
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Relfydess 100 Einheiten/ml Injektionslösung
1 ml Lösung enthält 100 Einheiten Botulinum-Toxin Typ A zur Injektion (Ph.Eur.), hergestellt von Clostridium botulinum, frei von Komplexproteinen.
Die Wirksamkeits-Einheiten sind spezifisch für Relfydess und nicht mit anderen Botulinum-Toxin-Arzneimitteln austauschbar.
Jede Durchstechflasche enthält 150 Einheiten in 1,5 ml Lösung.
Sonstiger Bestandteil mit bekannter Wirkung
1 ml Lösung enthält 1,1 mg Polysorbat 80 (E 433).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung
Klare, farblose bis blassgelbe Lösung.
Relfydess wird angewendet zur vorübergehenden Verbesserung des Aussehens von:
mittelstarken bis starken Glabellafalten bei maximalem Stirnrunzeln
mittelstarken bis starken seitlichen Kanthalfalten, sichtbar bei maximalem Lächeln
allein oder in Kombination, bei erwachsenen Patienten unter 65 Jahren, wenn das Ausmaß dieser Falten eine erhebliche psychische Belastung für den Patienten darstellt.
Das Behandlungsintervall sollte nicht kürzer als 12 Wochen sein.
Die Wirksamkeit und Sicherheit der wiederholten Anwendung dieses Arzneimittels über einen Zeitraum von mehr als 52 Wochen wurde nicht untersucht.
Wenn andere Botulinum-Toxin-haltige Arzneimittel zur Behandlung in anderen Anwendungsgebieten angewendet werden oder wurden, muss die kumulative Dosis berücksichtigt werden.
Dosierung
Die Wirksamkeits-Einheiten sind spezifisch für Relfydess und nicht mit anderen Botulinum-Toxin-Arzneimitteln austauschbar.
Relfydess ist eine gebrauchsfertige Injektionslösung mit einer Konzentration von 10 Einheiten pro 0,1 ml. Eine Rekonstitution ist nicht erforderlich.
Tabelle 1: Dosierungsanleitung für Relfydess
Behandlung(en) | Empfohlene Gesamtdosis | Dosis pro Injektion |
Glabellafalten (GL) | 50 Einheiten (0,5 ml) | 5 Injektionen von je 10 Einheiten (0,1 ml): |
Seitliche Kanthalfalten (LCL) | 60 Einheiten (0,6 ml) | 6 Injektionen von je 10 Einheiten (0,1 ml): |
Kombinierte Behandlung von Glabellafalten und seitlichen Kanthalfalten | 110 Einheiten (1,1 ml) | insgesamt 11 Injektionen von je 10 Einheiten (0,1 ml) für kombinierte GL und LCL |
Allgemeine Informationen
Im Falle eines Behandlungsversagens oder einer verminderten Wirkung nach wiederholten Injektionen sollten alternative Behandlungsmethoden angewendet werden. Im Falle eines Behandlungsversagens nach der ersten Behandlungssitzung können folgende Ansätze in Betracht gezogen werden:
Analyse der Ursachen für das Therapieversagen, z. B. Injektion in falsche Muskeln, ungeeignete Injektionstechnik und Bildung von Toxin-neutralisierenden Antikörpern.
Neubewertung der Relevanz der Behandlung mit Botulinum-Toxin Typ A.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Relfydess bei Kindern und Jugendlichen unter 18 Jahren sind nicht erwiesen. Die Anwendung von Relfydess bei Patienten unter 18 Jahren wird nicht empfohlen.
Ältere Patienten
Es liegen nur begrenzte klinische Daten der Phase III mit Relfydess bei Patienten ab 65 Jahren vor.
Art der Anwendung
Relfydess darf nur von Ärzten angewendet werden, die eine entsprechende Qualifikation und Erfahrung mit dieser Behandlung haben und die über die notwendige Ausstattung, entsprechend den nationalen Leitlinien und Gesetzen, verfügen.
Intramuskuläre Anwendung.
Die Dosierung und die Behandlungsintervalle sind von dem festgestellten individuellen Ansprechen des jeweiligen Patienten abhängig, die Dosierung sollte jedoch die zulässige Höchstdosis nicht überschreiten und das Behandlungsintervall sollte mindestens 12 Wochen betragen.
Jede Durchstechflasche Relfydess darf nur zur Behandlung eines einzelnen Patienten bei einer einzelnen Sitzung verwendet werden. Nicht verwendetes Arzneimittel muss nach der Behandlung verworfen werden.
Um eine Kreuzkontamination zu vermeiden, sind aseptische Technik und Standardverfahren anzuwenden.
Hinweise zur Handhabung und Beseitigung der Durchstechflaschen, siehe Abschnitt 6.6.
Die mediane Zeit bis zum Wirkungseintritt beträgt 2 bis 3 Tage, einige Patienten berichteten über eine Wirkung innerhalb eines Tages. Eine Wirkung der Behandlung wurde für 6 Monate nachgewiesen, wobei bis zu 75 % der Patienten nicht zum Ausgangswert zurückkehrten.
Glabellafalten
Die empfohlene Dosis zur Behandlung von Glabellafalten bei Erwachsenen beträgt insgesamt 50 Einheiten (0,5 ml), die durch intramuskuläre Injektion zu gleichen Teilen (10 Einheiten/0,1 ml pro Injektion) in jede der 5 intramuskulären Injektionsstellen verabreicht werden (siehe Abbildung 1): 2 Injektionen auf jeder Seite in den M. corrugator und 1 Injektion in den M. procerus in der Nähe des Nasofrontalwinkels. Die anatomischen Orientierungspunkte können schneller ermittelt werden, wenn sie bei maximalem Stirnrunzeln des Patienten beobachtet und palpiert werden. Vor und während der Injektion Daumen oder Zeigefinger fest unter den Augenhöhlenrand drücken, um eine Paravasation unter den Augenhöhlenrand zu vermeiden. Die Nadelöffnung sollte während der Injektion nach superior und medial ausgerichtet sein.
Um das Risiko einer Augenlidptosis zu reduzieren, sollten die folgenden Maßnahmen ergriffen werden:
Injektionen in der Nähe des M. levator palpebrae superioris sind zu vermeiden, insbesondere bei Patienten mit ausgeprägtem M. depressor supercilii.
Injektionen in den M. corrugator lateralis sollten mindestens 1 cm oberhalb des oberen knöchernen Orbitarandes erfolgen.
Es ist sicherzustellen, dass die injizierte Dosis (Volumen) korrekt ist.
Injektionen näher als 1 cm über der Augenbrauenmitte sind zu vermeiden.
Abbildung 1: Lage der Injektionsstellen für Glabellafalten

Seitliche Kanthalfalten
Die empfohlene Dosis für die Behandlung der seitlichen Kanthalfalten bei Erwachsenen beträgt insgesamt 60 Einheiten (0,6 ml), die durch intramuskuläre Injektion zu gleichen Teilen (10 Einheiten/0,1 ml pro Injektion) in jede der 6 intramuskulären Injektionsstellen verabreicht werden (siehe Abbildung 2: Option 1 und Option 2): 3 Injektionen (30 Einheiten/0,3 ml) auf jeder Seite in den M. orbicularis oculi. Die Injektionen sollten mit der Nadelöffnung schräg nach oben und vom Auge weg gerichtet in den M. orbicularis oculi lateralis gegeben werden. Wenn die Falten im seitlichen Kanthalbereich sowohl oberhalb als auch unterhalb des seitlichen Kanthus auftreten, ist gemäß Option 1 zu injizieren. Wenn die Falten im seitlichen Kanthalbereich hauptsächlich unterhalb des seitlichen Kanthus auftreten, ist gemäß Option 2 zu injizieren.
Abbildung 2: Lage der Injektionsstellen für die seitlichen Kanthalfalten
Option 1: Oberhalb und unterhalb des seitlichen Kanthus | Option 2: Unterhalb des seitlichen Kanthus |
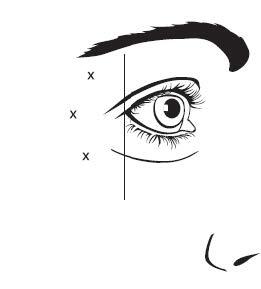 | 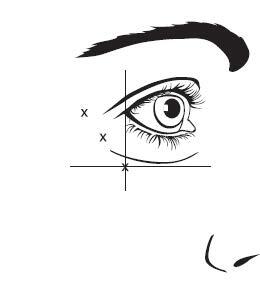 |
Die anatomischen Orientierungspunkte der seitlichen Kanthalfalten lassen sich leichter identifizieren, wenn sie beim maximalen Lächeln beobachtet und palpiert werden. Es muss darauf geachtet werden, dass nicht in den M. zygomatici major/minor injiziert wird, um ein seitliches Absinken des Mundes und ein asymmetrisches Lächeln zu vermeiden.
Kombinierte Behandlung von Glabellafalten/seitlichen Kanthalfalten
Für die Kombinationsbehandlung von Glabellafalten und seitlichen Kanthalfalten sollte die jeweilige Einzeldosierung und -anwendung für eine Gesamtdosis von 110 Einheiten (1,1 ml) Relfydess befolgt werden.
Die empfohlene Dosis für die Behandlung von Glabellafalten beträgt 50 Einheiten (0,5 ml) (10 Einheiten/0,1 ml pro Injektion) in jede der 5 intramuskulären Injektionsstellen und für die Behandlung der seitlichen Kanthalfalten 60 Einheiten (0,6 ml) (10 Einheiten/0,1 ml in jede der 6 intramuskulären Injektionsstellen).
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Bestehende Infektion an der vorgesehenen Injektionsstelle.
Bestehende Myasthenia gravis, Lambert-Eaton-Syndrom oder amyotrophe Lateralsklerose.
Allgemeine Hinweise
Relfydess darf nicht in ein Blutgefäß injiziert werden.
Wie bei allen intramuskulären Injektionen wird eine Behandlung mit Relfydess bei Patienten mit verlängerter Blutgerinnungszeit nicht empfohlen.
Bei Patienten, die mit den empfohlenen Dosen behandelt wurden, kann eine übermäßige Muskelschwäche auftreten.
Jede Durchstechflasche Relfydess darf nur zur Behandlung eines einzelnen Patienten bei einer einzelnen Sitzung verwendet werden.
Übrig gebliebenes, nicht verwendetes Arzneimittel muss wie in Abschnitt 6.6 beschrieben beseitigt werden. Bei der Inaktivierung und Beseitigung nicht verwendeter Lösung müssen besondere Vorsichtsmaßnahmen ergriffen werden (siehe Abschnitt 6.6).
Überempfindlichkeitsreaktionen
Schwere und/oder unmittelbare Überempfindlichkeitsreaktionen wurden für Botulinum-Toxin-haltige Arzneimittel berichtet, und sehr selten können anaphylaktische Reaktionen auftreten (siehe Abschnitt 4.8). Zu diesen Reaktionen gehören Anaphylaxie, Serumkrankheit, Urtikaria, Weichteilödeme und Dyspnoe. Ausrüstung und Arzneimittel (einschließlich Epinephrin (Adrenalin)), die zur Behandlung einer Anaphylaxie benötigt werden, müssen daher jederzeit verfügbar sein. Wenn eine solche Reaktion auftritt, muss die weitere Injektion von Relfydess abgebrochen und sofort eine angemessene medizinische Behandlung eingeleitet werden.
Ausbreitung der Toxinwirkung
Daten zur Sicherheit nach dem Inverkehrbringen von anderen zugelassenen Botulinum-Toxin-haltigen Arzneimitteln deuten darauf hin, dass die Wirkungen von Botulinum-Toxin (wie Doppeltsehen, verschwommenes Sehen und Ptosis) auch über die lokale Injektionsstelle hinaus beobachtet werden können (siehe Abschnitt 4.8). Darüber hinaus wurden sehr selten Nebenwirkungen, die möglicherweise mit der Ausbreitung der Toxinwirkung über die Injektionsstelle hinaus zusammenhängen, bei Botulinum-Toxin berichtet. Diese können Asthenie, generalisierte Muskelschwäche, Dysphagie, Dysphonie, Dysarthrie, Harninkontinenz und Atembeschwerden umfassen. Diese Symptome stimmen mit dem Wirkmechanismus von Botulinum-Toxinen überein und sind Stunden bis Wochen nach der Injektion aufgetreten.
Schluck- und Atembeschwerden können lebensbedrohlich sein, und es gibt Berichte über Todesfälle im Zusammenhang mit der Ausbreitung der Toxinwirkung. Patienten mit vorbestehenden Schluck- oder Atembeschwerden können anfälliger für diese Komplikationen sein. Insbesondere wurden nach der Behandlung mit Botulinum-Toxin sehr selten Todesfälle bei Patienten mit Dysphagie, Pneumopathie oder erheblicher Asthenie berichtet. Daher wird Relfydess bei solchen Patienten nicht empfohlen.
Patienten oder Betreuer sollten darauf hingewiesen werden, bei Schluck-, Sprach- oder Atembeschwerden sofort einen Arzt aufzusuchen.
Bestehende neuromuskuläre Erkrankungen
Relfydess sollte bei Patienten mit dem Risiko für oder klinischen Anzeichen einer ausgeprägten Störung der neuromuskulären Reizleitung mit Vorsicht angewendet werden. Diese Patienten können eine erhöhte Empfindlichkeit gegenüber Wirkstoffen wie Botulinum-Toxin aufweisen, was nach der Behandlung zu einer übermäßigen Muskelschwäche (einschließlich systemischer Auswirkungen wie schwerer Dysphagie und Atemwegsbeeinträchtigung) führen kann. In einigen dieser Fälle dauerte die Dysphagie mehrere Monate an und erforderte das Legen einer Magensonde.
Bestehende Erkrankungen an der Injektionsstelle
Vorsicht ist geboten, wenn Relfydess bei einer bestehenden Entzündung an der/den vorgesehenen Injektionsstelle(n) oder bei übermäßiger Schwäche oder Atrophie des/der betroffenen Muskels/Muskeln angewendet wird.
Vorsicht ist geboten, wenn die Relfydess-Behandlung bei Patienten mit ausgeprägter Gesichtsasymmetrie, Ptosis, übermäßiger Hauterschlaffung (z. B. Dermatochalasis, siehe Abschnitt 5.1), tiefer Narbenbildung auf der Haut oder dicker talghaltiger Haut angewendet wird.
Unerwünschte ophthalmische Reaktionen
Bei der Anwendung von Botulinum-Toxinen kann es zu trockenen Augen, verminderter Tränenproduktion, vermindertem Blinzeln und Erkrankungen der Hornhaut kommen. Bei anhaltenden Symptomen eines trockenen Auges (z. B. Augenreizung, Photophobie oder Sehveränderungen) ist eine Überweisung an einen Augenarzt zu erwägen. Bei der Anwendung von Botulinum-Toxinen kann vermehrter Tränenfluss auftreten.
Muskelatrophie
Nach einer wiederholten Botulinum-Behandlung ist eine Muskelatrophie zu erwarten, die auf eine schlaffe Lähmung der behandelten Muskeln zurückzuführen ist.
Antikörperbildung
Injektionen in kürzeren Abständen oder mit höheren Dosen können das Risiko einer Antikörperbildung gegen Botulinum-Toxin erhöhen. Klinisch kann die Bildung neutralisierender Antikörper die Wirksamkeit der nachfolgenden Behandlung verringern.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Kalium- und Natriumgehalt
Dieses Arzneimittel enthält Kalium, aber weniger als 1 mmol (39 mg) Kalium pro Durchstechflasche, d. h., es ist nahezu „kaliumfrei“.
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro Durchstechflasche, d. h., es ist nahezu „natriumfrei“.
Gehalt an Polysorbat 80 (E 433)
Dieses Arzneimittel enthält 1,6 mg Polysorbat 80 (E 433) pro Durchstechflasche entsprechend 1,1 mg/ml. Polysorbate können allergische Reaktionen hervorrufen.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Die gleichzeitige Behandlung mit Relfydess und Aminoglykosiden oder anderen Arzneimitteln, die auf die neuromuskuläre Reizleitung wirken (z. B. Curare-ähnliche Wirkstoffe oder andere Botulinum-Toxin-haltige Arzneimittel an anderen Stellen), sollte nur mit Vorsicht erfolgen, da die Wirkung des Botulinum-Toxins potenziert werden kann.
Schwangerschaft
Es liegen keine ausreichenden Daten zur Anwendung von Botulinum-Toxin Typ A bei schwangeren Frauen vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität, außer bei hohen Dosen, die eine maternale Toxizität verursachen (siehe Abschnitt 5.3). Das potenzielle Risiko beim Menschen ist nicht bekannt. Relfydess sollte während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, nicht angewendet werden.
Stillzeit
Es ist nicht bekannt, ob Relfydess in die Muttermilch ausgeschieden wird. Es wurden keine tierexperimentellen Studien zur Ausscheidung in die Milch durchgeführt. Relfydess sollte während der Stillzeit nicht angewendet werden.
Fertilität
Es liegen keine klinischen Daten zur Auswirkung von Relfydess auf die Fertilität vor. In tierexperimentellen Studien gab es keinen Hinweis auf eine direkte Auswirkung von Botulinum-Toxin Typ A auf die Fertilität (siehe Abschnitt 5.3)
Von anderen Botulinum-Toxin-haltigen Arzneimitteln wurde berichtet, dass sie einen geringen oder mäßigen Einfluss auf die Verkehrstüchtigkeit und/oder die Fähigkeit zum Bedienen von Maschinen haben. Es besteht ein potenzielles Risiko für eine lokalisierte Muskelschwäche oder Sehstörungen im Zusammenhang mit der Anwendung von Relfydess, was die Fähigkeit zum Führen eines Fahrzeugs oder zum Bedienen von Maschinen vorübergehend beeinträchtigen kann.
Zusammenfassung des Sicherheitsprofils
Die Mehrzahl der Nebenwirkungen nach einer einzelnen Behandlung mit Relfydess bei Probanden, die in allen zulassungsrelevanten placebokontrollierten Studien ≥ 50 Einheiten erhielten, waren von leichter bis mäßiger Intensität. Die am häufigsten berichteten Nebenwirkungen waren Reaktionen an der Injektionsstelle und Kopfschmerzen, die bei etwa 7 % bzw. 5 % der Probanden auftraten.
Im Allgemeinen traten behandlungs-/injektionstechnikbedingte Reaktionen innerhalb des ersten Monats nach der Injektion auf und waren vorübergehend.
Bei der kombinierten Behandlung von Glabellafalten und seitlichen Kanthalfalten waren Art und Häufigkeit der Nebenwirkungen vergleichbar mit denen, die bei der Behandlung in den einzelnen Anwendungsgebieten beobachtet wurden.
Tabellarische Übersicht der Nebenwirkungen
Die Häufigkeit von Nebenwirkungen wird wie folgt eingeteilt: Sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 2: Mittelstarke bis starke Glabellafalten
Die folgenden Nebenwirkungen wurden bei Patienten beobachtet, bei denen Relfydess zur vorübergehenden Verbesserung des Aussehens von mittelstarken bis starken Glabellafalten angewendet wurde.
Systemorganklasse | Häufigkeit | Nebenwirkungen |
Erkrankungen des Immunsystems | Gelegentlich | Überempfindlichkeit |
Erkrankungen des Nervensystems | Häufig | Kopfschmerzen |
Augenerkrankungen | Häufig | Augenlidptosis |
Gelegentlich | Sehverschlechterung, trockenes Auge, Asthenopie | |
Erkrankungen der Haut und des Unterhautgewebes | Gelegentlich | Brauenptose, Urtikaria |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | Gelegentlich | Muskuläre Schwäche, Muskelkrampf |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | Häufig | Reaktionen an der Injektionsstelle (z. B. blaue Flecken, Schwellung, Pruritus, Schmerz, Unbehagen, Hämatom, Überempfindlichkeit und Wärme) |
Tabelle 3: Mittelstarke bis starke seitliche Kanthalfalten
Die folgenden Nebenwirkungen wurden bei Patienten beobachtet, bei denen Relfydess zur vorübergehenden Verbesserung des Aussehens von mittelstarken bis starken seitlichen Kanthalfalten angewendet wurde.
Systemorganklasse | Häufigkeit | Nebenwirkungen |
Erkrankungen des Immunsystems | Gelegentlich | Überempfindlichkeit |
Erkrankungen des Nervensystems | Häufig | Kopfschmerzen |
Augenerkrankungen | Gelegentlich | Trockenes Auge, Asthenopie, Schwellung des Augenlids |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | Gelegentlich | Muskuläre Schwäche |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | Häufig | Reaktionen an der Injektionsstelle (z. B. Erythem, Schmerz und blaue Flecken) |
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen.
Überhöhte Dosen können zu weitreichenden und tiefgreifenden neuromuskulären Lähmungen mit einer Vielzahl von Symptomen führen. Wenn überhöhte Dosen zu einer Lähmung der Atemmuskulatur führen, kann eine unterstützende Beatmung erforderlich sein. Im Falle einer Überdosierung muss der Patient mehrere Wochen ärztlich auf Anzeichen/Symptome einer übermäßigen Muskelschwäche oder einer Muskellähmung überwacht werden. Eine symptomorientierte Behandlung kann nötig sein.
Symptome einer Überdosierung treten möglicherweise nicht unmittelbar nach einer Injektion auf.
Eine Einweisung in ein Krankenhaus sollte bei Patienten mit Symptomen einer Botulinum-Toxin-Überdosierung erwogen werden (z. B. eine Kombination aus Muskelschwäche, Ptosis, Diplopie, Schluck- und Sprechstörungen oder Lähmung der Atemmuskulatur).
Pharmakotherapeutische Gruppe: Andere Muskelrelaxanzien, peripher wirkende Mittel, ATC-Code: M03AX01
Wirkmechanismus
Die primäre pharmakodynamische Wirkung von Botulinum-Toxin Typ A ist die chemische Denervierung des behandelten Muskels, die zu einer messbaren Verringerung des Muskelaktionspotenzials führt. Dies bewirkt eine örtlich begrenzte Verringerung der Muskelaktivität.
Bei intramuskulärer Injektion führt das Toxin zu einer Lähmung des betroffenen Muskels, wodurch die Muskelaktivität vorübergehend reduziert wird. Die Wirkung hält so lange an, bis sich die neuromuskuläre Synapse regeneriert hat und die Muskelaktivität zurückkehrt.
Klinische Wirksamkeit und Sicherheit
Die im Folgenden beschriebenen Daten spiegeln die Ergebnisse der placebokontrollierten Phase-III-Studien READY‑1, READY‑2 und READY‑3 wider. In den drei Zulassungsstudien wurden insgesamt 1 012 Patienten behandelt, davon 806 Patienten mit Relfydess und 206 Patienten mit einem Placebo. Weitere 902 mit Relfydess behandelte Patienten nahmen an einer offenen Langzeitsicherheitsstudie teil (READY‑4). Insgesamt wurden in allen Phase-III-Studien 1 708 Patienten mit Relfydess behandelt.
Die Wirkung setzte innerhalb eines Tages ein (bis zu 39 % bei Glabellafalten und 34 % bei seitlichen Kanthalfalten), wobei die mittlere Zeit bis zum Einsetzen der Wirkung 2 bis 3 Tage betrug. Die Wirkung der Behandlung wurde für 6 Monate nachgewiesen, wobei bis zu 75 % der Patienten nicht zum Ausgangswert zurückkehrten.
Patienten (insgesamt 1 699), die ≥ 50 Einheiten Relfydess erhielten, wurden zu Beginn und nach jeder Behandlung auf Antikörperbildung (ADA) getestet. Klinische Daten deuten darauf hin, dass bei einigen Personen nach der Behandlung ein niedriger ADA-Titer auftreten kann. Insgesamt wurden 1,1 % der Probanden positiv auf ADA getestet. Für Relfydess kann auf eine geringe Immunogenität geschlossen werden.
Glabellafalten (READY‑1 und READY‑3)
In zwei zulassungsrelevanten, multizentrischen, doppelblinden, placebokontrollierten Phase-III-Studien wurden 451 Patienten mit Glabellafalten (glabellar lines, GL) mit der empfohlenen Dosis von 50 Einheiten behandelt. READY‑1 untersuchte die Behandlung mit Relfydess von GL allein; READY‑3 untersuchte die Kombinationsbehandlung von GL und seitlichen Kanthalfalten (lateral canthal lines, LCL).
Primärer Wirksamkeitsendpunkt war der Anteil der Probanden, die auf die Behandlung ansprachen, definiert als Erreichen eines Wertes von 0 oder 1 für den Schweregrad der Glabellafalten auf der GL-ILA-4-Punkte-Fotoskala (Investigator Live Assessment) bei maximalem Stirnrunzeln bei der Visite nach einem Monat. Die Mehrheit der Probanden, sowohl in der Relfydess- als auch in der Placebogruppe hatte zu Studienbeginn schwere Glabellafalten, wie vom Prüfarzt festgestellt (74,5 % bzw. 75,8 %). Patienten mit übermäßiger Hauterschlaffung im Behandlungsbereich oder im periorbitalen Bereich wurden von den Studien ausgeschlossen. Der Anteil der Probanden, die auf die Behandlung ansprachen, war in der Relfydess-Gruppe im Vergleich zur Placebo-Gruppe nach einem Monat statistisch signifikant größer (p < 0,001) (Tabelle 4).
Tabelle 4: Prüfärztliche Bewertung des Behandlungserfolgsa bei Glabellafalten (% und Anzahl der Probanden) nach 1 Monatb in doppelblinden, placebokontrollierten klinischen Studien, mITT-Populationc
Studie | Relfydess 50 Einheiten GL | Relfydess 50 Einheiten GL und 60 Einheiten LCL | Placebo |
READY‑1, nur GL | 96,3 % | - | 4,5 % |
READY‑3 LCL- & GL-Behandlung | 94,3 % | 96,3 % | 1,8 % |
a Auf der GL‑ILA wurde ein Schweregrad der GL von 0 (keine) oder 1 (leicht) erreicht.
b Primärer Wirksamkeitsendpunkt an Tag 30; p < 0,001
c Die modifizierte Intention-to-Treat-Population (mITT) umfasste alle Probanden, die randomisiert wurden und das Studienprodukt erhielten, und wurde gemäß dem Randomisierungsschema analysiert. Probanden mit einer fotografischen und kategorialen Skala zur Beurteilung nach 1 Monat über eine Televisite wurden aus der mITT-Population ausgeschlossen.
Bei den Probanden in der READY-1-Studie war das Ansprechen (Erreichen von 0 oder 1 auf der GL-ILA bei maximalem Stirnrunzeln) mit Relfydess im Vergleich zu Placebo vom Tag 7 bis zum sechsten Monat statistisch signifikant größer (p < 0,001), wie in Tabelle 5 dargestellt.
Tabelle 5: READY-1 Investigator Live Assessment (ILA) des Schweregrads der GL - Ansprechratena (%) nach der Injektion, ITT-Populationb
Zeitpunkt nach der Injektion | Relfydess (N = 223) | Placebo (N = 74) |
GL-ILA | GL-ILA | |
Tag 7 | 93,2 % | 4,3 % |
Tag 14 | 96,4 % | 6,3 % |
Monat 1 | 96,4 % | 4,7 % |
Monat 2 | 92,9 % | 8,9 % |
Monat 3 | 73,7 % | 7,9 % |
Monat 4 | 53,7 % | 6,3 % |
Monat 5 | 39,7 % | 6,3 % |
Monat 6 | 23,6 % | 1,5 % |
a Definiert als GL-Schweregrad von 2 (mittelstark) oder 3 (stark) bei Studienbeginn und von 0 (keine) oder 1 (leicht) bei der jeweiligen Visite, anhand der GL-ILA-Schweregradskala bewertet.
b Die Intention-to-Treat-Population (ITT) umfasste alle Probanden, die randomisiert wurden und das Studienprodukt erhielten, und wurde gemäß dem Randomisierungsschema analysiert.
Bei der kombinierten Behandlung mit LCL in der READY-3-Studie war das Ansprechen (Erreichen von 0 oder 1 auf dem GL‑ILA bei maximalem Stirnrunzeln) in der Relfydess-GL-/Relfydess-LCL-Gruppe im Vergleich zur Placebo-GL/Placebo-LCL-Gruppe während der gesamten sechs Monate nach der Behandlung statistisch signifikant höher (nominal p < 0,001).
Seitliche Kanthalfalten (READY‑2 und READY‑3)
In zwei zulassungsrelevanten multizentrischen, doppelblinden, placebokontrollierten Phase-III-Studien wurden 471 Patienten mit LCL mit der empfohlenen Dosis von 60 Einheiten behandelt. READY‑2 untersuchte nur die Behandlung von LCL mit Relfydess, READY‑3 untersuchte die Kombinationsbehandlung von GL und LCL.
Primärer Wirksamkeitsendpunkt war der Anteil der Probanden, die auf die Behandlung ansprachen, definiert als Erreichen eines Wertes von 0 oder 1 für den Schweregrad der seitlichen Kanthalfalten auf der LCL‑ILA-4‑Punkte-Fotoskala bei maximalem Lächeln bei der Visite nach 1 Monat. Patienten mit übermäßiger Hauterschlaffung im Behandlungsbereich oder im periorbitalen Bereich wurden von der Studie ausgeschlossen. Der Anteil der Probanden, die auf die Behandlung ansprachen, war in der Relfydess-Gruppe im Vergleich zur Placebo-Gruppe nach einem Monat statistisch signifikant größer (p < 0,001) (Tabelle 6).
Tabelle 6: Prüfärztliche Bewertung des Behandlungserfolgsa bei lateralen Kanthalfalten (% und Anzahl der Probanden) nach 1 Monatb in doppelblinden, placebokontrollierten klinischen Studien, mITT-Populationc
Studie | Relfydess 60 Einheiten LCL | Relfydess 60 Einheiten LCL & 50 Einheiten GL | Placebo |
READY‑2, nur LCL | 87,2 % | - | 11,9 % |
READY‑3, LCL- & GL-Behandlung | 78,1 % | 83,3 % | 19,3 % |
a Auf der LCL‑ILA wurde ein Schweregrad der LCL von 0 (keine) oder 1 (leicht) erreicht.
b Primärer Wirksamkeitsendpunkt an Tag 30; p < 0,001
c Die modifizierte Intention-to-Treat-Population (mITT) umfasste alle Probanden, die randomisiert wurden und das Studienprodukt erhielten, und wurde gemäß dem Randomisierungsschema analysiert. Probanden mit einer fotografischen und kategorialen Skala zur Beurteilung nach 1 Monat über eine Televisite wurden aus der mITT-Population ausgeschlossen
Bei den Probanden in der READY-2-Studie war das Ansprechen (Erreichen von 0 oder 1 auf der LCL-ILA bei maximalem Lächeln) mit Relfydess im Vergleich zu Placebo von Tag 7 bis zum sechsten Monat statistisch signifikant größer (p ≤ 0,002), wie in Tabelle 7 dargestellt.
Tabelle 7: READY-2 Investigator Live Assessment (ILA) des Schweregrads der LCL - Ansprechratena (%) nach der Injektion, ITT-Populationb
Zeitpunkt nach der Injektion | Relfydess (N = 230) | Placebo (N = 73) |
LCL-ILA | LCL-ILA | |
Tag 7 | 82,5 % | 8,5 % |
Tag 14 | 89,7 % | 11,4 % |
Monat 1 | 87,5 % | 11,8 % |
Monat 2 | 76,3 % | 14,3 % |
Monat 3 | 59,8 % | 14,9 % |
Monat 4 | 45,7 % | 10,9 % |
Monat 5 | 32,1 % | 6,2 % |
Monat 6 | 23,3 % | 7,2 % |
a Definiert als ein LCL-Schweregrad von 2 (mittelstark) oder 3 (schwer) bei Studienbeginn und 0 (keine) oder 1 (leicht) bei der jeweiligen Visite, anhand der LCL-ILA-Schweregradskala bewertet.
b Die Intention-to-Treat-Population (ITT) umfasste alle Probanden, die randomisiert wurden und das Studienprodukt erhielten, und wurde gemäß dem Randomisierungsschema analysiert.
Bei der kombinierten Behandlung mit GL in READY-3-Studie war das Ansprechen (Erreichen von 0 oder 1 auf dem LCL‑ILA bei maximalem Lächeln) in der Relfydess-GL-/Relfydess-LCL-Gruppe im Vergleich zur Placebo-GL/Placebo-LCL-Gruppe zu allen Zeitpunkten nach der Behandlung außer nach 6 Monaten statistisch signifikant höher (nominaler p ≤ 0,007).
Patientenzufriedenheit und psychologische Wirkung
Die psychologische Wirkung bei den Probanden wurde anhand der FACE-QTM-Skala zur psychologischen Befindlichkeit ermittelt.
Die FLTSQ-Skala (Facial Line Treatment Satisfaction Questionnaire) wurde verwendet, um die Zufriedenheit der Probanden mit dem Aussehen der GL und/oder LCL sowie die Zufriedenheit mit der Behandlung zu ermitteln.
Die Antworten auf die FACE-QTM-Skala zur psychologischen Wahrnehmung und die FLTSQ-Skala zeigten, dass die mit Relfydess behandelten Probanden zu allen Nachbehandlungszeitpunkten eine Verbesserung der psychologischen Befindlichkeit zeigten und mit ihrer Behandlung und ihrem Aussehen zufriedener waren als die Probanden unter Placebo. Wie anhand der FACE-QTM- und FLTSQ-Skala festgestellt wurde, blieben die positive psychologische Befindlichkeit und die Zufriedenheit der Probanden auch noch 6 Monate nach der Behandlung erhalten.
Offene Studie (READY‑4)
In der multizentrischen, offenen Phase-III-Studie READY‑4 wurde Relfydess mit bis zu 110 Einheiten pro Behandlung und bis zu 4 Wiederholungsbehandlungen in jeder Indikation verabreicht (insgesamt bis zu 7 GL- und/oder LCL-Behandlungen über 52 Wochen). Die vom Prüfarzt in Woche 4 ermittelten Ansprechraten wurden über wiederholte Zyklen bei Glabellafalten bei maximalem Stirnrunzeln in der Untergruppe von 175 Probanden, die 4 Behandlungszyklen erhielten, beibehalten (79,4 % in Behandlungszyklus 1 und 80,0 % in Behandlungszyklus 4). Die entsprechenden Ansprechraten bei 186 Probanden, die 4 Behandlungszyklen für die seitlichen Kanthalfalten erhielten, betrugen bei maximalem Lächeln 64,5 % im Behandlungszyklus 1 und 60,2 % im Behandlungszyklus 4.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Relfydess eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in der Behandlung zur vorübergehenden Verbesserung des Aussehens von mittelstarken bis starken Glabellafalten bei maximalem Stirnrunzeln und seitlichen Kanthalfalten sichtbar bei maximalem Lächeln (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen) gewährt.
Es ist nicht zu erwarten, dass Relfydess nach intramuskulärer Injektion in der empfohlenen Dosis im peripheren Blut in messbarer Menge vorhanden ist. Daher wurden keine pharmakokinetischen Studien durchgeführt.
Studien zur akuten Toxizität, Langzeit-Toxizität und lokalen Verträglichkeit an der Injektionsstelle ergaben bei klinisch relevanten Dosierungen keine ungewöhnlichen lokalen oder systemischen unerwünschten Wirkungen.
Literaturdaten weisen darauf hin, dass Botulinum-Toxine eine kurze Halbwertszeit im Blut und eine begrenzte Gewebediffusion, einschließlich durch die Plazenta, aufweisen. Bei Dosen, die unterhalb der eindeutigen parentalen Toxizität liegen, hatte Botulinum-Toxin bei Kaninchen keine nachteiligen Auswirkungen auf die Fertilität oder Reproduktionsfunktion. Die tägliche intramuskuläre Verabreichung von Botulinum-Toxin an trächtige Ratten oder Kaninchen während der Organogenese führte zu einem verringerten Körpergewicht des Fötus und einer verminderten Skelettverknöcherung, insbesondere bei höheren Dosen, die mit einer signifikanten maternalen Toxizität verbunden waren.
Natriummonohydrogenphosphat-Dihydrat (Ph.Eur.)
Natriumdihydrogenphosphat-Dihydrat
Kaliumchlorid
Natriumchlorid
Polysorbat 80 (E 433)
Tryptophan
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
18 Monate
Im Kühlschrank lagern (2 °C – 8 °C).
Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Die ungeöffnete Durchstechflasche kann lichtgeschützt auf Raumtemperatur (bis zu 25 °C) gebracht werden. Die Stabilität von Relfydess (ungeöffnete Durchstechflasche) bei Raumtemperatur wurde für bis zu 24 Stunden nachgewiesen.
Art des Behältnisses/Verschlusses
Durchstechflasche aus Typ-I-Glas, mit einem Brombutylgummi-Stopfen und einer Aluminium-Bördelkappe mit einer Schutzkappe aus Polypropylen.
Inhalt des Behältnisses
Jede Durchstechflasche enthält 1,5 ml Injektionslösung mit 150 Einheiten Botulinum-Toxin Typ A zur Injektion (Ph.Eur.).
Packungsgrößen
Packungen mit 1 oder 10 Durchstechflaschen.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Unmittelbar nach der Anwendung muss nicht verwendetes Relfydess (in der Durchstechflasche oder in der Spritze) mit verdünnter Natriumhypochloritlösung (0,1 %) oder Natriumhydroxidlösung (1 %) inaktiviert werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Empfehlungen für einen eventuellen Zwischenfall bei der Handhabung von Botulinum-Toxin
Verschüttetes Arzneimittel muss mit trockenem, saugfähigem Material aufgewischt werden. Das Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Kontaminierte Oberflächen müssen mit einer verdünnten Natriumhypochlorit- oder Natriumhydroxidlösung gereinigt und anschließend getrocknet werden.
Wenn eine Durchstechflasche zerbrochen ist, gehen Sie wie oben beschrieben vor, indem Sie die Scherben vorsichtig aufsammeln und das Arzneimittel aufwischen, wobei Schnittverletzungen der Haut zu vermeiden sind.
Bei Kontakt des Arzneimittels mit der Haut waschen Sie die betroffene Stelle mit Wasser und Seife.
Bei Kontakt des Arzneimittels mit den Augen gründlich mit viel Wasser oder einer Augenspüllösung spülen.
Bei Kontakt des Arzneimittels mit einer Wunde, einem Schnitt oder einer Hautverletzung spülen Sie gründlich mit viel Wasser und suchen Sie einen Arzt auf.
Diese Anweisungen zur Anwendung, Handhabung und Beseitigung müssen strikt eingehalten werden.
Ipsen Pharma
70 rue Balard
75015 Paris
Frankreich
Mitvertreiber
Galderma Laboratorium GmbH
Toulouser Allee 23a
40211 Düsseldorf
Deutschland
Tel.: +49 800 5888850
Fax: +49 211 6355 8270
E-Mail: patientenservice@galderma.com
7014449.00.00
Datum der Erteilung der Zulassung: 06. September 2024
04.2025
Verschreibungspflichtig