
Eylea 114,3 mg/ml Injektionslösung
Eylea 114,3 mg/ml Injektionslösung in einer Fertigspritze
1 ml Injektionslösung enthält 114,3 mg Aflibercept*.
Eylea 114,3 mg/ml Injektionslösung
Jede Durchstechflasche enthält 30,1 mg Aflibercept in 0,263 ml Lösung. Diese Menge reicht aus, um eine Einzeldosis von 0,07 ml, in denen 8 mg Aflibercept enthalten sind, anzuwenden.
Eylea 114,3 mg/ml Injektionslösung in einer Fertigspritze
Jede Fertigspritze enthält 21 mg Aflibercept in 0,184 ml Lösung. Diese Menge reicht aus, um eine Einzeldosis von 0,07 ml, in denen 8 mg Aflibercept enthalten sind, anzuwenden.
*Aflibercept ist ein Fusionsprotein aus Fragmenten der extrazellulären Domänen der humanen VEGF-Rezeptoren (vaskulärer endothelialer Wachstumsfaktor) 1 und 2 und dem Fc-Fragment des humanen IgG1, hergestellt in Ovarialzellen chinesischer Hamster (CHO) vom Typ K1 mit Hilfe rekombinanter DNA-Technologie.
Sonstiger Bestandteil mit bekannter Wirkung
Jeder ml Injektionslösung enthält 0,3 mg Polysorbat 20 (E 432).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung (Injektionszubereitung)
Klare bis leicht opaleszente, farblose bis blassgelbe, isoosmotische Lösung mit einem pH-Wert von 5,8.
Eylea wird angewendet bei Erwachsenen zur Behandlung
der neovaskulären (feuchten) altersabhängigen Makuladegeneration (nAMD) (siehe Abschnitt 5.1)
einer Visusbeeinträchtigung aufgrund eines diabetischen Makulaödems (DMÖ) (siehe Abschnitt 5.1).
einer Visusbeeinträchtigung aufgrund eines Makulaödems infolge eines retinalen Venenverschlusses (RVV) (Venenastverschluss [VAV], Zentralvenenverschluss [ZVV] und Hemi-Zentralvenenverschluss [HZVV]) (siehe Abschnitt 5.1).
Eylea darf nur von einem qualifizierten Arzt mit Erfahrung in der Durchführung intravitrealer Injektionen appliziert werden.
Dosierung
nAMD und DMÖ
Die empfohlene Dosis beträgt 8 mg Aflibercept, entsprechend 0,07 ml Lösung. Für die Anwendungsgebiete nAMD und DMÖ ist die Dosierung gleich. Die 8‑mg‑Dosierung erfordert die Anwendung von Eylea 114,3 mg/ml.
Bei Patienten, die eine Behandlung beginnen, wird Eylea mit 3 aufeinanderfolgenden monatlichen Injektionen angewendet. Danach kann der Arzt basierend auf dem funktionellen und/oder morphologischen Befund das Injektionsintervall auf bis zu 4 Monate verlängern. Anschließend können unter Aufrechterhaltung eines stabilen funktionellen und/oder morphologischen Befundes die Behandlungsintervalle, wie z.B. mit einem „Treat and Extend“-Dosierungsschema, weiter auf bis zu 6 Monate verlängert werden (siehe Abschnitt 5.1).
Bei Patienten, die zuvor mit Eylea 40 mg/ml oder anderen Anti-VEGF-Arzneimitteln behandelt wurden und auf Eylea 114,3 mg/ml umgestellt werden, kann sich das Behandlungsschema von dem für therapienaive Patienten unterscheiden. Behandlungsintervalle sollten basierend auf dem funktionellen und/oder morphologischen Befund festgelegt werden (siehe Abschnitt 5.1).
Bei Patienten mit stabilem funktionellem und morphologischem Befund können vorherige Behandlungsintervalle beibehalten oder nach der ersten Injektion von Eylea 114,3 mg/ml verlängert werden, wie z. B. mit einem „Treat and Extend“-Dosierungsschema.
Bei Patienten mit suboptimalem funktionellem und/oder morphologischem Befund kann die Behandlung mit Eylea 114,3 mg/ml mit bis zu 3 aufeinanderfolgenden monatlichen Dosen begonnen werden, gefolgt von einer Anpassung der Injektionsintervalle, wie z. B. mit einem „Treat and Extend“-Dosierungsschema.
Wenn sich der funktionelle und/oder morphologische Befund verschlechtert, sollte das Behandlungsintervall nach Ermessen des Arztes entsprechend verkürzt werden. Das Intervall zwischen 2 Injektionen sollte nicht kürzer als 1 Monat sein.
Wenn der funktionelle und/oder morphologische Befund darauf hinweist, dass der Patient nicht von einer Fortsetzung der Behandlung profitiert, sollte die Behandlung mit Eylea 114,3 mg/ml beendet werden.
Monatliche Injektionen von Eylea 8 mg für mehr als 3 aufeinanderfolgende Injektionen wurden in der PULSAR (nAMD)- und PHOTON (DMÖ)-Studie nicht untersucht. Verfügbare Daten unterstützen die Anwendung von mehr als 3 aufeinanderfolgenden monatlichen Dosen bei bestimmten Patienten, allerdings sind die Daten derzeit begrenzt.
Die Häufigkeit der Kontrolluntersuchungen sollte sich nach dem Zustand des Patienten und nach dem Ermessen des Arztes richten. Für Fälle, in denen eine Behandlung ausgesetzt werden sollte, siehe Abschnitt 4.4.
RVV
Die empfohlene Dosis beträgt 8 mg Aflibercept, entsprechend 0,07 ml Lösung. Die 8‑mg‑Dosierung erfordert die Anwendung von Eylea 114,3 mg/ml.
Bei Patienten, die eine Behandlung beginnen, wird Eylea mit 3 aufeinanderfolgenden monatlichen Injektionen angewendet. Danach kann der Arzt basierend auf dem funktionellen und/oder morphologischen Befund das Injektionsintervall verlängern (siehe Abschnitt 5.1).
Bei Patienten, die zuvor mit Eylea 40 mg/ml oder anderen Anti-VEGF-Arzneimitteln behandelt wurden und auf Eylea 114,3 mg/ml umgestellt werden, kann sich das Behandlungsschema von dem für therapienaive Patienten unterscheiden. Behandlungsintervalle sollten basierend auf dem funktionellen und/oder morphologischen Befund festgelegt werden (siehe Abschnitt 5.1).
Bei Patienten mit stabilem funktionellem und morphologischem Befund können vorherige Behandlungsintervalle beibehalten oder nach der ersten Injektion von Eylea 114,3 mg/ml verlängert werden, wie z. B. mit einem „Treat and Extend“-Dosierungsschema.
Bei Patienten mit suboptimalem funktionellem und/oder morphologischem Befund kann die Behandlung mit Eylea 114,3 mg/ml mit bis zu 3 aufeinanderfolgenden monatlichen Dosen begonnen werden, gefolgt von einer Anpassung der Injektionsintervalle, wie z. B. mit einem „Treat and Extend“-Dosierungsschema.
Wenn sich der funktionelle und/oder morphologische Befund verschlechtert, sollte das Behandlungsintervall nach Ermessen des Arztes entsprechend verkürzt werden (siehe Abschnitt 5.1). Das Intervall zwischen 2 Injektionen sollte nicht kürzer als 1 Monat sein.
Wenn der funktionelle und/oder morphologische Befund darauf hinweist, dass der Patient nicht von einer Fortsetzung der Behandlung profitiert, sollte die Behandlung mit Eylea 114,3 mg/ml beendet werden.
Die Häufigkeit der Kontrolluntersuchungen sollte sich nach dem Zustand des Patienten und nach dem Ermessen des Arztes richten. Für Fälle, in denen eine Behandlung ausgesetzt werden sollte, siehe Abschnitt 4.4.
Spezielle Patientengruppen
Patienten mit Nieren- oder Leberfunktionsstörung
Bei Patienten mit Nieren- oder Leberfunktionsstörung wurden keine speziellen Studien durchgeführt.
Verfügbare Daten weisen nicht darauf hin, dass bei diesen Patienten eine Anpassung der Eylea‑Dosis erforderlich ist (siehe Abschnitt 5.2).
Ältere Patienten
Verfügbare Daten weisen nicht darauf hin, dass bei diesen Patienten eine Anpassung der Eylea‑Dosis erforderlich ist.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Eylea 114,3 mg/ml bei Kindern und Jugendlichen unter 18 Jahren ist nicht erwiesen. Es gibt in den Anwendungsgebieten nAMD, DMÖ und RVV keinen relevanten Nutzen von Eylea 114,3 mg/ml bei Kindern und Jugendlichen.
Art der Anwendung
Eylea ist nur als intravitreale Injektion anzuwenden.
Intravitreale Injektionen sind entsprechend medizinischer Standards und geltenden Richtlinien nur von einem qualifizierten Arzt mit Erfahrung in der Durchführung intravitrealer Injektionen durchzuführen. Generell müssen eine adäquate Anästhesie und Asepsis, einschließlich des Einsatzes eines topischen Breitbandmikrobizids (z. B. Povidon-Iod, das auf die periokulare Haut, das Augenlid und die Augenoberfläche aufgetragen wird) gewährleistet werden. Die chirurgische Händedesinfektion, sterile Handschuhe, ein steriles Abdecktuch und ein steriler Lidsperrer (oder ein vergleichbares Instrument) werden empfohlen.
Die Injektionskanüle wird 3,5 bis 4,0 mm posterior zum Limbus in den Glaskörper eingebracht, dabei sollte der horizontale Meridian vermieden und in Richtung Bulbusmitte gezielt werden. Danach wird das Injektionsvolumen von 0,07 ml injiziert. Nachfolgende Injektionen sollten nicht an derselben skleralen Einstichstelle erfolgen.
Unmittelbar nach der intravitrealen Injektion sollten Patienten auf einen Anstieg des Augeninnendrucks kontrolliert werden. Eine angemessene Überwachung kann in einer Überprüfung der Perfusion des Sehnervenkopfes oder einer Tonometrie bestehen. Für den Bedarfsfall sollte steriles Besteck zur Durchführung einer Parazentese zur Verfügung stehen.
Nach einer intravitrealen Injektion sollten Patienten instruiert werden, unverzüglich alle Symptome zu melden, die auf eine Endophthalmitis hinweisen (z. B. Augenschmerzen, Augenrötung, Photophobie, verschwommenes Sehen).
Jede Durchstechflasche oder Fertigspritze sollte nur zur Behandlung eines einzigen Auges verwendet werden.
Nach der Injektion ist nicht verwendetes Arzneimittel oder Abfallmaterial entsprechend den nationalen Anforderungen zu beseitigen.
Zur Handhabung des Arzneimittels vor Anwendung, siehe Abschnitt 6.6.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Okulare oder periokulare Infektion.
Bestehende schwere intraokulare Entzündung.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Durch die intravitreale Injektion bedingte Reaktionen
Intravitreale Injektionen, einschließlich solcher mit Eylea, wurden mit Endophthalmitis, intraokularer Entzündung, Netzhautablösung, Einriss der Netzhaut und traumatischer Katarakt in Verbindung gebracht (siehe Abschnitt 4.8). Bei der Anwendung von Eylea sind immer angemessene aseptische Injektionsmethoden anzuwenden. Die Patienten sollten instruiert werden, unverzüglich alle Symptome zu melden, die auf eine Endophthalmitis oder auf eines der oben aufgeführten Ereignisse hinweisen, und sollten angemessen behandelt werden.
Erhöhter Augeninnendruck
Ein vorübergehendes Ansteigen des Augeninnendrucks wurde innerhalb von 60 Minuten nach einer intravitrealen Injektion, einschließlich derer mit Eylea, beobachtet (siehe Abschnitt 4.8). Daher müssen sowohl der Augeninnendruck als auch die Perfusion des Sehnervenkopfes überwacht und bei Bedarf angemessen behandelt werden. Besondere Vorsicht ist bei Patienten mit einem schlecht eingestellten Glaukom geboten (Eylea darf nicht injiziert werden, solange der Augeninnendruck bei ≥ 30 mmHg liegt).
Immunogenität
Da Aflibercept ein therapeutisches Protein ist, besteht die Möglichkeit einer Immunogenität (siehe Abschnitt 5.1). Patienten sollten dazu angehalten werden, alle Anzeichen oder Symptome einer intraokularen Entzündung, z. B. Schmerzen, Photophobie oder Rötung, zu berichten, da diese klinische Anzeichen einer Überempfindlichkeit sein könnten.
Systemische Effekte
Systemische Nebenwirkungen inklusive nicht-okularer Hämorrhagien und arterieller thromboembolischer Ereignisse wurden nach intravitrealer Injektion von VEGF-Hemmern berichtet. Es besteht ein theoretisches Risiko, dass diese in Zusammenhang mit der VEGF-Hemmung stehen können (siehe Abschnitt 4.8).
Es gibt begrenzte Daten zur Sicherheit bei der Behandlung von Patienten mit nAMD, DMÖ und RVV, die innerhalb der letzten 6 Monate einen Schlaganfall, transitorische ischämische Attacken oder einen Myokardinfarkt in der Vorgeschichte hatten. Die Behandlung entsprechender Patienten sollte mit Umsicht erfolgen.
Bilaterale Behandlung
Die Sicherheit und Wirksamkeit einer bilateralen Behandlung mit Eylea 114,3 mg/ml je Auge wurde nicht untersucht (siehe Abschnitt 5.1). Falls beide Augen gleichzeitig behandelt werden, kann die systemische Exposition und damit das Risiko systemischer Nebenwirkungen erhöht sein.
Gleichzeitige Anwendung anderer Anti-VEGF-Arzneimittel
Es liegen begrenzte Erfahrungen zur gleichzeitigen Anwendung von Eylea mit anderen Anti-VEGF Arzneimitteln (systemisch oder okular) vor.
Aussetzen der Behandlung
In folgenden Fällen sollte die Behandlung ausgesetzt werden:
bei Verminderung der bestmöglich korrigierten Sehschärfe (BCVA) von ≥ 30 Buchstaben im Vergleich zur zuletzt gemessenen Sehschärfe
bei rhegmatogener Netzhautablösung oder Makulaforamina Stadium 3 oder 4
bei einem Einriss der Retina
bei subretinaler Blutung, bei der das Zentrum der Fovea betroffen ist oder die Größe der Blutung ≥ 50 % der gesamten Läsion ausmacht
bei einem durchgeführten oder geplanten intraokularen Eingriff innerhalb der vergangenen oder kommenden 28 Tage.
Einriss des retinalen Pigmentepithels
Zu den Risikofaktoren, die nach einer Anti-VEGF-Therapie bei nAMD zur Entwicklung eines retinalen Pigmentepitheleinrisses führen können, gehören großflächige und/oder hohe Abhebungen des retinalen Pigmentepithels. Zu Beginn einer Therapie mit Aflibercept ist Vorsicht bei Patienten geboten, die diese Risikofaktoren für das Auftreten von retinalen Pigmentepitheleinrissen aufweisen.
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während der Behandlung und für mindestens 4 Monate nach der letzten intravitrealen Injektion von Eylea 114,3 mg/ml eine zuverlässige Verhütungsmethode anwenden (siehe Abschnitt 4.6).
Personengruppen mit begrenzten Daten
Es gibt nur begrenzte Erfahrungen mit Eylea bei der Behandlung von Diabetikern mit einem HbA1c über 12 % oder mit proliferativer diabetischer Retinopathie.
Eylea wurde nicht untersucht bei Patienten mit aktiven systemischen Infektionen oder bei Patienten, die gleichzeitig andere Augenerkrankungen wie eine Netzhautablösung oder ein Makulaforamen hatten. Es gibt ebenfalls keine Erfahrungen bei der Behandlung mit Eylea bei Diabetikern mit nicht eingestelltem Bluthochdruck. Der Arzt sollte diese fehlenden Informationen bei der Behandlung entsprechender Patienten berücksichtigen.
Information über sonstige Bestandteile
Dieses Arzneimittel enthält 0,021 mg Polysorbat 20 in jeder 0,07 ml Dosis, entsprechend 0,3 mg/ml. Polysorbate können allergische Reaktionen hervorrufen.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während der Behandlung und für mindestens 4 Monate nach der letzten intravitrealen Injektion von Eylea 114,3 mg/ml eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Es liegen begrenzte Daten zur Anwendung von Aflibercept bei Schwangeren vor.
Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3).
Eylea 114,3 mg/ml sollte während der Schwangerschaft nicht angewendet werden, es sei denn, der erwartete Nutzen überwiegt das potenzielle Risiko für den Fetus.
Stillzeit
Sehr begrenzte Daten beim Menschen weisen darauf hin, dass Aflibercept in geringen Mengen in die Muttermilch übergehen kann. Aflibercept ist ein großes Proteinmolekül und es ist zu erwarten, dass die Menge an Arzneimittel, die vom Säugling aufgenommen wird, gering ist. Die Auswirkungen von Aflibercept auf gestillte Neugeborene/Kinder sind nicht bekannt.
Als Vorsichtsmaßnahme wird das Stillen während der Anwendung von Eylea 114,3 mg/ml nicht empfohlen.
Fertilität
Es liegen keine Daten zur Fertilität beim Menschen vor. Ergebnisse aus tierexperimentellen Studien mit hohen systemischen Expositionen weisen darauf hin, dass Aflibercept die männliche und weibliche Fertilität beeinträchtigen kann (siehe Abschnitt 5.3).
Die Injektion von Eylea hat aufgrund möglicher, vorübergehender Sehstörungen im Zusammenhang mit der Injektion oder der Augenuntersuchung einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Patienten sollten kein Fahrzeug führen oder Maschinen bedienen, bis sich ihr Sehvermögen wieder ausreichend erholt hat.
Zusammenfassung des Sicherheitsprofils
Schwerwiegende Nebenwirkungen waren Katarakt (7,1 %), erhöhter Augeninnendruck (3,8 %), Einblutung in die Retina (2,8 %), Glaskörperblutung (1,1 %), subkapsuläre Katarakt (0,6 %), Netzhauteinriss (0,5 %), Kernkatarakt (0,4 %) und Netzhautablösung (0,4 %).
Die am häufigsten beobachteten Nebenwirkungen der mit Eylea 114,3 mg/ml behandelten Patienten waren Katarakt (7,1 %), verminderte Sehschärfe (4,3 %), Bindehautblutung (4,0 %), erhöhter Augeninnendruck (3,8 %), Glaskörperabhebung (3,5 %), Glaskörpertrübungen (3,2 %) und Einblutung in die Retina (2,8 %).
Das in den 4 klinischen Studien beobachtete Sicherheitsprofil war bei Patienten, die mit Eylea 114,3 mg/ml (N = 1 808) und Eylea 40 mg/ml (N = 857) behandelt wurden, sowie bei Patienten mit nAMD, DMÖ und RVV ähnlich.
Tabellarische Auflistung der Nebenwirkungen
Insgesamt 1 808 Patienten, die mit Eylea 114,3 mg/ml bis zu 96 Wochen behandelt wurden, bildeten die Sicherheitspopulation in 4 klinischen Phase-II/III-Studien (CANDELA, PULSAR, PHOTON, QUASAR).
Die unten aufgeführten Sicherheitsdaten schließen alle Nebenwirkungen ein, die aller Wahrscheinlichkeit nach auf den Injektionsvorgang oder das Arzneimittel zurückzuführen sind und berichtet wurden.
Die Nebenwirkungen werden entsprechend der Systemorganklasse und der Häufigkeit gemäß folgender Konvention aufgelistet: Sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1 000, <1/100), selten (≥1/10 000, <1/1 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 1: Alle während der Behandlung mit Eylea 114,3 mg/ml aufgetretenen Nebenwirkungen, über die bei Patienten mit nAMD, DMÖ oder RVV in Phase-II/III-Studien oder aus Beobachtungen der Anwendung nach Markteinführung berichtet wurde
Systemorganklasse |
Häufigkeit |
Nebenwirkung |
Erkrankungen des Immunsystems |
Häufig |
Überempfindlichkeit* |
Augenerkrankungen |
Häufig |
Katarakt, Anstieg des Augeninnendrucks, Glaskörpertrübung, Glaskörperabhebung, Glaskörperblutung, Einblutung in die Retina, verminderte Sehschärfe, Augenschmerzen, Bindehautblutung, Keratitis punctata, Hornhautabrasion |
Gelegentlich |
Netzhautablösung, Netzhauteinriss, Einriss des retinalen Pigmentepithels, Abhebung des retinalen Pigmentepithels, |
|
Selten |
Erblindung, Endophthalmitis, Reizung des Augenlids |
|
Nicht bekannt |
Skleritis** |
* Berichte von Überempfindlichkeit einschließlich Hautausschlag, Pruritus, Urtikaria.
** Aus Berichten nach Markteinführung.
Die folgenden Nebenwirkungen von Eylea 40 mg/ml sind auch bei Eylea 114,3 mg/ml zu erwarten: abnorme Empfindung im Auge, Hornhautepitheldefekt, Schwebeteilchen in der Vorderkammer, traumatische Katarakt, Hypopyon, schwere anaphylaktische/anaphylaktoide Reaktionen.
Beschreibung ausgesuchter Nebenwirkungen
Produktklassenbezogene Nebenwirkungen
Arterielle thromboembolische Ereignisse (ATE) sind Nebenwirkungen, die möglicherweise mit der systemischen VEGF-Hemmung in Verbindung stehen. Es besteht ein theoretisches Risiko von ATE inklusive Schlaganfall und Myokardinfarkt nach intravitrealer Anwendung von VEGF-Hemmern. Eine geringe Inzidenzrate von ATE wurde in klinischen Studien mit Aflibercept bei Patienten mit nAMD, DMÖ und RVV beobachtet. Indikationsübergreifend wurde kein nennenswerter Unterschied zwischen den mit Eylea 114,3 mg/ml behandelten Gruppen und den mit Eylea 40 mg/ml behandelten Vergleichsgruppen beobachtet.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem nationalen Meldesystem anzuzeigen.
Deutschland
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: http://www.bfarm.de
Österreich
Bundesamt für Sicherheit im Gesundheitswesen
Traisengasse 5
1200 WIEN
ÖSTERREICH
Fax: + 43 (0) 50 555 36207
Website: http://www.basg.gv.at/
Bei einer Überdosierung mit einem größeren Injektionsvolumen als üblich kann es zu einem Anstieg des Augeninnendrucks kommen. Daher sollte im Fall einer Überdosierung der Augeninnendruck überwacht werden und, falls dies vom behandelnden Arzt als notwendig erachtet wird, sollten geeignete Maßnahmen eingeleitet werden (siehe Abschnitte 4.4 und 6.6).
Pharmakotherapeutische Gruppe: Ophthalmika / Antineovaskuläre Mittel, ATC-Code: S01LA05
Aflibercept ist ein rekombinantes Fusionsprotein, bei dem Fragmente der extrazellulären Domänen der humanen VEGF-Rezeptoren 1 und 2 mit dem Fc-Fragment des humanen IgG1 fusioniert wurden.
Aflibercept wird in Ovarialzellen chinesischer Hamster (CHO) vom Typ K1 mit Hilfe rekombinanter DNA-Technologie hergestellt.
Wirkmechanismus
Der vaskuläre endotheliale Wachstumsfaktor A (VEGF-A) und der Plazenta-Wachstumsfaktor (PlGF) gehören zur VEGF-Familie der angiogenen Faktoren, die an den Endothelzellen als starke mitogene und chemotaktische Faktoren und als vaskuläre Permeabilitätsfaktoren wirken können. VEGF bindet an die beiden Rezeptor-Tyrosinkinasen VEGFR-1 und VEGFR-2, die sich an der Oberfläche von Endothelzellen befinden. PlGF bindet nur an VEGFR-1, welcher auch auf der Oberfläche von Leukozyten zu finden ist. Eine zu starke Aktivierung dieser Rezeptoren durch VEGF-A kann zu pathologischer Neovaskularisation und erhöhter vaskulärer Permeabilität führen. PlGF kann unabhängig davon den VEGFR‑1 aktivieren, um eine Entzündungsreaktion in der Netzhaut zu fördern. Weiter ist bekannt, dass PlGF bei pathologischen Zuständen wie nAMD, diabetischer Retinopathie (DR), DMÖ und retinalem Venenverschluss (RVV) erhöht ist.
Pharmakodynamische Wirkungen
Aflibercept wirkt als löslicher Köderrezeptor, der VEGF-A und PlGF mit höherer Affinität als deren natürliche Rezeptoren bindet und so die Bindung und Aktivierung dieser artverwandten VEGF-Rezeptoren hemmt.
In tierexperimentellen Studien kann Aflibercept die pathologische Neovaskularisation und die vaskuläre Leckage bei einer Reihe verschiedener Modelle von Augenerkrankungen verhindern.
nAMD
Die nAMD zeichnet sich durch eine pathologische choroidale Neovaskularisation (CNV) aus. Das Austreten von Blut und Flüssigkeit aus der CNV kann zu einem Netzhautödem und/oder sub-/intraretinalen Blutungen und damit zum Verlust der Sehschärfe führen.
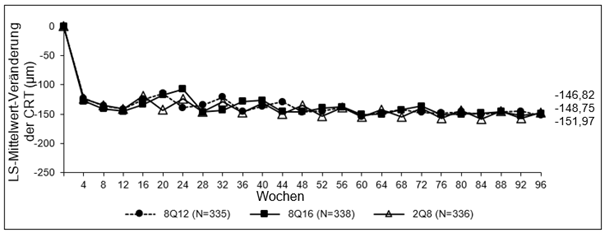
Die pharmakodynamischen Wirkungen von Aflibercept 114,3 mg/ml, angewendet alle 12 (8Q12) und alle 16 (8Q16) Wochen, werden im Vergleich zu Aflibercept 40 mg/ml, angewendet alle 8 Wochen (2Q8), für das Anwendungsgebiet nAMD beschrieben. Diese Wirkungen werden als Veränderung der CNV-Größe in Woche 12 im Vergleich zum Ausgangswert, als Veränderung der gesamten Läsionsfläche in Woche 48, 60 und 96 im Vergleich zum Ausgangswert und als Veränderung der zentralen Netzhautdicke (CRT) im Vergleich zum Ausgangswert dargestellt.
In der gepoolten Gruppe der mit 8Q12 oder 8Q16 behandelten Patienten betrug die Reduktion der CNV-Größe (LS-Mittelwert, basierend auf einem gemischten Modell für wiederholte Messungen [MMRM; mixed model repeated measures]) in Woche 12 ‑1,63 mm2 im Vergleich zu ‑1,17 mm2 bei den mit 2Q8 behandelten Patienten.
Pharmakodynamische Wirkungen wurden im Allgemeinen bis Woche 156 aufrechterhalten.
Tabelle 2: Pharmakodynamische Parameter (vollständiges Analyseset) in der PULSAR-Studie
Wirksamkeitsendpunkte |
Woche |
Eylea 8Q12 |
Eylea 8Q16 |
Eylea 2Q8 |
Veränderung der gesamten Läsionsfläche im Vergleich zum Ausgangswert [mm2] | ||||
LS-Mittelwert A |
12 |
-0,55 |
-0,30 |
|
Arithmetisches Mittel (SD), beobachtet |
48 |
‑0,4 (2,9) |
‑0,2 (3,1) |
0,1 (3,6) |
LS-Mittelwert (SE) A |
‑0,46 (0,19) |
‑0,35 (0,20) |
0,09 (0,22) |
|
Differenz der LS-Mittelwerte |
‑0,55 |
‑0,44 |
||
Arithmetisches Mittel (SD), beobachtet |
60 |
‑0,5 (2,8) |
‑0,4 (3,2) |
‑0,3 (3,2) |
LS-Mittelwert (SE) A |
‑0,48 (0,20) |
‑0,54 (0,21) |
‑0,24 (0,20) |
|
Differenz der LS-Mittelwerte |
‑0,24 |
‑0,29 |
||
Arithmetisches Mittel (SD), beobachtet |
96 |
‑0,3 (3,3) |
‑0,3 (3,2) |
‑0,2 (3,4) |
LS-Mittelwert (SE) A |
-0,43 (0,20) |
-0,42 (0,20) |
‑0,18 (0,20) |
|
Differenz der LS-Mittelwerte |
-0,25 |
-0,24 |
||
A LS-Mittelwert, KI und p-Wert basierend auf einem MMRM mit der Messung des Ausgangswerts als Kovariate, der Behandlungsgruppe als Faktor, der Visite und den bei der Randomisierung verwendeten Stratifizierungsvariablen (geographische Region, Kategorie BCVA-Ausgangswert) als feste Faktoren sowie Termen für die Interaktion zwischen der Messung des Ausgangswerts und der Visite und für die Interaktion zwischen Behandlung und Visite.
B Die absolute Differenz ist die Eylea 8Q12‑ bzw. 8Q16‑Gruppe minus die 2Q8‑Gruppe.
KI: Konfidenzintervall
LS: Kleinste Quadrate
SD: Standardabweichung
SE: Standardfehler
Abbildung 1: LS-Mittelwert-Veränderung der zentralen Netzhautdicke (CRT) vom Ausgangswert bis Woche 96 (vollständiges Analyseset) in der PULSAR-Studie
DMÖ
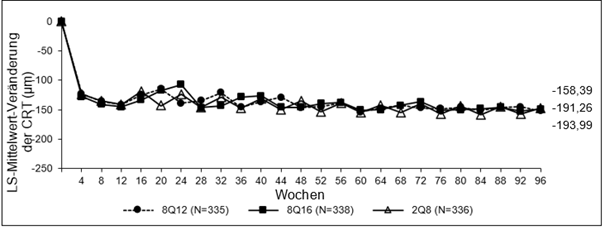
Das diabetische Makulaödem zeichnet sich durch eine erhöhte Gefäßpermeabilität und Schädigung der Netzhautkapillaren aus, was zu einem Verlust der Sehschärfe führen kann.
Die pharmakodynamischen Wirkungen von Aflibercept 114,3 mg/ml, angewendet alle 12 (8Q12) und alle 16 (8Q16) Wochen, werden im Vergleich zu Aflibercept 40 mg/ml, angewendet alle 8 Wochen (2Q8), für das Anwendungsgebiet DMÖ beschrieben. Diese Wirkungen werden als Veränderung der Leckage-Fläche in Woche 48, 60 und 96 im Vergleich zum Ausgangswert dargestellt.
Pharmakodynamische Wirkungen wurden im Allgemeinen bis Woche 156 aufrechterhalten.
Tabelle 3: Pharmakodynamische Parameter (vollständiges Analyseset) in der PHOTON-Studie
Wirksamkeitsendpunkte |
Woche |
Eylea 8Q12 |
Eylea 8Q16 |
Eylea 2Q8 |
Veränderung der Leckage-FlächeA im Vergleich zum Ausgangswert [mm2] | ||||
Arithmetisches Mittel (SD), beobachtet |
48 |
‑13,9 (13,91) |
‑9,4 (11,50) |
‑9,2 (12,11) |
60 |
‑13,9 (13,54) |
‑12,0 (13,26) |
‑14,4 (12,89) |
|
96 |
-12,8 (10,98) |
-9,4 (10,61) |
-11,9 (11,26) |
|
A basierend auf Fluoreszenzangiographie-Messungen
SD: Standardabweichung
Abbildung 2: LS-Mittelwert-Veränderung der zentralen Netzhautdicke (CRT) vom Ausgangswert bis Woche 96 (vollständiges Analyseset) in der PHOTON-Studie
Immunogenität
Nach einer bis zu 96-wöchigen Behandlung mit Eylea 114,3 mg/ml wurden bei 2,5 % bis 4,4 % der Patienten, die wegen DMÖ und nAMD behandelt wurden, therapiebedingte Antikörper gegen Eylea 114,3 mg/ml nachgewiesen. Es wurden keine Hinweise auf einen Einfluss von Anti-Drug-Antikörpern auf die Pharmakokinetik, Wirksamkeit oder Sicherheit beobachtet.
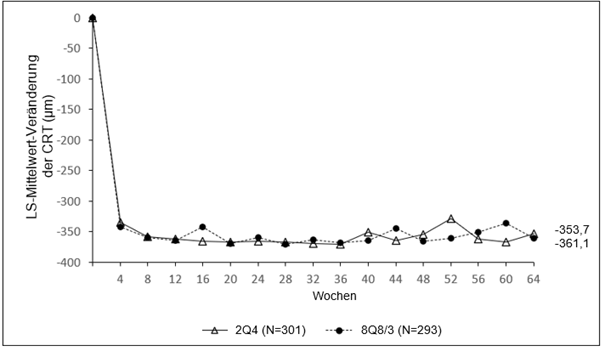
RVV
Beim RVV tritt eine Ischämie der Netzhaut auf, welche zur Freisetzung von VEGF führt. Dies wiederum bedingt eine Destabilisierung der tight junctions und fördert die Proliferation von Endothelzellen. Eine Hochregulierung von VEGF ist mit dem Zusammenbruch der Blut-Netzhaut-Schranke assoziiert und diese erhöhte vaskuläre Permeabilität führt zu einem Netzhautödem, einer Stimulation des endothelialen Zellwachstums und einer Neovaskularisation.
Tabelle 4: Pharmakodynamische Parameter (vollständiges Analyseset) in der QUASAR-Studie
Wirksamkeitsendpunkte |
Woche |
Eylea 8Q8/3 |
Eylea 2Q4 |
Veränderung der CRT im Vergleich zum Ausgangswert [µm] | |||
Arithmetisches Mittel (SD), beobachtet |
36 |
‑365,9 (239,9) |
‑397,3 (257,7) |
LS-Mittelwert (SE) A |
-370,9 (3,1) |
‑370,8 (3,9) |
|
Differenz der LS-Mittelwerte |
-0,1 (‑10,0; 9,8) |
||
Arithmetisches Mittel (SD), beobachtet |
64 |
‑355,5 (239,5) |
‑373,0 (252,1) |
LS-Mittelwert (SE) A |
-361,1 (4,3) |
‑353,7 (5,2) |
|
Differenz der LS-Mittelwerte |
-7,4 (‑20,7; 5,9) |
||
A LS-Mittelwert, KI und p-Wert basierend auf einem MMRM mit der Messung des CRT-Ausgangswerts als Kovariate, der Behandlungsgruppe als Faktor, der Visite und den bei der Randomisierung verwendeten Stratifizierungsvariablen (geographische Region, Kategorie BCVA-Ausgangswert und RVV‑Typ) als feste Faktoren sowie Termen für die Interaktion zwischen dem CRT‑Ausgangswert und der Visite und für die Interaktion zwischen Behandlung und Visite.
B Die absolute Differenz ist die Eylea 8Q8/3‑Gruppe minus die 2Q4‑Gruppe.
CRT: Zentrale Netzhautdicke
KI: Konfidenzintervall
LS: Kleinste Quadrate
SD: Standardabweichung
SE: Standardfehler
Abbildung 3: LS-Mittelwert-Veränderung der zentralen Netzhautdicke (CRT) vom Ausgangswert bis Woche 64 (vollständiges Analyseset) in der QUASAR-Studie
Klinische Wirksamkeit und Sicherheit
nAMD
Studienziele
Die Sicherheit und Wirksamkeit von Eylea 114,3 mg/ml wurden in einer randomisierten, multizentrischen, doppelmaskierten, aktiv kontrollierten Studie (PULSAR) bei Patienten mit therapienaiver nAMD untersucht.
Das primäre Ziel war die Untersuchung der Nicht-Unterlegenheit der Veränderung der bestkorrigierten Sehschärfe (BCVA) bei Patienten mit nAMD, die mit Eylea 114,3 mg/ml in Intervallen von 12 (8Q12) oder 16 (8Q16) Wochen im Vergleich zu Eylea 40 mg/ml alle 8 Wochen behandelt wurden.
Sekundäre Ziele waren die Untersuchung der Wirkung von Eylea 114,3 mg/ml auf morphologische und andere funktionelle Parameter im Vergleich zu Eylea 40 mg/ml sowie die Bewertung der Sicherheit, Immunogenität und Pharmakokinetik von Aflibercept.
Der primäre Wirksamkeitsendpunkt war die Veränderung der BCVA in Woche 48 im Vergleich zum Ausgangswert, gemessen anhand des Early Treatment Diabetic Retinopathy Study (ETDRS) Buchstaben-Scores.
Die wichtigsten sekundären Endpunkte waren die Veränderung der BCVA in Woche 60 im Vergleich zum Ausgangswert und der Anteil der Patienten ohne intraretinale Flüssigkeit (IRF) und ohne subretinale Flüssigkeit (SRF) im zentralen Teilfeld in Woche 16.
Weitere sekundäre Endpunkte waren unter anderen der Anteil der Patienten, die in Woche 48 mindestens 15 Buchstaben der BCVA im Vergleich zum Ausgangswert gewonnen haben, der Anteil der Patienten, die in Woche 48 einen ETDRS-Buchstaben-Score von mindestens 69 (ungefähres Snellen Äquivalent 20/40) erreichten und die Veränderung des Gesamtscores des National Eye Institute Visual Functioning Questionnaire‑25 (NEI-VFQ‑25) in Woche 48 im Vergleich zum Ausgangswert.
In der PULSAR-Studie wurden insgesamt 1 009 Patienten behandelt. Die Patienten wurden im Verhältnis 1:1:1 einer von 3 parallelen Behandlungsgruppen zugeordnet:
Anwendung von Eylea 114,3 mg/ml alle 12 Wochen (8Q12)
Anwendung von Eylea 114,3 mg/ml alle 16 Wochen (8Q16)
Anwendung von Eylea 40 mg/ml alle 8 Wochen (2Q8)
Alle Patienten erhielten 3 initiale Injektionen der ihnen zugewiesenen Dosis in 4-wöchigen Intervallen. Das Intervall der 8Q12- und der 8Q16-Gruppen war gemäß Studienprotokoll zu verkürzen, wenn beide der folgenden Kriterien erfüllt waren:
Verlust von > 5 Buchstaben der BCVA ab Woche 12 und
Zunahme der CRT um > 25 µm ab Woche 12 oder neue foveale Blutung oder neue foveale Neovaskularisation.
Unabhängig davon, ob die Intervalle der Patienten in Jahr 1 beibehalten oder verkürzt wurden, waren gemäß Studienprotokoll alle Patienten der 8Q12- und der 8Q16-Gruppen für eine Intervallverlängerung (in 4-wöchigen Schritten) ab Woche 52 geeignet, wenn die folgenden Kriterien erfüllt waren:
Verlust von < 5 Buchstaben der BCVA ab Woche 12 und
keine Flüssigkeit im zentralen Teilfeld in der optischen Kohärenztomographie (OCT) und
keine neu aufgetretene foveale Blutung oder foveale Neovaskularisation.
Bei Patienten, welche die Kriterien für eine Intervallverkürzung oder -verlängerung nicht erfüllten, wurde das Dosierungsintervall beibehalten. Der Mindestabstand zwischen den Injektionen betrug in allen Gruppen 8 Wochen.
Patienten mit bilateraler Erkrankung konnten für das andere Auge eine Behandlung mit Eylea 40 mg/ml oder einem anderen Anti-VEGF-Arzneimittel erhalten.
Patientenmerkmale bei Studienbeginn
Die Patienten waren 50 bis 96 Jahre alt, wobei der Mittelwert 74,5 Jahre betrug.
Ungefähr 92 % (309/335) und 87 % (295/338) der Patienten, die in die 8Q12- bzw. 8Q16-Gruppen randomisiert wurden, waren 65 Jahre oder älter, und ungefähr 51 % (172/335) und 51 % (171/338) waren 75 Jahre oder älter.
Ergebnisse
Patienten in den 8Q12-, 8Q16- und 2Q8-Gruppen, die Woche 48 abschlossen, erhielten im Median (Mittelwert) 6,0 (6,1), 5,0 (5,2) bzw. 7,0 (6,9) Injektionen.
In Woche 48 behielten 79,4 % der Patienten in der 8Q12-Gruppe die Q12-Intervalle bei, während 76,6 % der Patienten in der 8Q16-Gruppe die Q16-Intervalle beibehielten.
Patienten in den 8Q12-, 8Q16- und 2Q8-Gruppen, die Woche 60 abschlossen, erhielten im Median (Mittelwert) 7,0 (7,1), 6,0 (6,2) bzw. 9,0 (8,8) Injektionen.
In Woche 60 wurde das Behandlungsintervall bei 43,1 % der Patienten in der 8Q12-Gruppe auf 16 Wochen verlängert, und bei 38,5 % der Patienten in der 8Q16-Gruppe wurde das Behandlungsintervall auf 20 Wochen verlängert.
Patienten in den 8Q12-, 8Q16- und 2Q8-Gruppen, die Woche 96 abschlossen, erhielten im Median (Mittelwert) 9,0 (9,7), 8,0 (8,2) bzw. 13,0 (12,8) Injektionen.
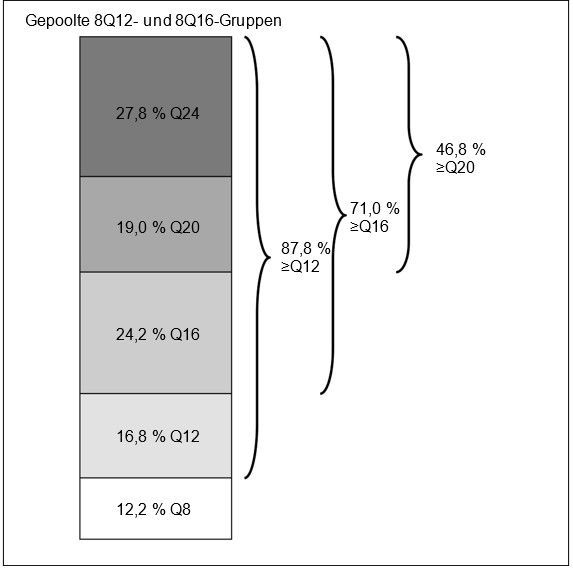
In Woche 96 hatten in den gepoolten 8Q12- und 8Q16-Gruppen 71,0 % der Patienten ein Behandlungsintervall von ≥16 Wochen, 46,8 % der Patienten ein Behandlungsintervall von ≥20 Wochen und 27,8 % der Patienten ein Behandlungsintervall von 24 Wochen erreicht, wobei der funktionelle und morphologische Befund erhalten blieben.
Die Behandlung mit 8Q12 und 8Q16 war der Behandlung mit 2Q8 in Bezug auf den primären Wirksamkeitsendpunkt „mittlere Veränderung der BCVA in Woche 48“ und den wichtigen sekundären Wirksamkeitsendpunkt „mittlere Veränderung der BCVA in Woche 60“ nicht unterlegen und klinisch gleichwertig. Der Behandlungseffekt von Eylea 114,3 mg/ml auf die mittlere Veränderung der BCVA blieb bis Woche 96 erhalten.
Darüber hinaus war die Behandlung mit Eylea (gepoolte 8Q12- und 8Q16-Gruppen) der Behandlung mit 2Q8 in Bezug auf den wichtigen sekundären Wirksamkeitsendpunkt „Anteil der Patienten ohne intraretinale Flüssigkeit (IRF) und ohne subretinale Flüssigkeit (SRF) im zentralen Teilfeld in Woche 16“ überlegen (siehe Tabelle 5).
Tabelle 5: Wirksamkeitsendpunkte der PULSAR-Studie
Wirksamkeitsendpunkte |
Woche |
Eylea 8Q12 |
Eylea 8Q16 |
Eylea 2Q8 |
Veränderung der BCVA, ermittelt durch ETDRS-Buchstaben-Score im Vergleich zum Ausgangswert D | ||||
Arithmetisches Mittel (SD), beobachtet |
48 |
6,7 (12,6) |
6,2 (11,7) |
7,6 (12,2) |
LS-Mittelwert (SE) A |
6,06 (0,77) |
5,89 (0,72) |
7,03 (0,74) |
|
Differenz der LS-Mittelwerte |
‑0,97 |
‑1,14 |
||
p‑Wert (einseitiger Nicht-Unterlegenheitstest mit einer Grenze von 4 Buchstaben) A,B |
0,0009 |
0,0011 |
||
Arithmetisches Mittel (SD), beobachtet |
60 |
6,6 (13,6) |
6,6 (11,7) |
7,8 (12,6) |
LS-Mittelwert (SE) A |
6,37 (0,74) |
6,31 (0,66) |
7,23 (0,68) |
|
Differenz der LS-Mittelwerte |
‑0,86 |
‑0,92 |
||
p‑Wert (einseitiger Nicht-Unterlegenheitstest mit einer Grenze von 4 Buchstaben) A,B |
0,0002 |
< 0,0001 |
||
Arithmetisches Mittel (SD), beobachtet |
96 |
5,9 (14,2) |
5,6 (13,7) |
7,4 (13,8) |
LS-Mittelwert (SE) A |
5,59 (0,77) |
5,52 (0,75) |
6,60 (0,73) |
|
Differenz der LS-Mittelwerte |
-1,01 |
-1,08 |
||
Patienten ohne IRF und ohne SRF im zentralen Teilfeld D | ||||
Anteil (LOCF) |
16 |
63,3 % |
51,6 % |
|
Adjustierte Differenz des Anteils |
11,7 % (5,3 %; 18,2 %) |
|||
p‑Wert (einseitiger Überlegenheitstest) B, C |
0,0002 |
|||
Anteil (LOCF) |
48 |
71,1 % |
66,8 % |
59,4 % |
Adjustierte Differenz des Anteils |
11,7 % |
7,5 % |
||
Anteil (LOCF) |
60 |
74,6 % |
72,2 % |
74,6 % |
Adjustierte Differenz des Anteils |
0,0 % |
‑2,2 % |
||
Anteil (LOCF) |
96 |
69,6 % |
63,6 % |
66,5 % |
Adjustierte Differenz des Anteils |
3,0 % |
‑3,0 % |
||
Patienten, die einen ETDRS-Buchstaben-Score von mindestens 69 (ungefähres Snellen Äquivalent 20/40) erreichten D | ||||
Anteil (LOCF) |
48 |
56,9 % |
54,3 % |
57,9 % |
Adjustierte Differenz des Anteils |
‑0,2 % |
‑2,2 % |
||
Anteil (LOCF) |
60 |
56,3 % |
54,6 % |
58,2 % |
Adjustierte Differenz des Anteils |
‑1,1 % |
‑2,3 % |
||
Anteil (LOCF) |
96 |
53,3 % |
53,1 % |
56,7 % |
Adjustierte Differenz des Anteils |
‑2,7 % |
‑2,4 % |
||
Patienten mit einem Gewinn von mindestens 15 Buchstaben der BCVA im Vergleich zum Ausgangswert D | ||||
Anteil (LOCF) |
48 |
20,7 % |
21,7 % |
22,1 % |
Adjustierte Differenz des Anteils |
‑1,7 % |
‑0,9 % |
||
Anteil (LOCF) |
60 |
23,7 % |
23,1 % |
23,3 % |
Adjustierte Differenz des Anteils |
0,1 % |
‑0,7 % |
||
Anteil (LOCF) |
96 |
22,2 % |
22,8 % |
24,2 % |
Adjustierte Differenz des Anteils |
‑2,4 % |
‑2,0 % |
||
Letzte vorgesehene Behandlungsintervalle | ||||
Patienten mit ≥Q12 Behandlungsintervall E | ||||
Anteil (gepoolte 8Q12- und 8Q16-Gruppen) |
96 |
87,8 % |
n/a |
|
Anteil |
86,6 % |
89,0 % |
n/a |
|
Patienten mit ≥Q16 Behandlungsintervall E | ||||
Anteil (gepoolte 8Q12- und 8Q16-Gruppen) |
96 |
71,0 % |
n/a |
|
Anteil |
63,6 % |
78,4 % |
n/a |
|
Patienten mit ≥Q20 Behandlungsintervall E | ||||
Anteil (gepoolte 8Q12- und 8Q16-Gruppen) |
96 |
46,8 % |
n/a |
|
Anteil |
40,5 % |
53,1 % |
n/a |
|
Patienten mit Q24 Behandlungsintervall E | ||||
Anteil (gepoolte 8Q12- und 8Q16-Gruppen) |
96 |
27,8 % |
n/a |
|
Anteil |
24,7 % |
30,8 % |
n/a |
|
A LS-Mittelwert, KI und p-Wert basierend auf einem MMRM mit der Messung des Ausgangswerts der bestkorrigierten Sehschärfe (BCVA) als Kovariate, der Behandlungsgruppe als Faktor, der Visite und den bei der Randomisierung verwendeten Stratifizierungsvariablen (geographische Region, Kategorie BCVA-Ausgangswert) als feste Faktoren sowie Termen für die Interaktion zwischen BCVA-Ausgangswert und Visite und für die Interaktion zwischen Behandlung und Visite.
B Die absolute Differenz ist die Eylea 8Q12‑ bzw. 8Q16‑Gruppe minus die 2Q8‑Gruppe.
C Mantel‑Haenszel gewichteter Behandlungsunterschied mit Stratifizierungsvariablen (geographische Region, Kategorie BCVA-Ausgangswert), die für die Randomisierung verwendet wurden; Berechnung des KI approximativ mit der Normalverteilung.
D Vollständiges Analyseset
E Sicherheitsanalyseset; Patienten, die zum jeweiligen Zeitpunkt als abgeschlossen betrachtet werden
KI: Konfidenzintervall
LOCF: Last Observation Carried Forward
LS: Kleinste Quadrate
SD: Standardabweichung
SE: Standardfehler
Behandlungsintervalle wurden in einer vorab festgelegten explorativen Weise analysiert.
Abbildung 4: LS-Mittelwert-Veränderung der BCVA ermittelt durch ETDRS-Buchstaben-Score vom Ausgangswert bis Woche 96 (vollständiges Analyseset) in der PULSAR-Studie
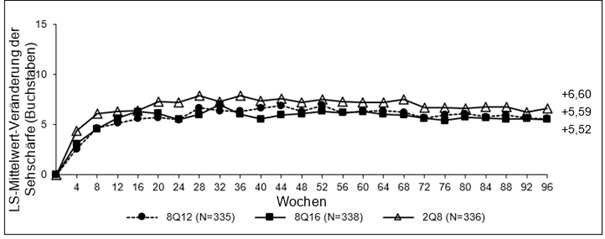
Abbildung 5: Letztes vorgesehenes Behandlungsintervall in Woche 96
Aflibercept zeigte in allen Dosierungen (8Q12, 8Q16, 2Q8) einen relevanten Anstieg im Vergleich zum Ausgangswert hinsichtlich des vorspezifizierten sekundären Wirksamkeitsendpunkts National Eye Institute Visual Function Questionnaire (NEI VFQ-25).
Es wurden keine klinisch relevanten Unterschiede zwischen den 8Q12-, 8Q16- und 2Q8-Gruppen bei den Veränderungen im Gesamtscore des NEI VFQ‑25 in Woche 48 und Woche 96 im Vergleich zum Ausgangswert festgestellt.
Die Ergebnisse zur Wirksamkeit in den auswertbaren Subgruppen für Alter, Geschlecht, geographische Region, ethnische Herkunft, BCVA-Ausgangswert und Läsionstyp entsprachen den Ergebnissen in der Gesamtpopulation.
Die Wirksamkeit wurde im Allgemeinen bis Woche 96 aufrechterhalten.
Ergebnisse - PULSAR-Extensionsphase
Am Ende der Hauptphase der Studie in Woche 96 konnten die Patienten in die 60-wöchige, offene Extensionsphase aufgenommen werden. 417 Patienten, die ursprünglich 8Q12 und 8Q16 zugeteilt wurden, erhielten weiterhin Eylea 114,3 mg/ml unter Beibehaltung ihrer letzten Intervalle. 208 Patienten, die zu Beginn der Studie ursprünglich 2Q8 zugeteilt wurden, wurden auf Eylea 114,3 mg/ml umgestellt und begannen mit 12-wöchigen Intervallen. Die Behandlungsintervalle konnten basierend auf dem funktionellen und/oder dem morphologischen Befund des Arztes weiter angepasst werden.
Bei den Patienten, die ursprünglich 8Q12 und 8Q16 zugeteilt wurden, wurde der Behandlungseffekt von Eylea 114,3 mg/ml im Allgemeinen über drei Jahre (Woche 156) aufrechterhalten. Die mittlere LS-Änderung gegenüber dem Ausgangswert in den gepoolten 8Q12- und 8Q16-Gruppen betrug +3,41 Buchstaben bei der BCVA und -148,05 Mikrometer bei der CRT in Woche 156.
Bei den Patienten, die ursprünglich 2Q8 zugeteilt wurden, war der Behandlungseffekt mit Eylea 114,3 mg/ml ähnlich. Die mittlere LS-Änderung der BCVA gegenüber dem Ausgangswert betrug +4,58 Buchstaben und -145,21 Mikrometer bei der CRT in Woche 156.
Patienten in den 8Q12- und 8Q16-Gruppen, die Woche 156 abschlossen, erhielten im Median (Mittelwert) 13,0 (13,5) bzw. 11,0 (12,2) Injektionen.
Patienten, die auf Eylea 114,3 mg/ml umgestellt wurden und Woche 156 abschlossen, erhielten im Median (Mittelwert) insgesamt 18,0 (17,7) Injektionen, von denen 5,0 (4,9) Injektionen nach der Umstellung auf Eylea 114,3 mg/ml innerhalb der 60 Wochen der Extensionsphase der Studie angewendet wurden.
Das Sicherheitsprofil in der Extensionsphase war insgesamt dem in der Hauptphase ähnlich.
Tabelle 6: Wirksamkeitsendpunkte der PULSAR-Extensionsphase in Woche 156
Wirksamkeitsendpunkte |
8Q12 fortgeführt mit Eylea 114,3 mg/ml (N = 185) |
8Q16 fortgeführt mit Eylea 114,3 mg/ml (N = 190) |
2Q8 umgestellt auf Eylea 114,3 mg/ml |
Veränderung der BCVA im Vergleich zum Ausgangswert (LS-Mittelwert) |
+3,57 Buchstaben |
+3,23 Buchstaben |
+4,58 Buchstaben |
Veränderung der CRT im Vergleich zum Ausgangswert (LS-Mittelwert) |
‑148,42 Mikrometer |
‑147,54 Mikrometer |
‑145,21 Mikrometer |
Letztes vorgesehenes Behandlungsintervall A | |||
≥12 Wochen |
76,2 % |
78,4 % |
78,5 % |
≥16 Wochen |
53,5 % |
62,1 % |
42,5 % |
≥20 Wochen |
37,8 % |
42,6 % |
16,1 % |
24 Wochen |
23,8 % |
24,2 % |
NA B |
A basierend auf Patienten, die Woche 156 abschlossen
B NA für Patienten, die ursprünglich für 2Q8 randomisiert wurden, aufgrund des Studiendesigns/der Studiendauer
DMÖ
Studienziele
Die Sicherheit und Wirksamkeit von Eylea 114,3 mg/ml wurden in einer randomisierten, multizentrischen, doppelmaskierten, aktiv kontrollierten Studie (PHOTON) bei Patienten mit DMÖ untersucht.
Das primäre Ziel war die Untersuchung der Nicht-Unterlegenheit der Veränderung der BCVA bei Patienten, die mit Eylea 114,3 mg/ml in Intervallen von 12 (8Q12) oder 16 (8Q16) Wochen im Vergleich zu Eylea 40 mg/ml alle 8 Wochen behandelt wurden.
Sekundäre Ziele waren die Untersuchung der Wirkung von Eylea 114,3 mg/ml auf morphologische und andere funktionelle Parameter im Vergleich zu Eylea 40 mg/ml sowie die Bewertung der Sicherheit, Immunogenität und Pharmakokinetik von Aflibercept.
Der primäre Wirksamkeitsendpunkt war die Veränderung der BCVA in Woche 48 im Vergleich zum Ausgangswert, gemessen anhand des Early Treatment Diabetic Retinopathy Study (ETDRS) Buchstaben-Scores.
Ein wichtiger sekundärer Endpunkt war die Veränderung der BCVA in Woche 60 im Vergleich zum Ausgangswert.
Weitere sekundäre Endpunkte waren unter anderen der Anteil der Patienten, die in Woche 48 mindestens 15 Buchstaben der BCVA im Vergleich zum Ausgangswert gewonnen haben, der Anteil der Patienten, die in Woche 48 einen ETDRS-Buchstaben-Score von mindestens 69 (ungefähres Snellen Äquivalent 20/40) erreichten und die Veränderung des Gesamtscores des National Eye Institute Visual Functioning Questionnaire‑25 (NEI-VFQ‑25) in Woche 48 im Vergleich zum Ausgangswert.
In der PHOTON-Studie wurden insgesamt 658 Patienten behandelt. Die Patienten wurden im Verhältnis 2:1:1 einer von 3 parallelen Behandlungsgruppen zugeordnet:
Anwendung von Eylea 114,3 mg/ml alle 12 Wochen (8Q12)
Anwendung von Eylea 114,3 mg/ml alle 16 Wochen (8Q16)
Anwendung von Eylea 40 mg/ml alle 8 Wochen (2Q8)
Patienten, die von anderen Anti-VEGF-Arzneimitteln auf Eylea 114,3 mg/ml umgestellt wurden, erhielten die letzte Injektion der vorherigen Behandlung mindestens 12 Wochen vor Beginn der Behandlung mit Eylea 114.3 mg/ml.
Alle Patienten in den 8Q12- und 8Q16-Gruppen erhielten 3 initiale Injektionen und alle Patienten in der 2Q8-Gruppe erhielten 5 initiale Injektionen in 4-wöchigen Intervallen.
Das Intervall der 8Q12- und der 8Q16-Gruppen war gemäß Studienprotokoll zu verkürzen, wenn beide der folgenden Kriterien erfüllt waren:
Verlust von > 10 Buchstaben der BCVA ab Woche 12 in Verbindung mit anhaltendem oder sich verschlechterndem DMÖ und
Zunahme der CRT um > 50 µm ab Woche 12.
Unabhängig davon, ob die Intervalle der Patienten in Jahr 1 beibehalten oder verkürzt wurden, waren gemäß Studienprotokoll alle Patienten der 8Q12- und der 8Q16-Gruppen für eine Intervallverlängerung (in 4-wöchigen Schritten) ab Woche 52 geeignet, wenn die folgenden Kriterien erfüllt waren:
Verlust von < 5 Buchstaben der BCVA ab Woche 12 und
CRT < 300 µm mit SD-OCT gemessen (oder < 320 µm bei Messung einschließlich RPE).
Bei Patienten, welche die Kriterien für eine Intervallverkürzung oder -verlängerung nicht erfüllten, wurde das Dosierungsintervall beibehalten. Der Mindestabstand zwischen den Injektionen betrug in allen Gruppen 8 Wochen.
Patienten mit bilateraler Erkrankung konnten für das andere Auge eine Behandlung mit Eylea 40 mg/ml erhalten.
Patientenmerkmale bei Studienbeginn
Die Patienten waren 24 bis 90 Jahre alt, wobei der Mittelwert 62,3 Jahre betrug.
Ungefähr 44 % (143/328) und 44 % (71/163) der Patienten, die in die 8Q12- bzw. 8Q16-Gruppen randomisiert wurden, waren 65 Jahre oder älter, und ungefähr 11 % (36/328) und 14 % (14/163) waren 75 Jahre oder älter.
Der Anteil der Patienten mit vorbehandeltem DMÖ war zwischen den Behandlungsgruppen ausgeglichen (43,6 % in der 8Q12‑, 43,6 % in der 8Q16‑ und 44,3 % in der 2Q8‑Gruppe).
Ergebnisse
Patienten in den 8Q12-, 8Q16- und 2Q8-Gruppen, die Woche 48 abschlossen, erhielten im Median (Mittelwert) 6,0 (6,0), 5,0 (5,0) bzw. 8,0 (7,9) Injektionen.
In Woche 48 behielten 91,0 % der Patienten in der 8Q12-Gruppe die Q12-Intervalle bei, während 89,1 % der Patienten in der 8Q16-Gruppe die Q16-Intervalle beibehielten.
Patienten in den 8Q12-, 8Q16- und 2Q8-Gruppen, die Woche 60 abschlossen, erhielten im Median (Mittelwert) 7,0 (7,0), 6,0 (6,0) bzw. 10,0 (9,8) Injektionen.
In Woche 60 wurde das Behandlungsintervall bei 42,6 % der Patienten in der 8Q12-Gruppe auf 16 Wochen verlängert, und bei 34,2 % der Patienten in der 8Q16-Gruppe wurde das Behandlungsintervall auf 20 Wochen verlängert.
Patienten in den 8Q12-, 8Q16- und 2Q8-Gruppen, die Woche 96 abschlossen, erhielten im Median (Mittelwert) 9,0 (9,5), 8,0 (7,8) bzw. 14,0 (13,8) Injektionen.
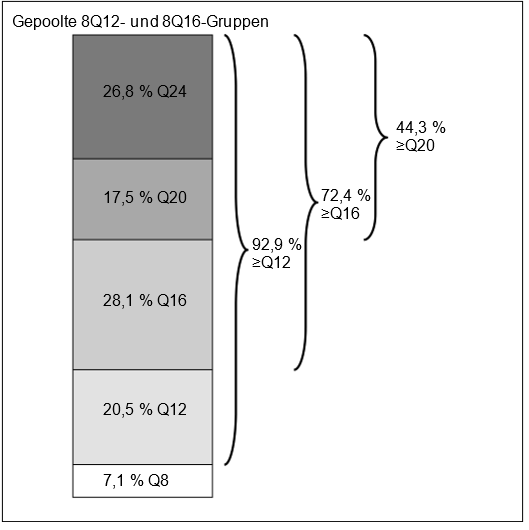
In Woche 96 hatten in den gepoolten 8Q12- und 8Q16-Gruppen 72,4 % der Patienten ein Behandlungsintervall von ≥16 Wochen, 44,3 % der Patienten ein Behandlungsintervall von ≥20 Wochen und 26,8 % der Patienten ein Behandlungsintervall von 24 Wochen erreicht, wobei der funktionelle und morphologische Befund erhalten blieben.
Die Behandlung mit Eylea (sowohl in der 8Q12- als auch der 8Q16-Gruppe) war der Behandlung mit 2Q8 in Bezug auf den primären Wirksamkeitsendpunkt „mittlere Veränderung der BCVA in Woche 48“ und den wichtigen sekundären Wirksamkeitsendpunkt „mittlere Veränderung der BCVA in Woche 60“ nicht unterlegen und klinisch gleichwertig. Der Behandlungseffekt von Eylea 114,3 mg/ml auf die mittlere Veränderung der BCVA blieb bis Woche 96 erhalten.
Tabelle 7: Wirksamkeitsendpunkte der PHOTON-Studie
Wirksamkeitsendpunkte |
Woche |
Eylea 8Q12 |
Eylea 8Q16 |
Eylea 2Q8 |
Veränderung der BCVA, ermittelt durch ETDRS-Buchstaben-Score im Vergleich zum Ausgangswert D | ||||
Arithmetisches Mittel (SD), beobachtet |
48 |
8,77 (8,95) |
7,86 (8,38) |
9,21 (8,99) |
LS-Mittelwert (SE) A |
8,10 (0,61) |
7,23 (0,71) |
8,67 (0,73) |
|
Differenz der LS-Mittelwerte |
‑0,57 |
‑1,44 |
||
p-Wert (einseitiger Nicht-Unterlegenheitstest mit einer Grenze von 4 Buchstaben) A,B |
< 0,0001 |
0,0031 |
||
Arithmetisches Mittel (SD), beobachtet |
60 |
9,05 (9,27) |
7,96 (9,14) |
9,62 (9,58) |
LS-Mittelwert (SE) A |
8,52 (0,63) |
7,64 (0,75) |
9,40 (0,77) |
|
Differenz der LS-Mittelwerte |
‑0,88 |
‑1,76 |
||
p-Wert (einseitiger Nicht-Unterlegenheitstest mit einer Grenze von 4 Buchstaben) A,B |
0,0003 |
0,0122 |
||
Arithmetisches Mittel (SD), beobachtet |
96 |
8,82 (9,93) |
7,50 (9,86) |
8,41 (11,10) |
LS-Mittelwert (SE) A |
8,15 (0,63) |
6,59 (0,77) |
7,70 (0,89) |
|
Differenz der LS-Mittelwerte |
0,45 |
‑1,11 |
||
Patienten, die einen ETDRS-Buchstaben-Score von mindestens 69 (ungefähres Snellen Äquivalent 20/40) erreichten D | ||||
Anteil (LOCF) |
48 |
65,3 % |
62,6 % |
63,0 % |
Adjustierte Differenz des Anteils |
2,45 % |
‑0,67 % |
||
Anteil (LOCF) |
60 |
64,7 % |
62,0 % |
60,6 % |
Adjustierte Differenz des Anteils |
4,34 % |
1,63 % |
||
Anteil (LOCF) |
96 |
66,9 % |
61,3 % |
63,0 % |
Adjustierte Differenz des Anteils |
4,01 % |
‑1,51 % |
||
Patienten mit einem Gewinn von mindestens 15 Buchstaben der BCVA im Vergleich zum Ausgangswert D | ||||
Anteil (LOCF) |
48 |
18,7 % |
16,6 % |
23,0 % |
Adjustierte Differenz des Anteils |
‑4,64 % |
‑7,14 % |
||
Anteil (LOCF) |
60 |
21,5 % |
16,0 % |
26,1 % |
Adjustierte Differenz des Anteils |
‑5,01 % |
‑10,78 % |
||
Anteil (LOCF) |
96 |
24,5 % |
19,6 % |
26,1 % |
Adjustierte Differenz des Anteils |
‑1,88 % |
‑7,07 % |
||
Letzte vorgesehene Behandlungsintervalle | ||||
Patienten mit ≥Q12 Behandlungsintervall E | ||||
Anteil (gepoolte 8Q12- und 8Q16-Gruppen) |
96 |
92,9 % |
n/a |
|
Anteil |
91,8 % |
95,0 % |
n/a |
|
Patienten mit ≥Q16 Behandlungsintervall E | ||||
Anteil (gepoolte 8Q12- und 8Q16-Gruppen) |
96 |
72,4 % |
n/a |
|
Anteil |
64,1 % |
87,8 % |
n/a |
|
Patienten mit ≥Q20 Behandlungsintervall E | ||||
Anteil (gepoolte 8Q12- und 8Q16-Gruppen) |
96 |
44,3 % |
n/a |
|
Anteil |
43,0 % |
46,8 % |
n/a |
|
Patienten mit Q24 Behandlungsintervall E | ||||
Anteil (gepoolte 8Q12- und 8Q16-Gruppen) |
96 |
26,8 % |
n/a |
|
Anteil |
23,8 % |
32,4 % |
n/a |
|
A LS-Mittelwert, KI und p-Wert basierend auf einem MMRM mit der Messung des Ausgangswerts der bestkorrigierten Sehschärfe (BCVA) als Kovariate, der Behandlungsgruppe als Faktor, der Visite und den bei der Randomisierung verwendeten Stratifizierungsvariablen (geographische Region, Kategorie BCVA-Ausgangswert) als feste Faktoren sowie Termen für die Interaktion zwischen BCVA-Ausgangswert und Visite und für die Interaktion zwischen Behandlung und Visite.
B Die absolute Differenz ist die Eylea 8Q12‑ bzw. 8Q16‑Gruppe minus die 2Q8‑Gruppe.
C Mantel‑Haenszel gewichteter Behandlungsunterschied mit Stratifizierungsvariablen (geographische Region, Kategorie BCVA-Ausgangswert), die bei der Randomisierung verwendet wurden; Berechnung des KI approximativ mit der Normalverteilung.
D Vollständiges Analyseset
E Sicherheitsanalyseset; Patienten, die zum jeweiligen Zeitpunkt als abgeschlossen betrachtet werden
KI: Konfidenzintervall
LOCF: Last Observation Carried Forward
LS: Kleinste Quadrate
SD: Standardabweichung
SE: Standardfehler
Behandlungsintervalle wurden in einer vorab festgelegten explorativen Weise analysiert.
Abbildung 6: LS-Mittelwert-Veränderung der BCVA ermittelt durch ETDRS-Buchstaben-Score vom Ausgangswert bis Woche 96 (vollständiges Analyseset) in der PHOTON-Studie
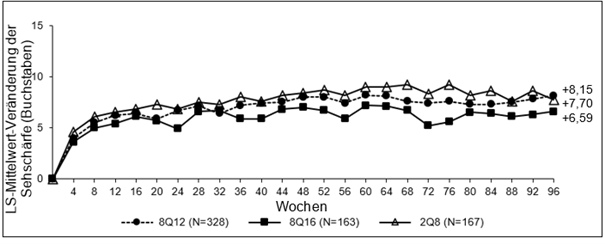
Abbildung 7: Letztes vorgesehenes Behandlungsintervall in Woche 96
Eylea zeigte in allen Dosierungen (8Q12, 8Q16, 2Q8) einen relevanten Anstieg im Vergleich zum Ausgangswert hinsichtlich des vorspezifizierten sekundären Wirksamkeitsendpunkts National Eye Institute Visual Function Questionnaire (NEI VFQ-25).
Es wurden keine klinisch relevanten Unterschiede zwischen den 8Q12-, 8Q16- und 2Q8-Gruppen bei den Veränderungen im Gesamtscore des NEI VFQ‑25 in Woche 48 und Woche 96 im Vergleich zum Ausgangswert festgestellt.
Die Ergebnisse zur Wirksamkeit in den auswertbaren Subgruppen für Alter, Geschlecht, geographische Region, ethnische Herkunft, BCVA-Ausgangswert, CRT-Ausgangswert und vorherige DMÖ-Behandlung entsprachen den Ergebnissen in der Gesamtpopulation.
Die Wirksamkeit wurde im Allgemeinen bis Woche 96 aufrechterhalten.
Die Behandlungseffekte in der Subgruppe vorbehandelter Patienten waren denen ähnlich, die bei therapienaiven Patienten beobachtet wurden.
Ergebnisse - PHOTON-Extensionsphase
Am Ende der Hauptphase der Studie in Woche 96 konnten die Patienten in die 60-wöchige, offene Extensionsphase aufgenommen werden. 195 Patienten, die ursprünglich 8Q12 und 8Q16 zugeteilt wurden, erhielten weiterhin Eylea 114,3 mg/ml unter Beibehaltung ihrer letzten Intervalle. 70 Patienten, die zu Beginn der Studie ursprünglich 2Q8 zugeteilt wurden, wurden auf Eylea 114,3 mg/ml umgestellt und begannen mit 12-wöchigen Intervallen. Die Behandlungsintervalle konnten basierend auf dem funktionellen und/oder dem morphologischen Befund des Arztes weiter angepasst werden.
Bei den Patienten, die ursprünglich 8Q12 und 8Q16 zugeteilt wurden, wurde der Behandlungseffekt von Eylea 114,3 mg/ml im Allgemeinen über drei Jahre (Woche 156) aufrechterhalten. Die mittlere LS-Änderung gegenüber dem Ausgangswert in den gepoolten 8Q12- und 8Q16-Gruppen betrug +7,2 Buchstaben bei der BCVA und -192,4 Mikrometer bei der CRT in Woche 156.
Bei den Patienten, die ursprünglich 2Q8 zugeteilt wurden, war der Behandlungseffekt mit Eylea 114,3 mg/ml ähnlich. Die mittlere LS-Änderung der BCVA gegenüber dem Ausgangswert betrug +6,5 Buchstaben und -197,4 Mikrometer bei der CRT in Woche 156.
Patienten in den 8Q12- und 8Q16-Gruppen, die Woche 156 abschlossen, erhielten im Median (Mittelwert) 13,0 (13,2) bzw. 11,0 (11,4) Injektionen.
Patienten, die auf Eylea 114,3 mg/ml umgestellt wurden und Woche 156 abschlossen, erhielten im Median (Mittelwert) insgesamt 19,0 (18,6) Injektionen, von denen 5,0 (4,8) Injektionen nach der Umstellung auf Eylea 114,3 mg/ml innerhalb der 60 Wochen der Extensionsphase der Studie angewendet wurden.
Das Sicherheitsprofil in der Extensionsphase war insgesamt dem in der Hauptphase ähnlich.
Tabelle 8: Wirksamkeitsendpunkte der PHOTON-Extensionsphase in Woche 156
Wirksamkeitsendpunkte |
8Q12 fortgeführt mit Eylea 114,3 mg/ml (N = 103) |
8Q16 fortgeführt mit Eylea 114,3 mg/ml (N = 49) |
2Q8 umgestellt auf Eylea 114,3 mg/ml |
Veränderung der BCVA im Vergleich zum Ausgangswert (LS-Mittelwert) |
+6,8 Buchstaben |
+8,1 Buchstaben |
+6,5 Buchstaben |
Veränderung der CRT im Vergleich zum Ausgangswert (LS-Mittelwert) |
‑190,3 Mikrometer |
‑198,1 Mikrometer |
‑197,4 Mikrometer |
Letztes vorgesehenes Behandlungsintervall A | |||
≥12 Wochen |
85,4 % |
91,8 % |
82,8 % |
≥16 Wochen |
62,1 % |
81,6 % |
50,0 % |
≥20 Wochen |
40,8 % |
63,3 % |
19,0 % |
24 Wochen |
20,4 % |
42,9 % |
NA B |
A basierend auf Patienten, die Woche 156 abschlossen
B NA für Patienten, die ursprünglich für 2Q8 randomisiert wurden, aufgrund des Studiendesigns/der Studiendauer
RVV
Studienziele
Die Sicherheit und Wirksamkeit von Eylea 114,3 mg/ml wurden in einer randomisierten, multizentrischen, doppelmaskierten, aktiv kontrollierten Studie (QUASAR) bei Patienten mit therapienaivem Makulaödem infolge eines RVV untersucht.
Das primäre Ziel war die Untersuchung der Nicht-Unterlegenheit der Veränderung der bestmöglich korrigierten Sehschärfe (BCVA) bei Patienten, die mit Eylea 114,3 mg/ml in Intervallen von 8 Wochen (8Q8) im Vergleich zu Eylea 40 mg/ml alle 4 Wochen (2Q4) behandelt wurden.
Sekundäre Ziele beinhalteten die Untersuchung, ob die Behandlung mit 8Q8 weniger Injektionen erfordert im Vergleich zu 2Q4, die Untersuchung der Wirkung von Eylea 114,3 mg/ml auf morphologische und andere funktionelle Parameter im Vergleich zu Eylea 40 mg/ml sowie die Bewertung der Sicherheit und Pharmakokinetik von Aflibercept.
Der primäre Wirksamkeitsendpunkt war die Veränderung der BCVA in Woche 36 im Vergleich zum Ausgangswert, gemessen anhand des Early Treatment Diabetic Retinopathy Study (ETDRS) Buchstaben-Scores.
Der wichtigste sekundäre Endpunkt war die Anzahl an aktiven Injektionen vom Ausgangswert bis Woche 64.
Weitere sekundäre Endpunkte waren unter anderen die Anzahl an aktiven Injektionen vom Ausgangswert bis Woche 36, der Anteil der Patienten, die in Woche 36 mindestens 15 Buchstaben der BCVA im Vergleich zum Ausgangswert gewonnen haben, der Anteil der Patienten, die in Woche 36 einen ETDRS-Buchstaben-Score von mindestens 69 (ungefähres Snellen Äquivalent 20/40) erreichten und die Veränderung des Gesamtscores des National Eye Institute Visual Functioning Questionnaire‑25 (NEI-VFQ‑25) in Woche 36 im Vergleich zum Ausgangswert.
In der QUASAR-Studie wurden insgesamt 892 Patienten behandelt. Die Patienten wurden im Verhältnis 1:1:1 einer von 3 parallelen Behandlungsgruppen zugeordnet:
Anwendung von Eylea 114,3 mg/ml alle 8 Wochen, nach 3 initialen Injektionen in 4‑wöchigen Intervallen (8Q8/3)
Anwendung von Eylea 114,3 mg/ml alle 8 Wochen, nach 5 initialen Injektionen in 4‑wöchigen Intervallen (8Q8/5)
Anwendung von Eylea 40 mg/ml alle 4 Wochen (2Q4)
Ab Woche 16 (8Q8/3), Woche 24 (8Q8/5) und Woche 40 (2Q4, falls zuvor auf Q8 verlängert) konnten Patienten ihr Dosierungsintervall um 4 Wochen verkürzen, wenn bei einem Injektionstermin beide der folgenden Kriterien erfüllt waren:
Verlust von > 5 Buchstaben der BCVA im Vergleich zum Referenztermin und
Zunahme der CRT um > 50 µm im Vergleich zum Referenztermin.
Eine Intervallverlängerung war ab Woche 32 (2Q4 und 8Q8/3) oder Woche 40 (8Q8/5) in 4‑wöchigen Schritten zulässig, wenn bei einem Injektionstermin beide der folgenden Kriterien erfüllt waren:
Verlust von < 5 Buchstaben der BCVA im Vergleich zum Referenztermin und
CRT < 320 µm mit SD-OCT gemessen (oder < 300 µm bei Messung ohne RPE).
Die Referenztermine fanden in Woche 12 für 8Q8/3 und in Woche 20 für 8Q8/5 und 2Q4 statt.
Bei Patienten, welche die Kriterien für eine Intervallverkürzung oder -verlängerung nicht erfüllten, wurde das Dosierungsintervall beibehalten. Der Mindestabstand zwischen den Injektionen betrug in allen Gruppen 4 Wochen.
Patienten mit bilateraler Erkrankung konnten für das andere Auge eine Behandlung mit Eylea 40 mg/ml oder einem anderen Anti-VEGF-Arzneimittel erhalten.
Patientenmerkmale bei Studienbeginn
Die Patienten waren 23 bis 95 Jahre alt, wobei der Mittelwert 65,9 Jahre betrug.
Etwa 57 % (168/293) und 57 % (170/298) der Patienten, die randomisiert den Gruppen 8Q8/3 und 8Q8/5 zugewiesen wurden, waren 65 Jahre oder älter, und etwa 26 % (76/293) und 25 % (74/298) waren 75 Jahre oder älter.
425 (48 %) der eingeschlossenen Patienten hatten einen Zentral-/Hemi-Zentralvenenverschluss und 467 (52 %) hatten einen Venenastverschluss. Die Anteile der Patienten pro Subtyp waren in allen Behandlungsgruppen ähnlich.
Ergebnisse
Die Behandlung mit Eylea 114,3 mg/ml war der Behandlung mit 2Q4 in Bezug auf den primären Wirksamkeitsendpunkt „Veränderung der BCVA in Woche 36 im Vergleich zum Ausgangswert“, gemessen anhand des ETDRS-Buchstaben-Scores, nicht unterlegen und klinisch gleichwertig.
Darüber hinaus erwies sich die Behandlung mit Eylea 114,3 mg/ml in Bezug auf den wichtigen sekundären Wirksamkeitsendpunkt „Anzahl der aktiven Injektionen vom Ausgangswert bis Woche 64“ gegenüber der Behandlung mit 2Q4 als überlegen. Die Eylea-8Q8/3‑Gruppe benötigte 3,2 Injektionen weniger als die 2Q4‑Gruppe.
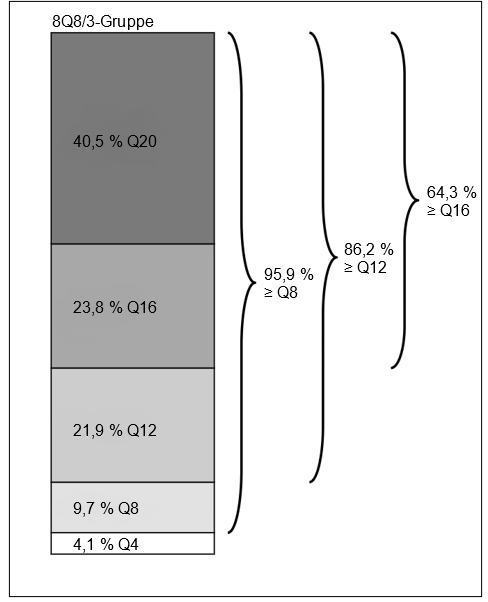
In Woche 36 hatten 93,9 % der Patienten in der 8Q8/3‑Gruppe Behandlungsintervalle von ≥ 8 Wochen erreicht, wobei der funktionelle und morphologische Befund erhalten blieben.
In Woche 64 hatten 56,1 % der Patienten in der 8Q8/3‑Gruppe Behandlungsintervalle von 16 Wochen abgeschlossen, wobei der funktionelle und morphologische Befund erhalten blieben.
In Woche 64 hatten 40,5 % der Patienten in der 8Q8/3‑Gruppe die letzten vorgesehenen Behandlungsintervalle von 20 Wochen erreicht, wobei der funktionelle und morphologische Befund erhalten blieben.
Tabelle 9: Wirksamkeitsendpunkte der QUASAR-Studie
Wirksamkeitsendpunkte |
Woche |
Eylea 8Q8/3 |
Eylea 2Q4 |
||
Veränderung der BCVA, ermittelt durch ETDRS-Buchstaben-Score im Vergleich zum Ausgangswert A | |||||
Arithmetisches Mittel (SD), beobachtet |
36 |
17,0 (11,8) |
17,8 (13,1) |
||
LS-Mittelwert (SE) B |
17,4 (0,7) |
17,5 (0,7) |
|||
Differenz der LS-Mittelwerte |
‑0,1 (‑2,0; 1,9) |
||||
p-Wert (einseitiger Nicht-Unterlegenheitstest mit einer Grenze von 4 Buchstaben) B, C |
< 0,0001 |
||||
Arithmetisches Mittel (SD), beobachtet |
64 |
17,3 (12,7) |
17,4 (14,6) |
||
LS-Mittelwert (SE) B |
17,8 (0,7) |
17,3 (0,8) |
|||
Differenz der LS-Mittelwerte |
0,5 (‑1,6; 2,7) |
||||
Zentral-/Hemizentralvenenverschluss D | |||||
Arithmetisches Mittel (SD), beobachtet |
36 |
16,5 (12,7) |
16,2 (14,7) |
||
LS-Mittelwert (SE) B |
16,6 (1,1) |
15,9 (1,2) |
|||
Differenz der LS-Mittelwerte |
0,6 (‑2,6, 3,9) |
||||
Arithmetisches Mittel (SD), beobachtet |
64 |
16,5 (13,8) |
14,8 (16,8) |
||
LS-Mittelwert (SE) B |
17,2 (1,2) |
15,2 (1,3) |
|||
Differenz der LS-Mittelwerte |
2,0 (‑1,5; 5,6) |
||||
Venenastverschluss D | |||||
Arithmetisches Mittel (SD), beobachtet |
36 |
17,4 (10,9) |
19,4 (11,0) |
||
LS-Mittelwert (SE) B |
18,3 (0,8) |
19,0 (0,8) |
|||
Differenz der LS-Mittelwerte |
‑0,8 (‑2,9; 1,4) |
||||
Arithmetisches Mittel (SD), beobachtet |
64 |
18,1 (11,8) |
20,1 (11,4) |
||
LS-Mittelwert (SE) B |
18,4 (0,9) |
19,6 (0,8) |
|||
Differenz der LS-Mittelwerte |
‑1,1 (‑3,5; 1,2) |
||||
Patienten, die einen ETDRS-Buchstaben-Score von mindestens 69 (ungefähres Snellen Äquivalent 20/40) erreichten A | |||||
Anteil (OC) |
36 |
72,7 % |
67,8 % |
||
64 |
70,4 % |
70,2 % |
|||
Patienten mit einem Gewinn von mindestens 15 Buchstaben der BCVA im Vergleich zum Ausgangswert A | |||||
Anteil (OC) |
36 |
58,8 % |
59,8 % |
||
64 |
61,7 % |
60,4 % |
|||
Patienten mit einem Verlust von mindestens 15 Buchstaben der BCVA im Vergleich zum Ausgangswert A | |||||
Anteil (OC) |
36 |
1,2 % |
1,5 % |
||
64 |
1,2 % |
2,4 % |
|||
Patienten ohne IRF und ohne SRF im zentralen Teilfeld A | |||||
Anteil (OC) |
36 |
81,2 % |
83,7 % |
||
64 |
76,3 % |
66,0 % |
|||
Anzahl an aktiven Injektionen | |||||
Arithmetisches Mittel (SD), beobachtet E |
36 |
6,1 (0,6) |
8,8 (0,8) |
||
LS-Mittelwert (SE) F |
6,1 (0,0) |
8,8 (0,0) |
|||
Differenz der LS-Mittelwerte |
‑2,7 (‑2,8; ‑2,6) |
||||
Arithmetisches Mittel (SD), beobachtet E |
64 |
8,4 (1,2) |
11,7 (1,6) |
||
LS-Mittelwert (SE) F |
8,5 (0,1) |
11,7 (0,1) |
|||
Differenz der LS-Mittelwerte |
‑3,2 (‑3,5; ‑3,0) |
||||
p-Wert (einseitiger Nicht-Unterlegenheitstest) G, C |
< 0,0001 |
||||
Aufrechterhaltung der Behandlungsintervalle | |||||
Patienten mit Aufrechterhaltung eines ≥ Q8 Behandlungsintervalls E | |||||
Anteil |
36 |
88,5 % |
n/a |
||
64 |
88,1 % |
70,0 % H |
|||
Letzte abgeschlossene Behandlungsintervalle | |||||
Patienten mit Q4 Behandlungsintervall E | |||||
Anteil |
64 |
4,8 % |
13,0 % |
||
Patienten mit ≥ Q8 Behandlungsintervall E | |||||
Anteil |
64 |
95,2 % |
87,0 % |
||
Patienten mit ≥ Q12 Behandlungsintervall E | |||||
Anteil |
64 |
81,4 % |
67,8 % |
||
Patienten mit Q16 Behandlungsintervall E | |||||
Anteil |
64 |
56,1 % |
n/a |
||
Letzte vorgesehene Behandlungsintervalle | |||||
Patienten mit ≥ Q8 Behandlungsintervall E | |||||
Anteil |
36 |
93,9 % |
75,6 % |
||
64 |
95,9 % |
92,2 % |
|||
Patienten mit Q12 Behandlungsintervall E | |||||
Anteil |
36 |
69,1 % |
n/a |
||
64 |
21,9 % |
27,8 % |
|||
Patienten mit ≥ Q12 Behandlungsintervall E | |||||
Anteil |
64 |
86,2 % |
77,8 % |
||
Patienten mit ≥ Q16 Behandlungsintervall E | |||||
Anteil |
64 |
64,3 % |
50,0 % |
||
Patienten mit Q20 Behandlungsintervall E | |||||
Anteil |
64 |
40,5 % |
n/a |
||
A Vollständiges Analyseset
B LS-Mittelwert, KI und p-Wert basierend auf einem MMRM mit der Messung des Ausgangswerts der bestkorrigierten Sehschärfe (BCVA) als Kovariate, der Behandlungsgruppe als Faktor, der Visite und den bei der Randomisierung verwendeten Stratifizierungsvariablen (geographische Region, Kategorie BCVA-Ausgangswert und RVV‑Typ) als feste Faktoren sowie Termen für die Interaktion zwischen BCVA-Ausgangswert und Visite und für die Interaktion zwischen Behandlung und Visite.
C Die absolute Differenz ist die Eylea 8Q8/3‑Gruppe minus die 2Q4‑Gruppe.
D Die Anzahl der Patienten mit Zentral-/Hemi-Zentralvenenverschluss betrug 134 bzw. 152 in den Behandlungsgruppen 8Q8/3 und 2Q4. Die Anzahl der Patienten mit Venenastverschluss betrug 159 bzw. 149 in den Behandlungsgruppen 8Q8/3 und 2Q4.
E Sicherheitsanalyseset; Patienten, die zum jeweiligen Zeitpunkt als abgeschlossen betrachtet werden.
F LS-Mittelwert und KI basierend auf einem multiplen Imputationsverfahren unter Anwendung eines linearen Regressionsmodells, das für den BCVA-Ausgangswert, den Ausgangswert der zentralen Teilfelddicke (central subfield thickness, CST) und die für die Randomisierung verwendeten Stratifizierungsvariablen (geographische Region, Kategorie BCVA-Ausgangswert und RVV‑Typ) auf jeden imputierten Datensatz und die Kombination der Ergebnisse unter Verwendung der Rubin-Regel angepasst wurde.
G p-Wert basierend auf einem multiplen Imputationsverfahren unter Anwendung einer nichtparametrischen Ranganalyse der Kovarianz, das für den BCVA-Ausgangswert, den CST-Ausgangswert und die für die Randomisierung verwendeten Stratifizierungsvariablen (geographische Region, Kategorie BCVA-Ausgangswert und RVV‑Typ) auf jeden imputierten Datensatz und die Kombination der Ergebnisse unter Verwendung der Rubin-Regel angepasst wurde.
H Patienten in der 2Q4-Behandlungsgruppe, die in Woche 32 verlängert wurden und bis Woche 64 bei ≥ Q8 gehalten wurden.
ETDRS: Early Treatment Diabetic Retinopathy Study
KI: Konfidenzintervall
LS: Kleinste Quadrate
MMRM: Mixed Model for Repeated Measurements
OC: Beobachtete Fälle (Observed Cases), Daten nach dem Auftreten eines interkurrenten Ereignisses gemäß der primären Schätzstrategie ausgeschlossen
SD: Standardabweichung
SE: Standardfehler
Abbildung 8: LS-Mittelwert-Veränderung der BCVA ermittelt durch ETDRS-Buchstaben-Score vom Ausgangswert bis Woche 64 (vollständiges Analyseset) in der QUASAR-Studie
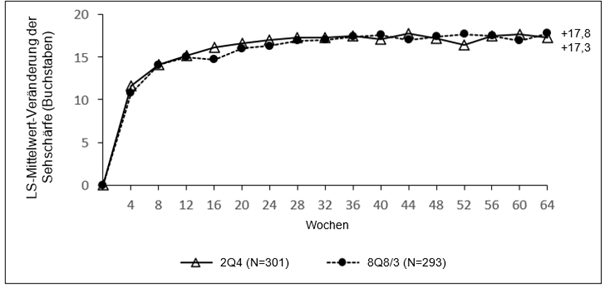
Eylea zeigte in allen Dosierungen (8Q8/3, 2Q4) einen relevanten Anstieg im Vergleich zum Ausgangswert hinsichtlich des vorab festgelegten sekundären Wirksamkeitsendpunkts National Eye Institute Visual Function Questionnaire (NEI VFQ-25). Das Ausmaß dieser Änderungen entsprach dem in veröffentlichten Studien beobachteten, was sich in Verbesserungen der visusbezogenen Lebensqualität widerspiegelte.
Es wurden keine klinisch relevanten Unterschiede zwischen den 8Q8/3- und 2Q4‑Gruppen bei den Veränderungen im Gesamtscore des NEI VFQ‑25 in Woche 36 und Woche 64 im Vergleich zum Ausgangswert festgestellt.
Die Ergebnisse zur Wirksamkeit in den vorab festgelegten auswertbaren Subgruppen für RVV‑Subtypen, Alter, Geschlecht, geographische Region, ethnische Herkunft, BCVA‑Ausgangswert und CRT-Ausgangswert entsprachen den Ergebnissen in der Gesamtpopulation.
Die Wirksamkeit wurde im Allgemeinen bis Woche 64 aufrechterhalten.
Beim vorab festgelegten sekundären Endpunkt „Teilnehmer, die bis Woche 36 nur 8Q8 in der Gruppe Eylea 114,3 mg/ml 8Q8 erhielten“ wurden 88,5 % der Patienten in der 8Q8/3‑Gruppe weiterhin mit ihrem ursprünglichen randomisierten Behandlungsintervall von 8 Wochen behandelt, wobei der funktionelle und morphologische Befund aufrechterhalten blieben.
Abbildung 9: Letztes vorgesehenes Behandlungsintervall in Woche 64
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Aflibercept eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen bei der nAMD, beim DMÖ und RVV gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Resorption/Verteilung
Aflibercept unterliegt nach intravitrealer Anwendung einer langsamen systemischen Resorption aus dem Auge und wird im systemischen Kreislauf überwiegend als inaktiver, stabiler Komplex mit VEGF beobachtet; allerdings ist nur "freies Aflibercept" in der Lage, endogenes VEGF zu binden.
Nach unilateraler intravitrealer Anwendung von 8 mg Aflibercept betrug die mittlere (SD) Cmax von freiem Aflibercept im Plasma 0,25 (0,21) mg/l und die mediane Zeit bis zur maximalen Konzentration 1 Tag für die kombinierte nAMD- und DMÖ-Population. Die Anreicherung von freiem Aflibercept im Plasma nach 3 initialen monatlichen Injektionen war minimal. Danach wurde keine weitere Anreicherung beobachtet. Diese Daten werden zusätzlich durch populationspharmakokinetische Analysen gestützt.
Diese pharmakokinetischen Ergebnisse stimmten mit den Daten überein, die bei Patienten mit RVV erhoben wurden.
Elimination
Aflibercept ist ein proteinbasiertes Therapeutikum. Es wurden keine Studien zur Verstoffwechselung durchgeführt.
Es ist davon auszugehen, dass Aflibercept sowohl über die Bindung an freiem endogenen VEGF ("target-mediated drug disposition") als auch durch proteolytische Verstoffwechselung eliminiert wird. Die mediane Zeit bis zum Erreichen der letzten messbaren Konzentration von freiem Aflibercept im Plasma betrug 3 Wochen bei intravitreal angewendetem Aflibercept 8 mg.
Nieren- oder Leberfunktionsstörung
Es wurden keine speziellen Studien bei Patienten mit Nieren- oder Leberfunktionsstörungen mit Eylea 114,3 mg/ml durchgeführt.
Die systemische Exposition gegenüber Aflibercept war bei Patienten mit leichter bis schwerer Nierenfunktionsstörung ähnlich wie bei Patienten mit normaler Nierenfunktion. Begrenzt verfügbare Daten bei Patienten mit leichter Leberfunktionsstörung weisen nicht auf einen Einfluss auf die systemische Exposition gegenüber Aflibercept im Vergleich zu Patienten mit normaler Leberfunktion hin.
Erosionen und Geschwürbildungen des respiratorischen Flimmerepithels der Nasenmuscheln wurden bei Affen beobachtet, die intravitreal mit Aflibercept behandelt wurden und einer systemischen Exposition ausgesetzt waren, die weit über der maximalen humanen Exposition lag. Die systemische Exposition gegenüber freiem Aflibercept, basierend auf Cmax und AUC, war im Vergleich zu den entsprechenden Werten bei erwachsenen Patienten nach einer intravitreal angewendeten Dosis von 8 mg ungefähr 26- und 33-mal höher. Beim No Observed Adverse Effect Level (NOAEL) von 0,5 mg/Auge war die systemische Exposition, basierend auf Cmax und AUC, bei Affen im Vergleich zu den entsprechenden Werten bei erwachsenen Patienten um das 3,2- und 3,8-Fache erhöht.
Es wurden keine Studien zum mutagenen oder kanzerogenen Potenzial von Aflibercept durchgeführt.
Es wurde ein Effekt von Aflibercept auf die intrauterine Entwicklung in Studien zur embryonalen und fetalen Entwicklung bei trächtigen Kaninchen, denen Aflibercept sowohl intravenös (3 bis 60 mg/kg) als auch subkutan (0,1 bis 1 mg/kg) angewendet wurde, gezeigt. Der mütterliche NOAEL war bei Dosen von 3 mg/kg bzw. 1 mg/kg. Ein entwicklungsbezogener NOAEL wurde nicht identifiziert. Bei der 0,1 mg/kg Dosis war die systemische Exposition gegenüber freiem Aflibercept ungefähr das 1,0- und 1,0-Fache, basierend auf Cmax und kumulativer AUC, im Vergleich zu den entsprechenden Werten bei erwachsenen Patienten nach einer intravitreal angewendeten Dosis von 8 mg.
Wirkungen auf die männliche und weibliche Fertilität wurden im Rahmen einer 6-monatigen Studie bei Affen, die Aflibercept intravenös in Dosen von 3 bis 30 mg/kg angewendet bekamen, untersucht. Bei allen Dosierungen wurden ausbleibende oder unregelmäßig auftretende Regelblutungen, die aufgrund der Änderungen der Spiegel weiblicher Fortpflanzungshormone auftraten, und Veränderungen der Morphologie und Motilität der Spermien beobachtet. Basierend auf Cmax und AUC des freien Aflibercept bei 3 mg/kg intravenös angewendeter Dosis waren die systemischen Expositionen ungefähr um das 377- bzw. 104-Fache höher als die Expositionen beim Menschen nach einer intravitreal angewendeten Dosis von 8 mg. Alle Änderungen waren reversibel.
Saccharose
Argininhydrochlorid
Histidinhydrochlorid-Monohydrat
Histidin
Polysorbat 20
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
3 Jahre
Eylea 114,3 mg/ml Injektionslösung
Im Kühlschrank lagern (2 °C ‑ 8 °C).
Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Vor der Anwendung darf die ungeöffnete Durchstechflasche außerhalb des Kühlschranks bis zu 24 Stunden unter 25 °C aufbewahrt werden.
Eylea 114,3 mg/ml Injektionslösung in einer Fertigspritze
Im Kühlschrank lagern (2 °C ‑ 8 °C).
Nicht einfrieren.
Die Fertigspritze in der Blisterpackung und im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Vor der Anwendung darf die ungeöffnete Blisterpackung außerhalb des Kühlschranks bis zu 24 Stunden unter 25 °C aufbewahrt werden.
Eylea 114,3 mg/ml Injektionslösung
Durchstechflasche (Typ I Glas) mit einem grauen Gummistopfen (Chlorbutyl), versiegelt mit einer Aluminiumschutzkappe mit weißem Deckel, und einer 18 G, 5 Mikrometer-Filternadel.
Jede Durchstechflasche enthält 0,263 ml Lösung.
Packungsgröße: 1 Durchstechflasche und 1 Filternadel.
Eylea 114,3 mg/ml Injektionslösung in einer Fertigspritze
Fertigspritze (Typ I Glas) mit einem grauen Kolbenverschluss (aus elastischem Gummi), einem weißen Luer-Lock-Adapter mit grauer Abdeckkappe (aus elastischem Gummi) und einem blauen OcuClick-Dosiersystem (PC/ABS-Kunststoff).
Jede Fertigspritze enthält 0,184 ml Lösung.
Packungsgröße: 1 Fertigspritze.
Eylea 114,3 mg/ml Injektionslösung
Die Durchstechflasche ist nur für den einmaligen Gebrauch in einem Auge bestimmt. Die Entnahme von mehr als einer Dosis aus einer einzelnen Durchstechflasche kann das Risiko einer Kontamination und nachfolgender Infektion erhöhen.
Nicht verwenden, wenn die Verpackung oder ihre Bestandteile abgelaufen oder beschädigt sind oder manipuliert wurden.
Das Etikett der Durchstechflasche überprüfen, um sicherzustellen, dass die für die Anwendung von Eylea vorgesehene Wirkstärke verwendet wird. Die 8‑mg‑Dosierung erfordert die Anwendung der Eylea 114,3 mg/ml Durchstechflasche.
18 G, 5 Mikrometer-Filternadel:
Stumpfe Filter-(Füll-)nadel, nicht zur Injektion in die Haut.
Die stumpfe Filter-(Füll-)nadel nicht autoklavieren.
Die Filternadel ist nicht pyrogen. Bei Beschädigung von Einzelverpackungen nicht verwenden.
Verwendete stumpfe Filter-(Füll-)nadel in einem geprüften Sicherheitsbehälter entsorgen.
Vorsicht: Erneute Verwendung der Filternadel kann zu Infektionen oder anderen Erkrankungen/Verletzungen führen.
Die intravitreale Injektion sollte mit einer 30 G × 1/2 Zoll Injektionsnadel durchgeführt werden (nicht enthalten). Die Verwendung einer kleineren Nadel (höhere Gauge-Werte) als der empfohlenen 30 G × 1/2 Zoll Injektionsnadel kann zu einem erhöhten Kraftaufwand bei der Injektion führen.
1. |
Vor der Anwendung ist die Injektionslösung visuell zu überprüfen. |
||
2. |
Die Kunststoffkappe entfernen und den Gummistopfen der Durchstechflasche von außen desinfizieren. |
 |
|
3. |
Unter sterilen Bedingungen die Schritte 3‑10 durchführen. |
 |
|
4. |
Die Filternadel durch die Mitte des Durchstechflaschen-Stopfens stechen, bis die Nadel vollständig in die Durchstechflasche eingeführt ist und die Spitze den Boden oder die Unterkante der Durchstechflasche berührt. |
||
5. |
Den gesamten Inhalt der Eylea-Durchstechflasche in die Spritze aufnehmen, indem die Durchstechflasche aufrecht in einer leicht geneigten Position gehalten wird, um das vollständige Entleeren zu erleichtern. Um das Aufziehen von Luft zu verhindern, sollte darauf geachtet werden, dass die abgeschrägte Kante der Filternadel in die Lösung eintaucht. Um dies auch während der Entnahme zu gewährleisten, ist die Durchstechflasche schräg zu halten. |
||
 |
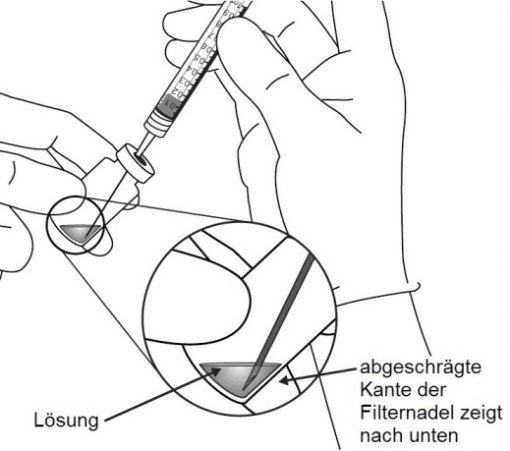 |
||
6. |
Bitte beachten, dass der Spritzenkolben beim Entleeren der Durchstechflasche ausreichend zurückgezogen wird, damit die Filternadel vollständig entleert wird. Überschüssiges Produkt ist nach der Injektion zu verwerfen. |
||
7. |
Die Filternadel entfernen und diese vorschriftsmäßig entsorgen. |
||
8. |
Die 30 G × 1/2 Zoll Injektionsnadel fest auf die Luer-Lock-Spitze der Spritze schrauben. |
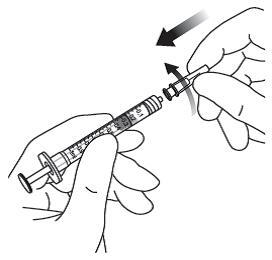 |
|
9. |
Die Spritze mit der Nadel nach oben halten und auf Bläschen hin prüfen. Wenn Bläschen zu sehen sind, leicht mit dem Finger gegen die Spritze klopfen, bis die Bläschen nach oben steigen. |
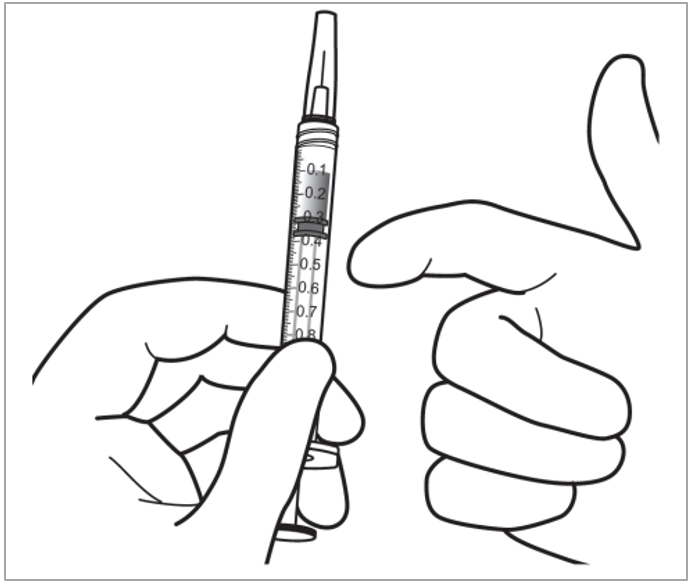 |
|
10. |
Um alle Bläschen und überschüssiges Arzneimittel zu entfernen, den Spritzenkolben langsam soweit eindrücken, bis der ebene Rand des Kolbens auf derselben Höhe ist wie die 0,07 ml-Linie der Spritze. |
||
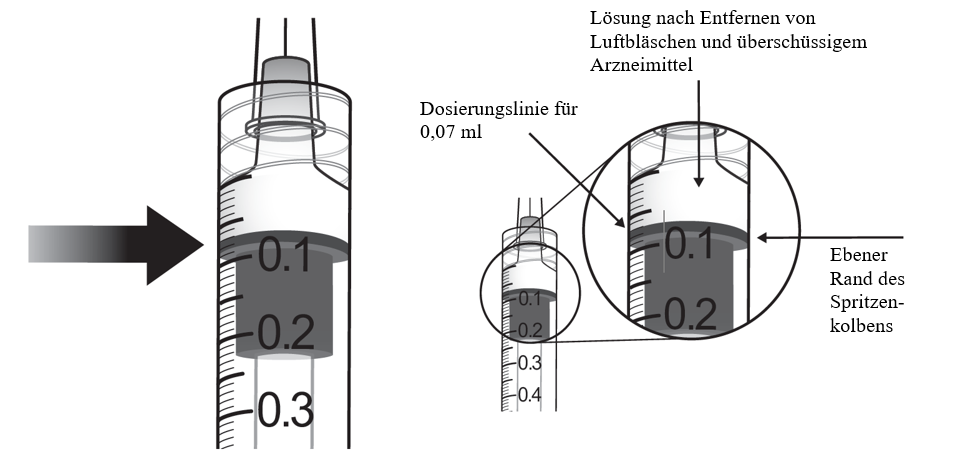 |
|||
Eylea 114,3 mg/ml Injektionslösung in einer Fertigspritze
Die Fertigspritze mit OcuClick-Dosiersystem ist nur für den einmaligen Gebrauch in einem Auge bestimmt. Die Entnahme von mehr als einer Dosis aus einer einzelnen Fertigspritze mit OcuClick-Dosiersystem kann das Risiko einer Kontamination und nachfolgender Infektion erhöhen.
Nicht verwenden, wenn die Verpackung oder ihre Bestandteile abgelaufen oder beschädigt sind oder manipuliert wurden.
Das Etikett der Fertigspritze mit OcuClick-Dosiersystem überprüfen, um sicherzustellen, dass die für die Anwendung von Eylea vorgesehene Wirkstärke verwendet wird. Die 8‑mg‑Dosierung erfordert die Anwendung der Eylea 114,3 mg/ml Fertigspritze.
Die intravitreale Injektion sollte mit einer 30 G × 1/2 Zoll Injektionsnadel durchgeführt werden (nicht enthalten).
Die Verwendung einer kleineren Nadel (höhere Gauge-Werte) als der empfohlenen 30 G × 1/2 Zoll Injektionsnadel kann zu einem erhöhten Kraftaufwand bei der Injektion führen.
Beschreibung der Fertigspritze mit integriertem OcuClick-Dosiersystem | ||
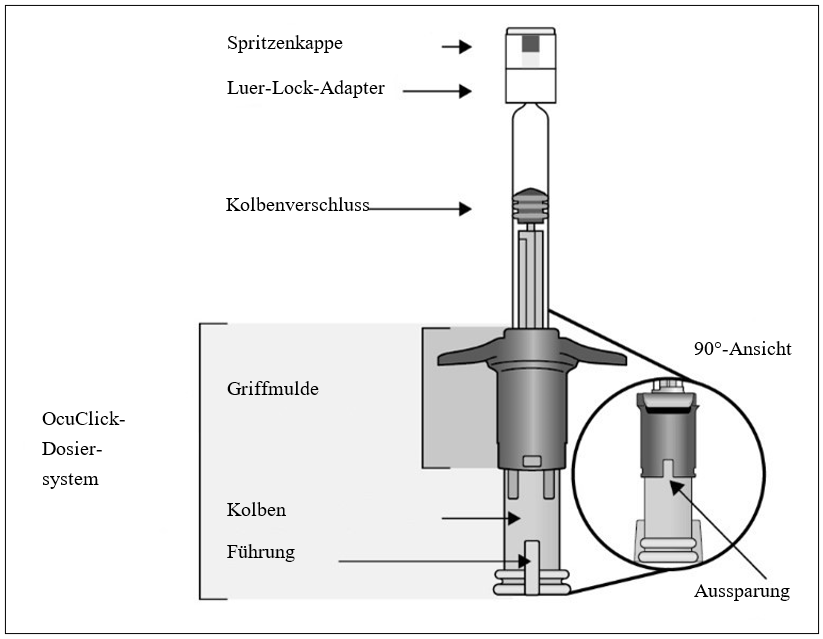 | ||
1. |
Vorbereiten |
|
|
Erst vor der Anwendung von Eylea 114,3 mg/ml den Umkarton öffnen und die sterile Blisterpackung entnehmen. Die Blisterpackung vorsichtig öffnen, so dass der Inhalt weiterhin steril bleibt. Unter sterilen Bedingungen die Schritte 2 bis 9 durchführen. | ||
2. |
Spritze entnehmen |
|
Die Spritze aus der sterilen Blisterpackung nehmen. | ||
3. |
Spritze und Injektionslösung überprüfen |
|
|
Die Fertigspritze nicht anwenden, wenn
| ||
4. |
Spritzenkappe abbrechen |
|
Zum Abbrechen (nicht Abschrauben) der Spritzenkappe die Spritze in einer Hand und die Spritzenkappe mit Daumen und Zeigefinger der anderen Hand festhalten. |
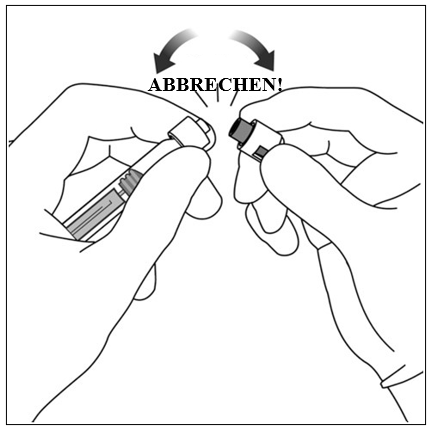 |
|
5. |
Injektionsnadel befestigen |
|
Die 30 G × 1/2 Zoll Injektionsnadel fest auf die Spitze des Luer-Lock-Adapters aufschrauben. |
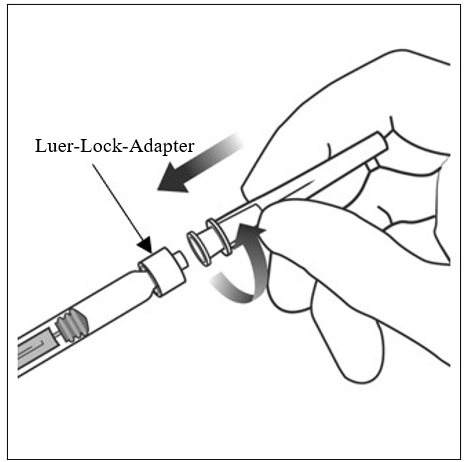 |
|
6. |
Luftbläschen entfernen |
|
Die Spritze mit der Nadel nach oben halten und auf Bläschen hin prüfen. Wenn Bläschen zu sehen sind, leicht mit dem Finger gegen die Spritze klopfen, bis die Bläschen nach oben steigen. |
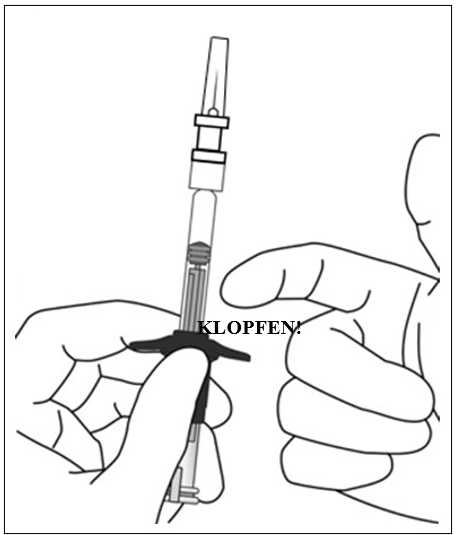 |
|
7. |
Zum Vorbereiten Luft und überschüssige Menge entfernen |
|
|
Die Spritze weist keine Dosierungslinie auf, da die Dosis mechanisch eingestellt wird, wie in den folgenden Schritten erläutert. Zur Vorbereitung und zum Einstellen der Dosis müssen folgende Schritte erfolgen. | ||
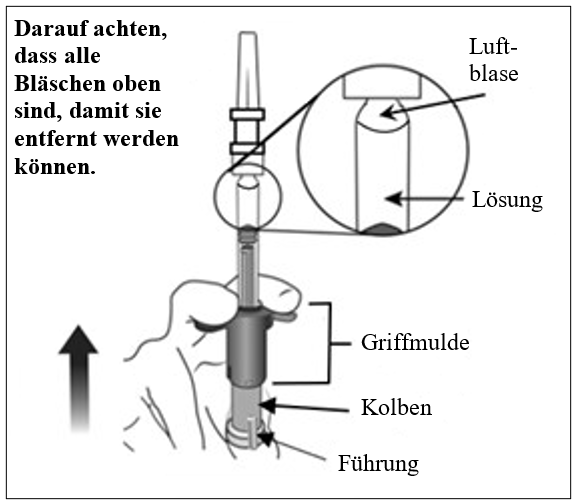 |
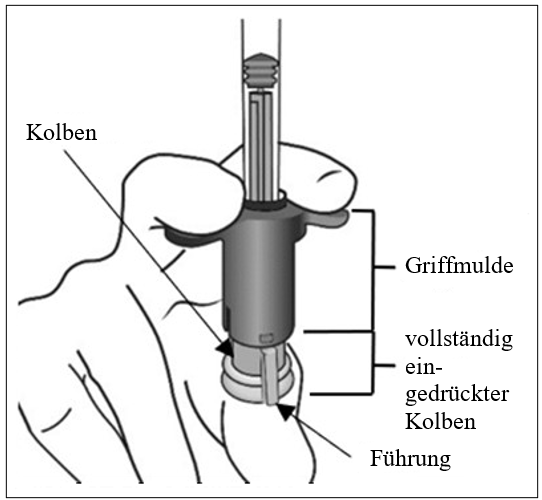 |
|
8. |
Dosis einstellen |
|
Das Kolbenende um 90 Grad im oder gegen den Uhrzeigersinn drehen, bis die Führung des Kolbens auf die Aussparung zeigt. Es kann ein Klickgeräusch zu hören sein. |
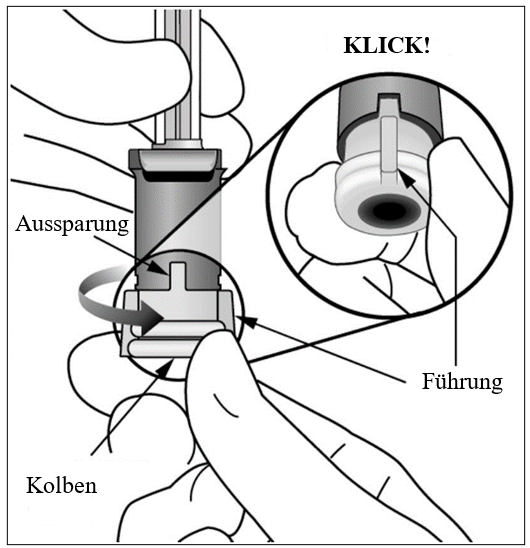 |
|
9. |
Injektion durchführen |
|
|
Nadel in die Injektionsstelle am Auge einführen. Injizieren der Lösung durch Eindrücken des Kolbens bis zum Anschlag, d. h., bis sich die Führung vollständig in der Aussparung befindet. Keinen zusätzlichen Druck ausüben, wenn sich die Führung vollständig in der Aussparung befindet. Es ist normal, wenn eine geringfügige Menge der Restlösung in der Spritze verbleibt. |
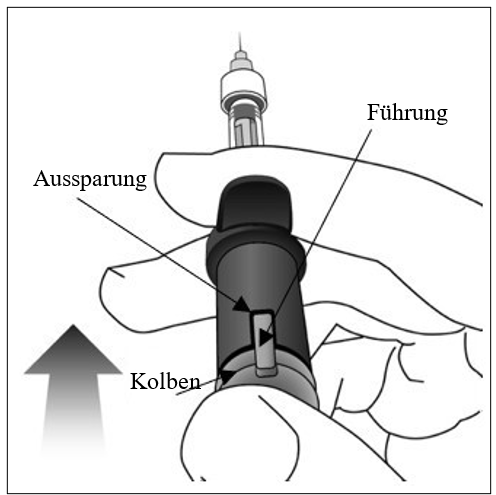 |
|
10. |
Die Fertigspritze ist nur zur einmaligen Abgabe einer Einzeldosis bestimmt. |
|
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Bayer AG
51368 Leverkusen
Deutschland
EU/1/12/797/003 - Eylea 114,3 mg/ml Injektionslösung
EU/1/12/797/004 - Eylea 114,3 mg/ml Injektionslösung in einer Fertigspritze
Datum der Erteilung der Zulassung: 22. November 2012
Datum der letzten Verlängerung der Zulassung: 13. Juli 2017
Januar 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
VERKAUFSABGRENZUNG IN DEUTSCHLAND
Verschreibungspflichtig
REZEPTPFLICHT/APOTHEKENPFLICHT IN ÖSTERREICH
Rezept- und apothekenpflichtig, wiederholte Abgabe verboten
Bayer Vital GmbH
51368 Leverkusen
Tel.: +49 (0)214-30 513 48
Fax: +49 (0)214-2605 516 03
E-Mail: medical-information@bayer.com
KONTAKTADRESSE IN ÖSTERREICH
Bayer Austria Ges.m.b.H.
Tel: +43-(0)1-711 46-0
DE/9