
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Kerendia 10 mg Filmtabletten
Kerendia 20 mg Filmtabletten
Kerendia 40 mg Filmtabletten
Kerendia 10 mg Filmtabletten
Jede Filmtablette enthält 10 mg Finerenon.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 45 mg Lactose (als Monohydrat), siehe Abschnitt 4.4.
Kerendia 20 mg Filmtabletten
Jede Filmtablette enthält 20 mg Finerenon.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 40 mg Lactose (als Monohydrat), siehe Abschnitt 4.4.
Kerendia 40 mg Filmtabletten
Jede Filmtablette enthält 40 mg Finerenon.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 25 mg Lactose (als Monohydrat), siehe Abschnitt 4.4.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
Filmtablette (Tablette)
Kerendia 10 mg Filmtabletten
Rosafarbene, länglich-ovale Filmtablette, 10 mm lang und 5 mm breit, mit der Prägung „10“ auf einer Seite und „FI“ auf der anderen Seite.
Kerendia 20 mg Filmtabletten
Hellgelbe, länglich-ovale Filmtablette, 10 mm lang und 5 mm breit, mit der Prägung „20“ auf einer Seite und „FI“ auf der anderen Seite.
Kerendia 40 mg Filmtabletten
Grau-orangefarbene, länglich-ovale Filmtablette, 11 mm lang und 5 mm breit, mit der Prägung „40“ auf einer Seite und „FI“ auf der anderen Seite.
Kerendia wird angewendet zur Behandlung von chronischer Nierenerkrankung (mit Albuminurie) in Verbindung mit Typ‑2-Diabetes bei Erwachsenen.
Für Studienergebnisse zu renalen und kardiovaskulären Ereignissen, siehe Abschnitt 5.1.
Kerendia wird angewendet zur Behandlung von symptomatischer chronischer Herzinsuffizienz mit linksventrikulärer Ejektionsfraktion (LVEF) ≥ 40 % bei Erwachsenen.
Dosierung
Chronische Nierenerkrankung in Verbindung mit Typ‑2‑Diabetes (T2D)
Die empfohlene Tagesdosis beträgt 20 mg Finerenon einmal täglich.
Die empfohlene Höchstdosis beträgt 20 mg Finerenon einmal täglich.
Einleitung der Behandlung
Zur Feststellung, ob mit der Finerenon-Behandlung begonnen werden darf, und zur Ermittlung der Anfangsdosis müssen das Serumkalium und die geschätzte glomeruläre Filtrationsrate (eGFR) bestimmt werden (siehe auch Abschnitt 4.4).
Für Angaben zur Überwachung des Serumkaliums siehe „Fortsetzung der Behandlung“ weiter unten.
Bei einem Serumkalium-Wert von ≤ 4,8 mmol/l kann die Behandlung mit Finerenon begonnen werden.
Bei einem Serumkalium-Wert von > 4,8 bis 5,0 mmol/l kann der Beginn der Finerenon-Behandlung unter zusätzlicher Überwachung des Serumkaliums während der ersten 4 Wochen auf Basis der Patientencharakteristika und des Serumkalium-Spiegels erwogen werden (siehe Abschnitt 4.4).
Bei einem Serumkalium-Wert von > 5,0 mmol/l sollte nicht mit einer Finerenon-Behandlung begonnen werden (siehe Abschnitt 4.4).
Die empfohlene Anfangsdosis von Finerenon ergibt sich aus der eGFR wie in Tabelle 1 aufgeführt.
Tabelle 1: Einleitung der Behandlung mit Finerenon und empfohlene Dosis
eGFR (ml/min/1,73 m2) |
Anfangsdosis (einmal täglich) |
≥ 60 |
20 mg |
≥ 25 bis < 60 |
10 mg |
< 25 |
Nicht empfohlen |
Fortsetzung der Behandlung
Serumkalium und eGFR müssen 4 Wochen nach Beginn bzw. Wiederaufnahme der Finerenon-Behandlung oder einer Dosiserhöhung von Finerenon (siehe Tabelle 2 zur Fortsetzung der Finerenon-Behandlung und Dosisanpassung) erneut gemessen werden.
Danach muss das Serumkalium in regelmäßigen Abständen und nach Bedarf auf Basis der Patientencharakteristika und des Serumkalium-Spiegels erneut gemessen werden.
Für weitere Informationen siehe Abschnitte 4.4 und 4.5.
Tabelle 2: Fortsetzung der Finerenon-Behandlung und Dosisanpassung
Derzeitige Finerenon-Dosis (einmal täglich) |
|||
10 mg |
20 mg |
||
Derzeitiger Serum–kalium-Wert (mmol/l) |
≤ 4,8 |
Auf 20 mg Finerenon einmal täglich erhöhen* |
20 mg einmal täglich beibehalten |
> 4,8 bis 5,5 |
10 mg einmal täglich beibehalten |
20 mg einmal täglich beibehalten |
|
> 5,5 |
Finerenon aussetzen. |
Finerenon aussetzen. |
|
* 10 mg einmal täglich beibehalten, wenn die eGFR gegenüber der vorherigen Messung um > 30% gesunken ist
Herzinsuffizienz mit LVEF ≥ 40 %
Die empfohlene Tagesdosis ist abhängig von der Nierenfunktion (eGFR) zum Zeitpunkt der Einleitung der Finerenon-Behandlung (siehe Tabelle 4):
40 mg einmal täglich bei eGFR ≥ 60 ml/min/1,73 m2
20 mg einmal täglich bei eGFR ≥ 25 bis ˂ 60 ml/min/1,73 m2
Die empfohlene Höchstdosis beträgt 40 mg Finerenon einmal täglich.
Einleitung der Behandlung
Bei einem Serumkalium-Wert von ≤ 5,0 mmol/l kann die Behandlung mit Finerenon begonnen werden.
Zur Feststellung, ob mit der Finerenon-Behandlung begonnen werden darf, und zur Ermittlung der Anfangsdosis müssen das Serumkalium und die geschätzte glomeruläre Filtrationsrate (eGFR) bestimmt werden (siehe auch Abschnitt 4.4).
Für Angaben zur Überwachung des Serumkaliums siehe „Fortsetzung der Behandlung“ weiter unten.
Die empfohlene Anfangsdosis von Finerenon ergibt sich aus der eGFR wie in Tabelle 3 aufgeführt.
Tabelle 3: Einleitung der Behandlung mit Finerenon und empfohlene Dosis
eGFR (ml/min/1,73 m2) |
Anfangsdosis (einmal täglich) |
≥ 60 |
20 mg |
≥ 25 bis < 60 |
10 mg |
< 25 |
Nicht empfohlen |
Fortsetzung der Behandlung
Serumkalium und eGFR müssen 4 Wochen nach Beginn bzw. Wiederaufnahme der Finerenon-Behandlung oder einer Dosisänderung von Finerenon (siehe Tabelle 4 zur Fortsetzung der Finerenon-Behandlung und Dosisanpassung) erneut gemessen werden.
Danach müssen das Serumkalium und die eGFR in regelmäßigen Abständen und nach Bedarf auf Basis der Patientencharakteristika erneut gemessen werden.
Für weitere Informationen siehe Abschnitte 4.4 und 4.5.
Tabelle 4: Fortsetzung der Finerenon-Behandlung und Dosisanpassung
Derzeitige Finerenon-Dosis (einmal täglich) |
||||
10 mg |
20 mg |
40 mg |
||
Derzeitiger Serum–kalium-Wert (mmol/l) |
< 5,0 |
Auf 20 mg Finerenon einmal täglich erhöhen, wenn die eGFR gegenüber der vorherigen Messung nicht um > 30 % gesunken ist. |
Auf 40 mg Finerenon einmal täglich erhöhen, wenn die eGFR gegenüber der vorherigen Messung nicht um > 30 % gesunken ist. 20 mg einmal täglich beibehalten bei eGFR < 60 ml/min/1,73 m2 bei Einleitung |
40 mg einmal täglich beibehalten Auf 20 mg Finerenon einmal täglich reduzieren, wenn die eGFR gegenüber der vorherigen Messung um > 30 % gesunken ist. |
5,0 bis ˂ 5,5 |
10 mg einmal täglich beibehalten |
20 mg einmal täglich beibehalten |
40 mg einmal täglich beibehalten Auf 20 mg Finerenon einmal täglich reduzieren, wenn die eGFR gegenüber der vorherigen Messung um > 30 % gesunken ist. |
|
5,5 bis ˂ 6,0 |
Finerenon aussetzen |
Auf 10 mg einmal täglich reduzieren |
Auf 20 mg einmal täglich reduzieren |
|
≥ 6,0 |
Finerenon aussetzen. |
|||
Wenn die eGFR gegenüber der vorherigen Messung um ≥ 40 % gesunken ist, sollte eine Dosisreduktion oder ein Aussetzen von Finerenon erwogen werden. Nachdem sich die eGFR-Werte stabilisiert haben, kann abhängig von den individuellen Patientencharakteristika eine Erhöhung der Dosis oder eine Wiederaufnahme der Behandlung erwogen werden.
Vergessene Dosis
Eine vergessene Dosis ist einzunehmen, sobald der Patient dies bemerkt, jedoch nur, wenn dies am selben Tag geschieht.
Der Patient darf nicht die doppelte Dosis einnehmen, wenn die vorherige Einnahme vergessen wurde.
Besondere Patientengruppen
Ältere Patienten (≥ 65 Jahre)
Bei älteren Patienten ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Eine regelmäßige Überwachung der Nierenfunktion wird empfohlen (siehe Abschnitt 4.4).
Eingeschränkte Nierenfunktion
Beginn der Behandlung
Bei Patienten mit einer eGFR von < 25 ml/min/1,73 m2 sollte nicht mit einer Finerenon-Behandlung begonnen werden, da nur begrenzte klinische Daten vorliegen (siehe Abschnitte 4.4 und 5.2).
Fortsetzung der Behandlung
Bei Patienten mit einer eGFR von ≥ 15 ml/min/1,73 m2 kann die Behandlung mit Finerenon mit Dosisanpassung auf Basis der Serumkalium-Werte fortgesetzt werden. Die eGFR ist 4 Wochen nach Behandlungsbeginn zu messen, um zu bestimmen, ob die Anfangsdosis erhöht werden kann (siehe „Dosierung, Fortsetzung der Behandlung“ und Tabellen 2 und 4).
Aufgrund begrenzter klinischer Daten sollte die Finerenon-Behandlung bei Patienten mit Progression zur terminalen Niereninsuffizienz (eGFR < 15 ml/min/1,73 m2) beendet werden (siehe Abschnitt 4.4).
Eingeschränkte Leberfunktion
Patienten mit
schwerer Leberfunktionsstörung:
Eine Behandlung mit Finerenon sollte nicht begonnen werden (siehe Abschnitte 4.4 und 5.2). Es liegen keine Daten vor.
mittelschwerer Leberfunktionsstörung:
Es ist keine anfängliche Dosisanpassung erforderlich. Eine zusätzliche Überwachung des Serumkaliums und Anpassung der Überwachung entsprechend der Patientencharakteristika sind zu erwägen (siehe Abschnitte 4.4 und 5.2).
leichter Leberfunktionsstörung:
Es ist keine anfängliche Dosisanpassung erforderlich.
Gleichzeitige Anwendung anderer Arzneimittel
Bei Patienten, die Finerenon zusammen mit moderaten oder schwachen CYP3A4‑Inhibitoren, Kaliumergänzungsmitteln, Trimethoprim oder Trimethoprim/Sulfamethoxazol einnehmen, ist eine zusätzliche Überwachung des Serumkaliums sowie die Anpassung der Überwachung auf Basis der Patientencharakteristika zu erwägen (siehe Abschnitt 4.4). Die Behandlungsentscheidungen für Finerenon sind anhand der Tabellen 2 und 4 zu treffen (siehe „Dosierung, Fortsetzen der Behandlung“).
Bei Patienten, die Trimethoprim oder Trimethoprim/Sulfamethoxazol einnehmen müssen, muss Finerenon unter Umständen vorübergehend abgesetzt werden. Für weitere Informationen siehe Abschnitte 4.4 und 4.5.
Körpergewicht
Eine Dosisanpassung auf Basis des Körpergewichts ist nicht erforderlich (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Finerenon bei Kindern und Jugendlichen unter 18 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Zum Einnehmen
Die Tabletten können mit einem Glas Wasser und mit oder ohne Nahrung eingenommen werden (siehe Abschnitt 5.2).
Die Tabletten dürfen nicht zusammen mit Grapefruit oder Grapefruitsaft eingenommen werden (siehe Abschnitt 4.5).
Zerstoßen der Tabletten
Für Patienten, die nicht in der Lage sind, ganze Tabletten zu schlucken, können Kerendia Tabletten unmittelbar vor der Einnahme zerstoßen und mit Wasser oder weicher Nahrung wie Apfelmus gemischt werden (siehe Abschnitt 5.2).
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Gleichzeitige Behandlung mit starken CYP3A4‑Inhibitoren (siehe Abschnitt 4.5), wie z. B.:
Itraconazol
Ketoconazol
Ritonavir
Nelfinavir
Cobicistat
Clarithromycin
Telithromycin
Nefazodon
Morbus Addison
Hyperkaliämie
Bei mit Finerenon behandelten Patienten wurde Hyperkaliämie beobachtet (siehe Abschnitt 4.8).
Einige Patienten haben ein höheres Risiko eine Hyperkaliämie zu entwickeln. Risikofaktoren sind unter anderem niedrige eGFR, erhöhte Serumkalium-Spiegel und frühere Episoden einer Hyperkaliämie. Bei diesen Patienten ist eine engmaschigere Überwachung zu erwägen.
Bei Patienten mit einem Serumkalium-Wert von > 5,0 mmol/l sollte nicht mit einer Finerenon-Behandlung begonnen werden.
Überwachung
Serumkalium und eGFR müssen 4 Wochen nach Beginn bzw. Wiederaufnahme der Finerenon-Behandlung oder einer Dosisanpassung von Finerenon erneut gemessen werden. Danach muss das Serumkalium in regelmäßigen Abständen und nach Bedarf auf Basis der Patientencharakteristika und des Serumkalium-Spiegels erneut gemessen werden (siehe Abschnitt 4.2).
Chronische Nierenerkrankung in Verbindung mit T2D
Beginn und Fortsetzen der Behandlung (siehe Abschnitt 4.2)
Bei einem Serumkalium-Wert von > 4,8 bis 5,0 mmol/l kann der Beginn der Finerenon-Behandlung unter zusätzlicher Überwachung des Serumkaliums während der ersten 4 Wochen auf Basis der Patientencharakteristika und des Serumkalium-Spiegels erwogen werden.
Bei einem Serumkalium-Wert von > 5,5 mmol/l muss die laufende Finerenon-Behandlung ausgesetzt werden. Lokale Leitlinien für das Management der Hyperkaliämie müssen befolgt werden.
Ab einem Serumkalium-Wert von ≤ 5,0 mmol/l kann die Behandlung mit 10 mg Finerenon einmal täglich wieder aufgenommen werden.
Herzinsuffizienz mit LVEF ≥ 40 %
Beginn und Fortsetzen der Behandlung (siehe Abschnitt 4.2)
Bei einem Serumkalium-Wert von ≥6,0 mmol/l muss die Finerenon-Behandlung ausgesetzt werden. Lokale Leitlinien für das Management von Hyperkaliämien müssen befolgt werden. Ab einem Serumkalium-Wert von < 5,5 mmol/l kann die Behandlung mit 10 mg Finerenon einmal täglich wieder aufgenommen werden. Bei wiederholter Messung eines Serumkalium-Werts von ≥5,5 mmol/l kann die Finerenon-Behandlung erst wieder aufgenommen werden, wenn der Serumkalium-Wert bei ˂ 5,0 mmol/l liegt.
Gleichzeitige Anwendung anderer Arzneimittel
Das Risiko für eine Hyperkaliämie kann auch ansteigen bei gleichzeitiger Einnahme anderer Arzneimittel, welche den Serumkalium-Spiegel erhöhen können (siehe Abschnitt 4.5). Siehe auch „Gleichzeitige Anwendung von Substanzen, welche die Finerenon-Exposition beeinflussen“.
Finerenon sollte nicht zusammen mit folgenden Arzneimitteln angewendet werden:
kaliumsparende Diuretika (z. B. Amilorid, Triamteren) und
andere Mineralokortikoid-Rezeptor-Antagonisten (MRA), wie z. B. Eplerenon, Esaxerenon, Spironolacton und Canrenon.
Bei gleichzeitiger Anwendung von Finerenon mit folgenden Arzneimitteln ist Vorsicht geboten und der Serumkalium-Wert ist zu überwachen:
Kaliumergänzungsmittel oder kaliumangereichtes Salz
Trimethoprim oder Trimethoprim/Sulfamethoxazol. Unter Umständen muss Finerenon vorübergehend abgesetzt werden.
Verschlechterung der Nierenfunktion bei Patienten mit Herzinsuffizienz mit LVEF ≥ 40 %
Bei Patienten mit Herzinsuffizienz mit LVEF ≥ 40 %, die mit Finerenon behandelt wurden, wurde eine erhöhte Häufigkeit von Verschlechterung der Nierenfunktion berichtet (siehe Abschnitt 4.8). Eine regelmäßige Überwachung der Nierenfunktion während der Behandlung sowie eine bedarfsorientierte Kontrolle auf Basis der Patientencharakteristika wird empfohlen. Ältere Patienten und Patienten mit eingeschränkter Nierenfunktion (eGFR < 60 ml/min/1,73 m2) haben ein höheres Risiko für eine Verschlechterung der Nierenfunktion und sollten häufiger überwacht werden (siehe Abschnitt 4.2).
Eingeschränkte Nierenfunktion
Das Risiko für eine Hyperkaliämie erhöht sich mit abnehmender Nierenfunktion. Die Nierenfunktion sollte laufend nach Bedarf gemäß der üblichen klinischen Praxis überwacht werden (siehe Abschnitt 4.2).
Beginn der Behandlung
Bei Patienten mit einer eGFR von < 25 ml/min/1,73 m2 sollte nicht mit einer Finerenon-Behandlung begonnen werden, da nur begrenzte klinische Daten vorliegen (siehe Abschnitte 4.2 und 5.2).
Fortsetzen der Behandlung
Aufgrund begrenzter klinischer Daten sollte die Finerenon-Behandlung bei Patienten mit Progression zum terminalen Nierenversagen (eGFR < 15 ml/min/1,73 m2) beendet werden (siehe Abschnitt 4.2).
Eingeschränkte Leberfunktion
Bei Patienten mit schwerer Leberfunktionsstörung sollte nicht mit einer Finerenon-Behandlung begonnen werden (siehe Abschnitt 4.2). Diese Patientengruppe wurde nicht untersucht (siehe Abschnitt 5.2), es wird jedoch ein signifikanter Anstieg der Finerenon-Exposition erwartet.
Bei Patienten mit mittelschwerer Leberfunktionsstörung erfordert die Anwendung von Finerenon aufgrund des Anstiegs der Finerenon-Exposition möglicherweise eine zusätzliche Überwachung. Eine zusätzliche Überwachung des Serumkaliums sowie die Anpassung der Überwachung entsprechend der Patientencharakteristika ist zu erwägen (siehe Abschnitte 4.2 und 5.2).
Patienten mit New York Heart Association (NYHA) Klasse IV
Die Erfahrungen mit Finerenon bei Patienten mit einer Herzinsuffizienz NYHA Klasse IV sind begrenzt (siehe Abschnitt 5.1).
Ältere Patienten
Bei älteren Patienten kommt eine eingeschränkte Nierenfunktion häufiger vor, und sie werden öfter mit Arzneimitteln behandelt, die die Nierenfunktion beeinflussen können. Daher wird eine regelmäßige Überwachung der Nierenfunktion empfohlen.
Gleichzeitige Anwendung von Substanzen, die die Finerenon-Exposition beeinflussen
Moderate und schwache CYP3A4-Inhibitoren
Bei Anwendung von Finerenon zusammen mit moderaten oder schwachen CYP3A4‑Inhibitoren sollte der Serumkalium-Spiegel überwacht werden (siehe Abschnitte 4.2 und 4.5).
Starke und moderate CYP3A4-Induktoren
Finerenon sollte nicht zusammen mit starken oder moderaten CYP3A4‑Induktoren angewendet werden (siehe Abschnitt 4.5).
Grapefruit
Grapefruits oder Grapefruitsaft sollten während der Behandlung mit Finerenon nicht verzehrt werden (siehe Abschnitte 4.2 und 4.5).
Embryofetale Toxizität
Finerenon darf während der Schwangerschaft nicht angewendet werden, es sei denn, der Nutzen für die Mutter und das Risiko für das ungeborene Kind wurden sorgfältig abgewogen. Wenn eine Frau während der Einnahme von Finerenon schwanger wird, ist sie über die potenziellen Risiken für das ungeborene Kind zu informieren.
Frauen im gebärfähigen Alter sind darauf hinzuweisen, dass sie während der Behandlung mit Finerenon eine zuverlässige Verhütungsmethode anwenden müssen.
Frauen sind darauf hinzuweisen, dass sie während der Behandlung mit Finerenon nicht stillen dürfen.
Für weitere Informationen siehe Abschnitte 4.6 und 5.3.
Informationen zu den sonstigen Bestandteilen
Kerendia enthält Lactose
Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactase-Mangel oder Glucose‑Galactose-Malabsorption sollten dieses Arzneimittel nicht anwenden.
Kerendia enthält Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Tablette, d. h. es ist nahezu „natriumfrei“.
Studien zur Erfassung von Wechselwirkungen wurden nur bei Erwachsenen durchgeführt.
Die Elimination von Finerenon erfolgt nahezu ausschließlich über den Cytochrom P450(CYP)‑vermittelten oxidativen Metabolismus (vorwiegend CYP3A4 [90 %], mit einem kleinen Anteil von CYP2C8 [10 %]).
Gleichzeitige Anwendung kontraindiziert
Starke CYP3A4‑Inhibitoren
Die gleichzeitige Anwendung von Kerendia mit Itraconazol, Clarithromycin und anderen starken CYP3A4‑Inhibitoren (z. B. Ketoconazol, Ritonavir, Nelfinavir, Cobicistat, Telithromycin oder Nefazodon) ist kontraindiziert (siehe Abschnitt 4.3), da ein beträchtlicher Anstieg der Finerenon-Exposition zu erwarten ist.
Gleichzeitige Anwendung nicht empfohlen
Starke und moderate CYP3A4‑Induktoren
Kerendia sollte nicht gleichzeitig mit Rifampicin und anderen starken CYP3A4‑Induktoren (z. B. Carbamazepin, Phenytoin, Phenobarbital, Johanniskraut) oder mit Efavirenz und anderen moderaten CYP3A4‑Induktoren angewendet werden. Diese CYP3A4‑Induktoren senken voraussichtlich die Plasmakonzentration von Finerenon beträchtlich, was zu einer Abschwächung der therapeutischen Wirkung führt (siehe Abschnitt 4.4).
Bestimmte Arzneimittel, die das Serumkalium erhöhen
Kerendia sollte nicht gleichzeitig mit kaliumsparenden Diuretika (z. B. Amilorid, Triamteren) und anderen MRA (z. B. Eplerenon, Esaxerenon, Spironolacton, Canrenon) angewendet werden. Es ist zu erwarten, dass diese Arzneimittel das Hyperkaliämie-Risiko erhöhen (siehe Abschnitt 4.4).
Grapefruit
Grapefruits oder Grapefruitsaft sollten während der Behandlung mit Finerenon nicht verzehrt werden, da dies voraussichtlich zu einem Anstieg der Plasmakonzentration von Finerenon durch Inhibition von CYP3A4 führt (siehe Abschnitte 4.2 und 4.4).
Gleichzeitige Anwendung mit Vorsicht
Moderate CYP3A4‑Inhibitoren
In einer klinischen Studie führte die gleichzeitige Anwendung von Erythromycin (500 mg dreimal täglich) zu einem 3,5‑fachen Anstieg der AUC und einem 1,9‑fachen Anstieg der Cₘₐₓ von Finerenon. In einer anderen klinischen Studie führte die Anwendung von Verapamil (240 mg Retardtablette einmal täglich) zu einem 2,7‑ bzw. 2,2‑fachen Anstieg der AUC bzw. Cₘₐₓ von Finerenon.
Da der Serumkalium-Spiegel ansteigen kann, wird eine Überwachung des Serumkaliums empfohlen, insbesondere bei Behandlungsbeginn und bei Dosierungsänderung von Finerenon oder des CYP3A4‑Inhibitors (siehe Abschnitte 4.2 und 4.4).
Schwache CYP3A4‑Inhibitoren
Die Physiologie-basierten pharmakokinetischen (PBPK) Simulationen deuten darauf hin, dass Fluvoxamin (100 mg zweimal täglich) die AUC (1,6‑fach) und Cₘₐₓ (1,4‑fach) von Finerenon erhöht.
Da der Serumkalium-Spiegel ansteigen kann, wird eine Überwachung des Serumkaliums empfohlen, insbesondere bei Behandlungsbeginn und bei Dosierungsänderung von Finerenon oder des CYP3A4‑Inhibitors (siehe Abschnitte 4.2 und 4.4).
Bestimmte Arzneimittel, die das Serumkalium erhöhen (siehe Abschnitt 4.4)
Bei Anwendung von Kerendia zusammen mit Kaliumergänzungsmitteln und Trimethoprim oder Trimethoprim/Sulfamethoxazol ist ein Anstieg des Hyperkaliämie-Risikos zu erwarten. Eine Überwachung des Serumkaliums ist erforderlich.
Unter Umständen muss Kerendia während der Behandlung mit Trimethoprim oder Trimethoprim/Sulfamethoxazol vorübergehend abgesetzt werden.
Blutdrucksenkende Arzneimittel
Das Risiko für eine Hypotension steigt bei gleichzeitiger Anwendung von mehreren anderen blutdrucksenkenden Arzneimitteln. Bei diesen Patienten wird eine Überwachung des Blutdrucks empfohlen.
Wirkung von 40 mg Finerenon auf CYP3A4- und CYP2C8-Substrate
Bei einer Dosierung von 40 mg einmal täglich ist Finerenon in vivo ein schwacher Hemmer des CYP3A4-Enzyms. Die gleichzeitige Andwendung mehrerer Dosen von 40 mg Finerenon mit dem CYP3A4-Prüfsubstrat Midazolam führte zu einem 1,31‑fachen Anstieg der mittleren Midazolam-AUC ohne Auswirkung auf die Cmax. Die potenziell erhöhte Exposition empfindlicher CYP3A4‑Substrate mit einem engen therapeutischen Fenster muss bei gleichzeitiger Anwendung mit Finerenon 40 mg einmal täglich berücksichtigt werden. Ein Mehrfachdosisregime mit 20 mg Finerenon einmal täglich über 10 Tage hatte keine relevante Wirkung auf die AUC des CYP3A4‑Prüfsubstrats Midazolam. Daher kann eine klinisch relevante Inhibition oder Induktion von CYP3A4 durch Finerenon in diesem Dosisbereich ausgeschlossen werden.
Bei einer Dosierung von 40 mg einmal täglich ist Finerenon in vivo ein schwacher Hemmer des CYP2C8-Enzyms. Die gleichzeitige Anwendung mehrerer Dosen von 40 mg Finerenon mit dem CYP2C8-Prüfsubstrat Repaglinid führte zu einem 1,59‑fachen Anstieg der mittleren Repaglinid-AUC und einem 1,30‑fachen Anstieg der Cmax. Die potenziell erhöhte Exposition von CYP2C8-Substraten mit einem engen therapeutischen Fenster muss bei gleichzeitiger Anwendung mit Finerenon 40 mg einmal täglich berücksichtigt werden.
Eine Einzeldosis von 20 mg Finerenon hatte keine klinisch relevante Wirkung auf die AUC und Cmax des CYP2C8‑Prüfsubstrats Repaglinid. Somit hemmt Finerenon in dieser Dosierung CYP2C8 nicht.
Kontrazeption bei Frauen
Frauen im gebärfähigen Alter müssen während der Behandlung mit Finerenon eine zuverlässige Verhütungsmethode anwenden (siehe Abschnitt 4.4).
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Finerenon bei Schwangeren vor.
Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3).
Kerendia darf während der Schwangerschaft nicht angewendet werden, es sei denn, eine Behandlung mit Finerenon ist aufgrund des klinischen Zustandes der Frau erforderlich. Wenn eine Frau während der Einnahme von Finerenon schwanger wird, ist sie über die potenziellen Risiken für das ungeborene Kind zu informieren (siehe Abschnitt 4.4).
Stillzeit
Es ist nicht bekannt ob Finerenon und/oder seine Metaboliten in die Muttermilch übergehen.
Die zur Verfügung stehenden pharmakokinetischen/toxikologischen Daten vom Tier zeigten, dass Finerenon und seine Metaboliten in die Milch übergehen. Rattenjunge zeigten bei diesbezüglicher Exposition Nebenwirkungen (siehe Abschnitt 5.3).
Ein Risiko für den Säugling kann nicht ausgeschlossen werden.
Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob die Behandlung mit Kerendia unterbrochen bzw. beendet werden soll. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen (siehe Abschnitt 4.4).
Fertilität
Bisher liegen keine Erfahrungen zu der Wirkung von Finerenon auf die menschliche Fertilität vor.
Tierexperimentelle Studien haben eine beeinträchtigte weibliche Fertilität bei Expositionen gezeigt, die höher als die maximale Exposition beim Menschen waren, was auf eine geringe klinische Relevanz hinweist (siehe Abschnitt 5.3).
Kerendia hat keinen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die am häufigsten gemeldete Nebenwirkung unter Finerenon war Hyperkaliämie (12,6 %). Siehe „Beschreibung ausgewählter Nebenwirkungen, Hyperkaliämie“ unten und Abschnitt 4.4.
Tabellarische Auflistung der Nebenwirkungen
Die Sicherheit von Finerenon bei Patienten mit chronischer Nierenerkrankung (Chronic Kidney Disease, CKD) und T2D wurde in den zwei pivotalen Phase‑III-Studien FIDELIO‑DKD (DKD: Diabetic Kidney Disease) und FIGARO‑DKD untersucht. In der Studie FIDELIO‑DKD erhielten 2 818 Patienten Finerenon (10 mg oder 20 mg einmal täglich). Die mittlere Behandlungsdauer betrug 2,2 Jahre. In der Studie FIGARO‑DKD erhielten 3 671 Patienten Finerenon (10 mg oder 20 mg einmal täglich). Die mittlere Behandlungsdauer betrug 2,9 Jahre.
Die Sicherheit von Finerenon bei Patienten mit Herzinsuffizienz (heart failure, HF) mit einer LVEF ≥ 40 % wurde in der Phase-III-Studie FINEARTS-HF untersucht. In dieser Studie erhielten 2 993 Patienten Finerenon (10 mg, 20 mg oder 40 mg einmal täglich). Die mittlere Behandlungsdauer betrug 2,1 Jahre.
Die beobachteten Nebenwirkungen sind in Tabelle 5 aufgelistet. Sie sind nach Systemorganklasse gemäß MedDRA und Häufigkeit klassifiziert.
Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Die Häufigkeiten sind wie folgt definiert:
Sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 5: Nebenwirkungen
Systemorganklasse |
Häufigkeit |
CKD mit T2D |
HF mit LVEF ≥ 40 % |
Stoffwechsel- und Ernährungsstörungen |
sehr häufig |
Hyperkaliämie |
- |
häufig |
Hyponatriämie |
Hyperkaliämie |
|
Gefäßerkrankungen |
häufig |
Hypotonie |
Hypotonie |
Erkrankungen des Gastrointestinaltrakts |
häufig |
- |
Durchfall |
Erkrankungen der Haut und des Unterhautzellgewebes |
häufig |
Pruritus |
- |
Erkrankungen der Nieren und Harnwege |
häufig |
- |
Nierenfunktionsstörung |
Untersuchungen |
häufig |
Kreatinin im Blut erhöht/Glomeruläre Filtrationsrate vermindert |
Kreatinin im Blut erhöht/Glomeruläre Filtrationsrate vermindert |
gelegentlich |
Hämoglobin vermindert |
- |
Beschreibung ausgewählter Nebenwirkungen
Hyperkaliämie
In der Finerenon-Gruppe wurde im ersten Monat der Behandlung eine Erhöhung der mittleren Serumkalium-Konzentration gegenüber Baseline in Höhe von bis zu 0,2 mmol/l verglichen mit Placebo beobachtet. Der Unterschied blieb danach stabil.
In den gepoolten Daten der Studien FIDELIO‑DKD und FIGARO-DKD traten Hyperkaliämie-Ereignisse bei 14,0 % der mit Finerenon behandelten Patienten auf. Im Vergleich dazu waren es bei Patienten unter Placebo 6,9 %. Schwerwiegende Hyperkaliämie-Ereignisse traten häufiger bei mit Finerenon behandelten Patienten auf (1,1 %) als bei Patienten unter Placebo (0,2 %). Serumkalium-Konzentrationen von > 5,5 mmol/l bzw. > 6,0 mmol/l wurden bei 16,8 % bzw. 3,3 % der Patienten unter Finerenon sowie bei 7,4 % bzw. 1,3 % der Patienten unter Placebo verzeichnet. Eine Hyperkaliämie, die zum dauerhaften Absetzen der Behandlung führte, trat bei 1,7 % der Patienten unter Finerenon auf. In der Placebo-Gruppe waren es 0,6 %. Zu Hospitalisierungen aufgrund von Hyperkaliämie kam es bei 0,9 % in der Finerenon-Gruppe und bei 0,2 % in der Placebo-Gruppe.
In der FINEARTS-HF-Studie traten Hyperkaliämie-Ereignisse bei 9,7 % der mit Finerenon behandelten Patienten auf. Im Vergleich dazu waren es bei Patienten unter Placebo 4,2 %. Eine Hyperkaliämie, die zum dauerhaften Absetzen der Behandlung führte, trat bei 0,4 % der Patienten unter Finerenon auf. In der Placebo-Gruppe waren es 0,2 %. Zu Hospitalisierungen aufgrund von Hyperkaliämie kam es bei 0,5 % in der Finerenon-Gruppe und bei 0,2 % in der Placebo-Gruppe.
In allen Studien war die Mehrzahl der Hyperkaliämie-Ereignisse leicht bis mittelschwer und klang bei den mit Finerenon behandelten Patienten wieder ab.
Für spezielle Empfehlungen siehe Abschnitte 4.2 und 4.4.
Verschlechterung der Nierenfunktion
In den gepoolten Daten der Studien FIDELIO‑DKD und FIGARO-DKD wurde bei 5,4 % der mit Finerenon behandelten Patienten eine Verminderung der eGFR beobachtet. Im Vergleich dazu waren es bei Patienten unter Placebo 4,2 %. Die Rate an Ereignissen von verminderter GFR, die zum dauerhaften Absetzen der Behandlung führten, war gleich bei Patienten in der Finerenon-Gruppe und in der Placebo-Gruppe (0,2 %). Die Rate an Hospitalisierungen aufgrund einer verminderten GFR war gleich bei Patienten in der Finerenon-Gruppe und in der Placebo-Gruppe (< 0,1 %).
Ereignisse von erhöhtem Kreatinin im Blut wurden bei 2,6 % der mit Finerenon behandelten Patienten berichtet. Im Vergleich dazu waren es bei Patienten unter Placebo 2,3 %.
Die Mehrzahl der Ereignisse von verminderter GFR/Kreatinin im Blut erhöht war leicht bis mittelschwer und klang bei den mit Finerenon behandelten Patienten wieder ab. Bei Patienten unter Finerenon trat zu Beginn eine Abnahme der eGFR (Mittelwert 2 ml/min/1,73 m2) auf, die sich im Zeitverlauf gegenüber Placebo wieder abschwächte. Diese Abnahme schien unter Fortführung der Behandlung reversibel.
In der FINEARTS‑HF‑Studie wurden im Vergleich zur Placebo‑Gruppe bei mit Finerenon behandelten Patienten häufiger Ereignisse im Zusammenhang mit einer Verschlechterung der Nierenfunktion berichtet (17,7 % vs. 10,9 %). Die berichteten Ereignisse umfassten Nierenfunktionsstörung (6,6 % vs. 3,9 %), GFR vermindert (5,2 % vs. 3,6 %), akutes Nierenversagen (3,7 % vs. 2,1 %), Niereninsuffizienz (2,6 % vs. 1,6 %) und Kreatinin im Blut erhöht (1,2 % vs. 0,8 %).
Insgesamt waren die meisten der gemeldeten Ereignisse bezüglich einer Verschlechterung der Nierenfunktion leicht bis mäßig und klangen bei den mit Finerenon behandelten Patienten wieder ab. In einigen Fällen, in denen Finerenon dauerhaft abgesetzt wurde, kehrte die eGFR nicht auf den Ausgangswert zurück. Ereignisse im Zusammenhang mit einer Verschlechterung der Nierenfunktion, die zu einem dauerhaften Absetzen führten, waren in beiden Gruppen identisch (0,3 %). Hospitalisierungen aufgrund einer Verschlechterung der Nierenfunktion wurden bei den mit Finerenon behandelten Patienten häufiger berichtet als in der Placebo‑Gruppe (2,0 % vs. 1,3 %), wobei akutes Nierenversagen das Ereignis war, das am häufigsten zu einer Hospitalisierung führte (1,6 % in der Finerenon-Gruppe vs. 0,8 % in der Placebo-Gruppe).
Patienten unter Finerenon zeigten eine initiale Abnahme der GFR, die sich während der fortgesetzten Behandlung als reversibel erwies. Der mittlere Unterschied in der GFR zwischen Finerenon und Placebo betrug von Monat 1 bis Monat 6 etwa 3-4 ml/min/1,73 m2. Nach Monat 6 war der GFR‑Abfall in der Placebo‑Gruppe leicht ausgeprägter, mit einem mittleren Unterschied zwischen Finerenon und Placebo von etwa 2-3 ml/min/1,73 m2.
Hypotonie
Unter Finerenon sank in Monat 1 der mittlere systolische Blutdruck um 2‑4 mmHg und der mittlere diastolische Blutdruck sank um 1‑2 mmHg, danach blieben die Werte stabil.
In den gepoolten Daten der Studien FIDELIO‑DKD und FIGARO-DKD traten Hypotonie-Ereignisse bei 4,7 % der mit Finerenon behandelten Patienten auf. Im Vergleich dazu waren es bei Patienten unter Placebo 3,0 %. Bei drei Patienten (< 0,1 %) wurde Finerenon aufgrund von Hypotonie dauerhaft abgesetzt. Die Hospitalisierungsrate aufgrund von Hypotonie war gleich bei Patienten in der Finerenon-Gruppe und der Placebo-Gruppe (0,1 %).
In der FINEARTS-HF-Studie traten Hypotonie-Ereignisse bei 7,6 % der mit Finerenon behandelten Patienten auf. Im Vergleich dazu waren es bei Patienten unter Placebo 4,7 %. Bei drei Patienten (0,1 %) wurde Finerenon aufgrund von Hypotonie dauerhaft abgesetzt. Zu Hospitalisierungen aufgrund von Hypotonie kam es bei 0,4 % in der Finerenon-Gruppe und bei 0,3 % in der Placebo-Gruppe.
In allen Studien war die Mehrzahl der Hypotonie-Ereignisse leicht bis mittelschwer und klang bei den mit Finerenon behandelten Patienten wieder ab.
Hyperurikämie
In den gepoolten Daten der Studien FIDELIO-DKD und FIGARO‑DKD trat Hyperurikämie bei 5,1 % der mit Finerenon behandelten Patienten und bei 3,9 % der Patienten unter Placebo auf. Eine Erhöhung der mittleren Serumharnsäure-Konzentration um 0,3 mg/dl gegenüber Baseline wurde bis zu Monat 16 in der Finerenon-Gruppe im Vergleich zur Placebo-Gruppe beobachtet. Dieser Unterschied verringerte sich mit der Zeit. Die Häufigkeit von Gicht war vergleichbar bei Patienten in der Finerenon-Gruppe (3,1 %) und in der Placebo-Gruppe (3,0 %).
In der FINEARTS-HF-Studie war die Häufigkeit von Hyperurikämie vergleichbar bei Patienten in der Finerenon-Gruppe (2,7 %) und in der Placebo-Gruppe (2,4 %). Die Häufigkeit von Gicht war vergleichbar bei Patienten in der Finerenon-Gruppe (2,4 %) und in der Placebo-Gruppe (2,8 %).
In allen Studien waren die Hyperurikämie-Ereignisse nicht schwerwiegend und führten bei den Patienten, die Finerenon erhielten, nicht zu einem dauerhaften Absetzen der Behandlung.
Gastrointestinale Beschwerden
In der FINEARTS-HF-Studie wurden Durchfall (5,7 % vs. 4,4 %) und Verstopfung (3,8 % vs. 2,7 %) in der Finerenon-Gruppe häufiger berichtet als in der Placebo-Gruppe.
Hämoglobin vermindert
In den gepoolten Daten der Studien FIDELIO‑DKD und FIGARO-DKD war Finerenon nach 4‑monatiger Behandlung mit einer placebokorrigierten absoluten Abnahme des mittleren Hämoglobins um 0,15 g/dl und eines mittleren Hämatokrits von 0,5 % verbunden. Die Häufigkeit von Anämie war vergleichbar bei Patienten in der Finerenon-Gruppe (6,5 %) und in der Placebo-Gruppe (6,1 %). Die Häufigkeit schwerwiegender Anämieereignisse war sowohl bei den mit Finerenon behandelten Patienten als auch bei den Patienten unter Placebo gering (0,5 %). Die Veränderungen von Hämoglobin und Hämatokrit waren vorübergehend und erreichten nach etwa 24‑32 Monaten vergleichbare Werte wie in der Placebo-Gruppe.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
Als wahrscheinlichste Manifestation einer Überdosierung ist Hyperkaliämie anzunehmen. Wenn sich eine Hyperkaliämie entwickelt, ist die übliche medizinische Behandlung einzuleiten.
Angesichts seiner Plasmaproteinbindung von etwa 90 % ist es unwahrscheinlich, dass Finerenon durch Hämodialyse wirksam entfernt werden kann.
Pharmakotherapeutische Gruppe: Diuretika, Aldosteronantagonisten, ATC‑Code: C03DA05
Wirkmechanismus
Finerenon ist ein nichtsteroidaler, selektiver Antagonist des Mineralokortikoid-Rezeptors (MR), welcher durch Aldosteron und Cortisol aktiviert wird und die Gentranskription reguliert. Durch seine Bindung an den MR entsteht ein spezifischer Rezeptor-Ligand-Komplex, der die Rekrutierung transkriptioneller Coaktivatoren blockiert, welche an der Expression proinflammatorischer und profibrotischer Mediatoren beteiligt sind.
Pharmakodynamische Wirkungen
In FIDELIO‑DKD und FIGARO‑DKD, randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase‑III-Studien an erwachsenen Patienten mit CKD und T2D, betrug die placebokorrigierte relative Reduktion des Albumin-Kreatinin-Quotienten im Urin (UACR) in Monat 4 bei den auf Finerenon randomisierten Patienten 31 % bzw. 32 %. Die UACR-Reduktion blieb über den Verlauf beider Studien bestehen.
In FINEARTS-HF, einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase‑III-Studie an erwachsenen Patienten mit HF mit einer LVEF ≥ 40 %, betrug die placebokorrigierte relative Reduktion des UACR bei den auf Finerenon randomisierten Patienten 30 % in Monat 6. Die UACR-Reduktion blieb bis zur letzten Bestimmung in Jahr 2 bestehen.
In ARTS‑DN, einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase‑IIb-Studie an erwachsenen Patienten mit CKD und T2D, betrug die placebokorrigierte relative Reduktion des UACR an Tag 90 bei den mit Finerenon 10 mg bzw. 20 mg einmal täglich behandelten Patienten 25 % bzw. 38 %.
Kardiale Elektrophysiologie
Eine spezielle QT‑Studie an 57 gesunden Probanden hat gezeigt, dass Finerenon die kardiale Repolarisation nicht beeinflusst. Nach Einzeldosen von 20 mg (therapeutisch) oder 80 mg (supratherapeutisch) gab es keinen Hinweis auf eine QT/QTc-verlängernde Wirkung von Finerenon.
Klinische Wirksamkeit und Sicherheit
Chronische Nierenerkrankung in Verbindung mit T2D
Die Studien FIDELIO‑DKD und FIGARO‑DKD untersuchten an erwachsenen Patienten mit CKD und T2D die Wirkung von Finerenon auf renale und kardiovaskuläre (CV) Endpunkte im Vergleich zu Placebo.
Patienten mussten eine Standardbehandlung, einschließlich der maximal tolerierten zugelassenen Dosis eines Inhibitors des Angiotensin-konvertierenden Enzyms (ACEi) oder eines Angiotensin-Rezeptorblockers (ARB), erhalten.
Patienten mit diagnostizierter Herzinsuffizienz mit reduzierter Ejektionsfraktion und NYHA-Klasse II‑IV waren aufgrund der Klasse-1A‑Empfehlung für MRA-Therapie von der Studie ausgeschlossen.
Für die Studie FIDELIO‑DKD waren die Patienten geeignet auf Basis einer nachgewiesenen persistenten Albuminurie (> 30 mg/g bis 5 000 mg/g), einer eGFR von 25 bis 75 ml/min/1,73 m2 und einem Serumkalium-Wert von ≤ 4,8 mmol/l beim Screening.
Der primäre Endpunkt war eine Kombination aus der Zeit bis zum ersten Auftreten von Nierenversagen (definiert als chronische Dialyse oder Nierentransplantation oder anhaltende Abnahme der eGFR auf < 15 ml/min/1,73 m2 über mindestens 4 Wochen), einer anhaltenden Abnahme der eGFR um 40 % oder mehr gegenüber Baseline über mindestens 4 Wochen oder renalem Tod. Der wichtigste sekundäre Endpunkt war eine Kombination aus der Zeit bis zu einem CV‑Tod, dem ersten Auftreten von nichttödlichem Myokardinfarkt (MI), nichttödlichem Schlaganfall oder Hospitalisierung aufgrund von Herzinsuffizienz.
Insgesamt 5 662 Patienten erhielten nach Randomisierung entweder Finerenon (n=2 824) oder Placebo (n=2 838) und wurden in die Analysen eingeschlossen. Die mediane Nachbeobachtungsdauer betrug 2,6 Jahre. Die Dosis von Finerenon bzw. Placebo konnte im Lauf der Studie zwischen 10 mg und 20 mg einmal täglich angepasst werden; diese Anpassung erfolgte vorwiegend auf Basis der Serumkalium-Konzentration. In Monat 24 erhielten 67 % der Patienten unter Finerenon 20 mg einmal täglich, 31 % erhielten 10 mg einmal täglich und bei 3 % war die Behandlung ausgesetzt worden.
Nach Studienende wurde der Vitalstatus bei 99,7 % der Patienten erhoben. Die Studienpopulation war folgendermaßen aufgeteilt: 63 % Weiß, 25 % Asiatisch und 5 % Schwarz. Das mittlere Alter bei der Aufnahme war 66 Jahre, und 70 % der Patienten waren männlich. Zu Baseline betrug die mittlere eGFR 44,4 ml/min/1,73 m2, wobei 55 % der Patienten eine eGFR von < 45 ml/min/1,73 m2 aufwiesen; der mediane UACR betrug 853 mg/g und der mittlere HbA1c-Wert betrug 7,7 %; bei 46 % lag eine atherosklerotische CV-Erkrankung, bei 30 % eine koronare Herzerkrankung und bei 8 % Herzinsuffizienz in der Anamnese vor; der mittlere Blutdruck betrug 138/76 mmHg. Die mittlere Dauer des T2D bei Baseline betrug 16,6 Jahre, und bei 47 % bzw. 26 % der Patienten wies die Anamnese diabetische Retinopathie bzw. diabetische Neuropathie auf. Fast alle Patienten waren bei Baseline unter einer Therapie mit ACEi (34 %) oder ARB (66 %), und 97 % der Patienten wendeten ein oder mehrere Antidiabetika an (Insulin [64 %], Biguanide [44 %], GLP[Glucagon‑like peptide]‑1-Rezeptoragonisten [7 %], SGLT2[Natrium-Glucose-Cotransporter 2]‑Inhibitoren [5 %]). Die anderen Arzneimittel, die bei Baseline am häufigsten eingenommen wurden, waren Statine (74 %) und Kalziumkanalblocker (63 %).
Für den primären kombinierten Endpunkt und den wichtigsten sekundären kombinierten Endpunkt wurde ein statistisch signifikanter Unterschied zugunsten von Finerenon gezeigt (siehe nachstehende Abbildung 1/ Tabelle 6). Der Behandlungseffekt für den primären und die wichtigsten sekundären Endpunkte war generell in allen Subgruppen (Region, eGFR, UACR, systolischer Blutdruck [SBD] und HbA1c bei Baseline) konsistent.
Für die Studie FIGARO‑DKD waren die Patienten geeignet auf Basis einer nachgewiesenen persistenten Albuminurie mit einem UACR von ≥ 30 mg/g bis < 300 mg/g und einer eGFR von 25 bis 90 ml/min/1,73 m2 oder einem UACR ≥ 300 mg/g und einer eGFR ≥ 60 ml/min/1,73 m2 beim Screening. Die Patienten mussten beim Screening einen Serumkalium-Wert von ≤ 4,8 mmol/l haben.
Der primäre Endpunkt war eine Kombination aus der Zeit bis zu einem CV‑Tod, dem ersten Auftreten von nichttödlichem MI, nichttödlichem Schlaganfall oder Hospitalisierung aufgrund von Herzinsuffizienz. Der sekundäre Endpunkt war eine Kombination aus der Zeit bis zum Auftreten von Nierenversagen, einer anhaltenden Abnahme der eGFR um 40 % oder mehr gegenüber Baseline über mindestens 4 Wochen oder renalem Tod.
Insgesamt 7 328 Patienten erhielten nach Randomisierung entweder Finerenon (n = 3 674) oder Placebo (n = 3 654) und wurden in die Analysen eingeschlossen. Die mediane Nachbeobachtungsdauer betrug 3,4 Jahre. Die Dosis von Finerenon bzw. Placebo konnte im Lauf der Studie zwischen 10 mg und 20 mg einmal täglich angepasst werden. Diese Anpassung erfolgte vorwiegend auf Basis der Serumkalium-Konzentration. In Monat 24 erhielten 82 % der Patienten unter Finerenon 20 mg einmal täglich, 15 % erhielten 10 mg einmal täglich und bei 3 % war die Behandlung ausgesetzt worden. Nach Studienende wurde der Vitalstatus bei 99,8 % der Patienten erhoben. Die Studienpopulation war folgendermaßen aufgeteilt: 72 % Weiß, 20 % Asiatisch und 4 % Schwarz. Das mittlere Alter bei der Aufnahme war 64 Jahre, und 69 % der Patienten waren männlich. Zu Baseline betrug die mittlere eGFR 67,8 ml/min/1,73 m2, wobei 62 % der Patienten eine eGFR ≥ 60 ml/min/1,73 m2 hatten. Der mediane UACR betrug 309 mg/g und der mittlere HbA1c-Wert lag bei 7,7 %. Bei 45 % der Patienten lag eine atherosklerotische CV-Erkrankung vor. 8 % der Patienten hatten Herzinsuffizienz in der Anamnese. Der mittlere Blutdruck betrug 136/77 mmHg. Die mittlere Dauer des T2D bei Baseline betrug 14,5 Jahre und bei 31 % bzw. 28 % der Patienten wies die Anamnese diabetische Retinopathie bzw. diabetische Neuropathie auf. Fast alle Patienten erhielten bei Baseline eine Therapie mit ACEi (43 %) oder ARB (57 %) und 98 % der Patienten wendeten ein oder mehrere Antidiabetika an (Insulin [54 %], Biguanide [69 %], GLP‑1-Rezeptoragonisten [8 %], SGLT2‑Inhibitoren [8 %]). Weitere Arzneimittel, die bei Baseline am häufigsten eingenommen wurden, waren Statine (71 %).
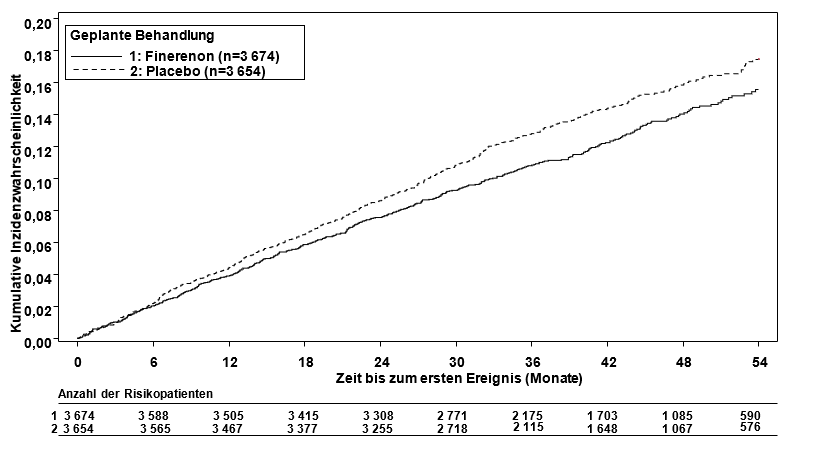
Für den primären kombinierten CV-Endpunkt wurde ein statistisch signifikanter Unterschied zugunsten von Finerenon gezeigt (siehe nachstehende Abbildung 2/ Tabelle 7). Der Behandlungseffekt für den primären Endpunkt war in allen Subgruppen (Region, eGFR, UACR, SBD und HbA1c bei Baseline) konsistent.
Eine niedrigere Inzidenzrate der sekundären kombinierten Endpunkte Nierenversagen, anhaltende Abnahme der eGFR von 40 % oder mehr oder renaler Tod wurde in der Finerenon-Gruppe im Vergleich mit der Placebo-Gruppe beobachtet. Dieser Unterschied erreichte jedoch keine statistische Signifikanz (siehe nachstehende Tabelle 7). Der Behandlungseffekt für den sekundären kombinierten renalen Endpunkt war in allen Subgruppen der eGFR zu Studienbeginn konsistent, aber für die Subgruppe der Patienten mit einer UACR < 300 mg/g betrug die HR 1,16 (95 % KI 0,91; 1,47) und für die Subgruppe der Patienten mit einer UACR ≥ 300 mg/g betrug die HR 0,74 (95 % KI 0,61; 0,89).
Präspezifizierte sekundäre Zeit-bis-zum-Ereignis-Endpunkte werden in Tabelle 7 aufgeführt.
Tabelle 6: Analyse der primären und sekundären Zeit-bis-zum-Ereignis-Endpunkte (und ihrer Einzelkomponenten) in der Phase‑III-Studie FIDELIO‑DKD
Kerendia* (n=2 824) |
Placebo (n=2 838) |
Behandlungseffekt |
||||
n (%) |
Ereignisse/ |
n (%) |
Ereignisse/ |
HR (95 %‑KI) |
||
Primärer kombinierter renaler Endpunkt und seine Komponenten | ||||||
Kombination aus Nierenversagen, anhaltender Abnahme der eGFR um ≥ 40 % oder renalem Tod |
498 (17,6) |
7,53 |
600 (21,1) |
9,09 |
0,82 (0,73; 0,92) |
|
Nierenversagen |
205 (7,3) |
2,96 |
235 (8,3) |
3,39 |
0,86 (0,72; 1,05) |
|
Anhaltende eGFR-Abnahme um ≥ 40 % |
473 (16,7) |
7,15 |
577 (20,3) |
8,74 |
0,81 (0,72; 0,91) |
|
Renaler Tod |
2 (< 0,1) |
- |
2 (< 0,1) |
- |
- |
|
Wichtigster sekundärer kombinierter CV‑Endpunkt und seine Komponenten | ||||||
Kombination aus CV‑Tod, nichttödlichem MI, nichttödlichem Schlaganfall oder Hospitalisierung aufgrund von Herzinsuffizienz |
366 (13,0) |
5,11 |
420 (14,8) |
5,93 |
0,86 (0,75; 0,99) |
|
CV‑Tod |
128 (4,5) |
1,70 |
150 (5,3) |
1,99 |
0,86 (0,68; 1,09) |
|
Nichttödlicher MI |
70 (2,5) |
0,94 |
87 (3,1) |
1,18 |
0,80 (0,58; 1,09) |
|
Nichttödlicher Schlaganfall |
90 (3,2) |
1,22 |
87 (3,1) |
1,18 |
1,03 (0,77; 1,38) |
|
Hospitalisierung aufgrund von Herzinsuffizienz |
138 (4,9) |
1,88 |
162 (5,7) |
2,22 |
0,85 (0,68; 1,07) |
|
Sekundäre Wirksamkeitsendpunkte | ||||||
Mortalität jeglicher Ursache |
219 (7,8) |
2,90 |
244 (8,6) |
3,24 |
0,90 (0,75; 1,08) ** |
|
Hospitalisierung jeglicher Ursache |
1 259 (44,6) |
22,59 |
1 321 (46,5) |
23,91 |
0,95 (0,88; 1,02)** |
|
Kombination aus Nierenversagen, anhaltender Abnahme der eGFR um ≥ 57 % oder renalem Tod |
248 (8,8) |
3,60 |
326 (11,5) |
4,74 |
0,75 (0,65; 0,90) ** |
|
* Behandlung mit 10 oder 20 mg einmal täglich als Zusatz zur maximal tolerierten zugelassenen Dosen eines ACEi oder ARB.
** p = nicht statistisch signifikant nach Adjustierung für Multiplizität
KI: Konfidenzintervall
HR: Hazard Ratio
PJ: Patientenjahre
Abbildung 1: Zeit bis zum ersten Auftreten von Nierenversagen, anhaltender Abnahme der eGFR um ≥ 40 % gegenüber Baseline oder renalem Tod in der Studie FIDELIO‑DKD
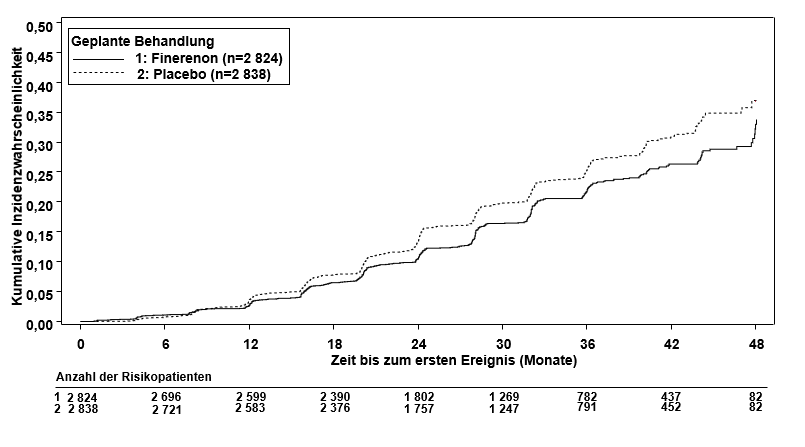
Tabelle 7: Analyse der primären und sekundären Zeit-bis-zum-Ereignis-Endpunkte (und ihrer Einzelkomponenten) in der Phase‑III-Studie FIGARO-DKD
Kerendia* (n=3 674) |
Placebo (n=3 654) |
Behandlungseffekt |
|||
n (%) |
Ereignisse/ |
n (%) |
Ereignisse/ |
HR (95 %‑KI) |
|
Primärer kombinierter CV-Endpunkt und seine Komponenten | |||||
Kombination aus CV-Tod, nichttödlichem MI, nichttödlichem Schlaganfall oder Hospitalisierung aufgrund von Herzinsuffizienz |
457 (12,4) |
3,88 |
518 (14,2) |
4,46 |
0,87 (0,76; 0,98) |
CV‑Tod |
193 (5,3) |
1,56 |
214 (5,9) |
1,75 |
0,89 (0,73; 1,08) |
Nichttödlicher MI |
103 (2,8) |
0,85 |
101 (2,8) |
0,84 |
1,00 (0,76; 1,32) |
Nichttödlicher Schlaganfall |
108 (2,9) |
0,89 |
111 (3,0) |
0,93 |
0,97 (0,74; 1,26) |
Hospitalisierung aufgrund von Herzinsuffizienz |
117 (3,2) |
0,97 |
163 (4,5) |
1,36 |
0,71 (0,56; 0,90) |
Sekundärer kombinierter renaler Endpunkt und seine Komponenten | |||||
Kombination aus Nierenversagen, anhaltender Abnahme der eGFR um ≥ 40 % oder renalem Tod |
350 (9,5) |
3,17 |
395 (10,8) |
3,59 |
0,87 (0,75; 1,01) |
Nierenversagen |
46 (1,3) |
0,40 |
62 (1,7) |
0,55 |
0,72 (0,49; 1,05) |
Anhaltende eGFR-Abnahme um ≥ 40 % |
338 (9,2) |
3,06 |
385 (10,5) |
3,50 |
0,86 (0,74; < 1,00) |
Renaler Tod |
0 |
- |
2 (< 0,1) |
- |
- |
Sekundäre Wirksamkeitsendpunkte | |||||
Mortalität jeglicher Ursache |
332 (9,0) |
2,69 |
370 (10,1) |
3,03 |
0,89 (0,77; 1,03) ** |
Hospitalisierung jeglicher Ursache |
1 569 (42,7) |
16,94 |
1 599 (43,8) |
17,54 |
0,97 (0,90; 1,04) ** |
Kombination aus Nierenversagen, anhaltender Abnahme der eGFR um ≥ 57 % oder renalem Tod |
108 (2,9) |
0,95 |
139 (3,8) |
1,23 |
0,77 (0,60; 0,99) ** |
* Behandlung mit 10 oder 20 mg einmal täglich als Zusatz zu maximal tolerierten zugelassenen Dosen eines ACEi oder ARB.
** p = nicht statistisch signifikant nach Adjustierung für Multiplizität
KI: Konfidenzintervall
HR: Hazard Ratio
PJ: Patientenjahre
Abbildung 2: Zeit bis zum CV‑Tod, ersten Auftreten von nichttödlichem Myokardinfarkt, nichttödlichem Schlaganfall oder Hospitalisierung aufgrund von Herzinsuffizienz in der Studie FIGARO‑DKD
Herzinsuffizienz mit LVEF ≥ 40 %
Die FINEARTS-HF-Studie untersuchte die Wirkung von Finerenon im Vergleich zu Placebo auf kardiovaskuläre Endpunkte bei erwachsenen Patienten mit Herzinsuffizienz.
Eingeschlossen wurden Patienten mit einer diagnostizierten Herzinsuffizienz der NYHA-Klasse II‑IV, die ambulant behandelt wurden oder primär wegen Herzinsuffizienz hospitalisiert waren und eine dokumentierte LVEF ≥ 40 % aufwiesen. Darüber hinaus wiesen die Patienten beim Screening und bei der Randomisierung einen eGFR-Wert von ≥ 25 ml/min/1,73 m2 und einen Serumkalium-Wert von ≤ 5,0 mmol/l auf und erhielten eine Hintergrundtherapie, einschließlich einer diuretischen Behandlung.
Der primäre Endpunkt war eine Kombination aus kardiovaskulärem Tod und allen (erstmaligen und wiederkehrenden) Herzinsuffizienzereignissen, bestehend aus Hospitalisierungen aufgrund von Herzinsuffizienz und Notfallbehandlungen wegen Herzinsuffizienz. Für die sekundären Endpunkte, darunter alle (erstmaligen und wiederkehrenden) Herzinsuffizienzereignisse sowie die Veränderung des Gesamt-Symptom-Scores (Total Symptom Score, TSS) des Kansas City Cardiomyopathy Questionnaire (KCCQ) (der die Häufigkeit und Schwere der Herzinsuffizienzsymptome quantifiziert) vom Ausgangswert bis zum 6., 9. und 12. Monat, wurde ein Mehrfach-Testverfahren angewendet.
Die Studie analysierte 6 001 Patienten, die randomisiert entweder Finerenon (n = 3 003) oder Placebo (n = 2 998) erhielten. Die mediane Nachbeobachtungsdauer betrug 2,7 Jahre. 3 247 (54 %) der in die Studie eingeschlossenen Patienten hatten ein Herzinsuffizienzereignis innerhalb von 3 Monaten vor Studieneinschluss, darunter 1 219 (20 %) Patienten, die während der Hospitalisierung oder innerhalb von 7 Tagen nach der Entlassung randomisiert wurden.
Abhängig von den Nierenwerten erhielten die Patienten während der Studie einmal täglich entweder 10 mg, 20 mg oder 40 mg Finerenon oder ein Placebo. In Monat 24 erhielten 35 % der Patienten unter Finerenon 40 mg einmal täglich, 32 % erhielten 20 mg einmal täglich, 12 % erhielten 10 mg einmal täglich und bei 1 % war die Behandlung ausgesetzt worden. Zu jedem Zeitpunkt während der Behandlung erreichten etwa 80 % der Patienten ihre Zieldosis.
Zum Studienende wurde der Vitalstatus bei 99,7 % der Patienten erhoben.
79 % der Studienpopulation waren Weiße, 17 % Asiaten und 1,5 % Schwarze/Afroamerikaner. Das mittlere Alter bei Einschluss war 72 Jahre und 46 % der Patienten waren weiblich. Zu Beginn der Studie betrug die mittlere LVEF 53 %, wobei 64 % der Patienten eine LVEF ≥ 50 % aufwiesen und 69 %, 30 % bzw. 1 % der Patienten der NYHA-Klasse II, III bzw. IV angehörten. Der mittlere Blutdruck betrug 129/75 mmHg, während der BMI bei 30 kg/m2 lag. Der Medianwert von NT-pro-BNP (N-terminal prohormone of brain natriuretic peptide) betrug 1 041 pg/ml, der mittlere eGFR-Wert lag bei 62 ml/min/1,73 m2, wobei 48 % der Patienten einen eGFR-Wert von ˂ 60 ml/min/1,73 m2 aufwiesen, und der UACR-Median betrug 18 mg/g. Bei 38 % der Patienten lag Vorhofflimmern vor, und 41 % hatten Diabetes mellitus. Die Mehrheit der Patienten erhielt Schleifendiuretika (87 %), ACE-Hemmer oder ARB (79 %) oder Angiotensin-Rezeptor-Neprilysin-Hemmer (9 %), und 14 % erhielten SGLT2-Hemmer.
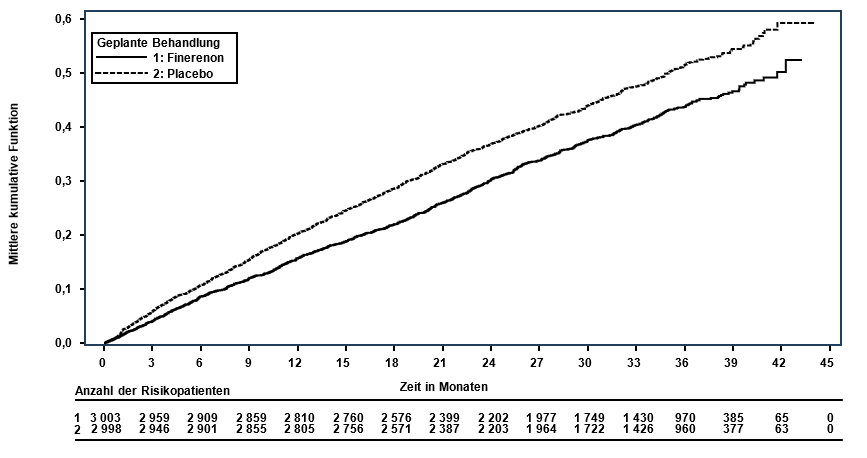
Für den primären kombinierten Endpunkt wurde ein statistisch signifikanter Unterschied zugunsten von Finerenon gezeigt (siehe nachstehende Tabelle 8). Der Effekt wurde früh beobachtet und blieb über die gesamte Dauer der Studie erhalten (siehe Abbildung 3 unten). Ein statistisch signifikanter Unterschied zugunsten von Finerenon zeigte sich auch bei den sekundären Endpunkten der Gesamt-Herzinsuffizienzereignisse. Die vorab spezifizierten sekundären Wirksamkeitsendpunkte sind ebenfalls nachstehend in Tabelle 8 aufgeführt. Die Behandlungseffekte für den primären und die wichtigsten sekundären Endpunkte waren in allen vorab spezifizierten Subgruppen konsistent, darunter Geschlecht, LVEF, NYHA-Klasse, eGFR, Zeit seit dem letzten Herzinsuffizienzereignis, SGLT2-Hemmer-Therapie und Diabetes-mellitus-Status.
Tabelle 8: Analyse der primären und sekundären Endpunkte (und ihrer Einzelkomponenten für die Zeit-bis-zum-Ereignis-Endpunkte) in der Phase‑III-Studie FINEARTS-HF
Kerendia* (n = 3 003) |
Placebo (n = 2 998) |
Behandlungseffekt |
|||
[Ereignis insgesamt] |
Ereignisse/ |
[Ereignis insgesamt] |
Ereignisse/ |
(95 %-KI) |
|
Primärer kombinierter CV-Endpunkt und seine Komponenten | |||||
Kombination aus CV-Tod und Gesamt-Herzinsuffizienzereignissen |
[1 083] |
14,88 |
[1 283] |
17,70 |
RR 0,84 |
Gesamt-Herzinsuffizienzereignisse** |
[842] |
11,57 |
[1 024] |
14,12 |
RR 0,82 |
CV‑Tod |
242 (8,1) |
3,33 |
260 (8,7) |
3,59 |
HR 0,93 |
Sekundäre Wirksamkeitsendpunkte | |||||
Veränderung des KCCQ-TSS gegenüber Baseline |
LSM |
- |
LSM |
- |
LSM-Differenz |
Verbesserung der NYHA-Klasse |
557 (18,6) |
- |
553 (18,4) |
- |
OR 1,01 |
Renaler kombinierter Endpunkt |
75 (2,5) |
1,16 |
55 (1,8) |
0,85 |
HR 1,33 |
Anhaltende eGFR-Abnahme um ≥ 50 % |
68 (2,3) |
1,05 |
51 (1,7) |
0,79 |
- |
Anhaltender Rückgang der eGFR auf ˂ 15 ml/min/1,73m2 |
5 (0,2) |
0,08 |
2 (< 0,1) |
0,03 |
- |
Einleitung der Dialyse |
2 (< 0,1) |
0,03 |
2 (˂ 0,1) |
0,03 |
- |
Nierentransplantation |
0 (0,0) |
0,00 |
0 (0,0) |
- |
- |
Mortalität jeglicher Ursache |
491 (16,4) |
6,71 |
522 (17,4) |
7,17 |
HR 0,93 |
Abkürzungen: KI: Konfidenzintervall; HR: Hazard Ratio; LSM: Mittelwert der kleinsten Quadrate; OR: Odds Ratio; RR: Ratenverhältnis; PJ: Patientenjahre
* Behandlung mit 10, 20 oder 40 mg einmal täglich zusätzlich zur Hintergrundtherapie, einschließlich Diuretikabehandlung
** Die Gesamtzahl der (erstmaligen und wiederkehrenden) Herzinsuffizienzereignisse war ebenfalls ein wichtiger sekundärer Endpunkt
† Nicht signifikant (Testverfahren abgebrochen)
†† Endpunkt nicht formal getestet (vorhergehender Endpunkt im Testverfahren nicht signifikant)
Abbildung 3: Primärer kombinierter Endpunkt aus CV-Tod und allen (erstmaligen und wiederkehrenden) Herzinsuffizienzereignissen in der FINEARTS-HF-Studie
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Kerendia eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in der Behandlung der chronischen Nierenerkrankung und der Herzinsuffizienz gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Resorption
Finerenon wird nach oraler Anwendung nahezu vollständig resorbiert. Die Resorption erfolgt schnell, die maximalen Plasmakonzentrationen (Cₘₐₓ) werden 0,5 bis 1,25 Stunden nach Einnahme der Tablette in nüchternem Zustand erreicht. Die absolute Bioverfügbarkeit von Finerenon beträgt aufgrund des First‑Pass-Metabolismus in Darmwand und Leber 43,5 %. Finerenon ist in vitro ein Substrat des Effluxtransporters P‑Glykoprotein, was aufgrund der hohen Diffusionsfähigkeit von Finerenon jedoch als nicht relevant für die Resorption in vivo angesehen wird.
Auswirkung von Nahrung
Bei Einnahme zusammen mit fetter, kalorienreicher Nahrung war die AUC von Finerenon um bis zu 21 % erhöht, die Cₘₐₓ um 19 % bis 23 % reduziert und die Zeit bis zum Erreichen der Cₘₐₓ auf bis zu 2,5 Stunden verlängert. Da dies nicht als klinisch relevant angesehen wird, kann Finerenon mit oder ohne Nahrung eingenommen werden.
Verteilung
Das Verteilungsvolumen von Finerenon im Steady State (Vₛₛ) beträgt 52,6 l. Die Plasmaproteinbindung von Finerenon beim Menschen beträgt in vitro 91,7 %, das primäre Bindungsprotein ist Serumalbumin.
Biotransformation
Der Finerenon-Metabolismus wird zu etwa 90 % durch CYP3A4 und zu 10 % durch CYP2C8 vermittelt. Im Plasma wurden vier Hauptmetaboliten gefunden. Alle Metaboliten sind pharmakologisch inaktiv.
Elimination
Finerenon wird mit einer Eliminationshalbwertszeit (t½) von etwa 2 bis 3 Stunden schnell aus dem Plasma eliminiert. Die systemische Clearance von Finerenon aus dem Blut beträgt etwa 25 l/h. Etwa 80 % der verabreichten Dosis wurden über den Urin und etwa 20 % über den Fäzes ausgeschieden. Die Ausscheidung erfolgte nahezu ausschließlich in Form von Metaboliten, während die Ausscheidung von unverändertem Finerenon (< 1 % der Dosis über den Urin aufgrund der glomerulären Filtration, < 0,2 % über den Fäzes) einen unbedeutenden Weg darstellt.
Linearität
Die Pharmakokinetik von Finerenon ist im untersuchten Dosisbereich von 1,25 mg bis 80 mg, verabreicht als Einzeldosis in Tablettenform, linear.
Besondere Patientengruppen
Ältere Patienten
Von den 2 818 Patienten, die Finerenon in der Studie FIDELIO‑DKD erhielten, waren 58 % mindestens 65 Jahre alt und 15 % mindestens 75 Jahre alt. Von den 3 671 Patienten, die Finerenon in der Studie FIGARO‑DKD erhielten, waren 53 % mindestens 65 Jahre alt und 14 % mindestens 75 Jahre alt. Von den 2 993 Patienten, die Finerenon in der Studie FINEARTS-HF erhielten, waren 79 % mindestens 65 Jahre alt und 43 % mindestens 75 Jahre alt.
In einer Phase‑I-Studie (n=48) wurden bei älteren gesunden Teilnehmern (≥ 65 Jahre) im Vergleich zu jüngeren gesunden Teilnehmern (≤ 45 Jahre) höhere Plasmakonzentrationen von Finerenon festgestellt, wobei die mittleren AUC- und Cₘₐₓ‑Werte bei den älteren Patienten um 34 % bzw. 51 % höher waren (siehe Abschnitt 4.2). In populationspharmakokinetischen Analysen wurde das Alter nicht als Kovariate für die AUC oder Cₘₐₓ von Finerenon identifiziert.
Eingeschränkte Nierenfunktion
Eine leichte Nierenfunktionsstörung (Kreatinin-Clearance [CLCR] 60 bis < 90 ml/min) hatte keine Auswirkung auf die AUC und Cₘₐₓ von Finerenon.
Im Vergleich zu Patienten mit normaler Nierenfunktion (CLCR ≥ 90 ml/min) war die Auswirkung einer mittelschweren (CLCR 30 bis < 60 ml/min) oder schweren (CLCR < 30 ml/min) Nierenfunktionsstörung auf die AUC von Finerenon mit einem Anstieg um 34‑36 % ähnlich. Eine mittelschwere oder schwere Nierenfunktionsstörung hatte keine Auswirkung auf die Cₘₐₓ (siehe Abschnitt 4.2).
Aufgrund der hohen Plasmaproteinbindung ist nicht zu erwarten, dass Finerenon dialysierbar ist.
Eingeschränkte Leberfunktion
Bei zirrhotischen Patienten mit leichter Leberfunktionsstörung war die Finerenon-Exposition unverändert (siehe Abschnitt 4.2).
Bei zirrhotischen Patienten mit mittelschwerer Leberfunktionsstörung war der AUC‑Wert für das gesamte und das ungebundene Finerenon im Vergleich zu gesunden Kontrollpatienten um 38 % bzw. 55 % erhöht, während die Cₘₐₓ keine Veränderung zeigte (Abschnitt 4.2).
Für Patienten mit schwerer Leberfunktionsstörung liegen keine Daten vor (siehe Abschnitte 4.2 und 4.5).
Körpergewicht
In populationspharmakokinetischen Analysen wurde das Körpergewicht als Kovariate für die Cₘₐₓ und AUC von Finerenon identifiziert. Die Cₘₐₓ und AUC von Personen mit einem Körpergewicht von unter 57 kg wurden im Durchschnitt um 52 % bzw. 30 % höher geschätzt, und von Personen mit einem Körpergewicht von über 122 kg 32 % bzw. 20 % niedriger geschätzt als bei Personen zwischen 57 und 122 kg. Eine Dosisanpassung auf Basis des Körpergewichts ist nicht gerechtfertigt (siehe Abschnitt 4.2).
Pharmakokinetische/pharmakodynamische Zusammenhänge
Die Dosis-Wirkungs-Beziehung für den UACR im Zeitverlauf wurde mithilfe eines Maximum-Effekt-Modells charakterisiert, das auf eine Sättigung bei hohen Expositionen schließen lässt. Die im Modell prognostizierte Zeit bis zum Erreichen der vollen (99 %) Wirkung des Arzneimittels auf den UACR‑Wert im Steady State betrug 138 Tage. Die pharmakokinetische (PK) Halbwertszeit betrug 2‑3 Stunden, und das pharmakokinetische Steady State wurde nach 2 Tagen erreicht, was auf eine indirekte und verzögerte Wirkung auf das pharmakodynamische Ansprechen hindeutet.
Klinische Studien ohne relevante Arzneimittelwechselwirkungen
Bei gleichzeitiger Anwendung des starken CYP2C8‑Inhibitors Gemfibrozil (600 mg zweimal täglich) waren die mittlere AUC und Cₘₐₓ von Finerenon um das 1,1‑ bzw. 1,2‑Fache erhöht. Dies wird nicht als klinisch relevant angesehen.
Die Vor- und Begleitbehandlung mit dem Protonenpumpeninhibitor Omeprazol (40 mg einmal täglich) hatte keine Wirkung auf die mittlere AUC und mittlere Cₘₐₓ von Finerenon.
Die begleitende Anwendung der Antazida Aluminiumhydroxid und Magnesiumhydroxid (70 mVal) hatte keine Wirkung auf die mittlere AUC von Finerenon und reduzierte seine mittlere Cₘₐₓ um 19 %. Dies wird nicht als klinisch relevant angesehen.
Das Fehlen einer gegenseitigen pharmakokinetischen Wechselwirkung zwischen Finerenon und dem CYP2C9‑Substrat Warfarin sowie zwischen Finerenon und dem P‑gp‑Substrat Digoxin wurde nachgewiesen.
Mehrere Dosen 40 mg Finerenon einmal täglich zeigten keine klinisch relevante Wirkung auf AUC und Cmax des breast cancer resistance protein (BCRP) und des Organo-Anion-Transporter-Polypeptid (OATP)-Substrats Rosuvastatin.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei Einmalgabe, Toxizität bei wiederholter Gabe, Genotoxizität, Phototoxizität, zum kanzerogenen Potential und zur männlichen und weiblichen Fertilität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Toxizität bei wiederholter Gabe
Bei Hunden wurde eine Abnahme des Gewichts und der Größe der Prostata bei einer AUCunbound festgestellt, die etwa dem 4- bis 60-Fachen des Wertes beim Menschen entspricht. Die Dosen ohne Befund stellten Sicherheitsgrenzen von etwa 1‑2 dar.
Kanzerogenes Potential
In 2‑jährigen Kanzerogenitätsstudien zeigte Finerenon bei männlichen und weiblichen Ratten sowie weiblichen Mäusen kein kanzerogenes Potential. Bei männlichen Mäusen führte Finerenon in Dosen, die dem 10- bis 26‑Fachen der AUCunbound beim Menschen entsprachen, zu einem vermehrten Auftreten von Leydig-Zell-Adenomen. Eine Dosis, die dem 7‑Fachen der AUCunbound der 40‑mg-Dosis und dem 17‑Fachen der AUCunbound der 20‑mg-Dosis beim Menschen entsprach, verursachte keine Tumore. Angesichts der bekannten Empfindlichkeit von Nagern für die Entwicklung dieser Tumoren und der pharmakologischen Mechanismen bei supratherapeutischen Dosen sowie der adäquaten Sicherheitsgrenzen ist der Anstieg von Leydig-Zell-Tumoren bei männlichen Mäusen klinisch nicht relevant.
Entwicklungstoxizität
In der Studie zur embryofetalen Toxizität an Ratten führte Finerenon in der maternal toxischen Dosis von 10 mg/kg/Tag (entspricht mindestens der 7‑fachen AUCunbound beim Menschen) zu einem reduzierten Plazentagewicht und Anzeichen einer fetalen Toxizität mit vermindertem Fetengewicht und verzögerter Ossifikation. Bei 30 mg/kg/Tag war die Inzidenz von viszeralen und skelettalen Variationen erhöht (leichtes Ödem, verkürzte Nabelschnur, geringfügig vergrößerte Fontanelle) und ein Fetus wies bei einer AUCunbound, die etwa dem 10‑fachen Wert beim Menschen bei einer Dosis von 40 mg und etwa dem 25‑fachen Wert beim Menschen bei einer Dosis von 20 mg entsprach, komplexe Fehlbildungen einschließlich einer seltenen Fehlbildung (doppelter Aortenbogen) auf. Die Dosen ohne Befund (niedrige Dosis bei Ratten, hohe Dosis bei Kaninchen) stellten für die AUCunbound Sicherheitsgrenzen vom 4‑ bis 13‑fachen Wert dar Daher lassen die Befunde bei Ratten nicht auf ein erhöhtes Risiko für das ungeborene Kind schließen.
Bei Ratten, die im Rahmen einer Studie zur prä- und postnatalen Entwicklungstoxizität während der Trächtigkeit und Laktation exponiert waren, wurde beim etwa 2- bzw. 4‑Fachen der beim Menschen unter der 40- bzw. 20‑mg-Dosis erwarteten AUCunbound bei den Jungtieren ein Anstieg der Mortalität und anderer Nebenwirkungen (geringeres Gewicht, verzögerte Auffaltung der Ohrmuschel) beobachtet. Zusätzlich zeigte der Nachwuchs bei einer Exposition ab dem etwa 2- bzw. 4‑Fachen der beim Menschen unter der 40- bzw. 20‑mg-Dosis erwarteten AUCunbound eine leicht erhöhte lokomotorische Aktivität, jedoch keine anderen Veränderungen des Neuroverhaltens. Die Dosen ohne Befund stellten für die AUCunbound Sicherheitsgrenzen von etwa 2 für die 20‑mg-Dosis dar; für die 40‑mg-Dosis liegen sie im therapeutischen Bereich. Die erhöhte lokomotorische Aktivität beim Nachwuchs könnte auf ein potentielles Risiko für das ungeborene Kind hindeuten. Angesichts der Befunde der Jungtiere kann ferner ein Risiko für das gestillte Neugeborene/den Säugling nicht ausgeschlossen werden.
Weibliche Fertilität
Bei Expositionen von dem etwa 9‑Fachen der AUCunbound beim Menschen für die 40‑mg-Dosis und dem 21‑Fachen der AUCunbound beim Menschen für die 20‑mg-Dosis reduzierte Finerenon die weibliche Fertilität (verminderte Anzahl von Corpora lutea und Implantationsstellen) und verursachte Anzeichen von früher embryonaler Toxizität (erhöhte postimplantative Verluste und abnehmende Anzahl lebensfähiger Feten). Überdies wurde bei Expositionen vom etwa 7‑Fachen der AUCunbound beim Menschen für die 40‑mg-Dosis und dem 17‑Fachen der AUCunbound beim Menschen für die 20‑mg-Dosis ein vermindertes Ovarialgewicht festgestellt. Expositionen vom 4‑Fachen der AUCunbound beim Menschen für die 40‑mg-Dosis und dem 10‑Fachen der AUCunbound beim Menschen für die 20‑mg-Dosis zeigten keine Wirkung auf die weibliche Fertilität und die frühe Embryonalentwicklung. Daher sind die Befunde in weiblichen Ratten von geringer klinischer Relevanz (siehe Abschnitt 4.6).
Beurteilung der Risiken für die Umwelt (Environmental Risk Assessment [ERA])
Studien zum Environmental Risk Assessment haben gezeigt, dass Finerenon ein Risiko für Oberflächengewässer und Grundwasser darstellen kann (siehe Abschnitt 6.6).
Tablettenkern
Mikrokristalline Cellulose (E 460)
Croscarmellose-Natrium
Hypromellose 2910 (E 464)
Lactose-Monohydrat
Magnesiumstearat (E 470b)
Natriumdodecylsulfat (E 487)
Filmüberzug
Hypromellose 2910 (E 464)
Titandioxid (E 171)
Talkum (E 553b)
Kerendia 10 mg Filmtabletten
Eisen(III)-oxid (E172)
Kerendia 20 mg Filmtabletten
Eisen(III)-hydroxid-oxid x H20 (E172)
Kerendia 40 mg Filmtabletten
Eisen(III)-oxid (E 172)
Eisen(III)-hydroxid-oxid x H20 (E 172)
Nicht zutreffend.
3 Jahre
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Transparente Kalender-Blisterpackungen aus PVC/PVDC/Aluminium mit 14 Filmtabletten. Packungsgrößen: 14, 28 oder 98 Filmtabletten.
Transparente, perforierte Einzeldosis-Blisterpackungen aus PVC/PVDC/Aluminium mit 10 x 1 Filmtabletten. Packungsgröße: 100 x 1 Filmtabletten.
Weiße, opake HDPE‑Flasche mit weißem, opaken, kindergesicherten Schraubdeckel aus Polypropylen mit Dichteinsatz. Packungsgröße: 100 Filmtabletten (nur Kerendia 10 mg und 20 mg Filmtabletten).
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Dieses Arzneimittel kann eine Gefahr für die Umwelt darstellen (siehe Abschnitt 5.3).
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Bayer AG
51368 Leverkusen
Deutschland
Kerendia 10 mg Filmtabletten
EU/1/21/1616/001-005
Kerendia 20 mg Filmtabletten
EU/1/21/1616/006-010
Kerendia 40 mg Filmtabletten
EU/1/21/1616/011-014
Datum der Erteilung der Zulassung: 16. Februar 2022
März 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
DE/3