
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Columvi® 2,5 mg Konzentrat zur Herstellung einer Infusionslösung
Columvi® 10 mg Konzentrat zur Herstellung einer Infusionslösung
Columvi 2,5 mg Konzentrat zur Herstellung einer Infusionslösung
Jede Durchstechflasche mit 2,5 ml Konzentrat enthält 2,5 mg Glofitamab in einer Konzentration von 1 mg/ml.
Columvi 10 mg Konzentrat zur Herstellung einer Infusionslösung
Jede Durchstechflasche mit 10 ml Konzentrat enthält 10 mg Glofitamab in einer Konzentration von 1 mg/ml.
Glofitamab ist ein humanisierter bispezifischer, monoklonaler Anti-CD20/Anti-CD3-Antikörper, der in Ovarialzellen des chinesischen Hamsters (CHO-Zellen) mittels rekombinanter Desoxyribonukleinsäure(DNA)-Technologie hergestellt wird.
Sonstige Bestandteile mit bekannter Wirkung
Jede 2,5‑ml-Durchstechflasche Columvi enthält 1,25 mg (0,5 mg/ml) Polysorbat 20.
Jede 10‑ml-Durchstechflasche Columvi enthält 5 mg (0,5 mg/ml) Polysorbat 20.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Konzentrat zur Herstellung einer Infusionslösung (steriles Konzentrat).
Farblose, klare Lösung mit einem pH-Wert von 5,5 und einer Osmolalität von 270 – 350 mosm/kg.
Columvi in Kombination mit Gemcitabin und Oxaliplatin wird angewendet zur Behandlung erwachsener Patienten mit rezidiviertem oder refraktärem diffusem großzelligem B-Zell-Lymphom, das nicht anderweitig spezifiziert ist (DLBCL NOS, not otherwise specified), die nicht für eine autologe Stammzelltransplantation (ASCT) geeignet sind.
Columvi als Monotherapie ist angezeigt für die Behandlung von erwachsenen Patienten mit rezidiviertem oder refraktärem diffusem großzelligem B-Zell-Lymphom (DLBCL) nach zwei oder mehr systemischen Behandlungslinien.
Columvi darf nur unter Aufsicht von medizinischem Fachpersonal mit Erfahrung in der Diagnose und Behandlung von Krebspatienten angewendet werden, das Zugang zu geeigneter medizinischer Versorgung hat, um schwere Reaktionen im Zusammenhang mit einem Zytokin-Freisetzungssyndrom (CRS, cytokine release syndrome) und des Immuneffektorzellen-assoziierten Neurotoxizitätssyndroms (ICANS, immune effector cell-associated neurotoxicity syndrome) zu behandeln.
Vor der Infusion von Columvi in Zyklus 1 und 2 muss mindestens 1 Dosis Tocilizumab für die Anwendung im Falle eines CRS zur Verfügung stehen. Der Zugang zu einer weiteren Dosis Tocilizumab innerhalb von 8 Stunden nach Anwendung der vorherigen Tocilizumab-Dosis muss sichergestellt sein (siehe Abschnitt 4.4).
Vorbehandlung mit Obinutuzumab
Alle Patienten in den Studien NP30179 und GO41944 (STARGLO) erhielten an Tag 1 von Zyklus 1 (7 Tage vor Beginn der Behandlung mit Columvi) eine Einzeldosis von 1 000 mg Obinutuzumab als Vorbehandlung, um die zirkulierenden und lymphoiden B-Zellen zu verringern (siehe Tabelle 2, Verspätete oder versäumte Dosen und Abschnitt 5.1).
Obinutuzumab wurde als intravenöse Infusion mit 50 mg/h verabreicht. Die Infusionsgeschwindigkeit wurde in Schritten von 50 mg/h alle 30 Minuten bis zu einem Maximum von 400 mg/h gesteigert.
Vollständige Informationen zur Prämedikation, Zubereitung, Verabreichung und Behandlung von Obinutuzumab-Nebenwirkungen sind der Zusammenfassung der Merkmale des Arzneimittels von Obinutuzumab zu entnehmen.
Prämedikation und Prophylaxe
Prophylaxe des Zytokin-Freisetzungssyndroms (CRS)
Columvi sollte gut hydrierten Patienten verabreicht werden. Die empfohlene Prämedikation für CRS (siehe Abschnitt 4.4) ist in Tabelle 1 dargestellt.
Tabelle 1: Prämedikation vor der Infusion von Columvi
Behandlungszyklus (Tag) |
Patienten, die eine Prämedikation benötigen |
Prämedikation |
Verabreichung |
Zyklus 1 (Tag 8, Tag 15); |
Alle Patienten |
20 mg Dexamethason1 intravenös |
Verabreichung mindestens 1 Stunde vor der Infusion von Columvi abgeschlossen |
Orale Analgetika/ |
Mindestens 30 Minuten vor der Infusion von Columvi |
||
Anti-Histamin3 | |||
Alle nachfolgenden Infusionen |
Alle Patienten |
Orale Analgetika/ |
Mindestens 30 Minuten vor der Infusion von Columvi |
Anti-Histamin3 | |||
Patienten, bei denen unter der vorherigen Dosis ein CRS aufgetreten ist |
20 mg Dexamethason1, 4 intravenös |
Mindestens 1 Stunde vor der Infusion von Columvi abgeschlossen |
1 Falls ein Patient eine Intoleranz gegenüber Dexamethason hat oder Dexamethason nicht verfügbar ist, 100 mg Prednison/Prednisolon oder 80 mg Methylprednisolon verabreichen.
2 Zum Beispiel 1 000 mg Paracetamol.
3 Zum Beispiel 50 mg Diphenhydramin.
4 Soll zusätzlich zur Prämedikation, die für alle Patienten erforderlich ist, verabreicht werden.
Infektionsprophylaxe
Zur Verringerung des Infektionsrisikos wird eine Prophylaxe empfohlen (siehe Abschnitt 4.4).
Bei Patienten mit erhöhtem Risiko ist eine Prophylaxe gegen Zytomegalievirus (CMV), Herpes, Pneumocystis jirovecii-Pneumonie und andere opportunistische Infektionen in Betracht zu ziehen (siehe Abschnitt 4.8).
Dosierung
Die Dosierung von Columvi beginnt mit einem Dosissteigerungsschema (das darauf ausgelegt ist, das Risiko eines CRS zu verringern), das zur empfohlenen Dosis von 30 mg führt.
Dosissteigerungsschema für Columvi Monotherapie
Columvi muss als intravenöse Infusion gemäß dem Dosissteigerungsschema verabreicht werden, das zur empfohlenen Dosis von 30 mg führt (wie in Tabelle 2 dargestellt), nachdem die Vorbehandlung mit Obinutuzumab an Tag 1 von Zyklus 1 abgeschlossen wurde. Jeder Zyklus dauert 21 Tage.
Tabelle 2: Dosissteigerungsschema für Columvi Monotherapie bei Patienten mit rezidiviertem oder refraktärem DLBCL
Behandlungszyklus, Tag |
Dosis von Columvi |
Infusionsdauer |
|
Zyklus 1 |
Tag 1 |
Vorbehandlung mit Obinutuzumab 1 000 mg1 |
|
Tag 8 |
2,5 mg |
4 Stunden2 |
|
Tag 15 |
10 mg |
||
Zyklus 2 |
Tag 1 |
30 mg |
|
Zyklus 3 bis 12 |
Tag 1 |
30 mg |
2 Stunden3 |
1 Siehe oben „Vorbehandlung mit Obinutuzumab“. | |||
Dosissteigerungsschema für Columvi in Kombination mit Gemcitabin und Oxaliplatin
Columvi muss als intravenöse Infusion gemäß dem Dosissteigerungsschema verabreicht werden, das zur empfohlenen Dosierung von 30 mg führt (wie in Tabelle 3 dargestellt), nachdem die Vorbehandlung mit Obinutuzumab an Tag 1 von Zyklus 1 abgeschlossen wurde.
Columvi wird in Kombination mit Gemcitabin und Oxaliplatin in den Zyklen 1 - 8 und als Monotherapie in den Zyklen 9 - 12 angewendet. Jeder Zyklus dauert 21 Tage.
Tabelle 3: Dosissteigerungsschema für Columvi in Kombination mit Gemcitabin und Oxaliplatin bei Patienten mit rezidiviertem oder refraktärem DLBCL
Behandlungszyklus, Tag |
Dosis von Columvi (Dauer der Infusion) |
Dosis von Gemcitabin |
Dosis von Oxaliplatin |
|
Zyklus 1 |
Tag 1 |
Vorbehandlung mit Obinutuzumab 1 000 mga |
||
Tag 2 |
– |
1 000 mg/m2 b |
100 mg/m2 b |
|
Tag 8 |
2,5 mg (4 Stunden)c |
– |
– |
|
Tag 15 |
10 mg (4 Stunden)c |
|||
Zyklus 2 |
Tag 1 |
30 mg (4 Stunden)c,d |
1 000 mg/m2 b, d |
100 mg/m2 b, d |
Zyklus 3 bis 8 |
Tag 1 |
30 mg (2 Stunden)d,e |
1 000 mg/m2 b, d |
100 mg/m2 b, d |
Zyklus 9 bis 12 |
Tag 1 |
30 mg (2 Stunden)e |
– |
– |
a Siehe oben „Vorbehandlung mit Obinutuzumab“.
b Zyklen 1 bis 8: Gemcitabin vor Oxaliplatin verabreichen.
c Bei Patienten, bei denen unter ihrer vorherigen Dosis Columvi ein CRS auftritt, kann die Infusionsdauer auf bis zu 8 Stunden verlängert werden (siehe Abschnitt 4.4).
d Zyklen 2 bis 8: Columvi vor Gemcitabin und Oxaliplatin verabreichen. Gemcitabin und Oxaliplatin können an Tag 1 oder 2 gegeben werden.
e Die Infusionszeit kann nach Ermessen des behandelnden Arztes auf 2 Stunden verkürzt werden, wenn die vorherige Infusion gut vertragen wurde. Wenn bei dem Patienten unter einer vorherigen Dosis ein CRS auftrat, sollte die Infusionsdauer bei 4 Stunden gehalten werden.
Patientenüberwachung
Wenn Columvi als Monotherapie gegeben wird, müssen die Patienten während aller Infusionen von Columvi und über mindestens 10 Stunden nach Abschluss der Infusion der ersten Dosis Columvi (2,5 mg an Tag 8 von Zyklus 1) auf Anzeichen und Symptome eines potenziellen CRS überwacht werden (siehe Abschnitt 4.8).
Wenn Columvi in Kombination mit Gemcitabin und Oxaliplatin gegeben wird, müssen die Patienten während aller Infusionen von Columvi und über 4 Stunden nach Beendigung der ersten Dosis von Columvi (2,5 mg an Tag 8 von Zyklus 1) auf Anzeichen und Symptome eines potenziellen CRS überwacht werden (siehe Abschnitt 4.8).
Patienten, bei denen es bei ihrer vorherigen Infusion zu einem CRS vom Grad ≥ 2 gekommen ist, sind nach Abschluss der Infusion zu überwachen (siehe Tabelle 4 in Abschnitt 4.2).
Alle Patienten müssen nach der Gabe von Columvi auf Anzeichen und Symptome des CRS und des Immuneffektorzellen-assoziierten Neurotoxizitätssyndroms (ICANS) überwacht werden.
Alle Patienten müssen über Risiko, Anzeichen und Symptome eines CRS und ICANS aufgeklärt und angewiesen werden, sofort das medizinische Fachpersonal zu kontaktieren, wenn zu irgendeinem Zeitpunkt Anzeichen und Symptome eines CRS und/oder ICANS auftreten (siehe Abschnitt 4.4).
Dauer der Behandlung
Die Behandlung mit Columvi Monotherapie wird für maximal 12 Zyklen oder bis zur Krankheitsprogression oder zu einer nicht beherrschbaren Toxizität empfohlen, je nachdem, was zuerst eintritt. Jeder Zyklus dauert 21 Tage.
Die Behandlung mit Columvi in Kombination mit Gemcitabin und Oxaliplatin wird für 8 Zyklen empfohlen, gefolgt von 4 Zyklen Columvi Monotherapie über insgesamt maximal 12 Zyklen Columvi oder bis zur Krankheitsprogression oder einer nicht beherrschbaren Toxizität, je nachdem, was zuerst eintritt. Jeder Zyklus dauert 21 Tage.
Verspätete oder versäumte Dosen
Während der Dosissteigerung (wöchentliche Dosierung):
Nach der Vorbehandlung mit Obinutuzumab, wenn die Dosis von 2,5 mg Columvi um mehr als 1 Woche verspätet ist, ist die Vorbehandlung mit Obinutuzumab zu wiederholen.
Wenn nach der Verabreichung von 2,5 mg oder 10 mg Columvi ein behandlungsfreies Intervall von 2 Wochen bis 6 Wochen besteht, ist die letzte verträgliche Dosis von Columvi zu wiederholen und die geplante Dosissteigerung fortzusetzen.
Nach der Verabreichung von 2,5 mg oder 10 mg Columvi muss bei einem behandlungsfreien Intervall von mehr als 6 Wochen die Vorbehandlung mit Obinutuzumab und die schrittweise Dosissteigerung mit Columvi wiederholt werden (siehe Zyklus 1 in Tabelle 2 und Tabelle 3).
Nach Zyklus 2 (Dosis von 30 mg):
Wenn zwischen den Behandlungszyklen mit Columvi ein behandlungsfreies Intervall von mehr als 6 Wochen besteht, ist die Vorbehandlung mit Obinutuzumab und die schrittweise Dosissteigerung von Columvi zu wiederholen (siehe Zyklus 1 in Tabelle 2 und Tabelle 3) und dann der vorgesehene Behandlungszyklus (30 mg Dosis) wieder aufzunehmen.
Dosisanpassung
Dosisreduktionen von Columvi werden nicht empfohlen.
Behandlung des Zytokin-Freisetzungssyndroms (CRS)
Ein CRS sollte aufgrund des klinischen Erscheinungsbildes identifiziert werden (siehe Abschnitte 4.4 und 4.8). Die Patienten sind auf andere Ursachen von Fieber, Hypoxie und Hypotonie wie Infektionen oder Sepsis zu untersuchen. Wenn ein CRS vermutet wird, ist es gemäß den Empfehlungen für die CRS-Behandlung, basierend auf den Konsensempfehlungen der American Society for Transplantation and Cellular Therapy (ASTCT) in Tabelle 4, zu behandeln.
Tabelle 4: ASTCT-Einstufung und Behandlungsleitfaden für CRS
Schweregrad1 |
CRS-Behandlung |
Für die nächste geplante Infusion von Columvi |
Grad 1 |
Wenn ein CRS während der Infusion auftritt:
Wenn ein CRS nach der Infusion auftritt:
Wenn das CRS nach symptomatischer Behandlung länger als 48 Stunden anhält:
Für CRS, das gleichzeitig mit ICANS auftritt, siehe Tabelle 5. |
|
Grad 2 |
Wenn ein CRS während der Infusion auftritt:
Wenn ein CRS nach der Infusion auftritt:
Für CRS, das gleichzeitig mit ICANS auftritt, siehe Tabelle 5. |
|
|
Bei Grad 2: Tocilizumab-Anwendung
Wenn innerhalb der letzten 6 Wochen 2 Dosen Tocilizumab angewendet wurden:
| ||
Grad 3 |
Bei Auftreten eines CRS während der Infusion:
Bei Auftreten eines CRS nach der Infusion:
Für CRS, das gleichzeitig mit ICANS auftritt, siehe Tabelle 5. |
|
Grad 4 |
Wenn das CRS während oder nach der Infusion auftritt:
Für CRS, das gleichzeitig mit ICANS auftritt, siehe Tabelle 5. |
|
|
Bei Grad 3 und Grad 4: Tocilizumab-Anwendung
Wenn innerhalb der letzten 6 Wochen 2 Dosen Tocilizumab angewendet wurden:
| ||
1 Konsensbewertungskriterien der American Society for Transplantation and Cellular Therapy (ASTCT) (Lee 2019). | ||
Behandlung des Immuneffektorzellen-assoziierten Neurotoxizitätssyndroms (ICANS)
Bei den ersten Anzeichen von ICANS ist, je nach Art und Schwere, eine unterstützende Therapie, eine neurologische Bewertung und ein Aussetzen der Anwendung von Columvi in Erwägung zu ziehen (siehe Tabelle 5). Andere Ursachen für neurologische Symptome sind auszuschließen. Bei Verdacht auf ICANS soll dieses entsprechend den Empfehlungen in Tabelle 5 behandelt werden.
Tabelle 5: ICANS-Einstufung und Behandlungsleitfaden
Schweregrad1 |
Auftreten von Symptomen2 |
ICANS Behandlung |
||
gleichzeitiges CRS |
kein gleichzeitiges CRS |
|||
Grad 1 |
ICE3-Score 7-9 Oder getrübter Bewusstseinszustand4: wacht spontan auf |
|
|
|
|
Die Behandlung mit Columvi aussetzen, bis ICANS abklingt. Nicht-sedierende Arzneimittel gegen Krampfanfälle (z. B. Levetiracetam) zur Prophylaxe von Anfällen in Erwägung ziehen. | ||||
Grad 2 |
ICE3-Score 3-6 Oder getrübter Bewusstseinszustand4: wacht bei Ansprache auf |
|
|
|
|
Die Behandlung mit Columvi aussetzen, bis ICANS abklingt. Nicht-sedierende Arzneimittel gegen Krampfanfälle (z. B. Levetiracetam) zur Prophylaxe von Anfällen in Erwägung ziehen. Neurologische und andere fachärztliche Beratung für eine weitere Bewertung nach Bedarf in Erwägung ziehen. | ||||
Grad 3 |
ICE3-Score 0-2 Oder getrübter Bewusstseinszustand4: wacht nur nach taktilem Reiz auf; Oder Krampfanfälle4, entweder:
Oder erhöhter intrakranieller Druck: fokale/lokale Ödeme in der Neurobildgebung4 |
|
|
|
|
Die Behandlung mit Columvi aussetzen, bis ICANS abklingt. Bei ICANS-Ereignissen des Grades 3, bei denen innerhalb von 7 Tagen keine Besserung eintritt, dauerhaftes Absetzen von Columvi in Erwägung ziehen. Nicht-sedierende Arzneimittel gegen Krampfanfälle (z. B. Levetiracetam) zur Prophylaxe von Anfällen in Erwägung ziehen. Neurologische und andere fachärztliche Beratung für eine weitere Bewertung nach Bedarf in Erwägung ziehen. | ||||
Grad 4 |
ICE3-Score 0 Oder ein getrübter Bewusstseinszustand4, entweder:
Oder Krampfanfälle4, entweder:
Oder motorische Befunde4:
Oder erhöhter intrakranialer Druck/zerebrales Ödem4, mit Anzeichen/Symptomen, wie z. B.:
|
|
|
|
|
Columvi dauerhaft absetzen. Nicht-sedierende Arzneimittel gegen Krampfanfälle (z. B. Levetiracetam) zur Prophylaxe von Anfällen in Erwägung ziehen. Neurologische und andere fachärztliche Beratung für eine weitere Bewertung nach Bedarf in Erwägung ziehen. Im Falle eines erhöhten intrakranialen Drucks/zerebralen Ödems die institutionellen Leitfäden für die Behandlung beachten. | ||||
1 Konsensbewertungskriterien der American Society for Transplantation and Cellular Therapy (ASTCT) (Lee 2019).
2 Die Behandlung wird durch das schwerwiegendste Ereignis bestimmt, das nicht auf eine andere Ursache zurückzuführen ist.
3 Wenn der Patient aufzuwecken und fähig ist, eine Immuneffektorzellen-assoziierte Enzephalopathie (ICE)-Beurteilung durchzuführen, Folgendes beurteilen:
Orientierung (bezogen auf Jahr, Monat, Stadt, Krankenhaus = 4 Punkte);
Benennung (Benennung von 3 Gegenständen, z. B. Zeigen Sie auf die Uhr, den Stift, den Knopf = 3 Punkte);
Befolgen von Befehlen (z. B. „zeigen Sie mir 2 Finger“ oder „schließen Sie die Augen und strecken Sie Ihre Zunge heraus“ = 1 Punkt);
Schreiben (Fähigkeit, einen Standardsatz zu schreiben = 1 Punkt);
Aufmerksamkeit (Rückrechnung von 100 bis 10 = 1 Punkt).
Wenn der Patient nicht aufzuwecken ist und nicht fähig ist, die ICE-Beurteilung durchzuführen (Grad 4 ICANS) = 0 Punkte.
4 Auf keine andere Ursache zurückzuführen.
5 Alle Verweise auf die Verabreichung von Dexamethason beziehen sich auf Dexamethason oder ein gleichwertiges Arzneimittel.
Besondere Patientengruppen
Ältere Patienten
Bei Patienten ab 65 Jahren ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Leberfunktionsstörung
Bei Patienten mit leichter Leberfunktionsstörung (Gesamtbilirubin > Obergrenze des Normalbereichs [Upper Limit of Normal - ULN] bis ≤ 1,5 x ULN oder Aspartat-Transaminase [AST] > ULN) ist keine Dosisanpassung erforderlich. Columvi wurde bei Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung nicht untersucht (siehe Abschnitt 5.2).
Nierenfunktionsstörung
Bei Patienten mit leichter oder mittelschwerer Nierenfunktionsstörung (Kreatinin-Clearance [CrCl] 30 bis < 90 ml/min) ist keine Dosisanpassung erforderlich. Columvi wurde bei Patienten mit schwerer Nierenfunktionsstörung nicht untersucht (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Columvi bei Kindern und Jugendlichen unter 18 Jahren wurde nicht ermittelt. Es liegen keine Daten vor.
Art der Anwendung
Columvi ist nur zur intravenösen Anwendung bestimmt.
Columvi muss vor der intravenösen Anwendung von einer medizinischen Fachkraft unter aseptischen Bedingungen verdünnt werden. Es muss als intravenöse Infusion über eine separate Infusionsleitung verabreicht werden.
Columvi darf nicht als intravenöse Druck- oder Bolus-Injektion verabreicht werden.
Hinweise zur Verdünnung von Columvi vor der Anwendung, siehe Abschnitt 6.6.
Überempfindlichkeit gegen den Wirkstoff, gegen Obinutuzumab, oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Bitte entnehmen Sie spezifische Gegenanzeigen für Obinutuzumab der Zusammenfassung der Merkmale des Arzneimittels für Obinutuzumab.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
CD20-negative Erkrankung
Es liegen nur begrenzte Daten über Patienten mit CD20-negativem DLBCL vor, die mit Columvi behandelt wurden, und es ist möglich, dass Patienten mit CD20-negativem DLBCL im Vergleich zu Patienten mit CD20-positivem DLBCL einen geringeren Nutzen haben. Die potenziellen Risiken und Vorteile verbunden mit der Behandlung von Patienten mit CD20-negativem DLBCL mit Columvi sollten abgewogen werden.
Zytokin-Freisetzungssyndrom (CRS)
Das Auftreten eines CRS, einschließlich lebensbedrohlicher Reaktionen, wurde bei Patienten berichtet, die Columvi erhielten (siehe Abschnitt 4.8).
Die häufigsten Manifestationen eines CRS waren Fieber, Tachykardie, Hypotonie, Schüttelfrost und Hypoxie. Infusionsbedingte Reaktionen sind klinisch möglicherweise nicht von Manifestationen eines CRS zu unterscheiden.
Die meisten CRS-Ereignisse traten nach der ersten Dosis von Columvi auf. Erhöhte Leberfunktionswerte (AST und Alanin-Aminotransferase [ALT] > 3 x ULN und/oder Gesamtbilirubin > 2 x ULN) in Verbindung mit CRS wurden nach Anwendung von Columvi berichtet (siehe Abschnitt 4.8).
Die Patienten in den Studien NP30179 und GO41944 (STARGLO) wurden 7 Tage vor Beginn der Behandlung mit Columvi mit Obinutuzumab vorbehandelt, um die zirkulierenden und lymphoiden B-Zellen zu verringern. Alle Patienten sind mit einer Prämedikation mit einem Antipyretikum, Antihistaminikum und einem Glucocorticoid zu behandeln (siehe Tabelle 1).
Vor der Infusion von Columvi in Zyklus 1 und 2 muss mindestens 1 Dosis Tocilizumab für die Anwendung im Falle eines CRS zur Verfügung stehen. Der Zugang zu einer weiteren Dosis Tocilizumab innerhalb von 8 Stunden nach Anwendung der vorherigen Tocilizumab-Dosis muss sichergestellt sein.
Wenn Columvi als Monotherapie angewendet wird, müssen die Patienten während aller Infusionen von Columvi und über mindestens 10 Stunden nach Abschluss der ersten Infusion überwacht werden.
Wenn Columvi in Kombination mit Gemcitabin und Oxaliplatin gegeben wird, müssen die Patienten während aller Infusionen von Columvi und über 4 Stunden nach Abschluss der ersten Infusion überwacht werden.
Vollständige Informationen zur Beobachtung siehe Abschnitt 4.2. Die Patienten müssen angewiesen werden, sofort einen Arzt aufzusuchen, falls zu irgendeinem Zeitpunkt Anzeichen oder Symptome von CRS auftreten (siehe Patientenpass unten).
Die Patienten sind auf andere Ursachen von Fieber, Hypoxie und Hypotonie wie Infektionen oder Sepsis zu untersuchen. Ein CRS sollte anhand des klinischen Bildes des Patienten und gemäß dem in Tabelle 4 (Abschnitt 4.2) aufgeführten CRS-Behandlungsleitfaden behandelt werden.
Immuneffektorzellen-assoziiertes Neurotoxizitätssyndrom
Nach der Behandlung mit Columvi sind schwerwiegende Fälle des Immuneffektorzellen-assoziierten Neurotoxizitätssyndroms (ICANS) aufgetreten, die lebensbedrohlich oder tödlich sein können (siehe Abschnitt 4.8).
Das Auftreten von ICANS kann gleichzeitig mit CRS, nach Abklingen von CRS oder ohne CRS, erfolgen. Zu den klinischen Anzeichen und Symptomen von ICANS können unter anderem Verwirrtheit, getrübter Bewusstseinszustand, Desorientiertheit, Krampfanfälle, Aphasie und Dysgraphie gehören.
Patienten sollten nach der Gabe von Columvi auf Anzeichen und Symptome von ICANS überwacht und unverzüglich behandelt werden. Die Patienten müssen angewiesen werden, sofort einen Arzt aufzusuchen, falls zu irgendeinem Zeitpunkt Anzeichen oder Symptome auftreten (siehe Patientenpass unten).
Bei den ersten Anzeichen oder Symptomen von ICANS gemäß dem ICANS-Behandlungsleitfaden in Tabelle 5 behandeln. Die Behandlung mit Columvi sollte wie empfohlen ausgesetzt oder dauerhaft abgesetzt werden.
Patientenpass
Der verschreibende Arzt muss den Patienten über das Risiko eines CRS und ICANS sowie über die Anzeichen und Symptome eines CRS und ICANS informieren. Die Patienten müssen angewiesen werden, sofort einen Arzt aufzusuchen, wenn bei ihnen Anzeichen und Symptome von CRS und ICANS auftreten. Den Patienten ist der Patientenpass auszuhändigen und sie sind anzuweisen, diesen stets bei sich zu tragen. Im Patientenpass werden die Symptome eines CRS und ICANS beschrieben, bei deren Auftreten der Patient umgehend einen Arzt aufsuchen sollte.
Wechselwirkung mit CYP450-Substraten
Die anfängliche Freisetzung von Zytokinen, die mit dem Beginn der Behandlung mit Columvi einhergeht, könnte CYP450-Enzyme unterdrücken und zu Schwankungen in der Konzentration von gleichzeitig verabreichten Arzneimitteln führen. Zu Beginn der Therapie mit Columvi sollten Patienten, die mit CYP450-Substraten mit engem therapeutischem Index behandelt werden, überwacht werden, da Schwankungen in der Konzentration von gleichzeitig verabreichten Arzneimitteln zu Toxizität, Wirkungsverlust oder unerwünschten Ereignissen führen können (siehe Abschnitt 4.5).
Schwerwiegende Infektionen
Bei mit Columvi behandelten Patienten sind schwerwiegende Infektionen, einschließlich opportunistischer Infektionen, aufgetreten (siehe Abschnitt 4.8).
Columvi darf Patienten mit einer aktiven Infektion nicht verabreicht werden. Vorsicht ist geboten, wenn die Anwendung von Columvi bei Patienten mit chronischen oder rezidivierenden Infektionen in der Anamnese, bei Patienten mit Grunderkrankungen, die sie für Infektionen prädisponieren können, oder bei Patienten, die zuvor eine intensive immunsuppressive Behandlung erhalten haben, erwogen wird. Gegebenenfalls sind antimikrobielle Mittel prophylaktisch zu verabreichen. Die Patienten sind vor und während der Behandlung mit Columvi auf das Auftreten möglicher bakterieller, Pilz- und neuer oder reaktivierter Virusinfektionen zu überwachen und angemessen zu behandeln.
Bei einer aktiven Infektion ist Columvi vorübergehend auszusetzen, bis die Infektion abgeklungen ist. Die Patienten sind anzuweisen, einen Arzt aufzusuchen, wenn Anzeichen oder Symptome auftreten, die auf eine Infektion hindeuten.
Unter der Behandlung mit Columvi wurde febrile Neutropenie berichtet. Patienten mit febriler Neutropenie sind auf eine Infektion zu untersuchen und umgehend zu behandeln.
Tumor Flare (Schub der Tumorerkrankung)
Bei Patienten, die Columvi erhielten, wurde über Tumor Flares berichtet (siehe Abschnitt 4.8). Zu den Manifestationen gehörten lokalisierte Schmerzen und Schwellungen.
Entsprechend dem Wirkmechanismus von Columvi sind Tumor Flares nach der Verabreichung von Columvi wahrscheinlich auf den Zustrom von T‑Zellen in die Tumorherde zurückzuführen und können ein Fortschreiten der Erkrankung vortäuschen. Ein Tumor Flare impliziert kein Therapieversagen und stellt auch kein Fortschreiten des Tumors dar.
Es wurden keine spezifischen Risikofaktoren für Tumor Flares identifiziert. Bei Patienten mit großen Tumoren, die sich in unmittelbarer Nähe der Atemwege und/oder eines lebenswichtigen Organs befinden, besteht jedoch ein erhöhtes Risiko für Beeinträchtigungen und Morbidität aufgrund des Masseeffekts infolge der Tumor Flares. Bei Patienten, die Columvi erhalten und wie klinisch indiziert behandelt werden, wird eine Überwachung und Beurteilung auf einen Tumor Flare an kritischen anatomischen Stellen empfohlen. Zur Behandlung eines Tumor Flares sind Corticosteroide und Analgetika in Erwägung zu ziehen.
Tumorlysesyndrom (tumor lysis syndrome - TLS)
Bei Patienten, die Columvi erhielten, wurde über TLS berichtet (siehe Abschnitt 4.8). Patienten mit großer Tumorlast, schnell proliferierenden Tumoren, Nierenfunktionsstörungen oder Dehydratation haben ein höheres Risiko für ein Tumorlysesyndrom.
Risikopatienten sind anhand geeigneter Labor- und klinischer Tests auf Elektrolytstatus, Hydrierung und Nierenfunktion engmaschig zu überwachen. Geeignete prophylaktische Maßnahmen mit antihyperurikämischen Wirkstoffen (z. B. Allopurinol oder Rasburicase) und eine angemessene Hydrierung sind vor der Obinutuzumab-Vorbehandlung und vor der Infusion von Columvi zu erwägen.
Die Behandlung eines TLS kann eine aggressive Hydrierung, die Korrektur von Elektrolytstörungen, eine antihyperurikämische Therapie und unterstützende Maßnahmen umfassen.
Immunisierung
Die Sicherheit einer Immunisierung mit Lebendimpfstoffen während oder nach einer Therapie mit Columvi wurde nicht untersucht. Eine Immunisierung mit Lebendimpfstoffen wird während einer Therapie mit Columvi nicht empfohlen.
Polysorbate
Dieses Arzneimittel enthält 1,25 mg Polysorbat 20 pro 2,5‑ml-Durchstechflasche und 5 mg Polysorbat 20 pro 10‑ml-Durchstechflasche, entsprechend 0,5 mg/ml.
Polysorbate können allergische Reaktionen hervorrufen.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt. Wechselwirkungen mit Columvi über Cytochrom-P450-Enzyme, andere metabolisierende Enzyme oder Transporter sind nicht zu erwarten.
Die initiale Freisetzung von Zytokinen in Zusammenhang mit dem Beginn der Behandlung mit Columvi könnte CYP450-Enzyme unterdrücken. Das höchste Risiko einer Arzneimittelwechselwirkung besteht im Zeitraum von einer Woche nach jeder der ersten beiden Dosen von Columvi (d. h. an Tag 8 und 15 von Zyklus 1) bei Patienten, die gleichzeitig CYP450-Substrate mit engem therapeutischem Index erhalten (z. B. Warfarin, Cyclosporin). Bei Einleitung einer Therapie mit Columvi sind Patienten, die mit CYP450-Substraten mit engem therapeutischem Index behandelt werden, engmaschig zu überwachen.
Die Pharmakokinetik (PK) von Glofitamab wird durch die gleichzeitige Anwendung von Gemcitabin oder Oxaliplatin nicht beeinflusst.
Frauen im gebärfähigen Alter/Kontrazeption
Patientinnen im gebärfähigen Alter müssen während der Behandlung mit Columvi und für mindestens 2 Monate nach der letzten Dosis von Columvi äußerst zuverlässige Verhütungsmethoden anwenden.
Schwangerschaft
Es liegen keine Daten zur Anwendung von Columvi bei Schwangeren vor. Es wurden keine tierexperimentellen Studien zur Reproduktionstoxizität durchgeführt (siehe Abschnitt 5.3).
Glofitamab ist ein Immunglobulin G (IgG). Es ist bekannt, dass IgG die Plazenta passiert. Aufgrund seines Wirkmechanismus ist es wahrscheinlich, dass Glofitamab bei Verabreichung an Schwangere zu einer B-Zell-Depletion beim Fötus führt.
Columvi wird während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die keine Kontrazeption anwenden, nicht empfohlen. Patientinnen, die Columvi erhalten, müssen über die potenzielle Schädigung des Fötus aufgeklärt werden. Patientinnen sind anzuweisen, bei Eintritt einer Schwangerschaft den behandelnden Arzt zu kontaktieren.
Stillzeit
Es ist nicht bekannt, ob Glofitamab in die Muttermilch übergeht. Es wurden keine Studien durchgeführt, um die Auswirkungen von Glofitamab auf die Milchproduktion oder sein Vorhandensein in der Muttermilch zu untersuchen. Es ist bekannt, dass humanes IgG in der Muttermilch vorhanden ist. Das Potenzial für eine Resorption von Glofitamab und das Potenzial für Nebenwirkungen beim gestillten Säugling sind nicht bekannt. Frauen sind anzuweisen, während der Behandlung mit Columvi und für die Dauer von 2 Monaten nach der letzten Dosis von Columvi nicht zu stillen.
Fertilität
Es liegen keine Daten zur Fertilität beim Menschen vor. Es wurden keine tierexperimentellen Untersuchungen zur Fertilität durchgeführt, um die Wirkung von Glofitamab auf die Fertilität zu beurteilen (siehe Abschnitt 5.3).
Columvi hat einen großen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Aufgrund des Potenzials für das Auftreten eines ICANS besteht bei Patienten, die Columvi erhalten, das Risiko eines getrübten Bewusstseinszustandes (siehe Abschnitt 4.4). Patienten sind anzuweisen, 48 Stunden nach jeder der ersten beiden Dosen während der Dosissteigerungs-Phase kein Fahrzeug zu führen oder keine Maschinen zu bedienen, dies gilt auch im Falle eines neuen Auftretens von Symptomen von ICANS (Verwirrtheit, Desorientiertheit, getrübter Bewusstseinszustand) und/oder CRS (Fieber, Tachykardie, Hypotonie, Schüttelfrost, Hypoxie), bis die Symptome abgeklungen sind (siehe Abschnitte 4.4 und 4.8).
Zusammenfassung des Sicherheitsprofils
Columvi Monotherapie
Die häufigsten Nebenwirkungen (≥ 20 %) waren Zytokin-Freisetzungssyndrom, Neutropenie, Anämie, Thrombozytopenie und Ausschlag.
Die häufigsten schwerwiegenden Nebenwirkungen, die bei ≥ 2 % der Patienten berichtet wurden, waren Zytokin-Freisetzungssyndrom (22,1 %), Sepsis (4,1 %), COVID-19 (3,4 %), Tumor Flare (3,4 %), COVID-19-Pneumonie (2,8 %), febrile Neutropenie (2,1 %), Neutropenie (2,1 %) und Pleuraerguss (2,1 %).
Bei 5,5 % der Patienten wurde Columvi aufgrund einer Nebenwirkung dauerhaft abgesetzt. Die häufigsten Nebenwirkungen, die zu einem dauerhaften Absetzen von Columvi führten, waren COVID-19 (1,4 %) und Neutropenie (1,4 %).
Columvi in Kombination mit Gemcitabin und Oxaliplatin
Die häufigsten Nebenwirkungen (≥ 20 %) waren Thrombozytopenie, Zytokin-Freisetzungssyndrom, Neutropenie, Anämie, Übelkeit, periphere Neuropathie, Diarrhö, erhöhte Aspartat-Aminotransferase, erhöhte Alanin-Aminotransferase, Ausschlag, Lymphopenie, Fieber und Erbrechen.
Die häufigsten schwerwiegenden Nebenwirkungen, die bei ≥ 2 % der Patienten berichtet wurden, waren Zytokin-Freisetzungssyndrom (20,3 %), Fieber (6,4 %), Pneumonie (5,8 %), COVID-19 (5,8 %), Thrombozytopenie (4,7 %), Atemwegsinfektion (3,5 %), Sepsis (2,3 %), febrile Neutropenie (2,3 %) und Diarrhö (2,3 %).
Bei 20,9 % der Patienten wurde Columvi aufgrund einer Nebenwirkung dauerhaft abgesetzt. Die häufigsten Nebenwirkungen, die zu einem dauerhaften Absetzen von Columvi führten, waren COVID‑19 (11,6 %), Sepsis (1,2 %) und Pneumonitis (1,2 %).
Tabellarische Auflistung der Nebenwirkungen
In Tabelle 6 sind Nebenwirkungen aufgeführt, die bei Patienten mit rezidiviertem oder refraktärem DLBCL auftraten, die in der Studie NP30179 mit Columvi als Monotherapie behandelt wurden (n = 145). Die Patienten erhielten im Median 5 Zyklen der Behandlung mit Columvi (Bereich: 1 – 13 Zyklen).
Nebenwirkungen, die bei Patienten mit rezidiviertem oder refraktärem DLBCL auftraten, die in der Studie GO41944 (STARGLO) mit Columvi in Kombination mit Gemcitabin und Oxaliplatin behandelt wurden (n = 172), sind in Tabelle 7 aufgeführt. Die Patienten erhielten im Median 11 Zyklen der Columvi Behandlung (Bereich: 1 bis 13 Zyklen).
Die Nebenwirkungen sind nach MedDRA-Systemorganklasse und Häufigkeitskategorien aufgeführt. Die folgenden Häufigkeitskategorien wurden verwendet: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 6: Nebenwirkungen bei Patienten mit rezidiviertem oder refraktärem DLBCL, die mit Columvi als Monotherapie behandelt wurden
Systemorganklasse |
Nebenwirkung |
Alle Grade |
Grade 3 – 4 |
Infektionen und parasitäre Erkrankungen |
Virusinfektionen1 |
Sehr häufig |
Häufig* |
Bakterielle Infektionen2 |
Häufig |
Häufig |
|
Infektionen der oberen Atemwege3 |
Häufig |
Sehr selten** |
|
Sepsis4 |
Häufig |
Häufig* |
|
Infektionen der unteren Atemwege5 |
Häufig |
Sehr selten** |
|
Pneumonie |
Häufig |
Gelegentlich |
|
Harnwegsinfektionen6 |
Häufig |
Gelegentlich |
|
Pilzinfektionen7 |
Häufig |
Sehr selten** |
|
Gutartige, bösartige und nicht spezifizierte Neubildungen (einschl. Zysten und Polypen) |
Tumor Flare |
Sehr häufig |
Häufig |
Erkrankungen des Blutes und des Lymphsystems |
Neutropenie |
Sehr häufig |
Sehr häufig |
Anämie |
Sehr häufig |
Häufig |
|
Thrombozytopenie |
Sehr häufig |
Häufig |
|
Lymphopenie |
Häufig |
Häufig |
|
Febrile Neutropenie8 |
Häufig |
Häufig |
|
Erkrankungen des Immunsystems |
Zytokin-Freisetzungssyndrom9 |
Sehr häufig |
Häufig |
Stoffwechsel- und Ernährungsstörungen |
Hypophosphatämie |
Sehr häufig |
Häufig |
Hypomagnesiämie |
Sehr häufig |
Sehr selten** |
|
Hypokalzämie |
Sehr häufig |
Sehr selten** |
|
Hypokaliämie |
Sehr häufig |
Gelegentlich |
|
Hyponatriämie |
Häufig |
Häufig |
|
Tumorlysesyndrom |
Häufig |
Häufig |
|
Psychiatrische Erkrankungen |
Verwirrtheitszustand |
Häufig |
Sehr selten** |
Erkrankungen des Nervensystems |
Kopfschmerzen |
Sehr häufig |
Sehr selten** |
Immuneffektorzellen-assoziiertes Neurotoxizitätssyndrom (ICANS)10 |
Häufig |
Gelegentlich* |
|
Somnolenz |
Häufig |
Gelegentlich |
|
Tremor |
Häufig |
Sehr selten** |
|
Myelitis11 |
Gelegentlich |
Gelegentlich |
|
Erkrankungen des Gastrointestinaltrakts |
Obstipation |
Sehr häufig |
Sehr selten** |
Diarrhö |
Sehr häufig |
Sehr selten** |
|
Übelkeit |
Sehr häufig |
Sehr selten** |
|
Gastrointestinale Blutung12 |
Häufig |
Häufig |
|
Erbrechen |
Häufig |
Sehr selten** |
|
Kolitis |
Gelegentlich |
Gelegentlich |
|
Erkrankungen der Haut und des Unterhautgewebes |
Ausschlag13 |
Sehr häufig |
Häufig |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber |
Sehr häufig |
Sehr selten** |
Untersuchungen |
Erhöhte Alanin-Aminotransferase |
Häufig |
Häufig |
Erhöhte Aspartat-Aminotransferase |
Häufig |
Häufig |
|
Erhöhte Alkalische Phosphatase im Blut |
Häufig |
Häufig |
|
Erhöhte Gamma-Glutamyltransferase |
Häufig |
Häufig |
|
Erhöhtes Bilirubin im Blut |
Häufig |
Gelegentlich |
|
Erhöhte Leberenzyme |
Häufig |
Häufig |
* Reaktionen von Grad 5 wurden berichtet. Siehe Beschreibung ausgewählter Nebenwirkungen.
** Es wurden keine Ereignisse von Grad 3 - 4 berichtet.
1 Einschließlich COVID-19, COVID-19-Pneumonie, Herpes Zoster, Influenza und ophthalmischen Herpes Zoster.
2 Einschließlich Infektionen durch vaskuläre Implantate, bakterielle Infektionen, Campylobacter-Infektionen, bakterielle Gallenwegsinfektionen, bakterielle Harnwegsinfektionen, Infektionen mit Clostridium difficile, Escherichia-Infektionen und Peritonitis.
3 Einschließlich Infektionen der oberen Atemwege, Sinusitis, Nasopharyngitis, chronische Sinusitis und Rhinitis.
4 Einschließlich Sepsis und septischen Schock.
5 Einschließlich Infektionen der unteren Atemwege und Bronchitis.
6 Einschließlich Harnwegsinfektionen und Harnwegsinfektionen mit Escherichia.
7 Einschließlich ösophageale Candidose und orale Candidose.
8 Einschließlich febrile Neutropenie und neutropenische Infektion.
9 Basierend auf der Konsensbewertung der ASTCT (Lee 2019).
10 ICANS auf der Grundlage von Lee 2019 und schließt Somnolenz, kognitive Störung, Verwirrtheitszustand, Delirium und Desorientiertheit ein.
11 Myelitis trat gleichzeitig mit CRS auf.
12 Einschließlich gastrointestinale Blutung, Dickdarmblutung und Magenblutung.
13 Einschließlich Ausschlag, juckenden Ausschlag, makulopapulösen Ausschlag, Dermatitis, akneähnliche Dermatitis, exfoliative Dermatitis, Erythem, Erythem der Handinnenflächen, Pruritis und erythematösen Ausschlag.
Tabelle 7: Nebenwirkungen bei Patienten mit rezidiviertem oder refraktärem DLBCL, die mit Columvi in Kombination mit Gemcitabin und Oxaliplatin behandelt wurden
Systemorganklasse |
Nebenwirkung |
Alle Grade |
Grad 3 – 4 |
Infektionen und parasitäre Erkrankungen |
COVID-191 |
Sehr häufig |
Häufig* |
Atemwegsinfektionen2 |
Sehr häufig |
Häufig* |
|
Pneumonie3 |
Sehr häufig |
Häufig* |
|
Zytomegalievirus-Infektionen4 |
Häufig |
Gelegentlich |
|
Herpes-Virusinfektionen5 |
Häufig |
Gelegentlich |
|
Harnwegsinfektionen6 |
Häufig |
Häufig |
|
Sepsis7 |
Häufig |
Häufig* |
|
Candida-Infektionen8 |
Häufig |
Sehr selten** |
|
Pneumocystis jirovecii-Pneumonie |
Gelegentlich |
Gelegentlich |
|
Gutartige, bösartige und nicht spezifizierte Neubildungen (einschl. Zysten und Polypen) |
Tumor Flare9 |
Häufig |
Sehr selten** |
Erkrankungen des Bluts und des Lymphsystems |
Thrombozytopenie |
Sehr häufig |
Sehr häufig |
Neutropenie |
Sehr häufig |
Sehr häufig |
|
Anämie |
Sehr häufig |
Sehr häufig |
|
Lymphopenie |
Sehr häufig |
Sehr häufig |
|
Febrile Neutropenie |
Häufig |
Häufig |
|
Erkrankungen des Immunsystems |
Zytokin-Freisetzungssyndrom10 |
Sehr häufig |
Häufig |
Stoffwechsel- und Ernährungsstörungen |
Hypokaliämie |
Sehr häufig |
Häufig |
Hyponatriämie |
Sehr häufig |
Gelegentlich |
|
Hypomagnesiämie |
Häufig |
Sehr selten** |
|
Hypokalzämie |
Häufig |
Gelegentlich |
|
Hypophosphatämie |
Häufig |
Häufig |
|
Tumorlysesyndrom |
Häufig |
Häufig |
|
Erkrankungen des Nervensystems |
Periphere Neuropathie11 |
Sehr häufig |
Häufig |
Immuneffektorzellen-assoziiertes Neurotoxizitätssyndrom12 |
Häufig |
Gelegentlich |
|
Kopfschmerzen |
Häufig |
Sehr selten** |
|
Tremor |
Gelegentlich |
Sehr selten** |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Pneumonitis |
Häufig |
Sehr selten*,** |
Erkrankungen des Gastrointestinaltrakts |
Übelkeit |
Sehr häufig |
Gelegentlich |
Diarrhö |
Sehr häufig |
Häufig |
|
Erbrechen |
Sehr häufig |
Gelegentlich |
|
Bauchschmerzen13 |
Sehr häufig |
Häufig |
|
Obstipation |
Sehr häufig |
Sehr selten** |
|
Kolitis14 |
Häufig |
Häufig |
|
Pankreatitis15 |
Häufig |
Häufig |
|
Erkrankungen der Haut und des Unterhautgewebes |
Ausschlag16 |
Sehr häufig |
Gelegentlich |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Schmerzen des Muskel- und Skelettsystems17 |
Sehr häufig |
Häufig |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber |
Sehr häufig |
Gelegentlich |
Untersuchungen |
Erhöhte Aspartat-Aminotransferase |
Sehr häufig |
Häufig |
Erhöhte Alanin-Aminotransferase |
Sehr häufig |
Häufig |
|
Erhöhte alkalische Phosphatase im Blut |
Sehr häufig |
Gelegentlich |
|
Erhöhte Gamma-Glutamyltransferase |
Sehr häufig |
Häufig |
|
Erhöhter Laktat-Dehydrogenase-Wert im Blut |
Sehr häufig |
Sehr selten** |
|
Erhöhtes Bilirubin im Blut18 |
Häufig |
Sehr selten** |
|
Erhöhte Leberenzymwerte |
Gelegentlich |
Sehr selten** |
* Reaktionen von Grad 5 wurden berichtet. Siehe Beschreibung ausgewählter Nebenwirkungen.
** Es wurden keine Ereignisse vom Grad 3 – 4 beschrieben.
1 Einschließlich COVID-19, COVID-19-Pneumonie und positiven SARS-CoV-2-Test.
2 Einschließlich Infektionen der oberen Atemwege, Infektionen der unteren Atemwege, Infektionen der Atemwege und bakterielle Infektionen der Atemwege.
3 Einschließlich Pneumonie, bakterielle Pneumonie und Pneumokokken-Pneumonie.
4 Neuauftreten oder Reaktivierung. Einschließlich Zytomegalievirus-Infektion, positiven Zytomegalievirustest, Reaktivierung einer Zytomegalievirus-Infektion und Zytomegalievirus-Virämie.
5 Neuauftreten oder Reaktivierung. Einschließlich Herpes zoster und Herpesvirus-Infektion.
6 Einschließlich Harnwegsinfektionen und Urosepsis.
7 Einschließlich Sepsis, Streptokokkensepsis, septischen Schock und Enterokokkensepsis.
8 Einschließlich orale Candidose und Candida-Infektion.
9 Einschließlich Tumor Flare und Tumorschmerzen.
10 Basierend auf der Konsensbewertung gemäß ASTCT (Lee 2019).
11 Einschließlich periphere Neuropathie, periphere sensorische Neuropathie, Dysästhesie, Parästhesie, Hypästhesie, periphere motorische Neuropathie und Polyneuropathie.
12 Einschließlich Verwirrtheitszustand, Delirium und ICANS.
13 Einschließlich Bauchschmerzen, abdominale Beschwerden, Schmerzen im Oberbauch, Schmerzen im Unterbauch und Schmerzen im Magen-Darm-Trakt.
14 Einschließlich Kolitis, ischämische Kolitis und Enterokolitis.
15 Einschließlich Pankreatitis und akute Pankreatitis.
16 Einschließlich Ausschlag, juckenden Ausschlag, makulopapulösen Ausschlag, Erythem, Pruritus, erythematösen Ausschlag, Urtikaria und Erythema multiforme.
17 Einschließlich Arthralgie, Schmerzen des Muskel- und Skelettsystems, Rückenschmerzen, Knochenschmerzen, Myalgie, Nackenschmerzen, Schmerzen in den Extremitäten, Schmerzen des Muskel- und Skelettsystems in der Brust und Thoraxschmerzen nicht kardialen Ursprungs.
18 Einschließlich erhöhtes Bilirubin im Blut und Hyperbilirubinämie.
Beschreibung ausgewählter Nebenwirkungen
Die folgenden Beschreibungen geben Informationen zu signifikanten Nebenwirkungen bei einer Columvi Monotherapie und/oder einer Kombinationstherapie. Einzelheiten zu den signifikanten Nebenwirkungen von Columvi bei kombinierter Anwendung sind separat aufgeführt, wenn klinisch relevante Unterschiede im Vergleich zur Columvi Monotherapie festgestellt wurden.
Zytokin-Freisetzungssyndrom (CRS)
Columvi Monotherapie
Ein CRS jeglichen Grades (nach ASTCT-Kriterien) trat bei 67,6 % der Patienten auf, die Columvi Monotherapie erhielten, wobei ein CRS vom Grad 1 bei 50,3 % der Patienten, ein CRS vom Grad 2 bei 13,1 % der Patienten, ein CRS vom Grad 3 bei 2,8 % der Patienten und ein CRS vom Grad 4 bei 1,4 % der Patienten berichtet wurde. Ein CRS trat bei 32,4 % (47/145) der Patienten mehr als einmal auf; bei 36/47 Patienten traten nur multiple CRS-Ereignisse vom Grad 1 auf. Es gab keine tödlichen Fälle von CRS. Das CRS klang bei allen Patienten bis auf einen ab. Ein Patient brach die Behandlung aufgrund von CRS ab.
Bei Patienten mit CRS waren die häufigsten Manifestationen von CRS Fieber (99,0 %), Tachykardie (25,5 %), Hypotonie (23,5 %), Schüttelfrost (14,3 %) und Hypoxie (12,2 %). Ereignisse von Grad 3 oder höher, die mit CRS assoziiert waren, schlossen Hypotonie (3,1 %), Hypoxie (3,1 %), Fieber (2,0 %) und Tachykardie (2,0 %) ein.
Ein CRS jeglichen Grades trat bei 54,5 % der Patienten nach der ersten 2,5-mg-Dosis von Columvi an Tag 8 von Zyklus 1 auf, mit einer medianen Zeit bis zum Auftreten (ab Beginn der Infusion) von 12,6 Stunden (Bereich: 5,2 – 50,8 Stunden) und einer medianen Dauer von 31,8 Stunden (Bereich: 0,5 – 316,7 Stunden); bei 33,3 % der Patienten nach der 10-mg-Dosis an Tag 15 von Zyklus 1 mit einer medianen Zeit bis zum Auftreten von 26,8 Stunden (Bereich: 6,7 – 125,0 Stunden) und einer medianen Dauer von 16,5 Stunden (Bereich: 0,3 – 109,2 Stunden); und bei 26,8 % der Patienten nach der 30-mg-Dosis in Zyklus 2 mit einer medianen Zeit bis zum Auftreten von 28,2 Stunden (Bereich: 15,0 – 44,2 Stunden) und einer medianen Dauer von 18,9 Stunden (Bereich: 1,0 – 180,5 Stunden). CRS wurde bei 0,9 % der Patienten in Zyklus 3 und bei 2 % der Patienten nach Zyklus 3 berichtet.
CRS vom Grad ≥ 2 trat bei 12,4 % der Patienten nach der ersten Dosis Columvi (2,5 mg) auf, mit einer medianen Zeit bis zum Auftreten von 9,7 Stunden (Bereich: 5,2 – 19,1 Stunden) und einer medianen Dauer von 50,4 Stunden (Bereich: 6,5 – 316,7 Stunden). Nach der Gabe von Columvi 10 mg an Tag 15 von Zyklus 1 verringerte sich die Inzidenz von CRS vom Grad ≥ 2 bei 5,2 % der Patienten mit einer medianen Zeit bis zum Auftreten von 26,2 Stunden (Bereich: 6,7 – 144,2 Stunden) und einer medianen Dauer von 30,9 Stunden (Bereich: 3,7 – 227,2 Stunden). Ein CRS vom Grad ≥ 2 trat nach einer Dosis von Columvi von 30 mg an Tag 1 von Zyklus 2 bei einem Patienten (0,8 %) auf, mit einer Zeit bis zum Auftreten von 15,0 Stunden und einer Dauer von 44,8 Stunden. Ein CRS vom Grad ≥ 2 wurde über Zyklus 2 hinaus nicht berichtet.
Bei 7 Patienten (4,8 %) der 145 Patienten traten erhöhte Leberfunktionswerte (AST und ALT > 3 x ULN und/oder Gesamtbilirubin > 2 x ULN) auf, die gleichzeitig mit einem CRS (n = 6) oder einer Progression der Erkrankung (n = 1) berichtet wurden.
Von den 25 Patienten, bei denen nach der Gabe von Columvi ein CRS vom Grad ≥ 2 auftrat, erhielten 22 (88,0 %) Tocilizumab, 15 (60,0 %) Corticosteroide und 14 (56,0 %) sowohl Tocilizumab als auch Corticosteroide. Zehn Patienten (40,0 %) erhielten Sauerstoff. Alle 6 Patienten (24,0 %) mit einem CRS vom Grad 3 oder 4 erhielten einen einzigen Vasopressor.
Eine Hospitalisierung aufgrund eines CRS nach der Verabreichung von Columvi trat bei 22,1 % der Patienten auf und die berichtete mediane Dauer der Hospitalisierung betrug 4 Tage (Bereich: 2 bis 15 Tage).
Columvi in Kombination mit Gemcitabin und Oxaliplatin
Ein CRS jeden Grades (nach ASTCT-Kriterien) trat bei 44,2 % der Patienten auf, die Columvi mit Gemcitabin und Oxaliplatin erhielten, wobei ein CRS vom Grad 1 bei 31,4 % der Patienten, ein CRS vom Grad 2 bei 10,5 % der Patienten und ein CRS vom Grad 3 bei 2,3 % der Patienten berichtet wurde. Ein CRS trat bei 21,5 % (37/172) der Patienten mehr als einmal auf; bei 30/37 Patienten traten nur multiple CRS-Ereignisse vom Grad 1 auf. Es gab keine CRS Grad 4 oder tödliche Fälle von CRS. Das CRS klang bei allen Patienten bis auf einen ab. Ein Patient brach die Behandlung aufgrund von CRS ab.
Bei Patienten mit CRS waren die häufigsten Manifestationen von CRS Fieber (98,7 %), Hypotonie (22,4 %), Schüttelfrost (17,1 %) und Hypoxie (14,5 %). Mit CRS assoziierte Ereignisse vom Grad 3 oder höher waren Hypotonie (6,6 %), Hypoxie (5,3 %), Fieber (3,9 %), Schüttelfrost (1,3 %) und Diarrhö (1,3 %).
Ein CRS jeglichen Grades trat bei 34,9 % der Patienten nach der ersten 2,5-mg-Dosis Columvi an Tag 8 von Zyklus 1 mit einer medianen Zeit bis zum Auftreten (ab Beginn der Infusion) von 12,6 Stunden (Bereich: 4,4 bis 54,7 Stunden) und einer medianen Dauer von 19,8 Stunden (Bereich: 2,0 bis 168,0 Stunden) auf; bei 14,4 % der Patienten nach der 10-mg-Dosis an Tag 15 von Zyklus 1 mit einer medianen Zeit bis zum Auftreten von 22,8 Stunden (Bereich: 7,4 bis 81,2 Stunden) und einer medianen Dauer von 10,6 Stunden (Bereich: 1,0 bis 248,5 Stunden); und bei 9,3 % der Patienten nach der 30-mg-Dosis in Zyklus 2 mit einer medianen Zeit bis zum Auftreten von 23,5 Stunden (Bereich: 14,7 bis 33,4 Stunden) und einer medianen Dauer von 18,4 Stunden (Bereich: 8,3 bis 137,0 Stunden). CRS wurde bei 6,7 % der Patienten in Zyklus 3 und bei 11,0 % der Patienten nach Zyklus 3 berichtet.
Ein CRS vom Grad ≥ 2 trat bei 10,5 % der Patienten nach der ersten Dosis Columvi (2,5 mg) auf, mit einer medianen Zeit bis zum Auftreten von 12,0 Stunden (Bereich: 4,4 bis 30,5 Stunden) und einer medianen Dauer von 42,3 Stunden (Bereich: 3,5 bis 143,7 Stunden). Bei der Mehrzahl (14/18) der Patienten, bei denen ein CRS vom Grad ≥ 2 auftrat, trat das CRS innerhalb von 8 Stunden nach Beginn der ersten Dosis Columvi (2,5 mg) auf oder sie hatten ≥ 1,5 Stunden vor dem Auftreten anderer Symptome eines CRS vom Grad ≥ 2 Fieber. Nach einer 10-mg-Dosis Columvi an Tag 15 von Zyklus 1 sank die Inzidenz eines CRS vom Grad ≥ 2 auf 1,8 % der Patienten mit einer medianen Zeit bis zum Auftreten von 22,3 Stunden (Bereich: 7,4 bis 22,8 Stunden) und einer medianen Dauer von 37,0 Stunden (Bereich: 34,8 bis 248,5 Stunden). Nach einer 30-mg-Dosis Columvi an Tag 1 von Zyklus 2 traten keine CRS-Ereignisse vom Grad ≥ 2 auf. Drei Patienten (2,0 %) hatten über Zyklus 2 hinaus ein CRS vom Grad ≥ 2 (alle Ereignisse vom Grad 2).
Von den 172 Patienten zeigten 2 Patienten (1,2 %) erhöhte Leberfunktionswerte (AST und ALT > 3 x ULN), die gleichzeitig mit einem CRS berichtet wurden.
Von den 76 Patienten mit CRS jeglichen Grades wurden 28 Patienten (36,8 %) mit Tocilizumab, 39 Patienten (51,3 %) mit Corticosteroiden und 18 Patienten (23,7 %) sowohl mit Tocilizumab als auch mit Corticosteroiden behandelt.
Von den 22 Patienten, bei denen nach der Behandlung mit Columvi ein CRS vom Grad ≥ 2 auftrat, erhielten 16 (72,7 %) Tocilizumab, 15 (68,2 %) Corticosteroide und 12 (54,5 %) sowohl Tocilizumab als auch Corticosteroide. Elf Patienten (50,0 %) erhielten Sauerstoff. Alle 4 Patienten (18,2 %) mit CRS Grad 3 erhielten einen einzigen Vasopressor.
Bei 19,8 % der Patienten kam es nach der Anwendung von Columvi zu Krankenhausaufenthalten aufgrund eines CRS, und die gemeldete mediane Dauer des Krankenhausaufenthalts betrug 5 Tage (Bereich: 2 bis 85 Tage).
Immuneffektorzellen-assoziiertes Neurotoxizitätssyndrom
ICANS, einschließlich Grad 3 und höher, wurde in klinischen Studien und Erfahrungen nach dem Inverkehrbringen gemeldet. Die häufigsten klinischen Manifestationen von ICANS waren Verwirrtheit, getrübter Bewusstseinszustand, Desorientiertheit, Krampfanfälle, Aphasie und Dysgraphie. Auf der Grundlage der verfügbaren Daten trat in den meisten Fällen der Beginn der neurologischen Toxizität gleichzeitig mit CRS auf.
Der beobachtete Zeitraum bis zum Einsetzen der meisten Fälle von ICANS betrug 1-7 Tage mit einem Median von 2 Tagen nach der letzten Dosis. Es wurde berichtet, dass nur wenige Ereignisse mehr als einen Monat nach Beginn der Behandlung mit Columvi auftraten.
Schwerwiegende Infektionen
Bei 15,9 % der Patienten, die Columvi Monotherapie erhielten, wurden schwerwiegende Infektionen berichtet. Die häufigsten schwerwiegenden Infektionen, die bei ≥ 2 % der Patienten berichtet wurden, waren Sepsis (4,1 %), COVID-19 (3,4 %) und COVID-19-Pneumonie (2,8 %). Infektionsbedingte Todesfälle wurden bei 4,8 % der Patienten berichtet (aufgrund von Sepsis, COVID-19-Pneumonie und COVID-19). Bei vier Patienten (2,8 %) traten schwerwiegende Infektionen gleichzeitig mit einer Neutropenie vom Grad 3 oder 4 auf.
Schwerwiegende Infektionen wurden bei 22,7 % der Patienten berichtet, die Columvi mit Gemcitabin und Oxaliplatin erhielten. Die häufigsten schwerwiegenden Infektionen, die bei ≥ 2 % der Patienten berichtet wurden, waren Pneumonie (5,8 %), COVID-19 (4,7 %) und Infektionen der unteren Atemwege (2,9 %). Infektionsbedingte Todesfälle wurden bei 3,5 % der Patienten berichtet (aufgrund von COVID-19, Pneumonie, Atemwegsinfektion und septischem Schock). Bei einem Patienten (0,6 %) trat gleichzeitig mit einer Neutropenie vom Grad 3 eine schwerwiegende Infektion (Pneumonie) auf.
Pneumonitis
Pneumonitis-Ereignisse (ausgenommen Pneumonie infektiöser Ätiologie) wurden bei 2 Patienten (1,2 %) berichtet, die Columvi mit Gemcitabin und Oxaliplatin erhielten, wobei beide Ereignisse tödlich verliefen. Die mediane Zeit bis zum Auftreten der Pneumonitis ab der ersten Gabe von Columvi betrug 168 Tage (Bereich: 102 bis 255 Tage).
Kolitis
Kolitis (Grad 4) wurde bei 1 Patienten (0,7 %) berichtet, der Columvi als Monotherapie erhielt, wobei die Zeit bis zum Auftreten ab der ersten Columvi Dosis 104 Tage betrug.
Kolitis-Ereignisse (ausgenommen infektiöse Ätiologie) wurden bei 4/172 Patienten (2,3 %) berichtet, die Columvi mit Gemcitabin und Oxaliplatin erhielten. Zwei Patienten (1,2 %) hatten Ereignisse vom Grad 3. Die mediane Zeit bis zum Auftreten der Kolitis nach der ersten Gabe von Columvi betrug 154 Tage (Bereich: 115 bis 187 Tage).
Opportunistische Infektionen
CMV-Ereignisse wurden bei 6/467 Patienten (1,3 %) berichtet, die Columvi als Monotherapie erhielten, wobei bei 1 Patienten (0,2 %) eine CMV-Chorioretinitis vom Grad 3 auftrat. Pneumocystis jirovecii-Pneumonie wurde bei 4/467 Patienten (0,9 %) berichtet, wobei 3 von ihnen (0,6 %) Ereignisse vom Grad 3 hatten.
CMV-Ereignisse wurden bei 11 Patienten (6,4 %) berichtet, die Columvi mit Gemcitabin und Oxaliplatin erhielten, wobei bei 1 Patienten (0,6 %) eine CMV-Virämie von Grad 3 auftrat. Eine orale Candidose wurde bei 3 Patienten (1,7 %) berichtet, die alle vom Grad 1 – 2 waren. Pneumocystis jirovecii-Pneumonie (Grad 3) wurde bei 1 Patienten (0,6 %) berichtet, derselbe Patient hatte eine CMV-Virämie von Grad 3. Borrelia-Meningitis (Grad 2) wurde bei 1 Patienten (0,6 %) berichtet.
Neutropenie
Neutropenie (einschließlich erniedrigter Neutrophilenzahl) wurde bei 40,0 % der Patienten und schwere Neutropenie (Grad 3 oder 4) wurde bei 29,0 % der Patienten berichtet, die Columvi Monotherapie erhielten. Die mediane Zeit bis zum Auftreten des ersten Neutropenie-Ereignisses betrug 29 Tage (Bereich: 1 – 203 Tage). Eine länger als 30 Tage anhaltende Neutropenie trat bei 11,7 % der Patienten auf. Die Mehrzahl der Patienten mit Neutropenie (79,3 %) wurde mit Granulozyten-Kolonie-stimulierendem Faktor (granulocyte colony stimulating factor - G-CSF) behandelt. Febrile Neutropenie wurde bei 3,4 % der Patienten berichtet.
Tumor Flare (Schub der Tumorerkrankung)
Ein Tumor Flare wurde bei 11,7 % der Patienten berichtet, die Columvi Monotherapie erhielten, einschließlich eines Tumor Flares von Grad 2 bei 4,8 % der Patienten und eines Tumor Flares von Grad 3 bei 2,8 % der Patienten. Es wurde über einen Tumor Flare berichtet, der bei Lymphknoten im Kopf- und Halsbereich mit Schmerzen und bei Lymphknoten im Thorax mit Symptomen von Atemnot aufgrund der Entwicklung eines Pleuraergusses einherging. Die meisten Tumor Flares (16/17) traten während Zyklus 1 auf, und nach Zyklus 2 wurden keine weiteren Tumor Flares berichtet. Die mediane Zeit bis zum Auftreten eines Tumor Flares jeglichen Grades betrug 2 Tage (Bereich: 1 – 16 Tage) und die mediane Dauer betrug 3,5 Tage (Bereich: 1 – 35 Tage).
Von den 11 Patienten, bei denen ein Tumor Flare mit einem Grad von ≥ 2 auftrat, erhielten 2 Patienten (18,2 %) Analgetika, 6 Patienten (54,5 %) erhielten Corticosteroide und Analgetika einschließlich Morphinderivate, 1 Patient (9,1 %) erhielt Corticosteroide und Antiemetika und 2 Patienten (18,2 %) benötigten keine Behandlung. Alle Tumor Flares klangen ab, außer bei einem Patienten mit einem Ereignis vom Schwergrad ≥ 2. Kein Patient brach die Behandlung aufgrund eines Tumor Flares ab.
Tumorlysesyndrom (TLS)
Ein TLS wurde bei 2 Patienten (1,4 %) berichtet, die Columvi Monotherapie erhielten, und war in beiden Fällen vom Grad 3. Die mediane Zeit bis zum Auftreten des TLS betrug 2 Tage, die mediane Dauer 4 Tage (Bereich: 3 – 5 Tage).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51‑59
63225 Langen
Tel: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
anzuzeigen.
Aus klinischen Studien liegen keine Erfahrungen mit Überdosierungen vor. Im Falle einer Überdosierung müssen die Patienten engmaschig auf Anzeichen oder Symptome von Nebenwirkungen überwacht werden und es muss eine angemessene symptomatische Behandlung eingeleitet werden.
Pharmakotherapeutische Gruppe: Antineoplastische Substanzen, andere monoklonale Antikörper und Antikörper-Wirkstoff-Konjugate, ATC-Code: L01FX28
Wirkmechanismus
Glofitamab ist ein bispezifischer monoklonaler Antikörper, der bivalent an CD20 bindet, das auf der Oberfläche von B-Zellen exprimiert wird, und monovalent an CD3 im T-Zell-Rezeptor-Komplex, das auf der Oberfläche von T-Zellen exprimiert wird. Durch gleichzeitige Bindung an CD20 auf der B-Zelle und CD3 auf der T-Zelle führt Glofitamab zur Bildung einer immunologischen Synapse mit nachfolgender T-Zell-Aktivierung und -Proliferation, Sekretion von Zytokinen und Freisetzung von zytolytischen Proteinen, was zur Lyse von CD20-exprimierenden B-Zellen führt.
Pharmakodynamische Wirkungen
In der Studie NP30179 hatten bereits 84 % (84/100) der Patienten vor der Vorbehandlung mit Obinutuzumab eine B-Zell-Depletion (< 70 Zellen/µl). Der Anteil der Patienten mit B-Zell-Depletion erhöhte sich nach der Vorbehandlung mit Obinutuzumab auf 100 % (94/94) der Patienten vor Beginn der Behandlung mit Columvi, und die B-Zell-Zahlen blieben während der Behandlung mit Columvi niedrig.
Während Zyklus 1 (Dosissteigerung) wurden 6 Stunden nach der Infusion von Columvi vorübergehende Anstiege der IL-6-Plasmaspiegel beobachtet, die 20 Stunden nach der Infusion erhöht blieben und vor der nächsten Infusion auf die Ausgangswerte zurückgingen.
In der Studie GO41944 (STARGLO) waren 63,9 % (115/180) der Patienten bereits vor der Vorbehandlung mit Obinutuzumab B-Zell-depletiert (< 70 Zellen/µl). Der Anteil an Patienten mit B-Zell-Depletion stieg nach Obinutuzumab-Vorbehandlung vor Beginn der Behandlung mit Columvi auf 79,4 % (143/180), und die B-Zell-Zahlen blieben während der Behandlung mit Columvi niedrig.
Kardiale Elektrophysiologie
In der Studie NP30179 kam es bei 16/145 Patienten, die Columvi erhielten, nach Baseline zu einem QTc-Wert von > 450 ms. Einer dieser Fälle wurde vom Prüfarzt als klinisch signifikant eingestuft. Kein Patient brach die Behandlung aufgrund einer QTc-Verlängerung ab.
In der Studie GO41944 (STARGLO) kam es bei 16/172 Patienten, die Columvi erhielten, nach Baseline zu einem QTc-Wert von > 450 ms. Kein Patient brach die Behandlung aufgrund einer QTc‑Verlängerung ab.
Klinische Wirksamkeit und Sicherheit
Rezidiviertes oder refraktäres DLBCL
Columvi Monotherapie
Es wurde eine unverblindete, multizentrische Multikohortenstudie (NP30179) durchgeführt, um Columvi bei Patienten mit rezidiviertem oder refraktärem B-Zell-Non-Hodgkin-Lymphom zu untersuchen. In der einarmigen DLBCL-Monotherapie-Kohorte (n = 108) mussten Patienten mit rezidiviertem oder refraktärem DLBCL zuvor mindestens zwei systemische Therapielinien, einschließlich eines monoklonalen Anti-CD20-Antikörpers und eines Anthrazyklin-Wirkstoffs, erhalten haben. Patienten mit FL3b- und Richter-Transformation waren nicht geeignet. Es wurde erwartet, dass die Patienten einen CD20-positiven DLBCL aufwiesen, aber die Erfüllung der Biomarker-Kriterien war keine Voraussetzung für den Studieneinschluss (siehe Abschnitt 4.4).
Von der Studie ausgeschlossen waren Patienten mit einem Eastern Cooperative Oncology Group (ECOG)-Performance-Status ≥ 2, signifikanter kardiovaskulärer Erkrankung (z. B. Herzinsuffizienz Klasse III oder IV der New York Heart Association, Myokardinfarkt innerhalb der letzten 6 Monate, instabile Arrhythmien oder instabile Angina pectoris), signifikanter aktiver Lungenerkrankung, eingeschränkter Nierenfunktion (CrCl < 50 ml/min mit erhöhtem Serumkreatininspiegel), aktiver Autoimmunerkrankung, die eine immunsuppressive Therapie erfordert, aktiven Infektionen (d. h. chronisch aktive Epstein-Barr-Virus(EBV)‑Infektion, akute oder chronische Hepatitis C, Hepatitis B, humanes Immundefizienz-Virus (HIV)), progressiver multifokaler Leukoenzephalopathie, aktuellem oder früherem Zentralnervensystem(ZNS)‑Lymphom oder einer ZNS‑Erkrankung, Makrophagenaktivierungssyndrom/hämophagozytischer Lymphohistiozytose in der Anamnese, früherer allogener Stammzelltransplantation, früherer Organtransplantation oder hepatischer Transaminasen ≥ 3 x ULN.
Alle Patienten erhielten eine Vorbehandlung mit Obinutuzumab an Tag 1 von Zyklus 1. Die Patienten erhielten 2,5 mg Columvi an Tag 8 von Zyklus 1, 10 mg Columvi an Tag 15 von Zyklus 1 und 30 mg Columvi an Tag 1 von Zyklus 2 gemäß dem Dosissteigerungsschema. Die Patienten erhielten weiterhin 30 mg Columvi an Tag 1 der Zyklen 3 bis 12. Die Dauer der einzelnen Zyklen betrug 21 Tage. Die Patienten erhielten im Median 5 Zyklen der Behandlung mit Columvi (Bereich: 1 – 13 Zyklen), wobei 34,7 % der Patienten 8 oder mehr Zyklen und 25,7 % der Patienten 12 Zyklen der Behandlung mit Columvi erhielten.
Die demographischen Merkmale und Krankheitscharakteristika bei Baseline waren wie folgt: medianes Alter 66 Jahre (Bereich: 21 – 90 Jahre), wobei 53,7 % 65 Jahre oder älter und 15,7 % 75 Jahre oder älter waren; 69,4 % waren Männer; 74,1 % waren Weiße, 5,6 % waren Asiaten und 0,9 % waren Schwarze oder Amerikaner afrikanischer Herkunft; 5,6 % waren hispanischer Herkunft oder Lateinamerikaner; und der ECOG-Performance-Status betrug 0 (46,3 %) oder 1 (52,8 %). Die meisten Patienten (71,3 %) hatten ein nicht näher spezifiziertes DLBCL, 7,4 % hatten DLBCL, das aus einem follikulären Lymphom hervorgegangen war, 8,3 % hatten ein hochgradiges B-Zell-Lymphom (HGBCL) oder eine andere Histologie, die aus einem follikulären Lymphom hervorgegangen war, 7,4 % hatten ein HGBCL und 5,6 % ein primär mediastinales großzelliges B-Zell-Lymphom (PMBCL). Die mediane Anzahl vorheriger Therapielinien betrug 3 (Bereich: 2 – 7), wobei 39,8 % der Patienten 2 vorherige Therapielinien und 60,2 % 3 oder mehr vorherige Therapielinien erhalten hatten. Alle Patienten hatten zuvor eine Chemotherapie erhalten (alle Patienten erhielten eine Alkylatortherapie und 98,1 % der Patienten eine Anthrazyklin-Therapie) und alle Patienten hatten zuvor eine monoklonale Anti-CD20-Antikörpertherapie erhalten; 35,2 % der Patienten hatten zuvor eine CAR-T-Zell-Therapie erhalten und 16,7 % der Patienten hatten eine autologe Stammzelltransplantation erhalten. Bei den meisten Patienten (89,8 %) lag eine refraktäre Erkrankung vor, 60,2 % der Patienten hatten eine primär refraktäre Erkrankung und 83,3 % der Patienten erwiesen sich als refraktär gegenüber ihrer letzten vorangegangenen Therapie.
Der primäre Wirksamkeitsendpunkt war die Rate des vollständigen Ansprechens (complete response - CR), beurteilt durch einen unabhängigen Prüfungsausschuss (independent review committee – IRC) anhand der Lugano-Kriterien von 2014. Die gesamte mediane Nachbeobachtungsdauer betrug 15 Monate (Bereich: 0 – 21 Monate). Die sekundären Wirksamkeitsendpunkte umfassten die Gesamtansprechrate (overall response rate - ORR), die Dauer des Ansprechens (duration of response - DOR), die Dauer des vollständigen Ansprechens (duration of complete response - DOCR) und die Zeit bis zum ersten vollständigen Ansprechen (time to first complete response - TFCR), beurteilt durch das IRC.
Die Ergebnisse zur Wirksamkeit sind in Tabelle 8 zusammengefasst.
Tabelle 8: Zusammenfassung der Wirksamkeit bei Patienten mit rezidiviertem oder refraktärem DLBCL
Wirksamkeitsendpunkte |
Columvi |
Vollständiges Ansprechen (CR) | |
Patienten mit vollständigem Ansprechen, n (%) |
38 (35,2) |
95-%-KI |
[26,24; 44,96] |
Gesamtansprechrate (ORR) | |
Patienten mit CR oder PR, n (%) |
54 (50,0) |
95-%-KI |
[40,22; 59,78] |
Dauer des vollständigen Ansprechens (DOCR)1 | |
Mediane DOCR, Monate [95-%-KI] |
NE [18,4; NE] |
Bereich, Monate |
02 – 202 |
12-Monats-DOCR, % [95-%-KI]3 |
74,6 [59,19; 89,93] |
Dauer des Ansprechens (DOR)4 | |
Mediane Dauer, Monate [95-%-KI] |
14,4 [8,6; NE] |
Bereich, Monate |
02 – 202 |
Zeit bis zum ersten vollständigen Ansprechen (TFCR) | |
Mediane TFCR, Tage [95-%-KI] |
42 [41; 47] |
Bereich, Tage |
31 – 308 |
KI = Konfidenzintervall; NE = nicht schätzbar (not estimable); PR = partial response – partielles Ansprechen.
Die Hypothese wurde am primären Endpunkt, der vom IRC beurteilten CR-Rate, getestet.
1 DOCR ist definiert als Datum des ersten vollständigen Ansprechens bis zur Krankheitsprogression oder bis zum Tod jeglicher Ursache.
2 Zensierte Beobachtungen.
3 Ereignisfreie Raten basierend auf Kaplan-Meier-Schätzungen.
4 DOR ist definiert als Datum des ersten Ansprechens (PR oder CR) bis zur Krankheitsprogression oder bis zum Tod jeglicher Ursache.
Die mediane Nachbeobachtungszeit für DOR betrug 12,8 Monate (Bereich: 0 – 20 Monate).
Columvi in Kombination mit Gemcitabin und Oxaliplatin
Die Wirksamkeit von Columvi in Kombination mit Gemcitabin und Oxaliplatin (Columvi+GemOx) wurde in der Studie GO41944 (STARGLO), einer offenen, multizentrischen, randomisierten klinischen Studie mit 274 Patienten mit rezidiviertem oder refraktärem DLBCL, das nicht anderweitig spezifiziert ist (DLBCL NOS), untersucht.
Die Studie schloss Patienten mit DLBCL NOS ein, die nur eine vorherige Therapielinie erhalten hatten und die nicht für eine autologe Stammzelltransplantation (ASCT) in Frage kamen oder die ≥ 2 vorherige Therapien erhalten hatten. Patienten mussten einen ECOG-Performance-Status ≤ 2 aufweisen, eine CrCl ≥ 30 ml/min, Lebertransaminasen ≤ 2,5 × ULN, keine signifikante kardiovaskuläre Erkrankung (wie Herzerkrankung nach New York Heart Association Klasse III oder IV, Myokardinfarkt innerhalb der letzten 3 Monate, instabile Arrhythmien oder instabile Angina) und kein aktuelles oder früheres ZNS-Lymphom oder ZNS-Erkrankung, keine aktive Autoimmunerkrankung, die eine immunsuppressive Therapie erfordert, keine aktive Infektionen (d. h. chronisches aktives EBV, aktive Hepatitis B, Hepatitis C) und keine der folgenden Erkrankungen in der Vorgeschichte: HIV, progressive multifokale Leukenzephalopathie, hämophagozytische Lymphohistiozytose, vorherige allogene Stammzelltransplantation oder vorherige Organtransplantation. Patienten mit HGBCL, PMBCL oder einer Transformation einer indolenten Erkrankung in ein DLBCL in der Vorgeschichte wurden ausgeschlossen.
Patienten, die nur eine vorherige Therapielinie erhalten hatten, wurden nicht als Kandidaten für eine Transplantation in Betracht gezogen, wenn mindestens eines der folgenden Kriterien auf sie zutraf: Alter ≥ 70 Jahre, ECOG-Performance-Status 2, linksventrikuläre Ejektionsfraktion ≤ 40 %, unzureichendes Ansprechen auf Salvage-Therapie vor einer ASCT, CrCl ≤ 45 ml/min, andere Komorbiditäten oder Kriterien, die eine Transplantation ausschließen, basierend auf lokalen Praxisstandards oder nach Ansicht des Prüfarztes, oder Ablehnung einer Hochdosis-Chemotherapie und/oder Transplantation durch den Patienten.
Die Patienten wurden im Verhältnis 2:1 randomisiert und erhielten 8 Zyklen lang Columvi+GemOx (n = 183) oder Rituximab in Kombination mit Gemcitabin plus Oxaliplatin (R-GemOx; n = 91), gefolgt von 4 zusätzlichen Zyklen einer Columvi Monotherapie für Patienten im Columvi+GemOx-Arm. Die Randomisierung wurde nach Anzahl der vorherigen systemischen Therapielinien für DLBCL (1 vs. ≥ 2) und dem Ergebnis der letzten systemischen Therapie (rezidiviert vs. refraktär) stratifiziert.
Im Behandlungsarm mit Columvi+GemOx erhielten die Patienten eine Vorbehandlung mit Obinutuzumab in Zyklus 1, Tag 1, gefolgt von 2,5 mg Columvi in Zyklus 1, Tag 8, 10 mg Columvi in Zyklus 1, Tag 15 und 30 mg Columvi in Zyklus 2, Tag 1, gemäß dem Dosissteigerungsschema. Die Patienten erhielten weiterhin 30 mg Columvi an Tag 1 der Zyklen 3 bis 12. Gemcitabin (1 000 mg/m2) und Oxaliplatin (100 mg/m2) wurden am Tag 2 von Zyklus 1 und dann am Tag 1 der nachfolgenden Zyklen bis Zyklus 8 intravenös verabreicht. Die Dauer jedes Zyklus betrug in beiden Armen 21 Tage. Die Patienten erhielten im Median 11 Zyklen der Columvi Behandlung (Bereich: 1 bis 13 Zyklen); 64,5 % erhielten 8 oder mehr Zyklen und 44,8 % erhielten 12 Zyklen der Columvi Behandlung.
Die Charakteristika bezüglich Demographie und Krankheitsbild zur Baseline waren: medianes Alter 68 Jahre (Bereich: 20 bis 88 Jahre), wobei 62,8 % 65 Jahre oder älter und 23,7 % 75 Jahre oder älter waren; 57,7 % waren Männer; 42 % Weiße, 50 % Asiaten und 1,1 % Schwarze oder Afroamerikaner; 5,8 % Hispano- oder Lateinamerikaner; und der ECOG-Performance-Status betrug 0 (43,3 %), 1 (46,6 %) oder 2 (10,1 %). Die Mehrzahl der Patienten (62,8 %) hatte eine vorherige systemische Therapielinie erhalten; 37,2 % der Patienten hatten 2 oder mehr vorherige Therapielinien erhalten. Alle Patienten hatten zuvor eine Chemotherapie erhalten und die meisten (98,5 %) hatten zuvor eine Therapie mit monoklonalen Anti-CD20-Antikörpern erhalten; 7,7 % der Patienten hatten zuvor eine CAR-T-Zell-Therapie erhalten und 4,0 % der Patienten hatten eine autologe Stammzelltransplantation erhalten. Die Mehrheit der Patienten (66,8 %) hatte eine refraktäre Erkrankung, 55,8 % der Patienten hatten eine primär refraktäre Erkrankung und 60,6 % der Patienten waren refraktär gegenüber ihrer letzten vorherigen Therapie. Die häufigsten Gründe, warum Patienten nicht als Kandidaten für eine Transplantation in Betracht gezogen wurden, waren Alter (42,3 %), Verweigerung einer Hochdosis-Chemotherapie und/oder einer Transplantation durch den Patienten (34,7 %) und unzureichendes Ansprechen auf die Salvage-Therapie (9,9 %).
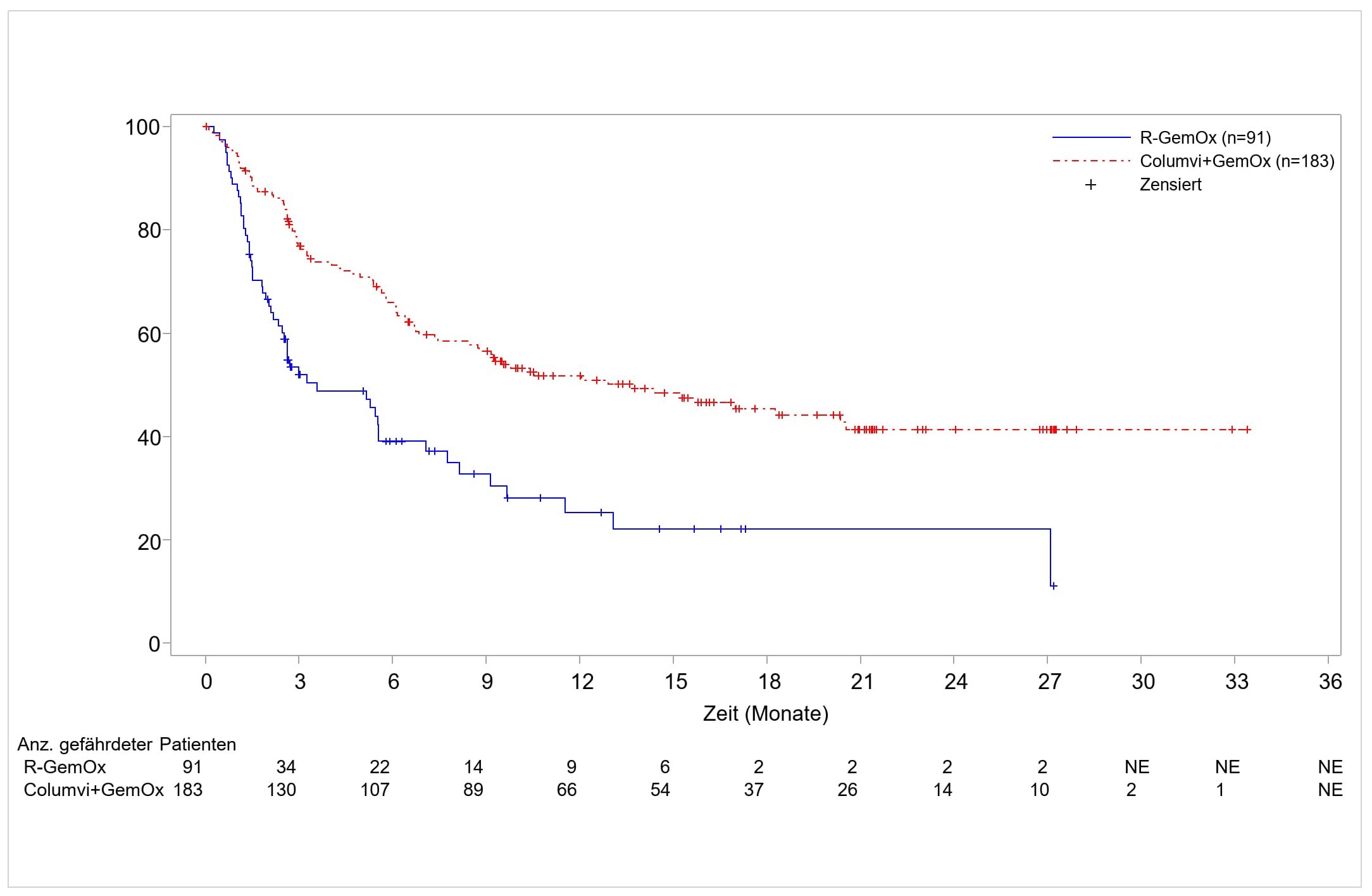
Der primäre Wirksamkeitsendpunkt war das Gesamtüberleben (OS). Zum Zeitpunkt der präspezifizierten primären Analyse wurde bei den in den Columvi+GemOx-Arm randomisierten Patienten im Vergleich zu den in den R-GemOx-Arm randomisierten Patienten eine statistisch signifikante Verbesserung des OS beobachtet. (HR 0,59, 95-%-KI: 0,40, 0,89; p-Wert = 0,011). Das mediane OS im R-GemOx-Arm betrug 9,0 Monate (95-%-KI: 7,3; 14,4) und wurde im Columvi+GemOx-Arm nicht erreicht (95-%-KI: 13,8; NE). Statistisch signifikante Verbesserungen des progressionsfreien Überlebens (PFS, progression free survival) und der CR-Rate, beurteilt durch eine IRC, wurden auch mit Columvi+GemOx gegenüber R-GemOx beobachtet. Das mediane PFS betrug 12,1 Monate (95-%-KI: 6,8; 18,3) im Columvi+GemOx-Arm gegenüber 3,3 Monaten (95-%-KI: 2,5; 5,6) im R-GemOx-Arm (HR 0,37; 95-%-KI: 0,25; 0,55; p-Wert < 0,001). Die Rate des vollständigen Ansprechens betrug unter Columvi+GemOx 50,3 % versus 22,0 % unter R-GemOx, ein Unterschied von 28,3 % (p-Wert < 0,001).
Die Ergebnisse zum Gesamtüberleben, zum PFS und zur CR aus einer aktualisierten Analyse, die nach einer weiteren Beobachtungszeit von 10,5 Monaten durchgeführt wurde, zeigen weiterhin einen Nutzen von Columvi+GemOx gegenüber R-GemOx. Die wichtigsten Ergebnisse sind in Tabelle 9 zusammengefasst. Kaplan-Meier-Diagramme für OS und PFS aus der aktualisierten Analyse sind in Abbildung 1 bzw. Abbildung 2 dargestellt. Eine explorative Subgruppenanalyse zum Zeitpunkt der aktualisierten Analyse zeigte bei den in Europa eingeschlossenen Patienten eine OS-Hazard-Ratio von 1,09 (95‑%‑KI: 0,54; 2,18) und eine PFS-Hazard-Ratio von 0,84 (95‑%‑KI: 0,44; 1,59).
Tabelle 9: Wirksamkeit bei Patienten mit rezidiviertem oder refraktärem DLBCL, die mit Columvi in Kombination mit Gemcitabin und Oxaliplatin (ITT) behandelt wurden
Wirksamkeitsendpunkte |
Aktualisierte Analyse |
|
Columvi+GemOx |
R-GemOx |
|
Gesamtüberleben | ||
Anzahl der Todesfälle (%) |
80 (43,7) |
52 (57,1) |
Median (95-%-KI), Monate |
25,5 (18,3; NE) |
12,9 (7,9; 18,5) |
HR (95-%-KI) |
0,62 (0,43; 0,88) |
|
Progressionsfreies Überleben – IRC-bewertet | ||
Anzahl (%) der Patienten mit Ereignissen |
90 (49,2) |
54 (59,3) |
Median (95-%-KI), Monate |
13,8 (8,7; 20,5) |
3,6 (2,5; 7,1) |
HR (95-%-KI) |
0,40 (0,28; 0,57) |
|
Komplette Ansprechrate - IRC-bewertet | ||
Patienten mit Ansprechen (%) |
107 (58,5) |
23 (25,3) |
Unterschied der Ansprechrate (95-%-KI), % |
33,2 (20,9; 45,5) |
|
Objektive Ansprechrate - IRC-bewertet | ||
Patienten mit Ansprechen (%) (CR, PR) |
125 (68,3) |
37 (40,7) |
Unterschied der Ansprechrate (95-%-KI), % |
27,7 (14,7; 40,6) |
|
KI = Konfidenzintervall; HR = Hazard Ratio; NE = nicht schätzbar.
Abbildung 1: Kaplan-Meier-Kurve des Gesamtüberlebens in Studie GO41944 (STARGLO, aktualisierte Analyse, ITT)
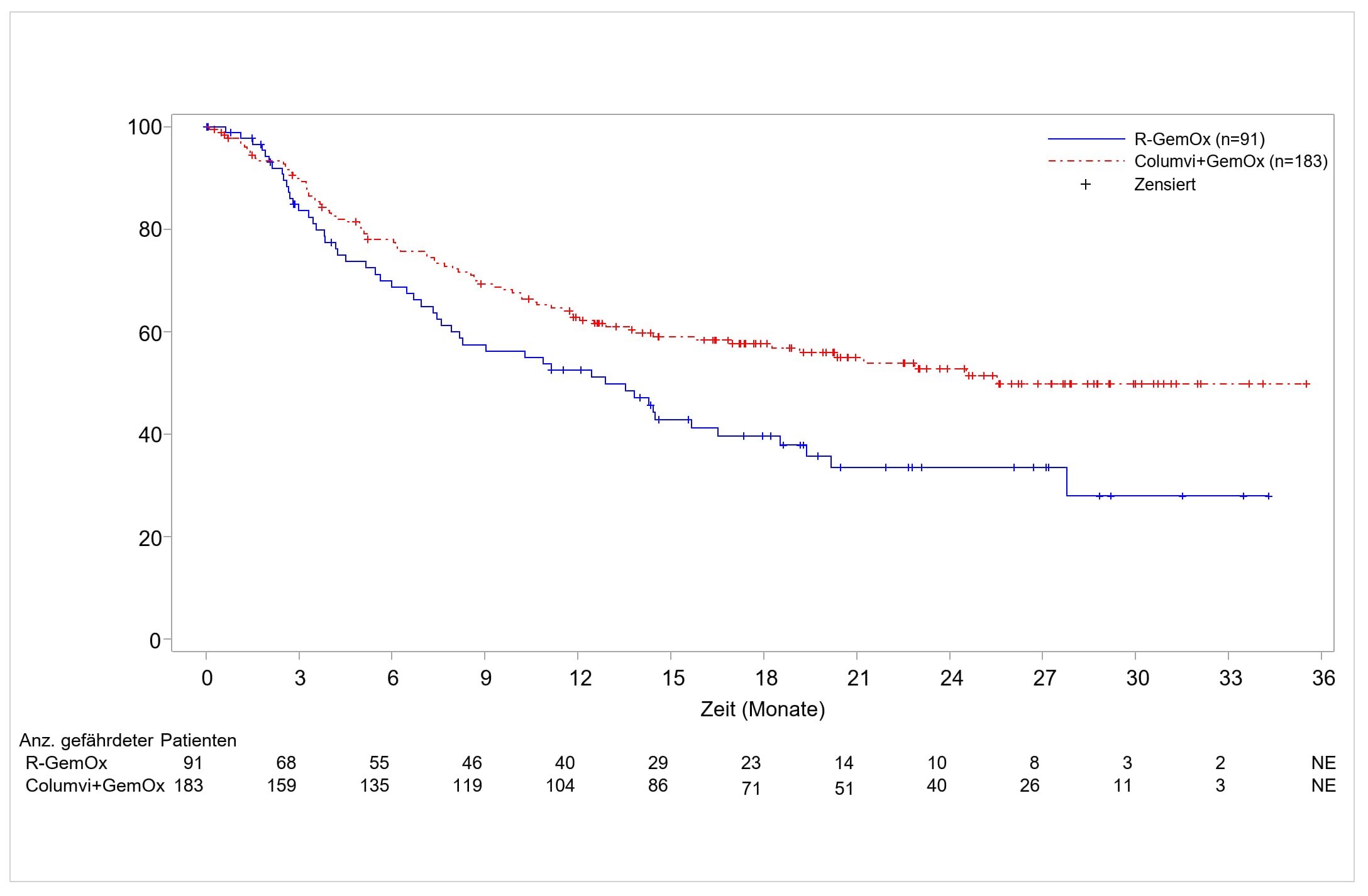
Abbildung 2: Kaplan-Meier-Kurve des vom IRC beurteilten progressionsfreien Überlebens in der Studie GO41944 (STARGLO, aktualisierte Analyse; ITT)
Immunogenität
Von den 608 Patienten in den Studien waren nur vier Patienten (0,7 %) zu Studienbeginn Anti-Glofitamab-Antikörper-negativ und wurden nach der Behandlung positiv. Aufgrund der begrenzten Anzahl von Patienten mit Antikörpern gegen Glofitamab können keine Schlussfolgerungen über einen möglichen Einfluss der Immunogenität auf die Wirksamkeit oder Sicherheit gezogen werden.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Columvi eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen zur Behandlung von reifen B-Zell-Neoplasien gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Nicht kompartimentelle Analysen weisen darauf hin, dass die Serumkonzentration von Glofitamab am Ende der Infusion den maximalen Wert (Cmax) erreicht und dass die Abnahme biexponentiell verläuft. Glofitamab zeigt über den untersuchten Dosisbereich (0,005 – 30 mg) hinweg und unabhängig vom Zeitpunkt eine lineare und zur applizierten Dosis proportionale Pharmakokinetik.
Resorption
Columvi wird als intravenöse Infusion verabreicht. Die maximale Konzentration von Glofitamab (Cmax) wurde am Ende der Infusion erreicht.
Verteilung
Nach intravenöser Verabreichung betrug das zentrale Verteilungsvolumen 3,34 l, und lag damit nahe am Gesamt-Serumvolumen. Das periphere Verteilungsvolumen betrug 2,35 l.
Biotransformation
Die Metabolisierung von Glofitamab wurde nicht untersucht. Antikörper werden hauptsächlich über den Katabolismus eliminiert.
Elimination
Die Serumkonzentration-Zeit-Daten von Glofitamab werden durch ein populationspharmakokinetisches Modell mit zwei Kompartimenten und sowohl einer zeitunabhängigen als auch einer zeitvariablen Clearance beschrieben.
Der zeitunabhängige Clearance-Pfad wurde auf 0,633 l/Tag und der initiale zeitvariable Clearance-Pfad auf 0,814 l/Tag geschätzt, mit einer exponentiellen Abnahme im Zeitverlauf (Kdes ~ 1,5/Tag). Die geschätzte Halbwertszeit der Abnahme vom anfänglichen Gesamtclearance-Wert bis zur zeitunabhängigen Clearance wurde auf 0,471 Tage geschätzt.
Basierend auf der populationspharmakokinetischen Analyse beträgt die effektive Halbwertszeit in der linearen Phase (d. h. nachdem der Beitrag der zeitvariablen Clearance auf einen vernachlässigbaren Wert geschrumpft ist) 7,92 Tage (geometrisches Mittel, 95-%-KI: 4,69; 11,90).
Besondere Patientengruppen
Ältere Patienten
Basierend auf der populationspharmakokinetischen Analyse wurden keine Unterschiede in der Glofitamab-Exposition von Patienten, die 65 Jahre oder älter waren, und der von Patienten, die jünger als 65 Jahre waren, festgestellt.
Nierenfunktionsstörung
Die populationspharmakokinetische Analyse von Glofitamab zeigte, dass die Kreatinin-Clearance keinen Einfluss auf die Pharmakokinetik von Glofitamab hat. Die Pharmakokinetik von Glofitamab bei Patienten mit leichter oder mittelschwerer Nierenfunktionsstörung (CrCl 30 bis < 90 ml/min) war ähnlich wie bei Patienten mit normaler Nierenfunktion. Columvi wurde bei Patienten mit schwerer Nierenfunktionsstörung nicht untersucht.
Leberfunktionsstörung
Populationspharmakokinetische Analysen zeigten, dass eine leichte Leberfunktionsstörung keinen Einfluss auf die Pharmakokinetik von Glofitamab hat. Die Pharmakokinetik von Glofitamab bei Patienten mit leichter Leberfunktionsstörung (Gesamtbilirubin > ULN bis ≤ 1,5 x ULN oder AST > ULN) war ähnlich wie bei Patienten mit normaler Leberfunktion. Columvi wurde bei Patienten mit mittelgradiger oder schwerer Leberfunktionsstörung nicht untersucht.
Auswirkungen von Alter, Geschlecht und Körpergewicht
Es wurden keine klinisch signifikanten Unterschiede in der Pharmakokinetik von Glofitamab in Bezug auf Alter (21 Jahre bis 90 Jahre), Geschlecht und Körpergewicht (31 kg bis 148 kg) beobachtet.
Es wurden keine Studien zum kanzerogenen und mutagenen Potenzial von Glofitamab durchgeführt.
Fertilität
Es wurden keine tierexperimentellen Untersuchungen zur Fertilität durchgeführt, um die Wirkung von Glofitamab zu beurteilen
Reproduktionstoxizität
Es wurden keine tierexperimentellen Studien zur Reproduktions- und Entwicklungstoxizität durchgeführt, um die Wirkung von Glofitamab zu beurteilen. Aufgrund der geringen plazentaren Übertragung von Antikörpern während des ersten Trimesters, des Wirkmechanismus von Glofitamab (B-Zell-Depletion, zielabhängige T-Zell-Aktivierung und Zytokin-Freisetzung), der verfügbaren Sicherheitsdaten zu Glofitamab und Daten zu anderen Anti-CD20-Antikörpern ist das Risiko einer Teratogenität gering. Eine anhaltende B-Zell-Depletion kann zu einem erhöhten Risiko für opportunistische Infektionen führen, die zu einem Verlust des Fötus führen können. Ein vorübergehendes CRS im Zusammenhang mit der Verabreichung von Columvi kann ebenfalls dem Fötus schaden (siehe Abschnitt 4.6).
Systemische Toxizität
In einer Studie an Cynomolgus-Affen kam es bei Tieren, die nach einmaliger intravenöser Gabe von Glofitamab (0,1 mg/kg) ohne Obinutuzumab-Vorbehandlung ein schweres CRS aufwiesen, zu Erosionen im Gastrointestinaltrakt und inflammatorischen Zellinfiltrationen in der Milz und in den Sinusoiden der Leber sowie gelegentlich in einigen anderen Organen. Diese inflammatorischen Zellinfiltrationen traten wahrscheinlich sekundär zu einer zytokininduzierten Immunzellaktivierung auf. Die Vorbehandlung mit Obinutuzumab führte zu einer Abschwächung der Glofitamab-induzierten Zytokinfreisetzung und der damit verbundenen unerwünschten Wirkungen durch B-Zell-Depletion im peripheren Blut und im Lymphgewebe. Dies ermöglichte mindestens 10-mal höhere Glofitamab-Dosierungen (1 mg/kg) bei Cynomolgus-Affen, was bei der empfohlenen Dosis von 30 mg zu einer Cmax von bis zum 3,74‑fachen der menschlichen Cmax führte.
Alle Feststellungen bei Glofitamab wurden als pharmakologisch vermittelte Wirkungen betrachtet und waren reversibel. Es wurden keine Studien über mehr als 4 Wochen durchgeführt, da Glofitamab bei Cynomolgus-Affen stark immunogen war und einen Expositionsverlust sowie einen Verlust der pharmakologischen Wirkung zur Folge hatte.
Da alle rezidivierten oder refraktären DLBCL-Patienten, die behandelt werden sollen, zuvor eine Anti-CD20-Behandlung erhalten haben, werden die meisten von ihnen vor der Behandlung mit Obinutuzumab wahrscheinlich niedrige Spiegel von zirkulierenden B-Zellen aufweisen, die auf die Restwirkung einer früheren Anti-CD20-Therapie zurückzuführen sind. Daher entspricht das Tiermodell ohne vorheriger Rituximab- (oder anderer anti-CD20-) Behandlung möglicherweise nicht vollständig dem klinischen Kontext.
Histidin
Histidinhydrochlorid-Monohydrat
Methionin
Saccharose
Polysorbat 20 (E 432)
Wasser für Injektionszwecke
Das Arzneimittel darf, außer mit den unter Abschnitt 6.6 aufgeführten, nicht mit anderen Arzneimitteln gemischt werden.
Ungeöffnete Durchstechflasche
30 Monate
Verdünnte Lösung zur intravenösen Infusion
Die chemische und physikalische Anbruchstabilität wurde für maximal 72 Stunden bei 2 °C bis 8 °C und für 24 Stunden bei 30 °C, gefolgt von einer maximalen Infusionszeit von 8 Stunden, nachgewiesen.
Aus mikrobiologischer Sicht soll die verdünnte Lösung sofort verwendet werden. Wenn sie nicht sofort verwendet wird, liegt die Verantwortung für die Lagerungszeiten und -bedingungen vor der Anwendung beim Anwender und diese sollten normalerweise 24 Stunden bei 2 °C bis 8 °C nicht überschreiten, es sei denn, die Verdünnung wurde unter kontrollierten und validierten, aseptischen Bedingungen vorgenommen.
Im Kühlschrank lagern (2 °C ‑ 8 °C).
Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Verdünnung des Arzneimittels, siehe Abschnitt 6.3.
Columvi 2,5 mg Konzentrat zur Herstellung einer Infusionslösung
2,5 ml Konzentrat zur Herstellung einer Infusionslösung in einer 6-ml-Durchstechflasche (farbloses Glas, Typ I) mit Stopfen (Butyl-Gummi).
Packungsgröße: 1 Durchstechflasche.
Columvi 10 mg Konzentrat zur Herstellung einer Infusionslösung
10 ml Konzentrat zur Herstellung einer Infusionslösung in einer 15-ml-Durchstechflasche (farbloses Glas, Typ I) mit Stopfen (Butyl-Gummi).
Packungsgröße: 1 Durchstechflasche.
Die verdünnte Lösung von Columvi kann über einen intravenösen Infusionsbeutel (alle Dosierungen) oder eine intravenöse Infusionsspritze (nur die 2,5-mg-Dosis) verabreicht werden.
Hinweise zur Verdünnung
Columvi enthält kein Konservierungsmittel und ist nur zur einmaligen Anwendung bestimmt
Columvi muss vor der intravenösen Verabreichung von medizinischem Fachpersonal unter aseptischen Bedingungen verdünnt werden.
Vor der Verabreichung muss die Durchstechflasche mit Columvi visuell auf Partikel oder Verfärbung überprüft werden. Columvi ist eine klare, farblose Lösung. Wenn die Lösung trüb oder verfärbt ist oder sichtbare Partikel enthält, die Durchstechflasche verwerfen.
Vorbereitung der intravenösen Infusion mit Infusionsbeutel
Mit einer sterilen Nadel und Spritze das entsprechende Volumen der 9-mg/ml-Natriumchlorid-Injektionslösung (0,9 %) oder 4,5-mg/ml-Natriumchlorid-Injektionslösung (0,45 %), wie in Tabelle 10 beschrieben, aus dem Infusionsbeutel entnehmen und verwerfen.
Für die vorgesehene Dosis das erforderliche Volumen des Konzentrats von Columvi mit einer sterilen Nadel und Spritze aus der Durchstechflasche entnehmen und im Infusionsbeutel (siehe Tabelle 10) verdünnen. In der Durchstechflasche verbliebene Reste entsorgen.
Die finale Glofitamab-Konzentration nach Verdünnung muss 0,1 mg/ml bis 0,6 mg/ml betragen.
Den Infusionsbeutel zum Mischen der Lösung vorsichtig umdrehen, um übermäßige Schaumbildung zu vermeiden. Nicht schütteln.
Den Infusionsbeutel auf Partikel inspizieren und gegebenenfalls verwerfen.
Vor Beginn der intravenösen Infusion sollte der Inhalt des Infusionsbeutels Raumtemperatur (25 °C) angenommen haben.
• Tabelle 10: Verdünnung von Columvi für die intravenöse Infusion mit Infusionsbeutel
Zu verabreichende Dosis von Columvi |
Größe des Infusionsbeutels |
Zu entnehmendes und zu verwerfendes Volumen der 9-mg/ml-Natriumchlorid-Injektionslösung (0,9 %) oder der 4,5-mg/ml-Natriumchlorid-Injektionslösung (0,45 %) für Injektionszwecke |
Menge des hinzuzufügenden Konzentrats von Columvi |
2,5 mg |
50 ml |
27,5 ml |
2,5 ml |
100 ml |
77,5 ml |
2,5 ml |
|
10 mg |
50 ml |
10 ml |
10 ml |
100 ml |
10 ml |
10 ml |
|
30 mg |
50 ml |
30 ml |
30 ml |
100 ml |
30 ml |
30 ml |
Vorbereitung der intravenösen Infusion mit Infusionsspritze (nur die 2,5-mg-Dosis)
Zur Vorbereitung der Dosis ist eine Zwei-Spritzen-Methode mit einem Verbindungsstück zu verwenden. Das Endvolumen der verdünnten Lösung beträgt 25 ml.
Ziehen Sie 22,5 ml einer 9-mg/ml-Natriumchlorid-Injektionslösung (0,9 %) oder 4,5-mg/ml-Natriumchlorid-Injektionslösung (0,45 %) aus einem Infusionsbeutel in eine Spritze mit geeigneter Größe (z. B. 30 ml) auf.
Ziehen Sie mit einer sterilen Nadel eine zweite Spritze 2,5 ml Columvi Konzentrat aus der Durchstechflasche auf. Verwerfen Sie die in der Durchstechflasche übrig gebliebene Lösung.
Verbinden Sie die beiden Spritzen mit einem Verbindungsstück und überführen Sie das Columvi Konzentrat in die Spritze, die 9-mg/ml-Natriumchlorid-Injektionslösung (0,9 %) oder 4,5-mg/ml-Natriumchlorid-Injektionslösung (0,45 %) enthält. Die Endkonzentration von Glofitamab nach Verdünnung sollte 0,1 mg/ml betragen.
Trennen Sie die Spritzen. Ziehen Sie Luft in die Spritze mit der verdünnten Columvi Lösung und verschließen Sie sie.
Drehen Sie die Spritze vorsichtig um, um die Lösung zu mischen und übermäßiges Schäumen zu vermeiden. Nicht schütteln.
Entfernen Sie vor der Verabreichung Luftblasen aus der Spritze.
Verabreichung
Ausschließlich als intravenöse Infusion verabreichen.
Nicht als intravenöse Druck- oder Bolus-Injektion verabreichen.
Über einen Zeitraum von maximal 8 Stunden als intravenöse Infusion in einer separaten Infusionsleitung mittels intravenöser Infusionspumpe oder Spritzenpumpe verabreichen.
Nachdem der Infusionsbeutel oder die Spritze von Columvi leer ist, stellen Sie sicher, dass die gesamte Dosis von Columvi verabreicht wurde, indem Sie die Infusionsleitung mit einer 9-mg/ml-Natriumchlorid-Injektionslösung (0,9 %) oder einer 4,5-mg/ml-Natriumchlorid-Injektionslösung (0,45 %) aus einem Infusionsbeutel oder einer Spritze spülen. Setzen Sie die Infusion gemäß Tabelle 2 mit der gleichen Geschwindigkeit fort.
Inkompatibilitäten
Da andere Lösungsmittel nicht geprüft wurden, darf Columvi nur mit 9-mg/ml-Natriumchlorid-Injektionslösung (0,9 %) oder 4,5-mg/ml-Natriumchlorid-Injektionslösung (0,45 %) verdünnt werden.
Nach Verdünnung mit 9-mg/ml-Natriumchlorid-Injektionslösung (0,9 %) ist Columvi mit intravenösen Infusionsbeuteln kompatibel, die aus Polyvinylchlorid (PVC), Polyethylen (PE), Polypropylen (PP) oder Polyolefin bestehen. Nach Verdünnung mit 4,5-mg/ml-Natriumchlorid-Injektionslösung (0,45 %) ist Columvi kompatibel mit intravenösen Infusionsbeuteln aus PVC.
Nach Verdünnung mit 9-mg/ml- (0,9 %) oder 4,5-mg/ml- (0,45 %) Natriumchlorid- Injektionslösung ist Columvi mit Spritzen aus PP kompatibel.
Es wurden keine Inkompatibilitäten bei Infusionssets mit Produktkontaktflächen aus Polyurethan (PUR), PVC, PE, Polybutadien (PBD), Polyetherurethan (PEU), Polycarbonat (PC), Silikon, Polytetrafluorethylen (PTFE) oder Acrylnitril-Butadien-Styrol (ABS) und Inline-Filtermembranen aus Polyethersulfon (PES) oder Polysulfon beobachtet. Die Verwendung von Inline-Filtermembranen ist optional.
Beseitigung
Die Columvi Durchstechflasche ist nur zur einmaligen Anwendung bestimmt.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Roche Registration GmbH
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Deutschland
EU/1/23/1742/001
EU/1/23/1742/002
Datum der Erteilung der Zulassung: 7. Juli 2023
Datum der letzten Verlängerung der Zulassung: 8. Mai 2025
Juli 2025
Verschreibungspflichtig
Roche Pharma AG
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Telefon (07624) 14-0
Telefax (07624) 1019
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.