
Skyrizi® 360 mg Injektionslösung in einer Patrone
Skyrizi® 180 mg Injektionslösung in einer Patrone
Skyrizi® 90 mg Injektionslösung in einer Fertigspritze
Skyrizi® 180 mg Injektionslösung in einer Fertigspritze
Skyrizi 360 mg Injektionslösung in einer Patrone
Jede Patrone enthält 360 mg Risankizumab in 2,4 ml Lösung.
Skyrizi 180 mg Injektionslösung in einer Patrone
Jede Patrone enthält 180 mg Risankizumab in 1,2 ml Lösung.
Skyrizi 90 mg Injektionslösung in einer Fertigspritze
Jede Fertigspritze enthält 90 mg Risankizumab in 1 ml Lösung.
Skyrizi 180 mg Injektionslösung in einer Fertigspritze
Jede Fertigspritze enthält 180 mg Risankizumab in 1,2 ml Lösung.
Risankizumab ist ein humanisierter monoklonaler Immunglobulin‑G1(IgG1)‑Antikörper, der mittels rekombinanter DNA‑Technologie in Ovarialzellen des chinesischen Hamsters hergestellt wird.
Sonstiger Bestandteil mit bekannter Wirkung
Nur 180 mg und 360 mg Injektionslösung
Dieses Arzneimittel enthält 0,24 mg Polysorbat 20 pro 180-mg-Dosis und 0,48 mg Polysorbat 20 pro 360-mg-Dosis.
Nur 90 mg Injektionslösung
Dieses Arzneimittel enthält 164 mg Sorbitol pro 360‑mg-Dosis.
Dieses Arzneimittel enthält 0,8 mg Polysorbat 20 pro 360-mg-Dosis.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung (Injektion)
Skyrizi 180 mg und 360 mg Injektionslösung in einer Patrone und 180 mg Injektionslösung in einer Fertigspritze
Die Lösung ist farblos bis gelblich und klar bis leicht opalisierend.
Skyrizi 90 mg Injektionslösung in einer Fertigspritze
Die Lösung ist farblos bis leicht gelblich und klar bis leicht opalisierend.
Morbus Crohn
Skyrizi wird angewendet zur Behandlung von erwachsenen Patienten mit mittelschwerem bis schwerem aktivem Morbus Crohn, die auf eine konventionelle Therapie oder eine Biologikatherapie unzureichend angesprochen, diese nicht vertragen haben oder nicht mehr darauf ansprechen.
Colitis ulcerosa
Skyrizi wird angewendet zur Behandlung von erwachsenen Patienten mit mittelschwerer bis schwerer aktiver Colitis ulcerosa, die auf eine konventionelle Therapie oder eine Biologikatherapie unzureichend angesprochen, diese nicht vertragen haben oder nicht mehr darauf ansprechen.
Dieses Arzneimittel ist zur Anwendung unter Anleitung und Überwachung eines Arztes mit Erfahrung in der Diagnose und Behandlung von Erkrankungen, für die Skyrizi indiziert ist, vorgesehen.
Dosierung
Morbus Crohn
Die empfohlene Dosis beträgt 600 mg als intravenöse Infusion in Woche 0, Woche 4 und Woche 8, gefolgt von 360 mg als subkutane Injektion in Woche 12 und danach alle 8 Wochen. Bei Patienten, die nach 24 Wochen keine Anzeichen eines therapeutischen Nutzens zeigen, ist ein Absetzen der Behandlung in Erwägung zu ziehen.
Die Dosierung des intravenösen Anfangsschemas ist Abschnitt 4.2 der Fachinformation zu Skyrizi 600 mg, Konzentrat zur Herstellung einer Infusionslösung, zu entnehmen.
Colitis ulcerosa
Die empfohlene Induktionsdosis beträgt 1 200 mg als intravenöse Infusion in Woche 0, Woche 4 und Woche 8. Ab Woche 12 und danach alle 8 Wochen basiert die empfohlene Erhaltungsdosis auf dem individuellen Ansprechen der Patienten:
Eine Dosis von 180 mg als subkutane Injektion wird für Patienten empfohlen, bei denen nach der Induktion eine ausreichende Verbesserung der Krankheitsaktivität festgestellt wurde.
Eine Dosis von 360 mg als subkutane Injektion wird für Patienten empfohlen, bei denen nach der Induktion keine ausreichende Verbesserung der Krankheitsaktivität festgestellt wurde.
Bei Patienten, die nach 24 Wochen keine Anzeichen eines therapeutischen Nutzens zeigen, ist ein Absetzen der Behandlung in Erwägung zu ziehen.
Die Dosierung des intravenösen Anfangsschemas ist Abschnitt 4.2 der Fachinformation zu Skyrizi 600 mg, Konzentrat zur Herstellung einer Infusionslösung, zu entnehmen.
Versäumte Einnahme
Wenn eine Dosis versäumt wurde, sollte diese so schnell wie möglich verabreicht werden. Danach sollte die Behandlung zu den regulär vorgesehenen Zeitpunkten fortgeführt werden.
Besondere Patientengruppen
Ältere Patienten
Es ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Bisher liegen nur begrenzte Erfahrungen zu Patienten ≥ 65 Jahre vor.
Eingeschränkte Nieren‑ oder Leberfunktion
Es wurden keine spezifischen Studien zur Beurteilung der Auswirkungen einer eingeschränkten Nieren‑ oder Leberfunktion auf die Pharmakokinetik von Skyrizi durchgeführt. Im Allgemeinen ist nicht zu erwarten, dass diese Einschränkungen einen signifikanten Einfluss auf die Pharmakokinetik monoklonaler Antikörper haben, sodass keine Dosisanpassungen als notwendig erachtet werden (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Skyrizi bei Kindern und Jugendlichen im Alter von 0 bis 17 Jahren zur Behandlung von Morbus Crohn und Colitis ulcerosa sind bisher noch nicht erwiesen. Die aktuell verfügbaren Daten werden in Abschnitt 5.1 und 5.2 beschrieben, es kann jedoch keine Dosierungsempfehlung abgegeben werden.
Übergewichtige Patienten
Es ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Art der Anwendung
Skyrizi wird als subkutane Injektion verabreicht.
Die Injektion ist in den Oberschenkel oder Bauch zu injizieren. Schmerzempfindliche, blutunterlaufene, gerötete, verhärtete oder geschädigte Haut oder Stellen sind zu vermeiden.
Skyrizi 180 mg und 360 mg Injektionslösung in einer Patrone
Nach entsprechender Schulung zur subkutanen Injektionstechnik mit dem On-Body-Injektor können Patienten Skyrizi selbst injizieren. Die Patienten sind anzuweisen, vor der Anwendung den Abschnitt 7 „Wie Skyrizi gespritzt wird“ im letzten Teil der Packungsbeilage zu lesen.
Skyrizi 90 mg Injektionslösung in einer Fertigspritze
Dieses Arzneimittel muss von medizinischem Fachpersonal verabreicht werden.
Um die vollständige 360-mg-Dosis zu verabreichen, müssen vier Fertigspritzen injiziert werden. Die vier Injektionen sind an anatomisch unterschiedlichen Stellen zu verabreichen (siehe Hinweise zur Verabreichung in der Packungsbeilage).
Skyrizi 180 mg Injektionslösung in einer Fertigspritze
Nach entsprechender Schulung zur subkutanen Injektionstechnik mit der Fertigspritze können Patienten Skyrizi selbst injizieren. Die Patienten sind anzuweisen, vor der Anwendung den Abschnitt 7 „Wie Skyrizi gespritzt wird“ im letzten Teil der Packungsbeilage zu lesen.
Um die 180-mg-Dosis zu verabreichen, muss eine Fertigspritze injiziert werden.
Um die 360-mg-Dosis zu verabreichen, müssen zwei Fertigspritzen injiziert werden. Die zwei Injektionen sind an anatomisch unterschiedlichen Stellen zu verabreichen.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Klinisch relevante aktive Infektionen (z. B. aktive Tuberkulose, siehe Abschnitt 4.4).
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Infektionen
Risankizumab kann das Infektionsrisiko erhöhen.
Bei Patienten mit einer chronischen Infektion, einer rezidivierenden Infektion in der Anamnese oder mit bekannten Risikofaktoren für eine Infektion sollte Risankizumab mit Vorsicht angewendet werden. Bei Patienten mit einer klinisch relevanten aktiven Infektion sollte die Behandlung mit Risankizumab nicht eingeleitet werden, bis die Infektion abgeklungen ist oder angemessen behandelt wird.
Mit Risankizumab behandelte Patienten sind anzuweisen, ärztlichen Rat einzuholen, wenn Anzeichen oder Symptome einer klinisch bedeutsamen chronischen oder akuten Infektion auftreten. Wenn ein Patient eine solche Infektion entwickelt oder auf eine Standardtherapie für die Infektion nicht anspricht, ist der Patient engmaschig zu überwachen und Risankizumab sollte bis zum Abklingen der Infektion nicht verabreicht werden.
Tuberkulose
Vor Beginn der Behandlung mit Risankizumab sind die Patienten auf eine Tuberkulose(TB)-Infektion zu untersuchen. Patienten, die Risankizumab erhalten, müssen auf Anzeichen und Symptome einer aktiven TB überwacht werden. Bei Patienten mit latenter oder aktiver TB in der Anamnese, bei denen nicht bestätigt werden kann, dass sie eine adäquate Behandlung erhalten haben, sollte vor Behandlungsbeginn mit Risankizumab eine Anti‑TB-Therapie in Erwägung gezogen werden.
Impfungen
Vor Einleitung der Therapie mit Risankizumab sollte in Übereinstimmung mit den aktuellen Impfempfehlungen die Durchführung aller vorgesehenen Impfungen erwogen werden. Wenn der Patient mit einem viralen oder bakteriellen Lebendimpfstoff geimpft wurde, wird empfohlen, mindestens 4 Wochen mit dem Beginn der Behandlung mit Risankizumab zu warten. Patienten, die mit Risankizumab behandelt werden, sollten während der Behandlung und für mindestens 21 Wochen nach der Behandlung keine Lebendimpfstoffe erhalten (siehe Abschnitt 5.2).
Überempfindlichkeit
Schwere Überempfindlichkeitsreaktionen, einschließlich Anaphylaxie, wurden bei der Anwendung von Risankizumab berichtet (siehe Abschnitt 4.8). Im Falle des Auftretens einer schwerwiegenden Überempfindlichkeitsreaktion muss die Anwendung von Risankizumab unverzüglich abgebrochen und eine geeignete Behandlung eingeleitet werden.
Sonstige Bestandteile mit bekannter Wirkung
Skyrizi 180 mg und 360 mg Injektionslösung in einer Patrone
Polysorbat
Dieses Arzneimittel enthält 0,24 mg Polysorbat 20 pro 180-mg-Dosis und 0,48 mg Polysorbat 20 pro 360-mg-Dosis. Polysorbate können allergische Reaktionen hervorrufen.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Patrone, d. h., es ist nahezu „natriumfrei“.
Skyrizi 90 mg Injektionslösung in einer Fertigspritze
Polysorbat
Dieses Arzneimittel enthält 0,8 mg Polysorbat 20 pro 360-mg-Dosis. Polysorbate können allergische Reaktionen hervorrufen.
Sorbitol
Dieses Arzneimittel enthält 164 mg Sorbitol pro 360-mg-Dosis.
Die additive Wirkung gleichzeitig angewendeter Sorbitol (oder Fructose)-haltiger Arzneimittel und die Einnahme von Sorbitol (oder Fructose) über die Nahrung ist zu berücksichtigen.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro 360-mg-Dosis, d. h., es ist nahezu „natriumfrei“.
Skyrizi 180 mg Injektionslösung in einer Fertigspritze
Polysorbat
Dieses Arzneimittel enthält 0,24 mg Polysorbat 20 pro 180-mg-Dosis und 0,48 mg Polysorbat 20 pro 360-mg-Dosis. Polysorbate können allergische Reaktionen hervorrufen.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro 180-mg-Dosis und 360-mg-Dosis, d. h., es ist nahezu „natriumfrei“.
Es ist nicht zu erwarten, dass Risankizumab durch Leberenzyme verstoffwechselt oder über die Niere ausgeschieden wird. Wechselwirkungen zwischen Risankizumab und Inhibitoren, Induktoren oder Substraten von Enzymen, die Arzneimittel verstoffwechseln, sind nicht zu erwarten und eine Dosisanpassung ist nicht erforderlich (siehe Abschnitt 5.2).
Begleitende Immunsuppressivatherapie
Die Sicherheit und Wirksamkeit von Risankizumab in Kombination mit Immunsuppressiva, einschließlich Biologika, wurden nicht untersucht.
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während der Behandlung und für mindestens 21 Wochen nach der Behandlung eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Bisher liegen keine oder nur sehr begrenzte Erfahrungen (weniger als 300 Schwangerschaftsausgänge) mit der Anwendung von Risankizumab bei Schwangeren vor. Tierexperimentelle Studien weisen nicht auf direkte oder indirekte schädliche Wirkungen hinsichtlich der Reproduktionstoxizität hin. Als Vorsichtsmaßnahme sollte eine Anwendung von Risankizumab während der Schwangerschaft vermieden werden.
Stillzeit
Es ist nicht bekannt, ob Risankizumab in die Muttermilch übergeht. Da humane Immunglobuline bekanntermaßen in den ersten Tagen nach der Geburt in die Muttermilch übergehen (kurz danach nur noch in geringer Konzentration), kann ein Risiko für den gestillten Säugling während dieses kurzen Zeitraums nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob die Behandlung mit Risankizumab unterbrochen bzw. abgesetzt wird. Dabei sind sowohl die Vorteile des Stillens für das Kind als auch der Nutzen der Therapie mit Risankizumab für die Mutter zu berücksichtigen.
Fertilität
Die Wirkung von Risankizumab auf die Fertilität des Menschen wurde nicht untersucht. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf die Fertilität.
Risankizumab hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die am häufigsten berichteten Nebenwirkungen waren Infektionen der oberen Atemwege (15,6 % bei Morbus Crohn und 26,2 % bei Colitis ulcerosa).
Tabellarische Auflistung der Nebenwirkungen
Die Nebenwirkungen von Risankizumab aus klinischen Studien (Tabelle 1) sind nach MedDRA-Systemorganklasse anhand folgender Häufigkeitskategorien gegliedert: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 1. Auflistung von Nebenwirkungen
Systemorganklasse |
Häufigkeit |
Nebenwirkungen |
Infektionen und parasitäre Erkrankungen |
Sehr häufig |
Infektionen der oberen Atemwegea |
Häufig |
Tinea-Infektionenb |
|
Gelegentlich |
Follikulitis |
|
Erkrankungen des Immunsystems |
Selten |
Anaphylaktische Reaktionen |
Erkrankungen des Nervensystems |
Häufig |
Kopfschmerzc |
Erkrankung der Haut und des Unterhautgewebes |
Häufig |
Pruritus |
Gelegentlich |
Urtikaria |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Häufig |
Fatigue/Ermüdungd |
a Einschließlich Atemwegsinfektion (viral, bakteriell oder nicht näher spezifiziert), Sinusitis (darunter akute Form), Rhinitis, Nasopharyngitis, Pharyngitis (darunter virale Form), Tonsillitis, Laryngitis, Tracheitis | ||
Beschreibung ausgewählter Nebenwirkungen
Psoriasis
Infektionen
Über das gesamte Psoriasis-Programm, einschließlich der Langzeitanwendung von Risankizumab lag die Rate der Infektionen bei 75,5 Ereignissen pro 100 Patientenjahre. Der Großteil der Fälle war nicht schwerwiegend, von leichtem bis moderatem Schweregrad und führte nicht zu einem Abbruch der Behandlung mit Risankizumab. Die Rate an schwerwiegenden Infektionen betrug 1,7 Ereignisse pro 100 Patientenjahre (siehe Abschnitt 4.4).
Morbus Crohn
Insgesamt entsprach das Sicherheitsprofil bei Patienten mit Morbus Crohn unter Risankizumab dem Sicherheitsprofil, das bei Patienten aller Indikationen beobachtet wurde.
Infektionen
Die Infektionsrate in den gepoolten Daten aus den 12‑wöchigen Induktionsstudien betrug 83,3 Ereignisse pro 100 Patientenjahre bei Patienten, die mit Risankizumab 600 mg intravenös behandelt wurden, im Vergleich zu 117,7 Ereignissen pro 100 Patientenjahre unter Placebo. Die Rate schwerwiegender Infektionen betrug 3,4 Ereignisse pro 100 Patientenjahre bei Patienten, die mit Risankizumab 600 mg intravenös behandelt wurden, im Vergleich zu 16,7 Ereignissen pro 100 Patientenjahre unter Placebo (siehe Abschnitt 4.4).
Die Infektionsrate in der 52‑wöchigen Erhaltungsstudie betrug 57,7 Ereignisse pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase mit Risankizumab 360 mg subkutan behandelt wurden, im Vergleich zu 76,0 Ereignissen pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase Placebo erhielten. Die Rate schwerwiegender Infektionen betrug 6,0 Ereignisse pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase mit Risankizumab 360 mg subkutan behandelt wurden, im Vergleich zu 5,0 Ereignissen pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase Placebo erhielten (siehe Abschnitt 4.4).
Colitis ulcerosa
Insgesamt entsprach das Sicherheitsprofil bei Patienten mit Colitis ulcerosa unter Risankizumab dem Sicherheitsprofil, das bei Patienten aller Indikationen beobachtet wurde.
Infektionen
Die Infektionsrate in den gepoolten Daten aus der 12‑wöchigen Induktionsstudie betrug 78,3 Ereignisse pro 100 Patientenjahre bei Patienten, die mit Risankizumab 1 200 mg intravenös behandelt wurden, im Vergleich zu 74,2 Ereignissen pro 100 Patientenjahre unter Placebo. Die Rate schwerwiegender Infektionen betrug 3,0 Ereignisse pro 100 Patientenjahre bei Patienten, die mit Risankizumab 1 200 mg intravenös behandelt wurden, im Vergleich zu 5,4 Ereignissen pro 100 Patientenjahre unter Placebo (siehe Abschnitt 4.4).
Die Infektionsrate in der 52‑wöchigen Erhaltungsstudie betrug 67,4 Ereignisse pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase mit Risankizumab 180 mg subkutan behandelt wurden, und 56,5 Ereignisse pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase mit Risankizumab 360 mg subkutan behandelt wurden, im Vergleich zu 64,6 Ereignissen pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase Placebo erhielten. Die Rate schwerwiegender Infektionen betrug 1,1 Ereignisse pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase mit Risankizumab 180 mg subkutan behandelt wurden, und 0,6 Ereignisse pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase mit Risankizumab 360 mg subkutan behandelt wurden, im Vergleich zu 2,3 Ereignissen pro 100 Patientenjahre bei Patienten, die nach der Risankizumab-Induktionsphase Placebo erhielten (siehe Abschnitt 4.4).
Immunogenität
Bei Patienten mit Morbus Crohn, die in klinischen Studien zu MC bis zu 64 Wochen Risankizumab in der empfohlenen intravenösen Induktions- und subkutanen Erhaltungsdosis erhielten, wurden bei 3,4 % (2/58) bzw. bei 0 % (0/58) der beurteilten Patienten unter Behandlung auftretende Antikörper gegen den Wirkstoff und neutralisierende Antikörper nachgewiesen.
Bei Patienten mit Colitis ulcerosa, die in klinischen Studien zu Colitis ulcerosa bis zu 64 Wochen Risankizumab in der empfohlenen intravenösen Induktions- und subkutanen Erhaltungsdosis (180 mg oder 360 mg) erhielten, wurden unter 180 mg subkutan bei 8,9 % (8/90) bzw. 6,7 % (6/90) und unter 360 mg subkutan bei 4,4 % (4/91) bzw. 2,2 % (2/91) der beurteilten Patienten unter Behandlung auftretende Antikörper gegen den Wirkstoff und neutralisierende Antikörper nachgewiesen.
Antikörper gegen Risankizumab, auch neutralisierende Antikörper, waren nicht mit Veränderungen des klinischen Ansprechens oder der Sicherheit assoziiert.
Ältere Patienten
Bisher liegen nur begrenzte Erfahrungen zu Patienten ≥ 65 Jahre vor.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem anzuzeigen:
Deutschland
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51 – 59
63225 Langen
Tel: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
Im Falle einer Überdosierung wird empfohlen, den Patienten auf Anzeichen und Symptome von Nebenwirkungen zu überwachen und umgehend eine geeignete symptomatische Behandlung einzuleiten.
Pharmakotherapeutische Gruppe: Immunsuppressiva, Interleukin-Inhibitoren, ATC‑Code: L04AC18
Wirkmechanismus
Risankizumab ist ein humanisierter monoklonaler IgG1-Antikörper, der selektiv mit hoher Affinität an die p19‑Untereinheit des humanen Interleukin 23 (IL‑23) bindet, ohne an IL‑12 zu binden, und dessen Interaktion mit dem IL‑23-Rezeptorkomplex hemmt. IL‑23 ist ein Zytokin, das an Entzündungs- und Immunreaktionen beteiligt ist. Durch die Hemmung der Bindung von IL‑23 an seinen Rezeptor hemmt Risankizumab die IL‑23-abhängige Signaltransduktion und die Freisetzung proinflammatorischer Zytokine.
Pharmakodynamische Wirkungen
In einer Studie mit Psoriasis-Patienten war die Expression von Genen in der Haut, die mit der IL‑23/IL‑17-Achse in Zusammenhang stehen, nach Einzeldosen von Risankizumab vermindert. Bei den psoriatischen Läsionen wurden auch eine Verringerung der Epidermisdicke, der Infiltration von Entzündungszellen und der Expression psoriatischer Krankheitsmarker beobachtet.
In einer Phase‑II-Studie mit MC‑Patienten war die Expression von Genen, die mit der IL‑23/Th17‑Achse assoziiert sind, im Darmgewebe nach Mehrfachgabe von Risankizumab vermindert. In Phase‑III-Induktionsstudien bei Morbus Crohn wurde nach Mehrfachgabe auch eine Verringerung von fäkalem Calprotectin (FCP), C‑reaktivem Protein (CRP) und IL‑22 beobachtet. Die Verringerung von FCP, CRP und IL‑22 im Serum wurde bis Woche 52 der Erhaltungsstudie beibehalten.
In einer Phase‑IIb/III-Studie mit Patienten mit Colitis ulcerosa wurde in Woche 12 der Induktionsstudie eine statistisch signifikante und klinisch bedeutsame Verringerung der entzündlichen Biomarker FCP und CRP sowie des mit dem IL‑23-Signalweg assoziierten Biomarkers IL‑22 im Serum gegenüber Baseline beobachtet. Die Verringerung von FCP, CRP und IL‑22 im Serum wurde bis Woche 52 der Erhaltungsstudie beibehalten.
Klinische Wirksamkeit und Sicherheit
Morbus Crohn
Die Wirksamkeit und Sicherheit von Risankizumab wurden bei 1 419 Patienten mit mittelschwerem bis schwerem aktivem Morbus Crohn in drei multizentrischen, randomisierten, doppelblinden, placebokontrollierten klinischen Studien untersucht. Die aufgenommenen Studienteilnehmenden waren mindestens 16 Jahre alt und wiesen einen Crohn’s Disease Activity Index (CDAI) von 220 bis 450, eine durchschnittliche tägliche Stuhlfrequenz (SF) von ≥ 4 und/oder einen durchschnittlichen täglichen Score für Bauchschmerzen (BS) von ≥ 2 sowie einen Simple Endoscopic Score for CD (SES‑CD) von ≥ 6 (bzw. ≥ 4 bei isolierter Erkrankung des Ileums) auf, abgesehen von einer vorliegenden Verengungskomponente und von einem zentralen Prüfer bestätigt.
Es gab zwei 12‑wöchige intravenöse Induktionsstudien (ADVANCE und MOTIVATE), die eine 12‑wöchige Verlängerungsphase für Patienten umfassten, die in Woche 12 kein klinisches SF/BS‑Ansprechen erreichten (≥ 30%ige Verringerung der SF und/oder ≥ 30%ige Verringerung der BS und beide nicht schlechter als bei Baseline). Auf ADVANCE und MOTIVATE folgte eine 52‑wöchige randomisierte Absetzstudie mit subkutaner Erhaltungstherapie (FORTIFY), in die Patienten mit einem klinischen SF/BS-Ansprechen auf die intravenöse Induktionstherapie eingeschlossen wurden, die somit auf eine Behandlungsdauer von mindestens 64 Wochen kamen.
ADVANCE und MOTIVATE
In den Studien ADVANCE und MOTIVATE wurden die Studienteilnehmenden randomisiert und erhielten in Woche 0, Woche 4 und Woche 8 entweder Risankizumab in einer Dosis von 600 mg (empfohlene Dosis) oder 1 200 mg oder Placebo.
In ADVANCE hatten 58 % (491/850) der Studienteilnehmenden nicht auf eine Behandlung mit einem oder mehreren Biologika angesprochen oder diese nicht vertragen (vorheriges Versagen der Biologikatherapie) und 42 % (359/850) hatten nicht auf eine Behandlung mit konventionellen Therapien, aber nicht mit Biologika (kein vorheriges Versagen der Biologikatherapie) angesprochen oder diese nicht vertragen. In ADVANCE waren von den Studienteilnehmenden ohne vorheriges Versagen der Biologikatherapie 87 % (314/359) noch nicht mit einem Biologikum behandelt worden und die restlichen 13 % hatten ein Biologikum erhalten, wiesen jedoch kein Behandlungsversagen und keine Unverträglichkeit auf. Alle Patienten in MOTIVATE hatten ein vorheriges Versagen der Biologikatherapie.
In beiden Studien erreichte ein größerer Anteil der mit Risankizumab behandelten Patienten in Woche 12 die koprimären Endpunkte einer klinischen Remission und eines endoskopischen Ansprechens als unter Placebo. Ein erweitertes klinisches SF/BS-Ansprechen und eine klinische Remission waren bei den mit Risankizumab behandelten Patienten bereits in Woche 4 signifikant und verbesserten sich bis Woche 12 weiter (Tabelle 2).
Tabelle 2. Wirksamkeitsergebnisse in den Studien ADVANCE und MOTIVATE
ADVANCE |
MOTIVATE |
||||||
|
Placebo intravenös (n = 175) % |
Risankizumab |
Behandlungs-unterschiedd (95 %‑CI) |
Placebo intravenös (n = 187) % |
Risankizumab 600 mg intravenös (n = 191) % |
Behandlungs-unterschiedd (95 %‑CI) |
|
Koprimäre Endpunkte | |||||||
Klinische Remission in Woche 12e |
22 % |
43 % |
22 % |
19 % |
35 % |
15 % |
|
Endoskopisches Ansprechen in Woche 12f |
12 % |
40 % |
28 % |
11 % |
29 % |
18 % |
|
Weitere Endpunkte | |||||||
Erweitertes klinisches SF/BS-Ansprechen in Woche 4g |
31 % |
46 % |
15 % |
32 % |
45 % |
14 % |
|
Erweitertes klinisches SF/BS-Ansprechen in Woche 12g |
42 % |
63 % |
21 % |
39 % |
62 % |
23 % |
|
CDAI < 150 in Woche 4 |
10 % |
18 % |
8 % |
11 % |
21 % |
10 % |
|
CDAI < 150 in Woche 12 |
25 % |
45 % |
21 % |
20 % |
42 % |
22 % |
|
Mukosaheilung in Woche 12h |
(n = 173) |
(n = 336) |
14 % |
(n = 186) |
(n = 190) |
9 % |
|
Endoskopische Remission in Woche 12i |
9 % |
24 % |
15 % |
4 % |
19 % |
15 % |
|
a Statistisch signifikant unter Multiplizitätskontrolle für den Vergleich von Risankizumab mit Placebo (p < 0,001) | |||||||
In Woche 12 erreichte ein größerer Anteil der mit Risankizumab behandelten Patienten eine Verringerung des CDAI um mindestens 100 Punkte gegenüber Baseline als unter Placebo (ADVANCE: Risankizumab = 60 %, Placebo = 37 %, p < 0,001; MOTIVATE: Risankizumab = 60 %, Placebo = 30 %, p < 0,001).
In Woche 12 erreichte ein größerer Anteil der mit Risankizumab behandelten Patienten sowohl ein erweitertes klinisches SF/BS-Ansprechen als auch ein endoskopisches Ansprechen als unter Placebo (ADVANCE: Risankizumab = 31 %, Placebo = 8 %, p < 0,001; MOTIVATE: Risankizumab = 21 %, Placebo = 7 %, p < 0,001).
Die Ergebnisse für die koprimären Endpunkte für die Untergruppen (ohne Berücksichtigung von Multiplizität) von Patienten mit und ohne vorheriges Versagen der Biologikatherapie sind in Tabelle 3 dargestellt.
Tabelle 3. Wirksamkeitsergebnisse in Woche 12 in den Untergruppen von Studienteilnehmenden mit vorherigem Versagen der Biologikatherapie und bei Patienten ohne vorheriges Versagen der Biologikatherapie in ADVANCE
ADVANCE |
|||
Placebo intravenös |
Risankizumab 600 mg |
Behandlungsunterschied (95 %‑CI) |
|
Klinische Remission gemäß SF/BS-Score | |||
Vorheriges Versagen der Biologikatherapie |
23 % (n = 97) |
41 % (n = 195) |
18 % [7 %, 29 %] |
Kein vorheriges Versagen der Biologikatherapie |
21 % (n = 78) |
48 % (n = 141) |
27 % [15 %, 39 %] |
Endoskopisches Ansprechen | |||
Vorheriges Versagen der Biologikatherapie |
11 % (n = 97) |
33 % (n = 195) |
21 % [12 %, 31 %] |
Kein vorheriges Versagen der Biologikatherapie |
13 % (n = 78) |
50 % (n = 141) |
38 % [27 %, 49 %] |
In ADVANCE erreichte ein größerer Anteil der mit Risankizumab behandelten Patienten mit und ohne vorheriges Versagen einer Biologikatherapie einen CDAI < 150 als unter Placebo (mit vorherigem Versagen der Biologikatherapie: Risankizumab = 42 %, Placebo = 26 %; kein vorheriges Versagen der Biologikatherapie: Risankizumab = 49 %, Placebo = 23 %).
MC‑bedingte Hospitalisierungen
Die Raten der MC‑bedingten Hospitalisierungen bis Woche 12 waren bei den mit Risankizumab behandelten Studienteilnehmenden niedriger als unter Placebo (ADVANCE, Risankizumab = 3 %, Placebo = 12 %, p < 0,001; MOTIVATE, Risankizumab = 3 %, Placebo = 11 %, p ≤ 0,01).
FORTIFY
In der Erhaltungsstudie FORTIFY wurden 462 Studienteilnehmende mit klinischem SF/BS-Ansprechen auf eine 12‑wöchige Induktionstherapie mit Risankizumab intravenös in den Studien ADVANCE und MOTIVATE untersucht. Die Studienteilnehmenden wurden randomisiert und erhielten entweder weiterhin ein Erhaltungsschema mit Risankizumab 360 mg subkutan (empfohlene Dosis) oder Risankizumab 180 mg subkutan alle 8 Wochen oder sie setzten die Induktionstherapie mit Risankizumab ab und erhielten bis zu 52 Wochen lang alle 8 Wochen Placebo subkutan.
Die koprimären Endpunkte waren die klinische Remission in Woche 52 und das endoskopische Ansprechen in Woche 52. Koprimäre Endpunkte wurden auch bei Studienteilnehmenden mit und ohne vorheriges Versagen einer Biologikatherapie beurteilt (siehe Tabelle 4).
Tabelle 4. Wirksamkeitsergebnisse in FORTIFY in Woche 52 (64 Wochen nach Einleitung der Induktionsdosis)
FORTIFY |
|||
|
Induktion mit Risankizumab intravenös/Placebo subkutanf |
Induktion mit Risankizumab intravenös/Risankizumab |
Behandlungsunterschied (95 %‑CI) |
Koprimäre Endpunkte | |||
Klinische Remission |
40 % |
52 % |
15 % [5 %, 25 %]a,g |
Vorheriges Versagen der Biologikatherapie |
34 % (n = 123) |
48 % (n = 102) |
14 % [1 %, 27 %] |
Kein vorheriges Versagen der Biologikatherapie |
56 % (n = 41) |
62 % (n = 39) |
5 % [-16 %, 27 %] |
Endoskopisches Ansprechen |
22 % |
47 % |
28 % [19 %, 37 %]b,g |
Vorheriges Versagen der Biologikatherapie |
20 % (n = 123) |
44 % (n = 102) |
23 % [11 %, 35 %] |
Kein vorheriges Versagen der Biologikatherapie |
27 % (n = 41) |
54 % (n = 39) |
27 % [6 %, 48 %] |
Weitere Endpunkte | |||
Erweitertes klinisches SF/BS-Ansprechen |
49 % |
59 % |
13 % [2 %, 23 %]e,g |
Aufrechterhaltung der klinischen Remissionh |
(n = 91) |
(n = 72) |
21 % [6 %, 35 %]d,g |
Endoskopische Remission |
13 % |
39 % |
28 % [20 %, 37 %]c,g |
Mukosaheilung |
(n = 162) |
(n = 141) |
22 % [14 %, 30 %]c,g |
a Statistisch signifikant unter Multiplizitätskontrolle für den Vergleich von Risankizumab mit Placebo (p ≤ 0,01). | |||
Eine tiefe Remission (klinische Remission und endoskopische Remission) wurde in Woche 52 bei den mit Risankizumab intravenös/Risankizumab subkutan behandelten Studienteilnehmenden häufiger beobachtet als bei Studienteilnehmenden, die Risankizumab intravenös/Placebo subkutan erhielten (28 % bzw. 10 %, nominal p < 0,001).
In Woche 52 erreichte ein größerer Anteil der mit Risankizumab intravenös/Risankizumab subkutan behandelten Patienten einen CDAI < 150 als unter Risankizumab intravenös/Placebo subkutan (52 % bzw. 41 %, nominal p ≤ 0,01). Ein größerer Anteil der mit Risankizumab intravenös/Risankizumab subkutan behandelten Studienteilnehmenden erreichte eine Verringerung des CDAI um mindestens 100 Punkte gegenüber Baseline im Vergleich zu den mit Risankizumab intravenös/Placebo subkutan behandelten Studienteilnehmenden (62 % bzw. 48 %, nominal p ≤ 0,01).
In den Studien ADVANCE und MOTIVATE erhielten 91 Patienten, die nach einer 12‑wöchigen Induktionstherapie mit Risankizumab kein klinisches SF/BS-Ansprechen zeigten, in Woche 12 und Woche 20 eine Dosis Risankizumab 360 mg subkutan Von diesen Studienteilnehmenden erreichten 64 % (58/91) in Woche 24 ein klinisches SF/BS-Ansprechen. 33 der Studienteilnehmenden, die ein klinisches SF/BS-Ansprechen erreichten, wurden in FORTIFY aufgenommen und erhielten bis zu 52 Wochen lang alle 8 Wochen Risankizumab 360 mg subkutan. Von diesen Studienteilnehmenden erreichten 55 % (18/33) in Woche 52 eine klinische Remission und 45 % (15/33) ein endoskopisches Ansprechen.
Während FORTIFY hatten 30 Studienteilnehmende einen Verlust des Ansprechens auf die Behandlung mit Risankizumab 360 mg subkutan und erhielten eine Notfallbehandlung mit Risankizumab (eine Einmaldosis 1 200 mg intravenös, gefolgt von 360 mg subkutan alle 8 Wochen). Von diesen Studienteilnehmenden erreichten 57 % (17/30) in Woche 52 ein klinisches SF/BS-Ansprechen. Darüber hinaus erreichten 20 % (6/30) bzw. 34 % (10/29) der Studienteilnehmenden in Woche 52 eine klinische Remission bzw. ein endoskopisches Ansprechen.
Ergebnisse zur (gesundheitsbezogenen) Lebensqualität
Die gesundheitsbezogene Lebensqualität wurde anhand des Inflammatory Bowel Disease Questionnaire (IBDQ) und des SF‑36-Fragebogens beurteilt. Die Verbesserung im Hinblick auf Fatigue wurde anhand der Skala des Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT‑F) gemessen. Die Arbeitsproduktivität wurde anhand des Work Productivity and Activity Impairment CD (WPAI-CD)-Fragebogens beurteilt.
In Woche 12 von ADVANCE und MOTIVATE erreichten die mit Risankizumab behandelten Studienteilnehmenden im Vergleich zu Placebo klinisch bedeutsame Verbesserungen gegenüber Baseline im Hinblick auf den IBDQ-Gesamtscore, alle IBDQ-Bereichsscores (Darmsymptome, systemische Funktion, emotionale Funktion und soziale Funktion), die körperlichen und psychischen Komponenten des SF‑36, FACIT‑F und WPAI-CD. Für WPAI-CD wurden größere Reduzierung der Beeinträchtigung während der Arbeit, der Beeinträchtigung der Arbeit insgesamt und der Beeinträchtigung der Aktivität in ADVANCE gezeigt. Eine stärkere Reduzierung der Beeinträchtigung der Aktivität wurde in MOTIVATE gezeigt. Diese Verbesserungen wurden bei den mit Risankizumab intravenös/Risankizumab subkutan behandelten Patienten in FORTIFY bis Woche 52 aufrechterhalten.
Colitis ulcerosa
Die Wirksamkeit und Sicherheit von Risankizumab wurden bei Patienten mit mittelschwerer bis schwerer aktiver Colitis ulcerosa in zwei multizentrischen, randomisierten, doppelblinden, placebokontrollierten klinischen Studien untersucht. Die aufgenommenen Studienteilnehmenden waren ≥ 18 und ≤ 80 Jahre alt und wiesen einen angepassten Mayo-Score (aMS) von 5 bis 9 (unter Verwendung des Mayo-Scoring-Systems, ohne die allgemeine Beurteilung durch den Arzt [Physician’s Global Assessment, PGA]) mit einem Endoskopie-Subscore (ES) von 2 oder 3 beim Screening auf, der durch ein zentrales Gutachten bestätigt wurde.
Die 12‑wöchige intravenöse Induktionsstudie (INSPIRE) umfasste eine 12‑wöchige Fortsetzungsphase für Studienteilnehmende, die in Woche 12 kein klinisches Ansprechen erreichten (definiert als Verringerung des aMS um ≥ 2 Punkte und ≥ 30 % gegenüber Baseline sowie Verringerung des Subscores zur Darmblutung [rectal bleeding score, RBS] um ≥ 1 oder absoluter RBS von ≤ 1). Auf INSPIRE folgte eine 52‑wöchige randomisierte Absetzstudie mit subkutaner Erhaltungstherapie (COMMAND), in die Patienten mit einem klinischen Ansprechen auf die 12‑wöchige intravenöse Induktionstherapie mit Risankizumab eingeschlossen wurden, die somit auf eine Behandlungsdauer von mindestens 64 Wochen kamen.
INSPIRE
In der Studie INSPIRE wurden 975 Studienteilnehmende randomisiert und erhielten in Woche 0, Woche 4 und Woche 8 entweder Risankizumab 1 200 mg oder Placebo.
In der INSPIRE-Studie zeigten 52 % (503/975) der Studienteilnehmenden ein Versagen (unzureichendes Ansprechen oder Unverträglichkeit) auf eine oder mehrere Biologikatherapien, JAK‑Inhibitoren und/oder S1P‑Rezeptormodulatoren. Von diesen 503 Studienteilnehmenden zeigten 488 (97 %) ein Versagen der Behandlung bei Biologika und 90 (18 %) bei JAK‑Inhibitoren.
In die Studie aufgenommenen Patienten war es gestattet, eine stabile Dosis oraler Corticosteroide (bis zu 20 mg/Tag Prednison oder Äquivalent), Immunsuppressiva und Aminosalizylate anzuwenden. Bei Baseline der INSPIRE-Studie erhielten 36 % der Studienteilnehmenden Corticosteroide, 17 % Immunmodulatoren und 73 % Aminosalizylate. Die Krankheitsaktivität war bei 58 % der Studienteilnehmenden mittelschwer (aMS ≤ 7) und bei 42 % schwer (aMS > 7).
In der INSPIRE-Studie erreichte in Woche 12 im Vergleich zu Placebo ein signifikant größerer Anteil der mit Skyrizi behandelten Patienten den primären Endpunkt einer klinischen Remission gemäß aMS (definiert als Subscore zur Stuhlfrequenz [SFS] ≤ 1 und nicht höher als bei Baseline, RBS = 0 und ES ≤ 1 ohne Hinweise auf Friabilität, Tabelle 5). Die Ergebnisse zum primären Endpunkt und zu den wichtigsten sekundären Endpunkten sind in Tabelle 5 aufgeführt.
Tabelle 5. Wirksamkeitsergebnisse in INSPIRE in Woche 12
Endpunkt |
Placebo intravenös |
Risankizumab |
Behandlungsunterschied |
Krankheitsaktivität und CU‑Symptome | |||
Klinische Remissiona,b |
6 % |
20 % |
14 %f |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
4 % (n = 170) |
11 % (n = 333) |
7 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
8 % (n = 155) |
30 % (n = 317) |
21 % |
Klinisches Ansprechenc |
36 % |
64 % |
29 %f |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
31 % (n = 170) |
55 % (n = 333) |
24 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
41 % (n = 155) |
74 % (n = 317) |
33 % |
Endoskopische und histologische Untersuchung | |||
Mukosaheilungd |
12 % |
37 % |
24 %f |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
10 % (n = 170) |
26 % (n = 333) |
16 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
14 % (n = 155) |
48 % (n = 317) |
33 % |
Histologisch-endoskopische Mukosaheilunge |
8 % |
24 % |
17 %f |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
7 % (n = 170) |
16 % (n = 333) |
9 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
8 % (n = 155) |
33 % (n = 317) |
25 % |
a Primärer Endpunkt | |||
Klinische Krankheitsaktivität und Symptome
Der partielle adaptierte Mayo-Score (paMS) setzt sich aus SFS und RBS zusammen. Klinisches Ansprechen gemäß paMS wird definiert als Verringerung um ≥ 1 Punkt und ≥ 30 % gegenüber Baseline und eine Verringerung des RBS um ≥ 1 oder ein absoluter RBS von ≤ 1. Die Ergebnisse zum klinischen Ansprechen gemäß paMS im Zeitverlauf in der INSPIRE-Studie sind in Abbildung 1 dargestellt. Die Wirksamkeit trat schnell ein, wobei ein größerer Anteil der mit Risankizumab behandelten Studienteilnehmenden bereits in Woche 4 ein klinisches Ansprechen erreichte als unter Placebo (52 % vs. 31 %, p < 0,00001).
Abbildung 1. Anteil der Patienten mit klinischem Ansprechen gemäß paMS im Zeitverlauf in der Induktionsstudie INSPIRE
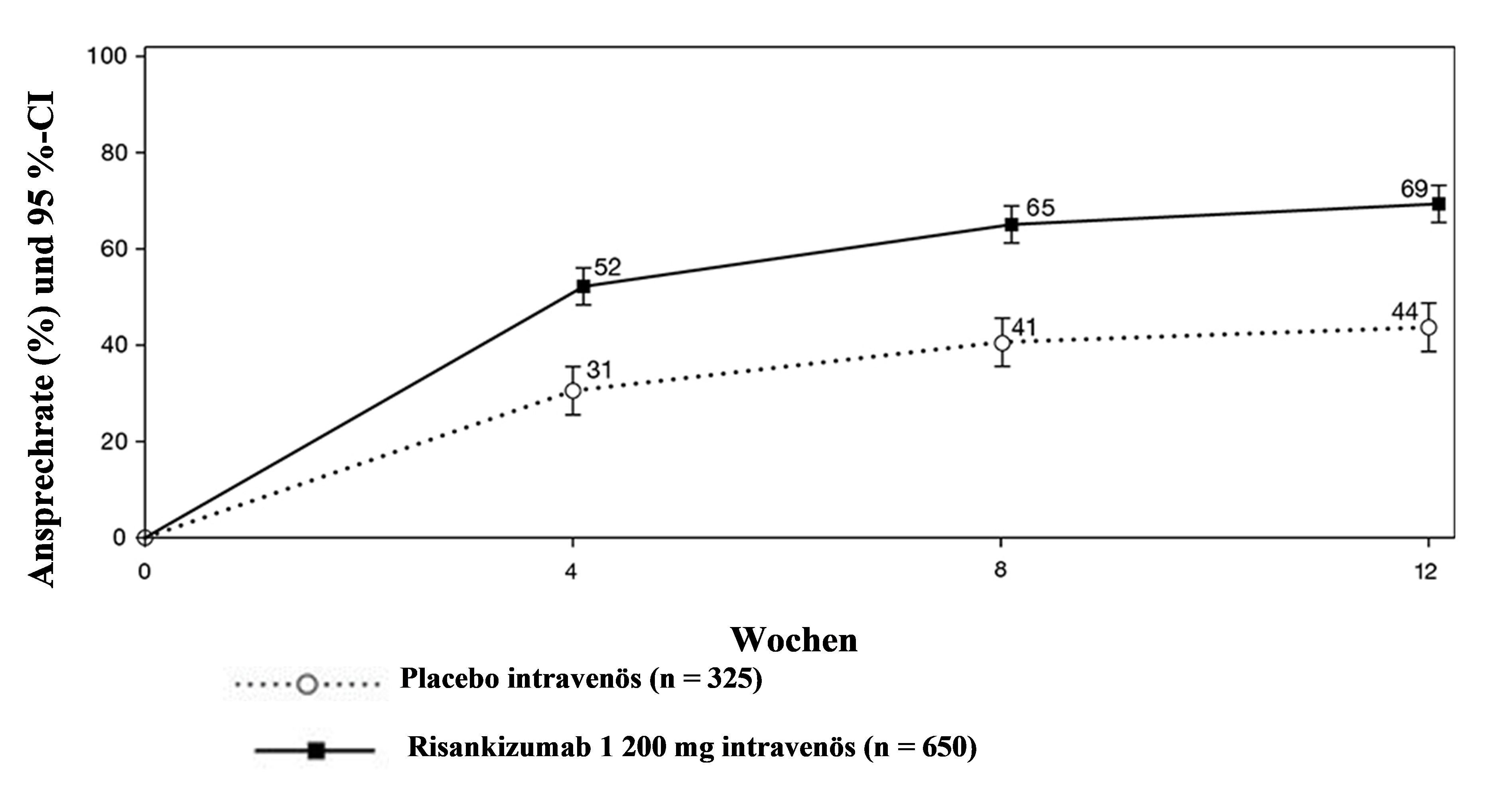
Ein signifikant größerer Anteil der mit Risankizumab behandelten Patienten hatte in Woche 12 im Vergleich zu Placebo keine Bauchschmerzen (36 % vs. 26 %, p < 0,01) und keinen Stuhldrang (44 % vs. 28 %, p < 0,00001).
Weitere CU‑Symptome
Die Anzahl der Stuhlinkontinenzepisoden pro Woche war in Woche 12 bei mit Risankizumab behandelten Patienten im Vergleich zu Placebo signifikant stärker reduziert (Veränderung gegenüber Baseline bei Risankizumab = ‑3,8, Placebo = ‑2,2, p = 0,00003).
Der Anteil der Patienten, die in Woche 12 keinen nächtlichen Stuhlgang hatten, war unter Risankizumab im Vergleich zu Placebo signifikant höher (67 % vs. 43 %, p < 0,00001).
Der Anteil der Patienten, die in Woche 12 keinen Tenesmus aufwiesen, war unter Risankizumab im Vergleich zu Placebo signifikant höher (49 % vs. 30 %, p < 0,00001).
Die Anzahl der Tage pro Woche mit Schlafstörungen aufgrund von CU‑Symptomen war in Woche 12 bei den mit Risankizumab behandelten Patienten im Vergleich zu Placebo signifikant stärker reduziert (Veränderung gegenüber Baseline bei Risankizumab = ‑2,5, Placebo = ‑1,5, p < 0,00001).
CU‑bedingte Hospitalisierung
Die Raten der CU‑bedingten Hospitalisierungen bis Woche 12 waren bei den mit Risankizumab behandelten Patienten signifikant niedriger als unter Placebo (1 % vs. 6 %, p < 0,00001).
Fortgesetzte Behandlung bei Non-Respondern in Woche 12
Insgesamt 141 Studienteilnehmende, die in Woche 12 der Risankizumab-Induktionsphase in der INSPIRE-Studie kein klinisches Ansprechen aufwiesen, erhielten in Woche 12 und Woche 20 subkutan entweder eine 180‑mg‑ oder 360‑mg-Dosis Risankizumab. Von den 71 Studienteilnehmenden, die Risankizumab 180 mg subkutan erhielten, bzw. von den 70, die 360 mg subkutan erhielten, erreichten in Woche 24 56 % bzw. 57 % ein klinisches Ansprechen.
COMMAND
In der Erhaltungsstudie COMMAND wurden 548 Studienteilnehmende mit klinischem Ansprechen nach 12‑wöchiger Induktionstherapie mit Risankizumab intravenös in der Studie INSPIRE untersucht. Die Studienteilnehmenden wurden randomisiert und erhielten ein Erhaltungsschema mit Risankizumab 180 mg subkutan oder 360 mg subkutan alle 8 Wochen oder sie setzten die Induktionstherapie mit Risankizumab ab und erhielten bis zu 52 Wochen lang alle 8 Wochen Placebo subkutan.
In der Studie COMMAND zeigten 75 % (411/548) der Studienteilnehmenden vor Beginn der Induktionstherapie ein Versagen (unzureichendes Ansprechen oder Unverträglichkeit) auf eine oder mehrere Biologikatherapien, JAK‑Inhibitoren und/oder S1P‑Rezeptormodulatoren. Von diesen 411 Studienteilnehmenden zeigten 407 (99 %) ein Versagen bei Biologika und 78 (19 %) bei JAK‑Inhibitoren.
In der Studie COMMAND erreichte in Woche 52 ein signifikant größerer Anteil der oben genannten 548 mit Risankizumab 180 mg subkutan bzw. Risankizumab 360 mg subkutan behandelten Studienteilnehmenden den primären Endpunkt der klinischen Remission gemäß aMS als unter Placebo (siehe Tabelle 6). Die Ergebnisse zum primären Endpunkt und zu den wichtigsten sekundären Endpunkten sind in Tabelle 6 aufgeführt.
Tabelle 6. Wirksamkeitsergebnisse in COMMAND in Woche 52 (64 Wochen nach Einleitung der Induktionsdosis)
Endpunkt |
Induktion mit Risankizumab intravenös/ |
Induktion mit Risankizumab intravenös/ |
Induktion mit Risankizumab intravenös/ |
Behandlungsunterschied |
|
Induktion mit Risankizumab intravenös/ |
Induktion mit Risankizumab intravenös/ |
||||
Krankheitsaktivität und CU‑Symptome | |||||
Klinische Remissiona,b |
25 % |
40 % |
38 % |
16 %h |
14 %h |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
23 % (n = 138) |
37 % (n = 134) |
29 % (n = 139) |
13 % |
6 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
31 % (n = 45) |
51 % (n = 45) |
62 % (n = 47) |
20 % |
31 % |
Aufrechterhaltung der klinischen Remissionc |
40 % (n = 53) |
70 % (n = 44) |
50 % (n = 40) |
29 %h |
13 %k |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
37 % (n = 35) |
65 % (n = 26) |
44 % (n = 25) |
28 % |
7 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
44 % (n = 18) |
77 % (n = 18) |
60 % (n = 15) |
33 % |
16 % |
Corticosteroidfreie klinische Remissiond |
25 % |
40 % |
37 % |
16 %h |
14 %h |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
23 % (n = 138) |
36 % (n = 134) |
29 % (n = 139) |
13 % |
6 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
31 % (n = 45) |
51 % (n = 45) |
60 % (n = 47) |
20 % |
28 % |
Klinisches Ansprechene |
52 % |
68 % |
62 % |
17 %i |
11 %j |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
46 % (n = 138) |
63 % (n = 134) |
57 % (n = 139) |
18 % |
11 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
71 % (n = 45) |
82 % (n = 45) |
79 % (n = 47) |
11 % |
8 % |
Endoskopische und histologische Untersuchung | |||||
Mukosaheilungf |
32 % |
51 % |
48 % |
20 %h |
17 %h |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
30 % (n = 138) |
48 % (n = 134) |
39 % (n = 139) |
17 % |
8 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
36 % (n = 45) |
60 % (n = 45) |
76 % (n = 47) |
24 % |
41 % |
Histologisch-endoskopische Mukosaheilungg |
23 % |
43 % |
42 % |
20 %h |
20 %h |
Mit Versagen von Biologika und/oder JAK-Inhibitoren |
22 % (n = 138) |
39 % (n = 134) |
33 % (n = 139) |
17 % |
11 % |
Ohne Versagen von Biologika und/oder JAK-Inhibitoren |
29 % (n = 45) |
55 % (n = 45) |
69 % (n = 47) |
26 % |
40 % |
+ Der Behandlungsarm, der nur die Induktionstherapie erhielt, bestand aus Studienteilnehmenden, die ein klinisches Ansprechen auf die Induktionstherapie mit Risankizumab erreichten und in der Erhaltungsstudie (COMMAND) zu Placebo randomisiert wurden. | |||||
Klinische Krankheitsaktivität und Symptome
Ein signifikant größerer Anteil der mit Risankizumab intravenös/Risankizumab 180 mg subkutan behandelten Patienten hatte in Woche 52 im Vergleich zu Risankizumab intravenös/Placebo keine Bauchschmerzen (47 % vs. 30 %, p < 0,001) und keinen Stuhldrang (54 % vs. 31 %, p < 0,00001). Ein größerer Anteil der mit Risankizumab intravenös/Risankizumab 360 mg subkutan behandelten Patienten hatte in Woche 52 im Vergleich zu Risankizumab intravenös/Placebo keinen Stuhldrang (49 % vs. 31 %, p < 0,001) und ein numerisch höherer Anteil keine Bauchschmerzen (38 % vs. 30 %, p = 0,0895).
Weitere CU‑Symptome
Der Anteil der Patienten, die in Woche 52 keinen nächtlichen Stuhlgang hatten, war unter Risankizumab intravenös/Risankizumab 180 mg subkutan bzw. Risankizumab intravenös/Risankizumab 360 mg subkutan im Vergleich zu Risankizumab intravenös/Placebo höher (42 % bzw. 43 % vs. 30 %, p < 0,01 bzw. p < 0,001).
Der Anteil der Patienten, die in Woche 52 keinen Tenesmus hatten, war unter Risankizumab intravenös/Risankizumab 180 mg subkutan bzw. Risankizumab intravenös/Risankizumab 360 mg subkutan im Vergleich zu Risankizumab intravenös/Placebo höher (37 % bzw. 37 % vs. 23 %, p < 0,01).
CU‑bedingte Hospitalisierung
Die Häufigkeit CU‑bedingter Hospitalisierungen bis Woche 52 war unter Risankizumab intravenös/Risankizumab 180 mg subkutan bzw. Risankizumab intravenös/Risankizumab 360 mg subkutan numerisch geringer als bei Studienteilnehmenden unter Risankizumab intravenös/Placebo (0,6 bzw. 1,2 pro 100 Patientenjahre vs. 3,1 pro 100 Patientenjahre, p = 0,0949 bzw. p = 0,2531).
Endoskopische und histologische Untersuchung
Die endoskopische Remission (Normalisierung des endoskopischen Erscheinungsbilds der Mukosa) wurde definiert als ES von 0. In Woche 12 der INSPIRE-Studie erreichte ein signifikant größerer Anteil der mit Risankizumab behandelten Patienten eine endoskopische Remission als unter Placebo (11 % vs. 3 %, p < 0,00001). In Woche 52 der COMMAND-Studie erreichte ein signifikant größerer Anteil der mit Risankizumab intravenös/Risankizumab 180 mg subkutan bzw. Risankizumab intravenös/Risankizumab 360 mg subkutan behandelten Patienten eine endoskopische Remission als unter Risankizumab intravenös/Placebo (23 % bzw. 24 % vs. 15 %, p < 0,05).
Tiefe Mukosaheilung wurde definiert als ES von 0 und Geboes-Score < 2,0 (weist darauf hin, dass keine Neutrophilen in Krypten oder der Lamina propria vorliegen und die Eosinophilen nicht erhöht sind, keine Zerstörung der Krypten und keine Erosionen, Ulzerationen oder Granulationsgewebe). In Woche 12 der INSPIRE-Studie erreichte ein signifikant größerer Anteil der mit Risankizumab behandelten Patienten eine tiefe Mukosaheilung als unter Placebo (6 % vs. 1 %, p < 0,00001). In Woche 52 der COMMAND-Studie erreichte ein numerisch höherer Anteil der mit Risankizumab intravenös/Risankizumab 180 mg subkutan bzw. Risankizumab intravenös/Risankizumab 360 mg subkutan behandelten Patienten eine tiefe Mukosaheilung als unter Risankizumab intravenös/Placebo (13 % bzw. 16 % vs. 10 %, p = 0,2062 bzw. p = 0,0618).
In der COMMAND-Studie wurde die Aufrechterhaltung der Mukosaheilung in Woche 52 (ES ≤ 1 ohne Friabilität) bei den Patienten, die am Ende der Induktionsphase eine Mukosaheilung erreichten, bei einem höheren Anteil der mit Risankizumab intravenös/Risankizumab 180 mg subkutan bzw. der mit Risankizumab intravenös/Risankizumab 360 mg subkutan behandelten Patienten beobachtet als unter Risankizumab intravenös/Placebo (74 % bzw. 54 % vs. 47 %, p < 0,01 bzw. p = 0,5629).
Rescue-Therapie
Während der COMMAND-Studie erhielten Studienteilnehmende mit einem Verlust des Ansprechens auf die Behandlung mit Risankizumab subkutan eine Notfallbehandlung mit Risankizumab (eine einzelne Induktionsdosis intravenös, gefolgt von 360 mg subkutan alle 8 Wochen). Von diesen Studienteilnehmenden erreichten in Woche 52 im Behandlungsarm mit Risankizumab 180 mg subkutan 85 % (17/20) und im Behandlungsarm mit Risankizumab 360 mg subkutan 74 % (26/35) ein klinisches Ansprechen. Darüber hinaus erreichten unter Risankizumab 180 mg subkutan bzw. Risankizumab 360 mg subkutan 24 % (6/25) bzw. 35 % (13/37) der Studienteilnehmenden in Woche 52 eine klinische Remission gemäß aMS und 38 % (10/26) bzw. 45 % (17/38) eine endoskopische Verbesserung.
Responder in Woche 24
Insgesamt 100 Studienteilnehmende zeigten nach 12‑wöchiger Induktionstherapie kein klinisches Ansprechen, erhielten entweder eine subkutane Dosis Risankizumab 180 mg (n = 56) oder 360 mg (n = 44) in Woche 12 und Woche 20, zeigten in Woche 24 ein klinisches Ansprechen und setzten die Behandlung mit Risankizumab 180 mg bzw. 360 mg subkutan alle 8 Wochen bis zu 52 Wochen lang in der COMMAND-Studie fort. Von diesen Studienteilnehmenden erreichten unter Risankizumab 180 mg bzw. 360 mg subkutan 46 % bzw. 45 % in Woche 52 ein klinisches Ansprechen gemäß aMS und 18 % bzw. 23 % in Woche 52 eine klinische Remission gemäß aMS.
Ergebnisse zur (gesundheitsbezogenen) Lebensqualität
Patienten, die mit Risankizumab behandelt wurden, erreichten im Vergleich zu Placebo seit Beginn der Behandlung (Baseline) klinisch bedeutsame Verbesserungen beim Inflammatory Bowel Disease Questionnaire (IBDQ) (Darmsymptome, systemische Funktion, emotionale Funktion und soziale Funktion). Veränderungen des IBDQ-Gesamtscores gegenüber Baseline betrugen in Woche 12 unter Risankizumab 42,6 gegenüber 24,3 unter Placebo. Veränderungen des IBDQ-Gesamtscores gegenüber Baseline betrugen in Woche 52 bei Patienten unter Risankizumab intravenös/Risankizumab 180 mg subkutan, Risankizumab intravenös/Risankizumab 360 mg subkutan und Risankizumab intravenös/Placebo 52,6, 50,3 bzw. 35,0.
In Woche 12 verzeichneten Patienten unter Risankizumab im Vergleich zu Patienten unter Placebo eine signifikant größere Verbesserung der Fatigue gegenüber Baseline, gemessen anhand des FACIT‑F-Scores. Veränderungen des FACIT‑F‐Scores gegenüber Baseline betrugen in Woche 12 unter Risankizumab 7,9 gegenüber 3,3 unter Placebo. Veränderungen des FACIT‑F-Gesamtscores gegenüber Baseline betrugen in Woche 52 bei Patienten unter Risankizumab intravenös/Risankizumab 180 mg subkutan, Risankizumab intravenös/Risankizumab 360 mg subkutan und Risankizumab intravenös/Placebo 10,9, 10,3 bzw. 7,0.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Skyrizi eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen zur Behandlung von Morbus Crohn und Colitis ulcerosa gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Pharmakokinetik von Risankizumab war vergleichbar zwischen Patienten mit Plaque-Psoriasis und Patienten mit Psoriasis-Arthritis sowie zwischen Patienten mit Morbus Crohn und Patienten mit Colitis ulcerosa.
Resorption
Risankizumab zeigte eine lineare Pharmakokinetik mit dosisproportionalem Anstieg bei Exposition über den Dosisbereich von 18 bis 360 mg und 0,25 bis 1 mg/kg subkutan sowie 200 bis 1 800 mg und 0,01 bis 5 mg/kg intravenös.
Nach subkutaner Gabe von Risankizumab wurde die maximale Plasmakonzentration 3–14 Tage nach der Anwendung erreicht, mit einer geschätzten absoluten Bioverfügbarkeit von 74–89 %. Bei einer Dosierung von 150 mg in Woche 0, Woche 4 und danach alle 12 Wochen beträgt die geschätzte maximale Plasmakonzentration im Steady State 12 µg/ml und der Talspiegel 2 µg/ml.
Bei Studienteilnehmenden mit Morbus Crohn, die mit einer Induktionsdosis von 600 mg intravenös in Woche 0, 4 und 8, gefolgt von einer Erhaltungsdosis von 360 mg subkutan in Woche 12 und danach alle 8 Wochen behandelt wurden, werden die maximalen Peak‑ und Talkonzentrationen im Median während der Induktionsphase (Woche 8–12) auf 156 bzw. 38,8 µg/ml und die Peak‑ und Talkonzentrationen im Median im Steady State während der Erhaltungsphase (Woche 40–48) auf 28,0 bzw. 8,13 µg/ml geschätzt.
Bei Studienteilnehmenden mit Colitis ulcerosa, die mit einer Induktionsdosis von 1 200 mg intravenös in Woche 0, 4, und 8 gefolgt von einer Erhaltungsdosis von 180 mg oder 360 mg subkutan in Woche 12 und danach alle 8 Wochen behandelt wurden, werden die maximalen Peak‑ und Talkonzentrationen im Median während der Induktionsphase (Woche 8–12) auf 350 bzw. 87,7 μg/ml und die Peak‑ und Talkonzentrationen im Median im Steady State während der Erhaltungsphase (Woche 40–48) auf 19,6 bzw. 4,64 μg/ml für die Dosis 180 mg subkutan und 39,2 bzw. 9,29 μg/ml für die Dosis 360 mg subkutan geschätzt.
Verteilung
Das mittlere Verteilungsvolumen (± Standardabweichung) im Steady State (Vss) von Risankizumab betrug in Phase‑III-Studien bei Patienten mit Psoriasis 11,4 (± 2,7) l. Dies deutet darauf hin, dass die Verteilung von Risankizumab hauptsächlich auf den vaskulären und den interstitiellen Raum beschränkt ist. Bei einem typischen Morbus-Crohn-Patienten mit einem Körpergewicht von 70 kg lag das Vss bei 7,68 l.
Biotransformation
Therapeutische monoklonale IgG‑Antikörper werden in der Regel analog zu endogenen IgGs über katabole Wege zu kleinen Peptiden und Aminosäuren abgebaut. Es ist nicht zu erwarten, dass Risankizumab durch Cytochrom-P450-Enzyme verstoffwechselt wird.
Elimination
Die mittlere (± Standardabweichung) systemische Clearance (CL) von Risankizumab betrug in Phase‑III-Studien bei Patienten mit Psoriasis 0,3 (± 0,1) l/Tag. Die mittlere terminale Eliminationshalbwertszeit von Risankizumab lag in Phase‑III-Studien bei Studienteilnehmenden mit Psoriasis zwischen 28 und 29 Tagen. Bei einem typischen Morbus-Crohn-Patienten mit einem Körpergewicht von 70 kg betrug die CL 0,30 l/Tag und die terminale Eliminationshalbwertszeit 21 Tage.
Es ist nicht zu erwarten, dass Risankizumab als monoklonaler IgG1-Antikörper durch glomeruläre Filtration in den Nieren filtriert oder als intaktes Molekül im Urin ausgeschieden wird.
Linearität/Nicht-Linearität
Risankizumab wies bei gesunden Probanden oder Studienteilnehmenden mit Psoriasis, Morbus Crohn oder Colitis ulcerosa eine lineare Pharmakokinetik mit annähernd dosisproportionalem Anstieg der systemischen Exposition (Cmax und area under the curve, AUC) im untersuchten Dosisbereich von 18 bis 360 mg bzw. 0,25 bis 1 mg/kg bei subkutaner Anwendung und 200 bis 1 800 mg bzw. 0,01 bis 5 mg/kg bei intravenöser Anwendung auf.
Wechselwirkungen
Es wurden Studien zu Wechselwirkungen bei Studienteilnehmenden mit Plaque-Psoriasis, Morbus Crohn und Colitis ulcerosa durchgeführt, um die Wirkung einer wiederholten Anwendung von Risankizumab auf die Pharmakokinetik von Cytochrom-P450(CYP)-sensitiven Prüfsubstraten zu untersuchen. Die Exposition von Koffein (CYP1A2-Substrat), Warfarin (CYP2C9-Substrat), Omeprazol (CYP2C19-Substrat), Metoprolol (CYP2D6-Substrat) und Midazolam (CYP3A-Substrat) nach Behandlung mit Risankizumab war mit deren Exposition vor der Behandlung mit Risankizumab vergleichbar. Dies deutet auf keine klinisch bedeutsamen Wechselwirkungen durch diese Enzyme hin.
In populationspharmakokinetischen Analysen zeigte sich, dass die Risankizumab-Exposition nicht durch Begleitmedikationen, die einige Patienten mit Plaque-Psoriasis während der klinischen Studien anwendeten, beeinflusst wurde. Ein ähnlicher fehlender Effekt durch Begleitmedikationen wurde auch bei populationspharmakokinetischen Analysen bei Morbus Crohn oder Colitis ulcerosa beobachtet.
Besondere Patientengruppen
Kinder und Jugendliche
Die Pharmakokinetik von Risankizumab bei Kindern und Jugendlichen unter 16 Jahren wurde nicht untersucht. 12 der 1 574 Studienteilnehmenden mit Morbus Crohn, die mit Risankizumab behandelt wurden, waren 16 bis 17 Jahre alt. Die Risankizumab-Exposition war bei 16‑ bis 17‑jährigen Patienten mit Morbus Crohn mit der von erwachsenen Patienten vergleichbar. Basierend auf den populationspharmakokinetischen Analysen wurde kein signifikanter Einfluss des Alters auf die Risankizumab-Exposition festgestellt.
Ältere Patienten
Von den 2 234 Studienteilnehmenden mit Plaque-Psoriasis, die Risankizumab erhielten, waren 243 ≥ 65 Jahre und 24 ≥ 75 Jahre alt. Von den 1 574 Studienteilnehmenden mit Morbus Crohn, die Risankizumab erhielten, waren 72 ≥ 65 Jahre und 5 ≥ 75 Jahre alt. Von den 1 512 Studienteilnehmenden mit Colitis ulcerosa, die Risankizumab erhielten, waren 103 ≥ 65 Jahre alt und 8 ≥ 75 Jahre alt. Zwischen den älteren und den jüngeren Studienteilnehmenden, die Risankizumab erhielten, wurden insgesamt keine Unterschiede hinsichtlich der Risankizumab-Exposition beobachtet.
Patienten mit eingeschränkter Nieren- oder Leberfunktion
Es wurden keine spezifischen Studien zur Ermittlung der Auswirkungen einer eingeschränkten Nieren- oder Leberfunktion auf die Pharmakokinetik von Risankizumab durchgeführt. Auf Basis populationspharmakokinetischer Analysen hatten der Serumkreatininspiegel, die Kreatinin-Clearance oder die Leberfunktionsmarker (ALT/AST/Bilirubin) bei Studienteilnehmenden mit Psoriasis, Morbus Crohn oder Colitis ulcerosa keinen bedeutsamen Einfluss auf die Risankizumab-Clearance.
Als monoklonaler IgG1-Antikörper erfolgt die Elimination von Risankizumab hauptsächlich über den intrazellulären Katabolismus und es ist nicht zu erwarten, dass Risankizumab durch hepatische Cytochrom-P450-Enzyme verstoffwechselt oder durch renale Elimination ausgeschieden wird.
Körpergewicht
Clearance und Verteilungsvolumen von Risankizumab steigen mit zunehmendem Körpergewicht. Dies kann bei Studienteilnehmenden mit einem hohen Körpergewicht (> 130 kg) zu einer verringerten Wirksamkeit führen. Allerdings beruht diese Beobachtung auf einer begrenzten Anzahl an Studienteilnehmenden mit Plaque-Psoriasis. Das Körpergewicht hatte bei Psoriasis-Arthritis, Morbus Crohn oder Colitis ulcerosa keinen klinisch bedeutsamen Einfluss auf die Exposition oder Wirksamkeit von Risankizumab. Daher wird momentan eine Dosisanpassung aufgrund des Körpergewichts nicht empfohlen.
Geschlecht und ethnische Zugehörigkeit
Bei erwachsenen Patienten mit Plaque-Psoriasis, Morbus Crohn oder Colitis ulcerosa hatte das Geschlecht oder die ethnische Abstammung keine bedeutenden Auswirkungen auf die Clearance von Risankizumab. In klinischen Studien zur Pharmakokinetik mit gesunden Probanden wurden zwischen chinesischen oder japanischen Probanden und kaukasischen Probanden keine klinisch bedeutsamen Unterschiede hinsichtlich der Risankizumab-Exposition beobachtet.
Basierend auf den Studien zur Toxizität bei wiederholter Gabe, einschließlich sicherheitspharmakologischer Untersuchungen und einer erweiterten Studie zur prä‑ und postnatalen Entwicklungstoxizität an Javaneraffen mit Dosen von bis zu 50 mg/kg/Woche, entsprechend einer Exposition in Höhe des 10‑Fachen der klinischen Exposition während der Induktionsphase (600 mg intravenös alle 4 Wochen) und des 39‑Fachen der klinischen Exposition in der Erhaltungsphase (360 mg subkutan alle 8 Wochen) für Morbus Crohn, ließen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Bei Colitis ulcerosa entsprachen diese Expositionen dem 5‑Fachen der klinischen Expositionen während der Induktionsphase mit einer Dosis von 1 200 mg intravenös alle 4 Wochen und dem 65‑ bzw. 32‑Fachen der klinischen Expositionen für die Erhaltungsphase mit einer Dosis von 180 mg bzw. 360 mg subkutan alle 8 Wochen.
Es wurden keine Studien zur Mutagenität oder Kanzerogenität von Risankizumab durchgeführt. In einer 26‑wöchigen Studie zur chronischen Toxikologie an Javaneraffen wurden bei Dosierungen von bis zu 50 mg/kg/Woche (das 7‑Fache der klinischen Exposition während der Induktionsphase (600 mg intravenös alle 4 Wochen) und das 28‑Fache der klinischen Exposition in der Erhaltungsphase (360 mg subkutan alle 8 Wochen) für Morbus Crohn und das 3‑Fache der klinischen Exposition während der Induktionsphase (1 200 mg intravenös alle 4 Wochen) und das 45‑ bzw. 23‑Fache der klinischen Exposition während der Erhaltungsphase (180 mg bzw. 360 mg subkutan alle 8 Wochen) für Colitis ulcerosa)) keine präneoplastischen oder neoplastischen Läsionen und keine unerwünschten immuntoxischen oder kardiovaskulären Wirkungen beobachtet.
Skyrizi 180 mg und 360 mg Injektionslösung in einer Patrone und Skyrizi 180 mg Injektionslösung in einer Fertigspritze
Natriumacetat-Trihydrat (E 262)
Essigsäure (E 260)
Trehalose-Dihydrat (Ph. Eur.)
Polysorbat 20 (E 432)
Wasser für Injektionszwecke
Skyrizi 90 mg Injektionslösung in einer Fertigspritze
Natriumsuccinat 6 H2O
Polysorbat 20 (E 432)
Sorbitol (E 420)
Bernsteinsäure (E 363)
Wasser für Injektionszwecke
Dieses Arzneimittel darf nicht mit anderen Arzneimitteln gemischt werden.
2 Jahre
Im Kühlschrank lagern (2 °C – 8 °C). Nicht einfrieren.
Die Patrone oder Fertigspritze(n) kann (können) bis zu 24 Stunden außerhalb des Kühlschranks (bei bis zu maximal 25 °C) aufbewahrt werden.
Die Patrone bzw. die Fertigspritze(n) im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Skyrizi 360 mg Injektionslösung in einer Patrone
Eine 360-mg-Lösung in einer Einwegpatrone aus zyklischem Olefinharz mit beschichtetem Chlorbutyl-Gummitrennstück und beschichtetem Chlorbutyl-Gummikolben als produktberührende Materialien und einer Kappe aus Harz. Der Patronenmontagesatz ist mit einem On-Body-Injektor (Applikationsgerät) zusammen verpackt. Der Flüssigkeitsweg im On-Body-Injektor besteht aus einem Polyvinylchloridrohr und einer 29-Gauge-Nadel aus Edelstahl. Der On-Body-Injektor enthält Silberoxid-Zink-Batterien und ein Klebepflaster aus Polyester mit einem Acrylkleber. Das Applikationsgerät ist für die Verwendung mit der mitgelieferten 360-mg-Patrone vorgesehen.
Skyrizi 360 mg ist in Packungen mit jeweils 1 Patrone und 1 On-Body-Injektor erhältlich.
Skyrizi 180 mg Injektionslösung in einer Patrone
Eine 180‑mg-Lösung in einer Einwegpatrone aus zyklischem Olefinharz mit beschichtetem Chlorbutyl-Gummitrennstück und mit beschichtetem Chlorbutyl-Gummikolben als produktberührende Materialien und einer Kappe aus Harz. Der Patronenmontagesatz ist mit einem On-Body-Injektor (Applikationsgerät) zusammen verpackt. Der Flüssigkeitsweg im On-Body-Injektor besteht aus einem Polyvinylchloridrohr und einer 29‑Gauge-Nadel aus Edelstahl. Der On-Body-Injektor enthält Silberoxid-Zink-Batterien und ein Klebepflaster aus Polyester mit einem Acrylkleber. Das Applikationsgerät ist für die Verwendung mit der mitgelieferten 180‑mg-Patrone vorgesehen.
Skyrizi 180 mg ist in Packungen mit jeweils 1 Patrone und 1 On-Body-Injektor erhältlich.
Skyrizi 90 mg Injektionslösung in einer Fertigspritze
Fertigspritze aus Glas mit fester Nadel und Nadelkappe, versehen mit einem automatischen Nadelschutz.
Skyrizi 90 mg ist in Packungen mit 4 Fertigspritzen erhältlich.
Skyrizi 180 mg Injektionslösung in einer Fertigspritze
Fertigspritze aus Glas mit fester Nadel und Nadelkappe, versehen mit einem automatischen Nadelschutz.
Skyrizi 180 mg ist in Packungen mit 1 und 2 Fertigspritzen erhältlich.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Skyrizi 180 mg und 360 mg Injektionslösung in einer Patrone
Vor der Injektion ist der Umkarton aus dem Kühlschrank zu nehmen und 45 bis 90 Minuten bei Raumtemperatur ohne direkte Sonneneinstrahlung stehen zu lassen, ohne die Patrone aus dem Umkarton zu entnehmen.
Vor der Verwendung wird eine visuelle Prüfung der Patrone empfohlen. Die Lösung ist frei von Fremdpartikeln und praktisch frei von produktbezogenen Partikeln. Skyrizi darf nicht angewendet werden, wenn die Lösung trüb oder verfärbt ist oder große Partikel enthält. Die Patrone darf nicht geschüttelt werden.
Die Lösung muss farblos bis gelblich und klar bis leicht opalisierend sein.
Skyrizi 90 mg und 180 mg Injektionslösung in einer Fertigspritze
Vor der Injektion ist der Umkarton aus dem Kühlschrank zu nehmen und 15 bis 30 Minuten bei Raumtemperatur ohne direkte Sonneneinstrahlung liegen zu lassen, ohne die Fertigspritzen aus dem Umkarton zu entnehmen.
Vor der Anwendung der Fertigspritze empfiehlt es sich, diese visuell zu prüfen. Es können sich wenige durchscheinende bis weiße produktbezogene Partikel in der Lösung befinden. Skyrizi darf nicht angewendet werden, wenn die Lösung trüb oder verfärbt ist oder große Partikel enthält. Die Fertigspritze darf nicht geschüttelt werden.
Die 90 mg Injektionslösung in einer Fertigspritze muss farblos bis leicht gelblich und klar bis leicht opalisierend sein.
Die 180 mg Injektionslösung in einer Fertigspritze muss farblos bis gelb und klar bis leicht opalisierend sein.
Generelle Vorsichtsmaßnahmen
Ausführliche Anweisungen zur Verabreichung sind in der Packungsbeilage enthalten.
Jeder On-Body-Injektor mit Patrone bzw. jede Fertigspritze ist nur zum einmaligen Gebrauch bestimmt.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
AbbVie Deutschland GmbH & Co. KG
Knollstraße
67061 Ludwigshafen
Deutschland
Skyrizi 360 mg Injektionslösung in einer Patrone
EU/1/19/1361/005
Skyrizi 180 mg Injektionslösung in einer Patrone
EU/1/19/1361/007
Skyrizi 90 mg Injektionslösung in einer Fertigspritze
EU/1/19/1361/006
Skyrizi 180 mg Injektionslösung in einer Fertigspritze
EU/1/19/1361/008
EU/1/19/1361/009
Datum der Erteilung der Zulassung: 26. April 2019
Datum der letzten Verlängerung der Zulassung: 05. Januar 2024
Juni 2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
|