborwert bei einem innerhalb von 7 Tagen durchgeführten erneuten Test bestätigt wird, sollte die Behandlung beendet werden. |
Bei Kindern und Jugendlichen mit einer absoluten Neutrophilenzahl (ANC) unter 1.200 Zellen/mm3 sollte keine Behandlung eingeleitet werden.
Tabelle 3: Niedrige absolute Neutrophilenzahl
Niedrige absolute Neutrophilenzahl (ANC) (siehe Abschnitt 4.4) | |
Laborwert |
Empfehlung |
ANC über 1.000 |
Dosis sollte beibehalten werden. |
ANC 500‑1.000 |
Bei einer anhaltenden Abnahme (2 Werte in Folge in diesem Bereich bei Routineuntersuchungen) sollte die Dosis reduziert oder die Behandlung unterbrochen werden, bis die ANC größer als 1.000 ist. Bei Patienten, die zweimal täglich 5 mg Tofacitinib erhalten, sollte die Behandlung unterbrochen werden. Bei einer ANC über 1.000 sollte die Behandlung im klinisch angemessenen Rahmen wieder aufgenommen werden. |
ANC unter 500 |
Wenn der Laborwert bei einem innerhalb von 7 Tagen durchgeführten erneuten Test bestätigt wird, sollte die Behandlung beendet werden. |
Bei Kindern und Jugendlichen mit einem Hämoglobinwert unter 10 g/dl sollte keine Behandlung eingeleitet werden.
Tabelle 4: Niedriger Hämoglobinwert
Niedriger Hämoglobinwert (siehe Abschnitt 4.4) | |
Laborwert |
Empfehlung |
Hb-Abfall bis 2 g/dl und ein Hb-Wert von 9,0 g/dl oder darüber |
Dosis sollte beibehalten werden. |
Hb-Abfall um mehr als 2 g/dl oder Hb-Wert von weniger als 8,0 g/dl |
Die Behandlung sollte unterbrochen werden, bis sich der Hämoglobinwert normalisiert hat. |
Wechselwirkungen
Die tägliche Tofacitinib-Gesamtdosis sollte bei Patienten, die 5 mg Filmtabletten oder eine äquivalente Dosis basierend auf dem Körpergewicht zweimal täglich und starke Inhibitoren von Cytochrom P450 (CYP) 3A4 (z. B. Ketoconazol) oder gleichzeitig mindestens ein Arzneimittel erhalten, das zu einer mittelstarken Hemmung von CYP3A4 und zu einer starken Hemmung von CYP2C19 führt (z. B. Fluconazol), auf 5 mg Filmtabletten einmal täglich oder eine äquivalente Dosis basierend auf dem Körpergewicht einmal täglich verringert werden (siehe Abschnitt 4.5).
Besondere Patientengruppen
Ältere Patienten
Die Sicherheit und Wirksamkeit von Tofacitinib Lösung zum Einnehmen bei älteren Patienten wurde nicht untersucht.
Eingeschränkte Leberfunktion
Tabelle 5: Dosisanpassung bei eingeschränkter Leberfunktion
Kategorie Leberfunktions-störung |
Klassifizierung |
Dosisanpassung bei eingeschränkter Leberfunktion für die Lösung zum Einnehmen |
Leicht |
Child Pugh A |
Keine Dosisanpassung erforderlich. |
Mittelschwer |
Child Pugh B |
Die Dosis sollte auf einmal täglich 5 mg oder eine äquivalente Dosis gemäß dem Körpergewicht reduziert werden, wenn die angezeigte Dosis bei normaler Leberfunktion zweimal täglich 5 mg oder eine äquivalente Dosis gemäß dem Körpergewicht beträgt (siehe Abschnitt 5.2). |
Schwer |
Child Pugh C |
Tofacitinib darf bei Patienten mit schwerer Leberfunktionsstörung nicht angewendet werden (siehe Abschnitt 4.3). |
Eingeschränkte Nierenfunktion
Tabelle 6: Dosisanpassung bei eingeschränkter Nierenfunktion
Kategorie Nierenfunktions-störung |
Kreatinin-Clearance |
Dosisanpassung bei eingeschränkter Nierenfunktion für die Lösung zum Einnehmen |
Leicht |
50–80 ml/min |
Keine Dosisanpassung erforderlich. |
Mittelschwer |
30–49 ml/min |
Keine Dosisanpassung erforderlich. |
Schwer (einschließlich Patienten, die sich einer Hämodialyse unterziehen) |
< 30 ml/min |
Die Dosis sollte auf einmal täglich 5 mg oder eine äquivalente Dosis gemäß dem Körpergewicht reduziert werden, wenn die angezeigte Dosis bei normaler Nierenfunktion zweimal täglich 5 mg oder eine äquivalente Dosis gemäß dem Körpergewicht beträgt. Bei Patienten mit schwerer Nierenfunktionsstörung sollte eine reduzierte Dosis auch nach der Hämodialyse beibehalten werden (siehe Abschnitt 5.2). |
Kinder (unter einem Alter von 2 Jahren)
Die Sicherheit und Wirksamkeit von Tofacitinib bei Kindern unter einem Alter von 2 Jahren sind nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Zum Einnehmen
Tofacitinib Lösung zum Einnehmen sollte unter Verwendung des Einpress-Flaschenadapters und der Applikationsspritze für Zubereitungen zum Einnehmen, die der Packung beiliegen, angewendet werden.
Tofacitinib kann mit oder ohne Nahrung eingenommen werden.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile
Aktive Tuberkulose (TB), schwerwiegende Infektionen wie z. B. Sepsis oder opportunistische Infektionen (siehe Abschnitt 4.4)
Schwere Leberfunktionsstörung (siehe Abschnitt 4.2)
Schwangerschaft und Stillzeit (siehe Abschnitt 4.6)
Bei folgenden Patienten sollte Tofacitinib nur angewendet werden, wenn keine geeigneten Behandlungsalternativen zur Verfügung stehen: |
Kombination mit anderen Therapien
Die Anwendung von Tofacitinib in Kombination mit Biologika, wie TNF-Antagonisten, Interleukin(IL)-1R-Antagonisten, IL-6R-Antagonisten, monoklonalen Anti-CD20-Antikörpern, IL‑17-Antagonisten, IL‑12/IL‑23-Antagonisten, Integrin-Antikörpern, selektiven Co-Stimulations-Modulatoren und starken Immunsuppressiva wie Azathioprin, 6-Mercaptopurin, Ciclosporin und Tacrolimus wurde nicht untersucht und ist aufgrund der Möglichkeit einer verstärkten Immunsuppression und eines erhöhten Infektionsrisikos zu vermeiden.
In klinischen RA-Studien traten bei der Kombination von Tofacitinib mit MTX Nebenwirkungen häufiger auf als bei der Monotherapie mit Tofacitinib.
Die Anwendung von Tofacitinib in Kombination mit Phosphodiesterase-4-Hemmern wurde in klinischen Tofacitinib-Studien nicht untersucht.
Venöse thromboembolische Ereignisse (VTE)
Schwerwiegende VTE-Ereignisse, einschließlich Lungenembolien (LE), einige davon mit tödlichem Verlauf, zerebrale Sinusvenenthrombosen (CVST) und tiefe Venenthrombosen (TVT) wurden bei Patienten beobachtet, die Tofacitinib einnahmen (sehen Sie Tabelle 7 in Abschnitt 4.8) . In einer randomisierten Unbedenklichkeitsstudie nach der Zulassung bei Patienten mit rheumatoider Arthritis, die 50 Jahre oder älter waren und mindestens einen zusätzlichen kardiovaskulären Risikofaktor aufwiesen, wurde ein dosisabhängig erhöhtes VTE-Risiko unter Tofacitinib im Vergleich zu TNF-Inhibitoren beobachtet (siehe Abschnitte 4.8 und 5.1).
In einer exploratorischen Post-hoc-Analyse im Rahmen dieser Studie bei Patienten mit bekannten VTE-Risikofaktoren wurden nachfolgende VTE häufiger bei mit Tofacitinib behandelten Patienten beobachtet, die nach 12-monatiger Behandlung D-Dimer-Werte vom 2‑Fachen des oberen Normal-Grenzwerts oder mehr (≥ 2× ULN, upper limit of normal) aufwiesen, als bei Patienten, die D-Dimer-Werte < 2× ULN hatten. Bei mit TNF-Inhibitoren behandelten Patienten wurde dies nicht festgestellt. Die Interpretation des Ergebnisses ist durch die geringe Anzahl an VTE-Ereignissen und die begrenzte Verfügbarkeit von D‑Dimer-Testergebnissen (Untersuchung nur bei Studienbeginn sowie in Monat 12 und am Ende der Studie) eingeschränkt. Bei Patienten ohne VTE während der Studie waren die mittleren D-Dimer-Werte in allen Behandlungsarmen in Monat 12 signifikant niedriger als bei Studienbeginn. D-Dimer-Werte ≥ 2× ULN in Monat 12 wurden jedoch bei etwa 30 % der Patienten ohne nachfolgende VTE-Ereignisse beobachtet, was als eingeschränkte Spezifität der D‑Dimer-Untersuchung in der Studie zu werten ist.
Bei Patienten mit kardiovaskulären Risikofaktoren oder Risikofaktoren für Malignome (siehe auch Abschnitt 4.4 „Schwerwiegende unerwünschte kardiovaskuläre Ereignisse [einschließlich Myokardinfarkt]“ und „Malignome und lymphoproliferative Erkrankungen“) sollte Tofacitinib nur angewendet werden, wenn keine geeigneten Behandlungsalternativen zur Verfügung stehen.
Bei Patienten mit anderen VTE-Risikofaktoren als solche für MACE oder Malignome sollte Tofacitinib mit Vorsicht angewendet werden. VTE-Risikofaktoren, die keine Risikofaktoren für MACE bzw. Malignome sind, umfassen: frühere VTE, Patienten, die sich einem größeren chirurgischen Eingriff unterziehen, Immobilisation, Anwendung von kombinierten hormonellen Kontrazeptiva oder einer Hormonersatztherapie, Vorliegen einer erblichen Gerinnungsstörung. Während der Behandlung mit Tofacitinib sollten Patienten in regelmäßigen Abständen auf Veränderungen des VTE-Risikos untersucht werden.
Bei RA-Patienten mit bekannten VTE-Risikofaktoren sollte eine Untersuchung der D-Dimer-Werte nach etwa 12 Monaten Behandlung in Betracht gezogen werden. Wenn das Ergebnis des D-Dimer-Tests ≥ 2× ULN beträgt, ist zu bestätigen, dass der klinische Nutzen die Risiken überwiegt, bevor eine Entscheidung über die Fortsetzung der Behandlung mit Tofacitinib getroffen wird.
Patienten mit Anzeichen und Symptomen einer VTE sind unverzüglich zu untersuchen. Bei Patienten mit VTE-Verdacht ist Tofacitinib unabhängig von Anwendungsgebiet oder Dosierung abzusetzen.
Retinale Venenthrombose
Retinale Venenthrombose (RVT) wurde bei Patienten berichtet, die mit Tofacitinib behandelt wurden (siehe Abschnitt 4.8). Patienten sollten angewiesen werden, bei Auftreten von Symptomen, die auf eine RVT hinweisen, unverzüglich einen Arzt aufzusuchen.
Schwerwiegende Infektionen
Schwerwiegende und bisweilen tödliche Infektionen aufgrund bakterieller, mykobakterieller, invasiv-fungaler, viraler oder anderer opportunistischer Krankheitserreger wurden bei Patienten berichtet, die Tofacitinib erhielten (siehe Abschnitt 4.8). Das Risiko für opportunistische Infektionen ist in asiatischen Regionen höher (siehe Abschnitt 4.8). Patienten mit rheumatoider Arthritis, die Kortikosteroide einnehmen, können anfällig für Infektionen sein.
Eine Tofacitinib-Therapie sollte nicht bei Patienten mit aktiven Infektionen, einschließlich lokalisierter Infektionen, eingeleitet werden.
Risiken und Nutzen der Behandlung sind vor der Behandlung mit Tofacitinib abzuwägen bei Patienten
mit wiederkehrenden Infektionen,
mit einer schwerwiegenden oder einer opportunistischen Infektion in der Vorgeschichte,
die in Gegenden mit endemischen Mykosen gelebt oder diese bereist haben,
mit Grunderkrankungen, die sie für Infektionen anfällig machen.
Die Patienten sind während und nach der Behandlung mit Tofacitinib engmaschig auf die Entwicklung von Anzeichen und Symptomen einer Infektion zu überwachen. Die Behandlung ist zu unterbrechen, wenn es bei einem Patienten zu einer schwerwiegenden Infektion, einer opportunistischen Infektion oder zu einer Sepsis kommt. Patienten, bei denen während der Behandlung mit Tofacitinib eine Neuinfektion auftritt, müssen umgehend vollständigen diagnostischen Tests unterzogen werden, die für immungeschwächte Patienten geeignet sind. Außerdem ist eine angemessene antimikrobielle Therapie einzuleiten, und die Patienten sind engmaschig zu überwachen.
Da bei älteren Patienten und Diabetes-Patienten generell eine höhere Infektionsrate vorliegt, ist bei der Behandlung dieser Patientengruppen Vorsicht geboten (siehe Abschnitt 4.8). Bei Patienten im Alter von 65 Jahren und älter sollte eine Behandlung mit Tofacitinib nur angewendet werden, wenn es keine geeigneten Behandlungsalternativen gibt (siehe Abschnitt 5.1).
Das Infektionsrisiko steigt möglicherweise mit dem Schweregrad einer Lymphopenie. Daher sollten für die Beurteilung des individuellen Infektionsrisikos die Lymphozytenzahlen berücksichtigt werden. Die Kriterien für den Behandlungsabbruch und die Lymphopenie-Überwachung werden in Abschnitt 4.2 erläutert.
Tuberkulose
Risiken und Nutzen der Behandlung sind vor der Behandlung mit Tofacitinib abzuwägen bei Patienten,
die Tuberkulose ausgesetzt waren,
die in Gegenden mit endemischer Tuberkulose gelebt oder diese bereist haben.
Die Patienten sind vor und während der Anwendung von Tofacitinib nach geltenden Leitlinien auf eine latente oder aktive Tuberkuloseinfektion zu untersuchen und zu testen.
Patienten mit latenter Tuberkulose, die positiv getestet werden, sollten vor der Einnahme von Tofacitinib mit einer antimykobakteriellen Standardtherapie behandelt werden.
Bei Patienten mit negativem Testergebnis, aber mit latenter oder aktiver Tuberkulose in der Vorgeschichte und bei denen eine adäquate Behandlung nicht bestätigt werden kann, und bei Patienten mit negativem Testergebnis, aber bestehenden Risikofaktoren für eine Tuberkuloseinfektion, sollte vor der Einnahme von Tofacitinib eine antituberkulöse Therapie erwogen werden. Es wird empfohlen, mit einem in der Tuberkulosebehandlung erfahrenen Arzt Rücksprache zu halten, um im Einzelfall zu entscheiden, ob die Einleitung einer antituberkulösen Therapie für einen Patienten geeignet ist. Die Patienten sind engmaschig auf Anzeichen und Symptome einer Tuberkulose zu überwachen. Dies gilt auch für Patienten, die vor Beginn der Therapie negativ auf eine latente Tuberkuloseinfektion getestet wurden.
Virusreaktivierung
Bei mit Tofacitinib behandelten Patienten wurden eine Virusreaktivierung und Fälle einer Reaktivierung von Herpesviren (z. B. Herpes zoster) beobachtet (siehe Abschnitt 4.8).
Bei Patienten, die mit Tofacitinib behandelt wurden, scheint das Auftreten von Herpes zoster erhöht zu sein bei:
Patienten japanischer oder koreanischer Herkunft.
Patienten mit einer ALC unter 1.000 Zellen/mm3 (siehe Abschnitt 4.2).
Patienten mit langjähriger RA, die zuvor mit 2 oder mehr biologischen krankheitsmodifizierenden Antirheumatika (bDMARD) behandelt wurden.
Die Wirkung von Tofacitinib auf die Reaktivierung einer chronischen Virushepatitis ist nicht bekannt. Patienten, die positiv auf Hepatitis B oder C getestet wurden, waren von der Teilnahme an den klinischen Studien ausgeschlossen. Vor Beginn der Therapie mit Tofacitinib sollte eine Untersuchung auf eine Virushepatitis entsprechend den klinischen Leitlinien durchgeführt werden.
Bei RA-Patienten, die Tofacitinib nach der Markteinführung erhielten, wurde mindestens ein bestätigter Fall von progressiver multifokaler Leukenzephalopathie (PML) berichtet. PML kann tödlich sein und soll bei immunsupprimierten Patienten mit neuen bzw. sich verschlimmernden neurologischen Symptomen bei der Differentialdiagnostik berücksichtigt werden.
Schwerwiegende unerwünschte kardiovaskuläre Ereignisse (einschließlich Myokardinfarkt)
Bei Patienten unter Tofacitinib wurden schwerwiegende unerwünschte kardiovaskuläre Ereignisse (MACE) beobachtet.
In einer randomisierten Unbedenklichkeitsstudie nach der Zulassung bei Patienten mit RA, die 50 Jahre oder älter waren und mindestens einen zusätzlichen kardiovaskulären Risikofaktor aufwiesen, wurde unter Tofacitinib im Vergleich zu TNF-Inhibitoren eine erhöhte Inzidenz von Myokardinfarkten beobachtet (siehe Abschnitte 4.8 und 5.1). Bei Patienten im Alter von 65 Jahren und älter, bei Patienten, die aktuelle oder ehemalige Langzeitraucher sind, und bei Patienten mit atherosklerotischer kardiovaskulärer Erkrankung in der Vorgeschichte oder anderen kardiovaskulären Risikofaktoren sollte Tofacitinib nur angewendet werden, wenn keine geeigneten Behandlungsalternativen verfügbar sind (siehe Abschnitt 5.1).
Malignome und lymphoproliferative Erkrankung
Tofacitinib kann die körpereigene Abwehr gegen Malignome beeinträchtigen.
In einer randomisierten Unbedenklichkeitsstudie nach der Zulassung bei Patienten mit RA, die 50 Jahre oder älter waren und mindestens einen zusätzlichen kardiovaskulären Risikofaktor aufwiesen, wurde unter Tofacitinib im Vergleich zu TNF-Inhibitoren eine erhöhte Inzidenz von malignen Erkrankungen, insbesondere NMSC, Lungenkarzinom und Lymphom, beobachtet (siehe Abschnitte 4.8 und 5.1).
NMSC, Lungenkarzinome und Lymphome bei mit Tofacitinib behandelten Patienten wurden auch in anderen klinischen Studien und bei der Anwendung nach der Zulassung beobachtet.
Andere Malignome bei mit Tofacitinib behandelten Patienten wurden in klinischen Studien und bei der Anwendung nach der Zulassung beobachtet, darunter, aber nicht ausschließlich, Brustkrebs, Melanom, Prostatakrebs und Bauchspeicheldrüsenkrebs.
Bei Patienten in einem Alter von 65 Jahren und älter, Patienten, die aktuelle oder ehemalige Langzeitraucher sind, und bei Patienten mit anderen Risikofaktoren für Malignome (z. B. aktuelles oder zurückliegendes Malignom, ausgenommen ein erfolgreich behandelter, nicht-melanozytärer Hautkrebs) sollte Tofacitinib nur angewendet werden, wenn keine geeigneten Behandlungsalternativen verfügbar sind (siehe Abschnitt 5.1).
Bei allen Patienten, insbesondere bei Patienten mit erhöhtem Risiko für Hautkrebs, werden regelmäßige Hautuntersuchungen empfohlen (siehe Tabelle 7 in Abschnitt 4.8).
Interstitielle Lungenerkrankung
Ebenfalls ist Vorsicht geboten bei Patienten mit einer chronischen Lungenerkrankung in der Vorgeschichte, da sie für Infektionen anfälliger sein können. In klinischen RA-Studien und in der Anwendungsbeobachtung nach Zulassung wurden Fälle von interstitieller Lungenerkrankung (einige davon mit tödlichem Ausgang) bei mit Tofacitinib behandelten Patienten berichtet, obwohl die Rolle der Januskinase(JAK)-Inhibition bei diesen Ereignissen nicht bekannt ist. Asiatische RA-Patienten unterliegen bekanntermaßen einem höheren Risiko für eine interstitielle Lungenerkrankung, weshalb bei der Behandlung dieser Patienten Vorsicht geboten ist.
Magen-Darm-Perforationen
In klinischen Studien wurden Fälle von Magen-Darm-Perforationen berichtet, obwohl die Rolle der JAK-Inhibition bei diesen Ereignissen nicht bekannt ist. Tofacitinib ist bei Patienten mit potenziell erhöhtem Risiko von Magen-Darm-Perforationen mit Vorsicht anzuwenden (z. B. bei Patienten mit Divertikulitis in der Vorgeschichte oder bei Patienten, die gleichzeitig Kortikosteroide und/oder nichtsteroidale entzündungshemmende Arzneimittel anwenden). Bei erstmaligem Auftreten von Anzeichen oder Symptomen abdomineller Komplikationen sind die Patienten unverzüglich zur Früherkennung einer Magen-Darm-Perforation zu untersuchen.
Frakturen
Frakturen wurden bei Patienten beobachtet, die mit Tofacitinib behandelt wurden.
Tofacitinib sollte bei Patienten mit bekannten Risikofaktoren für Frakturen, wie älteren Patienten, weiblichen Patienten und Patienten unter Kortikosteroidtherapie, unabhängig von Indikation und Dosierung, mit Vorsicht angewendet werden.
Leberenzyme
Die Behandlung mit Tofacitinib ging bei einigen Patienten mit einer erhöhten Rate von Leberwerterhöhungen einher (siehe Abschnitt 4.8 Leberenzymtests). Bei der Einleitung einer Tofacitinib-Behandlung von Patienten mit erhöhter Alanin-Aminotransferase (ALT) oder Aspartat-Aminotransferase (AST) ist Vorsicht geboten, besonders dann, wenn sie in Kombination mit potenziell hepatotoxischen Arzneimitteln, wie z. B. MTX, eingeleitet wird. Nach Beginn der Behandlung werden regelmäßige Kontrollen der Leberenzyme und eine sofortige Abklärung von beobachteten Leberenzymanstiegen empfohlen, um mögliche Fälle einer arzneimittelbedingten Leberschädigung zu erkennen. Bei Verdacht einer arzneimittelbedingten Leberschädigung sollte die Einnahme von Tofacitinib solange unterbrochen werden, bis diese Diagnose ausgeschlossen worden ist.
Überempfindlichkeit
Nach der Markteinführung wurden Fälle von Überempfindlichkeit im Zusammenhang mit der Anwendung von Tofacitinib berichtet. Allergische Reaktionen einschließlich Angioödem und Urtikaria sowie schwerwiegende Reaktionen traten auf. Wenn schwerwiegende allergische oder anaphylaktische Reaktionen auftreten, sollte Tofacitinib unverzüglich abgesetzt werden.
Laborparameter
Lymphozyten
Die Behandlung mit Tofacitinib war im Vergleich zu Placebo mit einer erhöhten Rate von Lymphozytopenien verbunden. Bei Lymphozytenzahlen unter 750 Zellen/mm3 wurden vermehrt schwerwiegende Infektionen beobachtet. Bei Patienten mit einer bestätigten Lymphozytenzahl unter 750 Zellen/mm3 sollte die Behandlung mit Tofacitinib nicht eingeleitet oder fortgeführt werden. Die Lymphozyten sollten zu Beginn der Therapie und danach alle 3 Monate kontrolliert werden. Empfehlungen zu Therapieänderungen auf Basis der Lymphozytenzahlen siehe Abschnitt 4.2.
Neutrophile
Die Behandlung mit Tofacitinib stand im Vergleich zu Placebo im Zusammenhang mit einer erhöhten Inzidenz von Neutropenie (weniger als 2.000 Zellen/mm³). Bei erwachsenen Patienten mit einer Neutrophilenzahl (ANC) unter 1.000 Zellen/mm3 und Kindern und Jugendlichen mit einer Neutrophilenzahl (ANC) unter 1.200 Zellen/mm3 sollte keine Behandlung mit Tofacitinib eingeleitet werden. Die ANC sollte bei Therapiebeginn, nach 4 bis 8-wöchiger Behandlung und danach alle 3 Monate kontrolliert werden. Empfehlungen zu Therapieänderungen aufgrund der ANC siehe Abschnitt 4.2.
Hämoglobin
Die Behandlung mit Tofacitinib wurde mit einer Abnahme der Hämoglobinwerte in Verbindung gebracht. Es wird empfohlen, die Tofacitinib-Behandlung bei erwachsenen Patienten mit einem Hb-Wert unter 9 g/dl und bei Kindern und Jugendlichen mit einem Hb-Wert unter 10 g/dl nicht zu beginnen. Der Hämoglobinwert sollte bei Therapiebeginn, nach 4 bis 8-wöchiger Behandlung und danach alle 3 Monate kontrolliert werden. Empfehlungen zu Therapieänderungen aufgrund des Hämoglobinwerts siehe Abschnitt 4.2.
Überwachung der Lipidwerte
Während der Behandlung mit Tofacitinib kam es zu einem Anstieg der Blutfettwerte, wie z. B. des Gesamtcholesterins, des Lipoprotein-Cholesterins niedriger Dichte (LDL) und des Lipoprotein-Cholesterins hoher Dichte (HDL). Maximale Effekte waren im Allgemeinen innerhalb von 6 Wochen zu beobachten. Acht Wochen nach Beginn der Tofacitinib-Therapie sollte eine Untersuchung der Blutfettwerte vorgenommen werden. Die Patienten sollten gemäß den klinischen Leitlinien für die Therapie der Hyperlipidämie behandelt werden. Erhöhte Gesamtcholesterin- und LDL-Werte im Zusammenhang mit Tofacitinib können mit einer Statin-Therapie auf die Werte vor der Behandlung gesenkt werden.
Hypoglykämie bei Patienten, die aufgrund eines Diabetes behandelt werden
Nach Einleitung einer Tofacitinib-Therapie bei Patienten, die Arzneimittel gegen Diabetes erhielten, wurden Fälle von Hypoglykämie berichtet. Bei Auftreten einer Hypoglykämie könnte eine Dosisanpassung der antidiabetischen Arzneimittel erforderlich sein.
Impfungen
Vor Beginn der Therapie mit Tofacitinib sollte der Impfstatus aller Patienten, insbesondere von pJIA- und jPsA-Patienten entsprechend den aktuellen Impfempfehlungen auf den neuesten Stand gebracht werden. Es wird empfohlen, Lebendimpfstoffe nicht gleichzeitig mit Tofacitinib anzuwenden. Bei der Entscheidung über die Anwendung von Lebendimpfstoffen vor Beginn der Therapie mit Tofacitinib sollte die vorbestehende Immunsuppression des jeweiligen Patienten berücksichtigt werden.
Eine prophylaktische Impfung gegen Herpes zoster sollte gemäß den Impfempfehlungen in Betracht gezogen werden. Patienten mit langjähriger RA, die zuvor 2 oder mehr bDMARD erhalten haben, sollte besondere Aufmerksamkeit geschenkt werden. Wenn der Herpes-Zoster-Lebendimpfstoff gegeben wird, sollte er nur Patienten mit bekannter Vorgeschichte von Windpocken oder Patienten, die seropositiv auf das Varicella-Zoster-Virus (VZV) getestet wurden, verabreicht werden. Sollte die Vorgeschichte von Windpocken als zweifelhaft oder unzuverlässig erachtet werden, so wird empfohlen, auf Antikörper gegen VZV zu testen.
Eine Impfung mit Lebendimpfstoffen sollte mindestens 2 Wochen, vorzugsweise aber 4 Wochen vor Beginn der Therapie mit Tofacitinib erfolgen, oder gemäß den aktuellen Impfempfehlungen zur Anwendung von immunmodulierenden Arzneimitteln. Bezüglich einer Sekundärübertragung von Infektionen durch Lebendimpfstoffe auf Patienten unter Tofacitinib liegen keine Daten vor.
Sonstige Bestandteile
Propylenglycol
Dieses Arzneimittel enthält 2,39 mg Propylenglycol pro ml.
Beispiele für eine Propylenglycol-Exposition basierend auf einer Tagesdosis (siehe Abschnitt 4.2) lauten wie folgt:
Eine Dosis von 3,2 mg zweimal täglich XELJANZ 1 mg/ml Lösung zum Einnehmen, eingenommen von einem Kind mit einem Körpergewicht von 10 kg bis < 20 kg, führt zu einer Propylenglycol-Exposition von 1,53 mg/kg/Tag.
Eine Dosis von 4 mg zweimal täglich XELJANZ 1 mg/ml Lösung zum Einnehmen, eingenommen von einem Kind mit einem Körpergewicht von 20 kg bis < 40 kg, führt zu einer Propylenglycol-Exposition von 0,96 mg/kg/Tag.
Eine Dosis von 5 mg zweimal täglich XELJANZ 1 mg/ml Lösung zum Einnehmen, eingenommen von einem Kind mit einem Körpergewicht von ≥ 40 kg führt zu einer Propylenglycol-Exposition von 0,60 mg/kg/Tag.
Natriumbenzoat
Dieses Arzneimittel enthält 0,9 mg Natriumbenzoat pro ml.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro ml, d. h. es ist nahezu „natriumfrei“.
Mögliche Beeinflussung der Pharmakokinetik (PK) von Tofacitinib durch andere Arzneimittel
Da Tofacitinib durch CYP3A4 metabolisiert wird, ist eine Wechselwirkung mit Arzneimitteln, die CYP3A4 hemmen oder induzieren, wahrscheinlich. Die Tofacitinib-Exposition ist erhöht, wenn gleichzeitig starke CYP3A4-Inhibitoren (z. B. Ketoconazol) angewendet werden oder wenn die gleichzeitige Anwendung mindestens eines Arzneimittels zu einer mittelstarken Hemmung von CYP3A4 und zu einer starken Hemmung von CYP2C19 (z. B. Fluconazol) führt (siehe Abschnitt 4.2).
Bei gleichzeitiger Anwendung mit starken CYP-Induktoren (z. B. Rifampicin) verringert sich die Tofacitinib-Exposition. CYP2C19-Inhibitoren alleine oder P-Glykoprotein-Inhibitoren beeinflussen die PK von Tofacitinib wahrscheinlich nur unwesentlich.
Bei gleichzeitiger Anwendung von Ketoconazol (starker CYP3A4-Inhibitor), Fluconazol (mittelstarker CYP3A4- und starker CYP2C19-Inhibitor), Tacrolimus (schwacher CYP3A4-Inhibitor) und Ciclosporin (mittelstarker CYP3A4-Inhibitor) erhöhte sich die AUC von Tofacitinib, während Rifampicin (ein starker CYP3A4-Induktor) die AUC erniedrigte. Die gleichzeitige Anwendung von Tofacitinib und starken CYP3A4-Induktoren (z. B. Rifampicin) kann zu einem Verlust oder zu einer Verringerung des klinischen Ansprechens führen (siehe Abbildung 1). Die gleichzeitige Anwendung starker CYP3A4-Induktoren mit Tofacitinib wird nicht empfohlen. Die gleichzeitige Anwendung von Ketoconazol und Fluconazol erhöhte den Cmax-Wert von Tofacitinib, während Tacrolimus, Ciclosporin und Rifampicin den Cmax von Tofacitinib senkten. Die gleichzeitige Anwendung von einmal wöchentlich 15‑25 mg MTX hatte keine Auswirkung auf die PK von Tofacitinib bei RA-Patienten (siehe Abbildung 1).
Abbildung 1 Auswirkung anderer Arzneimittel auf die PK von Tofacitinib
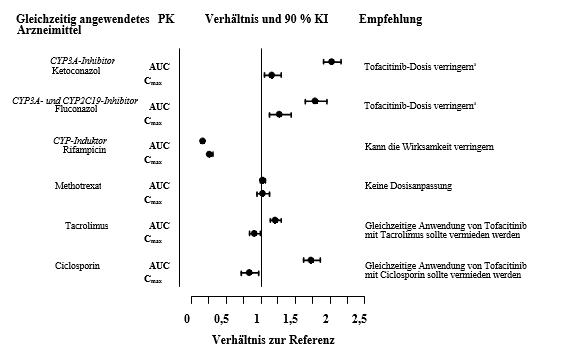
a Bei Patienten, die zweimal täglich 5 mg oder eine äquivalente Dosis gemäß ihrem Körpergewicht erhalten, sollte die Tofacitinib-Dosis auf einmal täglich 5 mg Filmtabletten oder eine äquivalente Dosis gemäß dem Körpergewicht verringert werden (siehe Abschnitt 4.2).
Mögliche Beeinflussung der PK anderer Arzneimittel durch Tofacitinib
Die gleichzeitige Anwendung von Tofacitinib hatte bei gesunden weiblichen Probanden keine Auswirkungen auf die Pharmakokinetik der oralen Empfängnisverhütungsmittel Levonorgestrel und Ethinylestradiol.
Bei gleichzeitiger Anwendung von Tofacitinib mit einmal wöchentlich 15‑25 mg MTX bei RA‑Patienten verringerten sich die AUC und Cmax von MTX um 10 % bzw. 13 %. Das Ausmaß der Verringerung der MTX-Exposition rechtfertigt keine Veränderungen der individuellen MTX-Dosierung.
Kinder und Jugendliche
Studien zur Erfassung von Wechselwirkungen wurden nur bei Erwachsenen durchgeführt.
Schwangerschaft
Es gibt keine adäquaten und gut kontrollierten Studien zur Anwendung von Tofacitinib bei schwangeren Frauen. Tofacitinib erwies sich bei Ratten und Kaninchen als teratogen, und es beeinträchtigte die Geburt und die peri-/postnatale Entwicklung (siehe Abschnitt 5.3).
Aus Vorsichtsgründen ist die Anwendung von Tofacitinib während der Schwangerschaft kontraindiziert (siehe Abschnitt 4.3).
Frauen im gebärfähigen Alter/Verhütung bei Frauen
Frauen im gebärfähigen Alter müssen angewiesen werden, während der Behandlung mit Tofacitinib und für mindestens 4 Wochen nach Einnahme der letzten Dosis eine zuverlässige Verhütungsmethode anzuwenden.
Stillzeit
Basierend auf veröffentlichten Daten geht Tofacitinib in die Muttermilch über. Die Auswirkung von Tofacitinib auf das gestillte Kind ist auf Grundlage von Daten aus veröffentlichter Literatur und Daten nach Markteinführung nicht bekannt. Diese Daten sind auf eine kleine Anzahl von Fällen ohne kausal bedingte unerwünschte Ereignisse beschränkt. Ein Risiko für das gestillte Kind kann nicht ausgeschlossen werden. Aus Vorsichtsgründen ist die Anwendung von Tofacitinib während der Stillzeit kontraindiziert (siehe Abschnitt 4.3).
Fertilität
Es wurden keine formalen Studien zur möglichen Wirkung auf die Fertilität beim Menschen durchgeführt. Tofacitinib beeinträchtigte bei Ratten die weibliche Fertilität, aber nicht die männliche Fertilität (siehe Abschnitt 5.3).
Tofacitinib hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Rheumatoide Arthritis
Die häufigsten schwerwiegenden Nebenwirkungen waren schwerwiegende Infektionen (siehe Abschnitt 4.4).
Bei der Langzeit-Sicherheits-Population über alle Expositionen waren die häufigsten im Zusammenhang mit Tofacitinib berichteten schwerwiegenden Infektionen Pneumonie (1,7 %), Herpes zoster (0,6 %), Harnwegsinfekt (0,4 %), Zellulitis (0,4 %), Divertikulitis (0,3 %) und Appendizitis (0,2 %). Zu den im Zusammenhang mit Tofacitinib berichteten opportunistischen Infektionen gehörten TB und andere mykobakterielle Infektionen, Infektionen mit Cryptococcus, Histoplasmose, ösophageale Candidose, multidermatomaler Herpes zoster, Cytomegalievirus-Infektion, BK-Virus-Infektionen und Listeriose. Einige Patienten wiesen eine disseminierte statt einer lokalisierten Erkrankung auf. Andere, nicht in klinischen Studien berichtete schwerwiegende Infektionen können ebenfalls auftreten (z. B. Kokzidiomykose).
Die in doppelblinden, Placebo- oder MTX-kontrollierten klinischen Studien während der ersten 3 Monate am häufigsten berichteten Nebenwirkungen waren Kopfschmerzen (3,9 %), Infektionen der oberen Atemwege (3,8 %), Virusinfektionen der oberen Atemwege (3,3 %), Diarrhö (2,9 %), Übelkeit (2,7 %) und Hypertonie (2,2 %).
Der Anteil der Patienten, die in den ersten 3 Monaten der doppelblinden, Placebo- oder MTX-kontrollierten Studien die Behandlung aufgrund von Nebenwirkungen abbrachen, betrug für die mit Tofacitinib behandelten Patienten 3,8 %. Die häufigsten Infektionen, die in kontrollierten klinischen Studien in den ersten 3 Monaten zu einem Therapieabbruch führten waren Herpes zoster (0,19 %) und Pneumonie (0,15 %).
Tabellarische Auflistung der Nebenwirkungen
Die nachfolgende Tabelle zeigt eine Auflistung der Nebenwirkungen aus klinischen Studien an erwachsenen RA-, PsA- und CU-Patienten nach Systemorganklassen und Häufigkeitskategorien, die wie folgt festgelegt sind: sehr häufig (≥ 1/10), häufig (≥ 1/100 bis < 1/10), gelegentlich (≥ 1/1.000 bis < 1/100), selten (≥ 1/10.000 bis < 1/1.000), sehr selten (< 1/10.000) oder nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad angeordnet.
Tabelle 7: Nebenwirkungen
Systemorgan-klasse |
Häufig |
Gelegentlich |
Selten |
Sehr selten |
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar) |
Infektionen und parasitäre Erkrankungen |
Pneumonie |
Tuberkulose |
Sepsis |
Tuberkulose des Zentralnervensystems |
|
Gutartige, bösartige und unspezifische Neubildungen (einschl. Zysten und Polypen) |
Lungenkrebs |
Lymphom |
|||
Erkrankungen des Blutes und des Lymphsystems |
Lymphopenie |
Leukopenie |
|||
Erkrankungen des Immunsystems |
Überempfindlichkeit* |
||||
Stoffwechsel- und Ernährungs-störungen |
Dyslipidämie |
||||
Psychiatrische Erkrankungen |
Insomnie |
||||
Erkrankungen des Nervensystems |
Kopfschmerzen |
Parästhesie |
|||
Herz-erkrankungen |
Myokardinfarkt |
||||
Gefäßerkrankungen |
Hypertonie |
Venöse thromboembolische Ereignisse ** |
|||
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Husten |
Dyspnoe |
|||
Erkrankungen des Gastrointestinaltrakts |
Bauchschmerzen |
||||
Leber- und Gallenerkran-kungen |
Lebersteatose |
Anormaler Leberfunktions-test |
|||
Erkrankungen der Haut und des Unterhautzell-gewebes |
Ausschlag |
Erythem |
|||
Skelettmuskula-tur‑, Bindegewebs- und Knochenerkrankungen |
Arthralgie |
Gelenkschwellung |
Muskuloskelettale Schmerzen |
||
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Peripheres Ödem |
Pyrexie |
|||
Untersuchungen |
Erhöhte Kreatinphosphokinase im Blut |
Erhöhter Blut-Kreatininspiegel |
|||
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen |
Bänderdehnung |
*Daten aus Spontanberichten
** Venöse thromboembolische Ereignisse umfassen LE, TVT, retinale Venenthrombose und zerebrale Sinusvenenthrombose
Beschreibung ausgewählter Nebenwirkungen
Venöse thromboembolische Ereignisse
Rheumatoide Arthritis
In einer groß angelegten (n = 4.362), randomisierten Sicherheitsstudie nach Zulassung an Patienten mit rheumatoider Arthritis im Alter von 50 Jahren und älter mit mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurde eine erhöhte und dosisabhängige Inzidenz von VTE bei Patienten beobachtet, die mit Tofacitinib behandelt wurden, im Vergleich zu Patienten, die TNF-Inhibitoren erhielten (siehe Abschnitt 5.1). Die Mehrzahl dieser Ereignisse war schwerwiegend und einige führten zum Tod. Die Inzidenzraten (95 % KI) für LE bei zweimal täglich 5 mg Tofacitinib, zweimal täglich 10 mg Tofacitinib und TNF-Inhibitoren betrugenjeweils 0,17 (0,08–0,33), 0,50 (0,32–0,74) und 0,06 (0,01–0,17) Patienten mit Ereignissen pro 100 Patientenjahre. Im Vergleich zu TNF-Inhibitoren betrug die Hazard Ratio (HR) für eine LE jeweils 2,93 (0,79–10,83) bzw. 8,26 (2,49–27,43) für zweimal täglich 5 mg Tofacitinib bzw. zweimal täglich 10 mg Tofacitinib (siehe Abschnitt 5.1). Die Mehrheit (97 %) der mit Tofacitinib behandelten Patienten, bei denen LE beobachtet wurde, hatten VTE-Risikofaktoren.
Allgemeine Infektionen
Rheumatoide Arthritis
In den kontrollierten klinischen Phase 3-Studien mit zweimal täglich 5 mg (insgesamt 616 Patienten) und zweimal täglich 10 mg (insgesamt 642 Patienten) betrugen die Infektionsraten über 0–3 Monate in den Tofacitinib-Monotherapie-Gruppen 16,2 % (100 Patienten) bzw. 17,9 % (115 Patienten) im Vergleich zu 18,9 % (23 Patienten) in der Placebogruppe (insgesamt 122 Patienten). In den kontrollierten klinischen Phase 3-Studien mit DMARD-Begleitmedikation mit zweimal täglich 5 mg (insgesamt 973 Patienten) und zweimal täglich 10 mg (insgesamt 969 Patienten) betrugen die Infektionsraten über 0–3 Monate in der Behandlungsgruppe Tofacitinib plus DMARD 21,3 % (207 Patienten) bzw. 21,8 % (211 Patienten) im Vergleich zu 18,4 % (103 Patienten) in der Behandlungsgruppe DMARD plus Placebo (insgesamt 559 Patienten).
Die am häufigsten berichteten Infektionen waren Infektionen der oberen Atemwege und Nasopharyngitis (3,7 % bzw. 3,2 %).
Die Gesamthäufigkeit von Infektionen unter Tofacitinib in der Langzeit-Sicherheits-Population über alle Expositionen (insgesamt 4.867 Patienten) betrug 46,1 Patienten mit Ereignissen pro 100 Patientenjahre (43,8 bzw. 47,2 Patienten mit Ereignissen für die Dosierungen zweimal täglich 5 mg bzw. 10 mg). Für Patienten unter Monotherapie (insgesamt 1.750) betrugen die Häufigkeiten für die Dosierungen zweimal täglich 5 mg bzw. 10 mg 48,9 bzw. 41,9 Patienten mit Ereignissen pro 100 Patientenjahre. Für Patienten unter DMARD-Begleittherapie (insgesamt 3.117) betrugen die Häufigkeiten für die Dosierungen zweimal täglich 5 mg bzw. 10 mg 41,0 bzw. 50,3 Patientenereignisse pro 100 Patientenjahre.
Schwerwiegende Infektionen
Rheumatoide Arthritis
In den kontrollierten klinischen Studien über 6 und 24 Monate betrug die Rate schwerwiegender Infektionen in der Monotherapie-Gruppe mit zweimal täglich 5 mg Tofacitinib 1,7 Patienten mit Ereignissen pro 100 Patientenjahre. In der Monotherapie-Gruppe mit zweimal täglich 10 mg Tofacitinib betrug die Inzidenzrate 1,6 Patienten mit Ereignissen pro 100 Patientenjahre, in der Placebo-Gruppe 0 Ereignisse pro 100 Patientenjahre und in der MTX-Gruppe 1,9 Patienten mit Ereignissen pro 100 Patientenjahre.
In den Studien mit einer Dauer von 6, 12, oder 24 Monaten betrugen die Inzidenzraten schwerwiegender Infektionen in den Tofacitinib plus DMARD-Behandlungsgruppen mit zweimal täglich 5 mg bzw. 10 mg 3,6 bzw. 3,4 Patienten mit Ereignissen pro 100 Patientenjahre im Vergleich zu 1,7 Patienten mit Ereignissen pro 100 Patientenjahre in der Placebo plus DMARD-Gruppe.
In der Langzeit-Sicherheits-Population über alle Expositionen betrug die Gesamthäufigkeit schwerwiegender Infektionen unter Tofacitinib 2,4 bzw. 3,0 Patienten mit Ereignissen pro 100 Patientenjahre für die Tofacitinib-Gruppen mit zweimal täglich 5 mg bzw. 10 mg. Die häufigsten schwerwiegenden Infektionen waren Pneumonie, Herpes zoster, Harnwegsinfekt, Zellulitis, Gastroenteritis und Divertikulitis. Es wurden Fälle opportunistischer Infektionen berichtet (siehe Abschnitt 4.4).
In einer großen (n = 4.362), randomisierten Sicherheitsstudie nach Zulassung an Patienten mit rheumatoider Arthritis im Alter von 50 Jahren oder älter und mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurde ein dosisabhängiger Anstieg schwerwiegender Infektionen unter Tofacitinib im Vergleich zu TNF-Inhibitoren beobachtet (siehe Abschnitt 4.4).
Die Inzidenzraten (95 % KI) für schwerwiegende Infektionen bei zweimal täglich 5 mg Tofacitinib, zweimal täglich 10 mg Tofacitinib und TNF-Inhibitoren betrugen jeweils 2,86 (2,41; 3,37), 3,64 (3,11; 4,23) und 2,44 (2,02; 2,92) Patienten mit Ereignissen pro 100 Patientenjahre. Im Vergleich zu TNF-Inhibitoren betrug die Hazard Ratio (HR) für schwerwiegende Infektionen 1,17 (0,92; 1,50) bzw. 1,48 (1,17; 1,87) für zweimal täglich 10 mg Tofacitinib bzw. zweimal täglich 5 mg Tofacitinib.
Virusreaktivierung
Bei mit Tofacitinib behandelten Patienten japanischer oder koreanischer Herkunft, bei Patienten mit langjähriger RA, die zuvor 2 oder mehr bDMARD erhalten hatten, bei Patienten mit einer ALC unter 1.000 Zellen/mm3 oder bei Patienten, die mit zweimal täglich 10 mg behandelt werden, ist das Risiko für Herpes zoster möglicherweise erhöht (siehe Abschnitt 4.4).
In einer großen (n = 4.362), randomisierten Sicherheitsüberwachungsstudie nach der Zulassung bei Patienten mit RA im Alter von 50 Jahren und älter und mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurde bei mit Tofacitinib behandelten Patienten im Vergleich zu TNF-Inhibitoren ein Anstieg von Herpes-zoster-Ereignissen beobachtet. Die Inzidenzraten (95 % KI) für Herpes zoster bei zweimal täglich 5 mg Tofacitinib, zweimal täglich 10 mg Tofacitinib bzw. TNF-Inhibitoren betrugen jeweils 3,75 (3,22; 4,34), 3,94 (3,38; 4,57) und 1,18 (0,90; 1,52) Patienten mit Ereignissen pro 100 Patientenjahre.
Laboruntersuchungen
Lymphozyten
In den kontrollierten klinischen RA-Studien kam es, bezogen auf die Dosierungen von zweimal täglich 5 mg und zweimal täglich 10 mg zusammengenommen, bei 0,3 % der Patienten zu bestätigten Abnahmen der ALC unter 500 Zellen/mm3 und bei 1,9 % der Patienten zu Abnahmen der ALC auf einen Wert zwischen 500 und 750 Zellen/mm3.
In der RA-Langzeit-Sicherheits-Population kam es, bezogen auf die Dosierungen von zweimal täglich 5 mg und zweimal täglich 10 mg zusammengenommen, bei 1,3 % der Patienten zu bestätigten Abnahmen der ALC unter 500 Zellen/mm3 und bei 8,4 % der Patienten zu Abnahmen der ALC auf einen Wert zwischen 500 und 750 Zellen/mm3.
Bestätigte ALC-Werte unter 750 Zellen/mm3 waren mit einem gehäuften Auftreten schwerwiegender Infektionen verbunden (siehe Abschnitt 4.4).
Neutrophile
In den kontrollierten klinischen RA-Studien kam es, bezogen auf die Dosierungen von zweimal täglich 5 mg und zweimal täglich 10 mg zusammengenommen, bei 0,08 % der Patienten zu bestätigten Abnahmen der ANC-Werte unter 1.000 Zellen/mm3. In keiner Patientengruppe wurden bestätigte Abnahmen der ANC unter 500 Zellen/mm3 beobachtet. Es war kein eindeutiger Zusammenhang zwischen Neutropenie und dem Auftreten schwerwiegender Infektionen zu erkennen.
In der RA-Langzeit-Sicherheits-Population stimmten Muster und Inzidenz der bestätigten ANC-Abnahmen mit den Befunden aus den kontrollierten klinischen Studien überein (siehe Abschnitt 4.4).
Leberenzymtests
Bestätigte Erhöhungen der Leberenzymwerte um mehr als das 3-Fache des oberen Normal-Grenzwerts (3x ULN, upper limit of normal) wurden bei RA-Patienten gelegentlich beobachtet. Bei RA-Patienten mit erhöhten Leberenzymwerten führte eine Therapieanpassung, wie z. B. Dosisreduktion gleichzeitig angewendeter DMARD, eine Unterbrechung der Behandlung oder eine Senkung der Tofacitinib-Dosis zu einer Abnahme oder Normalisierung der Leberenzymwerte.
Im kontrollierten Teil der RA-Monotherapie-Studie der Phase 3 (0‑3 Monate, Studie I, siehe Abschnitt 5.1) wurden ALT-Anstiege oberhalb 3x ULN bei 1,65 %, 0,41 % bzw. 0 % der Patienten beobachtet, die Placebo, zweimal täglich 5 mg oder 10 mg Tofacitinib erhielten. In dieser Studie wurden AST-Anstiege oberhalb 3x ULN bei 1,65 %, 0,41 % bzw. 0 % der Patienten beobachtet, die Placebo, zweimal täglich 5 mg oder 10 mg Tofacitinib erhielten.
In der RA-Monotherapie-Studie der Phase 3 (0‑24 Monate, Studie VI, siehe Abschnitt 5.1), wurden ALT-Anstiege oberhalb 3x ULN bei 7,1 %, 3,0 % bzw. 3,0 % der Patienten beobachtet, die MTX, zweimal täglich 5 mg oder 10 mg Tofacitinib erhielten. In dieser Studie wurden AST-Anstiege oberhalb 3x ULN bei 3,3 %, 1,6 % bzw. 1,5 % der Patienten beobachtet, die MTX, zweimal täglich 5 mg oder 10 mg Tofacitinib erhielten.
Im kontrollierten Teil der Phase 3-RA-Studien zur DMARD-Begleittherapie (0‑3 Monate, Studien II–V, siehe Abschnitt 5.1) wurden ALT-Anstiege oberhalb 3x ULN bei 0,9 %, 1,24 % bzw. 1,14 % der Patienten beobachtet, die Placebo, zweimal täglich 5 mg oder 10 mg Tofacitinib erhielten. In diesen Studien wurden AST-Anstiege oberhalb 3x ULN bei 0,72 %, 0,5 % bzw. 0,31 % der Patienten beobachtet, die Placebo, zweimal täglich 5 mg oder 10 mg Tofacitinib erhielten.
In den RA-Langzeit-Erweiterungsstudien zur Monotherapie wurden ALT-Anstiege oberhalb 3x ULN bei 1,1 % bzw. 1,4 % der Patienten beobachtet, die zweimal täglich 5 mg oder 10 mg Tofacitinib erhielten. AST-Anstiege oberhalb 3x ULN wurden sowohl in der Gruppe mit zweimal täglich 5 mg als auch in der mit zweimal täglich 10 mg Tofacitinib bei < 1,0 % der Patienten beobachtet.
In den RA-Langzeit-Erweiterungsstudien zur DMARD-Begleittherapie wurden ALT-Anstiege oberhalb 3x ULN bei 1,8 % bzw. 1,6 % der Patienten beobachtet, die zweimal täglich 5 mg oder 10 mg Tofacitinib erhielten. AST-Anstiege oberhalb 3x ULN wurden sowohl in der Gruppe mit zweimal täglich 5 mg als auch in der mit zweimal täglich 10 mg Tofacitinib bei < 1,0 % der Patienten beobachtet.
In einer großen (n = 4.362), randomisierten Sicherheitsüberwachungsstudie nach der Zulassung bei Patienten mit RA im Alter von 50 Jahren und älter und mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurden ALT-Anstiege größer oder gleich 3 x ULN bei jeweils 6,01 %, 6,54 % und 3,77 % der Patienten beobachtet, die zweimal täglich 5 mg Tofacitinib, zweimal täglich 10 mg Tofacitinib bzw. TNF-Inhibitoren erhielten. AST-Anstiege größer oder gleich 3 x ULN wurden bei jeweils 3,21 %, 4,57 % und 2,38 % der Patienten beobachtet, die zweimal täglich 5 mg Tofacitinib, zweimal täglich 10 mg Tofacitinib bzw. TNF-Inhibitoren erhielten.
Lipide
Erhöhungen der Lipidparameter (Gesamtcholesterin, LDL-Cholesterin, HDL-Cholesterin, Triglyzeride) wurden in den kontrollierten, doppelblinden klinischen Studien zur rheumatoiden Arthritis erstmals 1 Monat nach Beginn der Tofacitinib-Therapie untersucht. Die zu diesem Zeitpunkt beobachteten Erhöhungen blieben danach stabil.
Änderungen der Lipidparameter von den Ausgangswerten bis zum jeweiligen Studienende (6‑24 Monate) der kontrollierten, klinischen RA-Studien werden nachfolgend zusammengefasst:
Der mittlere LDL-Cholesterinwert stieg im Studienarm mit zweimal täglich 5 mg Tofacitinib bis Monat 12 um 15 % und im Studienarm mit zweimal täglich 10 mg Tofacitinib um 20 %. Nach 24 Monaten stieg der LDL-Cholesterinwert im Studienarm mit zweimal täglich 5 mg Tofacitinib um 16 % und im Studienarm mit zweimal täglich 10 mg Tofacitinib um 19 %.
Der mittlere HDL-Cholesterinwert stieg im Studienarm mit zweimal täglich 5 mg Tofacitinib bis Monat 12 um 17 % und im Studienarm mit zweimal täglich 10 mg Tofacitinib um 18 %. Nach 24 Monaten stieg der HDL-Cholesterinwert im Studienarm mit zweimal täglich 5 mg Tofacitinib um 19 % und im Studienarm mit zweimal täglich 10 mg Tofacitinib um 20 %.
Nach dem Absetzen von Tofacitinib gingen die Lipidspiegel auf die Ausgangswerte zurück.
Die mittleren LDL-Cholesterin/HDL-Cholesterin-Quotienten und die Apolipoprotein B (ApoB)/ApoA1-Quotienten blieben bei den mit Tofacitinib behandelten Patienten weitgehend unverändert.
In einer kontrollierten klinischen RA-Studie konnten die erhöhten LDL-Cholesterin- und ApoB-Werte mit einer Statin-Therapie auf die Werte vor der Behandlung gesenkt werden.
In den RA-Langzeit-Sicherheits-Populationen stimmten die Erhöhungen der Lipidparameter mit den Beobachtungen in den kontrollierten klinischen Studien überein.
In einer großen (n = 4.362), randomisierten Sicherheitsüberwachungsstudie nach der Zulassung bei Patienten mit RA im Alter von 50 Jahren und älter und mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurden die nachfolgend zusammengefassten Änderungen der Lipidparameter vom Ausgangswert bis Monat 24 beobachtet:
Der mittlere LDL-Cholesterinwert stieg bis Monat 12 um jeweils 13,80 %, 17,04 % und 5,50 % bei Patienten, die zweimal täglich 5 mg Tofacitinib, zweimal täglich 10 mg Tofacitinib bzw. TNF-Inhibitoren erhielten. Nach 24 Monaten betrug der Anstieg jeweils 12,71 %, 18,14 % bzw. 3,64 %.
Der mittlere HDL-Cholesterinwert stieg bis Monat 12 um jeweils 11,71 %, 13,63 % und 2,82 % bei Patienten, die zweimal täglich 5 mg Tofacitinib, zweimal täglich 10 mg Tofacitinib bzw. TNF-Inhibitoren erhielten. Nach 24 Monaten betrug der Anstieg jeweils 11,58 %, 13,54 % bzw. 1,42 %.
Myokardinfarkt
Rheumatoide Arthritis
In einer großen (n = 4.362) randomisierten Sicherheitsüberwachungsstudie nach Zulassung an Patienten mit rheumatoider Arthritis im Alter von 50 Jahren und älter und mindestens einem zusätzlichen kardiovaskulären Risikofaktor betrugen die Inzidenzraten (95 % KI) für nicht tödlichen Myokardinfarkt bei Tofacitinib 5 mg zweimal täglich, Tofacitinib 10 mg zweimal täglich und TNF-Inhibitoren 0,37 (0,22; 0,57), 0,33 (0,19; 0,53) und 0,16 (0,07, 0,31) Patienten mit Ereignissen pro 100 Patientenjahre. Es wurden wenige tödliche Myokardinfarkte berichtet, wobei die Raten bei mit Tofacitinib behandelten Patienten im Vergleich zu den mit TNF-Inhibitoren behandelten Patienten ähnlich waren (siehe Abschnitte 4.4 und 5.1). Für die Studie mussten mindestens 1.500 Patienten 3 Jahre lang nachbeobachtet werden.
Maligne Erkrankungen außer NMSC
Rheumatoide Arthritis
In einer großen (n = 4.362) randomisierten Sicherheitsüberwachungsstudie nach Zulassung an Patienten mit rheumatoider Arthritis im Alter von 50 Jahren und älter und mindestens einem zusätzlichen kardiovaskulären Risikofaktor betrugen die Inzidenzraten (95 % KI) für Lungenkrebs bei Tofacitinib 5 mg zweimal täglich, Tofacitinib 10 mg zweimal täglich und TNF-Inhibitoren 0,23 (0,12; 0,40), 0,32 (0,18; 0,51) und 0,13 (0,05; 0,26) Patienten mit Ereignissen pro 100 Patientenjahre (siehe Abschnitte 4.4 und 5.1). Für die Studie mussten mindestens 1.500 Patienten 3 Jahre lang nachbeobachtet werden.
Die Inzidenzraten (95 % KI) für Lymphome bei Tofacitinib 5 mg zweimal täglich, Tofacitinib 10 mg zweimal täglich und TNF-Inhibitoren betrugen 0,07 (0,02; 0,18), 0,11 (0,04; 0,24) und 0,02 (0,00; 0,10) Patienten mit Ereignissen pro 100 Patientenjahre (siehe Abschnitte 4.4 und 5.1).
Kinder und Jugendliche
Polyartikuläre juvenile idiopathische Arthritis und juvenile PsA
Die Nebenwirkungen bei JIA-Patienten in der Phase-3-Zulassungsstudie (Studie JIA-I) und in der Langzeit-Erweiterungsstudie, in die Kinder und Jugendliche mit pJIA, jPsA, ERA und sJIA aufgenommen wurden, stimmten im Allgemeinen in Art und Häufigkeit mit denjenigen überein, die bei erwachsenen RA-Patienten beobachtet wurden, mit Ausnahme einiger Infektionen (Infektion der oberen Atemwege und Virusinfektion) sowie gastrointestinaler oder allgemeiner Symptome (Bauchschmerzen und Fieber), die häufiger bei Kindern und Jugendlichen mit JIA auftraten. MTX war das am häufigsten gleichzeitig angewendete csDMARD (an Tag 1 nahmen 156 von 157 Patienten MTX als csDMARDs ein). Es liegen unzureichende Daten zum Sicherheitsprofil von Tofacitinib bei einer gleichzeitigen Anwendung mit anderen csDMARD vor.
Infektionen
In der doppelblinden Phase von Studie JIA-I waren Infektionen die am häufigsten gemeldeten Nebenwirkungen (44,3 %). Der Schweregrad der Infektionen war in der Regel leicht bis mittelschwer.
In Studie JIA-I wurden schwerwiegende Infektionen bei 3 Patienten (1,3 %), die mit Tofacitinib behandelt wurden, gemeldet. Dies entspricht einer Inzidenzrate von 2,43 Patienten mit Ereignissen pro 100 Patientenjahre. Herpes zoster wurde bei 2 Patienten (0,9 %) gemeldet, mit einer Inzidenzrate von 1,63 Patienten mit Ereignissen pro 100 Patientenjahre. Beide Ereignisse umfassten ein einzelnes Dermatom und waren nicht schwerwiegend.
In der Langzeit-Erweiterungsstudie traten schwerwiegende Infektionen bei 14 Patienten (4,6 %) auf, die mit Tofacitinib behandelt wurden. Dies entspricht einer Inzidenzrate von 1,50 Patienten mit Ereignissen pro 100 Patientenjahre. Die häufigsten schwerwiegenden Infektionen waren Herpes zoster und Pneumonie. Herpes-zoster-Ereignisse wurden bei 6 Patienten (2,0 %) gemeldet. Dies entspricht einer Inzidenzrate von 0,64 Patienten mit Ereignissen pro 100 Patientenjahre. Bei 2 dieser Patienten (0,7 %) traten schwerwiegende Ereignisse in Form von multidermatomalem Herpes zoster mit einer Inzidenzrate von 0,21 Ereignissen pro 100 Patientenjahre auf. Darüber hinaus traten 3 Herpes-zoster-Ereignisse (2 schwerwiegende und 1 nicht schwerwiegendes) auf, die auf ein einzelnes Dermatom begrenzt waren. Das übrige Herpes-zoster-Ereignis war nicht schwerwiegend, umfasste 2 nebeneinander liegende Dermatome und trat bei einem Patienten mit einem früheren nicht schwerwiegenden Varicella-Ereignis auf, das 5,8 Jahre zuvor in der Studie gemeldet wurde.
Nebenwirkungen der Leber
Patienten in der Zulassungsstudie zu JIA mussten AST- und ALT-Werte unter dem 1,5‑Fachen des oberen Normal-Grenzwerts (ULN) aufweisen, um in die Studie aufgenommen zu werden. In Studie JIA-I wurden ALT-Erhöhungen > 5 × ULN bei zwei aufeinanderfolgenden Besuchsterminen bei 1 Patienten (0,4 %), der Tofacitinib und eine Hintergrundtherapie mit Methotrexat erhielt, gemeldet; das Ereignis wurde als mit Tofacitinib im Zusammenhang stehend bewertet und klang nach Absetzen von Methotrexat und Tofacitinib ab. In der Langzeit-Erweiterungsstudie wurden AST- oder ALT-Erhöhungen > 5 × ULN bei 2 aufeinanderfolgenden Besuchsterminen bei 2 Patienten (0,66 %) gemeldet; beide Ereignisse wurden als nicht mit Tofacitinib im Zusammenhang stehend bewertet. Keines der Ereignisse in irgendeiner der Studien erfüllte die Kriterien nach „Hy’s Law“.
Laboruntersuchungen
Veränderungen von Laborwerte bei JIA-Patienten im klinischen Entwicklungsprogramm entsprachen denjenigen bei erwachsenen RA-Patienten. Patienten in der Zulassungsstudie zu JIA mussten für die Aufnahme in die Studie eine Thrombozytenzahl von ≥ 100.000 Zellen/mm3 aufweisen. Deshalb stehen keine Daten für JIA-Patienten mit Thrombozytenzahlen <100.000 Zellen/mm3 vor Beginn der Behandlung mit Tofacitinib zur Verfügung.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das aufgeführte nationale Meldesystem anzuzeigen.
Deutschland
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: https://www.bfarm.de
Österreich
Bundesamt für Sicherheit im Gesundheitswesen
Traisengasse 5
1200 WIEN
ÖSTERREICH
Fax: +43 (0) 50 555 36207
Website: https://www.basg.gv.at/
Bei einer Überdosierung wird empfohlen, den Patienten auf Anzeichen und Symptome von Nebenwirkungen zu überwachen. Es gibt kein spezifisches Gegenmittel bei einer Überdosierung von Tofacitinib. Die Behandlung sollte symptomatisch und unterstützend erfolgen.
Pharmakokinetische Daten gesunder Probanden mit einer Einzeldosis von bis zu 100 mg lassen erwarten, dass mehr als 95 % der angewendeten Dosis innerhalb von 24 Stunden ausgeschieden werden.
Pharmakotherapeutische Gruppen: Immunsuppressiva, Janus-assoziierte Kinase (JAK)-Inhibitoren; ATC-Code: L04AF01
Wirkmechanismus
Tofacitinib ist ein potenter, selektiver Inhibitor der JAK-Familie. In Enzym-Assays hemmt Tofacitinib JAK1, JAK2, JAK3 sowie in geringerem Maße TyK2. Im Gegensatz dazu zeichnet sich Tofacitinib durch eine hohe Selektivität gegenüber anderen Kinasen des menschlichen Genoms aus. In menschlichen Zellen hemmt Tofacitinib bevorzugt die Signalübertragung durch heterodimere Zytokin-Rezeptoren, die mit JAK3 und/oder JAK1 assoziieren, mit funktioneller Selektivität gegenüber Zytokin-Rezeptoren, deren Signalübertragung über JAK2-Dimere erfolgt. Die Hemmung von JAK1 und JAK3 durch Tofacitinib dämpft die Signalübertragung von Interleukinen (IL-2, IL-4, IL-6, IL-7, IL-9, IL-15 und IL-21) und von Typ-I- und Typ-II-Interferonen, was eine Modulation der immunologischen und inflammatorischen Antwort zur Folge hat.
Pharmakodynamische Wirkungen
Bei RA-Patienten war eine bis zu 6‑monatige Tofacitinib-Behandlung mit dosisabhängigen Verringerungen der CD16/56+ natürlichen Killerzellen (NK) verbunden, wobei maximale Verringerungen schätzungsweise etwa 8–10 Wochen nach Therapiebeginn auftraten. Nach Beendigung der Behandlung hatten sich diese Veränderungen innerhalb von 2–6 Wochen im Allgemeinen wieder normalisiert. Die Behandlung mit Tofacitinib ging mit einem dosisabhängigen Anstieg der B-Lymphozyten einher. Die Veränderungen der Anzahl zirkulierender T‑Lymphozyten und ihrer Untergruppen (CD3+, CD4+ und CD8+) waren gering und uneinheitlich.
Nach einer Langzeitbehandlung (mittlere Dauer der Tofacitinib-Behandlung ca. 5 Jahre) zeigten sich mittlere Verringerungen der CD4+ und CD8+ T-Lymphozytenzahl gegenüber dem Ausgangswert von 28 % bzw. 27 %. Im Gegensatz zur beobachteten Abnahme nach der kurzzeitigen Dosierung erhöhte sich die Anzahl der CD16/56+ natürlichen Killerzellen gegenüber dem Ausgangswert im Mittel um 73 %. Die CD19+ B-Zellzahlen zeigten nach der Langzeitbehandlung mit Tofacitinib keinen weiteren Anstieg. All diese Veränderungen der Lymphozyten-Untergruppen kehrten nach zeitweiser Unterbrechung der Behandlung auf den Ausgangswert zurück. Es gab keine Hinweise auf einen Zusammenhang zwischen schwerwiegenden oder opportunistischen Infektionen oder Herpes zoster und den Zellzahlen der Lymphozyten-Untergruppen (siehe Abschnitt 4.2 bezüglich der Überwachung der absoluten Lymphozytenzahl).
Veränderungen der IgG-, IgM- und IgA-Serumspiegel waren bei RA-Patienten nach 6-monatiger Tofacitinib-Behandlung gering, nicht dosisabhängig und ähnlich wie unter Placebo, was auf das Fehlen einer systemischen humoralen Suppression schließen lässt.
Nach Beginn der Behandlung mit Tofacitinib wurde bei Patienten mit rheumatoider Arthritis ein rascher Abfall des C‑reaktiven Proteins (CRP) im Serum festgestellt, der während der weiteren Behandlung bestehen blieb. Die unter Tofacitinib beobachteten Veränderungen des CRP normalisieren sich innerhalb von 2 Wochen nach Beendigung der Behandlung nicht vollständig, was auf eine im Vergleich zur Halbwertszeit längere Dauer der pharmakodynamischen Wirkung hindeutet.
Impfstoff-Studien
In einer kontrollierten klinischen Studie mit RA-Patienten, die die Behandlung mit zweimal täglich 10 mg Tofacitinib oder Placebo begannen, war das Ansprechen auf die Impfung mit Grippeimpfstoff in beiden Gruppen ähnlich: 57 % Tofacitinib und 62 % Placebo. Für die Impfung mit Pneumokokkenpolysaccharid-Impfstoff war das Ansprechen wie folgt: 32 % bei Patienten, die Tofacitinib und MTX erhielten, 62 % unter Tofacitinib-Monotherapie, 62 % unter MTX-Monotherapie und 77 % unter Placebo. Die klinische Relevanz dieser Daten ist nicht bekannt, jedoch lieferte eine separate Impfstoff-Studie mit Grippe- und Pneumokokkenpolysaccharid-Impfstoffen bei Patienten unter Langzeitbehandlung mit zweimal täglich 10 mg Tofacitinib ähnliche Ergebnisse.
Es wurde eine kontrollierte Studie an Patienten mit rheumatoider Arthritis und unter MTX-Basistherapie durchgeführt, in der die Patienten 2 bis 3 Wochen vor Einleitung einer 12-wöchigen Behandlung mit zweimal täglich 5 mg Tofacitinib oder Placebo mit einem attenuierten Lebendimpfstoff gegen Herpes immunisiert wurden. Nach 6 Wochen wurden sowohl bei den mit Tofacitinib als auch bei den mit Placebo behandelten Patienten humorale und zellvermittelte Immunantworten auf VZV nachgewiesen. Diese Immunantworten ähnelten denen, die bei gesunden Probanden im Alter ab 50 Jahren beobachtet wurden. Bei einem Patienten ohne Varizella-Infektion in der Vorgeschichte und ohne Varizella-Antikörper bei Studienbeginn wurde 16 Tage nach der Impfung eine Ausbreitung des Vakzinstamms festgestellt. Tofacitinib wurde abgesetzt und der Patient erholte sich nach der Behandlung mit Standarddosen eines antiviralen Arzneimittels. Dieser Patient zeigte anschießend eine robuste, wenn auch verzögerte humorale und zellvermittelte Immunantwort auf den Impfstoff (siehe Abschnitt 4.4).
Klinische Wirksamkeit und Sicherheit
Klinisches Ansprechen
Das Phase-3-Programm zu Tofacitinib bei JIA bestand aus einer abgeschlossenen Phase-3-Studie (Studie JIA-I [A3921104]) und einer noch laufenden Langzeit-Erweiterungsstudie (A3921145). In diese Studien wurden die folgenden JIA-Untergruppen eingeschlossen: Patienten mit RF+ oder RF- Polyarthritis, erweiterter Oligoarthritis, systemischer JIA mit aktiver Arthritis und aktuell keiner systemischen Symptomatik (sogenannter pJIA-Datensatz) und zwei separate Untergruppen mit juveniler PsA und Enthesitis-assoziierter Arthritis (enthesitis-related Arthritis, ERA). Allerdings umfasst die pJIA-Wirksamkeits-Population nur die Subgruppen mit RF+ oder RF- Polyarthritis oder erweiterter Oligoarthritis. In der Subgruppe mit systemischer JIA mit aktiver Arthritis und aktuell keiner systemischen Symptomatik wurden keine eindeutigen Ergebnisse erzielt. Patienten mit juveniler PsA werden als separate Subgruppe für die Wirksamkeit berücksichtigt. ERA-Patienten werden nicht in die Wirksamkeitsanalyse aufgenommen.
Alle infrage kommenden Patienten in der offenen Studie JIA-I erhielten 18 Wochen lang Tofacitinib 5 mg Filmtabletten zweimal täglich oder eine äquivalente Dosis Tofacitinib Lösung zum Einnehmen zweimal täglich gemäß ihrem Körpergewicht (Einleitungsphase). Patienten, die am Ende der offenen Phase mindestens ein Ansprechen von JIA ACR30 erreichten, wurden für die 26‑wöchige, doppelblinde, placebokontrollierte Phase im Verhältnis 1:1 entweder auf Tofacitinib 5 mg Filmtabletten oder Tofacitinib Lösung zum Einnehmen oder auf Placebo randomisiert. Patienten, die am Ende der offenen Einleitungsphase kein Ansprechen von JIA ACR30 erreichten oder zu einem beliebigen Zeitpunkt eine einzelne Episode eines Krankheitsschubs hatten, wurden aus der Studie ausgeschlossen. In die offene Einleitungsphase wurden insgesamt 225 Patienten aufgenommen, von denen 173 Patienten (76,9 %) in die doppelblinde Phase randomisiert werden konnten und entweder die wirkstoffhaltige Behandlung mit Tofacitinib 5 mg Filmtabletten oder eine äquivalente Dosis Tofacitinib Lösung zum Einnehmen zweimal täglich gemäß ihrem Körpergewicht (n = 88) oder Placebo (n = 85) erhielten. 58 Patienten (65,9 %) in der Tofacitinib-Gruppe und 58 Patienten (68,2 %) in der Placebogruppe nahmen während der doppelblinden Phase MTX ein. Dies war laut Prüfplan erlaubt, aber nicht erforderlich.
133 Patienten mit pJIA (RF+ oder RF- Polyarthritis und erweiterter Oligoarthritis) und 15 Patienten mit juveniler PsA wurden für die doppelblinde Studienphase randomisiert und wie unten beschrieben für die Analyse der Wirksamkeit berücksichtigt.
Krankheitszeichen und Symptome
Im Vergleich zu Patienten, die Placebo erhielten, kam es bei Patienten mit pJIA, die in Studie JIA-I mit Tofacitinib 5 mg Filmtabletten zweimal täglich oder einer äquivalenten Dosis Tofacitinib Lösung zum Einnehmen zweimal täglich gemäß ihrem Körpergewicht behandelt wurden, in Woche 44 bei einem signifikant geringeren Anteil zu einem Krankheitsschub. Im Vergleich zu Patienten, die Placebo erhielten, erreichte von den Patienten mit pJIA, die mit Tofacitinib 5 mg Filmtabletten oder Tofacitinib Lösung zum Einnehmen behandelt wurden, in Woche 44 ein signifikant größerer Anteil ein Ansprechen von JIA ACR30, 50 oder 70 (siehe Tabelle 8).
Die Ergebnisse zum Auftreten von Krankheitsschüben und zum JIA ACR30/50/70 fielen bei den JIA-Subtypen RF+ Polyarthritis, RF- Polyarthritis, erweiterte Oligoarthritis und jPsA im Vergleich zu Placebo zugunsten von Tofacitinib 5 mg zweimal täglich aus und stimmten mit den Ergebnissen für die Studienpopulation insgesamt überein.
Die Ergebnisse zum Auftreten von Krankheitsschüben und zum JIA ACR30/50/70 fielen bei pJIA-Patienten, die an Tag 1 Tofacitinib 5 mg zweimal täglich zusammen mit MTX erhielten (n = 101 [76 %]) und Patienten, die Tofacitinib als Monotherapie erhielten (n = 32 [24 %]), im Vergleich zu Placebo zugunsten von Tofacitinib 5 mg zweimal täglich aus. Des Weiteren fielen die Ergebnisse zum Auftreten von Krankheitsschüben und zum JIA ACR30/50/70 auch sowohl für pJIA-Patienten mit vorhergehender Behandlung mit bDMARD (n = 39 [29 %]) als auch bDMARD-naiven Patienten (n = 94 [71 %]) im Vergleich zu Placebo zugunsten von Tofacitinib 5 mg zweimal täglich aus.
In Studie JIA-I lag das Ansprechen von JIA ACR30 in Woche 2 der offenen Einleitungsphase bei Patienten mit pJIA bei 45,03 %.
Tabelle 8: Primäre und sekundäre Wirksamkeitsendpunkte bei Patienten mit pJIA in Woche 44* der Studie JIA-I (alle p-Werte < 0,05)
Primärer Endpunkt |
Behandlungs-gruppe |
Auftretensrate |
Unterschied (in %) zu Placebo (95% KI) |
Auftreten eines Krankheitsschubs |
Tofacitinib 5 mg zweimal täglich |
28 % |
-24,7 (-40,8; -8,5) |
Placebo |
53 % |
||
Sekundäre Endpunkte |
Behandlungs-gruppe |
Ansprechenrate |
Unterschied (in %) zu Placebo (95% KI) |
JIA ACR30 |
Tofacitinib 5 mg zweimal täglich |
72 % |
24,7 (-8,50; -40,8) |
Placebo |
47 % |
||
JIA ACR50 |
Tofacitinib 5 mg zweimal täglich |
67 % |
20,2 (-3,72; -36,7) |
Placebo |
47 % |
||
JIA ACR70 |
Tofacitinib 5 mg zweimal täglich |
55 % |
17,4 (-0,65; -34,0) |
Placebo |
38 % |
||
Sekundärer Endpunkt (Fehler 1. Art kontrolliert) |
Behandlungs-gruppe |
LS-Mittelwert (SEM) |
Unterschied zu Placebo (95% KI) |
Veränderung des CHAQ-Behinderungsindex gegenüber Ausgangswert der doppelblinden Phase |
Tofacitinib 5 mg zweimal täglich |
-0,11 (0,04) |
-0,11 (-0,22; -0,01) |
Placebo |
0,00 (0,04) |
ACR = American College of Rheumatology, CHAQ = Childhood Health Assessment Questionnaire, KI = Konfidenzintervall, LS = Kleinste Quadrate (least squares), n = Anzahl Patienten mit Beobachtung beim Termin, N = Gesamtanzahl Patienten, JIA = juvenile idiopathische Arthritis, SEM = Standardfehler des Mittelwerts (standard error of the mean)
* Die 26-wöchige, doppelblinde Phase geht von Woche 18 bis Woche 44 und nach dem Randomisierungstag.
Die Fehler 1. Art kontrollierten Endpunkte werden in der folgenden Reihenfolge getestet: Krankheitsschub, JIA ACR50, JIA ACR30, JIA ACR70, CHAQ Disability Index.
In der doppelblinden Phase zeigte sich in Woche 24 und Woche 44 für jede der Komponenten des JIA-ACR-Ansprechens eine größere Verbesserung gegenüber dem Ausgangswert der offenen Phase (Tag 1). Dies galt für pJIA-Patienten in Studie JIA-I, die Tofacitinib Lösung zum Einnehmen 5 mg zweimal täglich oder eine äquivalente Dosierung gemäß ihrem Körpergewicht zweimal täglich erhielten, im Vergleich zu Placebo.
Körperliche Funktionsfähigkeit und gesundheitsbezogene Lebensqualität
Veränderungen der körperlichen Funktionsfähigkeit in Studie JIA-I wurden mit dem CHAQ-Behinderungsindex gemessen. Die mittlere Veränderung des CHAQ-Behinderungsindex bei pJIA-Patienten gegenüber dem Ausgangswert der doppelblinden Phase war in Woche 44 in der Gruppe mit Tofacitinib 5 mg Filmtabletten zweimal täglich oder einer äquivalenten Dosis Tofacitinib Lösung zum Einnehmen gemäß Körpergewicht zweimal täglich signifikant geringer als bei Placebo (siehe Tabelle 8). Die mittlere Veränderung des CHAQ-Behinderungsindex gegenüber dem Ausgangswert der doppelblinden Phase fiel im Vergleich zu Placebo bei den JIA-Subtypen RF+ Polyarthritis, RF- Polyarthritis, erweiterte Oligoarthritis und jPsA zugunsten von Tofacitinib 5 mg 2x tägl. aus und entsprach derjenigen für die Gesamtpopulation der Studie.
Langzeit-Erweiterungsstudie
In der Langzeit-Erweiterungsstudie erhielten 152 Patienten mit pJIA aus Studie JIA-I bis zu 7,5 Jahre lang unverblindet Tofacitinib 5 mg zweimal täglich. Patienten, die während der unverblindeten (open-label, OL) Phase von Studie JIA-I (Tofacitinib OL) ausschieden, und jene, die in den doppelblinden (DB) Teil übertraten und Tofacitinib 5 mg zweimal täglich (Tofacitinib-Tofacitinib DB) oder Placebo (Tofacitinib-Placebo DB) erhielten, kamen für die Aufnahme in die Langzeit-Erweiterungsstudie infrage, wenn sie die Aufnahmekriterien erfüllten. Der Großteil der Patienten, die in die DB-Phase von Studie JIA-I übertraten, traten auch in die Langzeit-Erweiterungsstudie ein (64/67 aus Tofacitinib-Tofacitinib-DB und 64/66 aus Tofacitinib-Placebo DB). Die mediane Behandlungsdauer mit Tofacitinib betrug 46,3 Monate (Spanne: 1,0, 88,4) für die Gruppe Tofacitinib-Tofacitinib DB bzw. 43,8 Monate (Spanne: 1,5, 89,0) für die Gruppe Tofacitinib-Placebo DB. Die JIA ACR-Ansprechraten in Monat 45, ermittelt gemäß den Beobachtungen und mit einer modifizierten Imputation von Non-Respondern (wobei Patienten, die aufgrund eines unzureichenden klinischen Ansprechens oder eines unerwünschten Ereignisses ausschieden, als Non-Responder betrachtet wurden), sind in Tabelle 9 für die Gruppe Tofacitinib-Tofacitinib DB und die Gruppe Tofacitinib-Placebo DB dargestellt.
Tabelle 9: Wirksamkeitsendpunkte in Monat 45 der Langzeit-Erweiterungsstudie bei Patienten mit pJIA (Analysen gemäß den Beobachtungen und mit modifizierter Imputation von Non-Respondern)
Tofacitinib-Tofacitinib DB |
Tofacitinib-Placebo DB |
|||
Beobachtungen |
mNRI |
Beobachtungen |
mNRI |
|
JIA ACR30, n / N (%) |
33 / 34 (97,1) |
33 / 43 (76,7) |
34 / 37 (91,9) |
34 / 49 (69,4) |
JIA ACR50, n / N (%) |
32 / 34 (94,1) |
32 / 43 (74,4) |
34 / 37 (91,9) |
34 / 49 (69,4) |
JIA ACR70, n / N (%) |
29 / 34 (85,3) |
29 / 43 (67,4) |
32 / 37 (86,5) |
32 / 49 (65,3) |
JIA ACR90, n / N (%) |
19 / 34 (55,9) |
19 / 43 (44,2) |
26 / 37 (70,3) |
26 / 49 (53,1) |
Inaktive Erkrankung gemäß JIA ACR, n / N (%) |
13 / 36 (36,1) |
13 / 45 (28,9) |
17 / 39 (43,6) |
17 / 51 (33,3) |
ACR = American College of Rheumatology; DB = doppelblind; JIA = juvenile idiopathische Arthritis; mNRI = modifizierte Imputation von Non-Respondern; n = Anzahl Patienten mit Beobachtung beim Termin; N = Gesamtanzahl Patienten. | ||||
Langzeitsicherheitsdaten aus kontrollierten Studien zu RA
Bei der Studie ORAL Surveillance (A3921133) handelte es sich um eine große (n = 4.362), randomisierte, aktiv kontrollierte Studie zur Überwachung der Sicherheit nach Zulassung bei Patienten mit rheumatoider Arthritis, die 50 Jahre alt oder älter waren und mindestens einen zusätzlichen kardiovaskulären Risikofaktor aufwiesen (Kardiovaskuläre Risikofaktoren definiert als: gegenwärtiges Rauchen von Zigaretten, Diagnose einer Hypertonie, Diabetes mellitus, familiäre Vorbelastung für vorzeitige koronare Herzerkrankung, vorbestehende koronare Herzkrankheit, einschließlich revaskularisierender Maßnahmen in der Vorgeschichte, koronararterieller Bypass, Myokardinfarkt, Herzstillstand, instabile Angina pectoris, akutes Koronarsyndrom sowie bestehende extraartikuläre Manifestationen der RA, z. B. Noduli, Sjögren-Syndrom, Anämie bei chronischer Entzündung, Lungenmanifestationen). Die Mehrheit (über 90 %) der mit Tofacitinib behandelten Patienten, die aktuelle oder ehemalige Raucher waren, rauchten mehr als 10 Jahre lang und hatten im Median 35,0 bzw. 39,0 Raucherjahre. Die Patienten mussten bei Aufnahme in die Studie mit einer feststehenden Dosis Methotrexat behandelt sein; eine Dosisanpassung war während der Studie erlaubt.
In dieser offenen Studie wurden Patienten im Verhältnis 1:1:1 auf zweimal täglich 10 mg Tofacitinib, zweimal täglich 5 mg Tofacitinib oder einen TNF-Inhibitor (entweder 50 mg Etanercept einmal wöchentlich oder 40 mg Adalimumab alle zwei Wochen) randomisiert. Die ko-primären Endpunkte waren adjudizierte Krebserkrankungen (außer nicht-melanozytärer Hautkrebs, NMSC) und adjudizierte schwere kardiovaskuläre Ereignisse (major adverse cardiovascular events, MACE). Kumulative Inzidenz und statistische Auswertung der Endpunkte waren verblindet. Es handelte sich um eine ereignisgesteuerte Studie, in der mindestens 1.500 Patienten 3 Jahre lang nachbeobachtet werden mussten. Die Studienmedikation mit zweimal täglich 10 mg Tofacitinib wurde abgesetzt und die Patienten auf zweimal täglich 5 mg umgestellt, da venöse thromboembolische Ereignisse (VTE) als dosisabhängiges Signal festgestellt wurden. Für Patienten im Behandlungsarm mit Tofacitinib 10 mg zweimal täglich wurden die vor und nach dem Dosiswechsel erhobenen Daten in ihrer ursprünglich randomisierten Behandlungsgruppe analysiert.
Die Studie erfüllte nicht das Nichtunterlegenheitskriterium für den primären Vergleich der kombinierten Tofacitinib-Dosen mit dem TNF-Inhibitor, da die Obergrenze des 95 % KI für die HR das vorab festgelegte Nichtunterlegenheitskriterium von 1,8 für adjudizierte MACE und adjudizierte Malignome, außer NMSC, überschritt.
Die Ergebnisse für adjudizierte MACE, adjudizierte Malignome außer NMSC und andere ausgewählte Ereignisse sind unten angegeben.
MACE (einschließlich Myokardinfarkt) und venöse thromboembolische Ereignisse (VTE)
Bei mit Tofacitinib behandelten Patienten wurde im Vergleich zur Behandlung mit TNF-Inhibitor ein Anstieg von nicht tödlichem Myokardinfarkt beobachtet. Bei mit Tofacitinib behandelten Patienten wurde im Vergleich zur Behandlung mit TNF-Inhibitor ein dosisabhängiger Anstieg von VTE-Ereignissen beobachtet (siehe Abschnitte 4.4 und 4.8).
Tabelle 10: Inzidenzrate und Hazard Ratio für MACE, Myokardinfarkt und venöse thromboembolische Ereignisse
Tofacitinib 5 mg zweimal täglich |
Tofacitinib 10 mg zweimal täglicha |
Alle Tofacitinibb |
TNF-Inhibitor (TNFi) |
|||||
MACEc | ||||||||
IR (95 % KI) pro 100 PJ |
0,91 (0,67; 1,21) |
1,05 (0,78; 1,38) |
0,98 (0,79; 1,19) |
0,73 (0,52; 1,01) |
||||
HR (95 % KI) gegenüber TNFi |
1,24 (0,81; 1,91) |
1,43 (0,94; 2,18) |
1,33 (0,91; 1,94) |
|||||
Tödlicher MIc | ||||||||
IR (95 % KI) pro 100 PJ |
0,00 (0,00; 0,07) |
0,06 (0,01; 0,18) |
0,03 (0,01; 0,09) |
0,06 (0,01; 0,17) |
||||
HR (95 % KI) gegenüber TNFi |
0,00 (0,00; Inf) |
1,03 (0,21; 5,11) |
0,50 (0,10; 2,49) |
|||||
Nicht tödlicher MIc | ||||||||
IR (95 % KI) pro 100 PJ |
0,37 (0,22; 0,57) |
0,33 (0,19; 0,53) |
0,35 (0,24; 0,48) |
0,16 (0,07; 0,31) |
||||
HR (95 % KI) gegenüber TNFi |
2,32 (1,02; 5,30) |
2,08 (0,89; 4,86) |
2,20 (1,02; 4,75) |
|||||
VTEd | ||||||||
IR (95 % KI) pro 100 PJ |
0,33 (0,19; 0,53) |
0,70 (0,49; 0,99) |
0,51 (0,38; 0,67) |
0,20 (0,10; 0,37) |
||||
HR (95 % KI) gegenüber TNFi |
1,66 (0,76; 3,63) |
3,52 (1,74; 7,12) |
2,56 (1,30; 5,05) |
|||||
LEd | ||||||||
IR (95 % KI) pro 100 PJ |
0,17 (0,08; 0,33) |
0,50 (0,32; 0,74) |
0,33 (0,23; 0,46) |
0,06 (0,01; 0,17) |
||||
HR (95 % KI) gegenüber TNFi |
2,93 (0,79; 10,83) |
8,26 (2,49; 27,43) |
5,53 (1,70; 18,02) |
|||||
TVTd | ||||||||
IR (95 % KI) pro 100 PJ |
0,21 (0,11; 0,38) |
0,31 (0,17; 0,51) |
0,26 (0,17; 0,38) |
0,14 (0,06; 0,29) |
||||
HR (95 % KI) gegenüber TNFi |
1,54 (0,60; 3,97) |
2,21 (0,90; 5,43) |
1,87 (0,81; 4,30) |
|||||
a Die Behandlungsgruppe mit Tofacitinib 10 mg zweimal täglich umfasst Daten von Patienten, die infolge einer Studienänderung von Tofacitinib 10 mg zweimal täglich auf Tofacitinib 5 mg zweimal täglich umgestellt wurden. | ||||||||
Mit Hilfe eines multivariaten Cox-Modells mit Rückwärtsselektion wurden die folgenden prädiktiven Faktoren für die Entwicklung von MI (tödlich und nicht tödlich) ermittelt: Alter ≥ 65 Jahre, männlich, Raucher oder ehemaliger Raucher, Diabetes in der Anamnese und vorbestehende koronare Herzkrankheit (einschließlich Myokardinfarkt, koronare Herzkrankheit, stabile Angina pectoris oder koronare Gefäßeingriffe) (siehe Abschnitte 4.4 und 4.8).
Maligne Erkrankungen
Bei mit Tofacitinib behandelten Patienten wurde im Vergleich zur Behandlung mit TNF-Inhibitor ein Anstieg von malignen Erkrankungen, aus,genommen NMSC, insbesondere Lungenkrebs, Lymphom sowie ein Anstieg von NMSC beobachtet.
Tabelle 11: Inzidenzrate und Hazard Ratio für maligne Erkrankungena
Tofacitinib 5 mg zweimal täglich |
Tofacitinib 10 mg zweimal täglichb |
Alle Tofacitinibc |
TNF-Inhibitor (TNFi) |
|||||
Maligne Erkrankungen außer NMSC | ||||||||
IR (95 % KI) pro 100 PJ |
1,13 (0,87; 1,45) |
1,13 (0,86; 1,45) |
1,13 (0,94; 1,35) |
0,77 (0,55; 1,04) |
||||
HR (95 % KI) gegenüber TNFi |
1,47 (1,00; 2,18) |
1,48 (1,00; 2,19) |
1,48 (1,04; 2,09) |
|||||
Lungenkrebs | ||||||||
IR (95 % KI) pro 100 PJ |
0,23 (0,12; 0,40) |
0,32 (0,18; 0,51) |
0,28 (0,19; 0,39) |
0,13 (0,05; 0,26) |
||||
HR (95 % KI) gegenüber TNFi |
1,84 (0,74; 4,62) |
2,50 (1,04; 6,02) |
2,17 (0,95; 4,93) |
|||||
Lymphom | ||||||||
IR (95 % KI) pro 100 PJ |
0,07 (0,02; 0,18) |
0,11 (0,04; 0,24) |
0,09 (0,04; 0,17) |
0,02 (0,00; 0,10) |
||||
HR (95 % KI) gegenüber TNFi |
3,99 (0,45; 35,70) |
6,24 (0,75; 51,86) |
5,09 (0,65; 39,78) |
|||||
NMSC | ||||||||
IR (95 % KI) pro 100 PJ |
0,61 (0,41; 0,86) |
0,69 (0,47; 0,96) |
0,64 (0,50; 0,82) |
0,32 (0,18; 0,52) |
||||
HR (95 % KI) gegenüber TNFi |
1,90 (1,04; 3,47) |
2,16 (1,19; 3,92) |
2,02 (1,17; 3,50) |
|||||
a Für Malignome außer NMSC, Lungenkrebs und Lymphome auf Grundlage der Ereignisse, die während der Behandlung oder nach Absetzen der Behandlung bis Studienende auftraten. Für NMSC auf Grundlage der Ereignisse, die während der Behandlung oder innerhalb von 28 Tagen nach Beendigung der Behandlung auftraten. | ||||||||
Mit Hilfe eines multivariaten Cox-Modells mit Rückwärtsselektion wurden die folgenden prädiktiven Faktoren für die Entwicklung von malignen Erkrankungen mit Ausnahme von NMSC identifiziert: Alter ≥ 65 Jahre und aktuelles oder früheres Rauchen (siehe Abschnitt 4.4 und 4.8).
Mortalität
Erhöhte Mortalität wurde bei mit Tofacitinib behandelten Patienten im Vergleich zu Patienten unter TNF-Inhibitoren festgestellt. Die Mortalität war hauptsächlich durch kardiovaskuläre Ereignisse, Infektionen und Krebserkrankungen bedingt.
Tabelle 12: Inzidenzrate und Hazard Ratio für Mortalitäta
Tofacitinib 5 mg zweimal täglich |
Tofacitinib 10 mg zweimal täglichb |
Alle Tofacitinibc |
TNF-Inhibitor |
|
Mortalität (jegliche Ursache) |
||||
IR (95 % KI) pro 100 PJ |
0,50 (0,33; 0,74) |
0,80 (0,57; 1,09) |
0,65 (0,50; 0,82) |
0,34 (0,20; 0,54) |
HR (95 % KI) gegenüber TNFi |
1,49 (0,81; 2,74) |
2,37 (1,34; 4,18) |
1,91 (1,12; 3,27) |
|
Tödliche Infektionen |
||||
IR (95 % KI) pro 100 PJ |
0,08 (0,02; 0,20) |
0,18 (0,08; 0,35) |
0,13 (0,07; 0,22) |
0,06 (0,01; 0,17) |
HR (95 % KI) gegenüber TNFi |
1,30 (0,29; 5,79) |
3,10 (0,84; 11,45) |
2,17 (0,62; 7,62) |
|
Tödliche CV-Ereignisse |
||||
IR (95 % KI) pro 100 PJ |
0,25 (0,13; 0,43) |
0,41 (0,25; 0,63) |
0,33 (0,23; 0,46) |
0,20 (0,10; 0,36) |
HR (95 % KI) gegenüber TNFi |
1,26 (0,55; 2,88) |
2,05 (0,96; 4,39) |
1,65 (0,81; 3,34) |
|
Tödliche Malignome |
||||
IR (95 % KI) pro 100 PJ |
0,10 (0,03; 0,23) |
0,00 (0,00; 0,08) |
0,05 (0,02; 0,12) |
0,02 (0,00; 0,11) |
HR (95 % KI) gegenüber TNFi |
4,88 (0,57; 41,74) |
0 (0,00; Inf) |
2,53 (0,30; 21,64) |
a Auf Grundlage der Ereignisse, die während der Behandlung oder innerhalb von 28 Tagen nach Beendigung der Behandlung auftraten.
b Die Behandlungsgruppe mit Tofacitinib 10 mg zweimal täglich umfasst Daten von Patienten, die infolge einer Studienänderung von Tofacitinib 10 mg zweimal täglich auf Tofacitinib 5 mg zweimal täglich umgestellt wurden.
c Kombinierte Daten von Tofacitinib 5 mg zweimal täglich und Tofacitinib 10 mg zweimal täglich.
Abkürzungen: TNF = Tumornekrosefaktor, IR = Inzidenzrate, HR = Hazard Ratio, KI = Konfidenzintervall, PJ = Patientenjahre, CV = kardiovaskulär, Inf = unendlich.
Das PK-Profil von Tofacitinib ist von einer raschen Resorption (Maximalkonzentrationen im Plasma werden innerhalb von 0,5 bis 1 Stunde erreicht), einer raschen Elimination (Halbwertszeit ca. 3 Stunden) und einem dosisproportionalen Anstieg der systemischen Exposition gekennzeichnet. Steady-State-Konzentrationen werden innerhalb von 24–48 Stunden erreicht, die Akkumulation ist bei zweimal täglicher Einnahme vernachlässigbar.
Resorption und Verteilung
Tofacitinib wird gut resorbiert. Die orale Bioverfügbarkeit beträgt 74 %. Die gleichzeitige Einnahme von Tofacitinib mit einer fettreichen Mahlzeit bewirkte keine Veränderungen des AUC-Werts, aber die Cmax verringerte sich um 32 %. In klinischen Studien wurde Tofacitinib unabhängig von den Mahlzeiten eingenommen.
Nach der intravenösen Anwendung beträgt das Verteilungsvolumen 87 l. Das zirkulierende Tofacitinib wird zu etwa 40 % an Plasmaproteine gebunden. Tofacitinib wird in erster Linie an Albumin und offenbar nicht an α-1-saures Glykoprotein gebunden. Tofacitinib verteilt sich gleichmäßig zwischen Erythrozyten und Plasma.
Biotransformation und Elimination
Tofacitinib wird zu etwa 70 % über hepatische Metabolisierung und zu 30 % unverändert über die Nieren ausgeschieden. Der Metabolismus von Tofacitinib erfolgt hauptsächlich über CYP3A4, mit geringfügiger Beteiligung von CYP2C19. In einer Studie am Menschen mit radioaktiven Markern wurde über 65 % der im Blut vorhandenen Gesamtradioaktivität dem unveränderten Wirkstoff zugeschrieben. Die restlichen 35 % wurden 8 Metaboliten zugeschrieben, die jeweils mit weniger als 8 % zur Gesamtradioaktivität beitrugen. Alle Metaboliten wurden in Tierarten beobachtet und haben in Bezug auf die JAK1/3-Inhibition schätzungsweise weniger als 10 % der Wirksamkeit von Tofacitinib. In menschlichen Proben wurden keine Anzeichen einer Stereo-Konversion festgestellt. Die pharmakologische Aktivität von Tofacitinib wird dem Muttermolekül zugeschrieben. In vitro ist Tofacitinib ein Substrat für MDR1, jedoch nicht für Breast Cancer Resistance-Protein (BCRP), OATP1B1/1B3 oder OCT1/2.
Eingeschränkte Nierenfunktion
Studienteilnehmer mit leichter (Kreatinin-Clearance 50–80 ml/min), mittelschwerer (Kreatinin-Clearance 30–49 ml/min) und schwerer Nierenfunktionsstörung (Kreatinin-Clearance unter 30 ml/min) hatten im Vergleich zu Teilnehmern mit normaler Nierenfunktion eine um 37 %, 43 % bzw. 123 % höhere AUC (siehe Abschnitt 4.2). Bei Teilnehmern mit Nierenerkrankung im Endstadium (end‑stage renal disease, ESRD) war der Beitrag der Dialyse zur Gesamtclearance von Tofacitinib relativ gering. Nach einer Einzeldosis von 10 mg war die mittlere AUC bei Teilnehmern mit ESRD basierend auf den Konzentrationen, die an den Nicht-Dialyse-Tagen gemessen wurden, etwa 40 % (90 % Konfidenzintervalle: 1,5–95 %) höher als bei Teilnehmern mit normaler Nierenfunktion. In klinischen Studien wurde Tofacitinib nicht bei Patienten geprüft, deren Ausgangs-Kreatinin-Clearance (Schätzung nach Cockcroft-Gault-Gleichung) weniger als 40 ml/min betrug (siehe Abschnitt 4.2).
Eingeschränkte Leberfunktion
Bei Studienteilnehmern mit leichter (Child Pugh-Klasse A) und mittelschwerer (Child Pugh-Klasse B) Leberfunktionsstörung war die AUC im Vergleich zu Teilnehmern mit normaler Leberfunktion um 3 % bzw. 65 % höher. In klinischen Studien wurde Tofacitinib bei Patienten mit schwerer Leberfunktionsstörung (Child Pugh-Klasse C) (siehe Abschnitte 4.2 und 4.4) oder auf Hepatitis B oder C positiv getesteten Patienten nicht geprüft.
Wechselwirkungen
Tofacitinib ist weder ein Inhibitor, noch ein Induktor von CYP (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 und CYP3A4). Es ist kein Inhibitor von UGT (UGT1A1, UGT1A4, UGT1A6, UGT1A9 und UGT2B7). Tofacitinib ist in klinisch bedeutsamer Konzentration kein Inhibitor von MDR1, OATP1B1/1B3, OCT2, OAT1/3 oder MRP.
Pharmakokinetik bei Kindern und Jugendlichen mit juveniler idiopathischer Arthritis
Eine pharmakokinetische Populationsanalyse der Ergebnisse sowohl zu Tofacitinib 5 mg Filmtabletten zweimal täglich als auch zu Tofacitinib Lösung zum Einnehmen in äquivalenter Dosierung basierend auf dem Körpergewicht zweimal täglich zeigte, dass sowohl die Clearance als auch das Verteilungsvolumen von Tofacitinib sich mit abnehmendem Körpergewicht bei JIA-Patienten verringerten. Die verfügbaren Daten zeigten keine klinisch relevanten Unterschiede der Tofacitinib-Exposition (AUC) nach Alter, ethnischer Zugehörigkeit, Geschlecht, Patiententyp oder Ausgangsschwere der Erkrankung. Die interindividuelle Variabilität (prozentualer Variationskoeffizient) der AUC liegt schätzungsweise bei 24 %.
In nichtklinischen Studien wurden Auswirkungen auf das Immunsystem und das blutbildende System beobachtet, die den pharmakologischen Eigenschaften (JAK-Hemmung) von Tofacitinib zugeschrieben wurden. Bei klinisch relevanten Dosen wurden sekundäre Auswirkungen einer Immunsuppression wie etwa bakterielle und virale Infektionen und Lymphome festgestellt. Lymphome wurden bei 3 von 8 adulten Affen beim 6- oder 3-Fachen des klinischen Tofacitinib-Expositionsspiegels (AUC des ungebundenen Wirkstoffs beim Menschen bei einer Dosis von zweimal täglich 5 mg oder 10 mg) beobachtet und bei 0 von 14 juvenilen Affen beim Fünf- oder Zweieinhalbfachen des klinischen Expositionsspiegels bei zweimal täglich 5 mg oder 10 mg. Die Exposition von Affen, bei der keine schädigende Wirkung in Bezug auf Lymphome beobachtet wurde (no observed adverse effect level, NOAEL), entsprach ungefähr dem Ein- oder 0,5-Fachen des klinischen Expositionsspiegels bei zweimal täglich 5 mg oder 10 mg. Sonstige Befunde bei Dosen, die die humane Exposition überstiegen, schlossen Auswirkungen auf das hepatische und gastrointestinale System ein.
Tofacitinib ist, basierend auf den Ergebnissen einer Reihe von in vitro und in vivo-Tests auf Genmutationen und Chromosomenaberrationen, nicht mutagen oder genotoxisch.
Das karzinogene Potenzial von Tofacitinib wurde in einer 6-monatigen Studie zur Karzinogenität an transgenen rasH2-Mäusen und in einer 2-jährigen Studie zur Karzinogenität an Ratten untersucht. Bei Mäusen war Tofacitinib bei Expositionen bis zum 38- oder 19-Fachen des klinischen Expositionsspiegels bei zweimal täglich 5 mg oder 10 mg nicht karzinogen. Bei Ratten traten gutartige Tumoren der Leydigschen Zellen auf: Benigne Tumoren der Leydig-Zellen bei der Ratte sind nicht mit einem Risiko für Tumoren der Leydigschen Zellen beim Menschen verbunden. Bei weiblichen Ratten entwickelten sich bei Expositionen größer oder gleich dem 83- oder 41-Fachen des klinischen Expositionsspiegels bei zweimal täglich 5 mg oder 10 mg maligne Hibernome (Tumoren des braunen Fettgewebes). Gutartige Thymome traten bei weiblichen Ratten beim 187- oder 94-Fachen des klinischen Expositionsspiegels bei zweimal täglich 5 mg oder 10 mg auf.
Tofacitinib erwies sich bei Ratten und Kaninchen als teratogen und hatte bei Ratten Auswirkungen auf die weibliche Fertilität (geringere Trächtigkeitsquote, Abnahme der Zahl der Gelbkörper, der Implantationsstellen und lebensfähigen Föten sowie eine Zunahme frühzeitiger Resorptionen), die Geburt und die peri-/postnatale Entwicklung. Tofacitinib hatte keine Auswirkungen auf die männliche Fertilität, Spermien-Motilität oder Spermien-Konzentration. Tofacitinib wurde 1 bis 8 Stunden nach der Anwendung in die Muttermilch laktierender Ratten sezerniert in Konzentrationen, die ungefähr dem Zweifachen der Serumkonzentration entsprachen. In Studien an juvenilen Ratten und Affen hatte Tofacitinib bei Expositionen, die mit den für den Menschen zugelassenen Dosen vergleichbar waren, keine Auswirkungen auf die Knochenentwicklung bei männlichen oder weiblichen Tieren.
In Studien mit juvenilen Tieren wurden keine auf Tofacitinib bezogenen Befunde erhoben, die auf eine höhere Sensitivität pädiatrischer Populationen im Vergleich zu Erwachsenen hinweisen. In einer Fertilitätsstudie an juvenilen Ratten gab es keine Hinweise auf eine Entwicklungstoxizität, es wurden keine Auswirkungen auf die sexuelle Reifung festgestellt, und es gab keine Hinweise auf eine Reproduktionstoxizität (Paarung und Fertilität) nach der Geschlechtsreife. In einer 1-monatigen Studie an juvenilen Ratten und einer 39-wöchigen Studie an juvenilen Affen wurden durch Tofacitinib bedingte Auswirkungen auf immunologische und hämatologische Parameter beobachtet, die einer JAK1/3- und JAK2-Inhibition entsprechen. Diese Auswirkungen waren reversibel und stimmten mit denjenigen überein, die auch bei erwachsenen Tieren bei ähnlichen Expositionen beobachtet wurden.
Traubenaroma (enthält Propylenglycol [E 1520], Glycerin [E 422] und natürliche Aromen)
Salzsäure
Milchsäure (E 270)
Gereinigtes Wasser
Natriumbenzoat (E 211)
Sucralose (E 955)
Xylitol (E 967)
Nicht zutreffend
2 Jahre
Haltbarkeit nach Anbruch
60 Tage nach Anbruch verwerfen.
Für dieses Arzneimittel sind bezüglich der Temperatur keine besonderen Lagerungsbedingungen erforderlich.
In der Originalflasche und -verpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Anbruch des Arzneimittels, siehe Abschnitt 6.3.
Weiß gefärbte 250-ml-HDPE-Flaschen mit 240 ml Lösung zum Einnehmen mit kindergesichertem Verschluss (Polypropylen) mit PP-Liner und Aluminiumfolien-Wärmeinduktionsversiegelung und mit einer 5‑ml‑Applikationsspritze für Zubereitungen zum Einnehmen mit 3,2-ml-, 4-ml- und 5-ml-Skalierung.
Zum Verschlusssystem des Behältnisses gehört auch ein Flaschenadapter zum Einpressen aus Polyethylen niedriger Dichte (LDPE).
Packungsgröße: Jede Packung enthält eine Flasche, einen Einpress-Flaschenadapter und eine Applikationsspritze für Zubereitungen zum Einnehmen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Brüssel
Belgien
EU/1/17/1178/015
Datum der Erteilung der Zulassung: 22. März 2017
Datum der letzten Verlängerung der Zulassung: 04. März 2022
Juni 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
VERKAUFSABGRENZUNG IN DEUTSCHLAND
Verschreibungspflichtig
REZEPTPFLICHT/APOTHEKENPFLICHT IN ÖSTERREICH
Rezept- und apothekenpflichtig, wiederholte Abgabe verboten
PACKUNGSGRÖSSEN IN DEUTSCHLAND
240 ml Lösung zum Einnehmen in einer 250 ml Flasche
PACKUNGSGRÖSSEN IN ÖSTERREICH
240 ml Lösung zum Einnehmen in einer 250 ml Flasche
REPRÄSENTANT IN DEUTSCHLAND
PFIZER PHARMA GmbH
Friedrichstr. 110
10117 Berlin
Tel.: 030 550055-51000
Fax: 030 550054-10000
REPRÄSENTANT IN ÖSTERREICH
Pfizer Corporation Austria Ges.m.b.H.
Floridsdorfer Hauptstraße 1
A-1210 Wien
Tel.: +43 (0)1 521 15-0