
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Omlyclo™ 150 mg Injektionslösung in einer Fertigspritze
Omlyclo™ 300 mg Injektionslösung in einer Fertigspritze
Omlyclo™ 150 mg Injektionslösung im Fertigpen
Omlyclo 150 mg Injektionslösung in einer Fertigspritze
Jede Fertigspritze mit 1 ml Lösung enthält 150 mg Omalizumab*.
Omlyclo 300 mg Injektionslösung in einer Fertigspritze
Jede Fertigspritze mit 2 ml Lösung enthält 300 mg Omalizumab*.
Omlyclo 150 mg Injektionslösung im Fertigpen
Jeder Fertigpen mit 1 ml Lösung enthält 150 mg Omalizumab*.
*Omalizumab ist ein humanisierter monoklonaler Antikörper, der durch rekombinante DNA-Technologie in einer Säugetier-Zelllinie aus dem Ovar des chinesischen Hamsters (Chinese hamster ovary, CHO) hergestellt wird.
Sonstiger Bestandteil mit bekannter Wirkung
Dieses Arzneimittel enthält 0,40 mg Polysorbat 20 (E 432) pro ml Lösung.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung (Injektion).
Klare bis opalisierende, farblose bis schwach bräunlich-gelbe Lösung.
Allergisches Asthma
Omlyclo wird angewendet bei Erwachsenen, Jugendlichen und Kindern (6 bis <12 Jahre).
Die Behandlung mit Omlyclo sollte nur bei Patienten in Betracht gezogen werden, bei denen von einem IgE-(Immunglobulin E-)vermittelten Asthma ausgegangen werden kann (siehe Abschnitt 4.2).
Erwachsene und Jugendliche (ab 12 Jahren)
Omlyclo wird als Zusatztherapie zur verbesserten Asthmakontrolle bei Patienten mit schwerem persistierendem allergischem Asthma angewendet, die einen positiven Hauttest oder In-vitro-Reaktivität gegen ein ganzjährig auftretendes Aeroallergen zeigen und sowohl eine reduzierte Lungenfunktion (Forced expiratory voulme in 1 second, FEV1 <80 %) haben als auch unter häufigen Symptomen während des Tages oder nächtlichem Erwachen leiden und trotz täglicher Therapie mit hoch dosierten inhalativen Kortikosteroiden und einem langwirkenden inhalativen Beta2-Agonisten mehrfach dokumentierte, schwere Asthma-Exazerbationen hatten.
Kinder (6 bis <12 Jahre)
Omlyclo wird als Zusatztherapie zur verbesserten Asthmakontrolle bei Patienten mit schwerem persistierendem allergischem Asthma angewendet, die einen positiven Hauttest oder In-vitro-Reaktivität gegen ein ganzjährig auftretendes Aeroallergen zeigen und unter häufigen Symptomen während des Tages oder nächtlichem Erwachen leiden und trotz täglicher Therapie mit hoch dosierten inhalativen Kortikosteroiden und einem langwirkenden inhalativen Beta2-Agonisten mehrfach dokumentierte, schwere Asthma-Exazerbationen hatten.
Chronische Rhinosinusitis mit Nasenpolypen (Chronic rhinosinusitis with nasal polyps, CRSwNP)
Omlyclo wird als Zusatztherapie zu intranasalen Kortikosteroiden (Intranasal corticosteroids, INCS) zur Behandlung von Erwachsenen (ab 18 Jahren) mit schwerer CRSwNP angewendet, bei denen durch eine Therapie mit INCS keine ausreichende Krankheitskontrolle erzielt wird.
Chronische spontane Urtikaria (Chronic spontaneous urticaria, csU)
Omlyclo wird als Zusatztherapie für die Behandlung der chronischen spontanen Urtikaria bei Erwachsenen und Jugendlichen (ab 12 Jahren) mit unzureichendem Ansprechen auf eine Behandlung mit H1-Antihistaminika angewendet.
Die Behandlung sollte von Ärzten eingeleitet werden, die Erfahrung mit der Diagnose und der Behandlung von schwerem persistierendem Asthma, chronischer Rhinosinusitis mit Nasenpolypen (CRSwNP) oder chronischer spontaner Urtikaria haben.
Dosierung
Allergisches Asthma und chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
Die Dosierung bei allergischem Asthma und CRSwNP folgt den gleichen Dosierungsprinzipien. Die geeignete Dosierung und Häufigkeit der Anwendung von Omalizumab bei diesen Erkrankungen wird anhand des vor Behandlungsbeginn gemessenen IgE-Basiswertes (I.E./ml) und des Körpergewichts (kg) bestimmt. Zur Dosisfestlegung ist es erforderlich, vor der ersten Anwendung den IgE-Wert des Patienten mit einem handelsüblichen Gesamt-Serum-IgE-Test zu bestimmen. Basierend auf diesen Messungen können pro Verabreichung 75 bis 600 mg Omalizumab in Form von 1 bis 4 Injektionen benötigt werden.
Für Patienten mit allergischem Asthma mit einem IgE-Basiswert unter 76 I.E./ml war ein Nutzen weniger wahrscheinlich (siehe Abschnitt 5.1). Verschreibende Ärzte sollten vor Beginn der Therapie sicherstellen, dass erwachsene und jugendliche Patienten mit einem IgE-Wert unter 76 I.E./ml und Kinder (6 bis <12 Jahre) mit einem IgE-Wert unter 200 I.E./ml eine eindeutige In-vitro-Reaktivität (Radioallergosorbent test, RAST) gegenüber einem ganzjährig auftretenden Allergen zeigen.
Siehe Tabelle 1 für das Umrechnungsschema und Tabellen 2 und 3 zur Dosisbestimmung.
Patienten, deren IgE-Basiswert oder Körpergewicht in Kilogramm außerhalb der Grenzen der Dosierungstabelle liegen, sollten nicht mit Omalizumab behandelt werden.
Die empfohlene Maximaldosis beträgt 600 mg Omalizumab alle zwei Wochen.
Tabelle 1 Umrechnung der Dosierung auf die Anzahl der Fertigspritzen/Fertigpens*, die Anzahl der Injektionen** und die Gesamtinjektionsmenge pro Verabreichung
Dosis (mg) |
Anzahl der Spritzen/Pens* |
Anzahl der Injektionen** |
Gesamtinjektionsmenge (ml) |
||
75 mg |
150 mg |
300 mg* |
|||
75 |
1 |
0 |
0 |
1 |
0,5 |
150 |
0 |
1 |
0 |
1 |
1,0 |
225 |
1 |
1 |
0 |
2 |
1,5 |
300 |
0 |
0 |
1 |
1 |
2,0 |
375 |
1 |
0 |
1 |
2 |
2,5 |
450 |
0 |
1 |
1 |
2 |
3,0 |
525 |
1 |
1 |
1 |
3 |
3,5 |
600 |
0 |
0 |
2 |
2 |
4,0 |
* Omlyclo 300 mg Fertigspritze und alle Dosisstärken (75 mg und 150 mg) des Fertigpens sind nicht für die Anwendung bei Patienten unter 12 Jahren vorgesehen.
** Diese Tabelle stellt die geringste Anzahl von Injektionen für die Patienten dar, es sind jedoch auch andere Dosierkombinationen mit Spritze/Pen möglich, um die gewünschte Dosis zu erreichen.
Tabelle 2 VERABREICHUNG ALLE 4 WOCHEN. Dosierung von Omalizumab (Milligramm pro Dosis) bei subkutaner Injektion alle 4 Wochen
Körpergewicht (kg) |
||||||||||
IgE-Basiswert (I.E./ml) |
≥20-25* |
>25-30* |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90-125 |
>125-150 |
≥30-100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
>100-200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
>200-300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 |
|
>300-400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 |
||
>400-500 |
225 |
300 |
450 |
450 |
600 |
600 |
||||
>500-600 |
300 |
300 |
450 |
600 |
600 |
|||||
>600-700 |
300 |
450 |
600 |
|||||||
>700-800 |
||||||||||
>800-900 |
VERABREICHUNG ALLE 2 WOCHEN SIEHE TABELLE 3 |
|||||||||
>900-1 000 |
||||||||||
>1 000-1 100 |
||||||||||
*Körpergewichte unter 30 kg wurden in den Zulassungsstudien zu CRSwNP nicht untersucht.
Tabelle 3 VERABREICHUNG ALLE 2 WOCHEN. Dosierung von Omalizumab (Milligramm pro Dosis) bei subkutaner Injektion alle 2 Wochen
Körpergewicht (kg) |
||||||||||
IgE-Basiswert (I.E./ml) |
≥20- 25* |
>25- 30* |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90-125 |
>125-150 |
≥30-100 |
VERABREICHUNG ALLE 4 WOCHEN SIEHE TABELLE 2 |
|||||||||
>100-200 |
||||||||||
>200-300 |
375 |
|||||||||
>300-400 |
450 |
525 |
||||||||
>400-500 |
375 |
375 |
525 |
600 |
||||||
>500-600 |
375 |
450 |
450 |
600 |
||||||
>600-700 |
225 |
375 |
450 |
450 |
525 |
|||||
>700-800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 |
||
>800-900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 |
|||
>900-1 000 |
225 |
300 |
375 |
450 |
525 |
600 |
||||
>1 000-1 100 |
225 |
300 |
375 |
450 |
600 |
|||||
>1 100-1 200 |
300 |
300 |
450 |
525 |
600 |
Unzureichende Datenlage für eine Dosisempfehlung |
||||
>1 200-1 300 |
300 |
375 |
450 |
525 |
||||||
>1 300-1 500 |
300 |
375 |
525 |
600 |
||||||
*Körpergewichte unter 30 kg wurden in den Zulassungsstudien zu CRSwNP nicht untersucht.
Therapiedauer, Überwachung und Dosisanpassung
Allergisches Asthma
Omlyclo ist angezeigt für die Langzeitbehandlung. Klinische Studien zeigten, dass es mindestens 12-16 Wochen dauert, bis die Behandlung eine Wirkung zeigt. 16 Wochen nach Beginn der Therapie mit Omlyclo sollte bei den Patienten die Wirksamkeit der Behandlung durch den Arzt überprüft werden, bevor weitere Injektionen verabreicht werden. Nach dem 16-Wochen-Zeitpunkt oder auch später sollte die Entscheidung zur Weiterbehandlung auf einer merklichen Verbesserung der gesamten Asthma-Kontrolle basieren (siehe Abschnitt 5.1, Ärztliche Gesamtbewertung der Wirksamkeit der Behandlung).
Chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
In klinischen Studien zu CRSwNP wurden nach 4 Wochen Veränderungen im Nasenpolypenscore (Nasal polyp score, NPS) und im Score für nasale Kongestion (Nasal congestion score, NCS) beobachtet. Die Notwendigkeit, die Therapie fortzusetzen, sollte auf Grundlage des Schweregrades der Erkrankung des Patienten und des Grades der Symptomkontrolle in regelmäßigen Abständen neu bewertet werden.
Allergisches Asthma und chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
Ein Absetzen der Behandlung führt im Allgemeinen zu einer Rückkehr zu erhöhten Werten von freiem IgE und den damit verbundenen Symptomen. Der Gesamt-IgE-Spiegel ist während der Behandlung erhöht und bleibt bis zu einem Jahr nach Absetzen der Behandlung erhöht. Deshalb kann eine erneute Messung des IgE-Spiegels während der Behandlung nicht als Richtwert für die Dosisfestsetzung verwendet werden. Die Dosisfestsetzung nach Unterbrechungen der Behandlung um weniger als ein Jahr muss anhand der Serum-IgE-Spiegel erfolgen, die bei der ursprünglichen Dosisfestsetzung ermittelt wurden. Die Gesamt-Serum-IgE-Spiegel sollten für die Dosisfestsetzung erneut bestimmt werden, wenn die Behandlung für ein Jahr oder länger unterbrochen wurde.
Bei erheblichen Veränderungen des Körpergewichts sollte die Dosis angepasst werden (siehe Tabellen 2 und 3).
Chronische spontane Urtikaria (csU)
Die empfohlene Dosis beträgt 300 mg als subkutane Injektion alle vier Wochen. Jede 300-mg-Dosis wird als eine subkutane Injektion mit 300 mg oder als zwei subkutane Injektionen mit je 150 mg verabreicht.
Verordnenden Ärzten wird geraten, die Notwendigkeit der Fortsetzung der Therapie in regelmäßigen Abständen neu zu überprüfen.
Die Erfahrung aus klinischen Studien mit einer Langzeitbehandlung in diesem Anwendungsgebiet ist in Abschnitt 5.1 beschrieben.
Spezielle Patientengruppen
Ältere Patienten (ab 65 Jahren)
Es liegen begrenzte Daten zur Anwendung von Omalizumab bei Patienten über 65 Jahren vor, aber es gibt keine Hinweise, dass bei älteren Patienten eine andere Dosierung erforderlich ist als bei jüngeren erwachsenen Patienten.
Beeinträchtigung der Nieren- oder Leberfunktion
Es wurden keine Studien zum Einfluss einer eingeschränkten Nieren- oder Leberfunktion auf die Pharmakokinetik von Omalizumab durchgeführt. Da bei klinischen Dosen die Omalizumab-Clearance durch das retikuloendotheliale System (Reticular endothelial system, RES) bei Weitem überwiegt, ist eine Beeinflussung durch eine eingeschränkte Nieren- oder Leberfunktion unwahrscheinlich. Obwohl keine besondere Dosisanpassung für diese Patienten empfohlen wird, muss die Anwendung von Omalizumab mit Vorsicht erfolgen (siehe Abschnitt 4.4).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Omalizumab in der Behandlung von allergischem Asthma bei Patienten unter 6 Jahren sind nicht erwiesen. Es liegen keine Daten vor.
Die Sicherheit und Wirksamkeit von Omalizumab in der Behandlung von CRSwNP bei Patienten unter 18 Jahren sind nicht erwiesen. Es liegen keine Daten vor.
Die Sicherheit und Wirksamkeit von Omalizumab in der Behandlung von csU bei Patienten unter 12 Jahren sind nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Nur zur subkutanen Anwendung. Omalizumab darf nicht intravenös oder intramuskulär angewendet werden.
Die Omlyclo 300 mg Fertigspritze und alle Dosisstärken (75 mg und 150 mg) des Fertigpens sind nicht für die Anwendung bei Kindern unter 12 Jahren vorgesehen. Die Omlyclo 75 mg Fertigspritze und die Omlyclo 150 mg Fertigspritze können bei Kindern im Alter von 6 bis 11 Jahren mit allergischem Asthma angewendet werden.
Falls mehr als eine Injektion benötigt wird, um die erforderliche Dosierung zu erreichen, sollten die Injektionen auf zwei oder mehr Injektionsstellen verteilt werden (Tabelle 1).
Patienten mit keiner bekannten Anaphylaxie in der Vorgeschichte können sich ab der vierten Anwendung Omlyclo selbst injizieren oder von einer Betreuungsperson injizieren lassen, wenn ein Arzt dies für angemessen hält (siehe Abschnitt 4.4). Der Patient oder die Betreuungsperson muss in der korrekten Injektionstechnik und dem Erkennen von frühen Anzeichen und Symptomen schwerer allergischer Reaktionen geschult worden sein.
Patienten oder Betreuungspersonen sollten angewiesen werden, den gesamten Inhalt der Omlyclo-Spritze gemäß den Anwendungshinweisen in der Packungsbeilage zu injizieren.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Allgemein
Omalizumab ist nicht angezeigt für die Behandlung von akuten Asthma-Exazerbationen, akuten Bronchospasmen oder eines Status asthmaticus.
Omalizumab wurde nicht untersucht bei Patienten mit Hyper-Immunglobulin-E-Syndrom oder allergischer bronchopulmonarer Aspergillose oder zur Vorbeugung von anaphylaktischen Reaktionen, einschließlich durch Nahrungsmittelallergien, atopischer Dermatitis oder allergischer Rhinitis ausgelöster Anaphylaxien. Omalizumab ist für die Behandlung dieser Zustände nicht angezeigt.
Die Therapie mit Omalizumab wurde bei Patienten mit Autoimmunkrankheiten, immunkomplexvermittelten Erkrankungen sowie mit vorgeschädigter Niere oder Leber (siehe Abschnitt 4.2) nicht untersucht. Bei der Verabreichung von Omalizumab an diese Patienten ist Vorsicht geboten.
Nach Beginn der Therapie von allergischem Asthma oder CRSwNP mit Omalizumab wird ein plötzliches Absetzen von systemischen oder inhalativen Kortikosteroiden nicht empfohlen. Eine Reduktion der Kortikosteroide sollte unter ärztlicher Aufsicht erfolgen und muss gegebenenfalls stufenweise durchgeführt werden.
Erkrankungen des Immunsystems
Allergische Reaktionen Typ I
Bei der Anwendung von Omalizumab können lokale oder systemische allergische Reaktionen vom Typ I einschließlich Anaphylaxie und anaphylaktischem Schock auftreten. Diese können sogar nach einer längeren Behandlungsdauer eintreten. Allerdings traten die meisten dieser Reaktionen innerhalb von 2 Stunden nach der ersten und den folgenden Injektionen von Omalizumab auf, aber manche ereigneten sich nach mehr als 2 Stunden und sogar nach mehr als 24 Stunden nach der Injektion. Die meisten anaphylaktischen Reaktionen traten bei den ersten 3 Anwendungen von Omalizumab auf. Daher müssen die ersten 3 Anwendungen durch medizinisches Fachpersonal oder unter dessen Aufsicht verabreicht werden. Anaphylaxie in der Vorgeschichte, die nicht im Zusammenhang mit Omalizumab stand, kann einen Risikofaktor für das Auftreten von Anaphylaxie nach Verabreichung von Omalizumab darstellen. Aus diesem Grund muss Omalizumab bei Patienten mit bekannter Anaphylaxie in der Vorgeschichte durch medizinisches Fachpersonal verabreicht werden. Dabei sollten immer Arzneimittel für die Behandlung einer anaphylaktischen Reaktion zum sofortigen Einsatz nach der Verabreichung von Omalizumab vorhanden sein. Tritt eine anaphylaktische oder eine andere schwerwiegende allergische Reaktion auf, muss die Verabreichung von Omalizumab sofort abgebrochen und eine angemessene Therapie eingeleitet werden. Die Patienten müssen darüber informiert werden, dass solche Reaktionen möglich sind und sofortige medizinische Behandlung erforderlich ist, wenn allergische Reaktionen auftreten.
In klinischen Studien wurden bei einer geringen Anzahl von Patienten Antikörper gegen Omalizumab nachgewiesen (siehe Abschnitt 4.8). Die klinische Relevanz von Anti-Omalizumab-Antikörpern ist noch nicht gut verstanden.
Serumkrankheit
Bei Patienten, die mit humanisierten monoklonalen Antikörpern wie Omalizumab behandelt wurden, wurden Serumkrankheit und Serumkrankheit-ähnliche Reaktionen, die verzögerte allergische Typ-III-Reaktionen sind, festgestellt. Der vermutliche pathophysiologische Mechanismus beinhaltet die Bildung von Immunkomplexen und deren Ausfällung aufgrund der Entstehung von Antikörpern gegen Omalizumab. Das Auftreten der Symptome erfolgte typischerweise 1-5 Tage nach Verabreichung der ersten oder einer der folgenden Injektionen, aber auch nach längerer Dauer der Behandlung. Typische Symptome der Serumkrankheit sind Arthritis/Arthralgien, Ausschlag (Urtikaria oder andere Formen), Fieber und Lymphadenopathie. Zur Vorbeugung oder Behandlung dieser Erkrankung können Antihistaminika und Kortikosteroide verwendet werden. Patienten sollen angehalten werden, sämtliche vermuteten Symptome zu melden.
Churg-Strauss-Syndrom und hypereosinophiles Syndrom
Patienten mit schwerem allergischem Asthma können selten ein systemisches hypereosinophiles Syndrom oder eine allergische eosinophile granulomatöse Vaskulitis (Churg-Strauss-Syndrom) aufweisen, die beide üblicherweise mit systemischen Kortikosteroiden behandelt werden.
In seltenen Fällen können Patienten, die mit Arzneimitteln gegen Asthma einschließlich Omalizumab behandelt werden, eine systemische Eosinophilie oder Vaskulitis aufweisen oder entwickeln. Diese Ereignisse sind häufig mit der Reduktion einer oralen Kortikosteroid-Therapie vergesellschaftet.
Bei diesen Patienten sollten Ärzte mit der Entwicklung einer ausgeprägten Eosinophilie, eines vaskulitischen Exanthems, einer Verschlechterung pulmonaler Symptome, Anomalien der Nasennebenhöhlen, kardialen Komplikationen und/oder einer Neuropathie rechnen.
Das Absetzen von Omalizumab sollte bei allen schwerwiegenden Fällen der oben erwähnten Erkrankungen des Immunsystems in Erwägung gezogen werden.
Parasitäre (Wurm-)Infektionen
IgE kann in die Immunantwort auf manche Wurminfektionen involviert sein. In einer placebokontrollierten Studie an Patienten mit chronischem hohem Risiko für eine Wurminfektion zeigte sich bei allergischen Patienten ein geringer Anstieg der Infektionsrate unter Omalizumab, obgleich der Verlauf, die Schwere und das Ansprechen auf die Behandlung der Infektion unverändert waren. Die Häufigkeit der Wurminfektionen war im gesamten klinischen Programm, das nicht dafür ausgelegt war, solche Infektionen zu erfassen, kleiner als 1 von 1 000 Patienten. Bei Patienten mit einem hohen Risiko für eine Wurminfektion kann jedoch Vorsicht geboten sein, insbesondere bei Reisen in Gebiete mit endemischen Wurminfektionen. Wenn Patienten nicht auf die empfohlene Anti-Wurmbehandlung ansprechen, sollte ein Absetzen der Behandlung mit Omalizumab erwogen werden.
Sonstiger Bestandteil mit bekannter Wirkung
Jede 150-mg-Fertigspritze und jeder 150-mg-Fertigpen enthält 0,40 mg Polysorbat 20 (E 432) und jede 300-mg- Fertigspritze enthält 0,80 mg Polysorbat 20 (E 432), was 0,40 mg/ml entspricht. Polysorbate können allergische Reaktionen hervorrufen. Patienten mit einer Polysorbat-Allergie sollten dieses Arzneimittel nicht anwenden.
Da IgE an der Immunantwort auf manche Wurminfektionen beteiligt sein kann, kann Omalizumab indirekt die Wirksamkeit von Arzneimitteln zur Behandlung von Wurm- oder anderen parasitären Infektionen verringern (siehe Abschnitt 4.4).
Cytochrom-P450-Enzyme, Austausch-Pumpen und proteinbindende Mechanismen sind nicht an der Clearance von Omalizumab beteiligt; deshalb besteht nur eine geringe Wahrscheinlichkeit für Arzneimittel-Wechselwirkungen. Es wurden keine Studien zu Wechselwirkungen von Arzneimitteln oder Impfstoffen mit Omalizumab durchgeführt. Aus pharmakologischer Sicht besteht kein Grund, Wechselwirkungen von üblicherweise zur Behandlung von Asthma, CRSwNP oder csU verschriebenen Arzneimitteln mit Omalizumab zu erwarten.
Allergisches Asthma
In klinischen Studien wurde Omalizumab im Allgemeinen zusammen mit inhalativen und oralen Kortikosteroiden, inhalativen kurz- und langwirksamen Beta-Agonisten, Leukotrien-Rezeptorantagonisten, Theophyllinen und oralen Antihistaminika eingesetzt. Es gab keine Anzeichen, dass die Sicherheit von Omalizumab in Kombination mit diesen anderen, allgemein gegen Asthma verwendeten Arzneimitteln verändert war. Es sind begrenzte Daten zur Anwendung von Omalizumab in Kombination mit spezifischer Immuntherapie (Hyposensibilisierungstherapie) vorhanden. In einer klinischen Studie, in der Omalizumab zusammen mit einer Immuntherapie angewendet wurde, wurde festgestellt, dass die Sicherheit und Wirksamkeit von Omalizumab in Kombination mit einer spezifischen Immuntherapie nicht verschieden war von der von Omalizumab allein.
Chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
In klinischen Studien wurde Omalizumab in Verbindung mit intranasalem Mometasonspray gemäß Protokoll angewendet. Andere, häufig angewendete Begleitmedikationen umfassten andere intranasale Kortikosteroide, Bronchodilatatoren, Antihistaminika, Leukotrien-Rezeptorantagonisten, Adrenergika/Sympathomimetika und lokale Nasenanästhetika. Es gab keinen Hinweis darauf, dass die Sicherheit von Omalizumab durch die gleichzeitige Anwendung dieser häufig angewendeten Arzneimittel verändert war.
Chronische spontane Urtikaria (csU)
In klinischen Studien zur csU wurde Omalizumab gemeinsam mit Antihistaminika (Anti-H1, Anti-H2) und Leukotrien-Rezeptorantagonisten (Leukotriene receptor antagonists, LTRAs) angewendet. Es gab keinen Hinweis darauf, dass die Sicherheit von Omalizumab bei gemeinsamer Anwendung mit diesen Arzneimitteln gegenüber dem bekannten Sicherheitsprofil bei allergischem Asthma verändert war. Darüber hinaus zeigte sich in einer Analyse der Populationspharmakokinetik kein relevanter Effekt von H2-Antihistaminika und LTRAs auf die Pharmakokinetik von Omalizumab (siehe Abschnitt 5.2).
Kinder und Jugendliche
Klinische Studien zur csU schlossen einige Patienten im Alter von 12 bis 17 Jahren ein, die Omalizumab gemeinsam mit Antihistaminika (Anti-H1, Anti-H2) und LTRAs erhalten haben. Es wurden keine Studien mit Kindern unter 12 Jahren durchgeführt.
Schwangerschaft
Die überschaubare Menge an Daten über schwangere Frauen (zwischen 300 und 1 000 Schwangerschaftsverläufe), die auf einem Schwangerschaftsregister und Spontanberichten nach dem Inverkehrbringen basieren, zeigen keine Hinweise auf Fehlbildungen oder fötale/neonatale Toxizität. Eine prospektive Schwangerschafts-Registerstudie (EXPECT) an 250 schwangeren Frauen mit Asthma, die mit Omalizumab behandelt wurden, zeigte, dass die Prävalenz der wesentlichen kongenitalen Fehlbildungen zwischen EXPECT-Patientinnen und Asthma-Patientinnen allgemein (mittelschweres und schweres Asthma) vergleichbar war (8,1 % vs. 8,9 %). Die Interpretation der Daten kann aufgrund methodischer Einschränkungen, einschließlich einer geringen Stichprobengröße und eines nicht-randomisierten Designs, beeinträchtigt sein.
Omalizumab durchdringt die Plazentaschranke. Allerdings ergaben tierexperimentelle Studien keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen bezogen auf die Reproduktionstoxizität (siehe Abschnitt 5.3).
Omalizumab wurde mit altersabhängiger Abnahme von Blutplättchen bei nicht-humanen Primaten in Verbindung gebracht, mit einer höheren relativen Empfindlichkeit bei Jungtieren (siehe Abschnitt 5.3).
Bei klinischer Notwendigkeit kann die Anwendung von Omalizumab während der Schwangerschaft in Betracht gezogen werden.
Stillzeit
Immunglobuline G (IgGs) sind in der Muttermilch vorhanden und daher ist davon auszugehen, dass Omalizumab in die Muttermilch übergeht. Die zur Verfügung stehenden Daten von nicht-humanen Primaten zeigten, dass Omalizumab in die Milch übergeht (siehe Abschnitt 5.3).
Die EXPECT-Studie mit 154 Säuglingen, die während der Schwangerschaft und durch das Stillen Omalizumab ausgesetzt waren, zeigte keine negativen Auswirkungen auf das gestillte Kind. Die Interpretation der Daten kann aufgrund methodischer Einschränkungen, einschließlich einer geringen Stichprobengröße und eines nicht-randomisierten Designs, beeinträchtigt sein.
Oral verabreichte Immunglobulin-G-Proteine unterliegen im Darm einer Proteolyse und haben eine geringe Bioverfügbarkeit. Es sind keine Auswirkungen auf die gestillten Neugeborenen/Säuglinge zu erwarten. Folglich kann bei klinischer Notwendigkeit die Anwendung von Omalizumab während der Stillzeit in Betracht gezogen werden.
Fertilität
Es liegen keine Fertilitätsdaten für Omalizumab beim Menschen vor. In eigens geplanten präklinischen Fertilitätsstudien an nicht-humanen Primaten, einschließlich Fortpflanzungsstudien, wurde nach wiederholter Gabe von Omalizumab mit Dosierungen von bis zu 75 mg/kg keine Beeinträchtigung der männlichen oder weiblichen Fertilität beobachtet. Des Weiteren wurden in einer separaten präklinischen Genotoxizitätsstudie keine genotoxischen Effekte beobachtet.
Omalizumab hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Allergisches Asthma und chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
Zusammenfassung des Sicherheitsprofils
Während der klinischen Studien zu allergischem Asthma mit Erwachsenen und Jugendlichen ab 12 Jahren waren die am häufigsten berichteten Nebenwirkungen Kopfschmerzen und Reaktionen an der Injektionsstelle, einschließlich Schmerzen an der Injektionsstelle, Schwellungen, Erythem und Pruritus. In klinischen Studien mit Kindern im Alter von 6 bis <12 Jahren waren die am häufigsten berichteten Nebenwirkungen Kopfschmerzen, Fieber und Schmerzen im Oberbauch.
Die Schwere der meisten Reaktionen war leicht bis mittelschwer. In klinischen Studien zu CRSwNP mit Patienten ≥18 Jahren waren die am häufigsten berichteten Nebenwirkungen Kopfschmerzen, Schwindelgefühl, Arthralgie, Schmerzen im Oberbauch und Reaktionen an der Injektionsstelle.
Tabellarische Zusammenfassung von Nebenwirkungen
In Tabelle 4 sind gemäß MedDRA-Organklassen und Häufigkeiten die Nebenwirkungen aufgeführt, die in klinischen Studien in der mit Omalizumab behandelten gesamthaft überwachten Population mit allergischem Asthma und CRSwNP auftraten. Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben. Die Häufigkeitskategorien sind wie folgt definiert: Sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1 000, <1/100), selten (≥1/10 000, <1/1 000) und sehr selten (<1/10 000). Reaktionen, die nach der Markteinführung gemeldet wurden, sind mit der Häufigkeit nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar) aufgeführt.
Tabelle 4 Nebenwirkungen bei allergischem Asthma und CRSwNP
Infektionen und parasitäre Erkrankungen | |
Gelegentlich |
Pharyngitis |
Selten |
Parasitäre Infektion |
Erkrankungen des Blutes und des Lymphsystems | |
Nicht bekannt |
Idiopathische Thrombozytopenie, einschließlich schwerer Fälle |
Erkrankungen des Immunsystems | |
Selten |
Anaphylaktische Reaktion, andere schwerwiegende allergische Zustände, Bildung von Anti-Omalizumab-Antikörpern |
Nicht bekannt |
Serumkrankheit, eventuell mit Fieber und Lymphadenopathie |
Erkrankungen des Nervensystems | |
Häufig |
Kopfschmerzen* |
Gelegentlich |
Synkope, Parästhesie, Somnolenz, Schwindelgefühl # |
Gefäßerkrankungen | |
Gelegentlich |
Orthostasesyndrom, Flush |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
Gelegentlich |
Allergischer Bronchospasmus, Husten |
Selten |
Larynxödem |
Nicht bekannt |
Allergische granulomatöse Vaskulitis (Churg-Strauss-Syndrom) |
Erkrankungen des Gastrointestinaltrakts | |
Häufig |
Schmerzen im Oberbauch**,# |
Gelegentlich |
Klinische Zeichen und Symptome der Dyspepsie, Diarrhö, Übelkeit |
Erkrankungen der Haut und des Unterhautgewebes | |
Gelegentlich |
Lichtempfindlichkeit, Urtikaria, Ausschlag, Pruritus |
Selten |
Angioödem |
Nicht bekannt |
Haarausfall |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |
Häufig |
Arthralgie† |
Selten |
Systemischer Lupus erythematodes (Systemic lupus erythematosus, SLE) |
Nicht bekannt |
Muskelschmerzen, Gelenkschwellung |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Sehr häufig |
Fieber** |
Häufig |
Reaktionen an der Injektionsstelle wie Schwellung, Erythem, Schmerzen, Pruritus |
Gelegentlich |
Grippeähnliche Erkrankung, Anschwellen der Arme, Gewichtszunahme, Ermüdung/Fatigue |
*: Sehr häufig bei Kindern im Alter von 6 bis <12 Jahren
**: Bei Kindern im Alter von 6 bis <12 Jahren
#: Häufig in Studien zu Nasenpolypen
†: Häufigkeit nicht bekannt in Studien zu allergischem Asthma
Chronische spontane Urtikaria (csU)
Zusammenfassung des Sicherheitsprofils
Die Sicherheit und Verträglichkeit von Omalizumab wurden mit Dosen von 75 mg, 150 mg und 300 mg alle vier Wochen bei 975 csU-Patienten untersucht; 242 dieser Patienten erhielten Placebo. Insgesamt wurden 733 Patienten bis zu 12 Wochen lang mit Omalizumab behandelt und 490 Patienten bis zu 24 Wochen lang. Davon wurden 412 Patienten für bis zu 12 Wochen und 333 Patienten bis zu 24 Wochen lang mit der 300-mg-Dosis behandelt.
Tabellarische Zusammenfassung von Nebenwirkungen
Eine separate Tabelle (Tabelle 5) zeigt die Nebenwirkungen in der Indikation csU infolge von Unterschieden in der Dosis und den Behandlungsgruppen (mit deutlichen Unterschieden bezüglich Risikofaktoren, Komorbiditäten, Begleitmedikationen und Alter [z. B. schlossen Asthma-Studien Kinder im Alter von 6 bis 12 Jahren ein]).
Tabelle 5 enthält eine Auflistung der Nebenwirkungen (Ereignisse, die bei ≥1 % der Patienten in jeder Behandlungsgruppe auftraten und ≥2 % häufiger in den Omalizumab-Behandlungsgruppen beobachtet wurden als unter Placebo (nach medizinischer Auswertung)), die in den drei zusammengefassten Phase-III-Studien unter 300 mg verzeichnet wurden. Die aufgelisteten Nebenwirkungen sind in zwei Gruppen unterteilt: Nebenwirkungen, die während des 12-wöchigen bzw. des 24-wöchigen Behandlungszeitraums registriert wurden.
Die Nebenwirkungen sind nach MedDRA-Systemorganklassen aufgeführt. Innerhalb jeder Systemorganklasse sind die Nebenwirkungen nach abnehmender Häufigkeit geordnet. Die jeweiligen Häufigkeitskategorien der Nebenwirkungen beruhen auf der folgenden Konvention: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1 000, <1/100); selten (≥1/10 000, <1/1 000); sehr selten (<1/10 000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 5 Nebenwirkungen aus der zusammengefassten csU-Sicherheitsdatenbank (Tag 1 bis Woche 24) unter 300 mg Omalizumab
12-Wochen |
Zusammengefasste Omalizumab-Studien 1, 2 und 3 |
Häufigkeitskategorie |
||
Placebo n=242 |
300 mg n=412 |
|||
Infektionen und parasitäre Erkrankungen | ||||
Sinusitis |
5 (2,1 %) |
20 (4,9 %) |
Häufig |
|
Erkrankungen des Nervensystems | ||||
Kopfschmerzen |
7 (2,9 %) |
25 (6,1 %) |
Häufig |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | ||||
Arthralgie |
1 (0,4 %) |
12 (2,9 %) |
Häufig |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | ||||
Reaktionen an der Injektionsstelle* |
2 (0,8 %) |
11 (2,7 %) |
Häufig |
|
24-Wochen |
Zusammengefasste Omalizumab-Studien 1 und 3 |
Häufigkeitskategorie |
||
Placebo n=163 |
300 mg n=333 |
|||
Infektionen und parasitäre Erkrankungen | ||||
Infektionen der oberen Atemwege |
5 (3,1 %) |
19 (5,7 %) |
Häufig |
|
*Trotz eines nicht erreichten Unterschiedes von 2 % im Vergleich zu Placebo wurden alle Reaktionen an der Injektionsstelle einbezogen, da ein ursächlicher Zusammenhang zwischen der Studienbehandlung und den aufgetretenen Fällen angenommen wurde.
In einer 48-Wochen-Studie erhielten 81 csU-Patienten 300 mg Omalizumab alle 4 Wochen (siehe Abschnitt 5.1). Das Sicherheitsprofil bei Langzeitanwendung war ähnlich dem Sicherheitsprofil der 24-Wochen-csU-Studien.
Beschreibung ausgewählter Nebenwirkungen
Erkrankungen des Immunsystems
Weitere Informationen siehe Abschnitt 4.4.
Anaphylaxie
Anaphylaktische Reaktionen traten in klinischen Studien selten auf. Jedoch ergaben Daten nach der Markteinführung, basierend auf einer kumulativen Suche in der Sicherheitsdatenbank, eine Gesamtanzahl von 898 Anaphylaxie-Fällen. Basierend auf einer geschätzten Exposition von 566 923 Patientenbehandlungsjahren führt dies zu einer Melderate von ungefähr 0,20 %.
Arterielle thromboembolische Ereignisse (Arterial thromboembolic events, ATE)
In kontrollierten klinischen Studien und bei Interimsanalysen einer Beobachtungsstudie wurde ein numerisches Ungleichgewicht von ATEs beobachtet. Die Definition des kombinierten Endpunkts ATE beinhaltete Schlaganfall, transitorische ischämische Attacke, Herzinfarkt, instabile Angina Pectoris und kardiovaskulären Tod (einschließlich Tod unbekannter Ursache). In der Abschlussanalyse der Beobachtungsstudie war die Rate an ATEs pro 1 000 Patientenjahren 7,52 (115/15 286 Patientenjahre) für mit Omalizumab behandelte Patienten und 5,12 (51/9 963 Patientenjahre) für Kontrollpatienten. In einer multivariaten Analyse lag unter Berücksichtigung der vorliegenden kardiovaskulären Grund-Risikofaktoren die Hazard-Ratio bei 1,32 (95 %-Konfidenzintervall 0,91-1,91). In einer gesonderten gepoolten Analyse, die alle randomisierten, doppelblinden, placebokontrollierten klinischen Studien mit einer Dauer von 8 Wochen oder länger einschloss, betrug die Rate für ATE pro 1 000 Patientenjahre 2,69 (5/1 856 Patientenjahre) bei mit Omalizumab behandelten Patienten und 2,38 (4/1 680 Patientenjahre) für Placebo-Patienten (Verhältnis der Häufigkeiten 1,13, 95 %-Konfidenzintervall 0,24-5,71).
Blutplättchen
In den klinischen Studien war bei wenigen Patienten die Anzahl der Blutplättchen unterhalb des Normalbereiches. Nach der Markteinführung wurden einzelne Fälle von idiopathischer Thrombozytopenie, einschließlich schwerer Fälle, berichtet.
Parasitäre Infektionen
In einer placebokontrollierten Studie an allergischen Patienten mit chronischem, hohem Risiko für eine Wurminfektion zeigte sich ein geringer Anstieg der Infektionsrate mit Omalizumab, der nicht statistisch signifikant war. Der Verlauf, die Schwere und das Ansprechen auf die Behandlung der Infektion waren nicht beeinflusst (siehe Abschnitt 4.4).
Systemischer Lupus erythematodes
In klinischen Studien und nach der Markteinführung wurden Fälle von systemischem Lupus erythematodes (SLE) bei Patienten mit mittelschwerem bis schwerem Asthma und bei Patienten mit csU berichtet. Über die Pathogenese von SLE ist wenig bekannt.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de, anzuzeigen.
Die maximal tolerierte Dosis von Omlyclo wurde nicht ermittelt. Intravenöse Einzeldosen bis zu 4 000 mg wurden Patienten verabreicht ohne Anzeichen von dosislimitierender Toxizität. Die höchste an Patienten kumulativ verabreichte Dosis betrug 44 000 mg über einen Zeitraum von 20 Wochen und dies führte zu keinen ungünstigen Akuteffekten.
Falls eine Überdosierung vermutet wird, sollte der Patient hinsichtlich abnormer Anzeichen oder Symptome überwacht werden. Eine medizinische Behandlung sollte erfolgen und entsprechend eingeleitet werden.
Pharmakotherapeutische Gruppe: Mittel bei obstruktiven Atemwegserkrankungen, andere Mittel bei obstruktiven Atemwegserkrankungen zur systemischen Anwendung, ATC-Code: R03DX05.
Omlyclo ist ein biologisch / biotechnologisch hergestelltes Arzneimittel, das im Wesentlichen einem bereits zugelassenen Arzneimittel gleicht. Ausführliche Informationen sind auf den Internetseiten der Europäischen Arzneimittel-Agentur
https://www.ema.europa.eu verfügbar.
Allergisches Asthma und chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
Wirkmechanismus
Omalizumab ist ein rekombinanter, aus DNA abgeleiteter, humanisierter monoklonaler Antikörper, der selektiv an das menschliche Immunglobulin E (IgE) bindet und somit die Bindung von IgE an den FcεRI (hochaffiner IgE-Rezeptor) auf Basophilen und Mastzellen verhindert, wodurch die Menge an freiem IgE reduziert wird, das zum Auslösen der allergischen Kaskade verfügbar ist. Es handelt sich um einen IgG1kappa-Antikörper mit einem humanen Grundgerüst, dessen komplementaritätsbestimmende Regionen muriner Herkunft sind und an IgE binden.
Die Behandlung von atopischen Patienten mit Omalizumab führt zu einer merklichen Herabregulation der FcεRI-Rezeptordichte auf den Basophilen. Omalizumab hemmt die IgE-vermittelte Entzündung, belegt durch eine reduzierte Anzahl an Blut- und Gewebe-Eosinophilen und reduzierte Entzündungsmediatoren, einschließlich IL-4, IL-5 und IL-13 durch angeborene, adaptive und Nicht-Immunzellen.
Pharmakodynamische Wirkungen
Allergisches Asthma
Die In-vitro-Histamin-Freisetzung aus Basophilen, die von mit Omalizumab behandelten Patienten isoliert wurden, war nach Stimulation mit einem Allergen um etwa 90 % reduziert im Vergleich zu den Werten vor der Behandlung.
In klinischen Studien an Patienten mit allergischem Asthma wurde der Serum-Spiegel an freiem IgE dosisabhängig innerhalb 1 Stunde nach der ersten Dosis reduziert und zwischen den Verabreichungen beibehalten. Ein Jahr nach Absetzen von Omalizumab kehrten die IgE-Spiegel zu den Werten vor der Behandlung zurück, wobei nach dem Auswaschen des Arzneimittels kein Rebound beobachtet wurde.
Chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
In klinischen Studien an Patienten mit CRSwNP führte die Omalizumab-Behandlung zu einer Verringerung des freien IgE im Serum (ca. 95 %) und zu einem Anstieg des Gesamt-IgE-Spiegels im Serum, und zwar in einem ähnlichen Ausmaß, wie es bei Patienten mit allergischem Asthma beobachtet wurde. Die Gesamt-IgE-Spiegel im Serum stiegen aufgrund der Bildung von Omalizumab-IgE-Komplexen, die im Vergleich zum freien IgE eine langsamere Eliminationsrate aufweisen, an.
Chronische spontane Urtikaria (csU)
Wirkmechanismus
Omalizumab ist ein rekombinanter, aus DNA abgeleiteter, humanisierter monoklonaler Antikörper, der selektiv an das menschliche Immunglobulin E (IgE) bindet und die Menge an freiem IgE senkt. Es handelt sich um einen IgG1kappa-Antikörper mit einem humanen Grundgerüst, dessen komplementaritätsbestimmende Regionen muriner Herkunft sind und an IgE binden. Somit kommt es zur Herunterregulierung von IgE-Rezeptoren (FcεRI) auf den Zellen. Wie dies zu einer Linderung von csU-Symptomen führt, ist bislang noch nicht vollständig aufgeklärt.
Pharmakodynamische Wirkungen
In klinischen Studien mit csU-Patienten wurde die maximale Suppression des freien IgE 3 Tage nach der ersten subkutanen Dosis beobachtet. Nach wiederholter Verabreichung einmal alle 4 Wochen blieben die Spiegel des freien IgE vor der Gabe zwischen der 12. und 24. Behandlungswoche stabil. Nach dem Absetzen von Omalizumab stieg der Serumspiegel des freien IgE über einen 16-wöchigen behandlungsfreien Nachbeobachtungszeitraum wieder auf die prätherapeutischen Spiegel an.
Klinische Wirksamkeit und Sicherheit
Allergisches Asthma
Erwachsene und Jugendliche ab 12 Jahren
Die Wirksamkeit und Verträglichkeit von Omalizumab wurde in einer doppelblinden placebokontrollierten Studie über 28 Wochen (Studie 1) nachgewiesen, die 419 Patienten mit schwerem allergischem Asthma im Alter von 12-79 Jahren einschloss. Die Patienten hatten eine reduzierte Lungenfunktion (FEV1 40-80 % des Referenzwertes) und wiesen trotz einer Therapie mit hoch dosierten inhalativen Kortikosteroiden und einem langwirkenden inhalativen Beta2-Agonisten eine schlechte Kontrolle der Asthma-Symptome auf. Geeignete Patienten hatten im letzten Jahr trotz einer kontinuierlichen Behandlung mit hoch dosierten inhalativen Kortikosteroiden und einem langwirkenden inhalativen Beta2-Agonisten mehrere Asthma-Exazerbationen erfahren, die eine Behandlung mit systemischen Kortikosteroiden nötig machten, oder wurden wegen einer schweren Asthma-Exazerbation hospitalisiert oder waren in einer Notfallambulanz. Omalizumab oder Placebo wurde subkutan verabreicht als Zusatz-Therapie zu >1 000 Mikrogramm Beclometasondipropionat (oder Äquivalent) und einem langwirkenden Beta2-Agonisten. Erhaltungstherapien mit oralen Kortikoiden, Theophyllin und Leukotrien-Rezeptorantagonisten waren erlaubt (je 22 %, 27 % und 35 % der Patienten).
Den primären Endpunkt stellte die Rate der Asthma-Exazerbationen dar, bei denen eine Akut-Behandlung mit systemischen Kortikosteroiden nötig war. Omalizumab reduzierte die Rate der Asthma-Exazerbationen um 19 % (p = 0,153). Weitere Auswertungen, die statistische Signifikanz (p <0,05) zu Gunsten von Omalizumab zeigten, beinhalteten die Reduzierung von schweren Exazerbationen (bei denen die Lungenfunktion des Patienten auf weniger als 60 % des persönlichen Bestwertes reduziert war und systemische Kortikosteroide benötigt wurden) und asthmabedingtes Aufsuchen einer Notfallambulanz (einschließlich Hospitalisierungen, Notfallambulanz und nicht geplante Arztbesuche) sowie Verbesserungen der ärztlichen Gesamtbewertung der Wirksamkeit der Behandlung, der Lebensqualität bezüglich Asthma (Asthma quality of life questionnaire, AQL), der Asthmasymptome und der Lungenfunktion.
In einer Subgruppenanalyse bei Patienten mit einem IgE-Gesamtwert ≥76 I.E./ml vor der Behandlung war ein klinisch relevanter Nutzen von Omalizumab wahrscheinlicher. Bei diesen Patienten reduzierte Omalizumab in Studie 1 die Asthma-Exazerbationsrate um 40 % (p = 0,002). Zusätzlich zeigten im Studienprogramm zu Omalizumab bei schwerem Asthma in der Population mit einem IgE-Gesamtwert ≥76 I.E./ml mehr Patienten ein klinisch relevantes Ansprechen. Tabelle 6 beinhaltet die Ergebnisse für die Gesamtpopulation der Studie 1.
Tabelle 6 Ergebnisse der Studie 1
Gesamtpopulation der Studie 1 |
||
Omalizumab |
Placebo |
|
n=209 |
n=210 |
|
Asthma-Exazerbationen | ||
Häufigkeit pro 28 Wochen |
0,74 |
0,92 |
% Reduktion, p-Wert für Verhältnis der Häufigkeiten |
19,4 %, p = 0,153 |
|
Schwere Asthma-Exazerbationen | ||
Häufigkeit pro 28 Wochen |
0,24 |
0,48 |
% Reduktion, p-Wert für Verhältnis der Häufigkeiten |
50,1 %, p = 0,002 |
|
Notfallambulanzbesuche | ||
Häufigkeit pro 28 Wochen |
0,24 |
0,43 |
% Reduktion, p-Wert für Verhältnis der Häufigkeiten |
43,9 %, p = 0,038 |
|
Ärztliche Gesamtbewertung | ||
% Responder* |
60,5 % |
42,8 % |
p-Wert** |
<0,001 |
|
AQL-Verbesserungen | ||
% Patienten mit einer Verbesserung ≥0,5 |
60,8 % |
47,8 % |
p-Wert |
0,008 |
|
* merkliche Verbesserung oder vollständige Kontrolle
** p-Wert für die allgemeine Verteilung der Bewertung
In Studie 2 wurden die Wirksamkeit und Verträglichkeit von Omalizumab in einer Population von 312 Patienten mit schwerem allergischem Asthma untersucht, die der Population in Studie 1 entspricht. In dieser offenen Studie führte die Behandlung mit Omalizumab zu einer 61%igen Reduktion der klinisch relevanten Exazerbationsrate im Vergleich zur gängigen Asthma-Therapie alleine.
In vier weiteren großen placebokontrollierten unterstützenden Studien mit einer Dauer von 28 bis 52 Wochen mit 1 722 Erwachsenen und Jugendlichen (Studien 3, 4, 5, 6) wurden die Wirksamkeit und Verträglichkeit von Omalizumab bei Patienten mit schwerem persistierendem Asthma untersucht. Die meisten Patienten waren ungenügend kontrolliert, erhielten jedoch weniger Begleitmedikation für Asthma als Patienten in Studie 1 oder 2. Die Studien 3-5 hatten Exazerbationen als primären Endpunkt, wogegen Studie 6 primär das Einsparen von inhalativen Kortikosteroiden ermittelte.
In den Studien 3, 4 und 5 hatten die mit Omalizumab behandelten Patienten jeweils Reduktionen der Exazerbationsraten von 37,5 % (p = 0,027), 40,3 % (p <0,001) bzw. 57,6 % (p <0,001) im Vergleich zu Placebo.
In Studie 6 waren signifikant mehr Patienten mit schwerem allergischem Asthma unter Omalizumab in der Lage, ohne Verschlechterung der Asthma-Kontrolle ihre Fluticason-Dosis auf ≤500 Mikrogramm/Tag zu reduzieren (60,3 %), im Vergleich zur Placebo-Gruppe (45,8 %, p <0,05).
Die Lebensqualität wurde mit Hilfe des asthmabezogenen Lebensqualitäts-Fragebogens nach Juniper ermittelt. Bei allen sechs Studien zeigte sich bei Omalizumab-Patienten im Vergleich zur Placebo- oder Kontrollgruppe eine statistisch signifikante Verbesserung der Lebensqualität.
Ärztliche Gesamtbewertung der Wirksamkeit der Behandlung:
Eine ärztliche Gesamtbewertung wurde in fünf der oben angeführten Studien als eine umfassende Beurteilung der Asthma-Kontrolle durch den behandelnden Arzt durchgeführt. Der Arzt konnte exspiratorischer Peak-Flow (peak expiratory flow, PEF), Tages- und Nachtsymptome, Gebrauch von Notfallmedikation, Spirometrie und Exazerbationen berücksichtigen. In allen fünf Studien wurde im Vergleich zu Placebo-Patienten ein signifikant größerer Teil der mit Omalizumab behandelten Patienten eingeschätzt, entweder eine merkliche Verbesserung oder eine völlige Kontrolle ihrer Asthma-Symptome erreicht zu haben.
Kinder im Alter von 6 bis <12 Jahren
Die grundlegenden Daten für die Sicherheit und Wirksamkeit von Omalizumab in der Altersgruppe von 6 bis <12 Jahren stammen aus einer randomisierten, doppelblinden, placebokontrollierten multizentrischen Studie (Studie 7).
Studie 7 war eine placebokontrollierte Studie mit einer spezifischen Subgruppe (n=235) von Patienten nach derzeitiger Indikation, die mit hoch dosierten inhalativen Kortikosteroiden (≥500 µg Fluticason-Äquivalent/Tag) und langwirksamen Beta-Agonisten behandelt wurden.
Eine klinisch signifikante Exazerbation wurde definiert als eine vom Prüfarzt klinisch beurteilte Verschlechterung der Asthmasymptome, die eine Verdopplung der Ausgangsdosis des inhalativen Kortikosteroids für mindestens 3 Tage und/oder einer Notfallbehandlung mit systemischen (oral oder intravenös) Kortikosteroiden für mindestens 3 Tage erforderte.
Bei der spezifischen Subgruppe von Patienten, die hoch dosierte inhalative Kortikosteroide erhielten, zeigte die Omalizumab-Gruppe eine statistisch signifikant niedrigere Rate an klinisch signifikanten Asthma-Exazerbationen als die Placebo-Gruppe. Nach 24 Wochen wurde bei der Betrachtung der Differenzen der Raten für die Omalizumab-Patienten eine um 34 % (Verhältnis der Raten 0,662, p = 0,047) geringere Rate im Verhältnis zu Placebo erzielt. Im zweiten doppelblinden, 28-wöchigen Behandlungszeitraum wurde bei der Betrachtung der Differenzen der Raten für die Omalizumab-Patienten eine um 63 % (Verhältnis der Raten 0,37, p <0,001) geringere Rate im Verhältnis zu Placebo erzielt.
Während der 52-wöchigen, doppelblinden Behandlung (bestehend aus der 24-wöchigen Phase mit konstanter Steroid-Dosis und der 28-wöchigen Phase mit angepasster Steroid-Dosis) zeigten die Differenzen der Raten zwischen den Behandlungsgruppen eine 50%ige (Verhältnis der Raten 0,504, p <0,001) Abnahme der Exazerbationen für Omalizumab-Patienten.
Die Omalizumab-Gruppe zeigte am Ende der 52-wöchigen Behandlung eine größere Abnahme des Gebrauchs von Beta-Agonisten als Notfallmedikation als die Placebo-Gruppe, auch wenn der Unterschied zwischen den Behandlungsgruppen nicht statistisch signifikant war. In der Gesamtauswertung der Wirksamkeit nach 52-wöchiger, doppelblinder Behandlung war in der Untergruppe der schwer erkrankten Patienten mit hoch dosierten inhalativen Kortikosteroiden und gleichzeitigen langwirksamen Beta-Agonisten der Anteil der Patienten mit „exzellentem“ Behandlungserfolg bei der Omalizumab-Gruppe höher als bei der Placebo-Gruppe. Die Anteile der Patienten mit „moderatem“ oder „schlechtem“ Behandlungserfolg waren in der Omalizumab-Gruppe geringer als bei der Placebo-Gruppe. Die Unterschiede zwischen den Gruppen waren statistisch signifikant (p <0,001). Bei den subjektiven Patientenbewertungen ihrer Lebensqualität gab es keine Unterschiede zwischen der Omalizumab-Gruppe und der Placebo-Gruppe.
Chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
Die Sicherheit und Wirksamkeit von Omalizumab wurde in zwei randomisierten, doppelblinden, placebokontrollierten Studien an Patienten mit CRSwNP untersucht (Tabelle 8). Die Patienten erhielten Omalizumab oder Placebo subkutan alle 2 oder 4 Wochen (siehe Abschnitt 4.2). Alle Patienten erhielten während der gesamten Studie eine intranasale Mometason-Hintergrundtherapie. Ein vorheriger chirurgischer Eingriff an der Nasenhöhle oder den Nasennebenhöhlen oder die vorherige Behandlung mit systemischen Kortikosteroiden war für den Einschluss in die Studien nicht erforderlich. Die Patienten erhielten 24 Wochen lang Omalizumab oder Placebo, gefolgt von einer 4‑wöchigen Nachbeobachtungsphase. Demografische Angaben und Charakteristika zu Studienbeginn, einschließlich allergischer Komorbiditäten, sind in Tabelle 7 beschrieben.
Tabelle 7 Demografische Angaben und Charakteristika zu Studienbeginn in den Nasenpolypenstudien
Parameter |
Nasenpolypenstudie 1 |
Nasenpolypenstudie 2 |
n=138 |
n=127 |
|
Mittleres Alter (in Jahren) (SD) |
51,0 (13,2) |
50,1 (11,9) |
% Männlich |
63,8 |
65,4 |
Patienten mit systemischer Kortikosteroid-Behandlung im Vorjahr (%) |
18,8 |
26,0 |
Bilateraler endoskopischer Nasenpolypenscore (NPS): Mittelwert (SD), Bereich 0-8 |
6,2 (1,0) |
6,3 (0,9) |
Score für nasale Kongestion (NCS): Mittelwert (SD), Bereich 0-3 |
2,4 (0,6) |
2,3 (0,7) |
Score des Geruchssinns (sense of smell score): Mittelwert (SD), Bereich 0-3 |
2,7 (0,7) |
2,7 (0,7) |
SNOT-22 Gesamtscore: Mittelwert (SD) Bereich 0-110 |
60,1 (17,7) |
59,5 (19,3) |
Blut-Eosinophile (Zellen/µl): Mittelwert (SD) |
346,1 (284,1) |
334,6 (187,6) |
Gesamt-IgE I.E./ml: Mittelwert (SD) |
160,9 (139,6) |
190,2 (200,5) |
Asthma (%) |
53,6 |
60,6 |
leicht (%) |
37,8 |
32,5 |
mittelschwer (%) |
58,1 |
58,4 |
schwer (%) |
4,1 |
9,1 |
Aspirin-Intoleranz-Syndrom |
19,6 |
35,4 |
Allergische Rhinitis |
43,5 |
42,5 |
SD = Standardabweichung (Standard Deviation); SNOT-22 = Sino-nasaler Ergebnistest, bestehend aus 22 Fragen (Sino-Nasal Outcome Test 22 Questionnaire); IgE = Immunoglobulin E; I.E. = Internationale Einheiten. Bei NPS, NCS und SNOT-22 deuten höhere Werte auf einen höheren Schweregrad der Erkrankung hin.
Die co-primären Endpunkte waren der bilaterale Nasenpolypenscore (NPS) und der durchschnittliche tägliche Score für nasale Kongestion (NCS) in Woche 24. In den beiden Nasenpolypenstudien 1 und 2 zeigten sich bei Patienten, die Omalizumab erhielten, in Woche 24 statistisch signifikant größere Verbesserungen des NPS und des wöchentlichen Durchschnitts des NCS gegenüber dem Ausgangswert im Vergleich zu Patienten, die ein Placebo erhielten. Die Ergebnisse der Nasenpolypenstudien 1 und 2 sind in Tabelle 8 dargestellt.
Tabelle 8 Veränderung der klinischen Scores der Nasenpolypenstudie 1, der Nasenpolypenstudie 2 und der gepoolten Daten gegenüber den Ausgangswerten in Woche 24
Nasenpolypenstudie 1 |
Nasenpolypenstudie 2 |
Gepoolte Ergebnisse Nasenpolypen |
|||||||
Placebo |
Omalizumab |
Placebo |
Omalizumab |
Placebo |
Omalizumab |
||||
n |
66 |
72 |
65 |
62 |
131 |
134 |
|||
Nasenpolypenscore |
|||||||||
Mittelwert des Ausgangswertes |
6,32 |
6,19 |
6,09 |
6,44 |
6,21 |
6,31 |
|||
Änderung des LS-Mittelwertes in Woche 24 |
0,06 |
-1,08 |
-0,31 |
-0,90 |
-0,13 |
-0,99 |
|||
Differenz (95 %-KI) |
-1,14 (-1,59; -0,69) |
-0,59 (-1,05; -0,12) |
-0,86 (-1,18; -0,54) |
||||||
p-Wert |
<0,0001 |
0,0140 |
<0,0001 |
||||||
7-Tagesdurchschnitt des täglichen Scores für nasale Kongestion |
|||||||||
Mittelwert des Ausgangswertes |
2,46 |
2,40 |
2,29 |
2,26 |
2,38 |
2,34 |
|||
Änderung des LS-Mittelwertes in Woche 24 |
-0,35 |
-0,89 |
-0,20 |
-0,70 |
-0,28 |
-0,80 |
|||
Differenz (95 %-KI) |
-0,55 (-0,84; -0,25) |
-0,50 (-0,80; -0,19) |
-0,52 (-0,73; -0,31) |
||||||
p-Wert |
0,0004 |
0,0017 |
<0,0001 |
||||||
TNSS |
|||||||||
Mittelwert des Ausgangswertes |
9,33 |
8,56 |
8,73 |
8,37 |
9,03 |
8,47 |
|||
Änderung des LS-Mittelwertes in Woche 24 |
-1,06 |
-2,97 |
-0,44 |
-2,53 |
-0,77 |
-2,75 |
|||
Differenz (95 %-KI) |
-1,91 (-2,85; -0,96) |
-2,09 (-3,00; -1,18) |
-1,98 (-2,63; -1,33) |
||||||
p-Wert |
0,0001 |
<0,0001 |
<0,0001 |
||||||
SNOT‑22 |
|||||||||
Mittelwert des Ausgangswertes |
60,26 |
59,82 |
59,80 |
59,21 |
60,03 |
59,54 |
|||
Änderung des LS-Mittelwertes in Woche 24 |
-8,58 |
-24,70 |
-6,55 |
-21,59 |
-7,73 |
-23,10 |
|||
Differenz (95 %-KI) |
-16,12 (-21,86; -10,38) |
-15,04 (-21,26; -8,82) |
-15,36 (-19,57; -11,16) |
||||||
p-Wert |
<0,0001 |
<0,0001 |
<0,0001 |
||||||
(MID = 8,9) |
|||||||||
UPSIT |
|||||||||
Mittelwert des Ausgangswertes |
13,56 |
12,78 |
13,27 |
12,87 |
13,41 |
12,82 |
|||
Änderung des LS-Mittelwertes in Woche 24 |
0,63 |
4,44 |
0,44 |
4,31 |
0,54 |
4,38 |
|||
Differenz (95 %-KI) |
3,81 (1,38; 6,24) |
3,86 (1,57; 6,15) |
3,84 (2,17; 5,51) |
||||||
p-Wert |
0,0024 |
0,0011 |
<0,0001 |
||||||
LS = Methode der kleinsten Quadrate (least-square); KI = Konfidenzintervall; TNSS = Nasaler Gesamtsymptomscore (Total nasal symptom score); SNOT-22 = Sino-nasaler Ergebnistest, bestehend aus 22 Fragen (Sino-Nasal Outcome Test 22 Questionnaire); UPSIT = Riechtest der Universität von Pennsylvania (University of Pennsylvania Smell Identification Test); MID = Minimaler bedeutsamer Unterschied (minimal important difference).
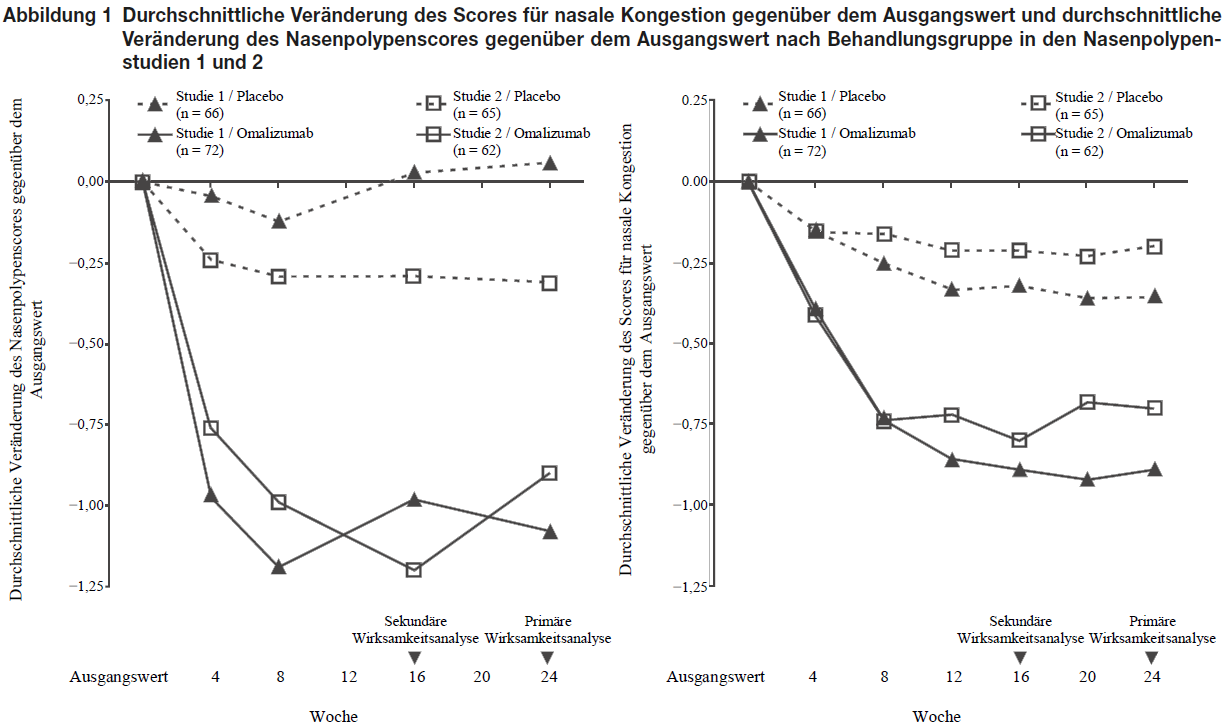
In einer präspezifizierten gepoolten Analyse der Notfallbehandlung (systemische Kortikosteroide für ≥3 aufeinander folgende Tage oder nasale Polypektomie) während der 24-wöchigen Behandlungsdauer war der Anteil der Patienten, die eine Notfallbehandlung benötigten, bei Omalizumab im Vergleich zu Placebo niedriger (2,3 % gegenüber 6,2 %). Das relative Risiko (odds ratio) einer Notfallbehandlung bei Omalizumab im Vergleich zu Placebo betrug 0,38 (95 %-KI: 0,10; 1,49). In beiden Studien wurden keine sino-nasalen Eingriffe berichtet.
Die langfristige Wirksamkeit und Sicherheit von Omalizumab bei Patienten mit CRSwNP, die an den Nasenpolypenstudien 1 und 2 teilgenommen hatten, wurden in einer Open-Label-Extension-Studie untersucht. Die Wirksamkeitsdaten aus dieser Studie deuten darauf hin, dass der klinische Nutzen, der sich in Woche 24 einstellte, bis Woche 52 anhielt. Die Sicherheitsdaten stimmten insgesamt mit dem bekannten Sicherheitsprofil von Omalizumab überein.
Chronische spontane Urtikaria (csU)
Die Wirksamkeit und Sicherheit von Omalizumab wurde in zwei randomisierten, placebokontrollierten Phase-III-Studien (Studie 1 und 2) an csU-Patienten nachgewiesen, die trotz Behandlung mit H1-Antihistaminika in der zugelassenen Dosis symptomatisch blieben. Eine dritte Studie (Studie 3) untersuchte primär die Sicherheit von Omalizumab bei csU-Patienten, die trotz Behandlung mit H1-Antihistaminika (bis zu dem Vierfachen der zugelassenen Dosis) und H2-Antihistaminika und/oder LTRAs symptomatisch blieben. In die drei Studien wurden 975 Patienten im Alter von 12 bis 75 Jahren eingeschlossen (mittleres Alter 42,3 Jahre; 39 Patienten 12-17 Jahre, 54 Patienten ≥65 Jahre; 259 Männer und 716 Frauen). Alle Patienten mussten an den 7 Tagen vor der Randomisierung eine unzureichende Symptomkontrolle aufweisen, trotz Anwendung eines Antihistaminikums während mindestens 2 vorangegangener Wochen. Als unzureichend wurde ein wöchentlicher Urtikaria-Aktivitäts-Wert (UAS7; Bereich 0-42) von ≥16 und ein wöchentlicher Wert für den Schweregrad des Juckreizes (der eine Komponente des UAS7 darstellt; Bereich 0-21) von ≥8 eingestuft.
In den Studien 1 und 2 wiesen die Patienten einen mittleren wöchentlichen Wert für den Schweregrad des Juckreizes zwischen 13,7 und 14,5 zu Studienbeginn sowie einen mittleren UAS7-Wert von 29,5 bzw. 31,7 auf. Patienten in der Sicherheitsstudie 3 zeigten zu Studienbeginn einen mittleren wöchentlichen Wert für den Schweregrad des Juckreizes von 13,8 und einen mittleren UAS7-Wert von 31,2. Die Patienten aller drei Studien gaben an, vor Aufnahme in die Studie im Durchschnitt 4 bis 6 Arzneimittel (einschließlich H1-Antihistaminika) zur Behandlung ihrer csU-Symptome erhalten zu haben. Die Patienten erhielten in den Studien 1 und 2 Omalizumab in einer Dosis von 75 mg, 150 mg oder 300 mg oder Placebo als subkutane Injektion alle 4 Wochen über 24 bzw. 12 Wochen und in Studie 3 300 mg oder Placebo als subkutane Injektion alle 4 Wochen über 24 Wochen. Alle Studien beinhalteten eine 16-wöchige behandlungsfreie Nachbeobachtungsphase.
Der primäre Endpunkt war die Veränderung des wöchentlichen Wertes für den Schweregrad des Juckreizes zwischen Studienbeginn und Woche 12. Unter der 300 mg Dosierung reduzierte Omalizumab den wöchentlichen Wert für den Schweregrad des Juckreizes um 8,55 bis 9,77 (p<0,0001) im Vergleich zu einer Reduktion um 3,63 bis 5,14 unter Placebo (siehe Tabelle 9). Statistisch signifikante Ergebnisse wurden darüber hinaus bei den Responder-Raten für UAS7≤6 (in Woche 12) erhalten, die für die Behandlung mit 300 mg bei 52-66 % (p<0,0001) lagen und somit im Vergleich zu 11-19 % in den Placebogruppen höher ausfielen. Ein vollständiges Ansprechen (UAS7=0) wurde bei 34-44 % (p<0,0001) der Patienten, die mit 300 mg behandelt wurden, erreicht, im Vergleich zu 5-9 % der Patienten in den Placebogruppen. Patienten mit einer 300 mg-Behandlung erreichten den höchsten mittleren Anteil an angioödemfreien Tagen von Woche 4 bis Woche 12 (91,0-96,1 %; p<0,001) verglichen mit der Placebogruppe (88,1-89,2 %). In den Behandlungsgruppen mit 300 mg war eine stärkere mittlere Veränderung des allgemeinen DLQI zwischen Studienbeginn und Woche 12 (p<0,001) zu verzeichnen als in den Placebogruppen; so zeigte sich in den Verumgruppen eine Verbesserung in einem Bereich von 9,7-10,3 Punkten, verglichen mit 5,1-6,1 Punkten in den entsprechenden Placebogruppen.
Tabelle 9 Veränderung des wöchentlichen Wertes für den Schweregrad des Juckreizes zwischen Studienbeginn und Woche 12; Studien 1, 2 und 3 (mITT-Population*)
Placebo |
Omalizumab 300 mg |
|
Studie 1 |
||
N |
80 |
81 |
Mittelwert (SD) |
−3,63 (5,22) |
−9,40 (5,73) |
Veränderung der LS-Mittelwerte vs. Placebo1 |
- |
−5,80 |
95 %-KI für den Unterschied |
- |
−7,49;−4,10 |
p-Wert vs. Placebo2 |
- |
<0,0001 |
Studie 2 |
||
N |
79 |
79 |
Mittelwert (SD) |
−5,14 (5,58) |
−9,77 (5,95) |
Veränderung der LS-Mittelwerte vs. Placebo1 |
- |
−4,81 |
95 %-KI für den Unterschied |
- |
−6,49;−3,13 |
p-Wert vs. Placebo2 |
- |
<0,0001 |
Studie 3 |
||
N |
83 |
252 |
Mittelwert (SD) |
−4,01 (5,87) |
−8,55 (6,01) |
Veränderung der LS-Mittelwerte vs. Placebo1 |
- |
-4,52 |
95 %-KI für den Unterschied |
- |
−5,97; −3,08 |
p-Wert vs. Placebo2 |
- |
<0,0001 |
*Modifizierte Intent-to-treat-(mITT-)Population: umfasste alle Patienten, die randomisiert wurden und mindestens eine Dosis der Studienmedikation erhalten haben.
Die Imputation fehlender Daten erfolgte mittels BOCF (Baseline Observation Carried Forward; Fortschreibung des zu Studienbeginn erfassten Wertes).
1 Der LS-Mittelwert wurde mittels ANCOVA-Modell geschätzt. Die Schichten waren der wöchentliche Wert für den Schweregrad des Juckreizes zu Studienbeginn (<13 vs. ≥13) sowie das Körpergewicht zu Studienbeginn (<80 kg vs. ≥80 kg).
2 Der p-Wert wurde aus dem ANCOVA-t-Test abgeleitet.
Abbildung 2 zeigt die mittleren wöchentlichen Werte für den Schweregrad des Juckreizes im Zeitverlauf in Studie 1. Die mittleren wöchentlichen Werte für den Schweregrad des Juckreizes nahmen signifikant ab; dabei wurde die maximale Wirkung um Woche 12 verzeichnet und über die 24-wöchige Behandlungsphase aufrechterhalten. Die Ergebnisse in Studie 3 waren ähnlich.
In allen drei Studien nahm der mittlere wöchentliche Wert für den Schweregrad des Juckreizes während der 16-wöchigen behandlungsfreien Nachbeobachtungsphase allmählich zu, was dem Wiederauftreten der Symptome entsprach. Die mittleren Werte am Ende der Nachbeobachtungsphase fielen ähnlich aus wie in der Placebogruppe, lagen jedoch unter den jeweiligen mittleren Ausgangswerten.
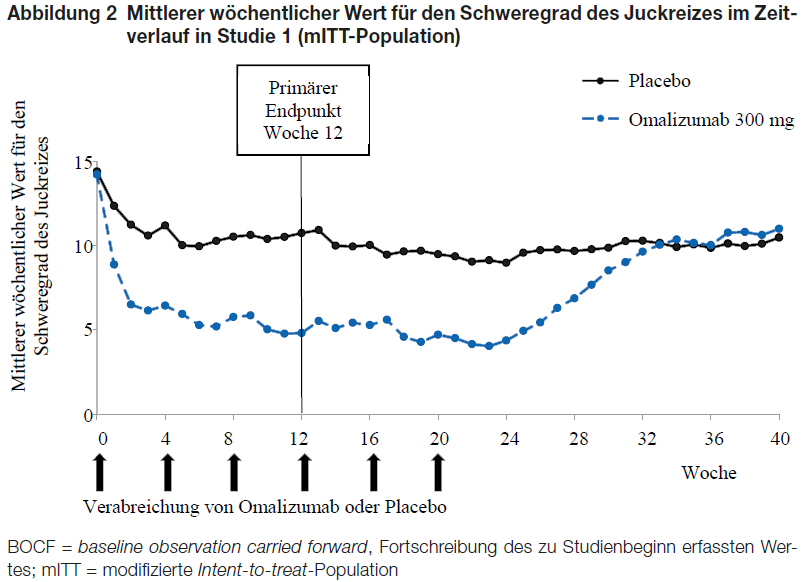
Die Größenordnung der in Behandlungswoche 24 verzeichneten Wirksamkeitsergebnisse war mit der in Woche 12 beobachteten vergleichbar:
Unter der Dosis von 300 mg in den Studien 1 und 3 lag die mittlere Verminderung des wöchentlichen Wertes für den Schweregrad des Juckreizes gegenüber Studienbeginn bei 9,8 bzw. 8,6, der Anteil von Patienten mit einem UAS7≤6 betrug 61,7 % bzw. 55,6 % und der Anteil von Patienten mit einem vollständigen Ansprechen (UAS7=0) belief sich auf 48,1 % bzw. 42,5 % (alle p<0,0001 im Vergleich mit Placebo).
Daten aus klinischen Studien an Jugendlichen (12 bis 17 Jahre) beinhalteten insgesamt 39 Patienten, von denen 11 die 300-mg-Dosis erhielten. Ergebnisse für die 300 mg sind für 9 Patienten in Woche 12 und 6 Patienten in Woche 24 verfügbar. Sie zeigen, im Vergleich zu Erwachsenen, ein ähnliches Ausmaß der Reaktion auf eine Omalizumab-Behandlung. Die gemittelte Abweichung vom Ausgangswert des wöchentlichen Wertes für den Schweregrad des Juckreizes reduzierte sich um 8,25 in Woche 12 und um 8,95 in Woche 24. Die Responder-Raten beliefen sich auf 33 % in Woche 12 und auf 67 % in Woche 24 für UAS7=0, und auf 56 % in Woche 12 und 67 % in Woche 24 für UAS7≤6.
In einer 48-wöchigen Studie wurden 206 Patienten im Alter von 12 bis 75 Jahren in eine 24-wöchige unverblindete Behandlungsphase mit 300 mg Omalizumab alle 4 Wochen eingeschlossen. Patienten, die auf die Behandlung in dieser unverblindeten Phase ansprachen, wurden anschließend randomisiert und erhielten für weitere 24 Wochen 300 mg Omalizumab (81 Patienten) oder Placebo (53 Patienten) alle 4 Wochen.
Von den Patienten, die 48 Wochen lang mit Omalizumab behandelt wurden, kam es bei 21 % zu einer klinischen Verschlechterung (UAS7-Score ≥12 für mindestens 2 aufeinander folgende Wochen nach der Randomisierung zwischen den Wochen 24 und 48), gegenüber 60,4 % der Patienten, die in Woche 48 mit Placebo behandelt wurden (Unterschied –39,4 %; p<0,0001; 95 %-KI: –54,5 %; – 22,5 %).
Die Pharmakokinetik von Omalizumab wurde an erwachsenen und jugendlichen Patienten mit allergischem Asthma, bei erwachsenen Patienten mit CRSwNP sowie bei erwachsenen und jugendlichen Patienten mit csU untersucht. Die allgemeinen pharmakokinetischen Eigenschaften von Omalizumab sind in diesen Patientengruppen vergleichbar.
Resorption
Nach subkutaner Verabreichung wird Omalizumab mit einer durchschnittlichen absoluten Bioverfügbarkeit von 62 % resorbiert. Nach einer einzelnen subkutanen Dosis bei erwachsenen und jugendlichen Patienten mit Asthma oder csU wurde Omalizumab langsam resorbiert und erreichte eine maximale Serumkonzentration nach durchschnittlich 6-8 Tagen. Bei Patienten mit Asthma waren nach mehreren Dosen Omalizumab die Flächen unter der Serumkonzentrations-Zeit-Kurve von Tag 0 bis Tag 14 unter Steady-State-Bedingungen bis zu 6‑fach höher als nach der ersten Dosis.
Die Pharmakokinetik von Omalizumab verläuft bei Dosen von mehr als 0,5 mg/kg linear. Nach Anwendung von Dosen von 75 mg, 150 mg oder 300 mg alle 4 Wochen bei Patienten mit csU stiegen die Talkonzentrationen von Omalizumab im Serum dosisproportional an.
Die Verabreichung von Omalizumab, hergestellt als lyophilisierte oder flüssige Formulierung, ergab ähnliche Serumkonzentrations-Kinetiken von Omalizumab.
Verteilung
In vitro bildet Omalizumab mit IgE Komplexe von begrenzter Größe. Komplexe, die ausfallen, und Komplexe mit einem Molekulargewicht von mehr als einer Million Dalton wurden weder in vitro noch in vivo beobachtet. Populationspharmakokinetische Untersuchungen zeigten, dass die Verteilung von Omalizumab bei Patienten mit allergischem Asthma und Patienten mit csU vergleichbar war. Das scheinbare Verteilungsvolumen betrug bei Asthma-Patienten nach einer subkutanen Verabreichung 78 ± 32 ml/kg.
Elimination
Die Clearance von Omalizumab ist sowohl mit Clearance-Prozessen von IgG als auch mit Clearance über spezifische Bindung und Komplexbildung mit seinem Zielliganden IgE verbunden. Elimination von IgG über die Leber umfasst den Abbau im retikuloendothelialen System und in Endothelzellen. Intaktes IgG wird auch in die Galle sezerniert. Bei Asthma-Patienten betrug die mittlere Halbwertszeit für die Elimination von Omalizumab aus dem Serum 26 Tage, mit einer scheinbaren mittleren Clearance von 2,4 ± 1,1 ml/kg/Tag. Ein doppeltes Körpergewicht führte näherungsweise zu einer doppelten scheinbaren Clearance. Populationspharmakokinetische Simulationen ergaben, dass bei csU-Patienten die Serumeliminationshalbwertszeit von Omalizumab im Steady-State im Mittel bei 24 Tagen lag und die scheinbare Clearance im Steady-State bei einem 80 kg schweren Patienten 3,0 ml/kg/Tag betrug.
Besondere Patientengruppen
Alter, ethnische Herkunft, Geschlecht, Body-Mass-Index
Patienten mit allergischem Asthma und chronischer Rhinosinusitis mit Nasenpolypen (CRSwNP)
Die Pharmakokinetik von Omalizumab wurde in verschiedenen Populationen analysiert, um Effekte von demografischen Besonderheiten zu bewerten. Die Analyse dieser begrenzten Daten deutet darauf hin, dass keine Dosisanpassungen bezüglich Alter (6 bis 76 Jahre für Patienten mit allergischem Asthma; 18 bis 75 Jahre für Patienten mit CRSwNP), ethnischer Herkunft, Geschlecht oder Body-Mass-Index benötigt werden (siehe Abschnitt 4.2).
Patienten mit csU
Die Effekte demografischer Merkmale und sonstiger Faktoren auf die Omalizumab-Exposition wurden auf Grundlage der Populationspharmakokinetik beurteilt. Darüber hinaus wurden die Effekte von Kovariablen durch eine Analyse des Zusammenhangs zwischen den Konzentrationen von Omalizumab und dem klinischen Ansprechen beurteilt. Diese Analysen deuten darauf hin, dass bei csU-Patienten keine Dosisanpassung auf Grundlage von Alter (12-75 Jahre), ethnischer Herkunft, Geschlecht, Körpergewicht, Body-Mass-Index, IgE-Ausgangswert, Anti-FcεRI-Autoantikörpern oder einer gleichzeitigen Anwendung von H2-Antihistaminika oder LTRAs erforderlich ist.
Renale und hepatische Störungen
Es gibt keine Daten zur Pharmakokinetik oder Pharmakodynamik bei Patienten mit allergischem Asthma oder csU mit renalen oder hepatischen Störungen (siehe Abschnitte 4.2 und 4.4).
Die Sicherheit von Omalizumab wurde an Cynomolgusaffen untersucht, da Omalizumab an Cynomolgus- und humanes IgE mit ähnlicher Affinität bindet. Antikörper gegen Omalizumab wurden bei manchen Affen nach einer wiederholten subkutanen oder intravenösen Verabreichung gefunden. Jedoch wurde keine offensichtliche Toxizität wie eine durch Immunkomplexe vermittelte Erkrankung oder eine Komplement-abhängige Zytotoxizität gesehen. Es gab bei Cynomolgusaffen keine Hinweise auf eine anaphylaktische Reaktion aufgrund von Mastzelldegranulation.
Die chronische Anwendung von Omalizumab in Dosierungen von bis zu 250 mg/kg (mindestens das 14-Fache der höchsten empfohlenen klinischen Dosierung in mg/kg gemäß der empfohlenen Dosistabelle) wurde von nicht-humanen Primaten gut vertragen (sowohl bei erwachsenen als auch bei juvenilen Tieren), mit Ausnahme einer dosis- und altersabhängigen Verminderung der Plättchenzahl, mit einer höheren Empfindlichkeit bei Jungtieren. Die Serumkonzentration, die bei erwachsenen Cynomolgusaffen für einen 50%igen Abfall der Plättchenzahl vom Basiswert nötig war, war etwa 4- bis 20‑fach höher als die zu erwartende maximale klinische Serumkonzentration. Außerdem wurden bei Cynomolgusaffen akute Hämorrhagie und Entzündung an der Injektionsstelle beobachtet.
Formale Karzinogenitätsstudien wurden mit Omalizumab nicht durchgeführt.
In Reproduktionsstudien an Cynomolgusaffen zeigten subkutane Dosen bis zu 75 mg/kg pro Woche (mindestens das 8-Fache der höchsten empfohlenen klinischen Dosierung in mg/kg über einen Zeitraum von 4 Wochen), die während der Organogenese verabreicht wurden, keine Toxizität bei den Muttertieren, Embryotoxizität oder Teratogenität. Außerdem wurden bei einer Verabreichung während der späten Schwangerschaftsphase, der Entbindung und des Stillens keine nachteiligen Effekte auf das fetale oder neonatale Wachstum beobachtet.
Omalizumab wird in die Muttermilch von Cynomolgusaffen sezerniert. Die Spiegel von Omalizumab in der Milch betrugen 0,15 % der mütterlichen Serumkonzentration.
L-Arginin-Hydrochlorid
L-Histidin-Hydrochlorid-Monohydrat
L-Histidin
Polysorbat 20 (E 432)
Wasser für Injektionszwecke
Das Arzneimittel darf nicht mit anderen Arzneimitteln gemischt werden.
2 Jahre.
Das Produkt darf für eine Gesamtdauer von 7 Tagen bei 25 °C aufbewahrt werden.
Im Kühlschrank lagern (2 °C – 8 °C).
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Omlyclo 150 mg Injektionslösung in einer Fertigspritze
Omlyclo 150 mg Injektionslösung in einer Fertigspritze ist erhältlich als 1 ml Lösung in einem vorgefüllten Spritzenzylinder (Typ-I-Glas) mit eingesetzter besonders dünner 27-Gauge-Nadel (Edelstahl), (Typ-I)-Kolbenstopfen (Elastomer) und Nadelschutzkappe (Elastomer und Polypropylen).
Eine Packung mit 1 Fertigspritze und Mehrfachpackungen mit 3 (3 x 1), 6 (6 x 1) oder 10 (10 x 1) Fertigspritzen.
Omlyclo 300 mg Injektionslösung in einer Fertigspritze
Omlyclo 300 mg Injektionslösung in einer Fertigspritze ist erhältlich als 2 ml Lösung in einem vorgefüllten Spritzenzylinder (Typ-I-Glas) mit eingesetzter besonders dünner 27-Gauge-Nadel (Edelstahl), (Typ-I)-Kolbenstopfen (Elastomer) und Nadelschutzkappe (Elastomer und Polypropylen).
Eine Packung mit 1 Fertigspritze und Mehrfachpackungen mit 2 (2 x 1), 3 (3 x 1) oder 6 (6 x 1) Fertigspritzen.
Omlyclo 150 mg Injektionslösung im Fertigpen
Omlyclo 150 mg Injektionslösung im Fertigpen ist erhältlich als 1 ml Lösung in einem vorgefüllten Penzylinder (Typ-I-Glas) mit eingesetzter besonders dünner 27-Gauge-Nadel (Edelstahl), (Typ-I)-Kolbenstopfen (Elastomer) und Nadelschutzkappe (Elastomer und Polypropylen).
Eine Packung mit 1 Fertigpen und Mehrfachpackungen mit 2 (2 x 1), 3 (3 x 1), 6 (6 x 1) oder 10 (10 x 1) Fertigpens.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Fertigspritze
Die Fertigspritze zum Einmalgebrauch ist für die individuelle Anwendung bestimmt. Sie sollte 30 bis 45 Minuten vor der Injektion aus dem Kühlschrank genommen werden, damit sie Raumtemperatur annehmen kann.
Fertigpen
Der Fertigpen zum Einmalgebrauch ist für die individuelle Anwendung bestimmt. Er sollte 30 bis 45 Minuten vor der Injektion aus dem Kühlschrank genommen werden, damit er Raumtemperatur annehmen kann.
Entsorgungsmaßnahme
Entsorgen Sie die benutzte Spritze oder den benutzten Pen unmittelbar in einem Behälter für spitze Gegenstände.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Celltrion Healthcare Hungary Kft.
1062 Budapest,
Váci út 1-3. WestEnd Office Building B torony
Ungarn
Omlyclo 150 mg Injektionslösung in einer Fertigspritze
EU/1/24/1817/002
EU/1/24/1817/003
EU/1/24/1817/004
EU/1/24/1817/011
Omlyclo 300 mg Injektionslösung in einer Fertigspritze
EU/1/24/1817/014
EU/1/24/1817/015
EU/1/24/1817/016
EU/1/24/1817/017
Omlyclo 150 mg Injektionslösung im Fertigpen
EU/1/24/1817/006
EU/1/24/1817/007
EU/1/24/1817/008
EU/1/24/1817/012
EU/1/24/1817/013
Datum der Erteilung der Zulassung: 16. Mai 2024
November 2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu/ verfügbar.
Verschreibungspflichtig
Celltrion Healthcare Deutschland GmbH
61348 Bad Homburg vor der Höhe
Tel: 030 346494150
E-Mail: infoDE@celltrionhc.com