
Ezetimib/Atorvastatin Viatris 10 mg/10 mg Filmtabletten
Ezetimib/Atorvastatin Viatris 10 mg/20 mg Filmtabletten
Ezetimib/Atorvastatin Viatris 10 mg/40 mg Filmtabletten
Ezetimib/Atorvastatin Viatris 10 mg/80 mg Filmtabletten
Ezetimib/Atorvastatin Viatris 10 mg/10 mg Filmtabletten
Jede Filmtablette enthält 10 mg Ezetimib und 10 mg Atorvastatin (als Calcium-Trihydrat).
Sonstige Bestandteile mit bekannter Wirkung
Jede 10 mg/10 mg Filmtablette enthält 2,74 mg Lactose.
Ezetimib/Atorvastatin Viatris 10 mg/20 mg Filmtabletten
Jede Filmtablette enthält 10 mg Ezetimib und 20 mg Atorvastatin (als Calcium-Trihydrat).
Sonstige Bestandteile mit bekannter Wirkung
Jede 10 mg/20 mg Filmtablette enthält 3,76 mg Lactose.
Ezetimib/Atorvastatin Viatris 10 mg/40 mg Filmtabletten
Jede Filmtablette enthält 10 mg Ezetimib und 40 mg Atorvastatin (als Calcium-Trihydrat).
Sonstige Bestandteile mit bekannter Wirkung
Jede 10 mg/40 mg Filmtablette enthält 5,81 mg Lactose.
Ezetimib/Atorvastatin Viatris 10 mg/80 mg Filmtabletten
Jede Filmtablette enthält 10 mg Ezetimib und 80 mg Atorvastatin (als Calcium-Trihydrat).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette
Ezetimib/Atorvastatin Viatris 10 mg/10 mg Filmtabletten
Weiße, runde, bikonvexe Filmtabletten mit einem Durchmesser von etwa 8,1 mm.
Ezetimib/Atorvastatin Viatris 10 mg/20 mg Filmtabletten
Weiße, ovale, bikonvexe Filmtabletten mit Abmessungen von etwa 11,6 x 7,1 mm.
Ezetimib/Atorvastatin Viatris 10 mg/40 mg Filmtabletten
Weiße, kapselförmige, bikonvexe Filmtabletten mit Abmessungen von etwa 16,1 x 6,1 mm.
Ezetimib/Atorvastatin Viatris 10 mg/80 mg Filmtabletten
Gelbe, längliche, bikonvexe Filmtabletten mit Abmessungen von etwa 19,1 x 7,6 mm.
Prävention kardiovaskulärer Ereignisse
Ezetimib/Atorvastatin Viatris ist angezeigt zur Risikoreduktion von kardiovaskulären Ereignissen (siehe Abschnitt 5.1) bei Patienten mit koronarer Herzkrankheit (KHK) und akutem Koronarsyndrom in der Vorgeschichte, unabhängig von einer Vorbehandlung mit einem Statin.
Hypercholesterinämie
Ezetimib/Atorvastatin Viatris ist begleitend zu einer Diät angezeigt zur Anwendung bei erwachsenen Patienten mit primärer (heterozygoter familiärer und nicht familiärer) Hypercholesterinämie oder gemischter Hyperlipidämie, für die eine Therapie mit einem Kombinationspräparat geeignet ist:
Patienten, bei denen eine Therapie mit einem Statin allein nicht ausreicht
Patienten, die bereits mit einem Statin und Ezetimib behandelt werden
Homozygote familiäre Hypercholesterinämie (HoFH)
Ezetimib/Atorvastatin Viatris ist begleitend zu einer Diät angezeigt zur Anwendung bei erwachsenen Patienten mit homozygoter familiärer Hypercholesterinämie. Die Patienten können weitere begleitende Therapien (wie Low Density Lipoprotein [LDL]‑Apherese) erhalten.
Dosierung
Hypercholesterinämie und/oder koronare Herzkrankheit (KHK) und akutes Koronarsyndrom in der Vorgeschichte
Der Patient sollte eine geeignete lipidsenkende Diät einhalten, die er auch während der Therapie mit Ezetimib/Atorvastatin Viatris fortsetzen sollte.
Der Dosierungsbereich von Ezetimib/Atorvastatin Viatris reicht von 10 mg/10 mg einmal täglich bis zu 10 mg/80 mg einmal täglich. Die übliche Dosis beträgt 10 mg/10 mg einmal täglich. Bei Therapiebeginn oder bei einer Dosisänderung sind die LDL‑Cholesterinwerte des Patienten, sein Risiko für die Entwicklung einer koronaren Herzkrankheit sowie sein Ansprechen auf die bisherige lipidsenkende Therapie zu berücksichtigen.
Die Dosis von Ezetimib/Atorvastatin Viatris sollte individuell auf Basis der bekannten Wirksamkeit der verschiedenen Stärken von Ezetimib/Atorvastatin Viatris (siehe Abschnitt 5.1, Tabelle 4) sowie dem Ansprechen auf die bisherige lipidsenkende Therapie gewählt werden. Dosisanpassungen sollten in Abständen von mindestens 4 Wochen durchgeführt werden.
Homozygote familiäre Hypercholesterinämie
Die Dosis für Patienten mit homozygoter familiärer Hypercholesterinämie beträgt 10 mg/10 mg bis 10 mg/80 mg Ezetimib/Atorvastatin Viatris einmal täglich. Ezetimib/Atorvastatin Viatris kann bei diesen Patienten sowohl begleitend zu anderen lipidsenkenden Maßnahmen (z. B. LDL‑Apherese) angewendet werden oder auch, wenn solche Behandlungsmaßnahmen nicht zur Verfügung stehen.
Gleichzeitige Therapie mit anderen Arzneimitteln
Die Einnahme von Ezetimib/Atorvastatin Viatris sollte mindestens 2 Stunden vor oder mindestens 4 Stunden nach der Einnahme eines Gallensäure-bindenden Arzneimittels erfolgen.
Bei Patienten, welche die Virostatika Elbasvir/Grazoprevir zur Behandlung einer Hepatitis‑C-Infektion gemeinsam mit Ezetimib/Atorvastatin Viatris einnehmen, sollte die Dosis von Ezetimib/Atorvastatin Viatris 10 mg/20 mg/Tag nicht überschritten werden (siehe Abschnitte 4.4 und 4.5).
Ältere Patienten
Für ältere Patienten ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Ezetimib/Atorvastatin Viatris bei Kindern und Jugendlichen ist nicht erwiesen (siehe Abschnitt 5.2). Es liegen keine Daten vor.
Eingeschränkte Leberfunktion
Ezetimib/Atorvastatin Viatris sollte bei Patienten mit eingeschränkter Leberfunktion mit Vorsicht angewendet werden (siehe Abschnitte 4.4 und 5.2). Ezetimib/Atorvastatin Viatris ist bei Patienten mit aktiver Lebererkrankung kontraindiziert (siehe Abschnitt 4.3).
Eingeschränkte Nierenfunktion
Für Patienten mit eingeschränkter Nierenfunktion ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Art der Anwendung
Zum Einnehmen.
Ezetimib/Atorvastatin Viatris wird oral eingenommen. Ezetimib/Atorvastatin Viatris kann als Einzeldosis zu jeder Tageszeit und unabhängig von der Nahrungsaufnahme eingenommen werden.
Überempfindlichkeit gegen die Wirkstoffe oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Ezetimib/Atorvastatin Viatris ist während der Schwangerschaft und Stillzeit, sowie bei Frauen im gebärfähigen Alter, die keine zuverlässige Verhütungsmethode anwenden, kontraindiziert (siehe Abschnitt 4.6).
Ezetimib/Atorvastatin Viatris ist bei Patienten mit aktiver Lebererkrankung oder bei unklarer, anhaltender Erhöhung der Transaminasen auf mehr als das Dreifache des oberen Normwertes kontraindiziert.
Ezetimib/Atorvastatin Viatris ist kontraindiziert bei Patienten, die mit den Virostatika Glecaprevir/Pibrentasvir zur Behandlung einer Hepatitis‑C-Infektion behandelt werden.
Myasthenia gravis oder okuläre Myasthenie
In wenigen Fällen wurde berichtet, dass Statine eine Myasthenia gravis oder eine Verschlechterung einer bereits bestehenden Myasthenia gravis oder okulärer Myasthenie auslösen (siehe Abschnitt 4.8). Ezetimib/Atorvastatin Viatris sollte bei einer Verschlimmerung der Symptome abgesetzt werden. Es wurde über Rezidive berichtet, wenn dasselbe oder ein anderes Statin (erneut) gegeben wurde.
Myopathie/Rhabdomyolyse
Nach Markteinführung von Ezetimib wurden Fälle von Myopathie und Rhabdomyolyse berichtet. Die meisten Patienten, die eine Rhabdomyolyse entwickelten, nahmen gleichzeitig mit Ezetimib ein Statin ein. Eine Rhabdomyolyse wurde jedoch sehr selten unter Monotherapie mit Ezetimib sowie sehr selten nach Zugabe von Ezetimib zu anderen Arzneimitteln berichtet, die für ein erhöhtes Rhabdomyolyserisiko bekannt sind.
Ezetimib/Atorvastatin Viatris enthält Atorvastatin. Wie andere HMG‑CoA-Reduktase-Inhibitoren kann auch Atorvastatin in seltenen Fällen die Skelettmuskulatur beeinträchtigen und eine Myalgie, Myositis oder Myopathie hervorrufen. Eine Myopathie kann sich zu einer Rhabdomyolyse entwickeln, einer potenziell tödlichen Erkrankung, die durch eine deutliche Erhöhung der Kreatinphosphokinase (CPK) (> das Zehnfache des oberen Normwertes), Myoglobinämie und Myoglobinurie gekennzeichnet ist und zum Nierenversagen führen kann.
Vor Beginn der Therapie
Ezetimib/Atorvastatin Viatris sollte mit Vorsicht bei Patienten mit Risikofaktoren für eine Rhabdomyolyse verordnet werden. Eine Bestimmung der CPK sollte bei Vorliegen folgender Risikofaktoren vor Behandlungsbeginn durchgeführt werden:
eingeschränkte Nierenfunktion,
Hypothyreose,
hereditäre Muskelerkrankungen in der eigenen oder in der Familienanamnese,
Muskelerkrankungen unter Behandlung mit Statinen oder Fibraten in der Anamnese,
Lebererkrankungen in der Anamnese und/oder bei starkem Alkoholkonsum,
ältere Patienten (> 70 Jahre); die Notwendigkeit einer solchen Untersuchung sollte abhängig davon, ob andere Risikofaktoren für eine Rhabdomyolyse vorhanden sind, in Betracht gezogen werden,
Umstände, die zu erhöhten Plasmaspiegeln führen können, wie Wechselwirkungen (siehe Abschnitt 4.5) und besondere Patientengruppen einschließlich genetischer Subpopulationen (siehe Abschnitt 5.2).
In solchen Fällen werden eine sorgfältige Nutzen-Risiko-Abwägung der Behandlung sowie eine klinische Überwachung der betroffenen Patienten empfohlen.
Wenn die Ausgangswerte der CPK signifikant erhöht sind (> das Fünffache des oberen Normwertes), sollte die Behandlung nicht begonnen werden.
Messungen der Kreatinphosphokinase (CPK)
Die Kreatinphosphokinase (CPK) sollte nicht nach körperlicher Anstrengung oder bei Vorliegen anderer plausibler Ursachen für eine CPK‑Erhöhung gemessen werden, da dies eine Interpretation der Werte erschwert. Wenn die Ausgangswerte der CPK signifikant erhöht sind (> das Fünffache des oberen Normwertes), sollte die Messung nach 5‑7 Tagen wiederholt werden, um die Ergebnisse zu bestätigen.
Während der Behandlung
Patienten müssen darüber aufgeklärt werden, Muskelschmerzen, Muskelkrämpfe und Muskelschwäche unverzüglich zu melden, besonders wenn diese mit Unwohlsein und Fieber einhergehen oder falls Anzeichen und Symptome muskulärer Nebenwirkungen nach dem Absetzen von Ezetimib/Atorvastatin Viatris fortbestehen.
Wenn solche Symptome während der Behandlung mit Ezetimib/Atorvastatin Viatris auftreten, sollten die CPK‑Werte der Patienten bestimmt werden. Wenn die CPK‑Werte signifikant erhöht sind (> das Fünffache des oberen Normwertes), sollte die Behandlung abgebrochen werden.
Bei schwerer muskulärer Symptomatik und täglichen Beschwerden sollte ein Abbrechen der Behandlung in Betracht gezogen werden, auch wenn die CPK‑Werte weniger als auf das Fünffache des oberen Normwertes erhöht sind.
Sofern die Symptome abklingen und die CPK‑Werte sich normalisieren, kann die erneute Behandlung mit Ezetimib/Atorvastatin Viatris oder mit einem alternativen statinhaltigen Produkt in der jeweils niedrigsten Dosis und unter engmaschiger Überwachung in Betracht gezogen werden.
Die Behandlung mit Ezetimib/Atorvastatin Viatris muss abgebrochen werden, wenn die CPK‑Werte klinisch signifikant erhöht sind (> das Zehnfache des oberen Normwertes) oder falls eine Rhabdomyolyse diagnostiziert oder vermutet wird.
In sehr seltenen Fällen wurde während oder nach der Behandlung mit einigen Statinen über eine immunvermittelte nekrotisierende Myopathie (immune-mediated necrotizing myopathy; IMNM) berichtet. Die klinischen Charakteristika einer IMNM sind persistierende proximale Muskelschwäche und erhöhte Serum-Kreatinkinase-Werte, die trotz Absetzen der Behandlung mit Statinen fortbestehen.
Aufgrund des in Ezetimib/Atorvastatin Viatris enthaltenen Wirkstoffs Atorvastatin ist das Rhabdomyolyserisiko erhöht, wenn Ezetimib/Atorvastatin Viatris zusammen mit bestimmten anderen Arzneimitteln eingenommen wird, die Wirkstoffe enthalten, welche den Plasmaspiegel von Atorvastatin erhöhen können, wie potente CYP3A4‑Inhibitoren oder Inhibitoren von Transportproteinen (z. B. Ciclosporin, Telithromycin, Clarithromycin, Delavirdin, Stiripentol, Ketoconazol, Voriconazol, Itraconazol, Posaconazol und HIV‑Protease-Inhibitoren wie Ritonavir, Lopinavir, Atazanavir, Indinavir, Darunavir, Tipranavir/Ritonavir etc.). Das Myopathierisiko kann ebenfalls erhöht sein, bei gemeinsamer Anwendung von Ezetimib/Atorvastatin Viatris mit Arzneimitteln mit den Wirkstoffen Gemfibrozil und anderen Fibraten, Virostatika zur Behandlung von Hepatitis C (HCV) (Boceprevir, Telaprevir, Elbasvir/Grazoprevir), Erythromycin oder Niacin. Alternative Therapien, ohne Wechselwirkungen, sind nach Möglichkeit anstelle dieser Arzneimittel in Betracht zu ziehen (siehe Abschnitt 4.8).
Falls eine gemeinsame Anwendung dieser Arzneimittel mit Ezetimib/Atorvastatin Viatris notwendig ist, wird eine sorgfältige Nutzen-Risiko-Abwägung der gleichzeitigen Behandlung empfohlen. Sofern die Patienten Arzneimittel erhalten, welche die Plasmaspiegel von Atorvastatin erhöhen, wird eine niedrigere Maximaldosis von Ezetimib/Atorvastatin Viatris empfohlen. Im Fall von potenten CYP3A4‑Inhibitoren sollten außerdem eine niedrigere Anfangsdosis von Ezetimib/Atorvastatin Viatris in Betracht gezogen und die Patienten angemessen klinisch überwacht werden (siehe Abschnitt 4.5).
Atorvastatin darf nicht zusammen mit systemischen Darreichungsformen von Fusidinsäure gegeben werden, auch nicht innerhalb von 7 Tagen nach Absetzen der Therapie mit Fusidinsäure. Sofern die systemische Gabe von Fusidinsäure bei Patienten als essenziell erachtet wird, ist die Statintherapie während der gesamten Behandlungsdauer mit Fusidinsäure abzusetzen. Bei Patienten, die Fusidinsäure und Statine in Kombination erhielten, wurde über das Auftreten einer Rhabdomyolyse berichtet (darunter einige Fälle mit Todesfolge) (siehe Abschnitt 4.5). Die Patienten sollten darüber informiert werden, sich umgehend an einen Arzt zu wenden, wenn sie jedwede Anzeichen von Muskelschwäche, ‑schmerzen oder ‑empfindlichkeit bemerken.
Die Statintherapie kann 7 Tage nach der letzten Dosis Fusidinsäure wieder aufgenommen werden.
Sofern in Ausnahmefällen eine längere systemische Gabe von Fusidinsäure notwendig ist, wie z. B. zur Behandlung von schweren Infektionen, sollte eine gemeinsame Gabe von Ezetimib/Atorvastatin mit Fusidinsäure nur im Einzelfall und unter engmaschiger medizinischer Überwachung in Betracht gezogen werden.
Daptomycin
Im Zusammenhang mit der gleichzeitigen Gabe von HMG‑CoA-Reduktase-Inhibitoren (z. B. Atorvastatin und Ezetimib/Atorvastatin) und Daptomycin wurden Fälle von Myopathie und/oder Rhabdomyolyse berichtet. Bei der Verschreibung von HMG‑CoA-Reduktase-Inhibitoren zusammen mit Daptomycin ist Vorsicht geboten, da beide Wirkstoffe bereits bei alleiniger Gabe Myopathien und/oder Rhabdomyolyse verursachen können. Es sollte in Erwägung gezogen werden, die Einnahme von Ezetimib/Atorvastatin Viatris bei Patienten, welche Daptomycin erhalten, vorübergehend auszusetzen, es sei denn, der Nutzen der gleichzeitigen Gabe überwiegt das Risiko. Konsultieren Sie die Fachinformation von Daptomycin, um weitere Informationen bezüglich dieser potenziellen Wechselwirkungen mit HMG‑CoA-Reduktase-Inhibitoren (z. B. Atorvastatin und Ezetimib/Atorvastatin) und weitere Handlungsempfehlungen bezüglich der Überwachung zu erhalten (siehe Abschnitt 4.5).
Leberenzyme
In kontrollierten klinischen Studien zur Koadministration wurden bei Patienten, die Ezetimib zusammen mit Atorvastatin erhielten, Erhöhungen der Transaminasenwerte (≥ dem Dreifachen des oberen Normwertes in Folge) beobachtet (siehe Abschnitt 4.8).
Leberfunktionstests sollten vor dem Beginn der Behandlung mit Ezetimib/Atorvastatin Viatris als auch anschließend in regelmäßigen Abständen durchgeführt werden. Leberfunktionstests sollten bei Patienten durchgeführt werden, die jedwede Anzeichen oder Symptome entwickeln, die auf eine Lebererkrankung hinweisen. Patienten, die erhöhte Transaminasenspiegel entwickeln, sollten überwacht werden bis sich die Werte wieder normalisieren. Sollte ein Anstieg der Transaminasenspiegel über das Dreifache des oberen Normwertes andauern, wird eine Dosisreduktion oder das Absetzen von Ezetimib/Atorvastatin Viatris empfohlen.
Ezetimib/Atorvastatin Viatris sollte mit Vorsicht bei Patienten angewendet werden, die in erheblichem Maße Alkohol zu sich nehmen und/oder Lebererkrankungen in der Vorgeschichte haben.
Leberinsuffizienz
Aufgrund fehlender Daten zu Auswirkungen einer erhöhten Exposition von Ezetimib bei Patienten mit mäßiger oder schwerer Leberinsuffizienz wird Ezetimib/Atorvastatin Viatris für diese Patienten nicht empfohlen (siehe Abschnitt 5.2).
Fibrate
Sicherheit und Wirksamkeit der gemeinsamen Anwendung von Ezetimib mit Fibraten sind nicht erwiesen. Deshalb wird eine gemeinsame Anwendung von Ezetimib/Atorvastatin Viatris und Fibraten nicht empfohlen (siehe Abschnitt 4.5).
Ciclosporin
Eine Behandlung von Ezetimib/Atorvastatin Viatris sollte im Rahmen einer Therapie mit Ciclosporin mit Vorsicht begonnen werden. Die Ciclosporinspiegel sollten bei Patienten überwacht werden, die Ezetimib/Atorvastatin Viatris und Ciclosporin erhalten (siehe Abschnitt 4.5).
Antikoagulanzien
Bei Koadministration von Ezetimib/Atorvastatin Viatris mit Warfarin, einem anderen Cumarin-Antikoagulans oder Fluindion sollte die „International Normalized Ratio“ (INR) entsprechend überwacht werden (siehe Abschnitt 4.5).
Schlaganfallprävention durch aggressive Senkung der Cholesterinspiegel (Stroke Prevention by Aggressive Reduction in Cholesterol Levels [SPARCL])
Im Rahmen einer Post-hoc-Analyse verschiedener Schlaganfallsubtypen bei Patienten ohne koronare Herzkrankheit (KHK), die kürzlich einen Schlaganfall oder eine transitorische ischämische Attacke (TIA) erlitten hatten, wurde eine höhere Inzidenz von hämorrhagischen Schlaganfällen bei Patienten, die initial mit 80 mg Atorvastatin behandelt wurden, im Vergleich zu Placebo festgestellt. Das erhöhte Risiko wurde insbesondere bei Patienten beobachtet, die vor Studieneintritt bereits einen hämorrhagischen Schlaganfall oder einen lakunären Infarkt hatten. Das Nutzen-Risiko-Verhältnis von 80 mg Atorvastatin bei Patienten mit vorausgegangenem hämorrhagischen Schlaganfall oder lakunären Infarkt ist ungewiss, und das potenzielle Risiko eines hämorrhagischen Schlaganfalls sollte sorgfältig vor dem Beginn der Behandlung abgewogen werden (siehe Abschnit 5.1).
Interstitielle Lungenkrankheit
Bei einigen Statinen wurden besonders bei Langzeittherapie Fälle einer interstitiellen Lungenkrankheit berichtet (siehe Abschnitt 4.8). Die auftretenden Beschwerden können dabei Dyspnoe, unproduktiven Husten und allgemeine Gesundheitsstörungen (Erschöpfung, Gewichtsverlust und Fieber) einschließen. Wenn vermutet wird, dass ein Patient eine interstitielle Lungenkrankheit entwickelt hat, sollte die Statintherapie abgebrochen werden.
Diabetes mellitus
Es gibt Hinweise darauf, dass Statine als Substanzklasse den Blutzuckerspiegel erhöhen und bei manchen Patienten, die ein hohes Risiko für die Entwicklung eines zukünftigen Diabetes mellitus haben, eine Hyperglykämie hervorrufen können, die eine adäquate Diabetesbehandlung erfordert. Dieses Risiko wird jedoch von der Reduktion des vaskulären Risikos durch Statine aufgewogen und sollte daher nicht zu einem Abbruch der Statinbehandlung führen. In Übereinstimmung mit nationalen Richtlinien sollten Risikopatienten (Nüchternblutzucker von 5,6 bis 6,9 mmol/l, BMI > 30 kg/m2, erhöhte Triglyzeridwerte, Hypertonie) sowohl klinisch als auch in Bezug auf die relevanten Laborwerte überwacht werden.
Sonstiger Bestandteil
Ezetimib/Atorvastatin Viatris 10 mg/10 mg, 10 mg/20 mg und 10 mg/40 mg enthalten Lactose.
Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten diese Arzneimittel nicht einnehmen.
Ezetimib/Atorvastatin Viatris enthält weniger als 1 mmol (23 mg) Natrium pro Tablette, d. h., es ist nahezu „natriumfrei“.
Mehrere Mechanismen können zu potenziellen Wechselwirkungen mit HMG‑CoA-Reduktase-Inhibitoren beitragen. Arzneimittel oder pflanzliche Präparate, die bestimmte Enzyme (z. B. CYP3A4) und/oder Transporterproteine (z. B. OATP1B) hemmen, können die Plasmaspiegel von Atorvastatin erhöhen und dadurch zu einem erhöhten Myopathie-/Rhabdomyolyserisiko führen.
Weitere Informationen hinsichtlich potenzieller Wechselwirkungen mit Atorvastatin und/oder möglicher Auswirkungen auf Enzyme oder Transporterproteine sowie möglicher Dosis- oder Therapieanpassungen sind den jeweiligen Fachinformationen von allen gemeinsam angewendeten Arzneimitteln zu entnehmen.
Pharmakodynamische Wechselwirkungen
Atorvastatin wird durch das Cytochrom P450 Isoenzym 3A4 (CYP3A4) metabolisiert und ist ein Substrat der hepatischen Transporter OATP1B1 (organic anion-transporting polypeptide 1B1) und OATP1B3 (organic anion-transporting polypeptide 1B3). Metaboliten von Atorvastatin sind Substrate von OATP1B1. Atorvastatin wird außerdem als Substrat von MDR1 (multi-drug resistance protein 1) und BCRP (breast cancer resistance protein) identifiziert, wodurch die intestinale Resorption und biliäre Ausscheidung von Atorvastatin begrenzt sein könnten (siehe Abschnitt 5.2). Die gemeinsame Anwendung von Arzneimitteln, die als CYP3A4-Inhibitoren oder Inhibitoren von Transportproteinen bekannt sind, kann zu erhöhten Plasmaspiegeln von Atorvastatin und damit zu einem erhöhten Myopathierisiko führen. Dieses Risiko kann ebenfalls durch die gemeinsame Anwendung von Ezetimib/Atorvastatin zusammen mit anderen Arzneimitteln erhöht werden, die potenziell eine Myopathie verursachen können, wie z. B. Fibrate und Ezetimib (siehe Abschnit 4.4).
Pharmakokinetische Wechselwirkungen
Ezetimib/Atorvastatin Viatris
Bei gemeinsamer Anwendung von Ezetimib und Atorvastatin wurden keine klinisch relevanten pharmakokinetischen Wechselwirkungen beobachtet.
Wirkungen anderer Arzneimittel auf Ezetimib/Atorvastatin Viatris
Ezetimib
Antazida: Die gemeinsame Anwendung mit Antazida verminderte die Resorptionsrate von Ezetimib, beeinflusste aber nicht die Bioverfügbarkeit von Ezetimib. Der verminderten Resorptionsrate wird keine klinische Bedeutung beigemessen.
Colestyramin: Die gemeinsame Anwendung mit Colestyramin verringerte die mittlere Fläche unter der Kurve (AUC) von Gesamt-Ezetimib (Ezetimib und glukuronidiertes Ezetimib) um ca. 55 %. Die zunehmende Senkung des LDL‑Cholesterins durch Kombination von Ezetimib/Atorvastatin Viatris mit Colestyramin könnte durch diese Interaktion vermindert werden (siehe Abschnitt 4.2).
Ciclosporin: Eine Studie mit acht Patienten, die nach einer Nierentransplantation mit einer Kreatinin-Clearance > 50 ml/min stabil auf eine Ciclosporin-Dosis eingestellt waren, zeigte nach Gabe einer Einzeldosis von 10 mg Ezetimib eine 3,4fache Zunahme der mittleren AUC von Gesamt-Ezetimib (Bereich von 2,3‑ bis 7,9fach) verglichen mit einer gesunden Kontrollpopulation einer anderen Studie (n = 17) unter Ezetimib allein. In einer weiteren Studie wies ein Patient nach einer Nierentransplantation mit schwerer Niereninsuffizienz, der Ciclosporin und zahlreiche andere Arzneimittel erhielt, eine 12fach größere Gesamt-Ezetimib-Exposition auf im Vergleich zu den anderen Kontrollpersonen unter Ezetimib allein. In einer zweiphasigen Crossover-Studie mit 12 gesunden Probanden führte die tägliche Anwendung von 20 mg Ezetimib über 8 Tage mit einer Einzeldosis von 100 mg Ciclosporin an Tag 7 zu einer mittleren 15 %igen Zunahme der AUC von Ciclosporin (Bereich von 10 %iger Verringerung bis 51 %iger Zunahme) verglichen mit einer Einzeldosis von 100 mg Ciclosporin allein. Es wurden keine kontrollierten Studien über die Wirkung einer gemeinsamen Anwendung mit Ezetimib bei Patienten nach einer Nierentransplantation auf die Ciclosporin-Exposition durchgeführt. Eine Behandlung mit Ezetimib/Atorvastatin Viatris sollte während einer Therapie mit Ciclosporin mit Vorsicht begonnen werden. Bei Patienten unter Ezetimib/Atorvastatin Viatris und Ciclosporin sollten die Ciclosporinspiegel überwacht werden (siehe Abschnitt 4.4).
Fibrate: Die gemeinsame Anwendung von Fenofibrat oder Gemfibrozil erhöhte die Konzentration von Gesamt-Ezetimib auf das ca. 1,5‑ bzw. 1,7‑Fache. Auch wenn diesen Erhöhungen keine klinische Bedeutung beigemessen wird, wird die gemeinsame Anwendung von Ezetimib/Atorvastatin Viatris mit Fibraten nicht empfohlen (siehe Abschnitt 4.4).
Atorvastatin
CYP3A4-Inhibitoren: Für potente CYP3A4-Inhibitoren wurde gezeigt, dass sie zu einer deutlichen Erhöhung der Atorvastatinspiegel führen (siehe Tabelle 1 und weitere spezielle Informationen unten). Die gemeinsame Anwendung von potenten CYP3A4-Inhibitoren (z. B. Ciclosporin, Telithromycin, Clarithromycin, Delavirdin, Stiripentol, Ketoconazol, Voriconazol, Itraconazol, Posaconazol, einigen Virostatika zur Behandlung von HCV [z. B. Elbasvir/Grazoprevir] und HIV‑Protease-Inhibitoren wie Ritonavir, Lopinavir, Atazanavir, Indinavir, Darunavir etc.) sollte nach Möglichkeit vermieden werden. Falls eine gemeinsame Anwendung mit diesen Arzneimitteln und Ezetimib/Atorvastatin Viatris nicht vermieden werden kann, sollten niedrigere Anfangs- und Maximaldosen von Ezetimib/Atorvastatin Viatris in Betracht gezogen und die Patienten angemessen klinisch überwacht werden (siehe Tabelle 1).
Moderate CYP3A4-Inhibitoren (z. B. Erythromycin, Diltiazem, Verapamil und Fluconazol) können die Plasmaspiegel von Atorvastatin erhöhen (siehe Tabelle 1). Ein erhöhtes Myopathierisiko wurde bei der Anwendung von Erythromycin in Kombination mit Statinen beobachtet. Interaktionsstudien zur Untersuchung der Wechselwirkungen von Amiodaron oder Verapamil mit Atorvastatin wurden nicht durchgeführt. Sowohl Amiodaron als auch Verapamil sind bekannt dafür, die Aktivität von CYP3A4 zu hemmen, so dass eine gemeinsame Anwendung von Ezetimib/Atorvastatin Viatris zu einer erhöhten Exposition an Atorvastatin führen kann. Deshalb sollte bei gemeinsamer Anwendung mit moderaten CYP3A4-Inhibitoren eine niedrigere Maximaldosis von Ezetimib/Atorvastatin Viatris in Betracht gezogen und die Patienten angemessen klinisch überwacht werden. Eine angemessene klinische Überwachung wird bei Behandlungsbeginn oder nachfolgenden Dosisanpassungen des Inhibitors empfohlen.
BCRP (Brustkrebs-Resistenz Protein – „Breast Cancer Resistance Protein“)-Inhibitoren: Die gemeinsame Anwendung von Ezetimib/Atorvastatin Viatris mit Arzneimitteln aus der Klasse der BCRP‑Inhibitoren (z. B. Elbasvir und Grazoprevir) kann zu erhöhten Plasmakonzentrationen von Atorvastatin und somit zu einem erhöhten Myopathierisiko führen. In Abhängigkeit von der verordneten Dosis sollte deshalb eine Dosisanpassung von Atorvastatin erwogen werden. Die gemeinsame Anwendung von Elbasvir und Grazoprevir mit Atorvastatin erhöht die Plasmakonzentrationen von Atorvastatin um das 1,9‑Fache (siehe Tabelle 1). Demzufolge sollte eine Tageshöchstdosis von Ezetimib/Atorvastatin Viatris 10 mg/20 mg bei Patienten, die gleichzeitig Arzneimittel mit den Wirkstoffen Elbasvir oder Grazoprevir erhalten, nicht überschritten werden (siehe Abschnitte 4.2 und 4.4).
CYP3A4-Induktoren: Die gemeinsame Anwendung von Atorvastatin mit CYP3A4-Induktoren (z. B. Efavirenz, Rifampicin, Johanniskraut) kann zu schwankenden (variablen) Erniedrigungen der Plasmaspiegel von Atorvastatin führen. Aufgrund des dualen Wechselwirkungsmechanismus von Rifampicin (CYP3A4-Induktion und Inhibition des hepatozytischen Aufnahmetransporters OATP1B1) wird die zeitgleiche Anwendung von Ezetimib/Atorvastatin Viatris mit Rifampicin empfohlen, da die verzögerte Gabe von Atorvastatin nach der Gabe von Rifampicin mit einer signifikanten Erniedrigung der Plasmaspiegel von Atorvastatin in Zusammenhang gebracht wurde. Die Auswirkungen von Rifampicin auf Atorvastatinspiegel in Hepatozyten sind jedoch nicht bekannt und falls eine gemeinsame Anwendung nicht vermieden werden kann, sollten die Patienten sorgfältig im Hinblick auf die Wirksamkeit überwacht werden.
Inhibitoren von Transportern: Inhibitoren von Transportproteinen (z. B. Ciclosporin) können die systemische Exposition von Atorvastatin erhöhen (siehe Tabelle 1). Die Auswirkungen der Inhibition von hepatischen Aufnahmetransportern auf die Atorvastatinspiegel in Hepatozyten sind nicht bekannt. Sofern eine gemeinsame Anwendung nicht vermieden werden kann, werden eine Dosisreduktion von Ezetimib/Atorvastatin Viatris und eine klinische Überwachung hinsichtlich der Wirksamkeit empfohlen (siehe Tabelle 1).
Gemfibrozil/Fibrate: Die Anwendung von Fibraten allein wird gelegentlich mit muskelbezogenen Nebenwirkungen einschließlich Rhabdomyolyse in Verbindung gebracht. Ein erhöhtes Risiko für derartige Nebenwirkungen kann durch die gemeinsame Anwendung von Fibraten mit Atorvastatin bestehen.
Ezetimib: Die Anwendung von Ezetimib allein wird mit muskelbezogenen Nebenwirkungen einschließlich Rhabdomyolyse in Verbindung gebracht. Daher kann durch die gemeinsame Anwendung von Ezetimib mit Atorvastatin ein erhöhtes Risiko für derartige Nebenwirkungen bestehen. Eine angemessene klinische Überwachung dieser Patienten wird empfohlen.
Colestipol: Bei der gemeinsamen Anwendung von Colestipol mit Atorvastatin waren die Plasmaspiegel von Atorvastatin und seiner aktiven Metaboliten erniedrigt (um ca. 25 %). Die lipidsenkende Wirkung war hingegen höher bei der gemeinsamen Anwendung von Colestipol und Atorvastatin im Vergleich zur jeweiligen alleinigen Anwendung der beiden Arzneimittel.
Fusidinsäure: Das Risiko einer Myopathie einschließlich Rhabdomyolyse kann bei gleichzeitiger systemischer Gabe von Fusidinsäure und Statinen erhöht sein. Der dieser Wechselwirkung zugrundeliegende Mechanismus (ob pharmakodynamisch oder pharmakokinetisch oder beides) ist derzeit noch nicht geklärt. Bei Patienten, die diese Kombination erhielten, wurde über das Auftreten einer Rhabdomyolyse berichtet (darunter einige Fälle mit Todesfolge). Sofern eine systemische Behandlung mit Fusidinsäure notwendig ist, ist die Behandlung mit Atorvastatin während der gesamten Behandlungsdauer mit Fusidinsäure abzusetzen. Siehe ebenfalls Abschnitt 4.4.
Colchicin: Es wurden keine Interaktionsstudien mit Atorvastatin und Colchicin durchgeführt. Da Fälle von Myopathie bei gemeinsamer Anwendung von Atorvastatin mit Colchicin berichtet wurden, ist bei der Verschreibung von Atorvastatin zusammen mit Colchicin Vorsicht angezeigt.
Daptomycin: Das Risiko einer Myopathie und/oder Rhabdomyolyse kann bei gleichzeitiger Gabe von HMG‑CoA-Reduktase-Inhibitoren und Daptomycin erhöht sein. Es sollte in Erwägung gezogen werden, die Einnahme von Ezetimib/Atorvastatin Viatris bei Patienten, welche Daptomycin erhalten, vorübergehend auszusetzen, es sei denn der Nutzen der gleichzeitigen Gabe überwiegt das Risiko (siehe Abschnitt 4.4).
Boceprevir: Bei gemeinsamer Anwendung mit Boceprevir war die Exposition mit Atorvastatin erhöht. Sofern eine gemeinsame Anwendung mit Ezetimib/Atorvastatin Viatris erforderlich ist, sollte die Behandlung mit der niedrigsten Dosis von Ezetimib/Atorvastatin Viatris begonnen werden. Die Dosis sollte unter sicherheitsbezogener Überwachung bis zur erwünschten klinischen Wirkung nach oben titriert werden, ohne jedoch eine Tageshöchstdosis von 10 mg/20 mg zu überschreiten. Bei Patienten, die bereits Ezetimib/Atorvastatin Viatris erhalten, sollte eine Tageshöchstdosis von 10 mg/20 mg während einer gemeinsamen Behandlung mit Boceprevir nicht überschritten werden.
Wirkungen von Ezetimib/Atorvastatin Viatris auf die Pharmakokinetik anderer Arzneimittel
Ezetimib
In präklinischen Studien wurde gezeigt, dass Ezetimib die Cytochrom‑P450-Stoffwechselenzyme nicht induziert. Es wurden keine klinisch bedeutenden pharmakokinetischen Wechselwirkungen zwischen Ezetimib und Arzneimitteln beobachtet, die bekanntermaßen über Cytochrom P450 1A2, 2D6, 2C8, 2C9 und 3A4 oder N‑Acetyltransferase metabolisiert werden.
Antikoagulanzien: In einer Studie mit 12 gesunden erwachsenen Männern hatte die gemeinsame Anwendung von Ezetimib (10 mg einmal täglich) keine signifikante Wirkung auf die Bioverfügbarkeit von Warfarin und auf die Prothrombinzeit. Nach Markteinführung wurde jedoch über Erhöhungen der „International Normalized Ratio“ (INR) bei Patienten unter Warfarin- oder Fluindion-Therapie berichtet, die zusätzlich Ezetimib erhielten. Bei Zugabe von Ezetimib/Atorvastatin Viatris zu Warfarin, einem anderen Cumarin-Antikoagulans oder Fluindion ist die „International Normalized Ratio“ (INR) entsprechend zu überwachen (siehe Abschnitt 4.4).
Atorvastatin
Digoxin: Bei gemeinsamer Gabe von mehreren Dosen Digoxin und 10 mg Atorvastatin stiegen die Steady-State-Plasmaspiegel von Digoxin leicht an. Patienten unter Digoxin sollten in geeigneter Weise überwacht werden.
Orale Kontrazeptiva: Die gemeinsame Anwendung von Atorvastatin mit einem oralen Kontrazeptivum führte zu erhöhten Plasmaspiegeln von Norethisteron und Ethinylestradiol.
Warfarin: Im Rahmen einer klinischen Studie mit Patienten unter chronischer Warfarintherapie führte die gemeinsame Gabe mit täglich 80 mg Atorvastatin während der ersten 4 Tage zu einer leichten Verkürzung der Prothrombinzeit um ca. 1,7 Sekunden, welche sich nach 15 Tagen Behandlung mit Atorvastatin wieder normalisierte. Obwohl nur in sehr seltenen Fällen über klinisch signifikante Wechselwirkungen mit der Gerinnungshemmung berichtet wurde, sollte bei Patienten unter Cumarin-Antikoagulanzien die Prothrombinzeit vor Behandlungsbeginn und ausreichend häufig in der frühen Therapiephase mit Ezetimib/Atorvastatin Viatris bestimmt werden, um sicherzustellen, dass keine signifikante Änderung der Prothrombinzeit auftritt. Bei nachgewiesener Stabilisierung der Prothrombinzeit kann diese in den üblichen, für Patienten unter Therapie mit Cumarin-Antikoagulanzien empfohlenen Zeitabständen gemessen werden. Sofern die Dosis von Ezetimib/Atorvastatin Viatris geändert oder die Behandlung abgebrochen wird, sollte in gleicher Weise wiederholt verfahren werden. Bei Patienten, die keine Antikoagulanzien erhalten, steht eine Therapie mit Atorvastatin in keiner Verbindung zu Blutungen oder Änderungen der Prothrombinzeit.
Tabelle 1: Auswirkungen von Arzneimitteln auf die Pharmakokinetik von Atorvastatin bei gemeinsamer Anwendung
Gemeinsam angewendetes Arzneimittel und Dosierung |
Atorvastatin |
Ezetimib/Atorvastatin |
|
Dosis (mg) |
Veränderung der AUC& |
Klinische Empfehlungen# |
|
Tipranavir 500 mg 2‑mal täglich/Ritonavir 200 mg 2‑mal täglich, 8 Tage (Tage 14 bis 21) |
40 mg an Tag 1, 10 mg an Tag 20 |
↑ 9,4fach |
Bei notwendiger gemeinsamer Gabe mit Ezetimib/Atorvastatin ist eine Tageshöchstdosis von Ezetimib/Atorvastatin 10 mg/10 mg nicht zu überschreiten. Klinische Überwachung der Patienten wird empfohlen. |
Ciclosporin 5,2 mg/kg/Tag, gleichbleibende Dosis |
10 mg einmal täglich über 28 Tage |
↑ 8,7fach |
|
Lopinavir 400 mg 2‑mal täglich/Ritonavir 100 mg 2‑mal täglich, 14 Tage |
20 mg einmal täglich über 4 Tage |
↑ 5,9fach |
Bei notwendiger gemeinsamer Gabe mit Ezetimib/Atorvastatin wird eine niedrigere Erhaltungsdosis von Ezetimib/Atorvastatin empfohlen. Bei Dosen von Ezetimib/Atorvastatin über 10 mg/20 mg wird die klinische Überwachung der Patienten empfohlen. |
Clarithromycin 500 mg 2‑mal täglich, 9 Tage |
80 mg einmal täglich über 8 Tage |
↑ 4,4fach |
|
Saquinavir 400 mg 2‑mal täglich/Ritonavir 300 mg 2‑mal täglich (von Tag 5 bis 7 mit Anstieg auf 400 mg an Tag 8), an den Tagen 5 bis 18 jeweils 30 Minuten nach der Gabe von Atorvastatin |
40 mg einmal täglich über 4 Tage |
↑ 3,9fach |
Bei notwendiger gemeinsamer Gabe mit Ezetimib/Atorvastatin wird eine niedrigere Erhaltungsdosis von Ezetimib/Atorvastatin empfohlen. Bei Dosen von Ezetimib/Atorvastatin über 10 mg/40 mg wird die klinische Überwachung der Patienten empfohlen. |
Darunavir 300 mg 2‑mal täglich/Ritonavir 100 mg 2‑mal täglich, 9 Tage |
10 mg einmal täglich über 4 Tage |
↑ 3,3fach |
|
Itraconazol 200 mg einmal täglich, 4 Tage |
40 mg als Einmalgabe |
↑ 3,3fach |
|
Fosamprenavir 700 mg 2‑mal täglich/Ritonavir 100 mg 2‑mal täglich, 14 Tage |
10 mg einmal täglich über 4 Tage |
↑ 2,5fach |
|
Fosamprenavir 1.400 mg 2‑mal täglich, 14 Tage |
10 mg einmal täglich über 4 Tage |
↑ 2,3fach |
|
Nelfinavir 1.250 mg 2‑mal täglich, 14 Tage |
10 mg einmal täglich über 28 Tage |
↑ 1,7fach^ |
Keine besondere Empfehlung |
Grapefruitsaft 240 ml einmal täglich* |
40 mg als Einmalgabe |
↑ 37 % |
Der Genuss von größeren Mengen Grapefruitsaft bei gleichzeitiger Einnahme von Ezetimib/Atorvastatin Viatris wird nicht empfohlen. |
Diltiazem 240 mg einmal täglich, 28 Tage |
40 mg als Einmalgabe |
↑ 51 % |
Nach Behandlungsbeginn oder nach Dosisanpassungen von Diltiazem wird eine angemessene klinische Überwachung der Patienten empfohlen. |
Erythromycin 500 mg 4‑mal täglich, 7 Tage |
10 mg als Einmalgabe |
↑ 33 %^ |
Ein Absenken der Maximaldosis und die klinische Überwachung der Patienten werden empfohlen. |
Amlodipin 10 mg einmalig |
80 mg als Einmalgabe |
↑ 18 % |
Keine besondere Empfehlung. |
Cimetidin 300 mg 4‑mal täglich, 2 Wochen |
10 mg einmal täglich über 4 Wochen |
↓ weniger als 1 %^ |
Keine besondere Empfehlung. |
Antazida-Suspension von Magnesium- und Aluminiumhydroxiden 30 ml 4‑mal täglich, 2 Wochen |
10 mg einmal täglich über 4 Wochen |
↓ 35 %^ |
Keine besondere Empfehlung. |
Efavirenz 600 mg einmal täglich, 14 Tage |
10 mg über 3 Tage |
↓ 41 % |
Keine besondere Empfehlung. |
Rifampicin 600 mg einmal täglich, 7 Tage (gemeinsame Gabe) |
40 mg als Einmalgabe |
↑ 30 % |
Sofern die gemeinsame Anwendung nicht vermieden werden kann, wird eine zeitgleiche Gabe von Ezetimib/Atorvastatin Viatris mit Rifampicin unter klinischer Überwachung empfohlen. |
Rifampicin 600 mg einmal täglich, 5 Tage (zeitversetzte Gabe) |
40 mg als Einmalgabe |
↓ 80 % |
|
Gemfibrozil 600 mg 2‑mal täglich, 7 Tage |
40 mg als Einmalgabe |
↑ 35 % |
Nicht empfohlen. |
Fenofibrat 160 mg einmal täglich, 7 Tage |
40 mg als Einmalgabe |
↑ 3 % |
Nicht empfohlen. |
Boceprevir 800 mg 3‑mal täglich, 7 Tage |
40 mg als Einmalgabe |
↑ 2,3 % |
Eine niedrigere Anfangsdosis und die klinische Überwachung der Patienten werden empfohlen. Bei gemeinsamer Anwendung mit Boceprevir ist eine Tageshöchstdosis von Ezetimib/Atorvastatin Viatris 10 mg/20 mg nicht zu überschreiten. |
Elbasvir 50 mg einmal täglich/Grazoprevir 200 mg einmal täglich, 13 Tage |
10 mg als Einmalgabe |
↑ 1,94 % |
Bei gemeinsamer Anwendung mit Arzneimitteln, die Elbasvir oder Grazoprevir enthalten, sollte die Tagesdosis von Ezetimib/Atorvastatin Viatris 10 mg/20 mg nicht überschreiten. |
Glecaprevir 400 mg einmal täglich/Pibrentasvir 120 mg einmal täglich, 7 Tage |
10 mg einmal täglich für 7 Tage |
↑ 8,3 % |
Eine gemeinsame Anwendung mit Präparaten, die Glecaprevir oder Pibrentasvir enthalten, ist kontraindiziert (siehe Abschnitt 4.3). |
& Daten, die als x‑fache Veränderung angegeben sind, beziehen sich auf ein einfaches Verhältnis aus gemeinsamer Anwendung und alleiniger Anwendung von Atorvastatin (d. h. 1fach = keine Veränderung). Daten, die als prozentuale Änderung angegeben sind, beziehen sich auf die prozentuale Veränderung in Bezug auf die alleinige Anwendung von Atorvastatin (d. h. 0 % = keine Veränderung). | |||
Tabelle 2: Auswirkungen von Atorvastatin auf die Pharmakokinetik von gemeinsam angewendeten Arzneimitteln
Atorvastatin und Dosierung |
Gemeinsam angewendetes Arzneimittel |
Ezetimib/Atorvastatin |
|
Arzneimittel und Dosierung (mg) ➔ |
Veränderung der AUC& |
Klinische Empfehlungen |
|
80 mg einmal täglich über 10 Tage |
Digoxin 0,25 mg einmal täglich, 20 Tage |
↑ 15 % |
Patienten, die Digoxin einnehmen, sollten angemessen klinisch überwacht werden. |
40 mg einmal täglich über 22 Tage |
Orales Kontrazeptivum einmal täglich, 2 Monate
|
↑ 28 % ↑ 19 % |
Keine besondere Empfehlung. |
80 mg einmal täglich über 15 Tage |
*Phenazon 600 mg als Einmalgabe |
↑ 3 % |
Keine besondere Empfehlung. |
10 mg einmal täglich über 4 Tage |
Fosamprenavir 1.400 mg zweimal täglich, 14 Tage |
↓ 27 % |
Keine besondere Empfehlung. |
& Daten, die als prozentuale Veränderung angegeben sind, beziehen sich auf die prozentuale Veränderung in Bezug auf die alleinige Anwendung von Atorvastatin (d. h. 0 % = keine Veränderung). | |||
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während der Behandlung eine zuverlässige Verhütungsmethode anwenden (siehe Abschnitt 4.3).
Schwangerschaft
Atherosklerose ist ein chronischer Prozess und das übliche Absetzen von Lipidsenkern während der Schwangerschaft sollte nur einen geringen Einfluss auf das Langzeitrisiko einer primären Hypercholesterinämie haben.
Ezetimib/Atorvastatin Viatris
Ezetimib/Atorvastatin Viatris ist während der Schwangerschaft kontraindiziert (siehe Abschnitt 4.3). Es liegen keine klinischen Daten zur Anwendung von Ezetimib/Atorvastatin Viatris bei Schwangeren vor. Ezetimib/Atorvastatin Viatris sollte nicht von Frauen eingenommen werden, die schwanger sind, eine Schwangerschaft planen oder vermuten schwanger zu sein. Die Behandlung mit Ezetimib/Atorvastatin Viatris muss für die Dauer der Schwangerschaft oder bis festgestellt ist, dass keine Schwangerschaft vorliegt, ausgesetzt werden (siehe Abschnitt 4.3).
Die gemeinsame Anwendung von Ezetimib und Atorvastatin bei trächtigen Ratten zeigte einen mit dem Prüfpräparat in Zusammenhang stehenden Anstieg der skeletalen Veränderung „verminderte Ossifikation der Sternebrae“ in der Ezetimib/Atorvastatin-Hochdosisgruppe. Dies könnte mit dem beobachteten verringerten fötalen Körpergewicht zusammenhängen. Bei trächtigen Kaninchen war die beobachtete Inzidenz skeletaler Deformationen gering (fusionierte Sternebrae, kaudale Wirbelfusion und asymmetrische Veränderungen der Sternebrae).
Atorvastatin
Die Sicherheit von Atorvastatin bei Schwangeren ist bisher nicht belegt. Bei schwangeren Frauen wurden keine kontrollierten klinischen Studien mit Atorvastatin durchgeführt. Es liegen seltene Berichte über angeborene Fehlbildungen nach intrauteriner Exposition mit HMG‑CoA-Reduktase-Inhibitoren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Eine Behandlung der Mutter mit Atorvastatin kann beim Fetus die Konzentration von Mevalonat, einem Vorprodukt der Cholesterinbiosynthese, verringern.
Ezetimib
Es liegen keine klinischen Daten zur Anwendung von Ezetimib bei Schwangeren vor. Tierstudien zur Monotherapie mit Ezetimib lassen keine direkt oder indirekt schädlichen Wirkungen auf Schwangerschaft, embryonale/fetale Entwicklung, Geburt oder postnatale Entwicklung erkennen (siehe Abschnitt 5.3).
Stillzeit
Ezetimib/Atorvastatin Viatris ist während der Stillzeit kontraindiziert. Aufgrund von möglichen schwerwiegenden Nebenwirkungen sollten Frauen, die Ezetimib/Atorvastatin Viatris erhalten, nicht stillen. Studien an Ratten haben gezeigt, dass Ezetimib in die Muttermilch übergeht. Bei Ratten wurden in der Milch und im Plasma ähnliche Konzentrationen von Atorvastatin und seinen aktiven Metaboliten beobachtet. Es ist nicht bekannt, ob die wirksamen Bestandteile von Ezetimib/Atorvastatin Viatris in die Muttermilch übergehen (siehe Abschnitt 4.3).
Fertilität
Es wurden keine Studien zur Fertilität mit Ezetimib/Atorvastatin durchgeführt.
Atorvastatin
In Tierstudien hatte Atorvastatin keine Auswirkungen auf die weibliche oder männliche Fertilität.
Ezetimib
Ezetimib hatte keine Auswirkungen auf die Fertilität von weiblichen oder männlichen Ratten.
Ezetimib/Atorvastatin hat einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Bei der Teilnahme am Straßenverkehr oder dem Bedienen von Maschinen ist jedoch zu berücksichtigen, dass über Schwindel berichtet wurde.
Zusammenfassung des Sicherheitsprofils
Die Sicherheit der gemeinsamen Anwendung von Ezetimib und Atorvastatin wurde bei mehr als 2.400 Patienten in 7 klinischen Studien untersucht.
Tabellarische Übersicht der Nebenwirkungen
Nebenwirkungen, die unter Ezetimib/Atorvastatin oder Ezetimib oder Atorvastatin in klinischen Studien beobachtet oder nach Markteinführung von Ezetimib/Atorvastatin berichtet wurden, sind in Tabelle 3 angegeben. Diese Nebenwirkungen sind nach Systemorganklasse und Häufigkeit aufgeführt. Die Häufigkeiten sind wie folgt definiert: Sehr häufig (≥ 1/10), Häufig (≥ 1/100, < 1/10), Gelegentlich (≥ 1/1.000, < 1/100), Selten (≥ 1/10.000, < 1/1.000), Sehr selten (< 1/10.000) und Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 3: Nebenwirkungen
Systemorganklasse |
Nebenwirkungen |
Infektionen und parasitäre Erkrankungen | |
Gelegentlich |
Grippe (Influenza) |
Nicht bekannt |
Nasopharyngitis |
Erkrankungen des Blutes und des Lymphsystems | |
Nicht bekannt |
Thrombozytopenie |
Erkrankungen des Immunsystems | |
Nicht bekannt |
Überempfindlichkeit einschließlich Anaphylaxie, Angioödem, Ausschlag und Urtikaria |
Stoffwechsel- und Ernährungsstörungen | |
Nicht bekannt |
verminderter Appetit; Anorexie; Hyperglykämie; Hypoglykämie |
Psychiatrische Erkrankungen | |
Gelegentlich |
Depression; Schlaflosigkeit; Schlafstörungen |
Nicht bekannt |
Alpträume |
Erkrankungen des Nervensystems | |
Gelegentlich |
Schwindelgefühl; Störung der Geschmackswahrnehmung; Kopfschmerzen; Parästhesie |
Nicht bekannt |
Hypästhesie; Amnesie; periphere Neuropathie; Myasthenia gravis |
Augenerkrankungen | |
Nicht bekannt |
verschwommenes Sehen; Sehstörung; okuläre Myasthenie |
Erkrankungen des Ohrs und des Labyrinths | |
Nicht bekannt |
Tinnitus; Hörverlust |
Herzerkrankungen | |
Gelegentlich |
Sinusbradykardie |
Gefäßerkrankungen | |
Gelegentlich |
Hitzewallung |
Nicht bekannt |
Hypertonie |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
Gelegentlich |
Dyspnoe |
Nicht bekannt |
Husten; Pharyngolaryngealschmerzen; Epistaxis |
Erkrankungen des Gastrointestinaltrakts | |
Häufig |
Diarrhö |
Gelegentlich |
Abdominale Beschwerden; aufgetriebener Bauch; Abdominalschmerzen; Schmerzen im Unterbauch; Schmerzen im Oberbauch; Obstipation; Dyspepsie; Flatulenz; häufige Darmentleerungen; Gastritis; Übelkeit; Magenbeschwerden |
Nicht bekannt |
Pankreatitis; gastroösophageale Refluxerkrankung; Aufstoßen; Erbrechen; Mundtrockenheit |
Leber- und Gallenerkrankungen | |
Nicht bekannt |
Hepatitis; Cholelithiasis; Cholezystitis; Cholestase; Leberversagen mit teils letalem Ausgang |
Erkrankungen der Haut und des Unterhautgewebes | |
Gelegentlich |
Akne; Urtikaria |
Nicht bekannt |
Alopezie; Hautausschlag; Pruritus; Erythema multiforme; angioneurotisches Ödem; bullöse Dermatitis einschließlich Erythema multiforme; Stevens-Johnson-Syndrom und Lyell-Syndrom (Epidermolysis acuta toxica) |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |
Häufig |
Myalgie |
Gelegentlich |
Arthralgie; Rückenschmerzen; Muskelermüdung; Muskelspasmen; Muskelschwäche; Schmerzen in den Extremitäten |
Selten |
Muskelruptur |
Sehr selten |
Lupus-ähnliches Syndrom |
Nicht bekannt |
Myopathie/Rhabdomyolyse; Muskelriss; Tendinopathie, gelegentlich bis hin zur Sehnenruptur; Nackenschmerzen; Schwellung an den Gelenken; Myositis; immunvermittelte nekrotisierende Myopathie (siehe Abschnitt 4.4) |
Erkrankungen der Geschlechtsorgane und der Brustdrüse | |
Nicht bekannt |
Gynäkomastie |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Gelegentlich |
Asthenie; Ermüdung (Fatigue); Unwohlsein; Ödem |
Nicht bekannt |
Schmerzen im Brustkorb; Schmerzen; peripheres Ödem; Fieber |
Untersuchungen | |
Gelegentlich |
ALT und/oder AST erhöht; alkalische Phosphatase erhöht; Kreatinphosphokinase (CPK) im Blut erhöht; Gamma-Glutamyltransferase erhöht; Leberenzyme erhöht; Leberfunktionstest anomal; Gewichtszunahme |
Nicht bekannt |
Leukozyten im Urin positiv |
Laborwerte
In kontrollierten klinischen Studien betrug die Inzidenz klinisch bedeutender Erhöhungen der Serum-Transaminasen (ALT und/oder AST ≥ dem Dreifachen des oberen Normwertes in Folge) 0,6 % unter Ezetimib/Atorvastatin. Diese Erhöhungen waren im Allgemeinen asymptomatisch, standen nicht im Zusammenhang mit einer Cholestase und kehrten nach Absetzen der Therapie oder bei Fortsetzung der Behandlung auf den Ausgangswert zurück (siehe Abschnitt 4.4).
Die folgenden Nebenwirkungen wurden bei einigen Statinen berichtet:
Störungen der Sexualfunktion
In Ausnahmefällen und besonders bei Langzeittherapie eine interstitielle Lungenkrankheit (siehe Abschnitt 4.4)
Diabetes mellitus: Die Häufigkeit ist abhängig von dem Vorhandensein oder dem Fehlen von Risikofaktoren (Nüchternblutzucker ≥ 5,6 mmol/l, BMI > 30 kg/m2, erhöhte Triglyzeridwerte, bestehende Hypertonie).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels.
Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D‑53175 Bonn, Website: https://www.bfarm.deanzuzeigen.
Ezetimib/Atorvastatin Viatris
Im Fall einer Überdosierung sollten symptomatische und unterstützende Maßnahmen ergriffen werden. Leberfunktionstests sollten durchgeführt und die CPK‑Serumspiegel überwacht werden.
Ezetimib
In klinischen Studien wurde die Gabe von 50 mg Ezetimib/Tag bei 15 Probanden über 14 Tage wie auch die Gabe von 40 mg/Tag bei 18 Patienten mit primärer Hypercholesterinämie über 56 Tage im Allgemeinen gut vertragen. Einige Fälle von Überdosierung wurden berichtet, die meist nicht von Nebenwirkungen begleitet waren. Die berichteten Nebenwirkungen waren nicht schwerwiegend. Bei Tieren wurden nach oral verabreichten Einzeldosen von 5.000 mg Ezetimib/kg an Ratten und Mäusen sowie von 3.000 mg Ezetimib/kg an Hunden keine toxischen Effekte beobachtet.
Atorvastatin
Aufgrund der beträchtlichen Plasmaproteinbindung von Atorvastatin ist eine signifikante Beschleunigung der Ausscheidung durch Hämodialyse nicht zu erwarten.
Pharmakotherapeutische Gruppe: Mittel, die den Lipidstoffwechsel beeinflussen, Kombinationen, Kombinationen verschiedener Mittel, die den Lipidstoffwechsel beeinflussen, ATC‑Code: C10BA05
Ezetimib/Atorvastatin ist ein Lipidsenker, der selektiv die intestinale Resorption von Cholesterin und verwandten Phytosterinen hemmt und die endogene Cholesterinsynthese reduziert.
Wirkmechanismus
Ezetimib/Atorvastatin Viatris
Das Cholesterin im Plasma stammt aus der intestinalen Resorption und der endogenen Synthese. Ezetimib/Atorvastatin Viatris enthält Ezetimib und Atorvastatin, zwei lipidsenkende Wirkstoffe mit komplementären Wirkmechanismen. Ezetimib/Atorvastatin Viatris senkt erhöhte Werte von Gesamtcholesterin, LDL‑Cholesterin, Apolipoprotein B, Triglyzeriden, non‑HDL‑Cholesterin und erhöht HDL‑Cholesterin durch die duale Hemmung der Cholesterinresorption und ‑synthese.
Ezetimib
Ezetimib hemmt die intestinale Cholesterinresorption. Ezetimib ist nach oraler Einnahme wirksam und sein Wirkmechanismus unterscheidet sich von dem anderer lipidsenkender Wirkstoffklassen (z. B. Statine, Gallensäure-bindenden Wirkstoffen [Harze], Fibrate und Phytosterine). Das molekulare Target von Ezetimib ist der Steroltransporter, das Niemann-Pick‑C1 Like 1 (NPC1L1) Protein, welches für die intestinale Aufnahme von Cholesterin und Phytosterinen verantwortlich ist.
Ezetimib lagert sich am Bürstensaum des Dünndarms an und hemmt die Cholesterinresorption, was zu einem verminderten Transport von Cholesterin aus dem Darm in die Leber führt. Statine reduzieren die Cholesterinsynthese in der Leber und gemeinsam führen diese unterschiedlichen Wirkungsmechanismen zu einer komplementären Cholesterinsenkung. In einer zweiwöchigen klinischen Studie an 18 Patienten mit Hypercholesterinämie hemmte Ezetimib im Vergleich zu Placebo die intestinale Cholesterinresorption um ca. 54 %.
Eine Reihe von präklinischen Studien wurde durchgeführt, um die Selektivität von Ezetimib für die Hemmung der Cholesterinresorption zu bestimmen. Ezetimib hemmte die Resorption von radioaktiv markiertem [14C]Cholesterin ohne Wirkung auf die Resorption von Triglyzeriden, Fettsäuren, Gallensäuren, Progesteron, Ethinylestradiol oder der fettlöslichen Vitamine A und D.
Atorvastatin
Atorvastatin ist ein selektiver und kompetitiver Inhibitor der HMG‑CoA-Reduktase. Dieses Enzym katalysiert geschwindigkeitsbestimmend die Umsetzung von 3‑Hydroxy‑3‑Methyl-Glutaryl-Coenzym‑A zu Mevalonsäure, einer Vorstufe bei der Synthese von Sterolen einschließlich Cholesterin. Triglyzeride und Cholesterin werden in der Leber in Very Low Density Lipoproteine (VLDL) eingebaut und zum weiteren Transport in periphere Gewebe an das Plasma abgegeben. Low Density Lipoproteine (LDL) entstehen aus VLDL und werden hauptsächlich über Rezeptoren mit hoher LDL-Affinität (LDL-Rezeptoren) abgebaut.
Atorvastatin senkt Plasmacholesterin- und Lipoprotein-Serumspiegel durch Hemmung von HMG‑CoA-Reduktase und als Folge davon die Cholesterinbiosynthese in der Leber und erhöht die Anzahl der hepatischen LDL‑Rezeptoren an der Zelloberfläche, wodurch die Aufnahme und der Abbau von LDL beschleunigt werden.
Atorvastatin senkt die LDL‑Produktion und die Anzahl der LDL‑Partikel. Atorvastatin führt zu einem umfassenden und anhaltenden Anstieg der LDL‑Rezeptoraktivität verbunden mit einer Veränderung zugunsten der Qualität der zirkulierenden LDL‑Partikel. Atorvastatin senkt wirksam das LDL‑Cholesterin bei Patienten mit homozygoter familiärer Hypercholesterinämie, einer Population, die üblicherweise nicht auf Lipidsenker anspricht.
Im Rahmen einer Dosis-Wirkungs-Studie wurde gezeigt, dass Atorvastatin zu einer Senkung von Gesamtcholesterin (30 %‑46 %), LDL‑Cholesterin (41 %‑61 %), Apolipoprotein B (34 %‑50 %) und Triglyzeriden (14 %‑33 %) führte bei gleichzeitiger Erhöhung von HDL‑Cholesterin und Apolipoprotein A1 in variablem Ausmaß. Diese Ergebnisse wurden in gleichem Maß bei Patienten mit heterozygoter familiärer Hypercholesterinämie, nicht familiären Formen der Hypercholesterinämie sowie gemischter Hyperlipidämie einschließlich Patienten mit nicht insulinabhängigem Diabetes mellitus beobachtet.
Klinische Wirksamkeit und Sicherheit
In kontrollierten klinischen Studien führte Ezetimib/Atorvastatin zu einer signifikanten Senkung von Gesamtcholesterin, LDL‑Cholesterin, Apolipoprotein B und Triglyzeriden und einer Erhöhung von HDL‑Cholesterin bei Patienten mit Hypercholesterinämie.
Primäre Hypercholesterinämie
Im Rahmen einer placebokontrollierten Studie erhielten 628 Patienten mit Hyperlipidämie für bis zu 12 Wochen randomisiert Placebo, Ezetimib (10 mg), Atorvastatin (10 mg, 20 mg, 40 mg oder 80 mg) oder gemeinsam Ezetimib und Atorvastatin (10 mg/10 mg, 10 mg/20 mg, 10 mg/40 mg und 10 mg/80 mg).
Patienten, die sämtliche Dosierungen von Ezetimib/Atorvastatin erhielten, wurden mit den Patienten, die sämtliche Dosierungen von Atorvastatin erhielten, verglichen. Ezetimib/Atorvastatin senkte die Spiegel von Gesamtcholesterin, LDL‑Cholesterin, Apolipoprotein B, Triglyzeriden und non-HDL‑Cholesterin und erhöhte HDL‑Cholesterin signifikant stärker als Atorvastatin allein (siehe Tabelle 4).
Tabelle 4: Ansprechen von Patienten mit primärer Hyperlipidämie auf Ezetimib/Atorvastatin (mittlerea prozentuale [%] Veränderung zum Ausgangswert [unbehandelt]b nach 12 Wochen)
Behandlung |
N |
Gesamt-cholesterin |
LDL-Cholesterin |
Apolipo-protein B |
Tri-glyzeridea |
HDL-Cholesterin |
Non-HDL-Cholesterin |
Gepoolte Daten (alle Dosen von Ezetimib/ |
255 |
-41 |
-56 |
-45 |
-33 |
+7 |
-52 |
Gepoolte Daten (alle Dosen von Atorvastatin)c |
248 |
-32 |
-44 |
-36 |
-24 |
+4 |
-41 |
Ezetimib 10 mg |
65 |
-14 |
-20 |
-15 |
-5 |
+4 |
-18 |
Placebo |
60 |
+4 |
+4 |
+3 |
-6 |
+4 |
+4 |
Ezetimib/Ator-vastatin nach Dosis |
|||||||
10 mg/10 mg |
65 |
-38 |
-53 |
-43 |
-31 |
+9 |
-49 |
10 mg/20 mg |
62 |
-39 |
-54 |
-44 |
-30 |
+9 |
-50 |
10 mg/40 mg |
65 |
-42 |
-56 |
-45 |
-34 |
+5 |
-52 |
10 mg/80 mg |
63 |
-46 |
-61 |
-50 |
-40 |
+7 |
-58 |
Atorvastatin nach Dosis |
|||||||
10 mg |
60 |
-26 |
-37 |
-28 |
-21 |
+6 |
-34 |
20 mg |
60 |
-30 |
-42 |
-34 |
-23 |
+4 |
-39 |
40 mg |
66 |
-32 |
-45 |
-37 |
-24 |
+4 |
-41 |
80 mg |
62 |
-40 |
-54 |
-46 |
-31 |
+3 |
-51 |
a Für Triglyzeride prozentuale (%) mediane Veränderung vom Ausgangswert | |||||||
Im Rahmen der kontrollierten TEMPO-Studie (Titration of Atorvastatin vs. Ezetimibe Add-On to Atorvastatin in Patients with Hypercholesterolaemia) wurden 184 Patienten mit moderatem Risiko einer koronaren Herzkrankheit (KHK) und LDL‑Cholesterinspiegeln zwischen ≥ 2,6 mmol/l und ≤ 4,1 mmol/l eingeschlossen. Alle Patienten erhielten 20 mg Atorvastatin über einen Mindestzeitraum von 4 Wochen vor Randomisierung. Die Patienten, welche einen LDL‑Cholesterinspiegel von < 2,6 mmol/l nicht erreicht hatten, erhielten randomisiert gemeinsam Ezetimib und Atorvastatin (äquivalent zu Ezetimib/Atorvastatin 10 mg/20 mg) oder Atorvastatin 40 mg über einen Zeitraum von 6 Wochen.
Ezetimib/Atorvastatin 10 mg/20 mg war signifikant wirksamer als die Verdopplung der Atorvastatindosis auf 40 mg im Hinblick auf eine weitere Senkung der Spiegel von Gesamtcholesterin (‑20 % vs. ‑7 %), LDL‑Cholesterin (‑31 % vs. ‑11 %), Apolipoprotein B (‑21 % vs. ‑8 %) und non‑HDL-Cholesterin (‑27 % vs. ‑10 %). Die Unterschiede der Ergebnisse hinsichtlich HDL‑Cholesterin und Triglyzeriden waren zwischen beiden Behandlungsarmen nicht signifikant. Des Weiteren erreichten signifikant mehr Patienten unter Ezetimib/Atorvastatin 10 mg/20 mg einen LDL‑Cholesterinspiegel von < 2,6 mmol/l im Vergleich zu den Patienten unter Atorvastatin 40 mg (84 % vs. 49 %).
Im Rahmen der kontrollierten EZ‑PATH-Studie (Ezetimibe Plus Atorvastatin Titration in Achieving Lower LDL‑C Targets in Hypercholesterolaemic Patients) wurden 556 Patienten mit hohem Risiko einer koronaren Herzkrankheit (KHK) und LDL‑Cholesterinspiegeln zwischen ≥ 1,8 mmol/l und ≤ 4,1 mmol/l eingeschlossen. Alle Patienten erhielten 40 mg Atorvastatin über einen Mindestzeitraum von 4 Wochen vor Randomisierung. Die Patienten, welche einen LDL‑Cholesterinspiegel von < 1,8 mmol/l nicht erreicht hatten, erhielten randomisiert gemeinsam Ezetimib und Atorvastatin (äquivalent zu Ezetimib/Atorvastatin 10 mg/40 mg) oder Atorvastatin 80 mg über einen Zeitraum von 6 Wochen.
Ezetimib/Atorvastatin 10 mg/40 mg war signifikant wirksamer als die Verdopplung der Atorvastatindosis auf 80 mg im Hinblick auf eine weitere Senkung der Spiegel von Gesamtcholesterin (‑17 % vs. - 7 %), LDL‑Cholesterin (‑27 % vs. ‑11 %), Apolipoprotein B (‑18 % vs. ‑8 %), Triglyzeriden (‑12 % vs. ‑6 %) und non‑HDL‑Cholesterin (‑23 % vs. ‑9 %). Die Unterschiede der Ergebnisse hinsichtlich HDL‑Cholesterin waren zwischen beiden Behandlungsarmen nicht signifikant. Darüber hinaus erreichten signifikant mehr Patienten unter Ezetimib/Atorvastatin 10 mg/40 mg einen LDL‑Cholesterinspiegel von < 1,8 mmol/l im Vergleich zu den Patienten unter Atorvastatin 80 mg (74 % vs. 32 %).
Im Rahmen einer placebokontrollierten, 8‑wöchigen Studie erhielten 308 Patienten mit Hypercholesterinämie, die bereits mit Atorvastatin behandelt wurden, aber nicht den LDL‑Cholesterin-Zielwert gemäß der NCEP-Richtlinie („National Cholesterol Education Program“) erreicht hatten (LDL‑Cholesterin-Zielwert basierend auf dem LDL‑Cholesterin-Ausgangswert und dem KHK‑Risiko), randomisiert entweder Ezetimib 10 mg oder Placebo als Zusatz zur Atorvastatin-Monotherapie.
Von den Patienten, deren LDL‑Cholesterin Ausgangswert nicht dem Zielwert entsprach (ca. 83 %), erreichten signifikant mehr Patienten unter gemeinsamer Gabe von Ezetimib und Atorvastatin ihren LDL‑Cholesterin-Zielwert im Vergleich zu den Patienten unter gemeinsamer Gabe von Placebo und Atorvastatin (67 % vs. 19 %). Ezetimib senkte nach Zugabe zur Behandlung mit Atorvastatin die Spiegel von LDL‑Cholesterin signifikant stärker als Placebo nach Zugabe zur Behandlung mit Atorvastatin (25 % vs. 4 %). Darüber hinaus senkte Ezetimib nach Zugabe zur Behandlung mit Atorvastatin die Spiegel von Gesamtcholesterin, Apolipoprotein B und Triglyzeriden signifikant im Vergleich zu Placebo nach Zugabe zur Behandlung mit Atorvastatin.
Im Rahmen einer kontrollierten, 12‑wöchigen, zweiphasigen Studie erhielten 1.539 mit Atorvastatin 10 mg vorbehandelte Patienten mit hohem kardiovaskulären Risiko und LDL‑Cholesterinspiegeln zwischen 2,6 mmol/l und 4,1 mmol/l randomisiert Atorvastatin 20 mg, Rosuvastatin 10 mg oder Ezetimib/Atorvastatin 10 mg/10 mg. Nach 6 Wochen Behandlungsdauer (Phase I) wechselten die Patienten unter Atorvastatin 20 mg, die einen LDL‑Cholesterinspiegel von < 2,6 mmol/l nicht erreicht hatten, auf eine Behandlung mit entweder Atorvastatin 40 mg oder Ezetimib/Atorvastatin 10 mg/20 mg über einen Zeitraum von 6 Wochen (Phase II). In analoger Weise wechselten die Patienten unter Rosuvastatin 10 mg auf entweder Rosuvastatin 20 mg oder Ezetimib/Atorvastatin 10 mg/20 mg. Die Daten zur Senkung der LDL‑Cholesterinspiegel sowie zu Vergleichen zwischen dem Ezetimib/Atorvastatin Behandlungsarm und den anderen untersuchten Behandlungsarmen sind in Tabelle 5 aufgeführt.
Tabelle 5: Ansprechen von Hochrisiko-Patienten auf Ezetimib/Atorvastatin* mit LDL‑Cholesterinspiegeln zwischen 2,6 mmol/l und 4,1 mmol/l und mit zu Beginn bestehender Vorbehandlung mit Atorvastatin 10 mg
Behandlung |
N |
Prozentuale (%) Veränderung zum Ausganswert† |
|||||
Gesamt-cholesterin |
LDL-Cholesterin |
Apolipo-protein B |
Triglyzeride‡ |
HDL-Cholesterin |
Non-HDL-Cholesterin |
||
Phase I |
|||||||
Ezetimib/Atorvastatin |
120 |
-13,5 |
-22,2 |
-11,3 |
-6,0 |
+0,6 |
-18,3 |
Atorvastatin 20 mg |
480 |
-6,4§ |
-9,5§ |
-6,0¶ |
-3,9 |
-1,1 |
-8,1§ |
Rosuvastatin 10 mg |
939 |
-7,7§ |
-13,0§ |
-6,9# |
-1,1 |
+1,1 |
-10,6§ |
Phase II |
|||||||
Ezetimib/Atorvastatin |
124 |
-10,7 |
-17,4 |
-9,8 |
-5,9 |
+0,7 |
-15,1 |
Atorvastatin 40 mg |
124 |
-3,8Þ |
-6,9Þ |
-5,4 |
-3,1 |
+1,7 |
-5,8Þ |
Wechsel von Rosuvastatin 10 mg |
|||||||
Ezetimib/Atorvastatin |
231 |
-11,8 |
-17,1 |
-11,9 |
-10,2 |
+0,1 |
-16,2 |
Rosuvastatin 20 mg |
205 |
-4,5Þ |
-7,5Þ |
-4,1Þ |
-3,2ß |
+0,8 |
-6,4Þ |
* Gemeinsame Gabe von Ezetimib und Atorvastatin äquivalent zu Ezetimib/Atorvastatin 10 mg/10 mg oder Ezetimib/Atorvastatin 10 mg/20 mg | |||||||
Tabelle 5 enthält keine Daten um die Wirkungen von Ezetimib/Atorvastatin 10 mg/10 mg oder 10 mg/20 mg mit höheren Dosen als Atorvastatin 40 mg oder Rosuvastatin 20 mg zu vergleichen.
Im Rahmen der placebokontrollierten MIRACL-Studie (Myocardial Ischaemia Reduction with Aggressive Cholesterol-Lowering) erhielten Patienten mit akutem Koronarsyndrom (Nicht‑Q-Wellen-Myokardinfarkt oder instabile Angina pectoris) randomisiert Atorvastatin 80 mg/Tag (n = 1.538) oder Placebo (n = 1.548). Die Behandlung wurde in der Akutphase nach Krankenhauseinweisung eingeleitet und dauerte 16 Wochen. Die Behandlung mit Atorvastatin 80 mg/Tag ergab eine Risikoreduktion von 16 % (p = 0,048) bezogen auf den kombinierten primären Endpunkt der Studie: Tod jeglicher Ursache, nicht-tödlicher Myokardinfarkt, Herzstillstand mit Wiederbelebung oder Angina pectoris mit Nachweis auf myokardiale Ischämie und Notwendigkeit stationärer Behandlung. Diese Risikoreduktion resultierte hauptsächlich aus der um 26 % gesenkten erneuten Krankenhauseinweisung aufgrund von Angina pectoris mit Nachweis auf myokardiale Ischämie (p = 0,018).
Ezetimib/Atorvastatin enthält Atorvastatin. Im Rahmen der placebokontrollierten ASCOTT-LLA-Studie (Anglo-Scandinavian Cardiac Outcomes Trial Lipid-Lowering Arm) wurde die Wirkung von Atorvastatin 10 mg im Hinblick auf tödliche und nicht-tödliche koronare Herzkrankheit bei 10.305 Patienten mit Bluthochdruck untersucht. Die Patienten waren 40‑80 Jahre alt, hatten Gesamtcholesterinspiegel von ≤ 6,5 mmol/l und mindestens 3 kardiovaskuläre Risikofaktoren. Die Patienten wurden über einen mittleren Zeitraum von 3,3 Jahren nachverfolgt. Atorvastatin 10 mg senkte signifikant (p < 0,001) die folgenden relativen Risiken: tödliche KHK sowie nicht-tödlicher Myokardinfarkt um 36 % (absolute Risikoreduktion = 1,1 %); gesamte kardiovaskuläre Ereignisse und Revaskularisierungsmaßnahmen um 20 % (absolute Risikoreduktion = 1,9 %); gesamte koronare Ereignisse um 29 % (absolute Risikoreduktion = 1,4 %).
Im Rahmen der placebokontrollierten CARDS-Studie (Collaborative Atorvastatin Diabetes Study) wurde die Wirkung von Atorvastatin 10 mg im Hinblick auf kardiovaskuläre Erkrankungen (KVE) bei 2.838 Patienten untersucht. Die Patienten waren 40‑75 Jahre alt, hatten Typ‑2-Diabetes und einen oder mehrere kardiovaskuläre Risikofaktoren und hatten Spiegel von LDL‑Cholesterin ≤ 4,1 mmol/l und Triglyzeriden ≤ 6,8 mmol/l. Die Patienten wurden über einen mittleren Zeitraum von 3,9 Jahren nachverfolgt. Atorvastatin 10 mg senkte signifikant (p < 0,05) sowohl die Rate schwerer (major) kardiovaskulärer Ereignisse um 37 % (absolute Risikoreduktion = 3,2 %) als auch das Schlaganfallrisiko um 48 % (absolute Risikoreduktion = 1,3 %) sowie das Myokardinfarktrisiko um 42 % (absolute Risikoreduktion = 1,9 %).
Prävention kardiovaskulärer Ereignisse
Im Rahmen einer multizentrischen, randomisierten, doppelblinden, aktiv-kontrollierten Ezetimib/Simvastatin-Studie wurden 18.144 Patienten untersucht, die innerhalb von 10 Tagen nach stationärer Einweisung aufgrund eines akuten Koronarsyndroms (entweder akuter Myokardinfarkt [MI] oder instabile Angina pectoris [UA]) in die Studie eingeschlossen wurden. Alle Patienten erhielten randomisiert 1:1 entweder Ezetimib/Simvastatin 10 mg/40 mg (n = 9.067) oder Simvastatin 40 mg (n = 9.077) und wurden im Median über 6,0 Jahre nachbeobachtet.
Die Patienten waren im Mittel 63,6 Jahre alt, 76 % waren Männer, 84 % waren kaukasischer Herkunft und 27 % waren Diabetiker. Der durchschnittliche LDL‑Cholesterinwert zum Zeitpunkt des Studieneinschlussereignisses lag bei den Patienten unter lipidsenkender Vortherapie (n = 6.390) bei 80 mg/dl (2,1 mmol/l) und bei den Patienten ohne lipidsenkende Vortherapie (n = 11.594) bei 101 mg/dl (2,6 mmol/l). Vor der stationären Aufnahme aufgrund von akutem Koronarsyndrom (Studieneinschlussereignis) erhielten 34 % der Patienten eine Vortherapie mit einem Statin. Zum Untersuchungszeitpunkt nach einem Jahr lag der durchschnittliche LDL‑Cholesterinwert unter fortlaufender Behandlung bei den Patienten in der Ezetimib/Simvastatin-Gruppe bei 53,2 mg/dl (1,4 mmol/l) und in der Simvastatin-Monotherapie-Gruppe bei 69,9 mg/dl (1,8 mmol/l).
Der primäre Endpunkt war eine Kombination der Ereignisse kardiovaskulärer Tod, schwere (major) koronare Ereignisse (MCE; definiert als nicht-tödlicher Myokardinfarkt, nachgewiesene instabile Angina pectoris mit erforderlicher stationärer Einweisung oder jegliche, mindestens 30 Tage nach Randomisierung erfolgte koronare Revaskularisierung) und nicht-tödlicher Schlaganfall. Die Studie zeigte, dass eine Behandlung mit Ezetimib/Simvastatin hinsichtlich der Reduktion von Ereignissen des primären kombinierten Endpunkts aus kardiovaskulärem Tod, schweren (major) koronaren Ereignissen (MCE) sowie nicht-tödlichem Schlaganfall im Vergleich zu einer Behandlung mit Simvastatin allein einen zusätzlichen Nutzen aufweist (relative Risikoreduktion um 6,4 %, p = 0,016). Der primäre Endpunkt trat bei 2.572 von 9.067 Patienten (Kaplan-Meier[KM]-Ereignisrate nach 7 Jahren von 32,72 %) in der Ezetimib/Simvastatin-Gruppe und bei 2.742 von 9.077 Patienten (Kaplan-Meier[KM]-Ereignisrate nach 7 Jahren von 34,67 %) in der Simvastatin-Monotherapie-Gruppe auf (siehe Abbildung 1 und Tabelle 6). Bei gemeinsamer Gabe von Ezetimib und Atorvastatin ist ein ähnlicher zusätzlicher Nutzen zu erwarten. Die Gesamtsterblichkeit war in dieser Hochrisikogruppe unverändert.
Insgesamt ergab sich ein Nutzen bei Betrachtung sämtlicher Schlaganfälle (unabhängig der Ursache), jedoch wurde ein geringer, nicht-signifikanter Anstieg hämorrhagischer Schlaganfälle in der Ezetimib/Simvastatin-Gruppe im Vergleich zur Simvastatin-Monotherapie-Gruppe beobachtet. Das Risiko für hämorrhagischen Schlaganfall bei gemeinsamer Anwendung von Ezetimib mit einem stärker wirksamen Statin wurde im Rahmen von langfristigen Endpunktstudien nicht untersucht.
Die Wirkung der Behandlung mit Ezetimib/Simvastatin entsprach in vielen Subgruppen im Allgemeinen den Gesamtergebnissen, einschließlich Geschlecht, Alter, ethnische Herkunft, Diabetes mellitus in der Vorgeschichte, Ausgangslipidwerte, vorhergehende Statintherapie, vorangegangener Schlaganfall und Bluthochdruck.
Abbildung 1 Effekt von Ezetimib/Simvastatin auf den primären kombinierten Endpunkt aus kardiovaskulärem Tod, schweren (major) koronaren Ereignissen (MCE) sowie nicht-tödlichem Schlaganfall
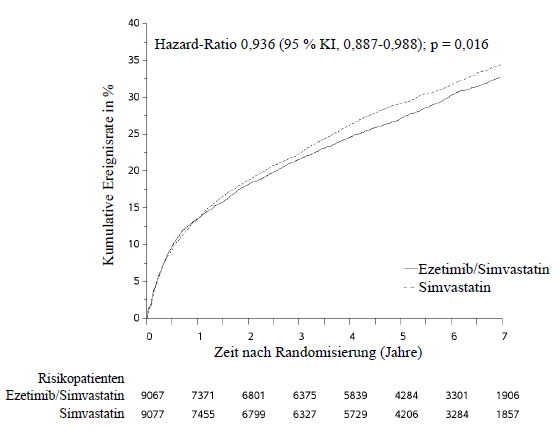
Tabelle 6: Schwere (major) kardiovaskuläre Ereignisse nach Behandlungsgruppe bei allen randomisierten Patienten der IMPROVE-IT-Studie
Outcome |
Ezetimib/Simvastatin |
Simvastatin |
Hazard-Ratio |
p‑Wert |
||
n |
K-M %‡ |
n |
K-M %‡ |
|||
Primärer kombinierter Wirksamkeitsendpunkt | ||||||
(Kardiovaskulärer Tod, schwere (major) koronare Ereignisse (MCE) und nicht-tödlicher Schlaganfall) |
2.572 |
32,72 % |
2.742 |
34,67 % |
0,936 |
0,016 |
Komponenten des primären kombinierten Endpunkts sowie ausgewählte Wirksamkeitsendpunkte | ||||||
Kardiovaskulärer Tod |
537 |
6,89 % |
538 |
6,84 % |
1,000 |
0,997 |
Schwere (major) koronare Ereignisse (MCE) | ||||||
Nicht-tödlicher Myokardinfarkt |
945 |
12,77 % |
1.083 |
14,41 % |
0,871 |
0,002 |
Instabile Angina pectoris mit erforderlicher stationärer Einweisung |
156 |
2,06 % |
148 |
1,92 % |
1,059 |
0,618 |
Koronare Revaskularisierung nach 30 Tagen |
1.690 |
21,84 % |
1.793 |
23,36 % |
0,947 |
0,107 |
Nicht-tödlicher Schlaganfall |
245 |
3,49 % |
305 |
4,24 % |
0,802 |
0,010 |
* 6 % wurden auf Ezetimib/Simvastatin 10 mg/80 mg hochtitriert | ||||||
Homozygote familiäre Hypercholesterinämie (HoFH)
Eine doppelblinde, randomisierte, 12‑wöchige Studie wurde mit Patienten mit klinischer und/oder genotypischer Diagnose einer HoFH durchgeführt. Die Daten einer Subgruppe von Patienten (n = 36) wurden analysiert, die bereits Atorvastatin 40 mg vor Studienbeginn erhielten. Die Erhöhung der Atorvastatindosis von 40 mg auf 80 mg (n = 12) führte zu einer Senkung von LDL‑Cholesterin um 2 % im Vergleich zur Ausgangssituation unter Atorvastatin 40 mg.
Die gemeinsame Gabe von Ezetimib und Atorvastatin äquivalent zu Ezetimib/Atorvastatin (10 mg/40 mg und 10 mg/80 mg gepoolt, n = 24) führte zu einer Senkung von LDL‑Cholesterin um 19 % im Vergleich zur Ausgangssituation unter Atorvastatin 40 mg. Die gemeinsame Gabe von Ezetimib und Atorvastatin äquivalent zu Ezetimib/Atorvastatin (10 mg/80 mg, n = 12) führte bei den Patienten zu einer Senkung von LDL‑Cholesterin um 25 % im Vergleich zur Ausgangssituation unter Atorvastatin 40 mg.
Nach Abschluss der 12‑wöchigen Studie erhielten geeignete Patienten (n = 35), die vor Studienbeginn Atorvastatin 40 mg erhielten, die gemeinsame Gabe von Ezetimib und Atorvastatin äquivalent zu Ezetimib/Atorvastatin 10 mg/40 mg über weitere 24 Monate Nach mindestens 4 Wochen Behandlungsdauer bestand die Möglichkeit, die Atorvastatindosis auf die Maximaldosis von 80 mg zu erhöhen. Ezetimib/Atorvastatin (10 mg/40 mg und 10 mg/80 mg gepoolt) führte nach Abschluss der 24 Monate zu einer Senkung von LDL‑Cholesterin, welche konsistent mit den Ergebnissen der 12‑wöchigen Studie war.
Die Europäische Arzneimittel-Agentur hat für Ezetimib/Atorvastatin eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in der Behandlung von Hypercholesterinämie und gemischter Hyperlipidämie gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Bioäquivalenz des Kombinationspräparats zur gemeinsamen Anwendung von vergleichbaren Dosen von Ezetimib- und Atorvastatin-Tabletten wurde nachgewiesen.
Resorption
Ezetimib/Atorvastatin
Die Auswirkungen von fettreichen Mahlzeiten auf die Pharmakokinetik von Ezetimib und Atorvastatin bei der Gabe von Ezetimib/Atorvastatin sind vergleichbar zur Gabe als Monopräparate.
Ezetimib
Nach oraler Gabe wird Ezetimib rasch resorbiert und weitgehend zu einem pharmakologisch aktiven Phenol-Glukuronid (Ezetimib-Glukuronid) konjugiert. Die mittlere maximale Plasmakonzentration (Cmax) wird nach 1‑2 Stunden für Ezetimib-Glukuronid und nach 4‑12 Stunden für Ezetimib erreicht. Die absolute Bioverfügbarkeit von Ezetimib kann nicht bestimmt werden, da die Substanz in wässrigen Lösungen, welche zur Injektion geeignet sind, praktisch unlöslich ist.
Eine gemeinsame Nahrungsaufnahme (fettreiche oder fettfreie Mahlzeiten) hatte keinen Einfluss auf die orale Bioverfügbarkeit von Ezetimib, wenn es in Form von 10 mg Tabletten eingenommen wurde.
Atorvastatin
Nach oraler Gabe wird Atorvastatin rasch resorbiert. Die maximale Plasmakonzentration (Cmax) wird nach 1‑2 Stunden erreicht. Das Ausmaß der Resorption steigt proportional zur Atorvastatindosis an. Nach oraler Gabe beträgt die relative Bioverfügbarkeit von Atorvastatin-Filmtabletten 95‑99 % im Vergleich zu einer Lösung zum Einnehmen. Die absolute Bioverfügbarkeit von Atorvastatin beträgt ca. 12 % und die systemisch verfügbare hemmende Wirkung auf die HMG‑CoA-Reduktase liegt bei ca. 30 %. Die niedrige systemische Verfügbarkeit wird der präsystemischen Elimination in der gastrointestinalen Mucosa und/oder dem hepatischen First-Pass-Effekt zugeschrieben.
Verteilung
Ezetimib
Ezetimib und Ezetimib-Glukuronid werden zu 99,7 % bzw. 88‑92 % an humane Plasmaproteine gebunden.
Atorvastatin
Das Hauptverteilungsvolumen von Atorvastatin beträgt ca. 381 l. Atorvastatin wird zu ≥ 98 % an Plasmaproteine gebunden.
Biotransformation
Ezetimib
Ezetimib wird vor allem im Dünndarm und in der Leber über Glukuronidkonjugation (eine Phase‑II-Reaktion) metabolisiert und anschließend über die Galle ausgeschieden. In allen untersuchten Spezies wurde ein minimaler oxidativer Metabolismus (eine Phase‑I-Reaktion) beobachtet. Ezetimib und Ezetimib-Glukuronid sind die im Plasma nachgewiesenen wirkstoffbezogenen Hauptkomponenten, wobei Ezetimib ca. 10‑20 % und Ezetimib-Glukuronid ca. 80‑90 % der Gesamtkonzentration des Wirkstoffs im Plasma ausmachen. Ezetimib und Ezetimib-Glukuronid werden langsam aus dem Plasma eliminiert mit Hinweis auf einen signifikanten enterohepatischen Kreislauf. Die Halbwertszeit von Ezetimib und Ezetimib-Glukuronid beträgt ca. 22 Stunden.
Atorvastatin
Atorvastatin wird mittels Cytochrom P450 3A4 zu ortho- und para-hydroxylierten Derivaten und verschiedenen Beta-Oxidationsprodukten metabolisiert. Neben anderen Stoffwechselwegen werden diese Stoffwechselprodukte durch Glukuronidierung weiter metabolisiert. Die Hemmung der HMG‑CoA-Reduktase durch die ortho- und para-hydroxylierten Metaboliten in vitro ist äquivalent zur Hemmung durch Atorvastatin. Annähernd 70 % der systemisch verfügbaren hemmenden Wirkung auf die HMG‑CoA-Reduktase wird den aktiven Metaboliten zugeschrieben.
Elimination
Ezetimib
Nach oraler Gabe von 20 mg radioaktiv markiertem [14C]Ezetimib an Probanden finden sich ca. 93 % der gesamten Radioaktivität im Plasma als Gesamt-Ezetimib. Über einen Beobachtungszeitraum von 10 Tagen wurden ca. 78 % der verabreichten radioaktiven Dosis in den Fäzes und 11 % im Urin wiedergefunden. Nach 48 Stunden war keine Radioaktivität mehr im Plasma nachweisbar.
Atorvastatin
Atorvastatin wird nach hepatischer und/oder extrahepatischer Metabolisierung hauptsächlich über die Galle eliminiert. Der Wirkstoff unterliegt scheinbar nicht ausgeprägt einem enterohepatischen Kreislauf. Die mittlere Eliminationshalbwertszeit von Atorvastatin aus dem Plasma beträgt beim Menschen ca. 14 Stunden. Die Halbwertszeit der Hemmaktivität auf die HMG‑CoA-Reduktase liegt aufgrund des Beitrags der aktiven Metaboliten bei ca. 20‑30 Stunden.
Atorvastatin ist ein Substrat der hepatischen Transporter OATP1B1 (organic anion-transporting polypeptide 1B1) und OATP1B3 (organic anion-transporting polypeptide 1B3). Metaboliten von Atorvastatin sind Substrate von OATP1B1. Atorvastatin wird außerdem als Substrat der Efflux-Transporter MDR1 (multi-drug resistance protein 1) und BCRP (breast cancer resistance protein) identifiziert, wodurch die intestinale Resorption und biliäre Ausscheidung von Atorvastatin begrenzt sein könnten.
Kinder und Jugendliche
Ezetimib
Die Pharmakokinetik von Ezetimib ist bei Kindern ab 6 Jahren ähnlich wie bei Erwachsenen. Pharmakokinetische Daten für Kinder unter 6 Jahren liegen nicht vor. Klinische Erfahrungen zur Behandlung von Kindern und Jugendlichen umfassen Patienten mit homozygoter familiärer Hypercholesterinämie, heterozygoter familiärer Hypercholesterinämie oder Sitosterinämie.
Atorvastatin
Im Rahmen einer 8‑wöchigen Open-Label-Studie wurden Kinder und Jugendliche (Tanner-Stadium I, n = 15; Tanner-Stadium II, n = 24) im Alter von 6‑17 Jahren mit heterozygoter familiärer Hypercholesterinämie und Ausgangswerten von LDL‑Cholesterin von ≥ 4 mmol/l behandelt und erhielten einmal täglich entweder Atorvastatin 5 mg oder 10 mg als Kautablette bzw. 10 mg oder 20 mg als Filmtablette. Das Körpergewicht war die einzige signifikante Kovariable des Population‑PK-Modells. Nach allometrischer Skalierung nach dem Körpergewicht schien die orale Clearance von Atorvastatin bei Kindern und Jugendlichen ähnlich zu sein wie die bei Erwachsenen. Über den gesamten Expositionsbereich von Atorvastatin und Ortho-Hydroxyatorvastatin wurde eine konsistente Abnahme von LDL‑Cholesterin und Gesamtcholesterin beobachtet.
Ältere Patienten
Ezetimib
Die Plasmakonzentrationen von Gesamt-Ezetimib sind bei älteren Patienten (ab 65 Jahren) etwa doppelt so hoch wie bei jüngeren Patienten (18‑45 Jahre). Die Senkung von LDL‑Cholesterin und das Sicherheitsprofil sind jedoch bei älteren und jüngeren mit Ezetimib behandelten Probanden vergleichbar.
Atorvastatin
Die Plasmaspiegel von Atorvastatin und der aktiven Metaboliten sind bei älteren Probanden höher als bei jüngeren erwachsenen Probanden, wobei die Wirkung auf die Lipidspiegel vergleichbar ist mit den Beobachtungen bei jüngeren Patienten.
Eingeschränkte Leberfunktion
Ezetimib
Nach einer Einzeldosis von 10 mg Ezetimib bei Patienten mit leichter Leberinsuffizienz (Child-Pugh-Score 5 oder 6) war die AUC für Gesamt-Ezetimib ca. 1,7‑mal größer als jene bei gesunden Probanden. In einer 14‑tägigen Mehrfachdosisstudie (10 mg pro Tag) bei Patienten mit moderater Leberinsuffizienz (Child-Pugh-Score 7‑9) war die mittlere AUC für Gesamt-Ezetimib am 1. und am 14. Tag ca. 4‑mal größer als die von gesunden Probanden. Für Patienten mit leichter Leberinsuffizienz ist keine Dosisanpassung erforderlich. Da die Folgen einer erhöhten Exposition mit Ezetimib bei Patienten mit moderater oder mit schwerer Leberinsuffizienz (Child-Pugh-Score > 9) nicht bekannt sind, wird Ezetimib für diese Patienten nicht empfohlen (siehe Abschnitte 4.2 und 4.4).
Atorvastatin
Die Plasmaspiegel von Atorvastatin und der aktiven Metaboliten sind bei Patienten mit chronischer, durch Alkohol bedingter Lebererkrankung (Child-Pugh B) deutlich erhöht (Cmax: ca. 16fach; AUC: ca. 11fach).
Eingeschränkte Nierenfunktion
Ezetimib
Nach einer Einzeldosis von 10 mg Ezetimib bei Patienten mit schwerer Niereninsuffizienz (n = 8; mittlere Kreatinin-Clearance ≤ 30 ml/min/1,73 m2) war die mittlere AUC für Gesamt-Ezetimib im Vergleich zu der bei gesunden Probanden (n = 9) um das ca. 1,5‑Fache vergrößert.
Ein Patient in dieser Studie (nach Nierentransplantation, unter multipler Arzneimitteltherapie, u. a. Ciclosporin) hatte eine 12fach höhere Exposition mit Gesamt-Ezetimib.
Atorvastatin
Nierenerkrankungen haben keinen Einfluss auf die Plasmakonzentrationen oder die Wirkung auf die Lipidspiegel von Atorvastatin und der aktiven Metaboliten.
Geschlecht
Ezetimib
Die Plasmakonzentrationen von Gesamt-Ezetimib sind bei Frauen etwas höher (ca. 20 %) als bei Männern. Bei der Behandlung mit Ezetimib sind sowohl die Senkung von LDL‑Cholesterin als auch das Sicherheitsprofil bei Männern und Frauen vergleichbar.
Atorvastatin
Die Plasmakonzentrationen von Atorvastatin und der aktiven Metaboliten sind bei Frauen (Frauen: Cmax: ca. 20 % höher; AUC: ca. 10 % niedriger) und Männern unterschiedlich. Diese Unterschiede sind klinisch nicht relevant und führen zu keinen klinisch signifikanten Unterschieden im Hinblick auf die Wirkung auf die Lipidspiegel zwischen Frauen und Männern.
SLCO1B1-Polymorphismus
Atorvastatin
Der OATP1B1-Transporter ist bei der hepatischen Aufnahme von allen HMG‑CoA-Reduktase-Inhibitoren einschließlich Atorvastatin beteiligt. Es besteht ein Risiko für eine erhöhte Exposition mit Atorvastatin bei Patienten mit SLCO1B1-Polymorphismus, welche zu einem erhöhten Rhabdomyolyserisiko führen kann (siehe Abschnitt 4.4). Ein Polymorphismus im Gen, welches für den OATP1B1-Transporter kodiert (SLCO1B1 c.521CC), steht im Zusammenhang mit einer 2,4fach erhöhten Exposition mit Atorvastatin (AUC) im Vergleich zu Personen ohne diese Genotyp-Variante (c.521TT). Eine genetisch bedingte eingeschränkte hepatische Aufnahme von Atorvastatin ist bei diesen Patienten ebenfalls möglich. Mögliche Auswirkungen auf die Wirksamkeit sind nicht bekannt.
Ezetimib/Atorvastatin
In 3‑monatigen Koadministrationsstudien mit Ezetimib und Atorvastatin an Ratten und Hunden wurden im Wesentlichen die toxischen Effekte beobachtet, die im Zusammenhang mit Statinen typisch sind. Die Statin-artigen histopathologischen Befunde waren auf die Leber beschränkt. Einige der toxischen Effekte waren ausgeprägter als diejenigen, die unter alleiniger Gabe von Statinen beobachtet wurden. Dies ist auf die pharmakokinetischen und/oder pharmakodynamischen Wechselwirkungen der gemeinsamen Anwendung zurückzuführen.
Die gemeinsame Anwendung von Ezetimib und Atorvastatin bei trächtigen Ratten zeigte einen mit der Testsubstanz in Zusammenhang stehenden Anstieg der skeletalen Veränderung „verminderte Ossifikation der Sternebrae“ in der Ezetimib/Atorvastatin (1.000/108,6 mg/kg)-Hochdosisgruppe. Dies könnte mit dem beobachteten verringerten fötalen Körpergewicht zusammenhängen. Bei trächtigen Kaninchen war die beobachtete Inzidenz skeletaler Deformationen niedrig (fusionierte Sternebrae, kaudale Wirbelfusion und asymmetrische Veränderungen der Sternebrae).
In einer Reihe von In‑vivo‑ und In‑vitro-Assays zeigte Ezetimib allein oder zusammen mit Atorvastatin kein genotoxisches Potenzial.
Ezetimib
In Tierstudien zur chronischen Toxizität von Ezetimib wurden keine Zielorgane für toxische Wirkungen identifiziert. Bei Hunden war nach 4‑wöchiger Behandlung mit Ezetimib (≥ 0,03 mg/kg/Tag) die Cholesterinkonzentration in der Blasengalle um das 2,5‑ bis 3,5‑Fache erhöht. In einer Studie an Hunden über ein Jahr wurde bei Dosen bis zu 300 mg/kg/Tag jedoch keine erhöhte Inzidenz von Cholelithiasis oder anderen hepatobiliären Effekten beobachtet. Die Bedeutung dieser Daten für den Menschen ist nicht bekannt. Ein lithogenes Risiko bei der therapeutischen Anwendung von Ezetimib kann nicht ausgeschlossen werden.
Langzeitstudien zur Kanzerogenität von Ezetimib verliefen negativ.
Ezetimib hatte weder einen Einfluss auf die Fertilität von männlichen oder weiblichen Ratten, noch erwies es sich bei Ratten und Kaninchen als teratogen, auch beeinflusste es nicht die prä- oder postnatale Entwicklung. Ezetimib war bei trächtigen Ratten und Kaninchen nach Mehrfachgabe von 1.000 mg/kg/Tag plazentagängig.
Atorvastatin
Bei vier In‑vitro-Tests und einem In‑vivo-Testsystem zeigte Atorvastatin kein mutagenes oder klastogenes Potenzial. Atorvastatin war in Ratten nicht karzinogen. Bei Mäusen kam es jedoch bei hohen Dosen (die zu einer 6‑ bis 11fach höheren AUC0‑24h führen, als mit der höchsten empfohlenen Dosis beim Menschen erreicht wird) zu hepatozellulären Adenomen bei den männlichen Tieren und bei den weiblichen Versuchstieren zu hepatozellulären Karzinomen. Aus tierexperimentellen Studien gibt es Hinweise, dass HMG‑CoA-Reduktase-Inhibitoren die embryonale oder fötale Entwicklung beeinflussen können. Bei Ratten, Kaninchen und Hunden beeinflusste Atorvastatin die Fertilität nicht und war nicht teratogen. Bei maternal toxischen Dosen wurde jedoch bei Ratten und Kaninchen eine fötotoxische Wirkung beobachtet. Bei einer Exposition des Muttertiers mit hohen Dosen von Atorvastatin kam es bei Ratten zu einer verzögerten Entwicklung und einer verringerten Überlebensrate des Nachwuchses. Bei Ratten ergaben sich Hinweise auf eine Plazentagängigkeit. Die Atorvastatin-Konzentrationen sind bei Ratten im Plasma und der Muttermilch ähnlich. Ob Atorvastatin oder seine Metaboliten in die menschliche Muttermilch übergehen, ist nicht bekannt.
Tablettenkern:
Mikrokristalline Cellulose
Mannitol (Ph.Eur.)
Calciumcarbonat
Croscarmellose-Natrium
Hydroxypropylcellulose (Ph.Eur.)
Polysorbat 80
Eisen(III)-hydroxid-oxid x H2O
Magnesiumstearat (Ph.Eur.) [pflanzlich]
Povidon K-30
Natriumdodecylsulfat
Ezetimib/Atorvastatin Viatris 10 mg/10 mg, 10 mg/20 mg und 10 mg/40 mg Filmtabletten
Filmüberzug:
Lactose-Monohydrat
Hypromellose
Titandioxid
Macrogol 4000
Ezetimib/Atorvastatin Viatris 10 mg/80 mg Filmtabletten
Filmüberzug:
Hypromellose
Titandioxid
Talkum
Macrogol 400
Eisen(III)-hydroxid-oxid x H2O
Nicht zutreffend.
2 Jahre
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Ezetimib/Atorvastatin Viatris 10 mg/10 mg, 10 mg/20 mg und 10 mg/40 mg Filmtabletten
OPA/Al/PVC//Al-Blisterpackungen mit 30, 60, 90 oder 100 Filmtabletten.
Ezetimib/Atorvastatin Viatris 10 mg/80 mg Filmtabletten
OPA/Al/PVC//Al-Blisterpackungen mit 30 Filmtabletten, 60 Filmtabletten oder Mehrfachpackungen mit 90 (2 Packungen zu 45) oder 100 (2 Packungen zu 50) Filmtabletten.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Viatris Germany GmbH
Lütticher Straße 5
53842 Troisdorf
Mitvertreiber:
Viatris Healthcare GmbH
Lütticher Straße 5
53842 Troisdorf
7012603.00.00
7012604.00.00
7012605.00.00
7012606.00.00
02.12.2024
Mai 2026
Verschreibungspflichtig