
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Jaypirca® 100 mg Filmtabletten
Jaypirca® 100 mg Filmtabletten
Jede Filmtablette enthält 100 mg Pirtobrutinib.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 77 mg Lactose (als Monohydrat).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette (Tablette).
Jaypirca® 100 mg Filmtabletten
Blaue, 10 mm, runde Tablette mit der Prägung „Lilly 100“ auf einer Seite und „7026“ auf der anderen Seite.
Jaypirca als Monotherapie ist indiziert zur Behandlung von erwachsenen Patienten mit rezidiviertem oder refraktärem Mantelzelllymphom (MCL), die zuvor mit einem Bruton-Tyrosinkinase (BTK)-Inhibitor behandelt wurden.
Jaypirca als Monotherapie ist indiziert zur Behandlung von erwachsenen Patienten mit chronischer lymphatischer Leukämie (CLL).
Die Behandlung mit Jaypirca sollte von einem Arzt eingeleitet und überwacht werden, der Erfahrung mit der Anwendung onkologischer Therapien hat.
Dosierung
Die empfohlene Dosis beträgt 200 mg Pirtobrutinib einmal täglich (QD).
Die Einnahme von Jaypirca sollte bis zur Erholung auf Grad 1 oder dem Ausgangszustand unterbrochen werden, wenn beim Patienten folgendes Ereignis auftritt:
Grad 3 Neutropenie mit Fieber und/oder Infektion
Grad 4 Neutropenie mit einer Dauer von ≥ 7 Tagen
Grad 3 Thrombozytopenie mit Blutungen
Grad 4 Thrombozytopenie
Grad 3 oder 4 nicht hämatologische Toxizität
Asymptomatische Lymphozytose wird nicht als Nebenwirkung angesehen. Patienten, bei denen dieses Ereignis auftritt, sollten Jaypirca weiterhin einnehmen.
In den klinischen Studien wurden bei einer begrenzten Anzahl von Patienten unerwünschte Ereignisse durch Dosisreduktion behandelt (siehe Abschnitt 5.1).
Die Behandlung sollte bis zur Krankheitsprogression oder bis zur inakzeptablen Toxizität fortgesetzt werden.
Vergessene Einnahme
Wenn mehr als 12 Stunden vergangen sind, nachdem ein Patient eine Dosis vergessen hat, weisen Sie den Patienten an, die nächste Dosis zum vorgesehenen Zeitpunkt einzunehmen; eine zusätzliche Dosis sollte nicht eingenommen werden. Wenn Erbrechen auftritt, sollte der Patient keine zusätzliche Dosis einnehmen, sondern mit der nächsten planmäßigen Dosis fortfahren.
Besondere Patientengruppen
Ältere Patienten
Dosisanpassungen aufgrund des Alters sind nicht erforderlich (siehe Abschnitt 5.2).
Nierenfunktionsstörungen
Bei Patienten mit leichter, mittelschwerer oder schwerer Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Es liegen keine Daten zu Dialysepatienten vor (siehe Abschnitt 5.2).
Leberfunktionsstörungen
Bei Patienten mit leichter, mittelschwerer oder schwerer Leberfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Jaypirca bei Kindern und Jugendlichen unter 18 Jahren ist nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Jaypirca ist zum Einnehmen bestimmt.
Die Tablette sollte als Ganzes geschluckt und mit einem Glas Wasser eingenommen werden, um eine gleichbleibende Wirkung zu gewährleisten (Patienten sollten die Tabletten vor dem Schlucken nicht kauen, zerkleinern oder teilen). Sie kann unabhängig von der Nahrung eingenommen werden.
Patienten sollten die Dosis jeden Tag, ungefähr zur gleichen Uhrzeit, einnehmen.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Infektionen
Bei mit Jaypirca behandelten Patienten sind schwere Infektionen, einschließlich tödlicher Fälle, aufgetreten. Die am häufigsten gemeldeten Infektionen vom Grad 3 oder höher waren Pneumonie, COVID-19-Pneumonie, COVID-19 und Sepsis. Bei Patienten mit erhöhtem Risiko für opportunistische Infektionen sollte eine prophylaktische antimikrobielle Therapie in Erwägung gezogen werden. Abhängig vom Grad der Infektion und davon, ob sie zusammen mit einer Neutropenie auftritt, kann eine Unterbrechung der Behandlung erforderlich sein (siehe Abschnitt 4.2).
Hämorrhagien
Bei mit Jaypirca behandelten Patienten sind Blutungsereignisse, einschließlich tödlicher Fälle, mit und ohne Thrombozytopenie aufgetreten. Schwere Blutungsereignisse von Grad 3 oder höher, einschließlich gastrointestinaler Blutungen und intrakranialer Blutungen, wurden beobachtet. Patienten sollten auf Anzeichen und Symptome von Blutungen überwacht werden. Bei Patienten, die Antikoagulanzien oder Thrombozytenaggregationshemmer erhalten, besteht möglicherweise ein erhöhtes Blutungsrisiko. Bei Anwendung von Jaypirca mit Antikoagulanzien oder Thrombozytenaggregationshemmern sollten Risiko und Nutzen abgewogen und eine zusätzliche Überwachung auf Anzeichen von Blutungen erwogen werden. Die Einnahme von Jaypirca mit Warfarin oder anderen Vitamin-K-Antagonisten wurde nicht untersucht.
Eine Dosisunterbrechung kann bei Blutungsereignissen 3. oder 4. Grades erforderlich sein (siehe Abschnitt 4.2).
Das Nutzen-Risiko-Verhältnis des Absetzens von Jaypirca für 3 bis 5 Tage vor und nach einer Operation sollte in Abhängigkeit von der Art der Operation und dem Blutungsrisiko abgewogen werden.
Zytopenien
Zytopenien 3. und 4. Grades, einschließlich Neutropenie, Anämie und Thrombozytopenie, traten bei mit Jaypirca behandelten Patienten auf. Wenn medizinisch indiziert, sollte während der Behandlung das große Blutbild überwacht werden. Abhängig vom Grad der Zytopenie kann ein Aussetzen der Dosis erforderlich sein (siehe Abschnitt 4.2).
Vorhofflimmern/-flattern
Bei mit Jaypirca behandelten Patienten wurden Vorhofflimmern und Vorhofflattern beobachtet, insbesondere bei Patienten mit Vorhofflimmern und/oder mehreren kardiovaskulären Komorbiditäten in der Vorgeschichte. Patienten sollten auf Anzeichen und Symptome von Vorhofflimmern und Vorhofflattern überwacht werden; gemäß medizinischer Indikation sollte ein Elektrokardiogramm erstellt werden. Abhängig vom Grad des Vorhofflimmerns/Vorhofflatterns kann ein Aussetzen der Dosis erforderlich sein (siehe Abschnitt 4.2).
Zweitmalignome
Bei mit Jaypirca behandelten Patienten traten häufig sekundäre Malignome auf, wobei nichtmelanozytäre Hautkrebsarten am häufigsten vorkamen. Patienten sollten auf das Auftreten von Hautkrebs überwacht werden und angewiesen werden, sich vor Sonneneinstrahlung zu schützen.
Tumorlysesyndrom
Ein Tumorlysesyndrom (TLS) wurde während der Therapie mit Jaypirca selten berichtet. Hochrisikopatienten für ein TLS sind diejenigen Patienten, die vor der Behandlung eine hohe Tumorlast hatten. Patienten sollten auf ein mögliches Risiko für TLS untersucht und entsprechend medizinischer Indikation engmaschig überwacht werden.
Empfängnisverhütung bei Frauen im gebärfähigen Alter und Männern
Basierend auf Beobachtungen bei Tieren und der Genotoxizität von Pirtobrutinib (siehe Abschnitt 5.3) kann Pirtobrutinib den Fötus schädigen, wenn es einer schwangeren Frau verabreicht wird. Frauen im gebärfähigen Alter müssen während der Behandlung und für 5 Wochen nach der letzten Jaypirca-Dosis eine zuverlässige Verhütungsmethode anwenden. Männer sollten angewiesen werden, während der Behandlung und für 3 Monate nach der letzten Jaypirca-Dosis eine zuverlässige Verhütungsmethode anzuwenden und kein Kind zu zeugen (siehe Abschnitt 4.6).
Lactose
Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactasemangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht anwenden.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro 200 mg Tagesdosis, d. h. es ist nahezu „natriumfrei“.
Pirtobrutinib wird primär über CYP3A4, UGT1A8 und UGT1A9 metabolisiert.
Auswirkungen anderer Arzneimittel auf die Pharmakokinetik von Pirtobrutinib
CYP3A-Inhibitoren
In einer klinischen Studie erhöhte Itraconazol, ein starker CYP3A4-Hemmer, die AUC von Pirtobrutinib um 48 %, erhöhte aber Cmax von Pirtobrutinib nicht. Diese Erhöhung der Pirtobrutinib-Exposition ist nicht klinisch relevant. Daher ist bei CYP3A-Inhibitoren keine Dosisanpassung von Jaypirca erforderlich.
CYP3A-Induktoren
In einer klinischen Studie verringerte Rifampicin, ein starker CYP3A-Induktor, die AUC und Cmax von Pirtobrutinib um 71 % bzw. 42 %. Obwohl zu erwarten ist, dass diese Verringerung der Pirtobrutinib-Exposition nicht klinisch relevant ist, vermeiden Sie nach Möglichkeit starke CYP3A-Induktoren (z. B. Rifampicin, Carbamazepin, Phenytoin).
Gleichzeitige Anwendung mit Protonenpumpen-Inhibitoren
Bei gleichzeitiger Anwendung mit Omeprazol, einem Protonenpumpen-Inhibitor, wurden keine klinisch signifikanten Unterschiede in der Pirtobrutinib-Pharmakokinetik beobachtet.
Auswirkungen von Pirtobrutinib auf die Pharmakokinetik anderer Arzneimittel (Anstieg der Plasmakonzentration)
CYP2C8-Substrate
Pirtobrutinib ist ein moderater Inhibitor von CYP2C8. Pirtobrutinib erhöhte die AUC und Cmax von Repaglinid (einem Substrat von CYP2C8) um 130 % bzw. 98 %. Da Pirtobrutinib die Plasmakonzentrationen von CYP2C8-Substraten erhöhen kann, ist bei gleichzeitiger Anwendung mit CYP2C8-Substraten (z. B. Repaglinid, Dasabuvir, Selexipag, Rosiglitazon, Pioglitazon und Montelukast) Vorsicht geboten.
BCRP-Substrate
Pirtobrutinib ist ein moderater Inhibitor von BCRP. Pirtobrutinib erhöhte die AUC und Cmax von Rosuvastatin (einem BCRP-Substrat) um 140 % bzw. 146 %. Da Pirtobrutinib die Plasmakonzentrationen von BCRP-Substraten erhöhen kann, ist bei der gleichzeitigen Verabreichung von BCRP-Substraten (z. B. Rosuvastatin) Vorsicht geboten. Wenn die gleichzeitige Anwendung mit BCRP-Substraten mit geringer therapeutischer Breite (z. B. hochdosiertes Methotrexat, Mitoxantron) nicht vermieden werden kann, sollte eine engmaschige klinische Überwachung in Erwägung gezogen werden.
P-gp-Substrate
Pirtobrutinib ist ein schwacher Inhibitor von P-gp. Pirtobrutinib erhöhte die AUC und Cmax von Digoxin (einem P-gp-Substrat) um 35 % bzw. 55 %. Demnach kann Pirtobrutinib die Plasmakonzentrationen von P-gp-Substraten erhöhen. Wenn die gleichzeitige Anwendung mit P-gp-Substraten mit geringer therapeutischer Breite (z. B. Dabigatranetexilat und Digoxin) nicht vermieden werden kann, sollte eine engmaschige klinische Überwachung in Erwägung gezogen werden.
CYP2C19-Substrate
Pirtobrutinib ist ein schwacher Inhibitor von CYP2C19. Pirtobrutinib erhöhte die AUC und Cmax von Omeprazol (einem CYP2C19-Substrat) um 56 % bzw. 49 %. Demnach kann Pirtobrutinib die Plasmakonzentrationen von CYP2C19-Substraten erhöhen. Wenn die gleichzeitige Anwendung mit CYP2C19-Substraten mit geringer therapeutischer Breite (z. B. Phenobarbital und Mephenytoin) nicht vermieden werden kann, sollte eine engmaschige klinische Überwachung in Erwägung gezogen werden.
CYP3A-Substrate
Pirtobrutinib ist ein schwacher Inhibitor von CYP3A. Pirtobrutinib erhöhte die AUC und Cmax von oral verabreichtem Midazolam (empfindliches CYP3A-Substrat) um 70 % bzw. 58 %. Pirtobrutinib hatte nach Exposition mit intravenös verabreichtem Midazolam keine klinisch relevante Auswirkung auf dieses. Demnach kann Pirtobrutinib die Plasmakonzentrationen von CYP3A-Substraten erhöhen. Wenn die gleichzeitige Verabreichung mit CYP3A-Substraten mit geringer therapeutischer Breite (z. B. Alfentanil, Midazolam, Tacrolimus) nicht vermieden werden kann, sollte eine engmaschige klinische Überwachung in Erwägung gezogen werden.
Frauen im gebärfähigen Alter / Empfängnisverhütung bei Männern und Frauen
Basierend auf Beobachtungen bei Tieren und der Genotoxizität von Pirtobrutinib (siehe Abschnitt 5.3) kann Pirtobrutinib den Fötus schädigen, wenn es einer schwangeren Frau verabreicht wird. Frauen im gebärfähigen Alter müssen während der Behandlung und für 5 Wochen nach der letzten Jaypirca-Dosis eine zuverlässige Verhütungsmethode anwenden. Männer sollten angewiesen werden, während der Behandlung und für 3 Monate nach der letzten Jaypirca-Dosis eine zuverlässige Verhütungsmethode anzuwenden und kein Kind zu zeugen (siehe Abschnitt 4.4).
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Jaypirca bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Jaypirca darf während der Schwangerschaft nicht angewendet werden.
Stillzeit
Es ist nicht bekannt, ob Pirtobrutinib in die Muttermilch übergeht. Ein Risiko für das gestillte Kind kann nicht ausgeschlossen werden. Das Stillen soll während der Behandlung mit Jaypirca und für eine Woche nach der letzten Dosis unterbrochen werden.
Fertilität
Es liegen keine Daten zur Wirkung von Pirtobrutinib auf die menschliche Fertilität vor.
Jaypirca hat einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Bei einigen Patienten wurde während der Behandlung mit Jaypirca von Fatigue, Schwindel und Asthenie berichtet. Dies soll berücksichtigt werden, wenn die Verkehrstüchtigkeit oder Fähigkeit zum Bedienen von Maschinen eines Patienten beurteilt wird.
Zusammenfassung des Sicherheitsprofils
Bei den 1 153 mit Jaypirca behandelten Patienten waren die häufigsten Nebenwirkungen jeden Schweregrades (bei ≥ 20 % der Patienten) Neutropenie (27,4 %) und Hämorrhagie (20,4 %).
Die häufigsten schweren (Grad ≥ 3) Nebenwirkungen (bei ≥ 5 % der Patienten) waren Neutropenie (22,8 %), Anämie (9,0 %), Pneumonie (8,8 %) und Thrombozytopenie (7,2 %).
Die Häufigkeit von Behandlungsabbrüchen aufgrund von Nebenwirkungen betrug 3,3 % und die Nebenwirkung, die am häufigsten zum Behandlungsabbruch führte, war Pneumonie (0,8 %). Die Häufigkeit von Dosisreduktionen aufgrund von Nebenwirkungen betrug 4,3 % und die Nebenwirkung, die am häufigsten zu einer Dosisreduktion führte, war Neutropenie (2,4 %).
Schwerwiegende Nebenwirkungen im Zusammenhang mit Jaypirca sind bei 18,0 % der Patienten aufgetreten, und die häufigsten schwerwiegenden Nebenwirkungen (bei ≥ 1 % der Patienten) waren Pneumonie (8,2 %), Hämorrhagie (2,9 %), Neutropenie (2,6 %), Anämie (2,4 %), Vorhofflimmern/Vorhofflattern (1,1 %) und Harnwegsinfektion (1,0 %).
Tödliche Nebenwirkungen wurden bei 0,7 % der Patienten mit Pneumonie, bei 0,3 % der Patienten mit Hämorrhagie und bei 0,1 % der Patienten mit Harnwegsinfektion beobachtet.
Tabellarische Auflistung der Nebenwirkungen
Tabelle 1 listet die Nebenwirkungen im Zusammenhang mit Jaypirca als Monotherapie aus klinischen Studiendaten und aus Erfahrungen nach Markteinführung. Die Nebenwirkungen aus klinischen Studiendaten basieren auf gepoolten Daten von 1 153 Patienten, die in einer klinischen Phase-1/2-Studie mit einer Anfangsdosis von 200 mg Jaypirca QD als Monotherapie, ohne Dosiseskalation, behandelt wurden, und von Patienten, die in Phase-3-Studien mit 200 mg Jaypirca QD als Monotherapie behandelt wurden. Die Patienten wurden wegen MCL, chronischer lymphatischer Leukämie/kleinzelligem lymphatischen Lymphom (CLL/SLL) und anderen Non-Hodgkin-Lymphomen (NHL) behandelt. Die Patienten wurden für eine mittlere Dauer von 18 Monaten mit Jaypirca behandelt. Die Nebenwirkungen sind nachfolgend nach MedDRA-Körpersystemorganklasse aufgelistet. Häufigkeitsgruppen werden durch die folgende Konvention definiert: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1 000, < 1/100); selten (≥ 1/10 000, < 1/1 000); sehr selten (< 1/10 000) und nicht bekannt (kann aus verfügbaren Daten nicht abgeschätzt werden). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad geordnet.
Tabelle 1: Nebenwirkungen von Patienten, die mit Jaypircaa behandelt wurden
Systemorganklasse |
Nebenwirkung |
Häufigkeitskategorie (%) |
Grad ≥ 3 c (%) |
Infektionen und parasitäre Erkrankungen |
Pneumonie |
Sehr häufig (13,9) |
8,8 |
Infektionen der oberen Atemwege |
Sehr häufig (13,4) |
0,3 |
|
Harnwegsinfekt |
Häufig (9,2) |
1,3 |
|
Erkrankungen des Blutes und des Lymphsystems |
Neutropenie b |
Sehr häufig (27,4) |
22,8 |
Anämie b |
Sehr häufig (18,4) |
9,0 |
|
Thrombozytopenie b |
Sehr häufig (14,7) |
7,2 |
|
Lymphozytose b |
Häufig (5,0) |
3,2 |
|
Erkrankungen des Nervensystems |
Kopfschmerzen |
Sehr häufig (10,7) |
0,5 |
Herzerkrankungen |
Vorhofflimmern/Vorhofflattern |
Häufig (3,1) |
1,6 |
Gefäßerkrankungen |
Hämorrhagie b |
Sehr häufig (20,4) |
2,9 |
Epistaxis |
Häufig (4,2) |
0,1 |
|
Hämaturie |
Häufig (4,8) |
0,2 |
|
Hämatom |
Häufig (2,4) |
0,3 |
|
Bindehautblutung |
Häufig (1,5) |
0,1 |
|
Bluterguss b |
Sehr häufig (17,0) |
0,2 |
|
Prellung |
Sehr häufig (14,2) |
0,1 |
|
Petechien |
Häufig (5,1) |
0 |
|
Erkrankungen des Gastrointestinaltraktes |
Diarrhö |
Sehr häufig (19,5) |
0,7 |
Übelkeit |
Sehr häufig (13,0) |
0,3 |
|
Bauchschmerzen |
Häufig (7,4) |
0,7 |
|
Leber- und Gallenerkrankungen |
Erhöhte Leberenzymwerte |
Nicht bekannt |
Nicht bekannt |
Erkrankungen der Haut und des Unterhautgewebes |
Ausschlag b |
Sehr häufig (19,2) |
1,2 |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Arthralgie |
Sehr häufig (12,5) |
0,8 |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fatigue |
Sehr häufig (17,9) |
1,4 |
Peripheres Ödem |
Häufig (9,0) |
0,3 |
a Die Häufigkeiten werden von der Jaypirca-Exposition bei Patienten mit malignen B-Zell-Erkrankungen abgeleitet.
b Umfasst mehrere Nebenwirkungsbegriffe
c Schweregradzuweisung basierend auf National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) Version 5.0
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem anzuzeigen:
Bundesinstitut für Arzneimittel und
Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: https://www.bfarm.de
In der Phase-1-Studie, in der die Patienten wiederholte Dosen von bis zu 300 mg einmal täglich erhielten, wurde keine maximal verträgliche Dosis erreicht. In Studien mit gesunden Freiwilligen wurde bei Verabreichung einer maximalen Einzeldosis von 900 mg keine dosisabhängige Toxizität beobachtet. Anzeichen und Symptome einer Pirtobrutinib-Überdosierung wurden nicht festgestellt. Es gibt keine spezifische Behandlung für eine Pirtobrutinib-Überdosierung.
Patienten, bei denen eine Überdosierung auftritt, müssen engmaschig überwacht und angemessen symptomatisch behandelt werden.
Pharmakotherapeutische Gruppe: Antineoplastische Mittel, Proteinkinase-Inhibitoren, ATC-Code: L01EL05
Wirkmechanismus
Pirtobrutinib ist ein reversibler, nicht-kovalenter Inhibitor von BTK. BTK ist ein Signalprotein des B‑Zell-Antigenrezeptors (BCR) und des Zytokinrezeptorweges. In B-Zellen führt die BTK-Signalgebung zur Aktivierung von Signalwegen, die für die Proliferation, den Transport, die Chemotaxis und die Adhäsion von B-Zellen erforderlich sind. Pirtobrutinib bindet sowohl an die Wildtyp-BTK als auch an die BTK mit C481-Mutationen, was zu einer Hemmung der BTK-Kinase-Aktivität führt.
Pharmakodynamische Wirkungen
Elektrophysiologie des Herzens
Die Wirkung einer Einzeldosis von 900 mg Pirtobrutinib auf das korrigierte QT (QTc)-Intervall wurde in einer Studie mit Placebo und Positivkontrollen bei 30 gesunden Probanden untersucht. Die gewählte Dosis entspricht etwa dem Zweifachen der Konzentrationen, die im Steady State bei der empfohlenen Dosierung von 200 mg einmal täglich erreicht werden. Pirtobrutinib hatte keinen klinisch bedeutsamen Einfluss auf die Veränderung des QT-Intervalls, korrigiert um die Herzfrequenz unter Verwendung der Fridericia-Formel (QTcF) (d. h. > 10 ms). Es gab keinen Zusammenhang zwischen der Pirtobrutinib-Exposition und der Veränderung des QTc-Intervalls.
Klinische Wirksamkeit und Sicherheit
Mantelzelllymphom
Die Wirksamkeit von Jaypirca wurde bei erwachsenen Patienten mit MCL in einer multizentrischen, unverblindeten, einarmigen klinischen Phase 1/2 Studie untersucht: Studie 18001 (BRUIN). Die Studie umfasste zwei Teile: eine Phase-1-Dosiseskalation, in der der Dosisbereich der Pirtobrutinib Monotherapie von 25 mg bis 300 mg einmal täglich untersucht wurde, und eine Phase-2-Dosiserweiterung. Das primäre Ziel von Phase-1 war die Bestimmung der empfohlenen Phase-2-Dosis von Pirtobrutinib, die bei 200 mg einmal täglich liegt. Eine maximal verträgliche Dosis wurde nicht erreicht. Das primäre Ziel des Phase-2-Teils war die Beurteilung der Anti-Tumor-Aktivität von Pirtobrutinib basierend auf der Gesamtansprechrate, wie sie von einer unabhängigen Prüfkommission bewertet wurde. Die Patienten erhielten Jaypirca täglich oral bis zum Fortschreiten der Krankheit oder einer unzumutbaren Toxizität.
In Studie 18001 wurden insgesamt 164 Patienten mit MCL-Diagnose eingeschlossen und behandelt. Das primäre Analyseset (PAS: primary analysis set) zur Bewertung der Wirksamkeit basierte auf den ersten 90 aufgenommenen Patienten mit MCL, die keine bekannte Beteiligung des Zentralnervensystems (ZNS) hatten, mit einem vorherigen BTK-Inhibitor behandelt wurden, eine oder mehrere Dosen von Jaypirca erhalten hatten und an mindestens 1 Stelle eine radiologisch messbare Erkrankung aufwiesen. Das mediane Alter betrug 70 Jahre (Spanne: 46 bis 87 Jahre), 80 % waren männlich, 84,4 % waren Weiße, 67,8 % hatten zu Beginn einen ECOG-Status (Eastern Cooperative Oncology Group) von 0 und 31,1 % hatten einen ECOG-Status von 1. Die Patienten hatten eine mediane Anzahl von 3 vorherigen Therapielinien (Spanne: 1 bis 8), wobei der Grund für das Absetzen der letzten vorangegangenen BTK-Inhibitor-Therapie bei 81,1 % der Patienten eine Progression und bei 13,3 % der Patienten eine Unverträglichkeit war. 95,6 % der Patienten erhielten eine vorherige Anti-CD20-Therapie, 87,8 % eine Chemotherapie, 18,9 % eine autologe Stammzelltransplantation, 4,4 % eine allogene Stammzelltransplantation, 15,6 % einen vorherigen BCL2-Inhibitor und 4,4 % eine vorherige Therapie mit chimären Antigenrezeptor-modifizierten T-Zellen (CAR-T). 38,9 % der Patienten hatten eine extranodale Beteiligung und 26,7 % hatten eine Tumorgröße von mindestens 5 cm. Der vereinfachte MCL International Prognostic Index (sMIPI)-Score war bei 22,2 % der Patienten niedrig, bei 55,6 % mittel und bei 22,2 % der Patienten hoch.
Von den 164 an MCL erkrankten Patienten, die in die Studie 18001 eingeschlossen wurden, wurde bei 9 Patienten die Dosis reduziert. Von diesen sprachen 6 Patienten an. Diese setzten die Therapie nach einer Dosisreduktion auf 150 mg QD (3), 100 mg QD (2) und 50 mg QD (1) fort und konnten damit ein dauerhaftes Ansprechen aufrechterhalten.
Die Wirksamkeit von Jaypirca wurde anhand der 2014 Lugano-Kriterien für maligne Lymphome bewertet. Die Wirksamkeitsergebnisse für Patienten, die zuvor mindestens einen BTK-Inhibitor erhalten hatten und in die PAS aufgenommen wurden, sind in Tabelle 2 zusammengefasst. Von den 90 Patienten in PAS erhielten 79 mindestens 1 Dosis von 200 mg QD. Von diesen 79 Patienten begannen 77 mit 200 mg QD, 1 Dosis wurde von einer niedrigeren Dosis eskaliert und 1 Dosis wurde von einer höheren Dosis reduziert. Die mediane Behandlungsdauer betrug 5,24 Monate (Spanne: 0,2 bis 39,6 Monate). Unter den 51 Patienten die ansprachen betrug die mediane Zeit bis zum Ansprechen 1,84 Monate (Spanne: 1,0 bis 7,5 Monate).
Während Subgruppenanalysen eine begrenzte Anzahl von Patienten darstellen, wurden klinisch bedeutsame Wirksamkeitsergebnisse in wichtigen Subgruppen beobachtet, einschließlich Patienten, die eine vorherige Behandlung mit BTK-Inhibitoren aufgrund von Unverträglichkeit oder Progression abgebrochen haben, unabhängig von Anzahl und Art der vorherigen Therapien.
Tabelle 2: Zusammenfassung der Wirksamkeitsdaten in Studie 18001 für MCL-Patienten, die zuvor mindestens einen BTK-Inhibitor erhielten
Pirtobrutinib |
|
Objektive Ansprechrate (vollständiges Ansprechen + teilweises Ansprechen) | |
Rate – Prozent (95 % KI) |
56,7 (45,8; 67,1) |
CR – Prozent |
18,9 |
PR – Prozent |
37,8 |
Ansprechdauer | |
Median - Monate (95 % KI) |
17,61 (7,29; 27,24) |
Abkürzungen: KI = Konfidenzintervall, NE = nicht schätzbar (not estimable), CR = vollständiges Ansprechen (complete response), PR = partielles Ansprechen (partial response).
Datenschnitt: 29. Juli 2022. Die mediane Nachbeobachtungszeit für die Dauer des Ansprechens war 12,68 Monate.
Chronische lymphatische Leukämie
Die Wirksamkeit von Jaypirca bei erwachsenen Patienten mit CLL wurde in drei randomisierten kontrollierten Phase-III-Studien untersucht.
BRUIN-CLL-313 (Studie 20023)
Die Wirksamkeit von Jaypirca wurde in einer randomisierten, multizentrischen, offenen, aktiv kontrollierten Studie an 282 Patienten mit behandlungsnaiver CLL/SLL ohne 17p-Deletion untersucht (BRUIN-CLL-313, Studie 20023). Die Patienten wurden im Verhältnis 1:1 randomisiert und erhielten entweder
Jaypirca einmal täglich oral in einer Dosis von 200 mg bis zum Krankheitsprogress oder inakzeptabler Toxizität, oder
Bendamustin plus ein Rituximab-Produkt (BR): Bendamustin 90 mg/m2 intravenös (Tag 1 und 2 jedes 28-tägigen Zyklus) in Kombination mit einem Rituximab-Produkt (375 mg/m2 intravenös am Tag 1 von Zyklus 1, gefolgt von 500 mg/m2 am Tag 1 der folgenden Zyklen), für bis zu 6 Zyklen.
Die Randomisierung wurde nach dem Mutationsstatus des variablen Abschnitts der schweren Immunglobulinkette (immunoglobulin variable region heavy chain, IGHV) (mutiert vs. nicht mutiert) und nach dem Rai-Stadium (niedriges/intermediäres vs. hohes Risiko) stratifiziert. Von den insgesamt 282 Patienten erhielten 141 eine Jaypirca-Monotherapie und 141 BR. Nach bestätigtem Krankheitsprogress hatten die Patienten, die BR randomisiert erhielten, die Möglichkeit, zur Jaypirca-Monotherapie zu wechseln. Von den 141 Patienten im BR-Arm wechselten 18 zu Jaypirca über.
Die Baseline-Charakteristika waren in allen Behandlungsarmen vergleichbar. Das mediane Alter betrug 66 Jahre (Spanne: 29 bis 88 Jahre), 61 % waren männlich und 73 % waren Weiße. 94 % der Patienten hatten zu Studienbeginn einen ECOG-Status von 0 oder 1 und 34 % der Patienten hatten eine fortgeschrittene klinische Erkrankung im Rai-Stadium III oder IV. Von den Patienten, für die eine zentrale Testung zur Verfügung stand, hatten 9 % (22 von 244 Patienten) eine TP53-Mutation, 56 % (149 von 268 Patienten) hatten eine unmutierte IGHV und 16 % (27 von 174) einen komplexen Karyotyp.
Der primäre Endpunkt für die Wirksamkeit war das progressionsfreie Überleben (progression-free survival, PFS) gemäß Beurteilung durch eine unabhängige Prüfkommission (Independent Review Committee, IRC). Die geschätzte mediane Nachbeobachtungszeit betrug 28,1 Monate. Wirksamkeitsergebnisse zugunsten von Pirtobrutinib wurden in allen Untergruppen beobachtet, einschließlich jenen mit beziehungsweise ohne IGHV-Mutation und unabhängig vom Alter. Die Ergebnisse zur Wirksamkeit für die primäre Analyse sind in Tabelle 3 angeführt. Die Kaplan-Meier-Kurve des PFS ist in Abbildung 1 dargestellt.
Tabelle 3: Ergebnisse zur Wirksamkeit gemäß IRC in BRUIN-CLL-313 (ITT-Population)
Parameter a |
Pirtobrutinib |
Bendamustin plus Rituximab |
Progressionsfreies Überleben |
||
Anzahl der Ereignisse, n |
13 (9 %) |
48 (34 %) |
Krankheitsprogression |
13 (9 %) |
40 (28 %) |
Todesfälle |
0 |
8 (6 %) |
Median PFS (95 %-KI), Monate b |
NE (NE, NE) |
33,5 (32,7; NE) |
HR (95 %-KI) c |
0,20 (0,11; 0,37) |
|
P-Wert d |
< 0,0001 |
|
PFS-Rate nach 24 Monaten (95 %-KI, %) b |
93 % (88; 96) |
71 % (61; 78) |
Gesamtansprechrate e |
||
ORR, n |
133 (94 %) |
114 (81 %) |
CR, n |
19 (13 %) |
29 (21 %) |
PR, n |
114 (81 %) |
85 (60 %) |
KI: Konfidenzintervall; CR: vollständiges Ansprechen; CRi: vollständiges Ansprechen mit unvollständiger hämatopoetischer Erholung; HR: Hazard Ratio; NE: nicht schätzbar; nPR: noduläres partielles Ansprechen; ORR: Gesamtansprechrate; PFS: progressionsfreies Überleben; PR: partielles Ansprechen.
a Die Wirksamkeit wurde anhand der Leitlinien des International Workshop for Chronic Lymphocytic Leukemia (iwCLL) 2018 bewertet.
b Basierend auf Kaplan-Meier-Schätzung.
c Basierend auf stratifiziertem Cox-Proportional-Hazards-Modell.
d 2-seitiger p-Wert basierend auf einem stratifizierten Log-Rank-Test.
e Definiert als CR + CRi + nPR + PR. Kein Patient hatte CRi oder nPR als bestes Ansprechen.
Abbildung 1: Kaplan-Meier-Kurve des PFS gemäß IRC-Bewertung in BRUIN-CLL-313 (ITT-Population)
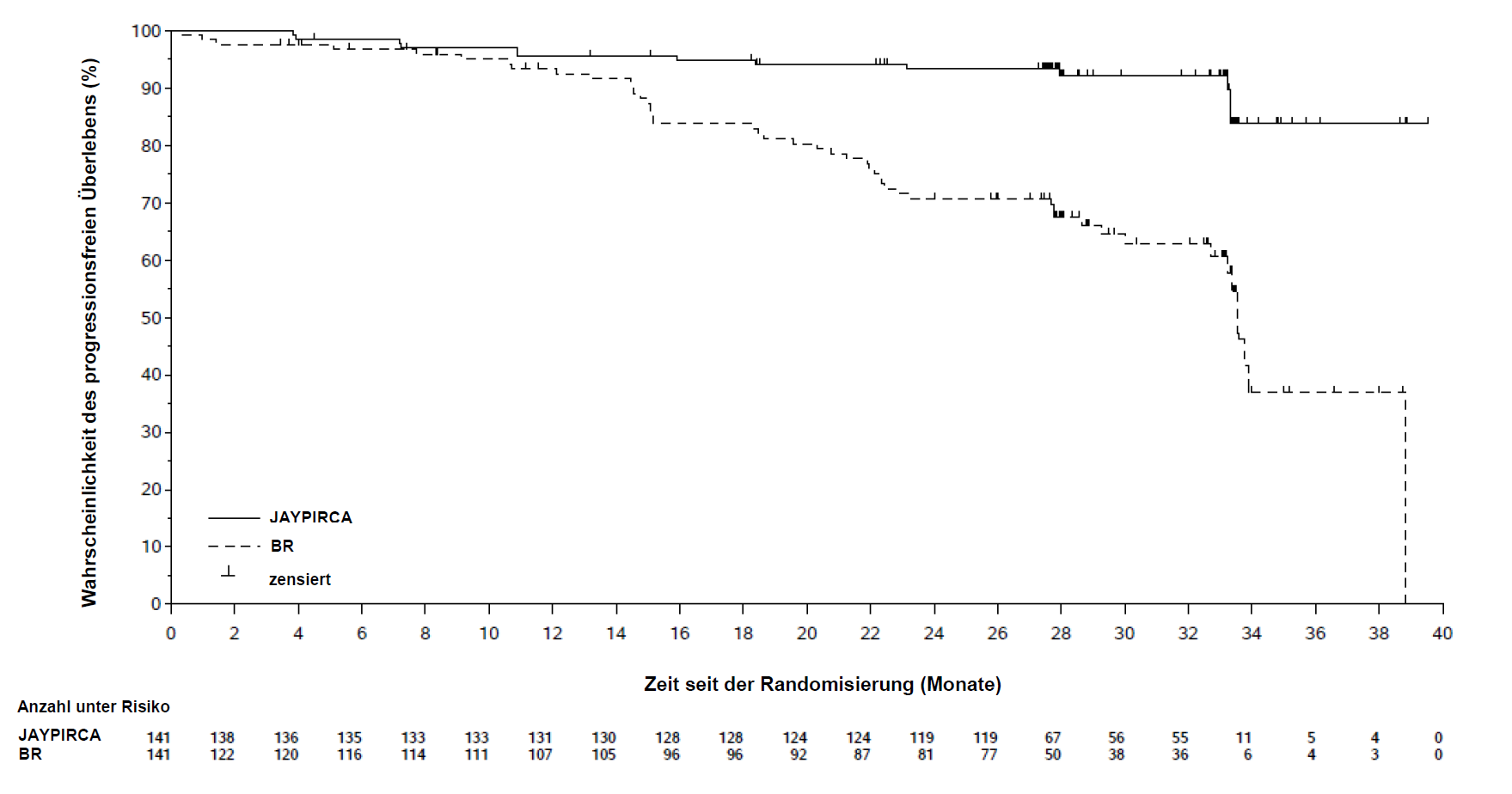
Zum Zeitpunkt der Analyse waren die Daten zum Gesamtüberleben (overall survival, OS) noch nicht reif. Bei einer geschätzten medianen Nachbeobachtung von 32,2 Monaten wurde das mediane OS in keinem Arm erreicht, wobei bei weniger als 5 % der Patienten ein Ereignis eintrat.
BRUIN-CLL-314 (Studie 20030)
Die Wirksamkeit von Jaypirca wurde in einer randomisierten, multizentrischen, offenen, aktiv kontrollierten Studie an 662 Patienten mit rezidivierter oder refraktärer CLL/SLL, die zuvor nicht mit einem BTK-Inhibitor behandelt worden waren, oder mit nicht vorbehandelter CLL/SLL untersucht (BRUIN-CLL-314, Studie 20030). Die Patienten wurden im Verhältnis 1:1 randomisiert und erhielten entweder
Jaypirca einmal täglich oral in einer Dosis von 200 mg bis zum Krankheitsprogress oder inakzeptabler Toxizität oder
Ibrutinib einmal täglich oral in einer Dosis von 420 mg bis zum Krankheitsprogress oder inakzeptabler Toxizität.
Die Randomisierung wurde nach dem 17p-Deletionsstatus (ja/nein) und Anzahl der vorherigen Therapielinien (0 vs. 1 vs. ≥ 2) stratifiziert. Von den insgesamt 662 Patienten erhielten 331 eine Jaypirca-Monotherapie und 331 eine Ibrutinib-Monotherapie.
Die Baseline-Charakteristika waren in allen Behandlungsarmen vergleichbar. Das mediane Alter betrug 67 Jahre (Spanne: 34 bis 90 Jahre), 65 % waren männlich und 76 % waren Weiße. 97 % der Patienten hatten zu Studienbeginn einen ECOG-Status von 0 oder 1 und 38 % der Patienten hatten eine fortgeschrittene klinische Erkrankung im Rai-Stadium III oder IV. Von den Patienten, für die eine zentrale Testung zur Verfügung stand, hatten 14 % (73 von 538) eine 17p-Deletion, 31 % (170 von 557) eine TP53-Mutation, 67 % (382 von 570 Patienten) hatten eine unmutierte IGHV und 37 % (182 von 486 Patienten) einen komplexen Karyotyp.
Die Patienten erhielten eine mediane Anzahl von 1 vorherigen Therapielinie (Spanne: 0 bis 9), wobei 34 % der Patienten nicht vorbehandelt waren und 66 % mindestens 1 vorherige Therapielinie hatten. Insgesamt hatten 62 % der Patienten eine vorherige Chemotherapie und 6 % eine vorherige BCL2-Inhibitor-Therapie erhalten.
Die Wirksamkeit wurde basierend auf der Gesamtansprechrate (overall response rate, ORR) und der Dauer des Ansprechens gemäß Bewertung durch eine unabhängige Prüfkommission (Independent Review Committee, IRC), in der ITT-Population und der rezidivierten oder refraktären Population ermittelt. Pirtobrutinib zeigte eine Nichtunterlegenheit gegenüber Ibrutinib beim primären Endpunkt ORR sowohl in der ITT- als auch in der rezidivierten oder refraktären Population (p < 0,0001). Die mediane Zeit bis zum Ansprechen bei allen ansprechenden Patienten betrug 3,8 Monate für Pirtobrutinib und 3,9 Monate für Ibrutinib. Über wichtige Subgruppen hinweg wurden konsistente Wirksamkeitsergebnisse beobachtet, auch bei jenen mit 17p-Deletion, bei behandlungsnaiven Patienten und unabhängig vom Alter. Die Ergebnisse zur Wirksamkeit für die primäre Analyse in der ITT-Population sind in Tabelle 4 angeführt.
Tabelle 4: Ergebnisse zur Wirksamkeit gemäß IRC in BRUIN-CLL-314 (ITT-Population)
Parameter a |
Pirtobrutinib |
Ibrutinib |
Gesamtansprechrate b |
||
ORR, n |
288 (87 %) |
260 (79 %) |
CR, n |
15 (5 %) |
8 (2 %) |
CRi, n |
1 (0,3 %) |
0 |
PR, n |
272 (82 %) |
252 (76 %) |
relative Ansprechrate (95 %-KI) c |
1,11 (1,03; 1,19) |
|
P-Wert d |
< 0,0001 |
|
Dauer des Ansprechens |
||
Median der DOR, Monate (95 %-KI) e |
24,7 (24,4; NE) |
NE (NE, NE) |
Rate des 12-monatigen DOR, % (95 %-KI) e |
88 (83; 92) |
85 (79; 89) |
KI: Konfidenzintervall; CR: vollständiges Ansprechen; CRi: vollständiges Ansprechen mit unvollständiger hämatopoetischer Erholung; DOR: Dauer des Ansprechens; NE: nicht schätzbar; nPR: noduläres partielles Ansprechen; ORR: Gesamtansprechrate; PR: partielles Ansprechen.
a Die Wirksamkeit wurde anhand der Leitlinien des International Workshop for Chronic Lymphocytic Leukemia (iwCLL) 2018 bewertet.
b Definiert als CR + CRi + nPR + PR. Kein Patient hatte nPR als bestes Ansprechen.
c Schätzung stratifiziert nach Stratifizierungsfaktoren der Randomisierung.
d 2-seitiger p-Wert basierend auf dem stratifizierten Wald-Test für Nichtunterlegenheit mit Nichtunterlegenheitsmarge von 0,88. Das 2-seitige Signifikanzniveau von 0,005 wurde zu Anfang für die ORR-Nichtunterlegenheitstests in der ITT-Population zugewiesen.
e Basierend auf Kaplan-Meier-Schätzung. Die geschätzte mediane Nachbeobachtung für DOR betrug 14,8 Monate.
Zum Zeitpunkt der Analyse waren die Daten zum Gesamtüberleben (overall survival, OS) noch nicht reif. Bei einer geschätzten medianen Nachbeobachtung von 22,3 Monaten wurde das mediane OS in keinem Arm erreicht, wobei bei 7 % der Patienten ein Ereignis eingetreten war.
BRUIN-CLL-321 (Studie 20020)
Die Wirksamkeit von Jaypirca bei Patienten mit zuvor mit BTK-Inhibitor behandelter CLL wurde in einer randomisierten, multizentrischen, internationalen, offenen, aktiv kontrollierten Studie (BRUIN CLL-321, Studie 20020) untersucht. An der Studie nahmen 238 Patienten mit CLL/SLL teil, die zuvor mit einem BTK-Inhibitor behandelt wurden. Die Patienten wurden im Verhältnis 1:1 randomisiert, und erhielten entweder Jaypirca einmal täglich oral in einer Dosis von 200 mg bis zur Krankheitsprogression oder bis zum Auftreten einer inakzeptablen Toxizität, oder sie erhielten je nach Wahl des Prüfarztes:
Idelalisib plus ein Rituximab-Produkt (IR): Idelalisib 150 mg oral zweimal täglich in Kombination mit 8 Infusionen eines Rituximab-Produkts (375 mg/m2 intravenös am Tag 1 von Zyklus 1, gefolgt von 500 mg/m2 alle 2 Wochen für 4 Dosen und anschließend alle 4 Wochen für 3 Dosen), mit einer Zykluslänge von 28 Tagen bis Krankheitsprogression oder eine inakzeptable Toxizität eintrat.
Bendamustin plus ein Rituximab-Produkt (BR): Bendamustin 70 mg/m2 intravenös (Tag 1 und 2 jedes 28-tägigen Zyklus) in Kombination mit einem Rituximab-Produkt (375 mg/m2 intravenös am Tag 1 von Zyklus 1, gefolgt von 500 mg/m2 am Tag 1 der folgenden Zyklen), für bis zu 6 Zyklen.
Die Randomisierung wurde nach dem 17p-Deletionsstatus (ja/nein) und dem Erhalt einer vorherigen Behandlung mit Venetoclax (ja/nein) stratifiziert. Von den insgesamt 238 Patienten erhielten 119 eine Jaypirca-Monotherapie, 82 IR und 37 BR. Nach bestätigtem Krankheitsprogress hatten die Patienten, die IR oder BR randomisiert erhielten, die Möglichkeit, zur Jaypirca-Monotherapie zu wechseln. Die Baseline-Charakteristika waren in allen Behandlungsarmen vergleichbar. Das mediane Alter betrug 67 Jahre (Spanne: 42 bis 90 Jahre), 70 % waren männlich und 81 % waren Weiße. 93 % der Patienten hatten zu Studienbeginn einen ECOG-Status von 0 oder 1 und 44 % der Patienten hatten eine fortgeschrittene klinische Erkrankung im Rai - Stadium III oder IV. Von den Patienten, für die eine zentrale Testung zur Verfügung stand, hatten 57 % (101 von 176 Patienten) eine 17p-Deletion und/oder eine TP53-Mutation, 86 % (164 von 190 Patienten) hatten eine unmutierte IGHV und 65 % (97 von 149) einen komplexen Karyotyp.
Die Patienten erhielten eine mediane Anzahl von 3 vorangegangenen Therapielinien (Spanne: 1 bis 13), wobei 57 % mindestens 3 Vortherapien und 51 % eine vorherige BCL2-Inhibitor Therapie hatten. Die häufigsten zuvor erhaltenen BTK-Inhibitoren waren Ibrutinib (87 %), Acalabrutinib (16 %) und Zanubrutinib (7 %). 70 % der Patienten setzten den vorherigen BTK-Inhibitor bei refraktärer oder progredienter Erkrankung ab, 14 % setzten wegen Toxizität und 15 % setzten aus anderen Gründen ab.
Die Wirksamkeit wurde anhand des progressionsfreien Überlebens (PFS) der Pirtobrutinib-Monotherapie im Vergleich zum vom Prüfarzt gewählten Arm bewertet, wobei die Beurteilung durch eine unabhängige Prüfkommission (Independent Review Committee, IRC) erfolgte. Die Studie erreichte ihren primären Endpunkt zum vordefinierten Zeitpunkt der finalen Analyse des IRC-bewerteten PFS (Datenschnitt 29. August 2023). Bei einer aktualisierten Analyse (Datenschnitt 29. August 2024) mit einer medianen Nachbeobachtungszeit von 19,4 Monaten (Spanne: 0,03 bis 33,3 Monate) für Pirtobrutinib und 17,7 Monaten (Spanne: 0,03 bis 27,9 Monate) für den vom Prüfarzt gewählten Arm wurde übereinstimmend mit der primären Analyse unter Pirtobrutinib eine Verbesserung des IRC-bewerteten PFS im Vergleich zum vom Prüfarzt gewählten Arm beobachtet. Klinisch relevante Wirksamkeitsergebnisse zugunsten von Pirtobrutinib wurden in wichtigen Subgruppen beobachtet, einschließlich Patienten, die eine vorherige BTK-Inhibitor-Therapie aufgrund von Unverträglichkeit oder Progression abbrachen, und unabhängig von der Anzahl und Art der vorherigen Therapien. Die Ergebnisse zur Wirksamkeit sind in Tabelle 5 angeführt. Die Kaplan-Meier-Kurve des PFS ist in Abbildung 2 dargestellt.
Tabelle 5: Ergebnisse zur Wirksamkeit aus Studie 20020 gemäß IRC-Bewertung bei CLL -Patienten, die zuvor mit einem BTK-Inhibitor behandelt wurden (ITT-Population)
Pirtobrutinib |
Idelalisib + Rituximab |
|
Progressionsfreies Überleben a | ||
Anzahl der Ereignisse, n |
74 (62 %) |
79 (66 %) |
Krankheitsprogression |
60 (50 %) |
66 (55 %) |
Todesfälle |
14 (12 %) |
13 (11 %) |
Median PFS (95 % KI), Monate b |
14,0 (11,2; 16,6) |
8,7 (8,1; 10,4) |
HR (95 % KI) c |
0,54 (0,39; 0,75) |
|
p-Wert d |
0,0002 |
|
KI, Konfidenzintervall; HR, Hazard Ratio.
Datenschnitt: 29. August 2024
a Die Wirksamkeit wurde anhand der Leitlinien des International Workshop for Chronic Lymphocytic Leukemia (iwCLL) 2018 bewertet.
b Basierend auf Kaplan-Meier-Schätzung.
c Basierend auf stratifiziertem Cox-Proportional-Hazards-Modell.
d 2-seitiger nominaler p-Wert basierend auf einem stratifizierten Log-Rank-Test.
Abbildung 2: Kaplan-Meier-Kurve des PFS aus Studie 20020 gemäß IRC-Bewertung bei CLL-Patienten, die zuvor mit einem BTK-Inhibitor behandelt wurden
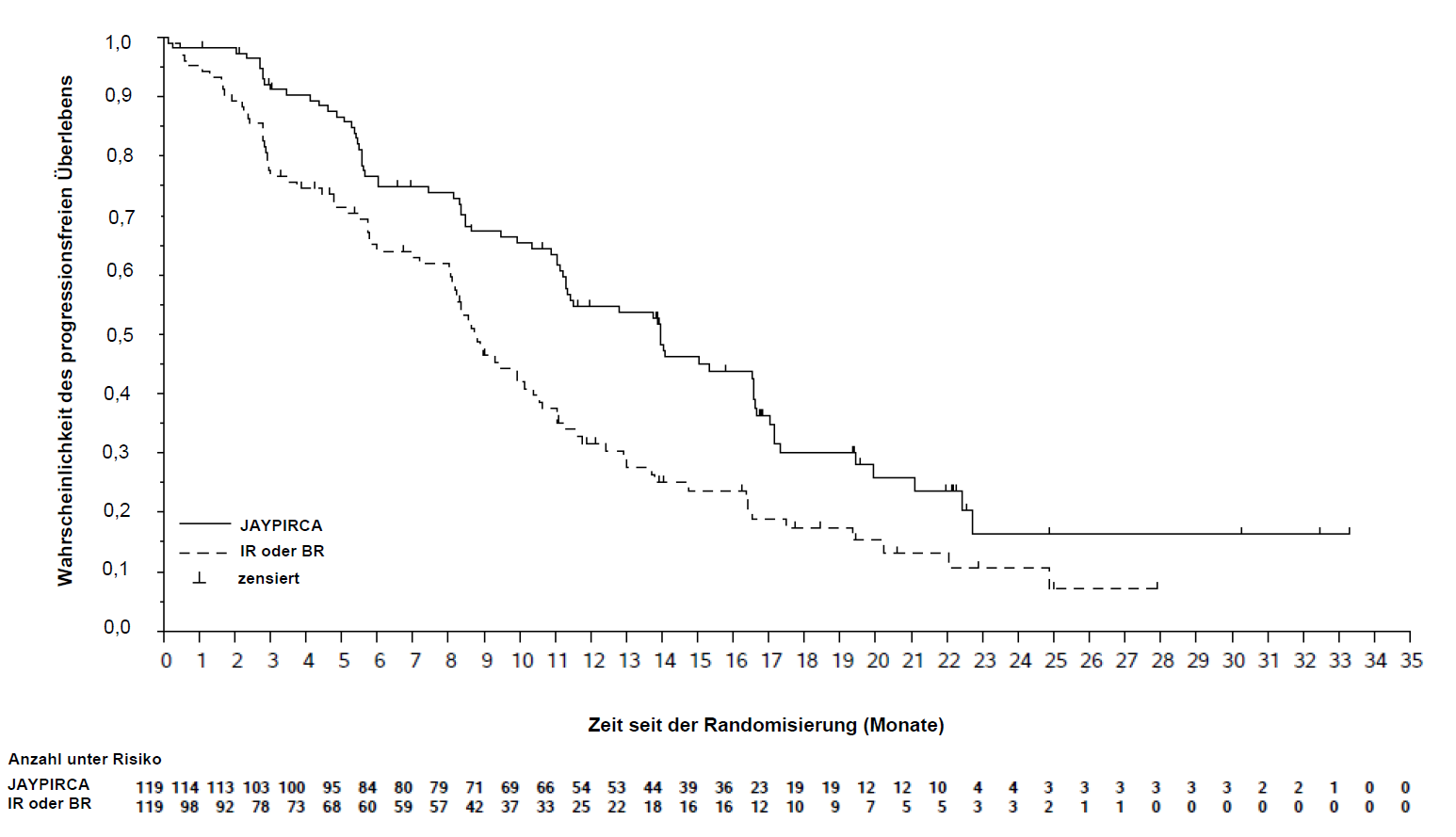
Bei einer medianen Nachbeobachtungszeit des Gesamtüberlebens (OS) von 20,4 Monaten für Pirtobrutinib und 19,2 Monaten im vom Prüfarzt gewählten Arm verstarben 38 Patienten (32,0 %) im Pirtobrutinib-Arm und 32 Patienten (27,0 %) im Vergleichsarm. Das mediane Gesamtüberleben betrug 29,7 Monate (95 % KI: 27,1, NE) im Pirtobrutinib-Arm und wurde im vom Prüfarzt gewählten Arm nicht erreicht. Die Hazard-Ratio betrug 1,090 (95 % KI: 0,679, 1,749; p = 0,7202). Die Analyse des Gesamtüberlebens könnte durch die 50 von 119 Patienten, die vom Vergleichsarm auf Pirtobrutinib gewechselt sind, verfälscht sein.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Jaypirca eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen bei malignen Erkrankungen der reifen B-Zellen gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Bedingte Zulassung
Dieses Arzneimittel wurde unter „Besonderen Bedingungen“ zugelassen. Das bedeutet, dass weitere Nachweise für den Nutzen des Arzneimittels erwartet werden. Die Europäische Arzneimittel-Agentur wird neue Informationen zu diesem Arzneimittel mindestens jährlich bewerten und, falls erforderlich, wird die Zusammenfassung der Merkmale des Arzneimittels aktualisiert werden.
Die Pharmakokinetik von Pirtobrutinib wurde bei gesunden Probanden und bei Patienten mit Krebs beschrieben. Die Dosen reichten von 25 mg bis 300 mg einmal täglich (das 0,125- bis 1,5- Fache der empfohlenen Dosis von 200 mg einmal täglich) bis hin zu Einzeldosen von 900 mg. Anstiege der Plasmaexposition waren ungefähr dosisproportional. Steady State wurde innerhalb von 5 Tagen nach einmal täglicher Gabe erreicht, und bei Krebspatienten betrug das mittlere Akkumulationsverhältnis [Variationskoeffizient (CV %)] nach Verabreichung von 200 mg einmal täglich 1,63 (26,7 %), basierend auf der AUC. Drei Faktoren wird Einfluss auf die Pirtobrutinib PK zugeschrieben: Körpergewicht, Serumalbumin und absolute eGFR. Eine Erhöhung des Körpergewichts von 70 kg auf 120 kg steigert die Pirtobrutinib Clearance voraussichtlich um 24 %; eine Absenkung der absoluten eGFR von 90 ml/min auf 30 ml/min führt voraussichtlich zu einer Verringerung der Pirtobrutinib-Clearance um 16 %; und eine Abnahme des Serumalbumins von 40 g/l auf 30 g/l erhöht die Pirtobrutinib-Clearance voraussichtlich um 21 %. Es ist unwahrscheinlich, dass diese Faktoren allein zu bedeutenden Änderungen der PK von Pirtobrutinib führen. Dosisanpassungen sind nicht empfohlen.
Die mittlere AUC und Cmax (CV %) im Steady State betrugen 97 100 h*ng/ml (35,5 %) bzw. 6 690 ng/ml (24 %) bei der empfohlenen Dosierung von 200 mg einmal täglich bei Krebspatienten.
Bei der empfohlenen Dosierung erreicht Pirtobrutinib pharmakokinetische Expositionen, die den BTK-IC96 Talwert überschreiten können, und führt so zu einer tonischen BTK-Zielinhibition während des einmal täglichen Dosierungszeitraums, unabhängig von der intrinsischen Rate des BTK Umsatzes.
Resorption
Die absolute Bioverfügbarkeit von Pirtobrutinib nach einer oralen Einzeldosis von 200 mg beträgt bei gesunden Probanden 85,5 %. Die mediane Zeit bis zum Erreichen der maximalen Plasmakonzentration (tmax) beträgt sowohl bei Krebspatienten als auch bei gesunden Probanden etwa 2 Stunden. Die Resorption ist nicht pH-abhängig.
Einfluss von Nahrung
Eine fettreiche, kalorienreiche Mahlzeit, die gesunden Probanden verabreicht wurde, verringerte die Cmax von Pirtobrutinib um 23 % und verzögerte tmax um 1 Stunde. Es gab keine Auswirkung auf die Pirtobrutinib-AUC. Pirtobrutinib kann unabhängig von Mahlzeiten eingenommen werden.
Verteilung
Das mittlere scheinbare zentrale Verteilungsvolumen von Pirtobrutinib beträgt bei Krebspatienten 35,4 l. Die Plasmaproteinbindung beträgt 96 % und war zwischen 0,5 und 50 µM konzentrationsunabhängig. Im Plasma von gesunden Probanden und Probanden mit schwerer Nierenfunktionsstörung betrug die Proteinbindung 96 %. Das mittlere Blut-zu-Plasma Verhältnis beträgt 0,79.
Biotransformation
Pirtobrutinib wird hauptsächlich über den hepatischen Metabolismus ausgeschieden. Pirtobrutinib wird über CYP3A4, UGT1A8 und UGT1A9 zu mehreren inaktiven Metaboliten metabolisiert. Die Modulation von CYP3A hatte keinen klinisch relevanten Einfluss auf die Pirtobrutinib-Exposition.
Pirtobrutinib hemmt CYP2C8, CYP2C9 und CYP3A4 in vitro und hemmt minimal CYP1A2, CYP2B6, CYP2C19 oder CYP2D6 bei 60 µM. In vitro induziert Pirtobrutinib CYP3A4, CYP3A5, CYP2C19 und CYP2B6.
Pirtobrutinib hemmt in vitro minimal UGT1A1 mit einem IC50 = 18 µM.
Gleichzeitige Verabreichung mit Transportsubstraten/Inhibitoren
In-vitro-Studien zeigten, dass Pirtobrutinib ein Substrat von P-gp und BCRP ist.
Pirtobrutinib ist ein In-vitro-Inhibitor von P-gp und BCRP. Pirtobrutinib beeinflusste in klinischen Studien die Pharmakokinetik von Digoxin, einem P-gp-Substrat, und Rosuvastatin, einem BCRP-Substrat (siehe Abschnitt 4.5).
Elimination
Die mittlere scheinbare Clearance von Pirtobrutinib beträgt 1,98 l/h bei einer effektiven Halbwertszeit von etwa 19,9 Stunden. Nach einer radioaktiv markierten Einzeldosis von Pirtobrutinib 200 mg an gesunden Probanden wurden 37 % der Dosis im Stuhl (18 % unverändert) und 57 % im Urin (10 % unverändert) wiedergefunden.
Besondere Patientengruppen
Alter, Geschlecht, Herkunft und Körpergewicht
Basierend auf einer populationspharmakokinetischen Analyse bei Krebspatienten hatten Alter (Spanne: 22 – 95 Jahre), Herkunft, Geschlecht und Körpergewicht (Spanne: 35,7 – 154 kg) keine klinisch bedeutsame Auswirkung auf die Pirtobrutinib-Exposition.
Nierenfunktionsstörung
In einer populationspharmakokinetischen Analyse von Krebspatienten war die Clearance von Pirtobrutinib bei Patienten mit leichter (eGFR 60 bis < 90 ml/min) oder moderater Nierenfunktionsstörung (eGFR 30 bis < 60 ml/min) um 16 % bis 27 % niedriger als die Clearance bei Patienten mit normaler Nierenfunktion. Dies führte zu einer erwarteten Exposition von AUC = 94 100 ng*h/ml und Cmax = 6 680 ng/ml bei Patienten mit leichter Nierenfunktionsstörung (16 bis 19 % höher im Vergleich zu Patienten mit normaler Nierenfunktion) und AUC = 108 000 ng*h/ml und Cmax = 7 360 ng/ml bei Patienten mit mäßig eingeschränkter Nierenfunktion (28 bis 36 % höher im Vergleich zu Patienten mit normaler Nierenfunktion).
In einer klinisch-pharmakologischen Studie mit ansonsten gesunden Probanden war die scheinbare Clearance bei vier Teilnehmern mit schwerer Nierenfunktionsstörung (eGFR 15 bis < 30 ml/min) um 35 % niedriger als bei acht Teilnehmern mit normaler Nierenfunktion (eGFR ≥ 90 ml/min). Dies führte zu Expositionen von AUC0 - inf = 115 000 ng*h/ml und Cmax = 2 980 ng/ml (62 % höher bzw. 7 % niedriger im Vergleich zur normalen Nierenfunktion).
Dialysepflichtige Patienten mit Nierenerkrankungen im Endstadium wurden nicht untersucht (siehe Abschnitt 4.2).
Leberfunktionsstörung
Es gab keine klinisch signifikanten Unterschiede in der PK von Pirtobrutinib unabhängig vom Grad der Leberfunktionsstörung (nach Child-Pugh A, B und C oder beliebiges Gesamtbilirubin und beliebiges AST). In einer speziellen Studie zu Leberfunktionsstörungen waren die mittleren AUC und Cmax von Pirtobrutinib bei Patienten mit leichter Leberfunktionsstörung (Child-Pugh A) und Patienten mit normaler Leberfunktion ähnlich. Bei Patienten mit mäßiger Leberfunktionsstörung (Child-Pugh B) war die AUC 15 % niedriger als bei normaler Leberfunktion und die Cmax war ähnlich. Bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh C) war die AUC von Pirtobrutinib um 21 % niedriger und die mittlere Cmax um 24 % niedriger als bei Patienten mit normaler Leberfunktion. Die ungebundene Fraktion (fu: fraction unbound) von Pirtobrutinib nahm im Allgemeinen mit zunehmender Schwere der Leberfunktionsstörung zu. Daher wurde nach Korrektur der Pirtobrutinib PK Expositionsparameter mit fu kein klinisch signifikanter Unterschied in den Pirtobrutinib PK Expositionsparametern (AUCu und Cmax,u) zwischen Patienten mit Leberfunktionsstörung jeglichen Grades und normaler Leberfunktion beobachtet.
Kinder und Jugendliche
Es wurden keine pharmakokinetischen Studien mit Pirtobrutinib bei Patienten unter 18 Jahren durchgeführt.
In Studien mit wiederholter Verabreichung wurden bei Ratten (bei 0,69‑facher humaner Exposition bei der empfohlenen Dosis von 200 mg, basierend auf der AUC) eine verminderte T-Zell-abhängige Antikörperreaktion und bei Hunden (bei 0,42‑facher humaner Exposition) minimale bis leichte Hornhautläsionen beobachtet. Leichte bis mittelschwere Gefäßnekrosen und vaskuläre/perivaskuläre Entzündungen in großen Lungenblutgefäßen wurden nur bei Ratten beobachtet. Diese Effekte traten bei klinisch relevanten Expositionsniveaus auf.
Genotoxizität / Karzinogenität
Pirtobrutinib war in einem bakteriellen Mutagenitätstest (Ames) nicht mutagen. Pirtobrutinib war in zwei in vitro Mikronukleus-Assays mit humanen peripheren Blutlymphozyten aneugen. Pirtobrutinib zeigte keine Wirkung in einem in vivo Mikronukleus-Assay im Knochenmark von Ratten bei Dosen von bis zu 2 000 mg/kg (Einzeldosis), was einer ungefähr 11‑fach höheren Exposition entspricht (unter Berücksichtigung des ungebundenen Cmax-Werts bei weiblichen Tieren) als die Exposition beim Menschen bei 200 mg.
Es wurden keine Karzinogenitätsstudien mit Pirtobrutinib durchgeführt.
Embryotoxizität / Teratogenität
In Reproduktionsstudien an Tieren führte die Verabreichung von Pirtobrutinib an trächtigen Ratten während der Organogenese zu verringertem fötalen Gewicht, zu embryofötaler Sterblichkeit und zu fötalen Missbildungen bei maternaler Exposition, die dem 3,0‑fachen der menschlichen Exposition bei der empfohlenen Dosis von 200 mg, basierend auf der AUC, entsprach.
Reproduktionstoxizität
Es wurden keine Fertilitätsstudien mit Pirtobrutinib durchgeführt. In Toxizitätsstudien mit wiederholter Gabe von bis zu 3-monatiger Dauer hatte Pirtobrutinib bei der empfohlenen Dosis von 200 mg, basierend auf der AUC, keine Wirkung auf die männlichen Fortpflanzungsorgane beim 0,69‑fachen bzw. 0,42‑fachen der humanen Exposition bei Ratten und Hunden. Pirtobrutinib hatte beim 4,0‑fachen bzw. 0,42‑fachen der humanen Exposition bei Ratten bzw. Hunden keine Wirkung auf die weiblichen Fortpflanzungsorgane.
Tablettenkern
Hypromelloseacetatsuccinat
Mikrokristalline Cellulose
Lactose-Monohydrat
Croscarmellose-Natrium
Magnesiumstearat
Siliciumdioxid-Hydrat
Filmüberzug
Hypromellose
Titandioxid
Triacetin
Indigocarmin (E 132)
Nicht zutreffend
3 Jahre
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Jaypirca® 100 mg Filmtabletten
Mit Aluminiumfolie versiegelte Polyvinylchlorid/Polychlortrifluorethylen-Blisterpackungen in Packungen mit 28, 30, 56, 60, 84 oder 168 Filmtabletten.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Eli Lilly Nederland B.V.
Orteliuslaan 1000
3528 BD Utrecht
Niederlande
EU/1/23/1738/004
EU/1/23/1738/005
EU/1/23/1738/006
EU/1/23/1738/007
EU/1/23/1738/008
EU/1/23/1738/009
Datum der Erteilung der Zulassung: 30. Oktober 2023
Datum der letzten Verlängerung der Zulassung: 08. September 2025
Juli 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig
Jaypirca® 100 mg Filmtabletten
Inhalt |
PZN |
56 Filmtabletten |
18468577 |
168 Filmtabletten |
19801062 |
Lilly Deutschland GmbH
Werner-Reimers-Straße 2 - 4
D-61352 Bad Homburg
Tel. +49-(0) 6172 273 2222