
OPDIVO 300 mg Injektionslösung
OPDIVO 600 mg Injektionslösung
Jeder ml Injektionslösung enthält 120 mg Nivolumab.
Eine 2,5‑ml‑Durchstechflasche enthält 300 mg Nivolumab.
Eine 5‑ml‑Durchstechflasche enthält 600 mg Nivolumab.
Nivolumab wird mittels rekombinanter DNA‑Technologie aus Ovarialzellen des Chinesischen Hamsters gewonnen.
Sonstiger Bestandteil mit bekannter Wirkung
Jede 2,5 ml-Durchstechflasche dieses Arzneimittels enthält 1,25 mg Polysorbat 80.
Jede 5 ml-Durchstechflasche dieses Arzneimittels enthält 2,5 mg Polysorbat 80.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung.
Klare bis opaleszierende, farblose bis gelbe Flüssigkeit, die praktisch frei von sichtbaren Partikeln ist. Die Lösung hat einen pH‑Wert von 5,5 – 6,5 und eine Osmolalität von 296 – 444 mOsm/kg.
Melanom
OPDIVO ist als Monotherapie bei Erwachsenen zur adjuvanten Behandlung des Melanoms im Stadium IIB oder IIC oder des Melanoms mit Lymphknotenbeteiligung oder Metastasierung nach vollständiger Resektion indiziert (siehe Abschnitt 5.1).
OPDIVO ist als Monotherapie oder in Kombination mit Ipilimumab bei Erwachsenen für die Behandlung des fortgeschrittenen (nicht resezierbaren oder metastasierten) Melanoms indiziert (siehe Abschnitt 4.2).
Im Vergleich zur Nivolumab-Monotherapie wurde in der Kombination Nivolumab mit Ipilimumab nur bei Patienten mit niedriger Tumor‑PD‑L1‑Expression ein Anstieg des progressionsfreien Überlebens (progression‑free survival, PFS) und des Gesamtüberlebens (overall survival, OS) gezeigt (siehe Abschnitte 4.4 und 5.1).
Nicht‑kleinzelliges Lungenkarzinom (non‑small cell lung cancer, NSCLC)
OPDIVO ist in Kombination mit platinbasierter Chemotherapie für die neoadjuvante Behandlung des resezierbaren nicht‑kleinzelligen Lungenkarzinoms mit Tumorzell-PD‑L1-Expression ≥ 1 % bei Erwachsenen mit hohem Rezidivrisiko indiziert (Auswahlkriterien siehe Abschnitt 5.1).
OPDIVO ist in Kombination mit platinbasierter Chemotherapie für die neoadjuvante Behandlung, gefolgt von OPDIVO als Monotherapie für die adjuvante Behandlung des resezierbaren nicht‑kleinzelligen Lungenkarzinoms mit Tumorzell-PD‑L1-Expression ≥ 1 % bei Erwachsenen mit hohem Rezidivrisiko indiziert (Auswahlkriterien siehe Abschnitt 5.1).
OPDIVO ist als Monotherapie zur Behandlung des lokal fortgeschrittenen oder metastasierten nicht‑kleinzelligen Lungenkarzinoms nach vorheriger Chemotherapie bei Erwachsenen indiziert.
Nierenzellkarzinom (renal cell carcinoma, RCC)
OPDIVO ist in Kombination mit Ipilimumab für die Erstlinientherapie des fortgeschrittenen Nierenzellkarzinoms bei Erwachsenen mit intermediärem/ungünstigem Risikoprofil indiziert (siehe Abschnitte 4.2 und 5.1).
OPDIVO ist in Kombination mit Cabozantinib für die Erstlinientherapie des fortgeschrittenen Nierenzellkarzinoms bei Erwachsenen indiziert (siehe Abschnitt 5.1).
OPDIVO ist als Monotherapie bei Erwachsenen zur Behandlung des fortgeschrittenen Nierenzellkarzinoms nach Vortherapie indiziert.
Plattenepithelkarzinom des Kopf‑Hals‑Bereichs (squamous cell cancer of the head and neck, SCCHN)
OPDIVO ist als Monotherapie zur Behandlung des rezidivierten oder metastasierten Plattenepithelkarzinoms des Kopf‑Hals‑Bereichs bei Erwachsenen mit einer Progression während oder nach einer platinbasierten Therapie indiziert (siehe Abschnitt 5.1).
Urothelkarzinom (urothel carcinoma, UC)
OPDIVO ist als Monotherapie zur adjuvanten Behandlung des muskelinvasiven Urothelkarzinoms (muscle invasive urothelial carcinoma, MIUC) mit Tumorzell‑PD‑L1‑Expression ≥ 1 % bei Erwachsenen mit hohem Rezidivrisiko nach radikaler Resektion des MIUC indiziert (siehe Abschnitt 5.1).
OPDIVO ist in Kombination mit Cisplatin und Gemcitabin für die Erstlinientherapie des nicht resezierbaren oder metastasierten Urothelkarzinoms bei Erwachsenen indiziert (siehe Abschnitte 4.2 und 5.1).
OPDIVO ist als Monotherapie zur Behandlung des lokal fortgeschrittenen nicht resezierbaren oder metastasierten Urothelkarzinoms bei Erwachsenen nach Versagen einer vorherigen platinhaltigen Therapie indiziert.
Kolorektalkarzinom (colorectal cancer, CRC) mit Mismatch‑Reparatur‑Defizienz (Mismatch repair deficient, dMMR) oder hoher Mikrosatelliteninstabilität (microsatellite instability high, MSI‑H)
OPDIVO ist in Kombination mit Ipilimumab zur Behandlung des Kolorektalkarzinoms mit Mismatch‑Reparatur‑Defizienz oder hoher Mikrosatelliteninstabilität bei Erwachsenen in den folgenden Fällen indiziert:
Erstlinientherapie des nicht resezierbaren oder metastasierten Kolorektalkarzinoms (siehe Abschnitte 4.2 und 5.1);
Behandlung des metastasierten Kolorektalkarzinoms nach vorheriger fluoropyrimidinbasierter Kombinationschemotherapie (siehe Abschnitte 4.2 und 5.1).
Plattenepithelkarzinom des Ösophagus (esophageal squamous cell carcinoma, ESCC)
OPDIVO ist in Kombination mit fluoropyrimidin‑ und platinbasierter Kombinationschemotherapie für die Erstlinienbehandlung des nicht resezierbaren fortgeschrittenen, rezidivierten oder metastasierten Plattenepithelkarzinoms des Ösophagus mit Tumorzell‑PD‑L1‑Expression ≥ 1 % bei Erwachsenen indiziert.
OPDIVO ist als Monotherapie zur Behandlung des nicht resezierbaren fortgeschrittenen, rezidivierten oder metastasierten Plattenepithelkarzinoms des Ösophagus bei Erwachsenen nach vorheriger fluoropyrimidin‑ und platinbasierter Kombinationschemotherapie indiziert.
Adjuvante Behandlung der Karzinome des Ösophagus (esophageal cancer, EC) oder des gastroösophagealen Übergangs (gastro‑esophageal junction cancer, GEJC)
OPDIVO ist als Monotherapie zur adjuvanten Behandlung der Karzinome des Ösophagus oder des gastroösophagealen Übergangs bei Erwachsenen mit pathologischer Resterkrankung nach vorheriger neoadjuvanter Chemoradiotherapie indiziert (siehe Abschnitt 5.1).
Adenokarzinome des Magens, des gastroösophagealen Übergangs (gastro‑esophageal junction, GEJ) oder des Ösophagus
OPDIVO ist in Kombination mit fluoropyrimidin‑ und platinbasierter Kombinationschemotherapie für die Erstlinienbehandlung der HER2‑negativen fortgeschrittenen oder metastasierten Adenokarzinome des Magens, des gastroösophagealen Übergangs oder des Ösophagus bei Erwachsenen indiziert, deren Tumoren PD‑L1 (Combined Positive Score [CPS] ≥ 5) exprimieren.
Hepatozelluläres Karzinom (hepatocellular carcinoma, HCC)
OPDIVO ist in Kombination mit Ipilimumab für die Erstlinientherapie des nicht resezierbaren oder fortgeschrittenen hepatozellulären Karzinoms bei Erwachsenen indiziert.
Die Behandlung muss von einem auf dem Gebiet der Krebsbehandlung erfahrenen Arzt eingeleitet und überwacht werden.
Patienten, die derzeit Nivolumab intravenös als Monotherapie oder in Kombination mit einer Chemotherapie oder Cabozantinib erhalten, können auf die OPDIVO-Injektionslösung umgestellt werden.
PD‑L1‑Testung
Falls im Anwendungsgebiet angegeben, sollen die Patienten für eine Behandlung mit OPDIVO basierend auf der durch einen Test mittels eines IVDs (In‑vitro‑Diagnostikum) mit CE-Kennzeichnung beurteilten Tumor‑PD‑L1‑Expression selektiert werden. Wenn das IVD mit CE-Kennzeichnung nicht verfügbar ist, sollte ein alternativer validierter Test verwendet werden (siehe Abschnitte 4.1, 4.4 und 5.1).
MSI‑/MMR‑Testung
Falls im Anwendungsgebiet angegeben, sollen die Patienten für eine Behandlung mit OPDIVO mittels eines IVDs (In-vitro-Diagnostikum) mit CE-Kennzeichnung und entsprechendem Verwendungszweck beurteilten MSI‑H-/dMMR‑Tumorstatus selektiert werden. Wenn das IVD mit CE-Kennzeichnung nicht verfügbar ist, sollte ein alternativer validierter Test verwendet werden (siehe Abschnitte 4.1, 4.4 und 5.1).
Dosierung
OPDIVO als Monotherapie
Die empfohlene Dosis OPDIVO‑Injektionslösung beträgt entweder Nivolumab 600 mg alle 2 Wochen oder 1 200 mg alle 4 Wochen (siehe Abschnitt 5.1).
Falls Patienten von der 2‑wöchentlichen Gabe von 600 mg umgestellt werden sollen auf 1 200 mg alle 4 Wochen, soll die erste 1 200 mg‑Dosis zwei Wochen nach der letzten 600 mg‑Dosis verabreicht werden. Dagegen sollen Patienten, die von der 4‑wöchentlichen Gabe von 1 200 mg umgestellt werden sollen auf 600 mg alle 2 Wochen, die erste 600 mg‑Dosis vier Wochen nach der letzten 1 200 mg‑Dosis verabreicht bekommen.
OPDIVO in Kombination mit Ipilimumab
Melanom
Tabelle 1: Empfohlene Dosierungen und Infusionszeiten für OPDIVO-Infusionslösung in Kombination mit Ipilimumab gefolgt von OPDIVO-Injektionslösung als Monotherapie für Melanom (siehe Abschnitt 5.1)
Kombinationsphase |
Monotherapiephase |
|
Nivolumab |
1 mg/kg alle 3 Wochen über 30 Minuten |
600 mg alle 2 Wochen oder 1 200 mg alle 4 Wochen
|
Ipilimumab |
3 mg/kg alle 3 Wochen über 30 Minuten |
- |
Nierenzellkarzinom (renal cell carcinoma, RCC)
Tabelle 2: Empfohlene Dosierungen und Infusionszeiten für OPDIVO-Infusionslösung in Kombination mit Ipilimumab gefolgt von OPDIVO-Injektionslösung als Monotherapie für RCC
Kombinationsphase |
Monotherapiephase |
|
Nivolumab |
3 mg/kg alle 3 Wochen über 30 Minuten |
600 mg alle 2 Wochen oder 1 200 mg alle 4 Wochen
|
Ipilimumab |
1 mg/kg alle 3 Wochen über 30 Minuten |
- |
dMMR‑ oder MSI‑H‑Kolorektalkarzinom (colorectal cancer, CRC)
Tabelle 3: Empfohlene Dosierungen und Infusionszeiten für OPDIVO-Infusionslösung in Kombination mit Ipilimumab gefolgt von OPDIVO-Injektionslösung als Monotherapie für dMMR oder MSI‑H CRC
Kombinationsphase, OPDIVO-Infusionslösung, intravenös (i.v.) und Ipilimumab, für bis zu 4 Dosierungszyklen |
Monotherapiephase |
||
Nivolumab |
Erstlinientherapie |
240 mg alle 3 Wochen über 30 Minuten |
600 mg alle 2 Wochen oder 1 200 mg alle 4 Wochen |
Behandlung nach vorheriger fluoropyrimidinbasierter Erstlinien-Kombinationschemotherapie |
3 mg/kg alle 3 Wochen über 30 Minuten |
600 mg alle 2 Wochen oder 1 200 mg alle 4 Wochen |
|
Ipilimumab |
1 mg/kg alle 3 Wochen über 30 Minuten |
- |
|
Hepatozelluläres Karzinom (HCC)
Tabelle 4: Empfohlene Dosierungen und Infusionszeiten für OPDIVO-Infusionslösung in Kombination mit Ipilimumab gefolgt von OPDIVO-Injektionslösung als Monotherapie für die Behandlung von HCC (siehe Abschnitte 5.1 und 5.2)
Kombinationsphase |
Monotherapiephase* |
|
Nivolumab |
1 mg/kg alle 3 Wochen über 30 Minuten |
600 mg alle 2 Wochen oder 1 200 mg alle 4 Wochen |
Ipilimumab |
3 mg/kg alle 3 Wochen über 30 Minuten |
- |
*Es wird empfohlen, die Behandlung bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate fortzusetzen. | ||
OPDIVO in Kombination mit Cabozantinib
Nierenzellkarzinom (RCC)
Tabelle 5: Empfohlene Dosierungen für OPDIVO-Injektionslösung in Kombination mit Cabozantinib für die Behandlung von RCC
Kombinationstherapie* |
|
Nivolumab |
600 mg alle 2 Wochen oder 1 200 mg alle 4 Wochen |
Cabozantinib |
40 mg täglich oral |
*Für die Behandlung mit OPDIVO in Kombination mit Cabozantinib soll OPDIVO bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate bei Patienten ohne Progression der Erkrankung gegeben werden. Die Behandlung mit Cabozantinib soll bis zur Progression der Erkrankung oder nicht akzeptabler Toxizität fortgesetzt werden. Lesen Sie die Fachinformation für Cabozantinib. | |
OPDIVO in Kombination mit Chemotherapie
Plattenepithelkarzinom des Ösophagus (esophageal squamous cell carcinoma, ESCC)
Tabelle 6: Empfohlene Dosierungen zur Verabreichung von OPDIVO-Injektionslösung in Kombination mit fluoropyrimidin- und platinbasierter Chemotherapie für die Behandlung von ESCC (siehe Abschnitt 5.1)*
Kombinationstherapie |
|
Nivolumab |
600 mg alle 2 Wochen oder 1 200 mg alle 4 Wochen |
Fluoropyrimidin- und platinbasierte Chemotherapie |
Alle 4 Wochen |
*Die Behandlung mit Nivolumab soll bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate bei Patienten ohne Progression der Erkrankung fortgesetzt werden. | |
Adenokarzinome des Magens, des gastroösophagealen Übergangs (gastro‑oesophageal junction, GEJ) oder des Ösophagus
Tabelle 7: Empfohlene Dosierungen zur Verabreichung von OPDIVO-Injektionslösung in Kombination mit fluoropyrimidin- und platinbasierter Chemotherapie für die Behandlung von Adenokarzinomen des Magens, des gastroösophagealen Übergangs oder des Ösophagus (siehe Abschnitt 5.1)*
Kombinationstherapie |
|
Nivolumab |
600 mg alle 2 Wochen oder 900 mg alle 3 Wochen |
Fluoropyrimidin- und platinbasierte Chemotherapie |
Alle 2 Wochen oder alle 3 Wochen, abhängig vom Nivolumab-Regime |
*Die Behandlung mit Nivolumab soll bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate bei Patienten ohne Progression der Erkrankung fortgesetzt werden. | |
Urothelkarzinom (urothelial carcinoma, UC)
Tabelle 8: Empfohlene Dosierungen für OPDIVO-Injektionslösung in Kombination mit Cisplatin und Gemcitabin gefolgt von OPDIVO-Monotherapie für UC (siehe Abschnitt 5.1)
Kombinationsphase |
Monotherapiephase* |
|
Nivolumab |
900 mg alle 3 Wochen |
600 mg alle 2 Wochen oder 1 200 mg alle 4 Wochen |
Cisplatin und Gemcitabin |
Alle 3 Wochen |
- |
*Die Behandlung mit Nivolumab soll bis zur Progression der Erkrankung, bis zu nicht akzeptabler Toxizität oder bis zu 24 Monate ab der ersten Dosis fortgesetzt werden, je nachdem, was zuerst eintritt. | ||
Neoadjuvante Behandlung des NSCLC
Tabelle 9: Empfohlene Dosierungen für OPDIVO-Injektionslösung in Kombination mit platinbasierter Chemotherapie für die neoadjuvante Behandlung des NSCLC (siehe Abschnitt 5.1)
Kombinationstherapie für 3 Zyklen |
|
Nivolumab |
900 mg alle 3 Wochen |
Platinbasierte Chemotherapie |
Alle 3 Wochen |
Neoadjuvante und adjuvante Behandlung des nicht‑kleinzelligen Lungenkarzinoms (NSCLC)
Tabelle 10: Empfohlene Dosierungen für OPDIVO-Injektionslösung in Kombination mit platinbasierter Chemotherapie für die neoadjuvante Behandlung gefolgt von OPDIVO-Monotherapie für die adjuvante Behandlung von NSCLC
Kombinationsphase |
Monotherapiephase* |
|
Nivolumab |
900 mg alle 3 Wochen |
1 200 mg alle 4 Wochen |
Platinbasierte Chemotherapie |
Alle 3 Wochen |
- |
*Die Behandlung soll bis zur Progression oder zum Rezidiv der Erkrankung, nicht akzeptabler Toxizität oder bis zu 13 Zyklen fortgesetzt werden (siehe Abschnitt 5.1). | ||
Dauer der Behandlung
Die Behandlung mit OPDIVO, entweder als Monotherapie oder in Kombination mit anderen Arzneimitteln, soll so lange fortgesetzt werden, wie ein klinischer Nutzen besteht oder bis die Behandlung vom Patienten nicht mehr vertragen wird (oder bis zur maximalen Therapiedauer, soweit diese für eine Indikation festgelegt ist).
Für die adjuvante Behandlung beträgt die maximale Behandlungsdauer mit OPDIVO 12 Monate.
Untypisches Ansprechen (d. h., eine initiale vorübergehende Zunahme der Tumorgröße oder kleine, neue Läsionen innerhalb der ersten Monate gefolgt von einer Schrumpfung des Tumors) wurde beobachtet. Bei klinisch stabilen Patienten mit initialen Anzeichen einer Krankheitsprogression wird empfohlen, die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab fortzusetzen, bis eine Krankheitsprogression bestätigt ist.
Eine Dosissteigerung oder ‑reduktion wird nicht empfohlen für OPDIVO als Monotherapie oder in Kombination mit anderen Arzneimitteln. Je nach individueller Sicherheit und Verträglichkeit ist möglicherweise ein Aufschieben einer Dosis oder ein dauerhafter Abbruch der Behandlung erforderlich. Richtlinien zum dauerhaften Absetzen oder Aufschieben von Dosen werden in Tabelle 10 beschrieben. Detaillierte Richtlinien zur Behandlung immunvermittelter Nebenwirkungen werden im Abschnitt 4.4 beschrieben. Bei einer Verabreichung von Nivolumab in Kombination mit anderen Arzneimitteln lesen Sie die Fachinformationen der entsprechenden Kombinationsarzneimittel bezüglich Dosierung.
Tabelle 11: Empfohlene Behandlungsmodifikationen für OPDIVO oder OPDIVO in Kombination
Immunvermittelte Nebenwirkung |
Schweregrad |
Behandlungsmodifikation |
Immunvermittelte Pneumonitis |
Pneumonitis Grad 2 |
Dosis(en) aufschieben, bis sich die Symptome zurückgebildet haben, radiologisch erkennbare Veränderungen sich gebessert haben und die Behandlung mit Corticosteroiden beendet ist |
Pneumonitis Grad 3 oder 4 |
Setzen Sie die Behandlung dauerhaft ab |
|
Immunvermittelte Kolitis |
Diarrhö oder Kolitis Grad 2 |
Dosis(en) aufschieben, bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden, falls erforderlich, beendet ist |
Diarrhö oder Kolitis Grad 3 |
||
OPDIVO‑Monotherapie |
Dosis(en) aufschieben, bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden beendet ist |
|
OPDIVO + Ipilimumaba |
Setzen Sie die Behandlung dauerhaft ab |
|
Diarrhö oder Kolitis Grad 4 |
Setzen Sie die Behandlung dauerhaft ab |
|
Immunvermittelte Hepatitis |
Erhöhung der Aspartat‑Aminotransferase (AST), Alanin‑Aminotransferase (ALT) oder Gesamtbilirubin Grad 2 |
Dosis(en) aufschieben, bis die Laborwerte auf den Ausgangswert zurückgegangen sind und die Behandlung mit Corticosteroiden, falls erforderlich, beendet ist |
Erhöhung von AST, ALT, oder Gesamtbilirubin Grad 3 oder 4 |
Setzen Sie die Behandlung dauerhaft ab |
|
Immunvermittelte Nephritis und Nierenfunktionsstörung |
Kreatinin‑Erhöhung Grad 2 oder 3 |
Dosis(en) aufschieben, bis das Kreatinin auf den Ausgangswert zurückgegangen ist und die Behandlung mit Corticosteroiden beendet ist |
Kreatinin‑Erhöhung Grad 4 |
Setzen Sie die Behandlung dauerhaft ab |
|
Immunvermittelte Endokrinopathien |
Symptomatische Grad 2 oder 3 Hypothyreose, Hyperthyreose, Hypophysitis |
Dosis(en) aufschieben, bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden (falls nötig bei Symptomen akuter Entzündung) beendet ist. Die Behandlung mit OPDIVO soll begleitend zur Hormonsubstitutionstherapieb fortgeführt werden, sofern keine Symptome auftreten |
Grad 4 Hypothyreose |
Setzen Sie die Behandlung dauerhaft ab |
|
Immunvermittelte Nebenwirkungen der Haut |
Ausschlag Grad 3 |
Dosis(en) aufschieben, bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden beendet ist |
Ausschlag Grad 4 |
Setzen Sie die Behandlung dauerhaft ab |
|
Stevens‑Johnson Syndrom (SJS) oder toxische epidermale Nekrolyse (TEN) |
Setzen Sie die Behandlung dauerhaft ab (siehe Abschnitt 4.4) |
|
Immunvermittelte Myokarditis |
Grad 2 Myokarditis |
Dosis(en) aufschieben, bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden beendet istc |
Grad 3 oder 4 Myokarditis |
Setzen Sie die Behandlung dauerhaft ab |
|
Andere immunvermittelte Nebenwirkungen |
Grad 3 (erstes Auftreten) |
Dosis(en) aufschieben |
Grad 4 oder wiederauftretender Grad 3; persistierender Grad 2 oder 3 trotz Behandlungsmodifikation; Fälle, in denen die Corticosteroiddosis nicht auf 10 mg Prednison oder das entsprechende Äquivalent pro Tag reduziert werden kann |
Setzen Sie die Behandlung dauerhaft ab |
|
Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndromd |
Grad 2 Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom |
Dosis(en) aufschieben, bis sich die Symptome zurückgebildet haben und die Behandlung mit Corticosteroiden beendet ist |
Grad 3 oder 4 Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom |
Setzen Sie die Behandlung dauerhaft ab |
|
Hinweis: Toxizitätsgrade entsprechen den Kriterien des National Cancer Institute (National Cancer Institute Common Terminology Criteria for Adverse Events), Version 4.0 (NCI‑CTCAE v4). | ||
OPDIVO als Monotherapie oder in Kombination mit anderen Arzneimitteln muss dauerhaft abgesetzt werden bei:
Grad 4 oder wieder auftretenden Grad 3 Nebenwirkungen,
Grad 2 oder 3 Nebenwirkungen, die trotz Behandlung persistieren.
Patienten, die mit OPDIVO behandelt werden, ist die Patientenkarte auszuhändigen und sie müssen über die Risiken von OPDIVO informiert werden (siehe auch Packungsbeilage).
Wenn OPDIVO intravenös in Kombination mit Ipilimumab angewendet wird, soll bei Aufschiebung des einen Wirkstoffes auch die Gabe des anderen Wirkstoffs aufgeschoben werden. Wenn die Behandlung nach einer Pause wieder aufgenommen wird, kann aufgrund der Beurteilung des individuellen Patienten entweder die intravenöse Kombinationsbehandlung oder die intravenöse oder subkutane OPDIVO‑Monotherapie wieder aufgenommen werden.
Wenn OPDIVO in Kombination mit Chemotherapie angewendet wird, lesen Sie die Fachinformationen der entsprechenden Kombinationsarzneimittel bezüglich Dosierung. Bei Aufschiebung eines Wirkstoffes können die anderen Wirkstoffe weiterhin verabreicht werden. Wenn die Behandlung nach einer Pause wieder aufgenommen wird, kann basierend auf der Beurteilung des individuellen Patienten entweder die Kombinationstherapie, OPDIVO als Monotherapie oder die Chemotherapie alleine wieder aufgenommen werden.
OPDIVO in Kombination mit Cabozantinib in RCC
Bei der Anwendung von OPDIVO in Kombination mit Cabozantinib gelten die oben genannten Behandlungsmodifikationen aus Tabelle 10 auch für die OPDIVO‑Komponente. Zusätzlich gilt bei einer Leberenzymerhöhung bei RCC‑Patienten, welche mit OPDIVO in Kombination mit Cabozantinib behandelt werden:
Falls ALT oder AST > 3‑mal ULN, aber ≤ 10‑mal ULN, ohne gleichzeitigem Gesamtbilirubin ≥ 2‑mal ULN beträgt, soll sowohl die Behandlung mit OPDIVO als auch mit Cabozantinib aufgeschoben werden, bis diese Nebenwirkungen auf Grad 0‑1 zurückgegangen sind. Die Behandlung mit Corticosteroiden soll erwogen werden. Die Wiederaufnahme der Behandlung mit nur einem Arzneimittel oder mit beiden Arzneimitteln nach Eintritt einer Besserung kann erwogen werden. Bei einer Wiederaufnahme von Cabozantinib, lesen Sie die Fachinformation für Cabozantinib.
Falls ALT oder AST > 10‑mal ULN oder > 3‑mal ULN mit gleichzeitigem Gesamtbilirubin ≥ 2‑mal ULN beträgt, soll sowohl die Behandlung mit OPDIVO als auch mit Cabozantinib dauerhaft abgesetzt und eine Behandlung mit Corticosteroiden erwogen werden.
Spezielle Patientenpopulationen
Ältere Menschen
Bei älteren Patienten (≥ 65 Jahre) ist keine Dosisanpassung erforderlich.
Eingeschränkte Nierenfunktion
Auf der Grundlage von Daten zur Populations‑Pharmakokinetik (PK) für intravenös verabreichtes Nivolumab ist bei Patienten mit leichter oder mäßiger Niereninsuffizienz keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Die Daten von Patienten mit schwerer Niereninsuffizienz sind begrenzt und lassen keine Schlussfolgerungen für diese Population zu.
Eingeschränkte Leberfunktion
Auf der Grundlage von Daten zur Populations‑PK für intravenös verabreichtes Nivolumab ist bei Patienten mit leicht oder mäßig eingeschränkter Leberfunktion keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Die Daten von Patienten mit stark eingeschränkter Leberfunktion sind begrenzt und lassen keine Schlussfolgerungen für diese Population zu. OPDIVO muss bei Patienten mit stark eingeschränkter Leberfunktion (Gesamtbilirubin > 3 × ULN und beliebige AST) mit Vorsicht angewendet werden.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von OPDIVO‑Injektionslösung bei Kindern unter 18 Jahren ist nicht erwiesen.
Art der Anwendung
OPDIVO‑Injektionslösung ist zur subkutanen Anwendung vorgesehen.
Es ist wichtig, das Etikett der Durchstechflasche zu prüfen, um sicherzustellen, dass dem Patienten die richtige Formulierung (intravenös oder subkutan) und Dosis gemäß Verschreibung verabreicht werden.
OPDIVO‑Injektionslösung ist nicht zur intravenösen Anwendung vorgesehen und darf nur als subkutane Injektion in den angegebenen Dosen verabreicht werden. Möglicherweise wird mehr als eine Durchstechflasche OPDIVO‑Injektionslösung benötigt, um die Gesamtdosis für den Patienten zu erhalten. Für Anweisungen zur Verwendung und Handhabung der OPDIVO‑Injektionslösung vor der Anwendung siehe Abschnitt 6.6.
Verabreichen Sie den gesamten Inhalt der Spritze mit der OPDIVO‑Injektionslösung über einen Zeitraum von 3 bis 5 Minuten in das subkutane Gewebe von Bauch oder Oberschenkel. Die Dosis sollte nicht auf zwei Spritzen und nicht auf zwei Injektionsstellen aufgeteilt werden. Wechseln Sie die Injektionsstelle bei nachfolgenden Injektionen. Injizieren Sie nicht in Bereiche, in denen die Haut empfindlich oder gerötet ist oder blaue Flecken aufweist, und nicht in Bereiche mit Narben oder Muttermalen. Wenn die Verabreichung der OPDIVO‑Injektionslösung unterbrochen wird, kann sie an derselben Stelle oder an einer anderen Stelle fortgesetzt werden.
Während der Behandlung mit OPDIVO‑Injektionslösung sollten andere Arzneimittel zur subkutanen Verabreichung vorzugsweise an anderen Stellen injiziert werden.
OPDIVO‑Infusionslösung (intravenöse Formulierung)
Für Informationen zur Dosierung und Art der Anwendung, siehe Zusammenfassung der Merkmale des Arzneimittels des OPDIVO Konzentrats zur Herstellung einer Infusionslösung.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Beurteilung des PD‑L1‑Status
Es ist wichtig, für die Beurteilung des PD‑L1‑Status eine gut validierte und robuste Methode zu verwenden.
Beurteilung des MSI‑/MMR‑Status
Es ist wichtig, für die Beurteilung des MSI‑H‑ und dMMR‑Tumorstatus eine gut validierte und robuste Methode zu verwenden.
Immunvermittelte Nebenwirkungen
Wenn Nivolumab in Kombination angewendet wird, lesen Sie vor Behandlungsbeginn die Fachinformationen der anderen Arzneimittel der Kombinationstherapie. Bei Anwendung von Nivolumab in Kombination mit Ipilimumab wurden höhere Häufigkeiten von immunvermittelten Nebenwirkungen beobachtet als bei der Nivolumab‑Monotherapie. Immunvermittelte Nebenwirkungen sind in der Kombination OPDIVO mit Cabozantinib vergleichbar häufig aufgetreten wie bei der Nivolumab‑Monotherapie. Daher gilt die folgende Richtlinie bei immunvermittelten Nebenwirkungen für die OPDIVO‑Komponente der Kombination, sofern nicht ausdrücklich anders angegeben. Die meisten immunvermittelten Nebenwirkungen verbesserten sich oder verschwanden bei geeignetem Nebenwirkungsmanagement, einschließlich Einleitung einer Corticosteroid‑Behandlung und Behandlungsmodifikationen (siehe Abschnitt 4.2).
Immunvermittelte Nebenwirkungen, die mehr als ein Körpersystem betreffen, können gleichzeitig auftreten.
Bei der Kombinationstherapie wurden auch kardiale und pulmonale Nebenwirkungen, einschließlich Lungenembolie, berichtet. Patienten sollen fortlaufend auf kardiale und pulmonale Nebenwirkungen hin überwacht werden sowie vor und regelmäßig während der Behandlung auf klinische Anzeichen und Symptome und Laborwertabweichungen, die Störungen des Elektrolythaushalts und Dehydrierung erkennen lassen. Nivolumab in Kombination mit Ipilimumab muss bei lebensbedrohlichen oder schweren wiederauftretenden kardialen und pulmonalen Nebenwirkungen abgesetzt werden (siehe Abschnitt 4.2).
Patienten sollen engmaschig überwacht werden (mindestens bis zu 5 Monate nach der letzten Dosis), da Nebenwirkungen unter Nivolumab oder Nivolumab in Kombination mit Ipilimumab jederzeit während oder nach der Behandlung auftreten können.
Bei vermuteten immunvermittelten Nebenwirkungen soll zur Bestätigung der Ätiologie oder zum Ausschluss anderer Ursachen eine angemessene Abklärung durchgeführt werden. In Abhängigkeit vom Schweregrad der Nebenwirkung soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben und die Patienten mit Corticosteroiden behandelt werden. Wenn eine Immunsuppression mit Corticosteroiden zur Behandlung von Nebenwirkungen eingesetzt wird, soll die Corticosteroidtherapie nach Besserung der Nebenwirkungen über mindestens einen Monat ausgeschlichen werden. Ein zu schnelles Ausschleichen kann zur Verschlechterung oder Wiederauftreten der Nebenwirkung führen. Wenn es trotz Corticosteroidanwendung zu einer Verschlechterung oder keiner Besserung kommt, sollen zusätzlich nicht‑steroidale Immunsuppressiva gegeben werden.
Bei Patienten mit einer bestehenden Autoimmunerkrankung (autoimmune disease, AID) deuten Daten aus Beobachtungsstudien darauf hin, dass das Risiko für immunvermittelte Nebenwirkungen nach einer Therapie mit Immun‑Checkpoint‑Inhibitoren im Vergleich zu Patienten ohne bestehende AID erhöht sein kann. Darüber hinaus traten häufig Schübe der zugrundeliegenden AID auf, die jedoch meistens leicht und beherrschbar waren.
Die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab soll nicht fortgesetzt werden, solange der Patient immunsuppressive Dosen von Corticosteroiden oder andere Immunsuppressiva erhält. Prophylaktisch sollen Antibiotika gegeben werden, um opportunistische Infektionen bei Patienten zu verhindern, die immunsuppressiv behandelt werden.
Nivolumab oder Nivolumab in Kombination mit Ipilimumab muss bei jeder schweren wiederauftretenden immunvermittelten Nebenwirkung und bei jeder lebensbedrohlichen immunvermittelten Nebenwirkung dauerhaft abgesetzt werden.
Immunvermittelte Pneumonitis
Unter Nivolumab‑Monotherapie oder Nivolumab in Kombination mit Ipilimumab wurden schwere Pneumonitis oder interstitielle Lungenerkrankung, auch mit tödlichem Verlauf, beobachtet (siehe Abschnitt 4.8). Die Patienten sollen auf Anzeichen und Symptome einer Pneumonitis wie beispielsweise radiologische Veränderungen (z. B. fokale milchglasartige Dichteanhebung, fleckige Infiltrate), Dyspnoe und Hypoxie überwacht werden. Infektionen und krankheitsbedingte Ursachen sollen ausgeschlossen werden.
Bei Pneumonitis Grad 3 oder 4 muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt und mit einer Behandlung mit Corticosteroiden in einer Dosierung von 2 bis 4 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden.
Bei (symptomatischer) Pneumonitis Grad 2 soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben und mit einer Behandlung mit Corticosteroiden in einer Dosierung von 1 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden. Bei einer Besserung kann die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab nach dem Ausschleichen der Corticosteroide fortgesetzt werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, soll die Corticosteroid‑Dosis auf 2 bis 4 mg/kg/Tag Methylprednisolon‑Äquivalent erhöht werden und Nivolumab oder Nivolumab in Kombination mit Ipilimumab muss dauerhaft abgesetzt werden.
Immunvermittelte Kolitis
Unter Nivolumab‑Monotherapie oder Nivolumab in Kombination mit Ipilimumab wurden schwere Diarrhö oder Kolitis beobachtet (siehe Abschnitt 4.8). Patienten sollen auf Diarrhö und weitere Symptome einer Kolitis wie Abdominalschmerz und Schleim oder Blut im Stuhl überwacht werden. Cytomegalievirus(CMV)‑Infektion/ ‑Reaktivierung wurde bei Patienten mit Corticosteroid‑refraktärer immunvermittelter Kolitis berichtet. Infektionen und andere Ursachen der Diarrhö sind deshalb durch geeignete Labortests und zusätzliche Untersuchungen auszuschließen. Falls sich die Diagnose der Corticosteroid‑refraktären immunvermittelten Kolitis bestätigt, soll zusätzlich zu dem Corticosteroid ein anderes Immunsuppressivum oder ein Austausch der Corticosteroidtherapie in Betracht gezogen werden.
Bei Diarrhö oder Kolitis Grad 4 muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt und es soll eine Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden.
Bei Diarrhö oder Kolitis Grad 3 soll die Nivolumab‑Monotherapie aufgeschoben und eine Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden. Bei einer Besserung kann die Nivolumab‑Monotherapie nach dem Ausschleichen der Corticosteroide fortgesetzt werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, muss die Nivolumab‑Monotherapie dauerhaft abgesetzt werden. Eine Diarrhö oder Kolitis Grad 3, die bei Nivolumab in Kombination mit Ipilimumab auftritt, erfordert ein dauerhaftes Absetzen der Behandlung und die Initiierung von Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent.
Bei Diarrhö oder Kolitis Grad 2 soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben werden. Bei anhaltender Diarrhö oder Kolitis soll mit Corticosteroiden in einer Dosierung von 0,5 bis 1 mg/kg/Tag Methylprednisolon‑Äquivalent behandelt werden. Bei einer Besserung kann die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab nach dem Ausschleichen der Corticosteroide (sofern erforderlich) fortgesetzt werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, soll die Dosis auf 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent erhöht werden und Nivolumab oder Nivolumab in Kombination mit Ipilimumab muss dauerhaft abgesetzt werden.
Immunvermittelte Hepatitis
Unter Nivolumab‑Monotherapie oder Nivolumab in Kombination mit Ipilimumab wurde schwere Hepatitis beobachtet (siehe Abschnitt 4.8). Patienten sollen auf Anzeichen und Symptome einer Hepatitis wie ein Anstieg der Transaminasen und des Gesamtbilirubins überwacht werden. Infektionen und krankheitsbedingte Ursachen sind auszuschließen.
Bei Erhöhung der Transaminasen oder des Gesamtbilirubins Grad 3 oder 4 muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt werden und es soll eine Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden.
Bei Erhöhung der Transaminasen oder des Gesamtbilirubins Grad 2 soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben werden. Bei anhaltenden Erhöhungen dieser Laborwerte soll mit Corticosteroiden in einer Dosierung von 0,5 bis 1 mg/kg/Tag Methylprednisolon‑Äquivalent behandelt werden. Bei einer Besserung kann die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab nach dem Ausschleichen der Corticosteroide (sofern erforderlich) fortgesetzt werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, soll die Dosis auf 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent erhöht werden, und Nivolumab oder Nivolumab in Kombination mit Ipilimumab ist dauerhaft abzusetzen.
Immunvermittelte Nephritis und Nierenfunktionsstörung
Unter der Behandlung mit Nivolumab‑Monotherapie oder Nivolumab in Kombination mit Ipilimumab wurden schwere Nephritis und Nierenfunktionsstörung beobachtet (siehe Abschnitt 4.8). Die Patienten sind auf Anzeichen und Symptome einer Nephritis oder Nierenfunktionsstörung zu überwachen. Bei den meisten Patienten tritt eine asymptomatische Kreatininerhöhung im Serum auf. Krankheitsbedingte Ursachen sind auszuschließen.
Bei einer Kreatininerhöhung im Serum Grad 4 muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt und es soll mit einer Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden.
Bei einer Kreatininerhöhung im Serum Grad 2 oder 3 soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben und mit einer Behandlung mit Corticosteroiden in einer Dosierung von 0,5 bis 1 mg/kg/Tag Methylprednisolon‑Äquivalent begonnen werden. Bei einer Besserung kann die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab nach dem Ausschleichen der Corticosteroide fortgesetzt werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, soll die Dosis auf 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent erhöht werden, und Nivolumab oder Nivolumab in Kombination mit Ipilimumab ist dauerhaft abzusetzen.
Immunvermittelte Endokrinopathien
Unter Nivolumab‑Monotherapie oder Nivolumab in Kombination mit Ipilimumab wurden schwere Endokrinopathien, einschließlich Hypothyreose, Hyperthyreose, Nebenniereninsuffizienz (einschließlich sekundäre Nebenniereninsuffizienz), Hypophysitis (einschließlich Hypophyseninsuffizienz), Diabetes mellitus und diabetische Ketoazidose beobachtet (siehe Abschnitt 4.8).
Patienten sollen hinsichtlich klinischer Anzeichen und Symptome von Endokrinopathien und Hyperglykämie und Veränderungen der Schilddrüsenfunktion überwacht werden (zu Beginn der Behandlung, regelmäßig während der Behandlung und wenn es nach klinischer Beurteilung angezeigt ist). Patienten können mit Ermüdung/Fatigue, Kopfschmerzen, psychischen Veränderungen, Abdominalschmerz, Veränderung der Stuhlgewohnheiten und Hypotonie oder unspezifischen Symptomen vorstellig werden, die anderen Ursachen, wie etwa Gehirnmetastasen oder der zugrundeliegenden Erkrankung, ähneln können. Bis eine andere Ätiologie identifiziert worden ist, sollen Anzeichen oder Symptome von Endokrinopathien als immunvermittelt betrachtet werden.
Bei symptomatischer Hypothyreose soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben und bei Bedarf mit einer Schilddrüsenhormonsubstitutionstherapie begonnen werden. Bei symptomatischer Hyperthyreose soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben und bei Bedarf mit einer Behandlung mit Thyreostatika begonnen werden. Bei Verdacht auf eine akute Entzündung der Schilddrüse soll auch eine Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent in Betracht gezogen werden. Bei einer Besserung kann die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab nach dem Ausschleichen der Corticosteroide (sofern erforderlich) fortgesetzt werden. Die Schilddrüsenfunktion soll weiterhin überwacht werden, um sicherzustellen, dass die passende Hormonsubstitutionstherapie angewandt wird. Bei lebensbedrohlicher Hyperthyreose oder Hypothyreose muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt werden.
Bei symptomatischer Nebenniereninsuffizienz Grad 2 soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben und bei Bedarf mit einer physiologischen Corticosteroid‑Ersatztherapie begonnen werden. Bei schwerwiegender (Grad 3) oder lebensbedrohlicher (Grad 4) Nebenniereninsuffizienz muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt werden. Die Nebennierenfunktion und Hormonspiegel sollen weiterhin überwacht werden, um sicherzustellen, dass die passende Corticosteroid‑Ersatztherapie angewandt wird.
Bei symptomatischer Hypophysitis von Grad 2 oder 3 soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben und bei Bedarf mit einer Hormonsubstitutionstherapie begonnen werden. Bei Verdacht auf akute Entzündung der Hypophyse soll auch eine Behandlung mit Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent in Betracht gezogen werden. Bei einer Besserung kann die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab nach dem Ausschleichen der Corticosteroide (sofern erforderlich) fortgesetzt werden. Bei lebensbedrohlicher (Grad 4) Hypophysitis muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt werden. Die Hypophysenfunktion und Hormonspiegel sollen weiterhin überwacht werden, um sicherzustellen, dass die passende Hormonsubstitutionstherapie angewandt wird.
Bei symptomatischem Diabetes soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben und bei Bedarf mit einer Insulinersatztherapie begonnen werden. Der Blutzuckerspiegel soll weiterhin überwacht werden, um sicherzustellen, dass die passende Insulinersatztherapie angewandt wird. Bei lebensbedrohlichem Diabetes muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt werden.
Immunvermittelte Nebenwirkungen der Haut
Unter Behandlung mit Nivolumab in Kombination mit Ipilimumab und, weniger häufig, bei Nivolumab‑Monotherapie wurden schwere Ausschläge beobachtet (siehe Abschnitt 4.8). Die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab soll bei Ausschlag Grad 3 aufgeschoben und bei Ausschlag Grad 4 abgesetzt werden. Schwerer Ausschlag soll mit hochdosierten Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg/Tag Methylprednisolon‑Äquivalent behandelt werden.
In seltenen Fällen wurden SJS und TEN berichtet, darunter waren auch einige Todesfälle. Wenn Symptome oder Anzeichen für SJS oder TEN auftreten, soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab abgesetzt und der Patient in eine spezialisierte Abteilung zur Beurteilung und Behandlung überwiesen werden. Wenn sich beim Patienten unter der Anwendung von Nivolumab oder Nivolumab in Kombination mit Ipilimumab SJS oder TEN entwickelt haben, wird die dauerhafte Absetzung der Behandlung empfohlen (siehe Abschnitt 4.2).
Vorsicht ist geboten, wenn für einen Patienten, der zuvor bei Behandlung mit anderen immunstimulierenden Arzneimitteln gegen Krebs eine schwere oder lebensbedrohliche Hautreaktion erlitten hat, die Anwendung von Nivolumab erwogen wird.
Andere immunvermittelte Nebenwirkungen
Folgende immunvermittelte Nebenwirkungen wurden bei weniger als 1 % der in klinischen Studien (in verschiedenen Dosierungen und bei diversen Tumorarten) mit Nivolumab‑Monotherapie oder Nivolumab in Kombination mit Ipilimumab behandelten Patienten berichtet: Pankreatitis, Uveitis, Demyelinisierung, autoimmune Neuropathie (einschließlich Gesichtsnerv‑ und Abduzensparese), Guillain‑Barré‑Syndrom, Myasthenia gravis, myasthenes Syndrom, aseptische Meningitis, Enzephalitis, Gastritis, Sarkoidose, Duodenitis, Myositis, Myokarditis, Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom, Rhabdomyolyse und Myelitis. Nach Markteinführung wurden Fälle von Vogt‑Koyanagi‑Harada‑Syndrom, Hypoparathyreoidismus und nicht‑infektiöser Zystitis berichtet (siehe Abschnitte 4.2 und 4.8).
Bei Verdacht auf immunvermittelte Nebenwirkungen soll eine adäquate Abklärung durchgeführt werden, um die Ursache zu bestätigen oder andere Gründe auszuschließen. Je nach Schweregrad der Nebenwirkung soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben und Corticosteroide gegeben werden. Bei einer Besserung kann die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab nach dem Ausschleichen der Corticosteroide fortgesetzt werden. Wenn eine schwere immunvermittelte Nebenwirkung erneut auftritt, sowie bei einer lebensbedrohlichen immunvermittelten Nebenwirkung ist Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abzusetzen.
Es wurden Fälle von Myotoxizität (Myositis, Myokarditis und Rhabdomyolyse) unter Nivolumab oder Nivolumab in Kombination mit Ipilimumab berichtet, manche davon mit tödlichem Ausgang. Wenn ein Patient Anzeichen und Symptome einer Myotoxizität entwickelt, soll er engmaschig überwacht und unverzüglich an einen Spezialisten zur Beurteilung und Behandlung überwiesen werden. Je nach Schweregrad der Myotoxizität soll Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben oder abgesetzt werden (siehe Abschnitt 4.2) und eine geeignete Behandlung eingeleitet werden.
Die Diagnose einer Myokarditis erfordert ein hohes Maß an Aufmerksamkeit. Patienten mit kardialen oder kardiopulmonalen Symptomen sollen auf eine mögliche Myokarditis untersucht werden. Falls eine Myokarditis vermutet wird, soll unverzüglich eine Hochdosistherapie mit Steroiden (Prednison 1 ‑ 2 mg/kg/Tag oder Methylprednisolon 1 ‑ 2 mg/kg/Tag) eingeleitet werden und unverzüglich eine kardiologische Untersuchung mit umfassender Diagnostik nach aktuellen klinischen Leitlinien veranlasst werden. Sobald die Diagnose einer Myokarditis bestätigt wurde, soll Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben oder dauerhaft abgesetzt werden (siehe Abschnitt 4.2).
Fälle von Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom (das sich als Überlappung von entweder zwei oder allen drei Erkrankungen äußert), einige davon mit tödlichem Ausgang, wurden unter Nivolumab und Nivolumab in Kombination mit anderen Arzneimitteln berichtet. Eine frühzeitige Erkennung und aggressive Behandlung sind unerlässlich, um der damit verbundenen Morbidität und dem Mortalitätsrisiko entgegenzuwirken.
Bei Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom Grad 3 oder 4 muss Nivolumab oder Nivolumab in Kombination mit Ipilimumab dauerhaft abgesetzt werden (siehe Abschnitt 4.2). Eine Behandlung mit Corticosteroiden soll gemäß klinischer Indikation eingeleitet werden.
Bei Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom Grad 2 soll die Behandlung mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab aufgeschoben werden und eine Behandlung mit Corticosteroiden gemäß klinischer Indikation eingeleitet werden (siehe Abschnitt 4.2). Bei einer Besserung kann die Fortsetzung der Behandlung mit Nivolumab nach dem Ausschleichen der Corticosteroide in Betracht gezogen werden. Wenn es trotz der Behandlung mit Corticosteroiden zu einer Verschlechterung oder keiner Besserung kommt, soll die Corticosteroid‑Dosis gemäß klinischer Indikation angepasst werden und Nivolumab oder Nivolumab in Kombination mit Ipilimumab muss dauerhaft abgesetzt werden.
Bei mit PD‑1‑Inhibitoren behandelten Patienten wurde im Postmarketing‑Umfeld eine Abstoßung von soliden Organtransplantaten beobachtet. Die Behandlung mit Nivolumab kann das Abstoßungsrisiko bei Empfängern solider Organtransplantate erhöhen. Bei diesen Patienten soll der Nutzen der Behandlung mit Nivolumab gegen das Risiko einer möglichen Organabstoßung abgewogen werden.
Eine hämophagozytische Lymphohistiozytose (HLH) wurde mit Nivolumab als Monotherapie und Nivolumab in Kombination mit Ipilimumab beobachtet. Vorsicht ist geboten, wenn Nivolumab als Monotherapie oder in Kombination mit Ipilimumab gegeben wird. Wenn HLH bestätigt wird, soll die Gabe von Nivolumab oder Nivolumab in Kombination mit Ipilimumab abgebrochen und die Behandlung von HLH eingeleitet werden.
In der Nachbeobachtungszeit von Patienten mit klassischem Hodgkin‑Lymphom, die nach der Behandlung mit intravenös verabreichtem Nivolumab eine allogene hämatopoetische Stammzelltransplantation (HSZT) erhalten hatten, wurden Fälle von akuter Graft‑versus‑Host‑Krankheit (GvHD, Spender‑gegen‑Empfänger‑Krankheit) und transplantatbezogener Mortalität (transplant related mortality, TRM) festgestellt. Die sorgfältige Abwägung des potenziellen Nutzens einer allogenen HSZT und des möglicherweise erhöhten Risikos von transplantatbezogenen Komplikationen soll einzelfallbezogen erbracht werden. Nach Markteinführung wurden bei Patienten, die nach allogener HSZT mit intravenös verabreichtem Nivolumab behandelt wurden, rasch einsetzende und schwere Ausprägungen der GvHD, einige mit tödlichem Ausgang, berichtet. Die Behandlung mit Nivolumab kann das Risiko schwerer GvHD und Todesfälle bei Patienten erhöhen, die zuvor eine allogene HSZT hatten, vor allem bei Patienten mit GvHD in der Vorgeschichte. Der Nutzen einer Behandlung mit Nivolumab soll bei diesen Patienten gegenüber dem möglichen Risiko abgewogen werden.
Infusionsreaktionen (intravenöse Formulierung)
In klinischen Studien mit intravenös verabreichtem Nivolumab oder intravenös verabreichtem Nivolumab in Kombination mit Ipilimumab wurden schwere Infusionsreaktionen berichtet (siehe Abschnitt 4.8). Falls eine schwere oder lebensbedrohliche Infusionsreaktion auftritt, muss die intravenöse Nivolumab‑Infusion bzw. die Infusion von intravenös verabreichtem Nivolumab in Kombination mit Ipilimumab abgesetzt und eine geeignete medizinische Behandlung eingeleitet werden. Patienten mit leichter oder mäßiger Infusionsreaktion können intravenös verabreichtes Nivolumab oder intravenös verabreichtes Nivolumab in Kombination mit Ipilimumab unter engmaschiger Überwachung und dem Einsatz von Prämedikation gemäß lokalen Behandlungsrichtlinien zur Prophylaxe von Reaktionen im Zusammenhang mit einer Infusion erhalten.
Krankheitsspezifische Vorsichtsmaßnahmen
Fortgeschrittenes Melanom
Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, aktiven Hirnmetastasen oder leptomeningealen Metastasen, Autoimmunerkrankung und Patienten, die vor Studienbeginn systemische Immunsuppressiva erhalten hatten, waren von den pivotalen klinischen Studien mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Patienten mit okulärem/uvealem Melanom waren von den pivotalen klinischen Studien zum Melanom ausgeschlossen. Zusätzlich wurden bei der Studie CA209037 Patienten ausgeschlossen, die eine Nebenwirkung vom Grad 4 hatten, die in Zusammenhang mit einer Anti‑CTLA‑4‑Therapie stand (siehe Abschnitt 5.1). Patienten mit einem anfänglichen ECOG‑Performance‑Status von 2, behandelten leptomeningealen Metastasen, okulärem/uvealem Melanom, Autoimmunerkrankungen und Patienten, die eine Nebenwirkung vom Grad 3‑4 hatten, die im Zusammenhang mit einer vorherigen Anti‑CTLA‑4‑Therapie stand, wurden in die Studie CA209172 eingeschlossen (siehe Abschnitt 5.1). Ohne weitere Daten für Patienten, die vor Studienteilnahme systemische Immunsuppresiva erhielten und für Patienten mit aktiven Hirnmetastasen oder leptomenigealen Metastasen, soll Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Im Vergleich zur Nivolumab‑Monotherapie wurde in der Kombination Nivolumab mit Ipilimumab nur bei Patienten mit niedriger Tumor‑PD‑L1‑Expression ein Anstieg des progressionsfreien Überlebens (PFS) gezeigt. Die Verbesserung des Gesamtüberlebens bei Patienten mit hoher Tumor‑PD‑L1‑Expression (PD‑L1 ≥ 1 %) war ähnlich bei der Behandlung mit Nivolumab in Kombination mit Ipilimumab und der Behandlung mit Nivolumab als Monotherapie. Bevor eine Behandlung mit der Kombination eingeleitet wird, wird den Ärzten empfohlen, die individuellen Patienten‑ und Tumorcharakteristika sorgfältig unter Berücksichtigung des beobachteten Nutzens und der Toxizität der Kombination relativ zur Nivolumab‑Monotherapie zu bewerten (siehe Abschnitte 4.8 und 5.1).
Anwendung von Nivolumab bei Melanom‑Patienten mit schnell fortschreitender Krankheit
Ärzte sollten das verzögerte Einsetzen der Wirkung von Nivolumab berücksichtigen, bevor sie eine Behandlung bei Patienten mit schnell fortschreitender Krankheit beginnen (siehe Abschnitt 5.1).
Adjuvante Behandlung des Melanoms
Es gibt keine Daten zur adjuvanten Behandlung bei Melanom‑Patienten mit folgenden Risikofaktoren (siehe Abschnitte 4.5 und 5.1):
Patienten mit vorheriger Autoimmunerkrankung und jeder Erkrankung, die eine systemische Behandlung mit Corticosteroiden (≥ 10 mg Prednison oder ‑Äquivalent täglich) oder anderen immunsuppressiven Arzneimitteln erfordert,
Patienten mit vorheriger Melanomtherapie (außer Patienten mit Operation, adjuvanter Strahlentherapie nach neurochirurgischer Resektion wegen Läsionen des Zentralnervensystems und zuvor adjuvanter Behandlung mit Interferon, welche ≥ 6 Monate vor der Randomisierung abgeschlossen wurde),
Patienten mit vorheriger Behandlung mit einem Anti‑PD‑1‑, Anti‑PD‑L1‑, Anti‑PD‑L2‑, Anti‑CD137‑ oder Anti‑CTLA‑4‑Antikörper (einschließlich Ipilimumab oder eines anderen Antikörpers oder Arzneimittels, das spezifisch auf T‑Zell‑Co‑Stimulation oder Checkpoint‑Wege abzielt),
Patienten unter 18 Jahren.
Ohne weitere Daten soll Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Nicht‑kleinzelliges Lungenkarzinom (NSCLC)
Behandlung von NSCLC nach vorheriger Chemotherapie
Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, aktiven Hirnmetastasen oder einer Autoimmunerkrankung, einer symptomatischen interstitiellen Lungenerkrankung und Patienten, die vor Studienbeginn eine systemische immunsuppressive Therapie erhalten hatten, waren von den pivotalen klinischen Studien bei NSCLC ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Patienten mit einem anfänglichen ECOG‑Performance‑Status von 2 wurden in die Studie CA209171 eingeschlossen (siehe Abschnitt 5.1). Ohne weitere Daten für Patienten mit Autoimmunerkrankungen, symptomatischen interstitiellen Lungenerkrankungen, aktiven Hirnmetastasen und Patienten, die vor Studienteilnahme systemische Immunsuppressiva erhielten, soll Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Ärzte sollten das verzögerte Einsetzen der Wirkung von Nivolumab berücksichtigen, bevor sie eine Behandlung bei Patienten mit schlechteren prognostischen Merkmalen und/oder aggressivem Krankheitsverlauf beginnen. Beim NSCLC mit nicht‑plattenepithelialer Histologie wurde innerhalb der ersten 3 Monate bei den mit Nivolumab behandelten Patienten eine höhere Anzahl an Todesfällen beobachtet verglichen mit den mit Docetaxel behandelten Patienten. Faktoren, die in Verbindung mit frühen Todesfällen stehen, waren schlechtere prognostische Merkmale und/oder ein aggressiverer Krankheitsverlauf in Kombination mit niedriger oder fehlender Tumor‑PD‑L1‑Expression (siehe Abschnitt 5.1).
Neoadjuvante Behandlung des NSCLC
Patienten mit einem anfänglichen Performance‑Status ≥ 2, einer aktiven Autoimmunerkrankung, einer symptomatischen interstitiellen Lungenerkrankung, mit Erkrankungen, die eine systemische immunsuppressive Therapie erforderlich machen, mit einer nicht‑resezierbaren oder metastasierten Erkrankung, die bereits zuvor eine Krebstherapie für die resezierbare Erkrankung erhalten haben, oder mit bekannten EGFR‑Mutationen oder ALK‑Translokationen waren von der pivotalen klinischen Studie zur neoadjuvanten Behandlung des resezierbaren NSCLC ausgeschlossen (siehe Abschnitt 5.1). Ohne weitere Daten soll Nivolumab in Kombination mit Chemotherapie bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Neoadjuvante und adjuvante Behandlung des NSCLC
Patienten mit einem anfänglichen Performance-Status ≥ 2, einer peripheren Neuropathie von Grad 2 oder höher, einer aktiven Autoimmunerkrankung, einer symptomatischen interstitiellen Lungenerkrankung, mit Erkrankungen, die eine systemische immunsuppressive Therapie erforderlich machen, mit einer nicht-resezierbaren oder metastasierten Erkrankung, die bereits zuvor eine Krebstherapie für die resezierbare Erkrankung erhalten haben, mit EGFR-Mutationen oder mit bekannten ALK-Translokationen oder die Hirnmetastasen hatten, waren von der pivotalen klinischen Studie zur neoadjuvanten und adjuvanten Behandlung des NSCLC ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten soll Nivolumab in Kombination mit Chemotherapie bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Nierenzellkarzinom (RCC)
Nivolumab oder Nivolumab in Kombination mit Ipilimumab
Patienten wurden bei einer Vorgeschichte gleichzeitig aufgetretener Hirnmetastasen, bei aktiver Autoimmunerkrankung oder bei Erkrankungen, die eine Behandlung mit einer systemischen Immunsuppression erfordern, von klinischen Studien mit Nivolumab oder Nivolumab in Kombination mit Ipilimumab ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten sollten Nivolumab oder Nivolumab in Kombination mit Ipilimumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Nivolumab in Kombination mit Cabozantinib
Patienten mit aktiven Gehirnmetastasen, Autoimmunerkrankung oder mit Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von den klinischen Studien mit Nivolumab in Kombination mit Cabozantinib ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Aufgrund der fehlenden Daten soll Nivolumab in Kombination mit Cabozantinib bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Im Vergleich zur Nivolumab‑Monotherapie wurden unter der Kombination Nivolumab mit Cabozantinib bei Patienten mit fortgeschrittenem RCC häufiger Grad 3 und 4 ALT‑ und AST‑Anstiege berichtet (siehe Abschnitt 4.8). Leberenzyme sollen vor Beginn und regelmäßig während der Behandlung beobachtet werden. Den Richtlinien für das medizinische Management für beide Arzneimittel soll gefolgt werden (siehe Abschnitt 4.2 und lesen Sie die Fachinformation von Cabozantinib).
Kopf‑Hals‑Tumoren
Patienten mit einem anfänglichen Performance‑Status ≥ 2, aktiven Hirnmetastasen oder leptomeningealen Metastasen, aktiver Autoimmunerkrankung, Erkrankungen, die eine systemische immunsuppressive Therapie erfordern oder Karzinomen mit primärer Lokalisation im Nasopharynx oder in der Speicheldrüse waren von der klinischen Studie bei SCCHN ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten soll Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Ärzte sollten das verzögerte Einsetzen der Wirkung von Nivolumab berücksichtigen, bevor sie eine Behandlung bei Patienten mit schlechteren prognostischen Merkmalen und/oder aggressivem Krankheitsverlauf beginnen. Bei Kopf‑Hals‑Tumoren wurde innerhalb der ersten 3 Monate bei den mit Nivolumab behandelten Patienten eine höhere Anzahl an Todesfällen beobachtet verglichen mit den mit Docetaxel behandelten Patienten. Faktoren, die in Verbindung mit frühen Todesfällen standen, waren ECOG‑Performance‑Status, schnelle Krankheitsprogression auf die vorherige Platintherapie und hohe Tumorlast.
Urothelkarzinom
Behandlung des fortgeschrittenen Urothelkarzinoms
Patienten mit einem anfänglichen Performance‑Status ≥ 2, aktiven Hirnmetastasen oder leptomeningealen Metastasen, aktiver Autoimmunerkrankung oder Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von den klinischen Studien beim Urothelkarzinom ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten soll Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Adjuvante Behandlung des Urothelkarzinoms
Patienten mit einem anfänglichen Performance‑Status ≥ 2 (ausgenommen Patienten mit einem anfänglichen Performance‑Status von 2, die keine neoadjuvante Cisplatin-basierte Chemotherapie erhalten haben und für eine adjuvante Cisplatin-basierte Chemotherapie nicht geeignet sind), Anzeichen der Erkrankung nach Operation, aktiver Autoimmunerkrankung, oder Erkrankungen, die eine systemische Immunsuppression erfordern, wurden von der klinischen Studie zur adjuvanten Behandlung des Urothelkarzinoms ausgeschlossen (siehe Abschnitt 4.5 und 5.1). Ohne weitere Daten soll Nivolumab bei diesen Patientenpopulationen nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall mit Vorsicht angewendet werden.
dMMR‑ oder MSI‑H‑Kolorektalkarzinom (CRC)
Patienten mit einem anfänglichen Performance‑Status ≥ 2, aktiven Hirnmetastasen oder leptomeningealen Metastasen, aktiver Autoimmunerkrankung oder Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von der klinischen Studie beim metastasierten dMMR‑ oder MSI‑H‑Kolorektalkarzinom ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten soll Nivolumab in Kombination mit Ipilimumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Plattenepithelkarzinom des Ösophagus (ESCC)
Erstlinientherapie des ESCC
Patienten mit einem anfänglichen Performance‑Status ≥ 2, mit einer Vorgeschichte von gleichzeitig aufgetretenen Hirnmetastasen, mit aktiver Autoimmunerkrankung, mit Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, oder mit erhöhtem Risiko für Blutungen oder Fisteln aufgrund von offensichtlicher Tumorinvasion in angrenzende Organe des ösophagealen Tumors waren von der klinischen Studie bei ESCC ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten soll Nivolumab in Kombination mit Chemotherapie bei diesen Patientenpopulationen nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall mit Vorsicht angewendet werden.
Behandlung des ESCC nach vorheriger Erstlinien‑Chemotherapie
Der Großteil der klinischen Daten, welche für das Plattenepithelkarzinom des Ösophagus zur Verfügung stehen, ist von Patienten asiatischer Herkunft (siehe Abschnitt 5.1).
Patienten mit einem anfänglichen Performance‑Status ≥ 2, mit symptomatischen oder behandlungsbedürftigen Hirnmetastasen, Patienten mit offensichtlicher Tumorinvasion in angrenzende Organe des Ösophagus (z. B. in die Aorta oder den Respirationstrakt), mit aktiver Autoimmunerkrankung oder mit Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von der klinischen Studie bei ESCC ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten soll Nivolumab bei diesen Patientenpopulationen mit Vorsicht und nur nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im Einzelfall angewendet werden.
Ärzte sollten das verzögerte Einsetzen der Wirkung von Nivolumab berücksichtigen, bevor sie eine Behandlung bei Patienten mit ESCC beginnen. Eine höhere Anzahl an Todesfällen innerhalb der ersten 2,5 Monate nach Randomisierung wurde bei Patienten beobachtet, die mit Nivolumab behandelt wurden, verglichen mit den mit Chemotherapie behandelten Patienten. Es konnten keine spezifischen Faktoren im Zusammenhang mit den frühen Todesfällen identifiziert werden (siehe Abschnitt 5.1).
Adjuvante Behandlung der Karzinome des Ösophagus oder des gastroösophagealen Übergangs
Patienten mit einem anfänglichen Performance‑Status ≥2, Patienten, die keine gleichzeitige Chemoradiotherapie (CRT) vor der Operation erhalten hatten, Patienten mit Stadium IV resezierbarer Erkrankung, aktiver Autoimmunerkrankung oder Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von der klinischen Studie bei Karzinomen des Ösophagus oder des gastroösophagealen Übergangs ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten soll Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Adenokarzinome des Magens, des gastroösophagealen Übergangs oder des Ösophagus
Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, unbehandelten Metastasen des zentralen Nervensystems, aktiver bekannter oder vermuteter Autoimmunerkrankung oder Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von der klinischen Studie bei Adenokarzinomen des Magens, des gastroösophagealen Übergangs oder des Ösophagus ausgeschlossen (siehe Abschnitte 4.5 und 5.1). Ohne weitere Daten soll Nivolumab in Kombination mit Chemotherapie bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
In der Studie CA209649 wurden Patienten mit bekanntem positiven HER2‑Status ausgeschlossen. Patienten mit unbekanntem Status waren in der Studie erlaubt und repräsentierten 40,3 % der Patienten (siehe Abschnitt 5.1).
Hepatozelluläres Karzinom (HCC)
Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, vorheriger Lebertransplantation, Child‑Pugh‑C‑Lebererkrankung, einer Vorgeschichte gleichzeitig aufgetretener Hirnmetastasen, einer Vorgeschichte von hepatischer Enzephalopathie (innerhalb von 12 Monaten vor der Randomisierung), klinisch signifikantem Aszites, einer HIV-Infektion oder aktiver Koinfektion mit Hepatitis-B-Virus (HBV) und Hepatitis-C-Virus (HCV) oder HBV und Hepatitis-D-Virus (HDV), aktiver Autoimmunerkrankung oder Erkrankungen, die eine systemische Immunsuppression erfordern, wurden von der klinischen Studie im HCC ausgeschlossen (siehe Abschnitt 4.5 und 5.1). Die Daten zu HCC‑Patienten mit Child‑Pugh B sind begrenzt. Ohne weitere Daten muss Nivolumab in Kombination mit Ipilimumab, gefolgt von Nivolumab bei diesen Patientenpopulationen mit Vorsicht nach sorgfältiger Abwägung des potenziellen Nutzen/Risikos im individuellen Einzelfall angewendet werden.
Beim HCC wurde eine höhere Anzahl an Todesfällen innerhalb der ersten 6 Monate bei Patienten beobachtet, die mit Nivolumab in Kombination mit Ipilimumab behandelt wurden, verglichen mit den mit Lenvatinib oder Sorafenib behandelten Patienten. Mit prognostisch ungünstigen Faktoren kann ein höheres Sterberisiko assoziiert sein. Ärzte sollten dieses Risiko bei Patienten mit prognostisch ungünstigen Faktoren vor Beginn der Therapie mit Nivolumab in Kombination mit Ipilimumab berücksichtigen.
OPDIVO enthält Polysorbat 80 (E 433)
Dieses Arzneimittel enthält 1,25 mg Polysorbat 80 pro 2,5-ml-Durchstechflasche und 2,5 mg Polysorbat 80 pro 5‑ml‑Durchstechflasche. Dies entspricht 5 mg/10 ml. Polysorbate können allergische Reaktionen hervorrufen.
Patientenkarte
Jeder Arzt, der OPDIVO verschreibt, muss sich mit der Fachinformation für Ärzte und den Behandlungsrichtlinien vertraut machen und die Risiken der Behandlung mit OPDIVO mit dem Patienten besprechen. Dem Patienten wird mit jeder Verschreibung eine Patientenkarte ausgehändigt.
Nivolumab ist ein humaner monoklonaler Antikörper. Es wurden keine pharmakokinetischen Wechselwirkungsstudien als solches durchgeführt. Da monoklonale Antikörper nicht von Cytochrom‑P450‑Enzymen (CYPs) oder anderen Enzymen des Arzneimittelmetabolismus abgebaut werden, ist nicht zu erwarten, dass die Pharmakokinetik von Nivolumab durch die Hemmung oder Induktion dieser Enzyme durch gleichzeitig verabreichte Arzneimittel beeinflusst wird.
Andere Arten von Wechselwirkungen
Systemische Immunsuppression
Vor Beginn der Nivolumab‑Behandlung soll die Anwendung systemischer Corticosteroide und anderer Immunsuppressiva wegen der potenziellen Beeinflussung der pharmakodynamischen Aktivität vermieden werden. Nach Beginn der Nivolumab‑Behandlung jedoch können systemische Corticosteroide und andere Immunsuppressiva zur Behandlung immunvermittelter Nebenwirkungen angewendet werden. Vorläufige Ergebnisse zeigen, dass die Anwendung systemischer Immunsuppressiva nach Beginn der Nivolumab‑Behandlung ein Ansprechen auf Nivolumab anscheinend nicht ausschließt.
Schwangerschaft
Über die Anwendung von Nivolumab bei Schwangeren liegen keine Daten vor. Bei tierexperimentellen Studien wurde embryofötale Toxizität festgestellt (siehe Abschnitt 5.3). Humanes IgG4 passiert die Plazentaschranke und Nivolumab ist ein IgG4; daher kann Nivolumab potenziell von der Mutter auf den wachsenden Fötus übertragen werden. Die Anwendung von Nivolumab wird während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht zuverlässig verhüten, nicht empfohlen, es sei denn, der klinische Nutzen überwiegt das potenzielle Risiko. Wirksame Verhütungsmethoden sind für mindestens 5 Monate nach der letzten Gabe von Nivolumab anzuwenden.
Stillzeit
Es ist nicht bekannt, ob Nivolumab in die Muttermilch übergeht. Da viele Arzneimittel, einschließlich Antikörper, in die Muttermilch ausgeschieden werden, ist ein Risiko für Neugeborene/ Kleinkinder nicht auszuschließen. Daher muss unter Abwägung des Nutzens des Stillens für das Kind und des Nutzens der Behandlung für die Mutter eine Entscheidung darüber getroffen werden, ob das Stillen oder die Behandlung mit Nivolumab unterbrochen werden soll.
Fertilität
Es wurden keine Studien durchgeführt, um die Auswirkung von Nivolumab auf die Fertilität zu untersuchen. Daher ist die Auswirkung von Nivolumab auf die männliche oder weibliche Fertilität unbekannt.
Nivolumab oder Nivolumab in Kombination mit Ipilimumab hat möglicherweise einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Aufgrund potenzieller Nebenwirkungen wie Ermüdung/Fatigue (siehe Abschnitt 4.8) sollen Patienten angewiesen werden, beim Autofahren oder beim Bedienen von Maschinen vorsichtig zu sein, bis sie sicher sind, nicht durch Nivolumab beeinträchtigt zu werden.
Nivolumab als Monotherapie (siehe Abschnitt 4.2)
Zusammenfassung des Sicherheitsprofils
Im zusammengefassten Datensatz zu intravenös verabreichtem Nivolumab als Monotherapie über die Tumorarten (n = 4 646) mit einem minimalen Nachbeobachtungszeitraum von 2,3 bis 28 Monaten waren die häufigsten Nebenwirkungen (≥ 10 %) Ermüdung/Fatigue (44 %), Schmerzen des Muskel‑ und Skelettsystems (28 %), Diarrhö (26 %), Ausschlag (24 %), Husten (22 %), Übelkeit (22 %) Pruritus (19 %), verringerter Appetit (17 %), Arthralgie (17 %), Obstipation (16 %), Dyspnoe (16 %), Abdominalschmerz (15 %), Infektion der oberen Atemwege (15 %), Fieber (13 %), Kopfschmerzen (13 %), Anämie (13 %) und Erbrechen (12 %). Die Mehrheit der Nebenwirkungen war leicht bis mäßig (Grad 1 oder 2). Die Häufigkeit von Grad‑3‑5‑Nebenwirkungen war 44 %, mit 0,3 % tödlichen Nebenwirkungen, welche auf die Studienmedikation zurückzuführen sind. In einer Nachbeobachtung von mindestens 63 Monaten bei NSCLC wurden keine neuen Sicherheitssignale identifiziert.
Das Sicherheitsprofil von subkutan verabreichtem Nivolumab war dem bekannten Sicherheitsprofil der intravenösen Formulierung von Nivolumab ähnlich, mit einer zusätzlichen Nebenwirkung in Form einer Reaktion an der Injektionsstelle (7 % im Nivolumab‑Arm mit subkutaner Verabreichung [n = 247] vs. 0 % im Nivolumab‑Arm mit intravenöser Verabreichung [n = 245]).
Tabellarische Aufstellung der Nebenwirkungen
In Tabelle 12 sind die Nebenwirkungen aufgeführt, die aus dem zusammengefassten Datensatz für die mit Nivolumab‑Monotherapie behandelten Patienten (n = 4 646) stammen. Die Nebenwirkungen sind nach Organklassen und Häufigkeit geordnet. Häufigkeiten sind wie folgt definiert: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1 000, < 1/100); selten (≥ 1/10 000, < 1/1 000); sehr selten (< 1/10 000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 12: Nebenwirkungen unter Nivolumab‑Monotherapie
Nivolumab‑Monotherapie |
|
Infektionen und parasitäre Erkrankungen | |
Sehr häufig |
Infektion der oberen Atemwege |
Häufig |
Pneumoniea, Bronchitis |
Selten |
Aseptische Meningitis |
Gutartige, bösartige und nicht spezifizierte Neubildungen (einschl. Zysten und Polypen) | |
Selten |
Histiozytäre nekrotisierende Lymphadenitis (Kikuchi‑Lymphadenitis) |
Erkrankungen des Blutes und des Lymphsystems | |
Sehr häufig |
Lymphopenieb, Anämieb,i, Leukopenieb, Neutropeniea,b, Thrombozytopenieb |
Gelegentlich |
Eosinophilie |
Nicht bekannt |
Hämophagozytische Lymphohistiozytose |
Erkrankungen des Immunsystems | |
Häufig |
Reaktion im Zusammenhang mit einer Infusion (einschließlich Zytokin-Freisetzungssyndrom), Überempfindlichkeit (einschl. anaphylaktische Reaktion) |
Gelegentlich |
Sarkoidose |
Nicht bekannt |
Abstoßung eines soliden Organtransplantatsf |
Endokrine Erkrankungen | |
Häufig |
Hypothyreose, Hyperthyreose, Thyroiditis |
Gelegentlich |
Nebenniereninsuffizienzj, Hypophyseninsuffizienz, Hypophysitis, Diabetes mellitus |
Selten |
Diabetische Ketoazidose, Hypoparathyreoidismus |
Stoffwechsel‑ und Ernährungsstörungen | |
Sehr häufig |
Verminderter Appetit, Hyperglykämieb |
Häufig |
Dehydrierung, Gewichtsverlust, Hypoglykämieb |
Gelegentlich |
Metabolische Azidose |
Nicht bekannt |
Tumorlyse‑Syndromg |
Erkrankungen des Nervensystems | |
Sehr häufig |
Kopfschmerzen |
Häufig |
Periphere Neuropathie, Schwindelgefühl |
Gelegentlich |
Polyneuropathie, autoimmune Neuropathie (einschließlich Gesichtsnerv‑ und Abduzensparese), Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndromn |
Selten |
Guillain‑Barré‑Syndrom, Demyelinisierung, myasthenes Syndrom, Enzephalitisa,k, Optikusneuritis |
Nicht bekannt |
Myelitis (einschließlich transverse Myelitis) |
Augenerkrankungen | |
Häufig |
Verschwommenes Sehen, trockene Augen |
Gelegentlich |
Uveitis |
Nicht bekannt |
Vogt‑Koyanagi‑Harada‑Syndromf |
Herzerkrankungen | |
Häufig |
Tachykardie, Vorhofflimmern |
Gelegentlich |
Myokarditisa, Perikardiale Erkrankungenh, Arrhythmie (einschließlich ventrikulärer Arrhythmie) |
Gefäßerkrankungen | |
Häufig |
Hypertonie |
Selten |
Vaskulitis |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
Sehr häufig |
Dyspnoea, Husten |
Häufig |
Pneumonitisa, Pleuraerguss |
Gelegentlich |
Lungeninfiltration |
Erkrankungen des Gastrointestinaltrakts | |
Sehr häufig |
Diarrhö, Erbrechen, Übelkeit, Abdominalschmerz, Obstipation |
Häufig |
Kolitisa, Stomatitis, trockener Mund |
Gelegentlich |
Pankreatitis, Gastritis |
Selten |
Zwölffingerdarmgeschwür, exokrine Pankreasinsuffizienz, Zöliakie |
Leber‑ und Gallenerkrankungen | |
Gelegentlich |
Hepatitis, Cholestase |
Erkrankungen der Haut und des Unterhautgewebes | |
Sehr häufig |
Ausschlagc, Pruritus |
Häufig |
Vitiligo, trockene Haut, Erythem, Alopezie |
Gelegentlich |
Psoriasis, Rosazea, Erythema multiforme, Urtikaria |
Selten |
Toxische epidermale Nekrolysea,d, Stevens‑Johnson‑Syndroma |
Nicht bekannt |
Lichen sclerosusg, andere Lichenerkrankungen |
Skelettmuskulatur‑, Bindegewebs‑ und Knochenerkrankungen | |
Sehr häufig |
Schmerzen des Muskel‑ und Skelettsystemse, Arthralgie |
Häufig |
Arthritis |
Gelegentlich |
Rheumatische Polymyalgie |
Selten |
Sjögren‑Syndrom, Myopathie, Myositis (einschließlich Polymyositis)a, Rhabdomyolysea,d |
Erkrankungen der Nieren und Harnwege | |
Häufig |
Nierenversagen (einschließlich akuter Nierenschädigung)a |
Selten |
Tubulointerstitielle Nephritis, nicht‑infektiöse Zystitis |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Sehr häufig |
Ermüdung/Fatigue, Fieber |
Häufig |
Schmerzen, Schmerzen in der Brust, Ödemel, Reaktion an der Injektionsstellem |
Untersuchungenb | |
Sehr häufig |
AST‑Anstieg, Hyponatriämie, Hypoalbuminämie, Anstieg der alkalischen Phosphatase, Kreatinin‑Anstieg, ALT‑Anstieg, Lipase‑Anstieg, Hyperkaliämie, Amylase‑Anstieg, Hypokalziämie, Hypomagnesiämie, Hypokaliämie, Hyperkalziämie |
Häufig |
Anstieg des Gesamtbilirubins, Hypernatriämie, Hypermagnesiämie |
Die Häufigkeiten der Nebenwirkungen in Tabelle 12 sind möglicherweise nicht vollständig Nivolumab zuzuordnen, sondern können auch Einflüsse der zugrundeliegenden Erkrankung beinhalten. |
Nivolumab in Kombination mit anderen Arzneimitteln (siehe Abschnitt 4.2)
Zusammenfassung des Sicherheitsprofils
Wenn Nivolumab in Kombination angewendet wird, lesen Sie für weitere Informationen zum Sicherheitsprofil die Fachinformation der anderen Arzneimittel, bevor Sie mit der Behandlung beginnen.
Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie)
Im zusammengefassten Datensatz von Nivolumab, verabreicht in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) über die Tumorarten (n = 2 626) mit einer minimalen Nachbeobachtungszeit von 6 bis 47 Monaten, waren die häufigsten Nebenwirkungen (≥ 10 %) Ermüdung/Fatigue (47 %), Diarrhö (35 %), Ausschlag (37 %), Übelkeit (27 %), Pruritus (29 %), Schmerzen des Muskel‑ und Skelettsystems (26 %), Fieber (23 %), verminderter Appetit (22 %), Husten (21 %), Abdominalschmerz (18 %), Erbrechen (18 %), Obstipation (18 %), Arthralgie (18 %), Dyspnoe (17 %), Hypothyreose (16 %), Kopfschmerzen (15 %), Infektion der oberen Atemwege (13 %), Ödeme (13 %) und Schwindelgefühl (10 %). Die Häufigkeit von Grad‑3‑5‑Nebenwirkungen war 66 % für Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie), mit 1,0 % tödlichen Nebenwirkungen, welche auf die Studienmedikation zurückzuführen sind. Bei Patienten, die mit Nivolumab 1 mg/kg in Kombination mit Ipilimumab 3 mg/kg für Melanom behandelt wurden, wurden die folgenden Nebenwirkungen, im Vergleich zu der Häufigkeitsrate, die im zusammengefassten Datensatz von Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) berichtet wurde, mit einer ≥ 10 % höheren Häufigkeitsrate berichtet: Ermüdung/Fatigue (62 %), Ausschlag (57 %), Diarrhö (52 %), Übelkeit (42 %), Pruritus (40 %), Fieber (36 %) und Kopfschmerzen (26 %). Bei Patienten, die mit Nivolumab 360 mg in Kombination mit Ipilimumab 1 mg/kg und Chemotherapie für NSCLC behandelt wurden, wurden die folgenden Nebenwirkungen, im Vergleich zu der Häufigkeitsrate, die im zusammengefassten Datensatz von Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) berichtet wurde, mit einer ≥ 10 % höheren Häufigkeitsrate berichtet: Anämie (32 %) und Neutropenie (15 %).
Nivolumab in Kombination mit Chemotherapie
Im zusammengefassten Datensatz von Nivolumab 240 mg alle 2 Wochen oder 360 mg alle 3 Wochen in Kombination mit Chemotherapie über die Tumorarten (n = 1 800) mit einer minimalen Nachbeobachtungszeit von 7,4 bis 23,6 Monaten oder nach 3 Behandlungszyklen bei resezierbarem NSCLC waren die häufigsten Nebenwirkungen (≥ 10 %) Übelkeit (48 %), Ermüdung/Fatigue (40 %), periphere Neuropathie (33 %), verminderter Appetit (31 %), Obstipation (31 %), Diarrhö (28 %), Erbrechen (24 %), Ausschlag (19 %), Abdominalschmerz (18 %), Stomatitis (18 %), Schmerzen des Muskel‑ und Skelettsystems (18 %), Fieber (16 %), Husten (13 %), Ödeme (einschließlich periphere Ödeme) (12 %) und Pruritus (11 %). Die Häufigkeiten von Grad‑3‑5‑Nebenwirkungen waren 69 % für Nivolumab in Kombination mit Chemotherapie, mit 1,2 % tödlichen Nebenwirkungen, welche auf Nivolumab in Kombination mit Chemotherapie zurückzuführen sind. Die mediane Behandlungsdauer war 6,14 Monate (95 % CI: 5,78, 6,60) für Nivolumab in Kombination mit Chemotherapie. Bei resezierbarem NSCLC erhielten dreiundneunzig Prozent (93 %) der Patienten 3 Zyklen Nivolumab in Kombination mit Chemotherapie.
Nivolumab in Kombination mit Cabozantinib
Im Datensatz zur intravenösen Formulierung von Nivolumab 240 mg alle 2 Wochen in Kombination mit Cabozantinib 40 mg einmal täglich bei RCC (n = 320), bei einer minimalen Nachbeobachtung von 16,0 Monaten, waren die häufigsten Nebenwirkungen (≥ 10 %) Diarrhö (64,7 %), Ermüdung/Fatigue (51,3 %), palmar‑plantares Erythrodysästhesiesyndrom (40,0 %), Stomatitis (38,8 %), Schmerzen des Muskel‑ und Skelettsystems (37,5 %), Hypertonie (37,2 %), Ausschlag (36,3 %), Hypothyreose (35,6 %), verminderter Appetit (30,3 %), Übelkeit (28,8 %), Abdominalschmerz (25,0 %), Dysgeusie (23,8 %), Infektionen der oberen Atemwege (20,6 %), Husten (20,6 %), Pruritus (20,6 %), Arthralgie (19,4 %), Erbrechen (18,4 %), Dysphonie (17,8 %), Kopfschmerzen (16,3 %), Dyspepsie (15,9 %), Schwindelgefühl (14,1 %), Obstipation (14,1 %), Fieber (14,1 %), Ödeme (13,4 %), Muskelspasmen (12,2 %), Dyspnoe (11,6 %), Proteinurie (10,9 %) und Hyperthyreose (10,0 %). Die Häufigkeit von Grad‑3‑5‑Nebenwirkungen war 78 %, mit 0,3 % tödlichen Nebenwirkungen, welche auf die Studienmedikation zurückzuführen sind.
Tabellarische Aufstellung der Nebenwirkungen
In Tabelle 13 sind die Nebenwirkungen aufgeführt, die aus dem zusammengefassten Datensatz für die mit Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) (n = 2 626), Nivolumab in Kombination mit Chemotherapie (n = 1 800) und Nivolumab in Kombination mit Cabozantinib behandelten Patienten (n = 320) stammen. Die Nebenwirkungen sind nach Organklassen und Häufigkeit geordnet. Häufigkeiten sind wie folgt definiert: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1 000, < 1/100); selten (≥ 1/10 000, < 1/1 000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 13: Nebenwirkungen unter Nivolumab in Kombination mit anderen Arzneimitteln
Kombination mit Ipilimumab (mit oder ohne Chemotherapie) |
Kombination mit Chemotherapie |
Kombination mit Cabozantinib |
|
Infektionen und parasitäre Erkrankungen | |||
Sehr häufig |
Infektion der oberen Atemwege |
Infektion der oberen Atemwege |
|
Häufig |
Pneumonie, Bronchitis, Konjunktivitis |
Infektion der oberen Atemwege, Pneumoniea |
Pneumonie |
Selten |
Aseptische Meningitis |
||
Erkrankungen des Blutes und des Lymphsystems | |||
Sehr häufig |
Anämieb,j, Thrombozytopenieb, Leukopenieb, Lymphopenieb, Neutropenieb |
Neutropenieb, Anämieb,j, Leukopenieb, Lymphopenieb, Thrombozytopenieb |
Anämieb, Thrombozytopenieb, Leukopenieb, Lymphopenieb, Neutropenieb |
Häufig |
Eosinophilie |
Febrile Neutropeniea |
Eosinophilie |
Gelegentlich |
Febrile Neutropenie |
Eosinophilie |
|
Nicht bekannt |
Hämophagozytische Lymphohistiozytose |
||
Erkrankungen des Immunsystems | |||
Häufig |
Reaktion im Zusammenhang mit einer Infusion (einschließlich Zytokin-Freisetzungssyndrom), Überempfindlichkeit |
Überempfindlichkeit, Reaktion im Zusammenhang mit einer Infusion (einschließlich Zytokin-Freisetzungssyndrom) |
Überempfindlichkeit (einschließlich anaphylaktische Reaktion) |
Gelegentlich |
Infusionsbedingte Überempfindlichkeitsreaktion |
||
Selten |
Sarkoidose |
||
Nicht bekannt |
Abstoßung eines soliden Organtransplantatsg |
||
Endokrine Erkrankungen | |||
Sehr häufig |
Hypothyreose |
Hypothyreose, Hyperthyreose |
|
Häufig |
Hyperthyreose, Thyroiditis, Nebenniereninsuffizienz, Hypophysitis, Hypophyseninsuffizienz, Diabetes mellitus |
Hypothyreose, Hyperthyreose, Diabetes mellitus |
Nebenniereninsuffizienz |
Gelegentlich |
Diabetische Ketoazidose |
Nebenniereninsuffizienz, Thyroiditis, Hypophyseninsuffizienz, Hypophysitis |
Hypophysitis, Thyroiditis |
Selten |
Hypoparathyreoidismus |
||
Stoffwechsel‑ und Ernährungsstörungen | |||
Sehr häufig |
Verminderter Appetit, Hyperglykämieb, Hypoglykämieb |
Verminderter Appetit, Hyperglykämieb, Hypoglykämieb |
Verminderter Appetit, Hypoglykämieb, Hyperglykämieb, Gewichtsverlust |
Häufig |
Dehydrierung, Hypoalbuminämie, Hypophosphatämie, Gewichtsverlust |
Hypoalbuminämie, Hypophosphatämie, |
Dehydrierung |
Gelegentlich |
Metabolische Azidose |
||
Selten |
Tumorlyse‑Syndrom |
||
Nicht bekannt |
Tumorlyse‑Syndromh |
||
Erkrankungen des Nervensystems | |||
Sehr häufig |
Kopfschmerzen |
Periphere Neuropathie |
Dysgeusie, Schwindelgefühl, Kopfschmerzen |
Häufig |
Schwindelgefühl, periphere Neuropathie |
Parästhesie, Schwindelgefühl, Kopfschmerzen |
Periphere Neuropathie |
Gelegentlich |
Polyneuropathie, Peroneuslähmung, autoimmune Neuropathie (einschließlich Gesichtsnerv‑ und Abduzensparese), Enzephalitis, Myasthenia gravis, Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndroml |
Guillain‑Barré-Syndrom |
autoimmune Enzephalitis, Guillain‑Barré‑Syndrom, myasthenes Syndrom |
Selten |
Guillain‑Barré‑Syndrom, Neuritis, Myelitis (einschließlich transverse Myelitis), Optikusneuritis |
Enzephalitis, Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndroml |
|
Nicht bekannt |
Myelitis (einschließlich transverse Myelitis), Optikusneuritis |
Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndroml |
|
Erkrankungen des Ohrs und des Labyrinths | |||
Häufig |
Tinnitus |
||
Augenerkrankungen | |||
Häufig |
Verschwommenes Sehen, trockene Augen |
Trockene Augen, verschwommenes Sehen |
Trockene Augen, verschwommenes Sehen |
Gelegentlich |
Uveitis, Episkleritis |
Uveitis |
Uveitis |
Selten |
Vogt‑Koyanagi‑Harada‑Syndrom |
||
Herzerkrankungen | |||
Häufig |
Tachykardie, Vorhofflimmern |
Tachykardie, Vorhofflimmern |
Vorhofflimmern, Tachykardie |
Gelegentlich |
Myokarditisa, Arrhythmie (einschließlich ventrikulärer Arrhythmie)a, Bradykardie |
Myokarditis |
Myokarditis |
Nicht bekannt |
Perikardiale Erkrankungeni |
||
Gefäßerkrankungen | |||
Sehr häufig |
Hypertonie |
||
Häufig |
Hypertonie |
Thrombosea,k, Hypertonie, Vaskulitis |
Thrombosek |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |||
Sehr häufig |
Husten, Dyspnoe |
Husten |
Dysphonie, Dyspnoe, Husten |
Häufig |
Pneumonitisa, Lungenemboliea, Pleuraerguss |
Pneumonitisa, Dyspnoe |
Pneumonitis, Lungenembolie, Pleuraerguss, Epistaxis |
Erkrankungen des Gastrointestinaltrakts | |||
Sehr häufig |
Diarrhö, Erbrechen, Übelkeit, Abdominalschmerz, Obstipation |
Diarrhöa, Stomatitis, Erbrechen, Übelkeit, Abdominalschmerz, Obstipation |
Diarrhö, Erbrechen, Übelkeit, Obstipation, Stomatitis, Abdominalschmerz, Dyspepsie |
Häufig |
Kolitisa, Pankreatitis, Stomatitis, Gastritis, trockener Mund |
Kolitis, trockener Mund |
Kolitis, Gastritis, Mundschmerzen, trockener Mund, Hämorrhoiden |
Gelegentlich |
Duodenitis |
Pankreatitis |
Pankreatitis, Dünndarmperforationa, Glossodynie |
Selten |
Darmperforationa, exokrine Pankreasinsuffizienz, Zöliakie |
||
Nicht bekannt |
Exokrine Pankreasinsuffizienz, Zöliakie |
Exokrine Pankreasinsuffizienz, Zöliakie |
|
Leber‑ und Gallenerkrankungen | |||
Häufig |
Hepatitis |
Hepatitis |
|
Gelegentlich |
Hepatitis |
||
Erkrankungen der Haut und des Unterhautgewebes | |||
Sehr häufig |
Ausschlagc, Pruritus |
Ausschlagc, Pruritus |
Palmar‑plantares Erythrodysästhesiesyndrom, Ausschlagc, Pruritus |
Häufig |
Alopezie, Vitiligo, Urtikaria, trockene Haut, Erythem |
Palmar‑plantares Erythrodysästhesiesyndrom, Hauthyperpigmentierung, Alopezie, trockene Haut, Erythem |
Alopezie, trockene Haut, Erythem, Änderung der Haarfarbe |
Gelegentlich |
Stevens‑Johnson‑Syndrom, Erythema multiforme, Psoriasis, andere Lichenerkrankungend |
Psoriasis, Urtikaria |
|
Selten |
Toxische epidermale Nekrolysea,e, Lichen sclerosus |
||
Nicht bekannt |
Lichen sclerosus, andere Lichenerkrankungen |
||
Skelettmuskulatur‑, Bindegewebs‑ und Knochenerkrankungen | |||
Sehr häufig |
Schmerzen des Muskel‑ und Skelettsystemsf, Arthralgie |
Schmerzen des Muskel‑ und Skelettsystemsf |
Schmerzen des Muskel‑ und Skelettsystemsf, Arthralgie, Muskelspasmen |
Häufig |
Muskelspasmen, muskuläre Schwäche, Arthritis |
Arthralgie, muskuläre Schwäche |
Arthritis |
Gelegentlich |
Polymyalgia rheumatica, Myopathie, Myositis (einschließlich Polymyositis)a |
Myopathie, Osteonekrose des Kiefers, Fistel |
|
Selten |
Spondyloarthropathie, Sjögren‑Syndrom, Rhabdomyolysea |
||
Erkrankungen der Nieren und Harnwege | |||
Sehr häufig |
Proteinurie |
||
Häufig |
Nierenversagen (einschließlich akuter Nierenschädigung)a |
Nierenversagena |
Nierenversagen, akute Nierenschädigung |
Gelegentlich |
Tubulointerstitielle Nephritis, Nephritis |
Nicht‑infektiöse Zystitis, Nephritis |
Nephritis |
Selten |
Nicht‑infektiöse Zystitis |
Nicht‑infektiöse Zystitish |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |||
Sehr häufig |
Ermüdung/Fatigue, Fieber, Ödeme (einschließlich peripheres Ödem) |
Ermüdung/Fatigue, Fieber, Ödeme (einschließlich peripheres Ödem) |
Ermüdung/Fatigue, Fieber, Ödeme |
Häufig |
Schmerzen in der Brust, Schmerzen, Schüttelfrost |
Unwohlsein |
Schmerzen, Schmerzen in der Brust |
Untersuchungen | |||
Sehr häufig |
Anstieg der alkalischen Phosphataseb, AST‑Anstiegb, ALT‑Anstiegb, Anstieg des Gesamtbilirubinsb, Kreatinin-Anstiegb, Amylase‑Anstiegb, Lipase‑Anstiegb, Hyponatriämieb, Hyperkaliämieb, Hypokaliämieb, Hyperkalziämieb, Hypokalziämieb, Hypomagnesiämieb |
Hypokalziämieb, AST‑Anstiegb, ALT‑Anstiegb, Hyponatriämieb, Amylase‑Anstiegb, Hypomagnesiämieb, Anstieg der alkalischen Phosphataseb, Hypokaliämieb, Kreatinin-Anstiegb, Lipase‑Anstiegb, Hyperkaliämieb, Anstieg des Gesamtbilirubinsb |
Anstieg der alkalischen Phosphataseb, ALT‑Anstiegb, AST‑Anstiegb, Anstieg des Gesamtbilirubinsb, Kreatinin-Anstiegb, Amylase‑Anstiegb, Lipase‑Anstiegb, Hypokaliämieb, Hypomagnesiämieb, Hyponatriämieb, Hypokalziämieb, Hyperkalziämieb, Hypophosphatämieb, Hyperkaliämieb, Hypermagnesiämieb, Hypernatriämieb |
Häufig |
Hypernatriämieb, Hypermagnesiämieb, Anstieg des Schilddrüsen‑stimulierenden Hormons, Anstieg der Gamma‑Glutamyltransferase |
Hypernatriämieb, Hyperkalziämieb, Hypermagnesiämieb |
Cholesterin‑Anstieg im Blut, Hypertriglyzeridämie |
Die in Tabelle 13 angegebenen Häufigkeiten der Nebenwirkungen sind möglicherweise nicht vollständig auf Nivolumab allein oder in Kombination mit anderen Arzneimitteln zurückzuführen, da auch die Grunderkrankung oder die in Kombination verwendeten Arzneimittel dazu beitragen können. |
Beschreibung einzelner Nebenwirkungen
Nivolumab oder Nivolumab in Kombination mit anderen Arzneimitteln ist mit immunvermittelten Nebenwirkungen assoziiert. Diese immunvermittelten Nebenwirkungen sind mit einer adäquaten medizinischen Behandlung meist reversibel. Das dauerhafte Absetzen der Therapie war im Allgemeinen bei Patienten, die Nivolumab in Kombination mit anderen Arzneimitteln erhielten im Vergleich zu Patienten, die Nivolumab als Monotherapie erhielten, häufiger nötig. Die Tabelle 14 zeigt den Prozentsatz an Patienten mit immunvermittelten Nebenwirkungen, bei welchen die Therapie dauerhaft abgesetzt werden musste, abhängig vom Dosierungsregime. Außerdem zeigt die Tabelle 14 für die Patienten, bei welchen eine Nebenwirkung auftrat, den Prozentsatz an Patienten, der mit hochdosierten Corticosteroiden (mindestens 40 mg Prednison‑Äquivalent täglich) behandelt werden musste, abhängig vom Dosierungsregime. Die Behandlungsrichtlinien für diese Nebenwirkungen werden im Abschnitt 4.4 beschrieben.
Tabelle 14: Immunvermittelte Nebenwirkungen, welche zum dauerhaften Absetzen der Therapie führen oder welche eine Behandlung mit hochdosierten Corticosteroiden erfordern, abhängig vom Dosierungsregime (Nivolumab‑Monotherapie oder Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie), Nivolumab in Kombination mit Chemotherapie oder Nivolumab in Kombination mit Cabozantinib)
Nivolumab‑Mono‑ |
Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) |
Nivolumab in Kombination mit Chemotherapie |
Nivolumab in Kombination mit Cabozantinib |
||
Immunvermittelte Nebenwirkung, welche zum dauerhaften Absetzen der Therapie führt | |||||
Pneumonitis |
1,4 |
2,1 |
2,0 |
2,5 |
|
Kolitis |
1,2 |
6 |
1,8 |
2,5 |
|
Hepatitis |
1,1 |
5 |
0,7 |
4,1 |
|
Nephritis und Nierenfunktionsstörung |
0,3 |
1,1 |
3,1 |
0,6 |
|
Endokrinopathien |
0,5 |
2,2 |
0,6 |
1,3 |
|
Haut |
0,8 |
1,0 |
0,9 |
2,2 |
|
Überempfindlichkeit/Reaktion im Zusammenhang mit einer Infusion |
0,1 |
0,3 |
1,7 |
0 |
|
Immunvermittelte Nebenwirkung, welche eine Behandlung mit hochdosierten Corticosteroiden erforderta,b | |||||
Pneumonitis |
65 |
59 |
59 |
56 |
|
Kolitis |
14 |
32 |
9 |
8 |
|
Hepatitis |
21 |
39 |
7 |
23 |
|
Nephritis und Nierenfunktionsstörung |
22 |
27 |
9 |
9 |
|
Endokrinopathien |
5 |
18 |
4,3 |
4,2 |
|
Haut |
3,3 |
8 |
6 |
8 |
|
Überempfindlichkeit/Reaktion im Zusammenhang mit einer Infusion |
18 |
18 |
22 |
0 |
|
a mindestens 40 mg Prednison‑Äquivalent täglich | |||||
Immunvermittelte Pneumonitis
Bei Patienten, die mit Nivolumab‑Monotherapie behandelt wurden, war die Häufigkeit von Pneumonitis, einschließlich einer interstitiellen Lungenerkrankung und Lungeninfiltration, 3,3 % (155/4 646). Der mehrheitliche Schweregrad der Fälle wurde mit Grad 1 bei 0,9 % (42/4 646) oder Grad 2 bei 1,7 % (77/4 646) der Patienten angegeben. Fälle von Grad 3 bzw. 4 wurden bei 0,7 % (33/4 646) bzw. < 0,1 % (1/4 646) der Patienten berichtet. Bei sechs Patienten (0,1 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 15,1 Wochen (Spanne: 0,7 ‑ 85,1). Bei 107 Patienten (69,0 %) kam es zu einer Rückbildung nach einer medianen Zeit von 6,7 Wochen (Spanne: 0,1+ ‑ 109,1+); + kennzeichnet eine zensierte Beobachtung.
Bei Patienten, die mit Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Pneumonitis, einschließlich einer interstitiellen Lungenerkrankung 6,0 % (157/2 626). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 3,0 % (78/2 626), 1,0 % (27/2 626) bzw. 0,3 % (8/2 626) der Patienten berichtet. Bei vier Patienten (0,2 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 2,7 Monate (Spanne: 0,1 ‑ 56,8). Bei 129 Patienten (82,2 %) kam es zu einer Rückbildung nach einer medianen Zeit von 6,1 Wochen (Spanne: 0,1+‑ 149,3+).
Bei Patienten, die mit Nivolumab in Kombination mit Chemotherapie behandelt wurden, war die Häufigkeit von Pneumonitis, einschließlich einer interstitiellen Lungenerkrankung, 4,4 % (80/1 800). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 2,2 % (40/1 800), 0,9 % (17/1 800) bzw. 0,2 % (3/1 800) der Patienten berichtet. Bei drei Patienten (0,2 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 24,6 Wochen (Spanne: 0,6 ‑ 96,9). Bei 58 Patienten (72,5 %) kam es zu einer Rückbildung nach einer medianen Zeit von 10,4 Wochen (Spanne: 0,3+ ‑ 171,4+).
Bei Patienten, die mit Nivolumab in Kombination mit Cabozantinib behandelt wurden, war die Häufigkeit von Pneumonitis, einschließlich einer interstitiellen Lungenerkrankung, 5,6 % (18/320). Grad 2 bzw. Grad 3 wurden für 1,9 % (6/320) bzw. 1,6 % (5/320) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 26,9 Wochen (Spanne: 12,3 ‑ 74,3 Wochen). Bei 14 Patienten (77,8 %) kam es zu einer Rückbildung nach einer medianen Zeit von 7,5 Wochen (Spanne: 2,1 ‑ 60,7+ Wochen).
Immunvermittelte Kolitis
Bei Patienten, die mit Nivolumab‑Monotherapie behandelt wurden, war die Häufigkeit von Diarrhö, Kolitis oder vermehrtem Stuhlgang 15,4 % (716/4 646). Der mehrheitliche Schweregrad der Fälle wurde mit Grad 1 bei 9,9 % (462/4 646) oder Grad 2 bei 4,0 % (186/4 646) der Patienten angegeben. Fälle mit Grad 3 und 4 wurden bei 1,4 % (67/4 646) bzw. bei < 0,1 % (1/4 646) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 8,3 Wochen (Spanne: 0,1 ‑ 115,6). Bei 639 Patienten (90,3 %) kam es zu einer Rückbildung nach einer medianen Zeit von 2,9 Wochen (Spanne: 0,1 ‑ 124,4+).
Bei Patienten, die mit Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Diarrhö oder Kolitis 26,0 % (682/2 626). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 8,1 % (212/2 626), 6,4 % (167/2 626) bzw. 0,2 % (4/2 626) der Patienten berichtet. Bei zwei Patienten (< 0,1 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 1,4 Monate (Spanne: 0,0 ‑ 48,9). Bei 618 Patienten (91 %) kam es zu einer Rückbildung in einer medianen Zeit von 2,9 Wochen (Spanne: 0,1 ‑ 170,0+). Bei Patienten, die mit Nivolumab 1 mg/kg in Kombination mit Ipilimumab 3 mg/kg für Melanom behandelt wurden, war die Häufigkeit von Diarrhö oder Kolitis 46,7 %, einschließlich Grad 2 (13,6 %), Grad 3 (15,8 %) und Grad 4 (0,4 %).
Bei Patienten, die mit Nivolumab in Kombination mit Chemotherapie behandelt wurden, war die Häufigkeit von Diarrhö oder Kolitis 22,5 % (405/1 800). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 7,2 % (130/1 800), 3,1 % (56/1 800) bzw. 0,3 % (6/1 800) der Patienten berichtet. Bei einem Patienten (< 0,1 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 4,4 Wochen (Spanne: 0,1 ‑ 93,6). Bei 357 Patienten (88,6 %) kam es zu einer Rückbildung nach einer medianen Zeit von 1,6 Wochen (Spanne: 0,1 ‑ 212,3+).
Bei Patienten, die mit Nivolumab in Kombination mit Cabozantinib behandelt wurden, war die Häufigkeit von Diarrhö, Kolitis, häufigen Darmentleerungen oder Enteritis 59,1 % (189/320). Grad 2 bzw. Grad 3, wurden bei 25,6 % (82/320) bzw. 6,3 % (20/320) der Patienten berichtet. Grad 4 wurden bei 0,6 % (2/320) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 12,9 Wochen (Spanne: 0,3 ‑ 110,9 Wochen). Bei 143 Patienten (76,1 %) kam es zu einer Rückbildung nach einer medianen Zeit von 12,9 Wochen (Spanne: 0,1 ‑ 139,7+ Wochen).
Immunvermittelte Hepatitis
Bei Patienten, die mit Nivolumab‑Monotherapie behandelt wurden, war die Häufigkeit von Anomalien bei Leberfunktionstests 8,0 % (371/4 646). Der mehrheitliche Schweregrad der Fälle wurde mit Grad 1 bei 4,3 % (200/4 646) oder Grad 2 bei 1,8 % (82/4 646) angegeben. Fälle mit Grad 3 bzw. 4 wurden bei 1,6 % (74/4 646) bzw. 0,3 % (15/4 646) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 10,6 Wochen (Spanne: 0,1 ‑ 132,0). Bei 298 Patienten (81,4 %) kam es zu einer Rückbildung nach einer medianen Zeit von 6,1 Wochen (Spanne: 0,1 ‑ 126,4+).
Bei Patienten, die mit Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Anomalien bei Leberfunktionstests 21,2 % (556/2 626). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 5,0 % (132/2 626), 8,3 % (218/2 626) bzw. 1,3 % (34/2 626) der Patienten berichtet. Bei sieben Patienten (0,3 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 1,5 Monate (Spanne: 0,0 ‑ 36,6). Bei 482 Patienten (87,0 %) kam es zu einer Rückbildung in einer medianen Zeit von 5,9 Wochen (Spanne: 0,1 ‑ 175,9+). Bei Patienten, die mit Nivolumab 1 mg/kg in Kombination mit Ipilimumab 3 mg/kg für Melanom behandelt wurden, war die Häufigkeit von Anomalien bei Leberfunktionstests 30,1 %, einschließlich Grad 2 (6,9 %), Grad 3 (15,8 %) und Grad 4 (1,8 %). Bei Patienten, die mit Nivolumab 1 mg/kg in Kombination mit Ipilimumab 3 mg/kg für HCC behandelt wurden, war die Häufigkeit von Anomalien bei Leberfunktionstests 34,3 %, einschließlich Grad 2 (8,4 %), Grad 3 (14,2 %) und Grad 4 (2,7 %).
Bei Patienten, die mit Nivolumab in Kombination mit Chemotherapie behandelt wurden, war die Häufigkeit von Anomalien bei Leberfunktionstests 18 % (322/1 800). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 5,1 % (92/1 800), 2,6 % (47/1 800) bzw. < 0,1 % (1/1 800) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 7,0 Wochen (Spanne: 0,1 ‑ 99,0). Bei 258 Patienten (81,1 %) kam es zu einer Rückbildung nach einer medianen Zeit von 7,4 Wochen (Spanne: 0,4 ‑ 240,0+).
Bei Patienten, die mit Nivolumab in Kombination mit Cabozantinib behandelt wurden, war die Häufigkeit von Anomalien bei Leberfunktionstests 41,6 % (133/320). Grad 2, Grad 3 bzw. Grad 4 wurden bei 14,7 % (47/320), 10,3 % (33/320) bzw. 0,6 % (2/320) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 8,3 Wochen (Spanne: 0,1 ‑ 107,9 Wochen). Bei 101 Patienten (75,9 %) kam es zu einer Rückbildung nach einer medianen Zeit von 9,6 Wochen (Spanne: 0,1 ‑ 89,3+ Wochen).
Immunvermittelte Nephritis und Nierenfunktionsstörung
Bei Patienten, die mit Nivolumab‑Monotherapie behandelt wurden, war die Häufigkeit von Nephritis oder Nierenfunktionsstörung 2,6 % (121/4 646). Der mehrheitliche Schweregrad der Fälle wurde mit Grad 1 bei 1,5 % (69/4 646) oder Grad 2 bei 0,7 % (32/4 646) angegeben. Fälle mit Grad 3 wurden bei 0,4 % (18/4 646) der Patienten und Fälle mit Grad 4 bei < 0,1 % (2/4 646) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 12,1 Wochen (Spanne: 0,1 ‑ 79,1). Bei 80 Patienten (69,0 %) kam es zu einer Rückbildung nach einer medianen Zeit von 8,0 Wochen (Spanne: 0,3 ‑ 79,1+).
Bei Patienten, die mit Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Nephritis oder Nierenfunktionsstörungen 5,4 % (141/2 626). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 2,0 % (52/2 626), 0,8 % (21/2 626) bzw. 0,4 % (11/2 626) der Patienten berichtet. Bei zwei Patienten (< 0,1 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 2,6 Monate (Spanne: 0,0 ‑ 34,8). Bei 110 Patienten (78,0 %) kam es zu einer Rückbildung in einer medianen Zeit von 5,9 Wochen (Spanne: 0,1 ‑ 172,1+).
Bei Patienten, die mit Nivolumab in Kombination mit Chemotherapie behandelt wurden, war die Häufigkeit von Nephritis oder Nierenfunktionsstörungen 10,9 % (196/1 800). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 3,7 % (66/1 800), 1,4 % (25/1 800) bzw. 0,2 % (3/1 800) der Patienten berichtet. Bei zwei Patienten (0,1 %) führte dies zum Tod. Die mediane Zeit bis zum Auftreten betrug 6,7 Wochen (Spanne: 0,1 ‑ 60,7). Bei 133 Patienten (67,9 %) kam es zu einer Rückbildung nach einer medianen Zeit von 9,1 Wochen (Spanne: 0,1 ‑ 226,0+).
Bei Patienten, die mit Nivolumab in Kombination mit Cabozantinib behandelt wurden, war die Häufigkeit von Nephritis, immunvermittelter Nephritis, Nierenversagen, akuter Nierenschädigung, erhöhtem Kreatinin im Blut oder erhöhtem Blutharnstoff 10,0 % (32/320). Grad 2 bzw. Grad 3 wurden bei 3,4 % (11/320) bzw. 1,3 % (4/320) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 14,2 Wochen (Spanne: 2,1 ‑ 87,1 Wochen). Bei 18 Patienten (58,1 %) kam es zu einer Rückbildung nach einer medianen Zeit von 10,1 Wochen (Spanne: 0,6 ‑ 90,9+ Wochen).
Immunvermittelte Endokrinopathien
Bei Patienten, die mit Nivolumab‑Monotherapie behandelt wurden, war die Häufigkeit von Schilddrüsenerkrankungen, einschließlich Hypothyreose oder Hyperthyreose, 13,0 % (603/4 646). Der mehrheitliche Schweregrad der Fälle wurde mit Grad 1 bei 6,6 % (305/4 646) oder Grad 2 bei 6,2 % (290/4 646) angegeben. Eine Grad‑3‑Schilddrüsenerkrankung wurde bei 0,2 % (8/4 646) der Patienten berichtet. Hypophysitis (3 von Grad 1, 7 von Grad 2, 9 von Grad 3 und 1 von Grad 4), Hypophyseninsuffizienz (6 von Grad 2 und 1 von Grad 3), Nebenniereninsuffizienz (einschließlich sekundärer Nebennierenrindeninsuffizienz, akuter Nebennierenrindeninsuffizienz und erniedrigten Corticotropins im Blut) (2 von Grad 1, 23 von Grad 2 und 11 von Grad 3), Diabetes mellitus (einschließlich Diabetes mellitus Typ 1 und diabetischer Ketoazidose) (1 von Grad 1, 3 von Grad 2, 8 von Grad 3 und 2 von Grad 4) wurden berichtet. Die mediane Zeit bis zum Auftreten dieser Endokrinopathien betrug 11,1 Wochen (Spanne: 0,1 ‑ 126,7). Bei 323 Patienten (48,7 %) kam es zu einer Rückbildung. Die mediane Zeit bis zur Rückbildung betrug 48,6 Wochen (Spanne: 0,4 ‑ 204,4+ Wochen).
Bei Patienten, die mit Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Schilddrüsenerkrankungen 23,2 % (608/2 626). Schilddrüsenerkrankungen mit dem Schweregrad 2 bzw. 3 wurden bei 12,7 % (333/2 626) bzw. 1,0 % (27/2 626) der Patienten berichtet. Hypophysitis (einschließlich lymphozytische Hypophysitis) von Grad 2 bzw. Grad 3 wurde bei 1,9 % (49/2 626) bzw. 1,5 % (40/2 626) der Patienten berichtet. Hypophyseninsuffizienz von Grad 2 bzw. 3 trat bei 0,6 % (16/2 626) bzw. 0,5 % (13/2 626) der Patienten auf. Nebenniereninsuffizienz (einschließlich sekundärer Nebennierenrindeninsuffizienz, akuter Nebennierenrindeninsuffizienz, Corticotropin im Blut erniedrigt und immunvermittelter Nebenniereninsuffizienz) von Grad 2, Grad 3 bzw. Grad 4 wurde bei 2,7 % (72/2 626), 1,6 % (43/2 626) bzw. 0,2 % (4/2 626) der Patienten berichtet. Diabetes mellitus (einschließlich Diabetes mellitus Typ 1 und diabetischer Ketoazidose) vom Schweregrad 1, 2, 3 bzw. 4 wurde bei < 0,1 % (1/2 626), 0,3 % (8/2 626), 0,3 % (7/2 626) bzw. 0,2 % (6/2 626) der Patienten berichtet. Die mediane Zeit bis zum Auftreten dieser Endokrinopathien betrug 2,1 Monate (Spanne: 0,0 ‑ 28,1). Bei 297 Patienten (40,0 %) kam es zu einer Rückbildung. Die Zeit bis zum Auftreten der Rückbildung betrug 0,3 bis 257,1+ Wochen.
Bei Patienten, die mit Nivolumab in Kombination mit Chemotherapie behandelt wurden, war die Häufigkeit von Schilddrüsenerkrankungen 12,8 % (230/1 800). Schilddrüsenerkrankungen vom Grad 2 und Grad 3 wurden bei 6,3 % (114/1 800) bzw. 0,1 % (2/1 800) der Patienten berichtet. Hypophysitis vom Grad 3 trat bei 0,1 % (2/1 800) der Patienten auf. Hypophyseninsuffizienz vom Grad 2 bzw. Grad 3 trat bei jeweils 0,2 % (4/1 800) der Patienten auf. Nebenniereninsuffizienz vom Grad 2, Grad 3 bzw. Grad 4 wurde bei 0,6 % (11/1 800), 0,2 % (3/1 800) bzw. < 0,1 % (1/1 800) der Patienten berichtet. Bei einem Patienten (< 0,1 %) führte dies zum Tod aufgrund einer Nebenniereninsuffizienz. Diabetes mellitus, einschließlich Typ‑1‑Diabetes‑mellitus und fulminanter Typ‑1‑Diabetes‑mellitus (4 von Grad 2, 2 von Grad 3 und 1 von Grad 4) und diabetische Ketoazidose (1 von Grad 2 und 1 von Grad 4) wurden berichtet. Die mediane Zeit bis zum Auftreten dieser Endokrinopathien betrug 15,3 Wochen (Spanne: 1,1 ‑ 124,3). Bei 101 Patienten (40,1 %) kam es zu einer Rückbildung. Die Zeit bis zum Eintreten der Rückbildung betrug 0,3+ bis 233,6+ Wochen.
Bei Patienten, die mit Nivolumab in Kombination mit Cabozantinib behandelt wurden, war die Häufigkeit von Schilddrüsenerkrankungen 43,1 % (138/320). Schilddrüsenerkrankungen mit dem Schweregrad 2 bzw. 3 wurden bei 23,1 % (74/320) bzw. 0,9 % (3/320) der Patienten berichtet. Hypophysitis wurde bei 0,6 % (2/320) der Patienten berichtet, alle vom Schweregrad 2. Nebenniereninsuffizienz (einschließlich sekundärer Nebennierenrindeninsuffizienz) wurde bei 4,7 % (15/320) der Patienten berichtet. Nebenniereninsuffizienz mit dem Schweregrad 2 bzw. 3 wurden bei 2,2 % (7/320) bzw. 1,9 % (6/320) der Patienten berichtet. Die mediane Zeit bis zum Auftreten dieser Endokrinopathien betrug 12,3 Wochen (Spanne: 2,0 ‑ 89,7 Wochen). Bei 50 Patienten (35,2 %) kam es zu einer Rückbildung. Die Zeit bis zum Auftreten der Rückbildung betrug 0,9 bis 132,0+ Wochen.
Immunvermittelte Nebenwirkungen der Haut
Bei Patienten, die mit Nivolumab‑Monotherapie behandelt wurden, war die Häufigkeit von Ausschlag 30,0 % (1 396/4 646). Der mehrheitliche Schweregrad der Fälle wurde mit Grad 1 bei 22,8 % (1 060/4 646) angegeben. Fälle mit Grad 2 wurden bei 5,9 % (274/4 646) der Patienten und Fälle mit Grad 3 bei 1,3 % (62/4 646) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 6,7 Wochen (Spanne: 0,1 ‑ 121,1). Bei 896 Patienten (64,6 %) kam es zu einer Rückbildung nach einer medianen Zeit von 20,1 Wochen (0,1‑192,7+).
Bei Patienten, die mit Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Ausschlag 46,1 % (1 210/2 626). Fälle mit Grad 2, Grad 3 bzw. Grad 4 wurden bei 14,3 % (375/2 626), 4,6 % (120/2 626) bzw. 0,1 % (3/2 626) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 0,7 Monate (Spanne: 0,0 ‑ 33,8). Bei 843 Patienten (70 %) kam es zu einer Rückbildung in einer medianen Zeit von 12,1 Wochen (Spanne: 0,1 ‑ 268,7+). Bei Patienten, die mit Nivolumab 1 mg/kg in Kombination mit Ipilimumab 3 mg/kg für Melanom behandelt wurden, war die Häufigkeit von Ausschlag 65,2 %, einschließlich Grad 2 (20,3 %) und Grad 3 (7,8 %).
Bei Patienten, die mit Nivolumab in Kombination mit Chemotherapie behandelt wurden, war die Häufigkeit von Ausschlag 25,4 % (457/1 800). Fälle von Grad 2 bzw. Grad 3 wurden bei 6,2 % (111/1 800) bzw. 2,3 % (42/1 800) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 6,4 Wochen (Spanne: 0,1 ‑ 97,4). Bei 320 Patienten (70,2 %) kam es zu einer Rückbildung nach einer medianen Zeit von 12,1 Wochen (Spanne: 0,1 ‑ 258,7+).
Bei Patienten, die mit Nivolumab in Kombination mit Cabozantinib behandelt wurden, war die Häufigkeit von Ausschlag 62,8 % (201/320). Grad 2 bzw. Grad 3 wurde bei 23,1 % (74/320) bzw. 10,6 % (34/320) der Patienten berichtet. Die mediane Zeit bis zum Auftreten betrug 6,14 Wochen (Spanne: 0,1 ‑ 104,4 Wochen). Bei 137 Patienten (68,2 %) kam es zu einer Rückbildung in einer medianen Zeit von 18,1 Wochen (Spanne: 0,1 ‑ 130,6+ Wochen).
Seltene Fälle von SJS und TEN wurden beobachtet, manche davon mit tödlichem Ausgang (siehe Abschnitte 4.2 und 4.4).
Infusionsreaktionen (intravenöse Formulierung)
Bei Patienten, die mit Nivolumab‑Monotherapie behandelt wurden, war die Häufigkeit von Überempfindlichkeit/Reaktion im Zusammenhang mit einer Infusion 4,0 % (188/4 646), einschließlich 9 Fälle mit Grad 3 und 3 Fälle mit Grad 4.
Bei Patienten, die mit Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) behandelt wurden, war die Häufigkeit von Überempfindlichkeit/Reaktion im Zusammenhang mit einer Infusion 4,5 % (118/2 626). Fälle mit Grad 1, Grad 2, Grad 3 bzw. Grad 4 wurden bei 1,9 % (49/2 626), 2,4 % (62/2 626), 0,2 % (6/2 626) bzw. < 0,1 % (1/2 626) der Patienten berichtet. Bei Patienten mit MPM, die mit Nivolumab 3 mg/kg in Kombination mit Ipilimumab 1 mg/kg behandelt wurden, war die Häufigkeit von Überempfindlichkeit/Reaktion im Zusammenhang mit einer Infusion 12 %.
Bei Patienten, die mit Nivolumab in Kombination mit Chemotherapie behandelt wurden, war die Häufigkeit von Überempfindlichkeit/Reaktion im Zusammenhang mit einer Infusion 8,2 % (148/1 800). Fälle von Grad 2, Grad 3 bzw. Grad 4 wurden bei 4,6 % (83/1 800), 1,1 % (20/1 800) bzw. 0,2 % (3/1 800) der Patienten berichtet.
Bei Patienten, die mit Nivolumab in Kombination mit Cabozantinib behandelt wurden, war die Häufigkeit von Überempfindlichkeit/Reaktion im Zusammenhang mit einer Infusion 2,5 % (8/320), hiervon waren alle Fälle vom Schweregrad 1 oder 2. Fälle mit Grad 2 wurden bei 0,3 % (1/320) der Patienten berichtet.
Erhöhte Leberenzyme bei Nivolumab in Kombination mit Cabozantinib bei RCC
In einer klinischen Studie bei zuvor unbehandelten RCC‑Patienten wurde bei der Kombination Nivolumab mit Cabozantinib eine höhere Häufigkeit von Grad‑3‑ und ‑4‑ALT‑Anstieg (10,1 %) und ‑AST‑Anstieg (8,2 %) gegenüber der Nivolumab‑Monotherapie bei Patienten mit fortgeschrittenem RCC beobachtet. Bei Patienten mit Grad ≥ 2 erhöhten ALT oder AST (n = 85): Die mediane Zeit bis zum Auftreten betrug 10,1 Wochen (Spanne: 2,0 ‑ 106,6 Wochen), 26 % erhielten Corticosteroide für eine mediane Dauer von 1,4 Wochen (Spanne: 0,9 ‑ 75,3). Bei 91 % der Patienten kam es zu einer Rückbildung zu Grad 0 ‑ 1 in einer medianen Zeit von 2,3 Wochen (Spanne: 0,4 ‑ 108,1+ Wochen). Bei 45 Patienten mit einem ALT‑ oder AST‑Anstieg von Grad ≥ 2, bei welchen die Behandlung mit Nivolumab (n = 10) oder Cabozantinib (n = 10) als Monotherapie oder Nivolumab plus Cabozantinib als Kombinationstherapie (n = 25) wieder aufgenommen wurde, traten bei 3 Patienten, die OPDIVO erhielten, bei 4 Patienten, die Cabozantinib erhielten, bzw. bei 8 Patienten, die sowohl OPDIVO als auch Cabozantinib erhielten, erneut Grad ≥ 2 ALT‑ oder AST‑Anstiege auf.
Laborwertanomalien
Der Anteil der Patienten, bei denen es unter der Nivolumab‑Monotherapie zu einer Laborwertanomalie Grad 3 oder 4 gegenüber dem Ausgangswert zu Studienbeginn kam, war 3,4 % für Anämie (alle Grad 3), 0,7 % für Thrombozytopenie, 0,7 % für Leukopenie, 8,7 % für Lymphozytopenie, 0,9 % für Neutropenie, 1,7 % für Anstieg der alkalischen Phosphatase, 2,6 % für AST‑Anstieg, 2,3 % für ALT‑Anstieg, 0,8 % für Anstieg des Gesamtbilirubins, 0,7 % für Kreatinin-Anstieg, 2,0 % für Hyperglykämie, 0,7 % für Hypoglykämie, 3,8 % für Amylase‑Anstieg, 6,9 % für Lipase‑Anstieg, 4,7 % für Hyponatriämie, 1,6 % für Hyperkaliämie, 1,3 % für Hypokaliämie, 1,1 % für Hyperkalziämie, 0,6 % für Hypermagnesiämie, 0,4 % für Hypomagnesiämie, 0,6 % für Hypokalziämie, 0,6 % für Hypoalbuminämie und < 0,1 % für Hypernatriämie.
Der Anteil der Patienten, bei denen es unter der Therapie mit Nivolumab in Kombination mit Ipilimumab (mit oder ohne Chemotherapie) zu einer Verschlechterung der Laborwerte gegenüber dem Ausgangswert auf Grad 3 oder 4 kam, war 4,8 % für Anämie, 1,8 % für Thrombozytopenie, 2,2 % für Leukopenie, 6,9 % für Lymphozytopenie, 3,3 % für Neutropenie, 2,7 % für Anstieg der alkalischen Phosphatase, 9,8 % für AST‑Anstieg, 9,3 % für ALT‑Anstieg, 2,3 % für Anstieg des Gesamtbilirubins, 1,8 % für Kreatinin-Anstieg, 1,4 % für Hypoalbuminämie, 7,1 % für Hyperglykämie, 0,7 % für Hypoglykämie, 7,8 % für Amylase‑Anstieg, 16,3 % für Lipase‑Anstieg, 0,8 % für Hypokalziämie, 0,2 % für Hypernatriämie, 0,8 % für Hyperkalziämie, 2,0 % für Hyperkaliämie, 0,8 % für Hypermagnesiämie, 0,4 % für Hypomagnesiämie, 3,0 % für Hypokaliämie und 8,7 % für Hyponatriämie.
Bei Patienten, die mit Nivolumab 1 mg/kg in Kombination mit Ipilimumab 3 mg/kg für Melanom behandelt wurden, kam es bei einem höheren Anteil von Patienten zu einer Verschlechterung der ALT-Anstiege auf Grad 3 oder 4 (15,3 %) gegenüber dem Ausgangswert.
Der Anteil an Patienten, bei denen es unter der Therapie mit Nivolumab in Kombination mit Chemotherapie zu einer Verschlechterung der Laborwerte auf Grad 3 oder 4 gegenüber dem Ausgangswert kam, war 14,7 % für Anämie, 6,2 % für Thrombozytopenie, 11,7 % für Leukopenie, 13,6 % für Lymphozytopenie, 26,3 % für Neutropenie, 2,0 % für Anstieg der alkalischen Phosphatase, 3,3 % für AST‑Anstieg, 2,6 % für ALT Anstieg, 1,9 % für Anstieg des Gesamtbilirubins, 1,3 % für Kreatinin-Anstieg, 4,5 % für Amylase‑Anstieg, 5,2 % für Lipase‑Anstieg, 0,4 % für Hypernatriämie, 8,1 % für Hyponatriämie, 1,8 % für Hyperkaliämie, 5,1 % für Hypokaliämie, 0,7 % für Hyperkalziämie, 1,8 % für Hypokalziämie, 1,5 % für Hypermagnesiämie, 2,9 % für Hypomagnesiämie, 3,7 % für Hyperglykämie und 0,6 % für Hypoglykämie.
Der Anteil an Patienten, bei denen es unter der Therapie mit Nivolumab in Kombination mit Cabozantinib zu einer Verschlechterung der Laborwerte auf Grad 3 oder 4 gegenüber dem Ausgangswert kam, war 3,5 % für Anämie (alle Grad 3), 0,3 % für Thrombozytopenie, 0,3 % für Leukopenie, 7,5 % für Lymphozytopenie, 3,5 % für Neutropenie, 3,2 % für Anstieg der alkalischen Phosphatase, 8,2 % für AST‑Anstieg, 10,1 % für ALT‑Anstieg, 1,3 % für Anstieg des Gesamtbilirubins, 1,3 % für Kreatinin-Anstieg, 11,9 % für Amylase‑Anstieg, 15,6 % für Lipase‑Anstieg, 3,5 % für Hyperglykämie, 0,8 % für Hypoglykämie, 2,2 % für Hypokalziämie, 0,3 % für Hyperkalziämie, 5,4 % für Hyperkaliämie, 4,2 % für Hypermagnesiämie, 1,9 % für Hypomagnesiämie, 3,2 % für Hypokaliämie, 12,3 % für Hyponatriämie und 21,2 % für Hypophosphatämie.
Immunogenität
Subkutane Formulierung
Von den 202 Patienten, die mit Nivolumab‑Injektionslösung behandelt wurden und deren Daten hinsichtlich des Auftretens von gegen Nivolumab gerichteten Antikörpern auswertbar waren, wurden circa 23 % (46/202) mittels eines Elektrochemilumineszenz(ECL)-Immunoassays positiv auf das Vorliegen von während der Behandlung aufgetretenen Antikörpern gegen Nivolumab getestet. Bei 1 % (2/202) der Patienten zeigten sich neutralisierende Antikörper gegen Nivolumab. Die Häufigkeit von während der Behandlung aufgetretenen Antikörpern gegen rekombinante humane Hyaluronidase PH20 (Anti-rHuPH20-Antikörper) bei Patienten, die mit Nivolumab‑Injektionslösung behandelt wurden, lag bei 8,8 % (19/215).
Intravenöse Formulierung
Von den 3 529 Patienten, die mit Nivolumab‑Monotherapie in einer Dosis von 3 mg/kg oder 240 mg alle 2 Wochen behandelt wurden und deren Daten hinsichtlich des Auftretens von gegen den Wirkstoff gerichteten Antikörpern auswertbar waren, wurden 328 Patienten (9,3 %) positiv auf das Vorliegen von während der Behandlung aufgetretenen Antikörpern gegen den Wirkstoff getestet. Bei 21 Patienten (0,6 %) zeigten sich neutralisierende Antikörper.
Die kombinierte Gabe mit Chemotherapie hatte keinen Einfluss auf die Immunogenität von Nivolumab. Von den 1 407 Patienten, die mit Nivolumab 240 mg alle 2 Wochen oder 360 mg alle 3 Wochen in Kombination mit Chemotherapie behandelt wurden und deren Daten hinsichtlich des Auftretens von gegen den Wirkstoff gerichteten Antikörpern auswertbar waren, wurden 7,2 % positiv auf das Vorliegen von während der Behandlung aufgetretenen Antikörpern gegen den Wirkstoff getestet. Bei 0,5 % der Patienten zeigten sich neutralisierende Antikörper.
Von den Patienten, die mit Nivolumab in Kombination mit Ipilimumab behandelt wurden und deren Daten hinsichtlich des Auftretens von gegen Nivolumab gerichteten Antikörpern auswertbar waren, betrug die Inzidenz der gegen Nivolumab gerichteten Antikörper 26,0 % bei Nivolumab 3 mg/kg und Ipilimumab 1 mg/kg alle 3 Wochen, 24,9 % bei Nivolumab 3 mg/kg alle 2 Wochen und Ipilimumab 1 mg/kg alle 6 Wochen, bzw. 37,8 % bei Nivolumab 1 mg/kg und Ipilimumab 3 mg/kg alle 3 Wochen. Die Inzidenz von neutralisierenden Antikörpern gegen Nivolumab betrug 0,8 % bei Nivolumab 3 mg/kg und Ipilimumab 1 mg/kg alle 3 Wochen, 1,5 % bei Nivolumab 3 mg/kg alle 2 Wochen und Ipilimumab 1 mg/kg alle 6 Wochen, bzw. 4,6 % bei Nivolumab 1 mg/kg und Ipilimumab 3 mg/kg alle 3 Wochen. Bei Patienten, die hinsichtlich des Vorhandenseins von gegen Ipilimumab gerichteten Antikörpern auswertbar waren, lag die Inzidenz von Anti‑Ipilimumab Antikörpern zwischen 6,3 und 13,7 % und die der neutralisierenden Antikörper gegen Ipilimumab zwischen 0 und 0,4 %.
Von den Patienten, die mit Nivolumab in Kombination mit Ipilimumab und Chemotherapie behandelt wurden und deren Daten hinsichtlich des Auftretens von gegen Nivolumab gerichteten Antikörpern oder neutralisierenden Antikörpern gegen Nivolumab auswertbar waren, betrug die Inzidenz von gegen Nivolumab gerichteten Antikörpern 33,8 % und die Inzidenz von neutralisierenden Antikörpern 2,6 %. Von den Patienten, die mit Nivolumab in Kombination mit Ipilimumab und Chemotherapie behandelt wurden und deren Daten hinsichtlich des Auftretens von gegen Ipilimumab gerichteten Antikörpern oder neutralisierenden Antikörpern gegen Ipilimumab auswertbar waren, betrug die Inzidenz von gegen Ipilimumab gerichteten Antikörpern 7,5 % und die Inzidenz von neutralisierenden Antikörpern1,6 %.
Obwohl die Clearance von Nivolumab um 20 % erhöht war, wenn Anti‑Nivolumab‑Antikörper detektiert wurden, ergaben pharmakokinetische sowie Exposure‑Response‑Analysen, dass dies sowohl bei Mono- wie auch bei Kombinationstherapie nicht mit einem Wirksamkeitsverlust oder einem veränderten Toxizitätsprofil verbunden war.
Ältere Menschen
Insgesamt wurden in Bezug auf die Sicherheit keine Unterschiede zwischen älteren (≥ 65 Jahre) und jüngeren Patienten (< 65 Jahre) berichtet. Daten von Patienten ab 75 Jahren mit SCCHN, adjuvanter Behandlung des Melanoms und adjuvanter Behandlung der Karzinome des Ösophagus oder des gastroösophagealen Übergangs sind zu limitiert, um Schlussfolgerungen für diese Patientengruppe ziehen zu können (siehe Abschnitt 5.1). Daten von dMMR‑ oder MSI‑H‑CRC‑Patienten ab 75 Jahren sind limitiert (siehe Abschnitt 5.1). Bei Patienten ab 75 Jahren mit HCC traten höhere Raten an schwerwiegenden Nebenwirkungen und an Therapieabbrüchen aufgrund von Nebenwirkungen auf (67 % bzw. 35 %) im Vergleich zu allen Patienten, die Nivolumab mit Ipilimumab erhalten hatten (53 % bzw. 27 %).
Für Patienten, die mit Nivolumab in Kombination mit Cabozantinib behandelt wurden, sind Daten von RCC‑Patienten ab 75 Jahren zu limitiert, um Schlussfolgerungen für diese Patientengruppe ziehen zu können (siehe Abschnitt 5.1).
Leber‑ oder Nierenfunktionsstörung
In der nicht‑plattenepithelialen NSCLC‑Studie (CA209057) war das Sicherheitsprofil der Patienten mit vorbestehender Nieren‑ oder Leberfunktionsstörung zu Studienbeginn mit der Gesamtpopulation vergleichbar. Aufgrund des geringen Stichprobenumfangs innerhalb der Subgruppen sollten diese Ergebnisse mit Vorsicht interpretiert werden.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen‑Risiko‑Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung anzuzeigen am:
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51-59
63225 Langen
Tel: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
In klinischen Studien wurden keine Fälle von Überdosierung berichtet. Bei Überdosierung müssen die Patienten sorgfältig auf Anzeichen oder Symptome von Nebenwirkungen beobachtet und es muss unverzüglich eine adäquate symptomatische Behandlung eingeleitet werden.
Pharmakotherapeutische Gruppe: Antineoplastische Mittel, monoklonale Antikörper und Antikörper‑Wirkstoff‑Konjugate, PD‑1/PDL‑1 (Programmed Cell Death Protein 1‑Rezeptor/Death‑Ligand 1)‑Inhibitoren, ATC‑Code: L01FF01.
OPDIVO‑Injektionslösung enthält den Wirkstoff Nivolumab, der für die therapeutische Wirkung dieses Arzneimittels verantwortlich ist, sowie rekombinante humane Hyaluronidase (rHuPH20), ein Enzym, das bei subkutaner Verabreichung die Verteilung und Resorption der Stoffe in der Co-Formulierung verbessert.
Wirkmechanismus
Nivolumab ist ein humaner Immunoglobulin‑G4‑(IgG4) monoklonaler Antikörper (HuMAb), der an den "Programmed Death"‑1‑(PD‑1)‑Rezeptor bindet und die Interaktion des Rezeptors mit den Liganden PD‑L1 und PD‑L2 blockiert. Der PD‑1‑Rezeptor ist ein negativer Regulator der T‑Zellaktivität, der erwiesenermaßen an der Kontrolle der T‑Zell-Immunantwortreaktionen beteiligt ist. Die Bindung von PD‑1 an die Liganden PD‑L1 und PD‑L2, die von Antigen‑präsentierenden Zellen exprimiert werden und von Tumoren oder anderen Zellen aus dem Mikromilieu des Tumors exprimiert werden können, führt zur Hemmung der T‑Zellproliferation und Zytokinausschüttung. Nivolumab potenziert die T‑Zellreaktionen, einschließlich der Tumorabwehrreaktion, durch Blockade der Bindung von PD‑1 an die PD‑L1‑ und PD‑L2‑Liganden. In genidentischen Mausmodellen führte eine Blockade der PD‑1‑Aktivität zu einer Verringerung des Tumorwachstums.
Die Kombination einer Nivolumab (Anti‑PD‑1) und Ipilimumab (Anti‑CTLA‑4) ‑ vermittelten Hemmung resultiert in einer verbesserten Anti‑Tumor‑Aktivität beim metastasierten Melanom. In genidentischen Mausmodellen führte die duale Blockade von PD‑1 und CTLA‑4 zu synergistischer Tumoraktivität.
Klinische Wirksamkeit und Sicherheit
Subkutane Formulierung
Die Ergebnisse einer simulationsbasierten pharmakokinetischen Bridging-Analyse zeigten, dass über alle untersuchten soliden Tumorarten subkutane Nivolumab-Dosierungsregimes (600 mg alle 2 Wochen und 1 200 mg alle 4 Wochen) zu Expositionen führten, die denen der zugelassenen intravenösen Nivolumab-Dosierungsregimes (240 mg alle 2 Wochen und 480 mg alle 4 Wochen) nichtunterlegen waren (Quotient des geometrischen Mittels: > 1). Die geometrischen mittleren Expositionen lagen auch unter denen von intravenös verabreichtem Nivolumab 10 mg/kg alle 2 Wochen, einem Regime, das sich in klinischen Studien als sicher erwiesen hat.
Das klinische Sicherheitsprofil von subkutan verabreichtem Nivolumab war mit dem von intravenös verabreichtem Nivolumab vergleichbar.
Melanom
Adjuvante Behandlung des Melanoms
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie von Nivolumab vs. Placebo (CA20976K)
Die Sicherheit und Wirksamkeit von Nivolumab 480 mg als Monotherapie zur Behandlung von Patienten mit vollständig reseziertem Melanom wurden in einer randomisierten, doppelblinden Phase‑III‑Studie (CA20976K) untersucht. In die Studie wurden Patienten mit einem ECOG‑Performance‑Status von 0 oder 1 eingeschlossen, die gemäß der Einstufung des American Joint Committee on Cancer (AJCC), 8. Ausgabe, ein histologisch bestätigtes Melanom im Stadium IIB oder IIC hatten, das vollständig chirurgisch reseziert wurde. Die Aufnahme erforderte eine vollständige Resektion des primären Melanoms mit negativen Rändern und eine negative Wächterlymphknotenbiopsie innerhalb von 12 Wochen vor Randomisierung. Der Einschluss von Patienten erfolgte unabhängig von ihrem Tumor‑PD‑L1‑Status. Von der Studie ausgeschlossen waren Patienten mit okulärem/uvealem Melanom oder Schleimhautmelanom, einer aktiven Autoimmunerkrankung, jeder Erkrankung, die eine systemische Behandlung mit Corticosteroiden (≥ 10 mg Prednison oder ‑Äquivalent täglich) oder anderen immunsuppressiven Arzneimitteln erforderte, sowie Patienten mit vorheriger Melanomtherapie (ausgenommen chirurgische Eingriffe).
Insgesamt wurden 790 Patienten entweder für Nivolumab (n = 526), das in einer Dosierung von 480 mg alle 4 Wochen über 30 Minuten intravenös verabreicht wurde, oder Placebo (n = 264) für bis zu 1 Jahr oder bis zum Wiederauftreten der Erkrankung oder bis zu nicht akzeptabler Toxizität (2:1) randomisiert. Die Randomisierung wurde gemäß der T‑Kategorie (T3b vs. T4a vs. T4b) des AJCC, 8. Ausgabe, stratifiziert. Tumorbeurteilungen wurden in den Jahren 1–3 alle 26 Wochen und nach 3 Jahren bis 5 Jahren alle 52 Wochen durchgeführt. Der primäre Endpunkt für die Wirksamkeit war das rezidivfreie Überleben (recurrence‑free survival, RFS). Das vom Prüfarzt beurteilte RFS wurde definiert als die Zeit zwischen dem Datum der Randomisierung und dem Datum des ersten Rezidivs (lokale, regionale oder Fernmetastasen), neuer primärer Melanome oder Tod jeglicher Ursache, je nachdem, was zuerst auftrat. Die sekundären Endpunkte umfassten das OS und das fernmetastasenfreie Überleben (distant metastatis‑free survival, DMFS).
Die Ausgangsmerkmale der beiden Gruppen waren etwa gleich. Das mediane Alter war 62 Jahre (Spanne: 19 – 92), 61 % der Patienten waren männlich und 98 % waren weiß. Der ECOG‑Performance‑Status zu Studienbeginn war 0 (94 %) oder 1 (6 %). 60 % hatten eine Erkrankung im Stadium IIB und 40 % im Stadium IIC.
Bei einer präspezifizierten primären Zwischenanalyse (minimale Nachbeobachtungszeit 7,8 Monate) wurde eine statistisch signifikante Verbesserung des RFS mit Nivolumab im Vergleich zu Placebo mit einer HR von 0,42 (95 % CI: 0,30; 0,59; p < 0,0001) gezeigt. Bei einer aktualisierten deskriptiven RFS‑Analyse (minimale Nachbeobachtungszeit von 15,6 Monaten) zeigte Nivolumab weiterhin eine RFS‑Verbesserung mit einer HR von 0,53 (95 % CI: 0,40; 0,71). Das OS war nicht ausgereift. Bei einer aktualisierten deskriptiven RFS‑Analyse (minimale Nachbeobachtungszeit von 26,9 Monaten) zeigte Nivolumab weiterhin eine RFS‑Verbesserung mit einer HR von 0,62 (95 % CI: 0,47; 0,80). Die mediane Nachbeobachtungszeit betrug im Nivolumab-Arm 34,25 Monate und im Placebo-Arm 33,92 Monate. Die Ergebnisse stimmten mit der formalen Zwischenanalyse überein. Die Ergebnisse der Analysen mit einer minimalen Nachbeobachtungszeit von 15,6 Monaten sind in Tabelle 15 und Abbildung 1 zusammengefasst.
Tabelle 15: Wirksamkeitsergebnisse (CA20976K)
Nivolumab |
Placebo |
|
Rezidivfreies Überleben mit einer minimalen Nachbeobachtungszeit von 15,6 Monaten | ||
Rezidivfreies Überleben | ||
Ereignisse |
102 (19,4 %) |
84 (31,8 %) |
Hazard‑Ratioa |
0,53 |
|
95 % CI |
(0,40; 0,71) |
|
Median (95 % CI) Monate |
NR |
36,14 (24,77; NR) |
Rate (95 % CI) nach 12 Monatenb |
88,8 (85,6; 91,2) |
81,1 (75,7; 85,4) |
Rate (95 % CI) nach 18 Monatenb |
83,9 (80,3; 86,9) |
70,7 (64,5; 76,1) |
a Mit einem stratifizierten Cox‑Modell für proportionale Hazards berechnet. | ||
Der RFS-Vorteil war in allen wichtigen Untergruppen konsistent, einschließlich Erkrankungsstadium, T‑Kategorie und Alter.
Abbildung 1: Rezidivfreies Überleben (CA20976K)
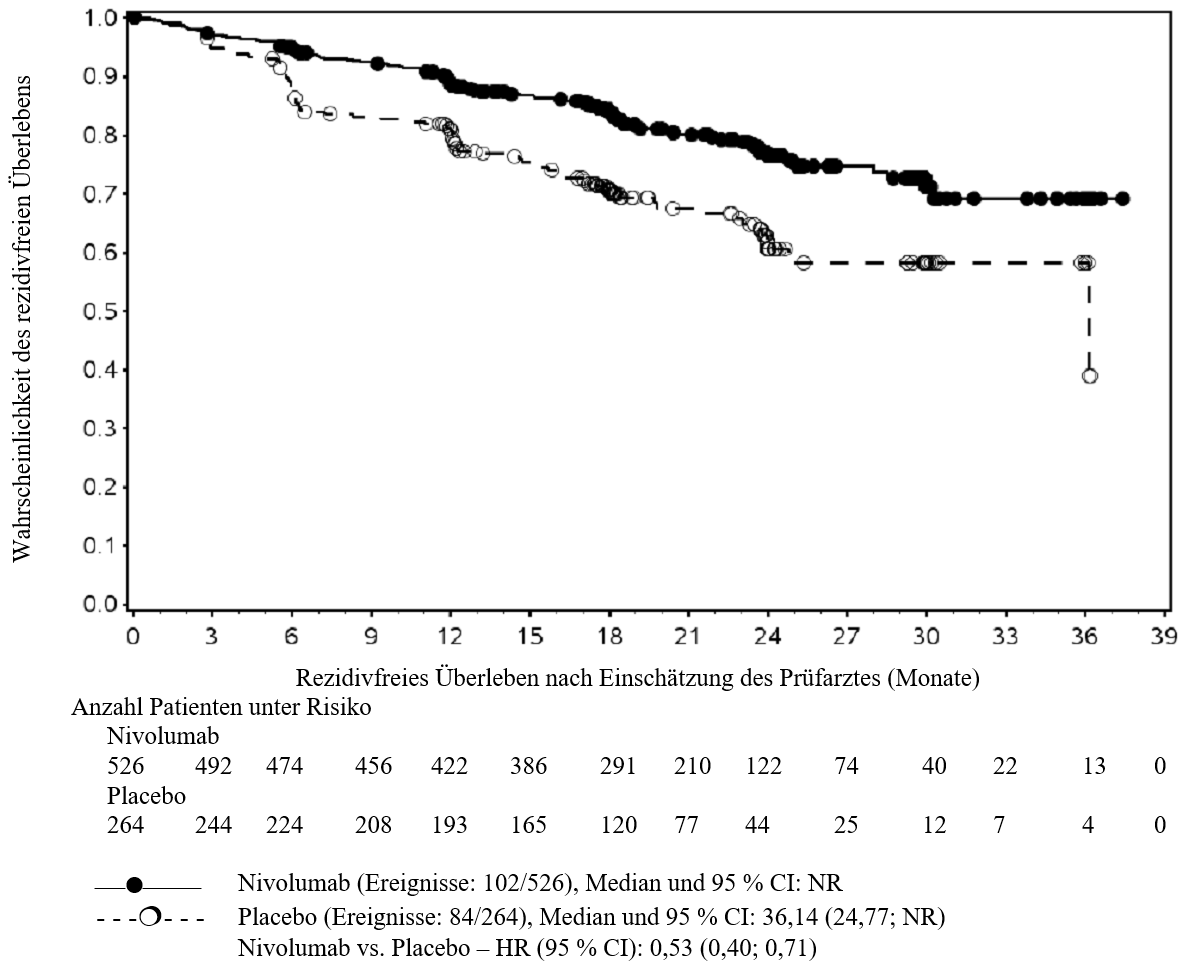
Basierend auf Datenschnitt: 21. Februar 2023, minimale Nachbeobachtungszeit von 15,6 Monaten
Daten zur Tumor‑PD‑L1‑Expression lagen für 302/790 (38,2 %) randomisierte Patienten (36,3 % im Nivolumab‑Arm bzw. 42,0 % im Placebo‑Arm) vor, da die PD‑L1‑Expression kein Stratifizierungsfaktor für die Randomisierung war. Die explorativen RFS‑Analysen nach PD‑L1‑Expression zeigten für Nivolumab im Vergleich zu Placebo eine HR von 0,43 (95 % CI: 0,22; 0,84) bei Patienten (N = 167) mit PD‑L1‑Expression ≥ 1 %, 0,82 (95 % CI: 0,44; 1,54) bei Patienten (N = 135) mit PD‑L1‑Expression < 1 % und 0,50 (95 % CI: 0,34; 0,73) bei Patienten (N = 488) mit unbestimmter/nicht gemeldeter/nicht auswertbarer PD‑L1‑Expression.
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie mit Nivolumab vs. Ipilimumab 10 mg/kg (CA209238)
Sicherheit und Wirksamkeit von 3 mg/kg Nivolumab als Einzelsubstanz zur Behandlung von Patienten mit vollständig reseziertem Melanom wurden in einer randomisierten, doppelblinden Phase‑III‑Studie (CA209238) untersucht. In die Studie wurden erwachsene Patienten eingeschlossen, die einen ECOG‑Performance‑Status von 0 oder 1, einen Krankheitsstatus von IIIB/C oder IV gemäß der Einstufung des American Joint Committee on Cancer (AJCC), 7. Ausgabe, und ein histologisch bestätigtes Melanom hatten, das vollständig chirurgisch reseziert wurde. Laut der 8. Ausgabe der AJCC‑Klassifikation entspricht dies Patienten mit Lymphknotenbeteiligung oder Metastasen. Der Einschluss von Patienten erfolgte unabhängig von ihrem Tumor‑PD‑L1‑Status. Von der Studie ausgeschlossen wurden Patienten mit vorheriger Autoimmunerkrankung und jeder Erkrankung, die eine systemische Behandlung mit Corticosteroiden (≥ 10 mg Prednison oder ‑Äquivalent täglich) oder anderen immunsuppressiven Arzneimitteln erforderte, sowie Patienten mit vorheriger Melanomtherapie (ausgenommen chirurgische Eingriffe, adjuvante Strahlentherapie nach neurochirurgischer Resektion von Läsionen des Zentralnervensystems und zuvor adjuvante Behandlung mit Interferon, welche ≥ 6 Monate vor der Randomisierung abgeschlossen war), vorheriger Behandlung mit einem Anti‑PD‑1‑, Anti‑PD‑L1‑, Anti‑PD‑L2‑, Anti‑CD137‑ oder Anti‑CTLA‑4‑Antikörper (einschließlich Ipilimumab oder eines anderen Antikörpers oder Arzneimittels, das spezifisch auf T‑Zell‑Co‑Stimulation oder Checkpoint‑Wege abzielt).
Insgesamt wurden 906 Patienten entweder für Nivolumab (n = 453) 3 mg/kg alle 2 Wochen oder für Ipilimumab (n = 453) 10 mg/kg alle 3 Wochen für 4 Dosen, anschließend alle 12 Wochen beginnend ab Woche 24 für bis zu 1 Jahr, randomisiert. Die Randomisierung wurde mittels PD‑L1‑Expressionsstatus stratifiziert (≥ 5 % gegenüber ≤ 5 %/unbestimmt) und Krankheitsstadium gemäß der AJCC‑Klassifikation. Tumorbeurteilungen wurden in den ersten 2 Jahren alle 12 Wochen, anschließend alle 6 Monate durchgeführt. Der primäre Endpunkt war das rezidivfreie Überleben (RFS). Das vom Prüfarzt beurteilte RFS wurde definiert als die Zeit zwischen dem Datum der Randomisierung und dem Datum des ersten Rezidivs (lokale, regionale oder Fernmetastasen), neuer primärer Melanome oder Tod jeglicher Ursache, je nachdem, was zuerst auftrat.
Die Ausgangsmerkmale der beiden Gruppen waren etwa gleich. Das mediane Alter war 55 Jahre (Spanne: 18 ‑ 86), 58 % der Patienten waren männlich und 95 % waren weiß. Der ECOG‑Performance‑Status zu Studienbeginn war 0 (90 %) oder 1 (10 %). Die Mehrheit der Patienten entsprach gemäß AJCC dem Krankheitsstadium III (81 %) und 19 % dem Stadium IV. 48 % der Patienten hatten makroskopische Lymphknoten und 32 % hatten Tumorgeschwüre. 42 % der Patienten waren BRAF‑V600‑Mutation‑positiv, 45 % waren BRAF‑Wildtyp und bei 13 % war der BRAF‑Status unbekannt. Hinsichtlich der Tumor‑PD‑L1‑Expression, die mit einem in dieser Studie durchgeführten Test (clinical trial assay) bestimmt wurde, hatten 34 % der Patienten eine Tumor‑PD‑L1‑Expression von ≥ 5 % und 62 % eine von < 5 %. Bei Patienten mit quantifizierbarer Tumor‑PD‑L1‑Expression war die Verteilung der Patienten über die Behandlungsgruppen hinweg ausgeglichen. Die Tumor‑PD‑L1‑Expression wurde unter Verwendung des PD‑L1‑IHC‑28‑8‑pharmDx‑Assays bestimmt.
Bei einer präspezifizierten primären Zwischenanalyse (minimale Nachbeobachtungszeit 18 Monate) wurde eine statistisch signifikante Verbesserung des rezidivfreien Überlebens (RFS) mit Nivolumab im Vergleich zu Ipilimumab mit einer HR von 0,65 (97,56 % CI: 0,51; 0,83; stratifizierter Log‑Rank p < 0,0001) gezeigt. Bei einer aktualisierten deskriptiven RFS‑Analyse mit einer minimalen Nachbeobachtungszeit von 24 Monaten wurde eine RFS‑Verbesserung mit einer HR von 0,66 (95 % CI: 0,54; 0,81; p < 0,0001) bestätigt, das Gesamtüberleben (Overall Survival = OS) war nicht ausgereift. Die Wirksamkeitsergebnisse mit einer minimalen Nachbeobachtungszeit von 36 Monaten (präspezifizierte finale RFS‑Analyse) und 48 Monaten (präspezifizierte finale OS‑Analyse) sind in Tabelle 16 sowie in Abbildung 2 und 3 (alle randomisierten Populationen) dargestellt.
Tabelle 16: Wirksamkeitsergebnisse (CA209238)
Nivolumab |
Ipilimumab 10 mg/kg |
|
Präspezifizierte finale Analyse | ||
Ereignisse |
188 (41,5 %) |
239 (52,8 %) |
Hazard‑Ratioa |
0,68 |
|
95 % CI |
(0,56; 0,82) |
|
p‑Wert |
p < 0,0001 |
|
Median (95 % CI) (Monate) |
NR (38,67; NR) |
24,87 (16,62; 35,12) |
Rezidivfreies Überleben mit einer minimalen Nachbeobachtungszeit von 48 Monaten | ||
Ereignisse |
212 (46,8 %) |
253 (55,8 %) |
Hazard‑Ratioa |
0,71 |
|
95 % CI |
(0,60; 0,86) |
|
Median (95 % CI) Monate |
52,37 (42,51; NR) |
24,08 (16,56; 35,09) |
Rate (95 % CI) nach 12 Monaten |
70,4 (65,9; 74,4) |
60,0 (55,2; 64,5) |
Rate (95 % CI) nach 18 Monaten |
65,8 (61,2; 70,0) |
53,0 (48,1; 57,6) |
Rate (95 % CI) nach 24 Monaten |
62,6 (57,9; 67,0) |
50,2 (45,3; 54,8) |
Rate (95 % CI) nach 36 Monaten |
57,6 (52,8; 62,1) |
44,4 (39,6; 49,1) |
Rate (95 % CI) nach 48 Monaten |
51,7 (46,8; 56,3) |
41,2 (36,4; 45,9) |
Präspezifizierte finale Analyse | ||
Ereignisse |
100 (22,1 %) |
111 (24,5 %) |
Hazard‑Ratioa |
0,87 |
|
95,03 % CI |
(0,66; 1,14) |
|
p‑Wert |
0,3148 |
|
Median (95 % CI) (Monate) |
NR |
NR |
Rate (95 % CI) nach 12 Monaten |
96,2 (93,9; 97,6) |
95,3 (92,8; 96,9) |
Rate (95 % CI) nach 18 Monaten |
91,9 (88,9; 94,1) |
91,8 (88,8; 94,0) |
Rate (95 % CI) nach 24 Monaten |
88,0 (84,6; 90,7) |
87,8 (84,4; 90,6) |
Rate (95 % CI) nach 36 Monaten |
81,7 (77,8; 85,1) |
81,6 (77,6; 85,0) |
Rate (95 % CI) nach 48 Monaten |
77,9 (73,7; 81,5) |
76,6 (72,2; 80,3) |
a Mit einem stratifizierten Modell für proportionale Hazards berechnet. | ||
Bei einer minimalen Nachbeobachtungszeit von 36 Monaten zeigte die Studie eine statistisch signifikante Verbesserung des RFS für Patienten, die in den Nivolumab‑Arm randomisiert waren, gegenüber dem Ipilimumab‑10‑mg/kg‑Arm. Der RFS‑Vorteil wurde über alle Untergruppen hinweg konsistent gezeigt, einschließlich der Tumor‑PD‑L1‑Expression, des BRAF‑Status und des Stadiums der Erkrankung. Bei einer minimalen Nachbeobachtungszeit von 48 Monaten, dargestellt in Abbildung 2, zeigt die Studie weiterhin einen RFS‑Vorteil des Nivolumab‑Arms gegenüber dem Ipilimumab‑Arm. Der RFS‑Vorteil wurde über alle Untergruppen hinweg beibehalten.
Abbildung 2: Rezidivfreies Überleben (CA209238)
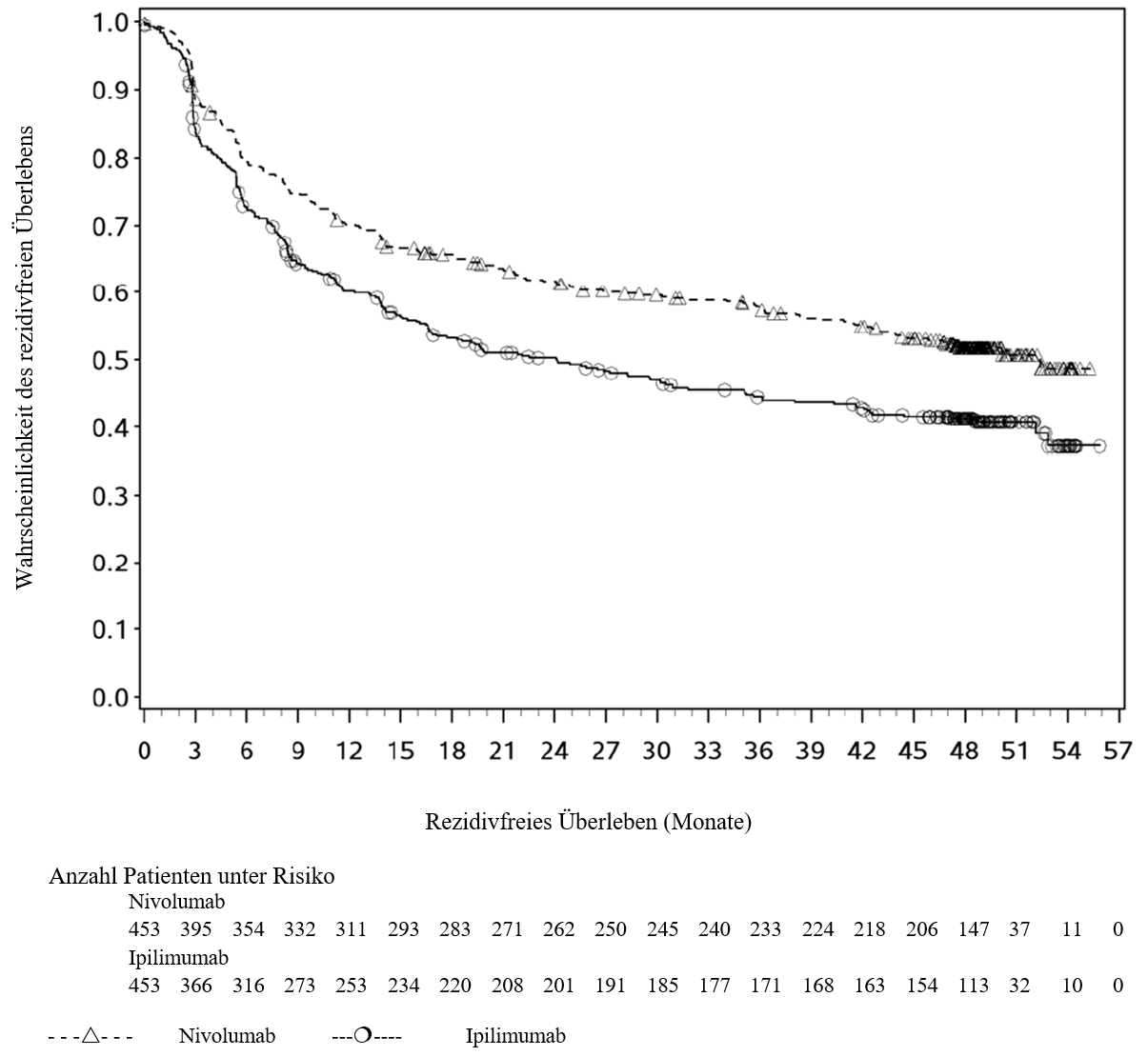
Abbildung 3: Gesamtüberleben (CA209238)
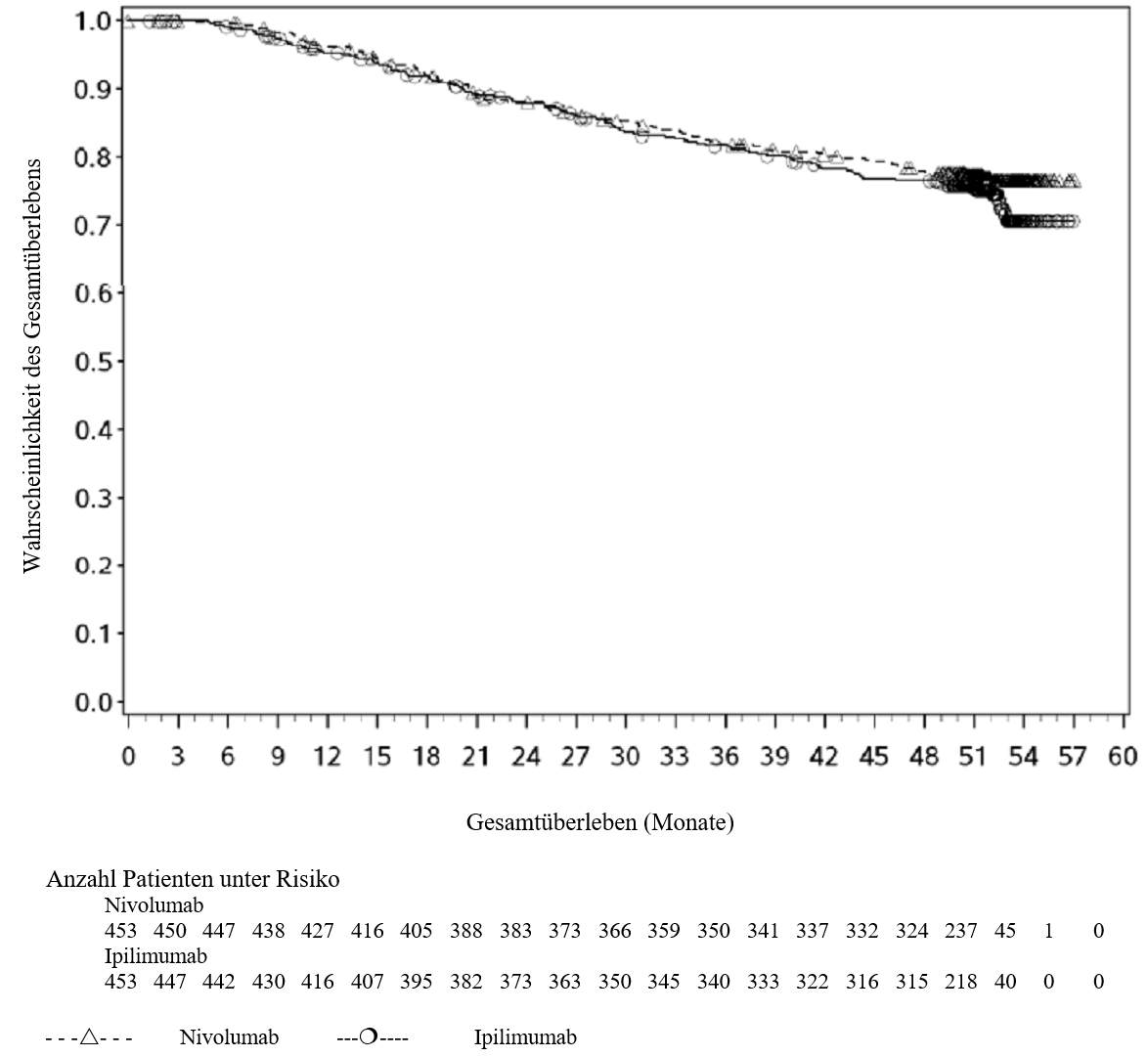
Bei einer minimalen Nachbeobachtungszeit von 48 Monaten, dargestellt in Abbildung 3, wurde das mediane OS in keiner Gruppe erreicht (HR = 0,87; 95,03 % CI: 0,66; 1,14; p‑Wert: 0,3148). Die Gesamtüberlebensdaten sind durch die Auswirkungen der anschließenden wirksamen Krebstherapien verzerrt. Eine anschließende systemische Therapie erhielten 33 % der Patienten im Nivolumab‑Arm bzw. 42 % im Ipilimumab‑Arm. Eine anschließende Immuntherapie (einschließlich Anti‑PD1‑Therapie, Anti‑CTLA‑4‑Antikörper oder eine andere Immuntherapie) erhielten 23 % der Patienten im Nivolumab‑Arm bzw. 34 % im Ipilimumab‑Arm.
Die Lebensqualität (Quality of life = QoL) mit Nivolumab blieb während der Behandlung stabil und nahe bei den Ausgangswerten, wie anhand valider und verlässlicher Skalen wie der QLQ‑C30‑Skala der Europäischen Organisation für Forschung und Behandlung von Krebs (European Organisation for Research and Treatment of Cancer = EORTC) und dem EQ‑5D Utility Index und der Visuellen Analogskala (VAS) gezeigt werden konnte.
Behandlung des fortgeschrittenen Melanoms
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie vs. Dacarbazin (CA209066)
Sicherheit und Wirksamkeit von 3 mg/kg Nivolumab zur Behandlung des fortgeschrittenen (nicht resezierbaren oder metastasierten) Melanoms wurden in einer randomisierten, doppelblinden Phase‑III‑Studie (CA209066) untersucht. In die Studie wurden erwachsene Patienten (ab 18 Jahren) eingeschlossen mit bestätigtem, behandlungsnaivem Melanom vom BRAF‑Wildtyp im Stadium III oder IV und mit einem ECOG‑Performance‑Status von 0 oder 1. Patienten mit akuter Autoimmunerkrankung, okulärem Melanom, aktiven Hirnmetastasen oder leptomeningealen Metastasen waren von der Studie ausgeschlossen.
Insgesamt wurden 418 Patienten entweder für Nivolumab (n = 210), das in einer Dosierung von 3 mg/kg Körpergewicht alle 2 Wochen über 60 Minuten intravenös verabreicht wurde oder für Dacarbazin (n = 208), das zu 1 000 mg/m2 Körperoberfläche alle 3 Wochen verabreicht wurde, randomisiert. Die Randomisierung wurde nach Tumor‑PD‑L1‑Status und M‑Stadium (M0/M1a/M1b versus M1c) stratifiziert. Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen bestand oder bis die Behandlung nicht mehr vertragen wurde. Eine Behandlung nach Krankheitsprogression wurde für Patienten zugelassen, die, nach Ermessen des Prüfarztes, klinisch profitierten und keine erheblichen Nebenwirkungen durch die Studienmedikation zeigten. Tumorbeurteilungen wurden gemäß den „Response Evaluation Criteria in Solid Tumours“ (RECIST), Version 1.1, zum ersten Mal 9 Wochen nach Randomisierung und dann im ersten Jahr alle 6 Wochen und anschließend alle 12 Wochen durchgeführt. Das primäre Wirksamkeitskriterium war das Gesamtüberleben (Overall Survival = OS). Sekundäre Wirksamkeitskriterien waren das von den Prüfärzten bewertete PFS und die objektive Ansprechrate (Objective Response Rate = ORR).
Die Ausgangsmerkmale der beiden Gruppen waren etwa gleich. Das mittlere Alter betrug 65 Jahre (Spanne: 18 ‑ 87), 59 % waren männlich und 99,5 % waren weiß. Die meisten Patienten hatten einen ECOG‑Performance‑Status von 0 (64 %) oder 1 (34 %). 61 % der Patienten hatten bei Studienbeginn einen Krankheitsstatus von M1c. 74 % der Patienten hatten ein kutanes Melanom und 11 % ein Melanom der Schleimhaut; 35 % der Patienten hatten PD‑L1‑positives Melanom (≥ 5 % Tumorzellmembranexpression). 16 % der Patienten hatten zuvor eine adjuvante Therapie erhalten; die häufigste adjuvante Behandlung war Interferon (9 %). 4 % der Patienten hatten Hirnmetastasen in der Vorgeschichte und 37 % der Patienten hatten zu Studienbeginn einen LDH‑Ausgangsspiegel über dem oberen Normbereich (ULN).
Die Kaplan‑Meier‑Kurven des OS sind in Abbildung 4 dargestellt.
Abbildung 4: Kaplan‑Meier‑Kurven des Gesamtüberlebens (CA209066)
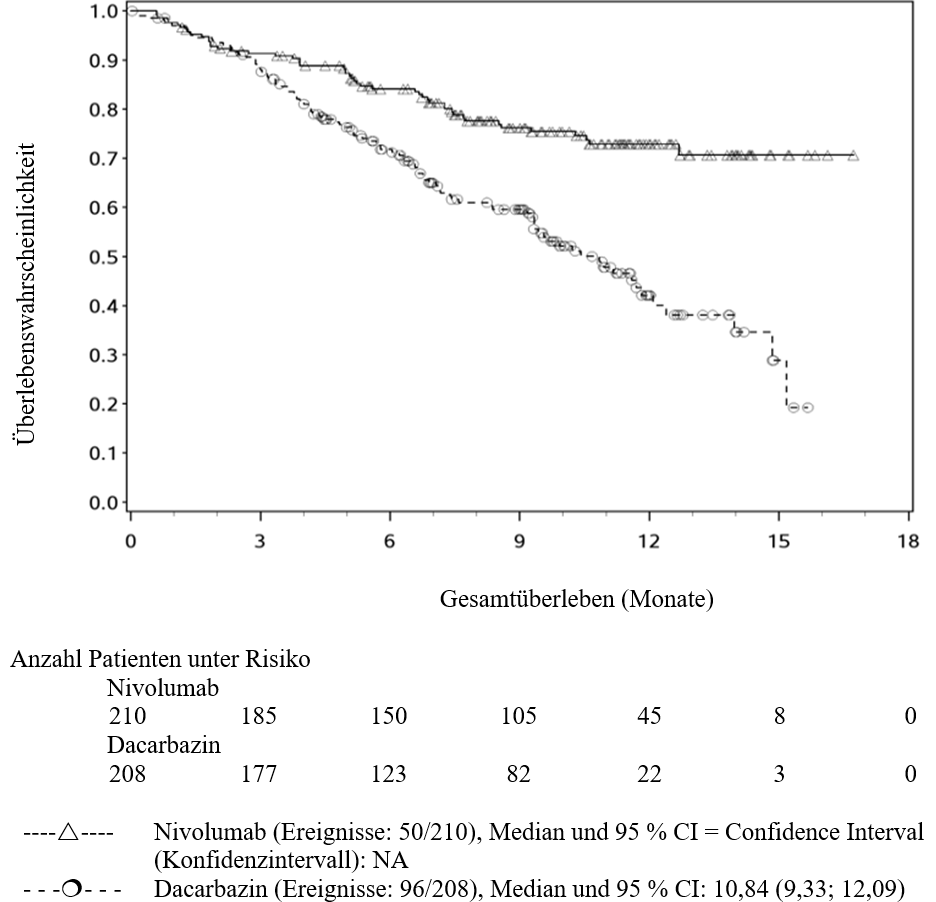
Der beobachtete Gesamtüberlebensvorteil wurde durchgehend in verschiedenen Patienten‑Untergruppen nachgewiesen, einschließlich Ausgangs‑ECOG‑Performance‑Status, M‑Stadium, Hirnmetastasen in der Vorgeschichte und Ausgangs‑LDH‑Werten. Der Überlebensvorteil wurde unabhängig davon beobachtet, ob die Tumoren der Patienten als PD‑L1-negativ oder PD‑L1‑positiv eingestuft wurden (Tumormembranexpressionsgrenze von 5 % oder 10 %).
Die verfügbaren Daten zeigen, dass die Wirkung von Nivolumab mit Verzögerung einsetzt, so dass es 2‑3 Monate dauern kann bis der Vorteil von Nivolumab gegenüber Chemotherapie zum Tragen kommt.
Die Wirksamkeitsergebnisse sind in Tabelle 17 dargestellt.
Tabelle 17: Wirksamkeitsergebnisse (CA209066)
Nivolumab |
Dacarbazin |
||
Gesamtüberleben | |||
Ereignisse |
50 (23,8 %) |
96 (46,2 %) |
|
Hazard-Ratio |
0,42 |
||
99,79 % CI |
(0,25; 0,73) |
||
95 % CI |
(0,30; 0,60) |
||
p‑Wert |
< 0,0001 |
||
Median (95 % CI) |
Nicht erreicht |
10,8 (9,33; 12,09) |
|
Rate (95 % CI) |
|||
Nach 6 Monaten |
84,1 (78,3; 88,5) |
71,8 (64,9; 77,6) |
|
Nach 12 Monaten |
72,9 (65,5; 78,9) |
42,1 (33,0; 50,9) |
|
Progressionsfreies Überleben |
|||
Ereignisse |
108 (51,4 %) |
163 (78,4 %) |
|
Hazard-Ratio |
0,43 |
||
95 % CI |
(0,34; 0,56) |
||
p‑Wert |
< 0,0001 |
||
Median (95 % CI) |
5,1 (3,48; 10,81) |
2,2 (2,10; 2,40) |
|
Rate (95 % CI) |
|||
Nach 6 Monaten |
48,0 (40,8; 54,9) |
18,5 (13,1; 24,6) |
|
Nach 12 Monaten |
41,8 (34,0; 49,3) |
NA |
|
Objektives Ansprechen |
84 (40,0 %) |
29 (13,9 %) |
|
(95 % CI) |
(33,3; 47,0) |
(9,5; 19,4) |
|
Odds Ratio (95 % CI) |
4,06 (2,52; 6,54) |
||
p‑Wert |
< 0,0001 |
||
Vollständiges Ansprechen (Complete Response = CR) |
16 (7,6 %) |
2 (1,0 %) |
|
Teilweises Ansprechen (Partial Response = PR) |
68 (32,4 %) |
27 (13,0 %) |
|
Stabile Erkrankung (Stable Disease = SD) |
35 (16,7 %) |
46 (22,1 %) |
|
Mediane Ansprechdauer | |||
Monate (Spanne) |
Nicht erreicht |
(0+ ‑ 12,5+) |
6,0 (1,1 ‑ 10,0+) |
Mediane Zeit bis zum Ansprechen |
|||
Monate (Spanne) |
2,1 (1,2 ‑ 7,6) |
2,1 (1,8 ‑ 3,6) |
|
„+“ kennzeichnet eine zensierte Beobachtung. | |||
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie vs. Chemotherapie (CA209037)
Sicherheit und Wirksamkeit von 3 mg/kg Nivolumab zur Behandlung des fortgeschrittenen (nicht resezierbaren oder metastasierten) Melanoms wurden in einer randomisierten, offenen Phase‑III‑Studie (CA209037) untersucht. In die Studie wurden erwachsene Patienten eingeschlossen, bei denen es unter oder nach Ipilimumab und, bei positiver BRAF‑V600‑Mutation, auch unter oder nach einer Behandlung mit einem BRAF‑Kinase‑Inhibitor zu einer Progression kam. Patienten mit akuter Autoimmunerkrankung, okulärem Melanom, aktiven Hirnmetastasen oder leptomeningealen Metastasen oder früheren unter Ipilimumab aufgetretenen schwerwiegenden (Grad 4 nach CTCAE v4.0) Nebenwirkungen (ausgenommen zurückgebildete Übelkeit, Ermüdung/Fatigue, Infusionsreaktionen oder Endokrinopathien) waren von der Studie ausgeschlossen.
Insgesamt wurden 405 Patienten entweder für Nivolumab (n = 272), das in einer Dosierung von 3 mg/kg Körpergewicht alle 2 Wochen über 60 Minuten intravenös verabreicht wurde, oder für Chemotherapie (n = 133) randomisiert. Die Chemotherapie erfolgte nach Ermessen des Prüfarztes mit Dacarbazin (1 000 mg/m2 alle 3 Wochen) oder Carboplatin (AUC 6 alle 3 Wochen) und Paclitaxel (175 mg/m2 alle 3 Wochen). Die Randomisierung wurde nach BRAF‑ und Tumor‑PD‑L1‑Status und bestem Ansprechen auf zuvor erhaltenes Ipilimumab stratifiziert.
Die koprimären Wirksamkeitskriterien waren bestätigtes ORR bei den ersten 120 Patienten, die mit Nivolumab behandelt wurden, beurteilt nach RECIST, Version 1.1, durch ein unabhängiges radiologisches Bewertungskomitee (independent radiology review committee, IRRC) und Vergleich des OS unter Nivolumab mit dem unter Chemotherapie. Weitere Wirksamkeitskriterien beinhalteten Zeit bis zum Ansprechen und Dauer des Ansprechens.
Das mediane Alter betrug 60 Jahre (Spanne: 23 ‑ 88). 64 % der Patienten waren männlich und 98 % weiß. Der ECOG‑Performance‑Status war 0 bei 61 % der Patienten und 1 bei 39 % der Patienten. Die meisten (75 %) Patienten hatten bei Studienbeginn einen Krankheitsstatus von M1c. 73 % der Patienten hatten ein kutanes Melanom, 10 % ein Melanom der Schleimhaut. 27 % der Patienten hatten eine systemische Vorbehandlung, 51 % 2 Vorbehandlungen und 21 %>2 Vorbehandlungen erhalten. 22 % der Patienten hatten Tumoren, die positiv auf eine BRAF‑Mutation getestet worden waren und 50 % der Patienten hatten Tumoren, die als PD‑L1‑positiv betrachtet wurden. 64 % der Patienten haben klinisch nicht von einer Vorbehandlung mit Ipilimumab profitiert (CR/PR oder SD). Die Ausgangsmerkmale waren in den Gruppen etwa gleich, mit Ausnahme des Anteils von Patienten mit Hirnmetastasen in der Vorgeschichte (19 % in der Nivolumab‑Gruppe und 13 % in der Chemotherapie-Gruppe) und Patienten mit einem LDH oberhalb des Normwertes zu Studienbeginn (51 % bzw. 35 %).
Zum Zeitpunkt dieser finalen ORR‑Analyse wurden die Ergebnisse von 120 mit Nivolumab und 47 mit Chemotherapie behandelten Patienten ausgewertet, die mindestens 6 Monate nachbeobachtet worden waren. Die Wirksamkeitsergebnisse sind in Tabelle 18 dargestellt.
Tabelle 18: Bestes Gesamtansprechen, Zeit bis zum Ansprechen und Dauer des Ansprechens (CA209037)
Nivolumab |
Chemotherapie |
|
Bestätigtes objektives Ansprechen (IRRC) |
38 (31,7 %) |
5 (10,6 %) |
(95 % CI) |
(23,5; 40,8) |
(3,5; 23,1) |
Vollständiges Ansprechen (Complete response = CR) |
4 (3,3 %) |
0 |
Teilweises Ansprechen (Partial Response = PR) |
34 (28,3 %) |
5 (10,6 %) |
Stabile Erkrankung (Stable Disease = SD) |
28 (23,3 %) |
16 (34,0 %) |
Mediane Ansprechdauer |
||
Monate (Spanne) |
Nicht erreicht |
3,6 (Nicht vorhanden) |
Mediane Zeit bis zum Ansprechen |
||
Monate (Spanne) |
2,1 (1,6 ‑ 7,4) |
3,5 (2,1 ‑ 6,1) |
Die verfügbaren Daten zeigen, dass die Wirkung von Nivolumab mit Verzögerung einsetzt, so dass es 2 ‑ 3 Monate dauern kann, bis der Vorteil von Nivolumab gegenüber Chemotherapie zum Tragen kommt.
Aktualisierte Analyse (24 Monate Nachverfolgung)
Die ORR unter allen randomisierten Patienten betrug in der Nivolumab‑Gruppe 27,2 % (95 % CI: 22,0; 32,9) und 9,8 % (95 % CI: 5,3; 16,1) in der Chemotherapie‑Gruppe. Die mediane Dauer des Ansprechens war 31,9 Monate (Spanne: 1,4+ ‑ 31,9) in der Nivolumab‑ und 12,8 Monate (Spanne: 1,3+ ‑ 13,6+) in der Chemotherapie‑Gruppe. Die PFS Hazard Ratio für Nivolumab vs. Chemotherapie war 1,03 (95 % CI: 0,78; 1,36). ORR und PFS wurden vom IRRC nach RECIST, Version 1.1 beurteilt.
Bei der finalen OS‑Analyse gab es zwischen Nivolumab und Chemotherapie keinen statistisch signifikanten Unterschied. Die primäre OS‑Analyse war nicht für die Auswirkungen nachfolgender Therapien adjustiert, wobei 54 (40,6 %) Patienten im Chemotherapie-Arm nachfolgend eine Anti‑PD1‑Behandlung erhielten. OS kann durch Studienabbruch, Ungleichgewicht der Folgetherapien und Unterschiede bei den Charakteristika zu Beginn der Studie verzerrt sein. Im Nivolumab‑Arm hatten mehr Patienten schlechte prognostische Faktoren (erhöhtes LDH und Hirnmetastasen) als im Chemotherapie‑Arm.
Wirksamkeit in Abhängigkeit von BRAF‑Status: Objektives Ansprechen auf Nivolumab (gemäß der Definition des koprimären Endpunkts) wurde bei Patienten mit oder ohne positiver BRAF‑Mutation des Melanoms beobachtet. Die ORR in der Untergruppe mit BRAF‑Mutation betrug 17 % (95 % CI: 8,4; 29,0) für Nivolumab und 11 % (95 % CI: 2,4; 29,2) für Chemotherapie. In der Untergruppe mit BRAF‑Wildtyp war die ORR 30 % (95 % CI: 24,0; 36,7) für Nivolumab und 9 % (95 % CI: 4,6; 16,7) für Chemotherapie.
Die PFS Hazard Ratios für Nivolumab vs. Chemotherapie waren 1,58 (95 % CI: 0,87; 2,87) für Patienten mit BRAF‑Mutation und 0,82 (95 % CI: 0,60; 1,12) für Patienten mit BRAF‑Wildtyp. Die OS Hazard Ratios für Nivolumab vs. Chemotherapie betrugen 1,32 (95 % CI: 0,75; 2,32) für Patienten mit BRAF-Mutation und 0,83 (95 % CI: 0,62; 1,11) für Patienten mit BRAF‑Wildtyp.
Wirksamkeit in Abhängigkeit von Tumor‑PD‑L1‑Expression: Objektives Ansprechen auf Nivolumab wurde unabhängig von der Tumor‑PD‑L1‑Expression beobachtet. Die Rolle dieses Biomarkers (Tumor‑PD‑L1‑Expression) ist jedoch nicht abschließend geklärt.
Bei Patienten mit einer Tumor‑PD‑L1‑Expression ≥ 1 % betrug die ORR 33,5 % für Nivolumab (n = 179; 95 % CI: 26,7; 40,9) und 13,5 % für Chemotherapie (n = 74; 95 % CI: 6,7; 23,5). Bei Patienten mit einer Tumor‑PD‑L1‑Expression < 1 % war die vom IRRC bestimmte ORR jeweils 13,0 % (n = 69; 95 % CI: 6,1; 23,3) bzw. 12,0 % (n = 25; 95 % CI: 2,5; 31,2).
Die PFS Hazard Ratios für Nivolumab vs. Chemotherapie waren 0,76 (95 % CI: 0,54; 1,07) bei Patienten mit einer Tumor‑PD‑L1 Expression ≥ 1 % und 1,92 (95 % CI: 1,05; 3,5) bei Patienten mit einer Tumor‑PD‑L1‑Expression < 1 %.
Die OS Hazard Ratios für Nivolumab vs. Chemotherapie waren 0,69 (95 % CI: 0,49; 0,96) bei Patienten mit einer Tumor‑PD‑L1 Expression ≥ 1 % und 1,52 (95 % CI: 0,89; 2,57) bei Patienten mit Tumor‑PD‑L1‑Expression < 1 %.
Diese Untergruppen‑Analysen sollten aufgrund der kleinen Gruppengröße und des Fehlens eines signifikanten Unterschiedes im OS in der randomisierten Gesamtpopulation mit Vorsicht interpretiert werden.
Intravenöse Formulierung
Offene Dosiseskalationsstudie der Phase I (MDX1106‑03)
Sicherheit und Verträglichkeit von Nivolumab wurden in einer offenen Dosiseskalationsstudie der Phase I bei verschiedenen Tumorarten (einschließlich malignes Melanom) untersucht. Von den 306 vorbehandelten Patienten, die in die Studie eingeschlossen wurden, hatten 107 ein Melanom und erhielten maximal 2 Jahre lang Nivolumab in einer Dosierung von 0,1 mg/kg, 0,3 mg/kg, 1 mg/kg, 3 mg/kg oder 10 mg/kg. In dieser Patientengruppe wurde bei 33 Patienten (31 %) über ein objektives Ansprechen mit einer medianen Ansprechdauer von 22,9 Monaten berichtet (95 % CI: 17,0; NR). Das mediane PFS betrug 3,7 Monate (95 % CI: 1,9; 9,3). Das mediane OS war 17,3 Monate (95 % CI: 12,5; 37,8) und die berechneten OS‑Raten waren 42 % (95 % CI: 32; 51) nach 3 Jahren, 35 % (95 % CI: 26; 44) nach 4 Jahren und 34 % (95 % CI: 25; 43) nach 5 Jahren (Minimum 45 Monate Nachverfolgung).
Intravenöse Formulierung
Einarmige Phase‑II‑Studie (CA209172)
Die Studie CA209172 war eine einarmige, offene Studie mit Nivolumab‑Monotherapie bei Patienten mit Stadium‑III‑ (nicht‑resezierbarem) oder Stadium‑IV‑ metastasiertem Melanom nach einer vorherigen Therapie mit einem monoklonalen Anti‑CTLA‑4‑Antikörper. Der primäre Endpunkt war Sicherheit und ein sekundärer Endpunkt war Wirksamkeit. Von den 1 008 behandelten Patienten hatten 103 (10 %) ein okuläres/uveales Melanom, 66 (7 %) einen ECOG‑Performance‑Status von 2, 165 (16 %) asymptomatische behandelte oder unbehandelte ZNS‑Metastasen, 13 (1,3 %) behandelte leptomeningeale Metastasen, 25 (2 %) eine Autoimmunerkrankung und 84 (8 %) Grad 3‑4 immunvermittelte Nebenwirkungen unter vorheriger Anti‑CTLA‑4‑Therapie. Es wurden keine neuen Sicherheitssignale identifiziert und das Gesamtsicherheitsprofil von Nivolumab war über die Subgruppen hinweg vergleichbar. Die Wirksamkeitsergebnisse, basierend auf den vom Prüfarzt bewerteten Ansprechraten, in Woche 12 sind in Tabelle 19 dargestellt.
Tabelle 19: Ansprechrate in Woche 12 – Alle Patienten und Subgruppen mit auswertbarem Ansprechen (CA209172)
Gesamtanzahl |
Okuläres/ |
ECOG PS 2 |
ZNS-Metastasen |
Autoimmunerkrankungen |
Grad 3 - 4 |
|
N |
161/588 |
4/61 |
4/20 |
20/73 |
3/16 |
13/46 |
a Das Ansprechen wurde nach RECIST, Version 1.1 für 588/1 008 (58,3 %) Patienten ausgewertet, die bis Woche 12 behandelt wurden und einen Follow‑up Scan in Woche 12 erhielten. | ||||||
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie mit Nivolumab in Kombination mit Ipilimumab oder Nivolumab als Monotherapie im Vergleich gegen Ipilimumab als Monotherapie (CA209067)
Die Sicherheit und Wirksamkeit von Nivolumab 1 mg/kg in Kombination mit Ipilimumab 3 mg/kg oder Nivolumab 3 mg/kg vs. Ipilimumab‑Monotherapie 3 mg/kg zur Behandlung des fortgeschrittenen (nicht resezierbaren oder metastasierten) Melanoms wurden in einer randomisierten, doppelblinden Phase‑III‑Studie (CA209067) untersucht. Die Unterschiede zwischen den beiden Nivolumab‑enthaltenden Gruppen wurden deskriptiv untersucht. In die Studie wurden erwachsene Patienten mit bestätigtem nicht resezierbaren Melanom im Stadium III oder IV eingeschlossen. Die Patienten mussten einen ECOG‑Performance‑Status 0 oder 1 haben und durften keine systemischen Vortherapien zur Behandlung des nicht resezierbaren oder metastatischen Melanoms erhalten haben. Adjuvante oder neoadjuvante Vortherapie war erlaubt, wenn diese mindestens 6 Wochen vor Einschluss in die Studie abgeschlossen worden war. Patienten mit akuter Autoimmunerkrankung, okulärem/uvealem Melanom, aktiven Hirnmetastasen oder leptomeningealen Metastasen waren von der Studie ausgeschlossen.
Insgesamt wurden 945 Patienten entweder für Nivolumab in Kombination mit Ipilimumab (n = 314), Nivolumab als Monotherapie (n = 316) oder Ipilimumab als Monotherapie (n = 315) randomisiert. Patienten im Kombinationsarm erhielten Nivolumab in einer Dosierung von 1 mg/kg Körpergewicht über 60 Minuten und Ipilimumab in einer Dosierung von 3 mg/kg Körpergewicht über 90 Minuten jeweils als intravenös verabreichte Infusion alle 3 Wochen für die ersten vier Gaben, gefolgt von Nivolumab in einer Dosierung von 3 mg/kg Körpergewicht als Monotherapie alle 2 Wochen. Patienten im Nivolumab‑Monotherapie‑Arm erhielten Nivolumab in einer Dosierung von 3 mg/kg Körpergewicht alle 2 Wochen. Patienten im Vergleichsarm erhielten Ipilimumab in einer Dosierung von 3 mg/kg Körpergewicht und Placebo für Nivolumab als intravenöse Infusion alle 3 Wochen für 4 Gaben und anschließend Placebo alle 2 Wochen. Die Randomisierung wurde mittels PD‑L1‑Expressionsstatus stratifiziert (≥ 5 % gegenüber ≤ 5 % Expression auf der Tumorzellmembran), BRAF‑Status und M‑Stadium gemäß der Einstufung des American Joint Committee on Cancer (AJCC). Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen bestand oder bis die Behandlung nicht mehr vertragen wurde. Tumorbeurteilungen wurden zum ersten Mal 12 Wochen nach Randomisierung und dann im ersten Jahr alle 6 Wochen und anschließend alle 12 Wochen durchgeführt. Die primären Wirksamkeitskriterien waren das progressionsfreie Überleben (Progression‑Free Survival = PFS) sowie Gesamtüberleben (Overall Survival = OS). Die objektive Ansprechrate (Objective Response Rate = ORR) und die Dauer des Ansprechens wurden ebenfalls bewertet.
Die Ausgangsmerkmale der drei Gruppen waren etwa gleich verteilt. Das mittlere Alter betrug 61 Jahre (Spanne: 18 ‑ 90), 65 % waren Männer und 97 % waren weiß. Der ECOG‑Performance‑Status war 0 bei 73 % der Patienten und 1 bei 27 % der Patienten. 93 % der Patienten hatten einen Krankheitsstatus von IV gemäß AJCC, 58 % hatten einen Krankheitsstatus von M1c bei Studienbeginn. 22 % der Patienten hatten eine adjuvante Vortherapie erhalten, 32 % der Patienten hatten ein BRAF‑Mutations‑positives Melanom und 26,5 % der Patienten hatten eine PD‑L1‑Expression in der Tumorzellmembran von ≥ 5 %. 4 % der Patienten hatten Hirnmetastasen in der Vorgeschichte und 36 % der Patienten hatten zu Studienbeginn einen LDH‑Ausgangsspiegel über dem oberen Normbereich (ULN). Bei den Patienten mit quantifizierbarer Tumor‑PD‑L1‑Expression war die Verteilung der Patienten zwischen den 3 Behandlungsgruppen ausgeglichen. Die Tumor‑PD‑L1‑Expression wurde mittels PD‑L1‑IHC‑28‑8‑PharmDx‑Assay ermittelt.
In der primären Analyse (minimale Nachbeobachtungszeit 9 Monate) betrug das mediane PFS 6,9 Monate in der Nivolumab‑Gruppe verglichen mit 2,9 Monaten in der Ipilimumab‑Gruppe (HR = 0,57; 99,5 % CI: 0,43; 0,76; p < 0,0001). Das mediane PFS betrug 11,5 Monate in der Ipilimumab‑in‑Kombination‑mit‑Nivolumab‑Gruppe verglichen mit 2,9 Monaten in der Ipilimumab‑Gruppe (HR = 0,42; 99,5 % CI: 0,31; 0,57; p < 0,0001).
Die PFS‑Ergebnisse aus einer deskriptiven Analyse (mit einer minimalen Nachbeobachtungszeit von 90 Monaten) sind in Abbildung 5 dargestellt (gesamte randomisierte Population), Abbildung 6 zeigt einen Tumor‑PD‑L1‑5‑%‑Cut‑off und Abbildung 7 einen Tumor‑PD‑L1‑1‑%‑Cut‑off.
Abbildung 5: Progressionsfreies Überleben (CA209067)
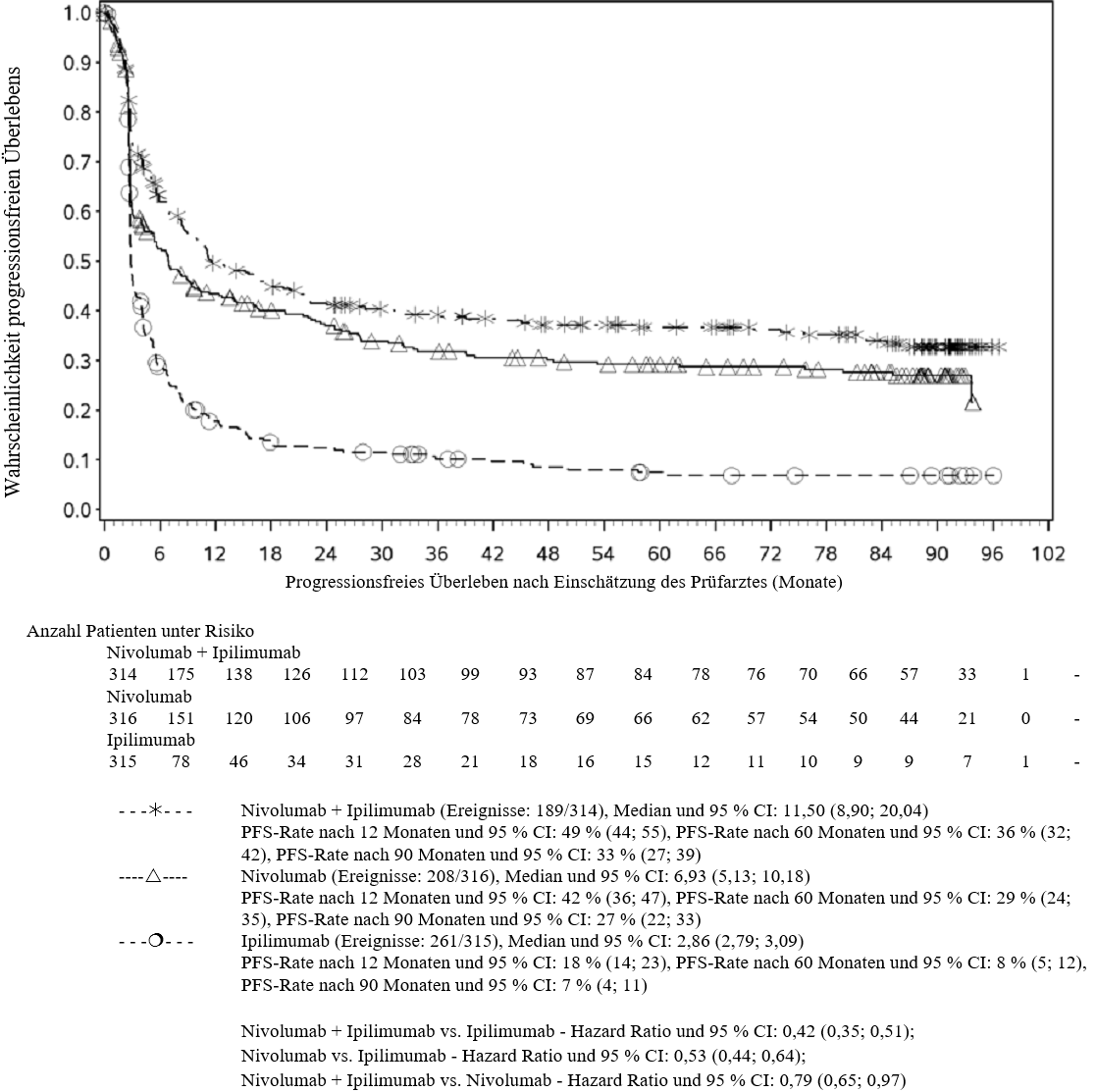
Abbildung 6: Progressionsfreies Überleben bei PD‑L1‑Expression: 5‑%‑Cut‑off (CA209067)
PD‑L1‑Expression < 5 %
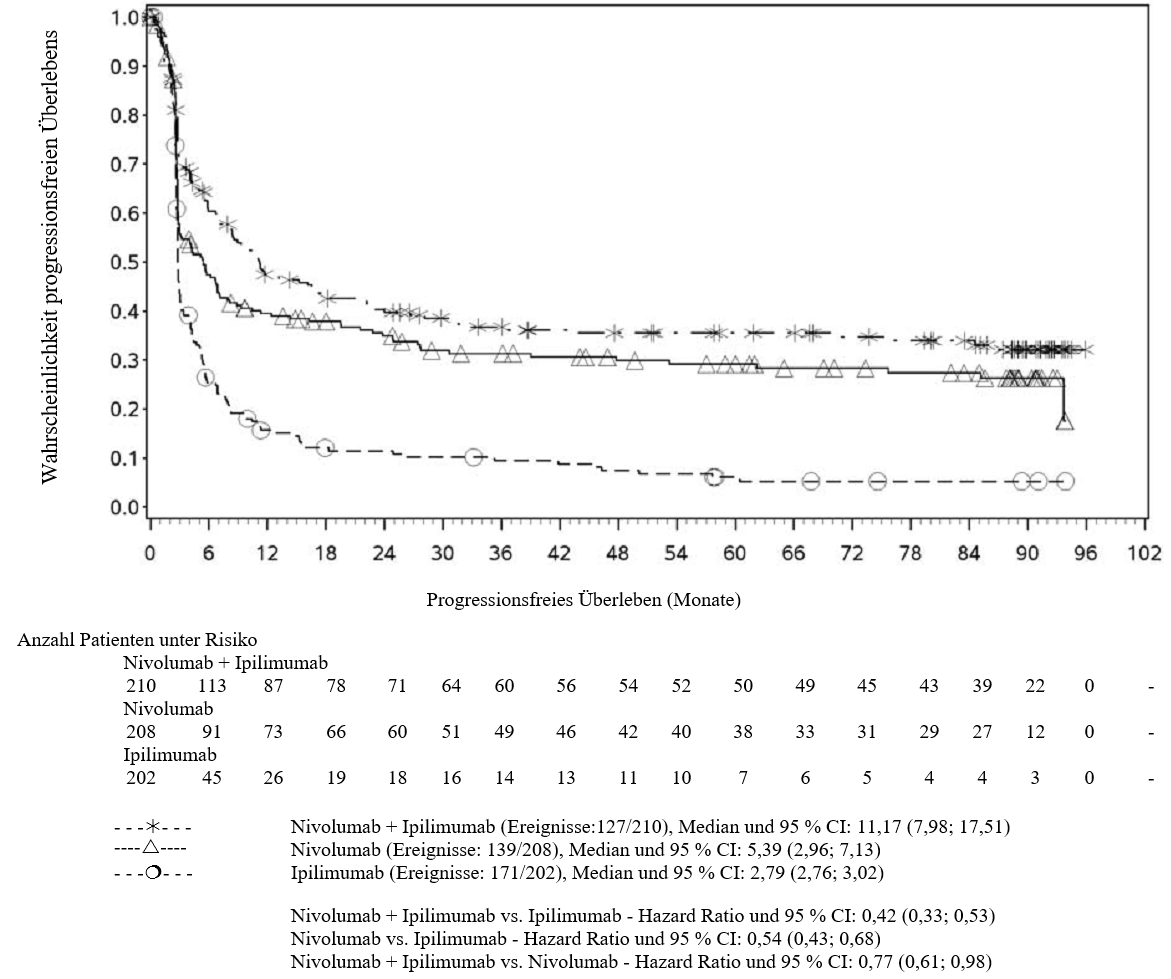
PD‑L1‑Expression ≥ 5 %
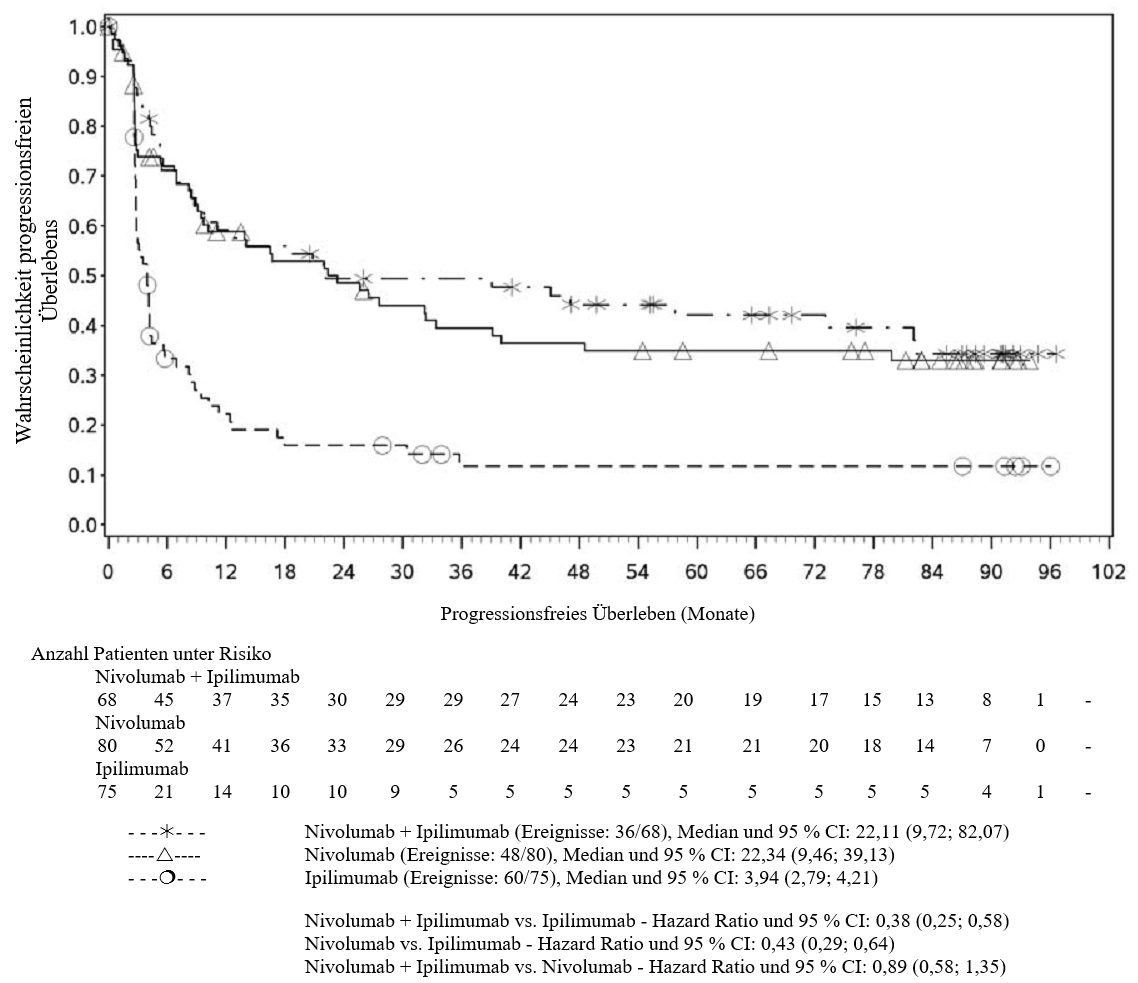
Abbildung 7: Progressionsfreies Überleben bei PD‑L1‑Expression: 1‑%‑Cut‑off (CA209067)
PD‑L1‑Expression < 1 %
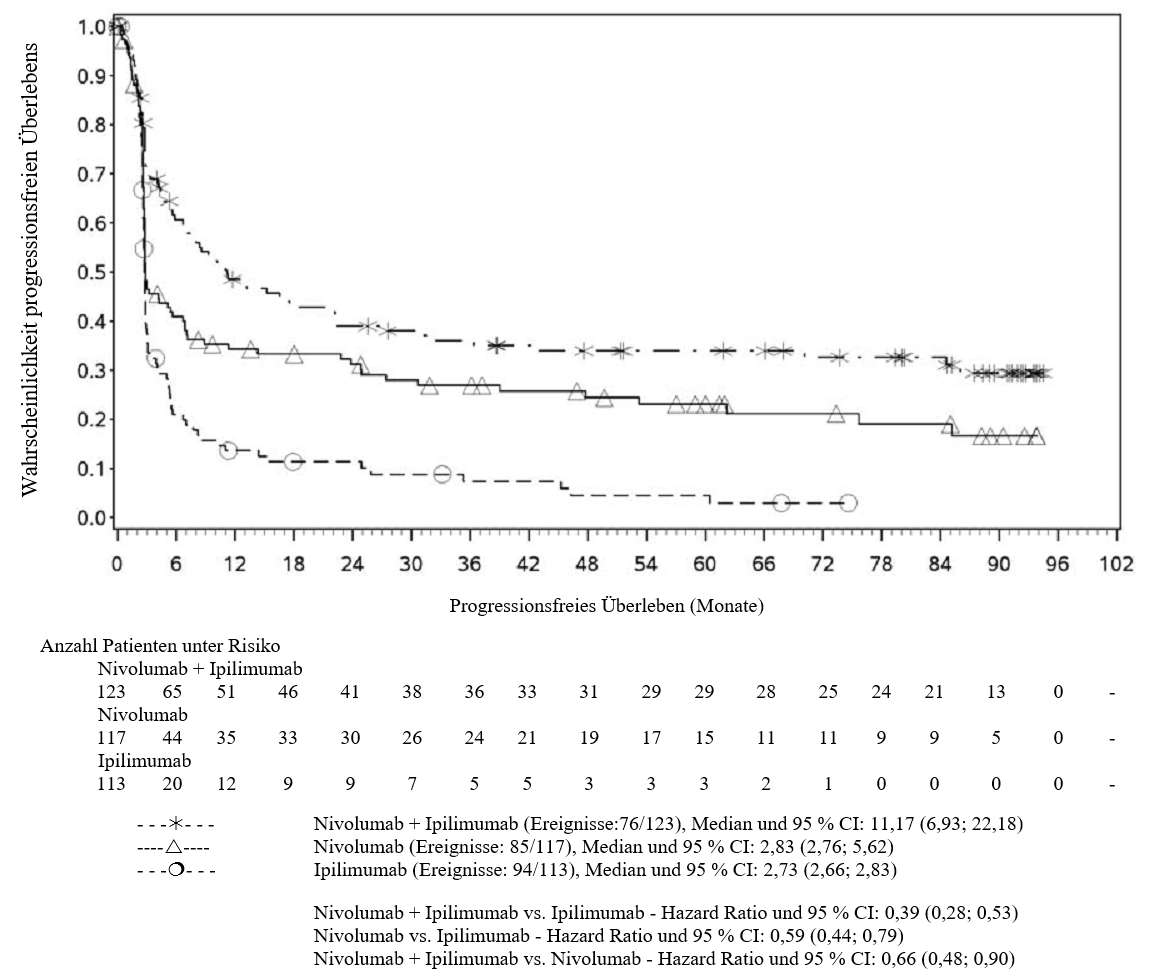
PD‑L1‑Expression ≥ 1 %
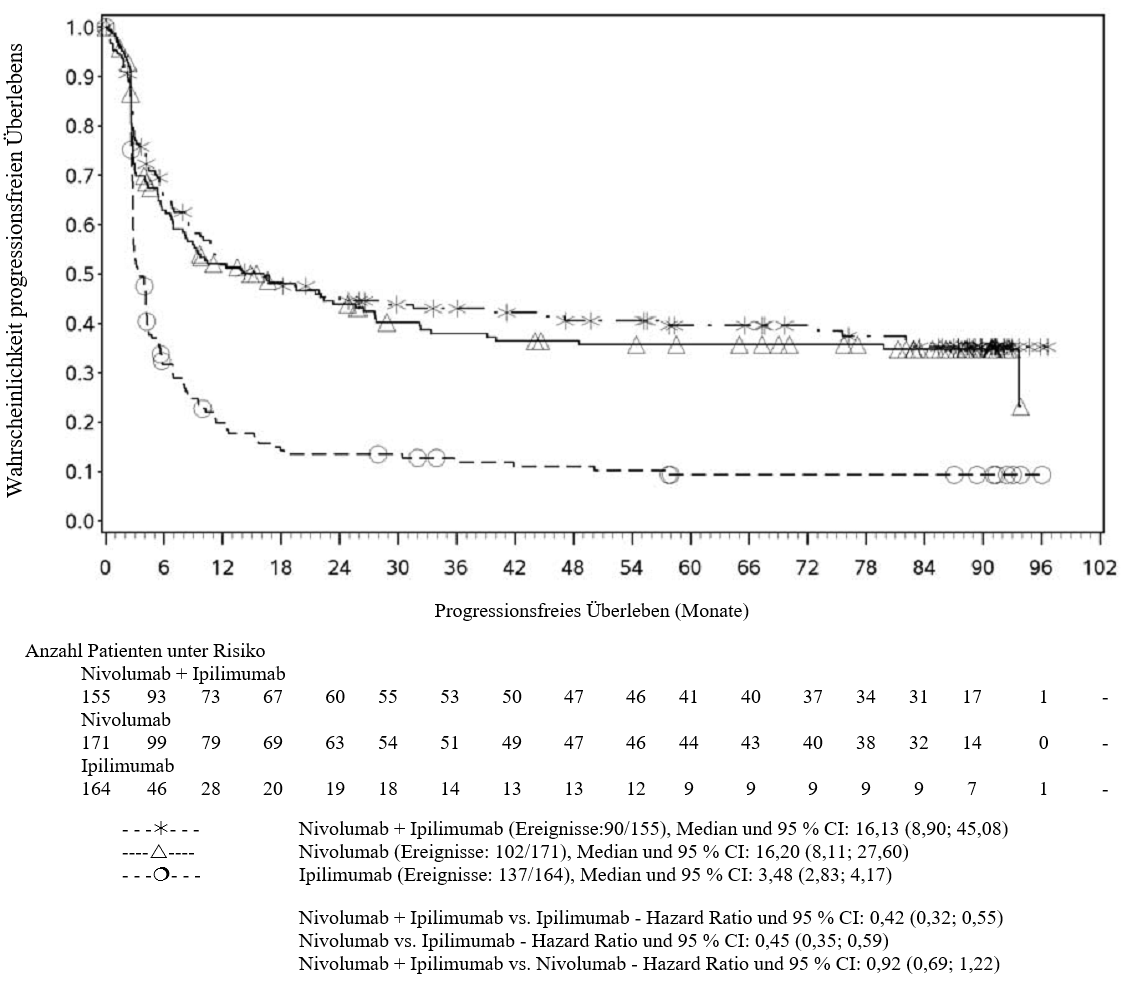
Die finale (primäre) OS‑Analyse wurde durchgeführt, nachdem alle Patienten eine minimale Nachbeobachtungszeit von 28 Monaten erreicht hatten. Nach 28 Monaten wurde das mediane OS in der Nivolumab‑Gruppe nicht erreicht verglichen mit 19,98 Monaten in der Ipilimumab‑Gruppe (HR = 0,63; 98 % CI: 0,48; 0,81; p‑Wert: < 0,0001). Das mediane OS wurde in der Patientengruppe, die mit der Kombination aus Nivolumab und Ipilimumab behandelt wurde, nicht erreicht. Im Vergleich zur Ipilimumab‑Gruppe beträgt die HR = 0,55 (98 % CI: 0,42; 0,72; p‑Wert < 0,0001).
Die OS‑Ergebnisse einer zusätzlichen deskriptiven Analyse nach einer minimalen Nachbeobachtungszeit von 90 Monaten waren konsistent mit den Ergebnissen der ursprünglichen primären Analyse. Die OS‑Ergebnisse dieser Nachbeobachtungsanalyse sind in Abbildung 8 (alle randomisierten Populationen), Abbildungen 9 und 10 (Tumor‑PD‑L1‑5‑%‑und‑1‑%‑Cut‑off) dargestellt.
Die OS‑Analyse war nicht darauf ausgerichtet, die nachfolgend erhaltenen Therapien zu erfassen. Anschließende systemische Therapien erhielten 36,0 % der Patienten, die die Kombination erhalten hatten, 49,1 % der Nivolumab‑Monotherapie‑Patienten und 66,3 % der Ipilimumab‑Patienten. Anschließende Immuntherapien (einschließlich Anti‑PD1‑Therapie, Anti‑CTLA‑4‑Antikörper oder andere Immuntherapien) erhielten 19,1 % der Patienten, die die Kombination erhalten hatten, 34,2 % der Nivolumab‑Monotherapie‑Patienten und 48,3 % der Ipilimumab‑Patienten.
Abbildung 8: Gesamtüberleben (CA209067) ‑ Minimale Nachbeobachtungszeit 90 Monate
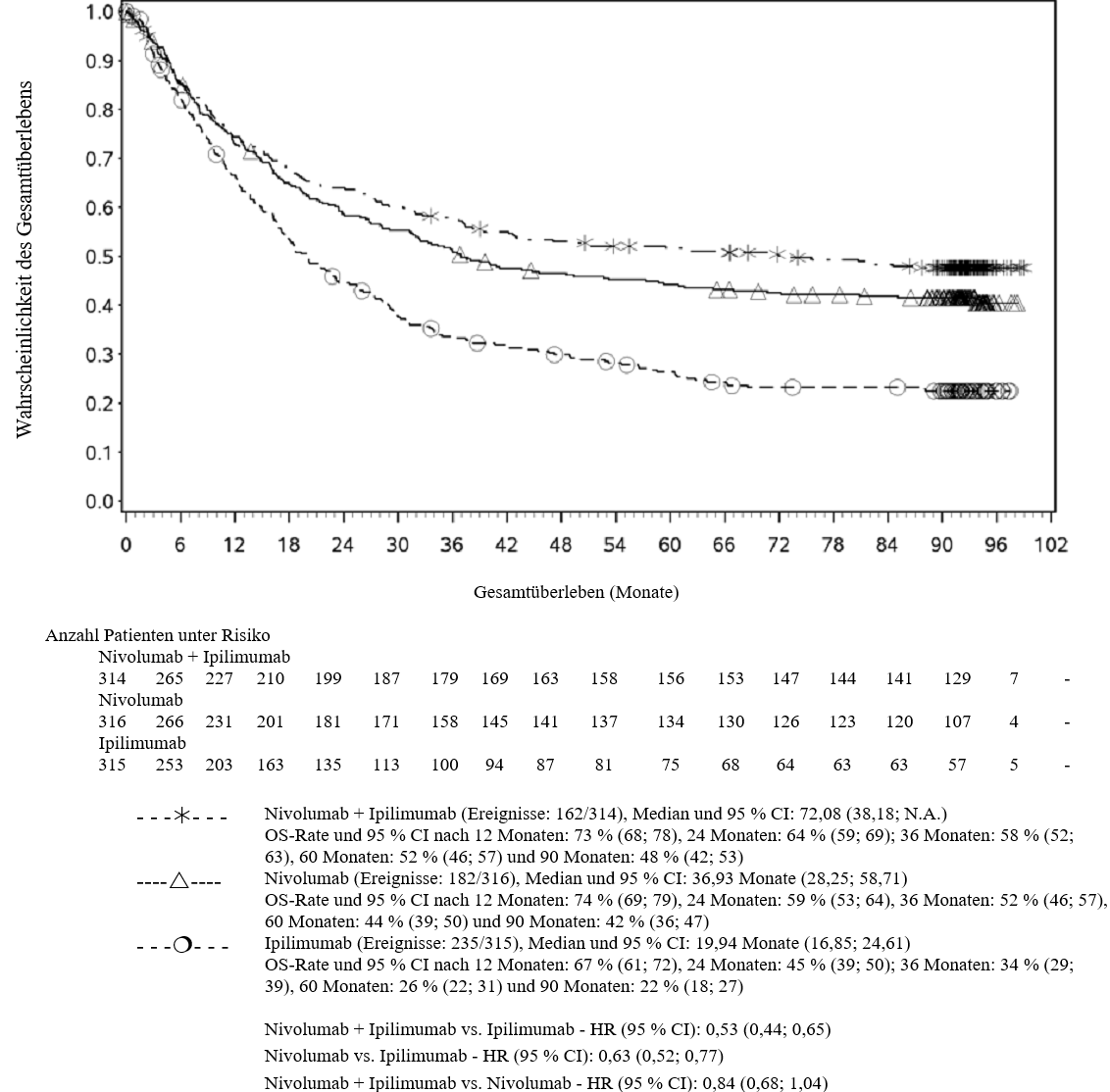
Abbildung 9: Gesamtüberleben nach PD‑L1‑Expression: 5‑%‑Cut‑off (CA209067) ‑ Minimale Nachbeobachtungszeit 90 Monate
PD‑L1‑Expression < 5 %
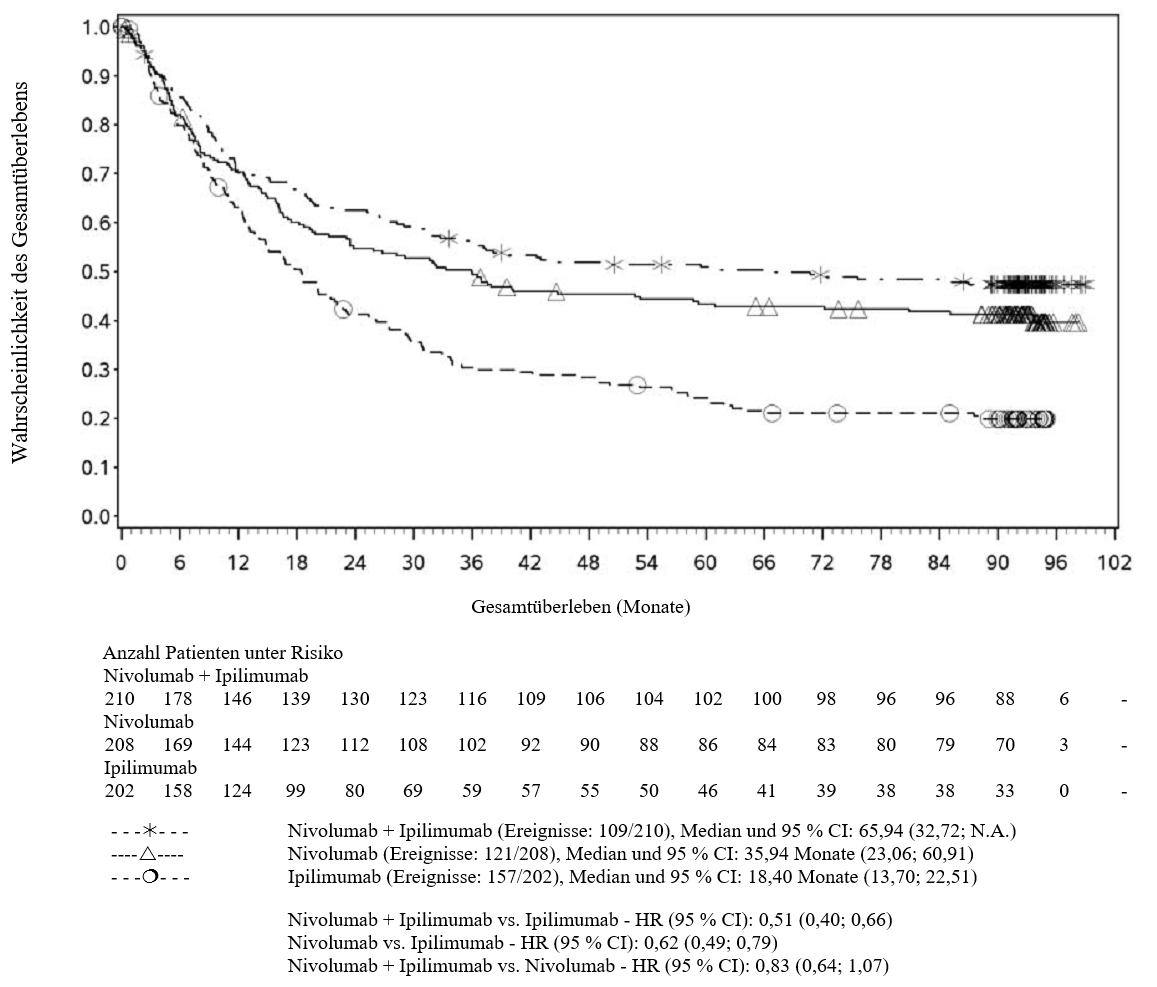
PD‑L1‑Expression ≥ 5 %
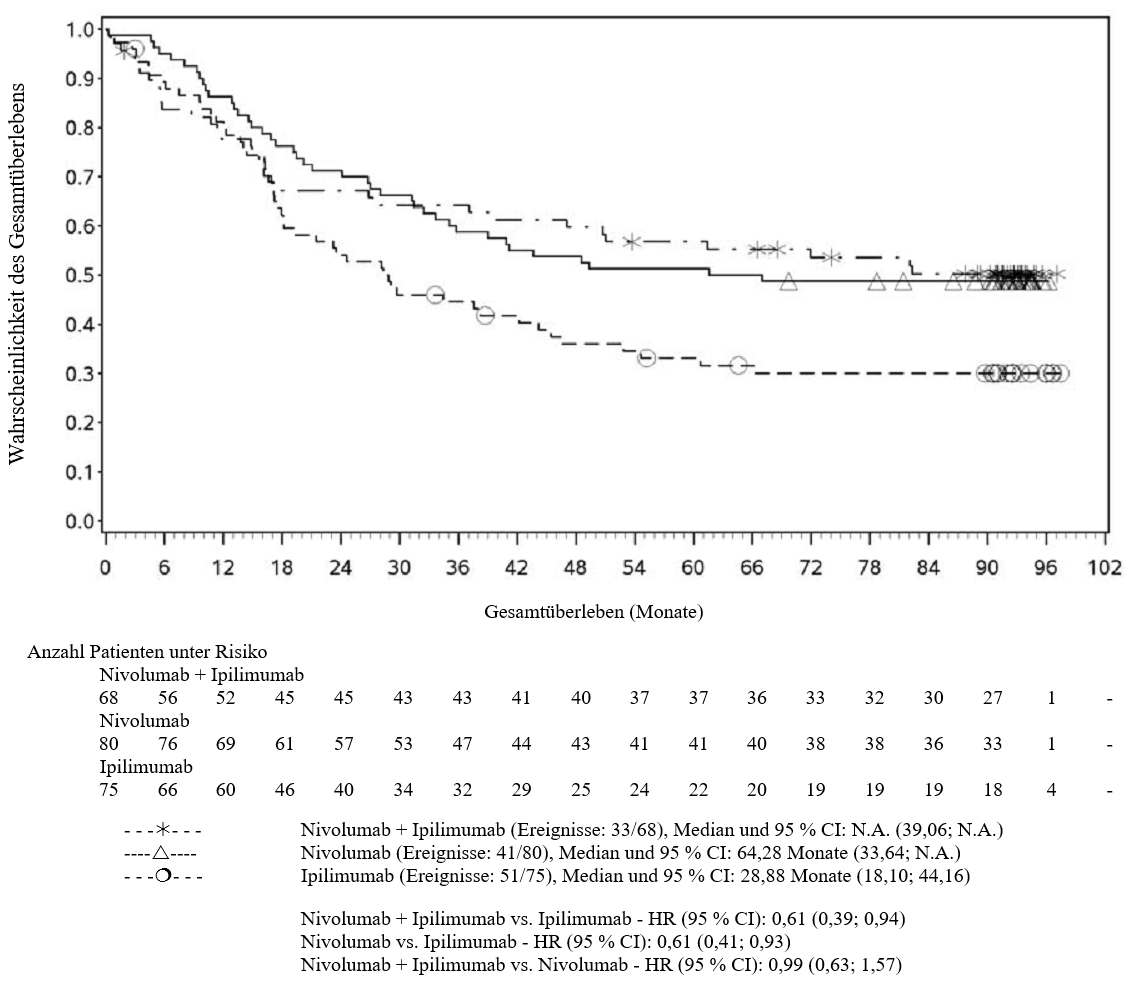
Abbildung 10: Gesamtüberleben nach PD‑L1‑Expression: 1‑%‑Cut‑off (CA209067) ‑ Minimale Nachbeobachtungszeit 90 Monate
PD‑L1‑Expression < 1 %
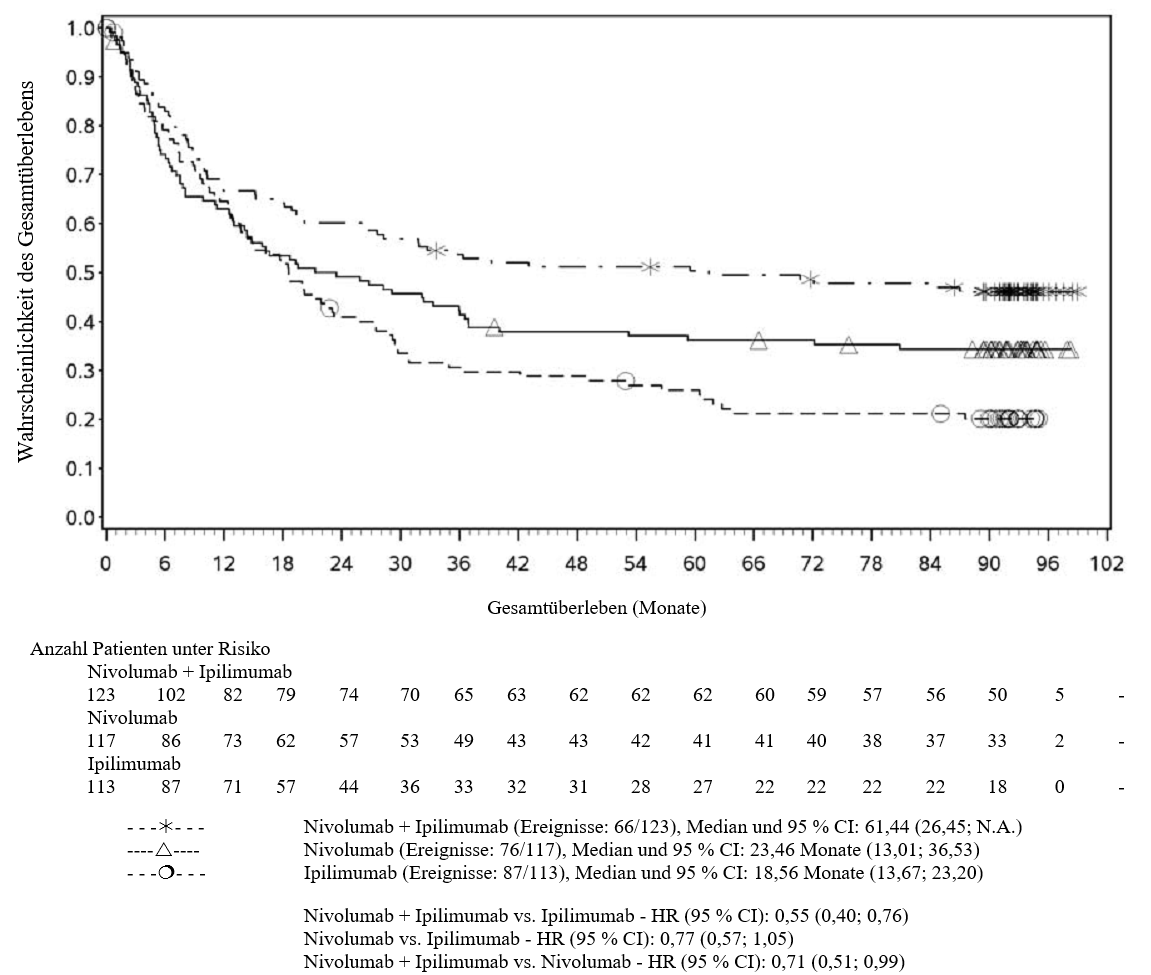
PD‑L1‑Expression ≥ 1 %
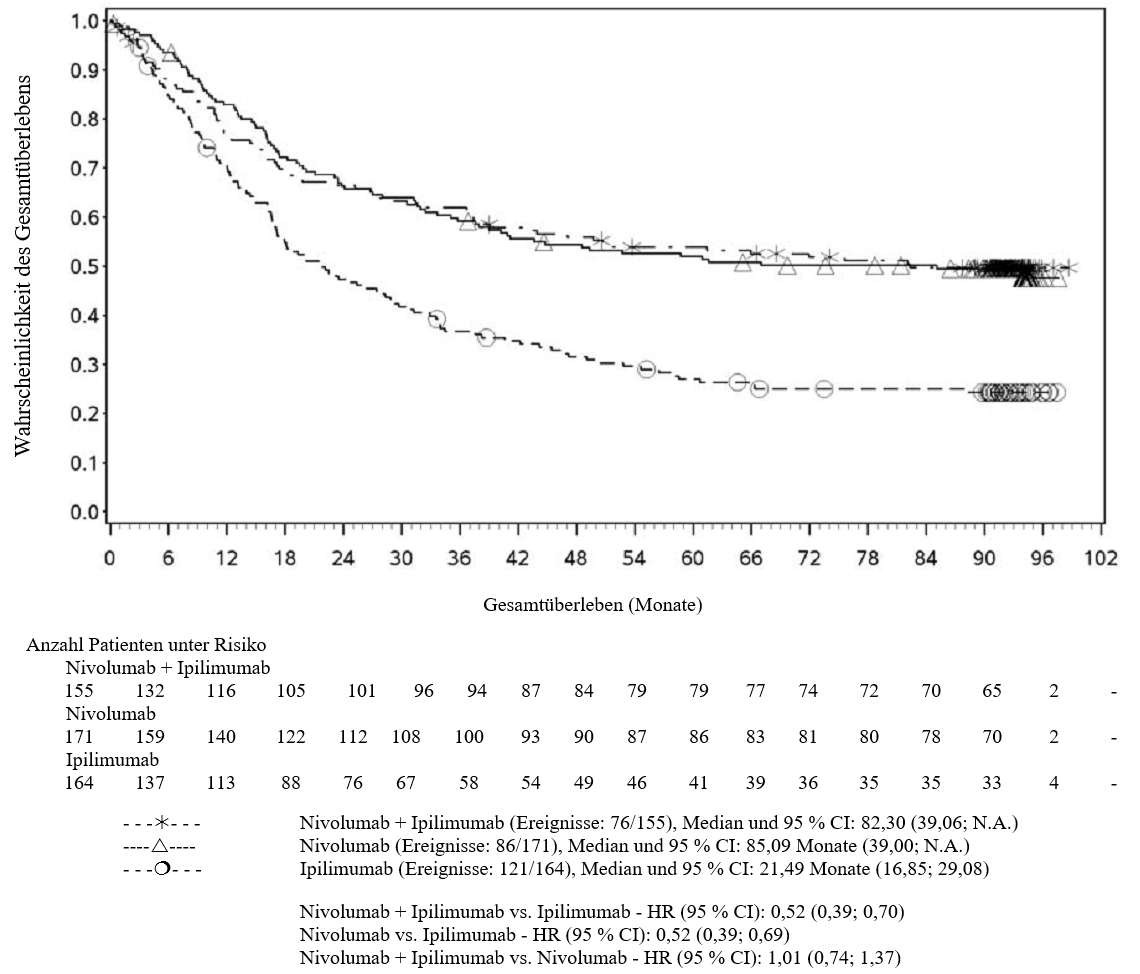
Minimale Nachbeobachtungszeit für die ORR‑Analyse waren 90 Monate. Das Ansprechen ist in Tabelle 20 zusammengefasst.
Tabelle 20: Objektives Ansprechen (CA209067)
Nivolumab + Ipilimumab |
Nivolumab |
Ipilimumab |
|
Objektives Ansprechen |
183 (58 %) |
142 (45 %) |
60 (19 %) |
(95 % CI) |
(52,6; 63,8) |
(39,4; 50,6) |
(14,9; 23,8) |
Odds Ratio (vs. Ipilimumab) |
6,35 |
3,5 |
|
(95 % CI) |
(4,38; 9,22) |
(2,49; 5,16) |
|
Vollständiges Ansprechen (Complete Response = CR) |
71 (23 %) |
59 (19 %) |
19 (6 %) |
Teilweises Ansprechen (Partial Response = PR) |
112 (36 %) |
83 (26 %) |
41 (13 %) |
Stabile Erkrankung (Stable Disease = SD) |
38 (12 %) |
29 (9 %) |
69 (22 %) |
Ansprechdauer |
|||
Mediane Zeitspanne, Monate |
N.A. |
90,8 |
19,3 |
Anteil ≥ 12 Monate Ansprechdauer |
68 % |
73 % |
44 % |
Anteil ≥ 24 Monate Ansprechdauer |
58 % |
63 % |
30 % |
ORR (95 % CI) bei Tumor‑PD‑L1‑Expression | |||
< 5 % |
56 % (48,7; 62,5) |
43 % (36; 49,8) |
18 % (12,8; 23,8) |
≥ 5 % |
72 % (59,9; 82,3) |
59 % (47,2; 69,6) |
21 % (12,7; 32,3) |
< 1 % |
54 % (44,4; 62,7) |
36 % (27,2; 45,3) |
18 % (11,2; 26,0) |
≥ 1 % |
65 % (56,4; 72) |
55 % (47,2; 62,6) |
20 % (13,7; 26,4) |
Patienten in beiden Nivolumab‑Armen zeigten einen signifikanten Nutzen bzgl. PFS und OS und ein größeres ORR verglichen mit Ipilimumab-Monotherapie. Die Resultate bezüglich des progressionsfreien Überlebens nach 18 Monaten Nachbeobachtungszeit und ORR‑ und OS‑Ergebnisse nach 28 Monaten Nachbeobachtungszeit waren in den verschiedenen Patienten‑Subgruppen konsistent, einschließlich bei Patienten mit unterschiedlichem ECOG‑Status, BRAF‑Mutationsstatus, M‑Stadium, Alter, Hirnmetastasen in der Anamnese und LDH‑Ausgangsspiegel. Diese Beobachtungen wurden auch mit den OS‑Ergebnissen nach einer minimalen Nachbeobachtungszeit von 90 Monaten beibehalten.
Bei den 131 Patienten, die die Kombinationstherapie aufgrund von Nebenwirkungen nach 28 Monaten Nachbeobachtungszeit abgebrochen haben, war die Ansprechrate 71 % (93/131). Von diesen 71 % erreichten 20 % (26/131) der Patienten ein vollständiges Ansprechen. Das mediane Gesamtüberleben wurde nicht erreicht.
Patienten in beiden Nivolumab‑Armen hatten ein größeres objektives Ansprechen als Patienten im Ipilimumab‑Arm, unabhängig vom PD‑L1‑Expressionsstatus. Nach 90 Monaten Nachbeobachtungszeit war die objektive Ansprechrate für die Kombination aus Nivolumab und Ipilimumab über alle Tumor‑PD‑L1‑Expressionsgrade höher als für die Nivolumab‑Monotherapie (siehe Tabelle 19), wobei die beste Gesamtansprechrate für das vollständige Ansprechen mit einer verbesserten Überlebensrate korrelierte.
Nach 90 Monaten Nachbeobachtungszeit betrug die mediane Ansprechdauer bei Patienten mit Tumor‑PD‑L1‑Expressionsstatus ≥ 5 % 78,19 Monate im Kombinations‑Arm (Spanne: 18,07 ‑ N.A.) und 77,21 Monate im Nivolumab‑Monotherapie‑Arm (Spanne: 26,25 ‑ N.A.). Sie betrug 31,28 Monate (Spanne: 6,08 ‑ N.A.) im Ipilimumab‑Arm. Bei einer Tumor‑PD‑L1‑Expression < 5 % wurde die mediane Ansprechdauer im Kombinations‑Arm (Spanne: 61,93 ‑ N.A.) nicht erreicht und betrug 90,84 Monate im Nivolumab‑Monotherapie‑Arm (Spanne: 50,43 ‑ N.A.). Sie betrug 19,25 Monate im Ipilimumab‑Monotherapie‑Arm (Spanne: 5,32 ‑ 47,44).
Bezüglich der relevanten Endpunkte Tumoransprechen, PFS und OS konnte kein klarer Grenzwert für die PD‑L1‑Expression verlässlich definiert werden. Ergebnisse von exploratorischen multivarianten Analysen zeigten, dass auch andere Patienten‑ und Tumorcharakteristika (z. B. ECOG‑Status, M‑Stadium, Ausgangs‑LDH, BRAF‑Mutationsstatus, PD‑L1‑Status und Geschlecht) zum Überlebensresultat beitragen könnten.
Wirksamkeit bei BRAF‑Status:
BRAF‑V600‑Mutation‑positive und BRAF‑Wildtyp‑Patienten, welche zu Nivolumab in Kombination mit Ipilimumab randomisiert wurden, hatten nach 90 Monaten Nachbeobachtungszeit ein medianes PFS von 16,76 Monaten (95 % CI: 8,28; 32,0) bzw. 11,7 Monaten (95 % CI: 7,0; 19,32) während die in den Nivolumab‑Monotherapie‑Arm randomisierten Patienten ein medianes PFS von 5,62 Monaten (95 % CI: 2,79; 9,46) bzw. 8,18 Monaten (95 % CI: 5,13; 19,55) hatten. BRAF‑V600‑Mutations‑positive bzw. BRAF‑Wildtyp‑Patienten, welche zur Ipilimumab‑Monotherapie randomisiert wurden, hatten ein medianes PFS von 3,09 Monaten (95 % CI: 2,79; 5,19) bzw. 2,83 Monaten (95 % CI: 2,76; 3,06).
Nach 90 Monaten Nachbeobachtungszeit hatten BRAF‑V600‑Mutations‑positive bzw. BRAF‑Wildtyp‑Patienten, welche zu Nivolumab in Kombination mit Ipilimumab randomisiert wurden, ein ORR von 67,0 % (95 % CI: 57,0; 75,9; n = 103) bzw. 54,0 % (95 % CI: 47,1; 60,9; n = 211) während die in den Nivolumab‑Monotherapie‑Arm randomisierten Patienten ein ORR von 37,87 % (95 % CI: 28,2; 48,1; n = 98) bzw. 48,2 % (95 % CI: 41,4; 55,0; n = 218) hatten. BRAF‑V600‑Mutations‑positive bzw. BRAF‑Wildtyp‑Patienten, welche zur Ipilimumab‑Monotherapie randomisiert wurden, hatten ein ORR von 23,0 % (95 % CI: 15,2; 32,5; n = 100) bzw. 17,2 % (95 % CI: 12,4; 22,9; n = 215).
Nach 90 Monaten Nachbeobachtungszeit wurde für BRAF‑V600‑Mutations‑positive Patienten das mediane OS im Kombinations‑Arm nicht erreicht und betrug 45,5 Monate im Nivolumab‑Monotherapie‑Arm. Das mediane OS für BRAF‑V600‑Mutations‑positive Patienten im Ipilimumab‑Monotherapie‑Arm betrug 24,6 Monate. Für BRAF‑Wildtyp‑Patienten betrug das mediane OS 39,06 Monate im Kombinations‑Arm, 34,37 Monate im Nivolumab‑Monotherapie‑Arm und 18,5 Monate im Ipilimumab‑Monotherapie‑Arm. Die Hazard Ratios des Gesamtüberlebens waren im Nivolumab‑in‑Kombination‑mit‑Ipilimumab‑Arm versus Nivolumab‑Monotherapie‑Arm für BRAF‑V600‑Mutations‑positive Patienten 0,66 (95 % CI: 0,44; 0,98) und für BRAF‑Wildtyp‑Patienten 0,95 (95 % CI: 0,74; 1,22).
Intravenöse Formulierung
Randomisierte Phase‑II‑Studie mit Nivolumab in Kombination mit Ipilimumab und Ipilimumab (CA209069)
Die Studie CA209069 war eine randomisierte, doppelblinde Phase‑II‑Studie, in der Nivolumab mit Ipilimumab im Vergleich zu Ipilimumab allein bei 142 Patienten mit fortgeschrittenem (nicht‑resezierbarem oder metastasiertem) Melanom evaluiert wurde. Die Einschlusskriterien dieser Studie waren denen der Studie CA209067 ähnlich und die primäre Analyse erfolgte bei Patienten mit BRAF‑Wildtyp‑Melanom (77 % der Patienten). Die vom Prüfarzt bewertete Ansprechrate betrug 61 % (95 % CI: 48,9; 72,4) im Kombinations‑Arm (n = 72) versus 11 % (95 % CI: 3,0; 25,4) im Ipilimumab‑Monotherapie‑Arm (n = 37). Die geschätzten OS‑Raten nach 2 bzw. 3 Jahren betrugen 68 % (95 % CI: 56; 78) bzw. 61 % (95 % CI: 49; 71) für die Kombination (n = 73) und 53 % (95 % CI: 36; 68) bzw. 44 % (95 % CI: 28; 60) für Ipilimumab‑Monotherapie (n = 37).
Nicht‑kleinzelliges Lungenkarzinom
Neoadjuvante Behandlung des NSCLC
Intravenöse Formulierung
Randomisierte, offene Phase‑III‑Studie zu Nivolumab in Kombination mit platinbasierter Chemotherapie vs. platinbasierte Chemotherapie (CA209816)
Die Sicherheit und Wirksamkeit von Nivolumab in Kombination mit platinbasierter Chemotherapie für 3 Zyklen wurde in einer randomisierten, offenen Phase‑III‑Studie (CA209816) untersucht. In die Studie wurden Patienten mit einem ECOG‑Performance‑Status von 0 oder 1, messbarer Erkrankung (gemäß RECIST, Version 1.1), deren Tumoren resezierbar waren, und mit einem histologisch bestätigten NSCLC des Stadiums IB (≥ 4 cm), II oder IIIA (gemäß 7. Auflage der Staging‑Kriterien der AJCC/Union for International Cancer Control [UICC]) eingeschlossen.
Die folgenden Auswahlkriterien definieren Patienten mit hohem Rezidivrisiko, die in das Anwendungsgebiet eingeschlossen sind und die eine Patientenpopulation mit einer Erkrankung des Stadiums II bis IIIA gemäß der 7. Auflage der Staging‑Kriterien der AJCC/UICC widerspiegeln: Alle Patienten mit einer Tumorgröße ≥ 5 cm; alle Patienten mit N1‑ oder N2‑Erkrankung (unabhängig von der Größe des Primärtumors); Patienten mit multiplen Tumorknoten entweder im selben Lungenlappen oder in verschiedenen ipsilateralen Lungenlappen; Patienten mit Tumoren, die invasiv in thorakale Strukturen eindringen (direkte Invasion in viszerale Pleura, parietale Pleura, Brustwand, Diaphragma, Nervus phrenicus, mediastinale Pleura, parietales Perikard, Mediastinum, Herz, große Gefäße, Trachea, Nervus laryngeus recurrens, Ösophagus, Wirbelkörper, Carina); oder Tumoren, die den Hauptbronchus befallen; oder Tumoren, die mit Atelektase oder obstruktiver Pneumonitis verbunden sind, die sich bis in die Hilusregion ausdehnt oder die gesamte Lunge betrifft.
In die Studie wurden keine Patienten mit N2‑Status und Tumoren mit zusätzlicher Invasion in das Mediastinum, Herz, große Gefäße, Luftröhre, Nervus laryngeus recurrens, Ösophagus, Wirbelkörper, Carina oder mit separaten Tumorknoten in einem anderen ipsilateralen Lungenlappen eingeschlossen.
Patienten mit nicht‑resezierbarem oder metastasiertem NSCLC, bekannten EGFR‑Mutationen oder ALK‑Translokationen (ein Test auf EGFR‑Mutationen oder ALK‑Translokationen war bei Studienbeginn nicht obligatorisch), peripherer Neuropathie von Grad 2 oder höher, aktiver Autoimmunerkrankung oder mit einer Erkrankung, die eine Behandlung mit einer systemischen Immunsuppression erfordert, waren von der Studie ausgeschlossen. Die Randomisierung wurde nach Tumor‑PD‑L1‑Expressionslevel (≥ 1 % vs. < 1 % oder nicht‑quantifizierbar), Krankheitsstadium (IB/II vs. IIIA) und Geschlecht (männlich vs. weiblich) stratifiziert. Patienten wurden unabhängig vom PD‑L1‑Status ihres Tumors eingeschlossen. Die Tumor‑PD‑L1‑Expression wurde mithilfe des PD‑L1‑IHC‑28‑8‑pharmDx‑Tests bestimmt.
Insgesamt wurden 358 Patienten entweder zu Nivolumab in Kombination mit platinbasierter Chemotherapie (n = 179) oder zu platinbasierter Chemotherapie (n = 179) randomisiert. Patienten im Nivolumab‑plus‑Chemotherapie‑Arm erhielten Nivolumab 360 mg intravenös über 30 Minuten in Kombination mit platinbasierter Chemotherapie alle 3 Wochen für bis zu 3 Zyklen. Patienten im Chemotherapie‑Arm erhielten eine platinbasierte Chemotherapie alle 3 Wochen für bis zu 3 Zyklen. Die platinbasierte Chemotherapie bestand nach Wahl des Prüfarztes aus Paclitaxel 175 mg/m2 oder 200 mg/m2 und Carboplatin AUC 5 oder AUC 6 (alle Histologien); Pemetrexed 500 mg/m2 und Cisplatin 75 mg/m2 (nicht‑plattenepitheliale Histologie); oder Gemcitabin 1 000 mg/m2 oder 1 250 mg/m2 und Cisplatin 75 mg/m2 (plattenepitheliale Histologie). Im Chemotherapie-Arm bestanden als zwei zusätzliche Therapieoptionen Vinorelbin 25 mg/m2 oder 30 mg/m2 und Cisplatin 75 mg/m2; oder Docetaxel 60 mg/m2 oder 75 mg/m2 und Cisplatin 75 mg/m2 (alle Histologien).
Tumorbeurteilungen wurden bei Studienbeginn, innerhalb von 14 Tagen nach der Operation, 2 Jahre lang nach der Operation alle 12 Wochen, danach 3 Jahre lang alle 6 Monate und dann 5 Jahre lang jährlich bis zum Rezidiv oder zur Progression der Erkrankung durchgeführt. Die primären Wirksamkeitskriterien waren das ereignisfreie Überleben (Event-Free Survival = EFS) auf Basis der BICR-Beurteilung und die pathologische Komplettremission (pathological Complete Response = pCR) durch eine verblindete unabhängige pathologische Beurteilung (Blinded Independent Pathology Review = BIPR). Das OS war ein wichtiges sekundäres Wirksamkeitskriterium, und die explorativen Endpunkte umfassten die Durchführbarkeit der Operation.
Die bei Studieneintritt vorliegenden Merkmale der Patienten waren im Allgemeinen über die Behandlungsgruppen hinweg gleich verteilt. Das mediane Alter betrug 65 Jahre (Spanne: 34 ‑ 84); 51 % der Patienten waren ≥ 65 Jahre und 7 % waren ≥ 75 Jahre, 50 % der Patienten waren Asiaten, 47 % waren Kaukasier und 71 % waren männlich. Der Ausgangs‑ECOG‑Performance‑Status war 0 (67 %) oder 1 (33 %); 50 % der Patienten hatten eine PD‑L1‑Expression ≥ 1 % und 43 % eine PD‑L1‑Expression < 1 %; 5 % hatten eine Erkrankung im Stadium IB, 17 % im Stadium IIA, 13 % im Stadium IIB und 64 % im Stadium IIIA; 51 % wiesen eine plattenepitheliale Histologie und 49 % eine nicht‑plattenepitheliale Histologie auf; 89 % waren früher oder sind derzeit Raucher. Eine definitive Operation erfolgte bei 83 % der Patienten im Nivolumab‑plus‑Chemotherapie‑Arm und bei 75 % der Patienten im Chemotherapie‑Arm. 14,8 % der Patienten im Nivolumab‑plus‑Chemotherapie‑Arm erhielten eine adjuvante systemische Therapie und 25 % der Patienten im Chemotherapie‑Arm.
Bei der finalen pCR‑Analyse und der vordefinierten EFS‑Zwischenanalyse (mit minimaler Nachbeobachtungszeit von 21 Monaten) über alle randomisierten Patienten wurde im Vergleich zur Chemotherapie allein eine statistisch signifikante Verbesserung von pCR und EFS bei Patienten nachgewiesen, die zu Nivolumab in Kombination mit Chemotherapie randomisiert wurden. Die pCR‑Ansprechrate betrug 24 % im Nivolumab‑plus‑Chemotherapie‑Arm und 2,2 % im Chemotherapie‑Arm (Differenz des pCR 21,6; 99 % CI: 13,0; 30,3; Odds Ratio des pCR 13,9; 99 % CI: 3,49; 55,75; stratifizierter p‑Wert < 0,0001). Das mediane EFS war 31,6 Monate im Nivolumab‑plus‑Chemotherapie‑Arm und 20,8 Monate im Chemotherapie‑Arm (HR = 0,63; 97,38 % CI: 0,43; 0,91; Log‑Rank‑stratifizierter p‑Wert 0,0052). Bei einer präspezifizierten Zwischenanalyse war die HR für das OS (minimale Nachbeobachtungszeit von 21 Monaten) 0,57 (99,67 % CI: 0,30; 1,07) für Nivolumab in Kombination mit Chemotherapie vs. Chemotherapie.
Bei der finalen OS‑Analyse mit einer minimalen Nachbeobachtungszeit von 59,9 Monaten war die HR für OS 0,72 (95,18 % CI: 0,52; 1,00), mit einem statistisch signifikanten p‑Wert von 0,0479 (stratifizierter Log‑Rank‑Test) für Nivolumab in Kombination mit Chemotherapie vs. Chemotherapie.
Explorative Subgruppenanalyse nach Tumor‑PD‑L1‑Expression und Krankheitsstadium
Die wichtigsten Wirksamkeitsergebnisse für die Subgruppe von Patienten mit Tumor‑PD‑L1‑Expression ≥ 1% und Krankheitsstadium II‑IIIA einer explorativen Analyse mit einer minimalen Nachbeobachtungszeit von 32,9 Monaten sind in Tabelle 21 zusammengefasst.
Tabelle 21: Wirksamkeitsergebnisse bei Patienten mit Tumor PD‑L1 ≥ 1% und Krankheitsstadium II‑IIIA* (CA209816)
Nivolumab + Chemotherapie |
Chemotherapie |
||
Ereignisfreies Überleben nach BICR | |||
Ereignisse |
22 (27,2 %) |
39 (45,3 %) |
|
Hazard Ratioa |
0,49 |
||
(95 % CI) |
(0,29; 0,83) |
||
Median (Monate)b |
NR |
26,71 |
|
Pathologische Komplettremission nach BIPR | |||
Ansprechen |
26 (32,1 %) |
2 (2,3 %) |
|
(95 % CI)c |
(22,2; 43,4) |
(0,3; 8,1) |
|
Differenz des pCR (95 % CI)d |
29,8 % (19,0; 40,7) |
||
a Mit einem nicht stratifizierten Cox‑Modell für proportionale Hazards berechnet. | |||
Die Kaplan‑Meier‑Kurven für EFS bei der Subgruppe von Patienten mit Tumor‑PD‑L1-Expression ≥ 1% und Krankheitsstadium II‑IIIA mit einer minimalen Nachbeobachtungszeit von 32,9 Monaten sind in Abbildung 11 dargestellt.
Abbildung 11: Kaplan‑Meier‑Kurven für EFS bei Patienten mit Tumor PD‑L1 ≥ 1% und Krankheitsstadium II‑IIIA (CA209816)
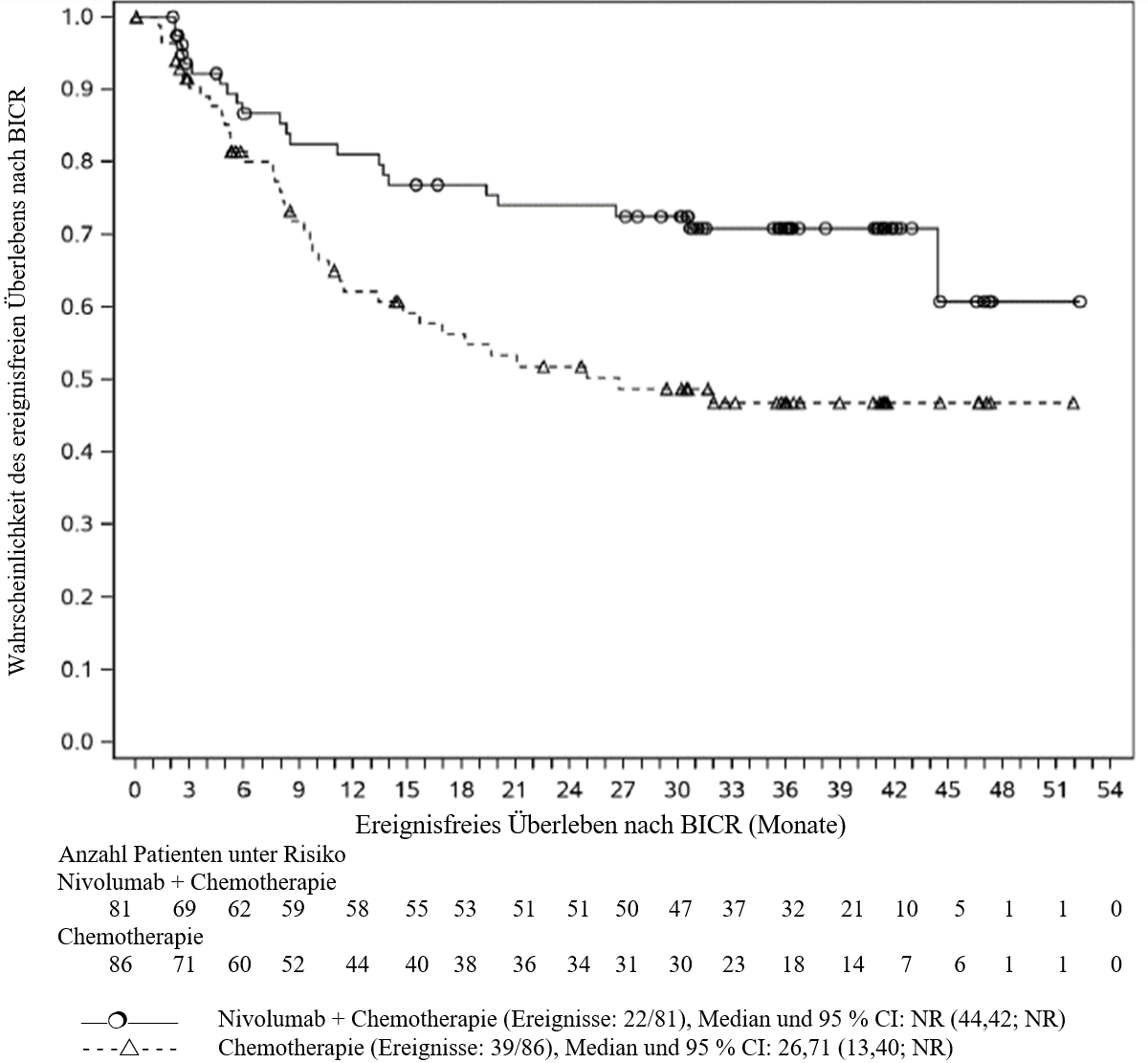
Basierend auf Datenschnitt: 06‑Sep‑2022, minimale Nachbeobachtungszeit von 32,9 Monaten
Zum Zeitpunkt der aktualisierten EFS‑Analyse wurde eine Zwischenanalyse für das OS durchgeführt (minimale Nachbeobachtungszeit von 32,9 Monaten). Die explorative, deskriptive HR für OS bei Patienten mit Tumor PD‑L1-Expression ≥ 1% und Krankheitsstadium II‑IIIA war 0,43 (95 % CI: 0,22; 0,83) für Nivolumab in Kombination mit Chemotherapie vs. Chemotherapie.
Bei der finalen Analyse (minimale Nachbeobachtungszeit von 59,9 Monaten) war die HR für OS bei Patienten mit Tumor PD‑L1-Expression ≥ 1% und Krankheitsstadium II‑IIIA 0,59 (95 % CI: 0,35; 0,98) für Nivolumab in Kombination mit Chemotherapie vs. Chemotherapie. Die korrespondierenden Kaplan‑Meier‑Kurven für OS sind in Abbildung 12 dargestellt.
Abbildung 12: Kaplan‑Meier‑Kurven für OS bei Patienten mit Tumor PD‑L1 ≥ 1 % und Krankheitsstadium II‑IIIA (CA209816)
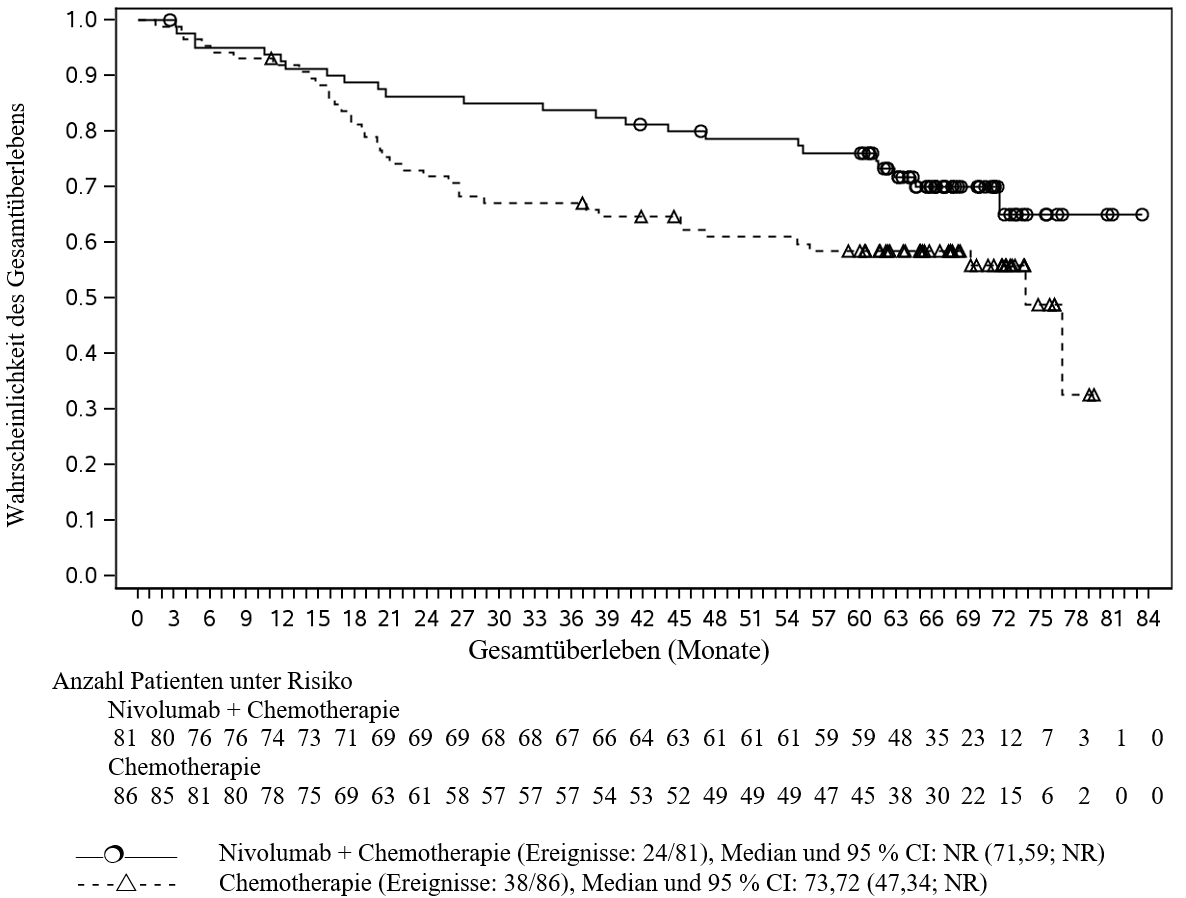
Basierend auf Datenschnitt: 23-Jan-2025, minimale Nachbeobachtungszeit von 59,9 Monaten
Neoadjuvante und adjuvante Behandlung des NSCLC
Intravenöse Formulierung
Randomisierte, doppelblinde Phase-III-Studie zur neoadjuvanten Behandlung mit Nivolumab in Kombination mit platinbasierter Chemotherapie vs. platinbasierte Chemotherapie und adjuvanter Nivolumab‑Monotherapie vs. Placebo (CA20977T)
Die Sicherheit und Wirksamkeit von Nivolumab in Kombination mit platinbasierter Chemotherapie für 4 Zyklen, gefolgt von einer Nivolumab‑Monotherapie wurden in einer randomisierten, doppelblinden Studie (CA20977T) untersucht. In die Studie wurden Patienten mit einem ECOG-Performance-Status von 0 oder 1, deren Tumoren resezierbar waren, und mit einem vermuteten oder histologisch bestätigtem NSCLC des Stadiums IIA (> 4 cm) bis IIIB (T3‑T4 N2) eingeschlossen (gemäß der 8. Auflage des American Joint Committee on Cancer (AJCC) Staging Manual). Der Einschluss von Patienten erfolgte unabhängig von ihrem Tumor-PD‑L1-Status.
Die folgenden Auswahlkriterien definieren Patienten mit hohem Rezidivrisiko, die in das Anwendungsgebiet eingeschlossen sind und die eine Patientenpopulation mit einer Erkrankung des Stadiums IIA bis IIIB gemäß der 8. Auflage der Staging‑Kriterien der AJCC/UICC widerspiegeln: Alle Patienten mit einer Tumorgröße > 4 cm; alle Patienten mit N1- oder N2-Erkrankung (unabhängig von der Größe des Primärtumors); Patienten mit multiplen Tumorknoten entweder im selben Lungenlappen oder in verschiedenen ipsilateralen Lungenlappen; Patienten mit Tumoren, die invasiv in thorakale Strukturen eindringen (direkte Invasion in viszerale Pleura, parietale Pleura, Brustwand, Diaphragma, Nervus phrenicus, mediastinale Pleura, parietales Perikard, Mediastinum, Herz, große Gefäße, Trachea, Nervus laryngeus recurrens, Ösophagus, Wirbelkörper, Carina); oder Tumoren, die den Hauptbronchus befallen; oder Tumoren, die mit Atelektase oder obstruktiver Pneumonitis verbunden sind, die sich bis in die Hilusregion ausdehnt oder die gesamte Lunge betrifft.
Patienten mit nicht resezierbarem oder metastasiertem NSCLC, EGFR-Mutationen oder bekannten ALK-Translokationen (ein Test auf ALK-Translokationen war bei Studienbeginn nicht obligatorisch), Hirnmetastasen, peripherer Neuropathie von Grad 2 oder höher, interstitieller Lungenerkrankung oder aktiver, nicht infektiöser Pneumonitis (symptomatisch und/oder Behandlung erforderlich), aktiver Autoimmunerkrankung oder mit einer Erkrankung, die eine Behandlung mit einer systemischen Immunsuppression erfordert, waren von der Studie ausgeschlossen.
Insgesamt wurden 461 Patienten entweder zu Nivolumab in Kombination mit platinbasierter Chemotherapie gefolgt von einer Nivolumab‑Monotherapie (n = 229) oder zu platinbasierter Chemotherapie gefolgt von Placebo (n = 232) randomisiert. In der neoadjuvanten Phase erhielten die Patienten entweder 360 mg Nivolumab intravenös über 30 Minuten und eine platinbasierte Kombinationschemotherapie alle 3 Wochen oder Placebo und eine platinbasierte Kombinationschemotherapie alle 3 Wochen bis zur Progression der Erkrankung oder bis zu nicht akzeptabler Toxizität, für bis zu 4 Zyklen. Die Patienten in beiden Behandlungsarmen konnten eine postoperative Strahlentherapie (Post-Operative Radiation Therapy, PORT) als Behandlungsstandard erhalten. In der adjuvanten Phase erhielten die Patienten innerhalb von 90 Tagen nach der Operation entweder 480 mg Nivolumab intravenös über 30 Minuten alle 4 Wochen oder Placebo alle 4 Wochen bis zur Progression der Erkrankung, bis zum Rezidiv oder bis zu nicht akzeptabler Toxizität, für bis zu 13 Zyklen. Die platinbasierte Chemotherapie bestand aus Paclitaxel 175 mg/m2 oder 200 mg/m2 und Carboplatin AUC 5 oder AUC 6 (alle Histologien); Pemetrexed 500 mg/m2 und Cisplatin 75 mg/m2 oder Carboplatin AUC 5 oder AUC 6 (nicht-plattenepitheliale Histologie); oder Cisplatin 75 mg/m2 und Docetaxel 75 mg/m2 (plattenepitheliale Histologie).
Stratifikationsfaktoren für die Randomisierung waren Tumor-PD‑L1‑Expressionswerte (≥ 1 % vs. < 1 % vs. unbestimmt/nicht auswertbar), Krankheitsstadium (Stadium II vs. Stadium III) und Tumorhistologie (plattenepithelial vs. nicht-plattenepithelial). Tumor‑PD‑L1‑Expressionswerte wurden mittels PD‑L1‑IHC‑28‑8‑PharmDx‑Assay beurteilt. Tumorbeurteilungen wurden bei Studienbeginn, innerhalb von 14 Tagen nach der letzten Dosis der neoadjuvanten Behandlung und vor der Operation, innerhalb von 7 Tagen vor Beginn der adjuvanten Behandlung nach der Operation, alle 12 Wochen nach der ersten Dosis der adjuvanten Behandlung für 2 Jahre, dann alle 24 Wochen für bis zu 5 Jahre durchgeführt, bis ein Rezidiv oder die Progression der Erkrankung durch BICR bestätigt wurde.
Von den 442 Patienten in der Studie CA20977T hatten 256 (58 %) eine mittels PD‑L1‑IHC‑28‑8‑PharmDx Assay ermittelte PD‑L1‑Expression ≥ 1 %. Das mediane Alter betrug 66 Jahre (Spanne: 35 bis 86); 55 % der Patienten waren ≥ 65 Jahre und 7 % der Patienten waren ≥ 75 Jahre, 69 % der Patienten waren Kaukasier, 28 % waren Asiaten, 2 % waren Schwarze und 75 % waren männlich. Der Ausgangs-ECOG‑Performance‑Status war 0 (59 %) oder 1 (41 %); 36 % hatten eine Erkrankung im Stadium II und 63 % im Stadium III; 24 % waren N1 und 39 % N2; 25 % waren auf einem einzelnen Level und 14 % auf mehreren Leveln; 61 % wiesen Tumoren mit plattenepithelialer Histologie und 39 % mit nicht‑plattenepithelialer Histologie auf; 91 % waren früher oder sind derzeit Raucher.
Achtundsiebzig Prozent der Patienten im Arm mit neoadjuvantem Nivolumab in Kombination mit platinbasierter Kombinationschemotherapie gefolgt von adjuvantem Nivolumab hatten eine definitive Operation im Vergleich zu 77 % der Patienten im Arm mit neoadjuvantem Placebo und platinbasierter Kombinationschemotherapie gefolgt von Placebo. Etwa 5 % der Patienten in jedem Behandlungsarm erhielten eine PORT.
Der primäre Wirksamkeitsendpunkt war das ereignisfreie Überleben (Event-Free Survival = EFS) auf Grundlage der BICR-Beurteilung. Zusätzliche Wirksamkeitsendpunkte umfassten das Gesamtüberleben (Overall Survival = OS), die pathologische Komplettremission (pathologic Complete Response = pCR) und das bedeutende pathologische Ansprechen durch eine verblindete unabhängige pathologische Beurteilung (Blinded Independent Pathology Review = BIPR).
Bei einer präspezifizierten Zwischenanalyse mit allen randomisierten Patienten mit einer medianen Nachbeobachtungszeit von 25,4 Monaten (Bereich: 15,7 ‑ 44,2 Monate) zeigte die Studie statistisch signifikante Verbesserungen im Bereich des EFS. Das mediane EFS wurde im Arm mit Nivolumab in Kombination mit Chemotherapie/Nivolumab nicht erreicht (95 % CI: 28,94; NE) und betrug 18,43 Monate (95 % CI: 13,63; 28,06) im Arm mit Placebo mit Chemotherapie/Placebo (HR = 0,58; 97,36 % CI: 0,42; 0,81; Log-Rank-stratifizierter p‑Wert 0,00025). In einer präspezifizierten Zwischenanalyse mit allen randomisierten Patienten mit einer medianen Nachbeobachtungszeit von 41 Monaten (Bereich: 31,3 ‑ 59,8 Monate) wurde das mediane OS sowohl im Arm mit Nivolumab in Kombination mit Chemotherapie/Nivolumab als auch im Arm mit Placebo mit Chemotherapie/Placebo nicht erreicht (HR = 0,85, 97,63 % CI: 0,58; 1,25).
Explorative Subgruppenanalyse nach Tumor-PD‑L1-Expression
Das EFS für die Subgruppe der Patienten mit Tumor-PD‑L1-Expression ≥ 1% mit einer medianen Nachbeobachtungszeit von 41 Monaten (Bereich: 31,3 ‑ 59,8 Monate) ist in Tabelle 22 und Abbildung 13 dargestellt.
Tabelle 22: Wirksamkeitsergebnisse bei Patienten mit Tumor-PD‑L1 ≥ 1 % (CA20977T)
Nivolumab mit Chemotherapie/Nivolumab |
Placebo mit Chemotherapie/Placebo |
|
Ereignisfreies Überleben (EFS) nach BICR | ||
Ereignisse (%) |
47 (37 %) |
70 (55 %) |
Median (Monate)a |
46,55 |
15,08 |
Hazard Ratiob |
0,53 |
|
NE = nicht abschätzbar (non-estimable) |
Abbildung 13: Kaplan‑Meier-Kurven des EFS bei Patienten mit Tumor-PD‑L1 ≥ 1 % (CA20977T)
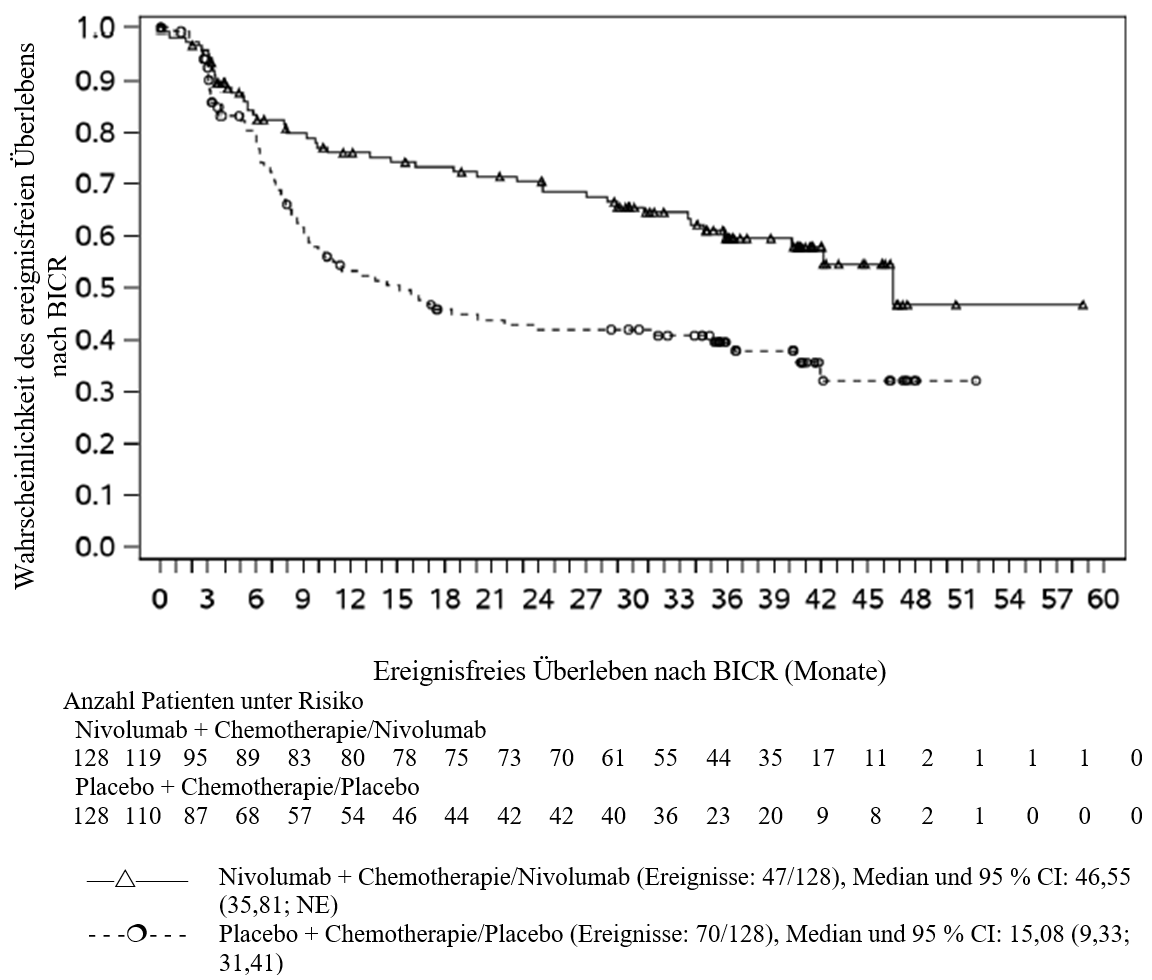
Basierend auf Datenschnitt: 11‑Nov‑2024, minimale Nachbeobachtungszeit von 31,3 Monaten
Zum Zeitpunkt der aktualisierten EFS-Analyse wurde eine Zwischenanalyse für das OS durchgeführt (minimale Nachbeobachtungszeit von 31,3 Monaten). Die explorative, deskriptive HR für OS bei Patienten mit Tumor-PD‑L1-Expression ≥ 1% war 0,61 (95 % CI: 0,39; 0,97) für den Arm mit Nivolumab in Kombination mit Chemotherapie/Nivolumab vs. den Arm mit Placebo mit Chemotherapie/Placebo. Die Kaplan‑Meier-Kurven für das OS für die Subgruppe der Patienten mit Tumor-PD‑L1-Expression ≥ 1 % sind in Abbildung 14 dargestellt.
Abbildung 14: Kaplan‑Meier-Kurven des OS bei Patienten mit Tumor-PD‑L1 ≥ 1 % (CA20977T)
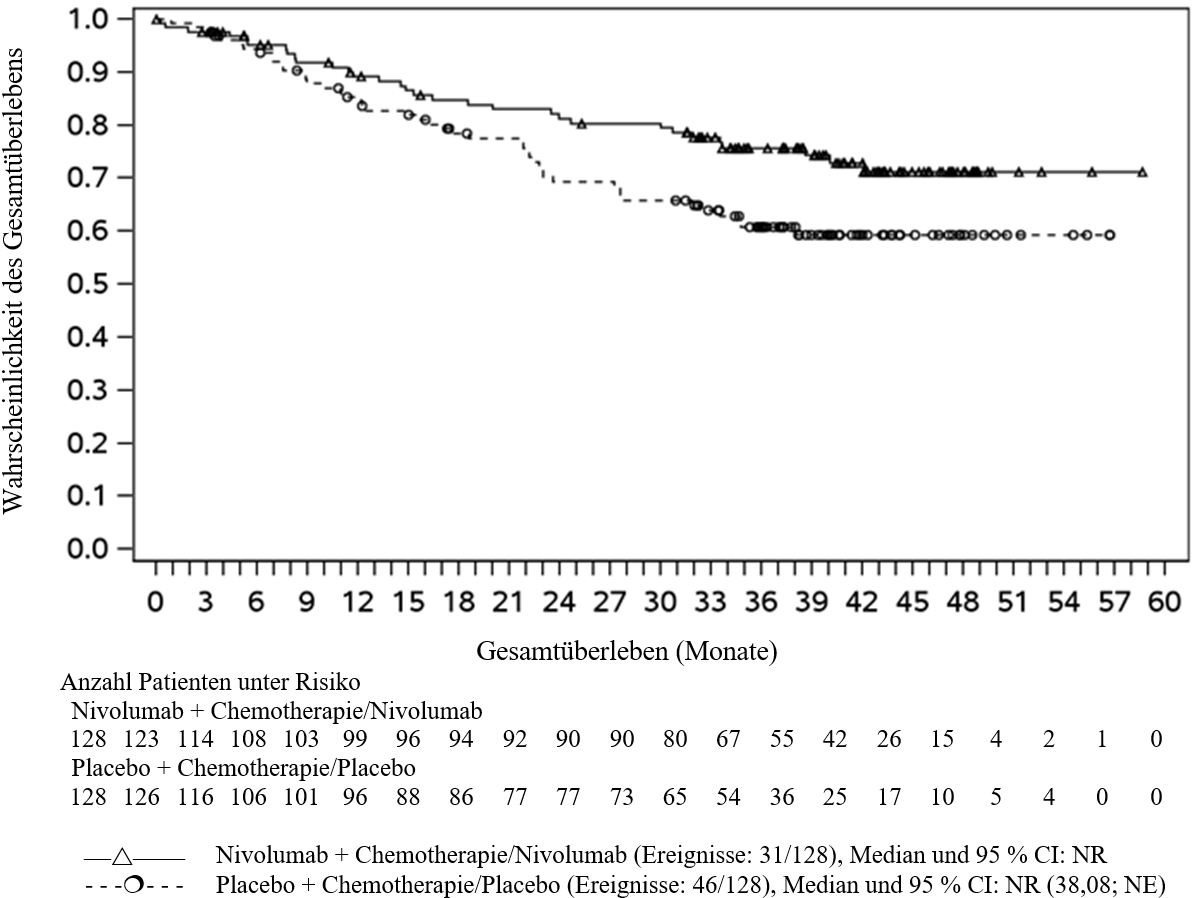
Basierend auf Datenschnitt: 11‑Nov‑2024, minimale Nachbeobachtungszeit von 31,3 Monaten
Behandlung des NSCLC nach vorheriger Chemotherapie
NSCLC mit plattenepithelialer Histologie
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie vs. Docetaxel (CA209017)
Sicherheit und Wirksamkeit von 3 mg/kg Nivolumab als Einzelsubstanz zur Behandlung des fortgeschrittenen oder metastasierten NSCLC mit plattenepithelialer Histologie wurden in einer randomisierten, offenen Phase‑III‑Studie (CA209017) untersucht. In die Studie wurden Patienten (ab 18 Jahren) eingeschlossen, bei denen es während oder nach einer Vorbehandlung mit einer platinbasierten Kombinationschemotherapie zu einer Progression kam und die einen ECOG‑Performance‑Status von 0 oder 1 hatten. Der Einschluss von Patienten erfolgte unabhängig von ihrem Tumor‑PD‑L1‑Status. Patienten mit aktiver Autoimmunerkrankung, symptomatischer interstitieller Lungenerkrankung oder aktiven Hirnmetastasen waren von der Studie ausgeschlossen. Patienten mit behandelten Hirnmetastasen konnten in die Studie eingeschlossen werden, wenn sich die neurologische Symptomatik mindestens 2 Wochen vor Einschluss in die Studie auf den Ausgangsbefund zurückgebildet hatte und die Patienten entweder Corticosteroide abgesetzt hatten oder eine stabile oder abnehmende Dosierung von < 10 mg Prednison‑Äquivalent pro Tag erhielten.
Insgesamt wurden 272 Patienten entweder für Nivolumab, das in einer Dosierung von 3 mg/kg alle 2 Wochen über 60 Minuten intravenös verabreicht wurde (n = 135), oder für Docetaxel, das zu 75 mg/m2 alle 3 Wochen verabreicht wurde (n = 137), randomisiert. Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen ersichtlich war oder bis die Behandlung nicht mehr vertragen wurde. Tumorbeurteilungen wurden gemäß den RECIST, Version 1.1, 9 Wochen nach Randomisierung und anschließend alle 6 Wochen durchgeführt. Das primäre Wirksamkeitskriterium war OS. Sekundäre Wirksamkeitskriterien waren die von den Prüfärzten bewertete ORR und PFS. Zusätzlich wurden die Verbesserung der Symptome und der allgemeine Gesundheitszustand anhand des durchschnittlichen Symptombelastungsindex des „Lung Cancer Symptom Score (LCSS)“ bzw. mit der „EQ‑5D Visual Analogue Scale (EQ‑VAS)“ bewertet.
Die Ausgangsmerkmale der beiden Gruppen waren etwa gleich. Das mediane Alter war 63 Jahre (Spanne: 39‑85), darunter 44 % ≥ 65 Jahre und 11 % ≥ 75 Jahre. Die Mehrheit der Patienten war weiß (93 %) und männlich (76 %). Bei 31 % wurde Krankheitsprogression als das beste Ansprechen auf ihre letzte vorherige Behandlung berichtet und 45 % erhielten Nivolumab innerhalb von 3 Monaten nach Abschluss ihrer letzten Vorbehandlung. Der ECOG‑Performance‑Status zu Studienbeginn war 0 (24 %) oder 1 (76 %).
Die Kaplan‑Meier‑Kurven des OS sind in Abbildung 15 dargestellt.
Abbildung 15: Kaplan‑Meier‑Kurven des Gesamtüberlebens (CA209017)
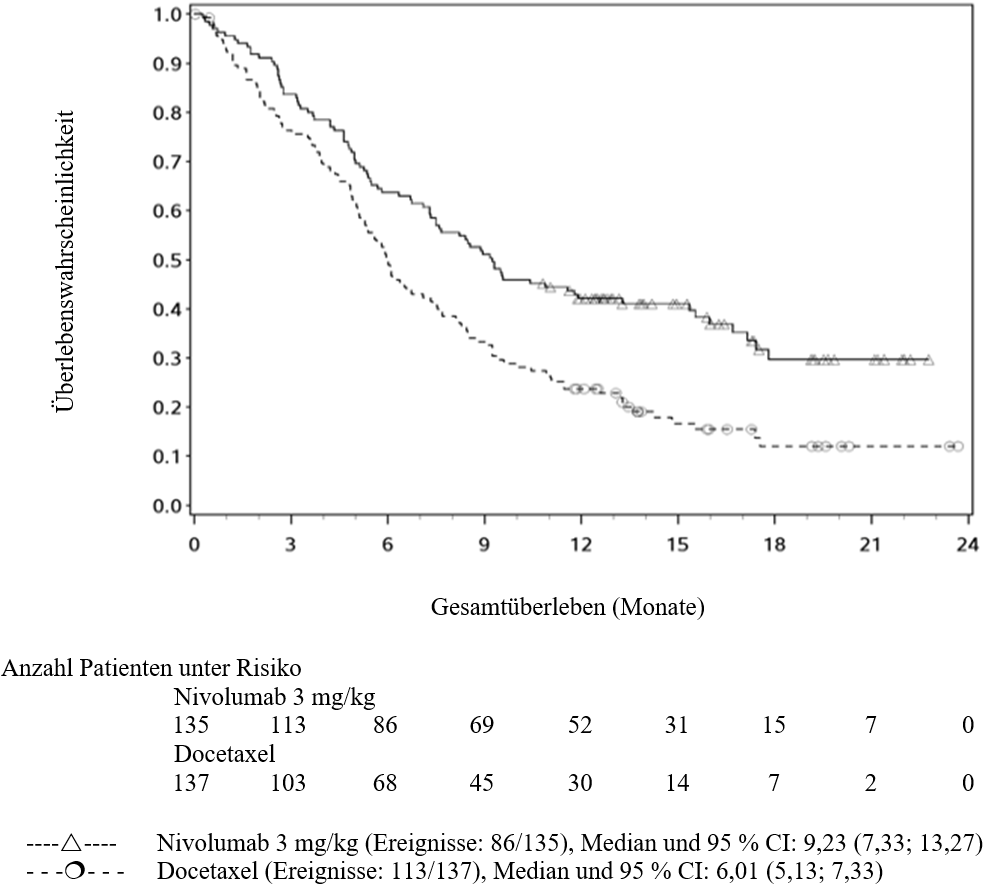
Der beobachtete Gesamtüberlebensvorteil wurde durchgehend in verschiedenen Patientenuntergruppen nachgewiesen. Der Überlebensvorteil wurde unabhängig davon beobachtet, ob die Patienten Tumoren hatten, die als PD‑L1‑negativ oder PD‑L1‑positiv bestimmt worden waren (Tumormembranexpressionsgrenze bei 1 %, 5 % oder 10 %). Die Rolle dieses Biomarkers (Tumor‑PD‑L1‑Expression) konnte jedoch nicht vollständig aufgeklärt werden. In einer Nachbeobachtung von mindestens 62,6 Monaten blieb der Überlebensvorteil durchweg in allen Untergruppen erhalten.
In der Studie CA209017 wurde eine geringe Anzahl Patienten ≥ 75 Jahre eingeschlossen (11 in der Nivolumab‑Gruppe und 18 in der Docetaxel‑Gruppe). Nivolumab zeigt hier numerisch weniger Einfluss auf OS (HR = 1,85; 95 % CI: 0,76; 4,51), PFS (HR=1,76; 95 % CI: 0,77; 4,05) und ORR (9,1 % vs. 16,7 %). Aufgrund der geringen Anzahl können aus diesen Daten keine endgültigen Schlussfolgerungen gezogen werden.
Die Wirksamkeitsergebnisse sind in Tabelle 23 dargestellt.
Tabelle 23: Wirksamkeitsergebnisse (CA209017)
Nivolumab |
Docetaxel |
|
Primäre Analyse | ||
Gesamtüberleben | ||
Ereignisse |
86 (63,7 %) |
113 (82,5 %) |
Hazard Ratio |
0,59 |
|
96,85 % CI |
(0,43; 0,81) |
|
p‑Wert |
0,0002 |
|
Median (95 % CI) (Monate) |
9,23 (7,33; 13,27) |
6,01 (5,13; 7,33) |
Rate (95 % CI) nach 12 Monaten |
42,1 (33,7; 50,3) |
23,7 (16,9; 31,1) |
Bestätigtes objektives Ansprechen |
27 (20,0 %) |
12 (8,8 %) |
(95 % CI) |
(13,6; 27,7) |
(4,6; 14,8) |
Odds Ratio (95 % CI) |
2,64 (1,27; 5,49) |
|
p‑Wert |
0,0083 |
|
Vollständiges Ansprechen |
1 (0,7 %) |
0 |
Teilweises Ansprechen |
26 (19,3 %) |
12 (8,8 %) |
Stabile Erkrankung |
39 (28,9 %) |
47 (34,3 %) |
Mediane Ansprechdauer |
||
Monate (Spanne) |
Nicht erreicht (2,9 ‑ 20,5+) |
8,4 (1,4+ ‑ 15,2+) |
Mediane Zeit bis zum Ansprechen |
||
Monate (Spanne) |
2,2 (1,6 ‑ 11,8) |
2,1 (1,8 ‑ 9,5) |
Progressionsfreies Überleben |
||
Ereignisse |
105 (77,8 %) |
122 (89,1 %) |
Hazard Ratio |
0,62 |
|
95 % CI |
(0,47; 0,81) |
|
p‑Wert |
< 0,0004 |
|
Median (95 % CI) (Monate) |
3,48 (2,14; 4,86) |
2,83 (2,10; 3,52) |
Rate (95 % CI) nach 12 Monaten |
20,8 (14,0; 28,4) |
6,4 (2,9; 11,8) |
Aktualisierte Analyse | ||
Gesamtüberlebena |
||
Ereignisse |
110 (81,4 %) |
128 (93,4 %) |
Hazard Ratio |
0,62 |
|
95 % CI |
(0,47; 0,80) |
|
Rate (95 % CI) nach 24 Monaten |
22,9 (16,2; 30,3) |
8 (4,3; 13,3) |
Bestätigtes objektives Ansprechen |
20,0 % |
8,8 % |
(95 % CI) |
(13,6; 27,7) |
(4,6; 14,8) |
Mediane Ansprechdauer |
||
Monate (Spanne) |
25,2 (2,9 ‑ 30,4) |
8,4 (1,4+ ‑ 18,0+) |
Progressionsfreies Überleben |
||
Rate (95 % CI) nach 24 Monaten |
15,6 (9,7; 22,7) |
Alle Patienten hatten entweder eine Progression, wurden zensiert oder konnten nicht mehr nachbeobachtet werden |
Aktualisierte Analyse | ||
Gesamtüberlebena |
||
Ereignisse |
118 (87,4 %) |
133 (97,1 %) |
Hazard Ratio |
0,62 |
|
95 % CI | ||
Rate (95 % CI) nach 60 Monaten |
12,3 (7,4; 18,5) |
3,6 (1,4; 7,8) |
Bestätigtes objektives Ansprechen |
20,0 % |
8,8 % |
(95 % CI) |
(13,6; 27,7) |
(4,6; 14,8) |
Mediane Ansprechdauer |
||
Monate (Spanne) |
25,2 (2,9 ‑ 70,6+) |
7,5 (0,0+ ‑ 18,0+) |
Progressionsfreies Überleben |
||
Rate (95 % CI) nach 60 Monaten |
9,4 (4,8; 15,8) |
Alle Patienten hatten entweder eine Progression, wurden zensiert oder konnten nicht mehr nachbeobachtet werden |
a Sechs Patienten (4 %), die auf Docetaxel randomisiert worden waren, haben zu irgendeinem Zeitpunkt auf eine Nivolumab‑Behandlung gewechselt. | ||
Die Häufigkeit der Verbesserung von krankheitsbezogenen Symptomen, gemessen am LCSS, war bei der Nivolumab‑Gruppe (18,5 %) und der Docetaxel‑Gruppe (21,2 %) ähnlich. Der durchschnittliche EQ‑VAS stieg in beiden Behandlungsgruppen mit der Zeit an, was auf einen besseren allgemeinen Gesundheitszustand für Patienten hindeutet, die die Behandlung beibehalten.
Intravenöse Formulierung
Einarmige Phase‑II‑Studie (CA209063)
Studie CA209063, eine einarmige, offene Studie, wurde bei 117 Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC mit plattenepithelialer Histologie nach zwei oder mehr Therapielinien durchgeführt; ansonsten wurden ähnliche Einschlusskriterien wie in der Studie CA209017 angewendet. Nivolumab 3 mg/kg zeigte ein Gesamtansprechen von 14,5 % (95 % CI: 8,7; 22,2 %), ein medianes OS von 8,21 Monaten (95 % CI: 6,05; 10,9) und ein medianes PFS von 1,87 Monaten (95 % CI 1,77; 3,15). Das PFS wurde anhand von RECIST, Version 1.1 bestimmt. Die berechnete Ein‑Jahres‑Überlebensrate war 41 %.
Intravenöse Formulierung
Einarmige Phase‑II‑Studie (CA209171)
Die Studie CA209171 war eine einarmige, offene Studie mit Nivolumab‑Monotherapie bei Patienten mit zuvor behandeltem fortgeschrittenem oder metastasiertem NSCLC mit plattenepithelialer Histologie. Der primäre Endpunkt war Sicherheit und der sekundäre Endpunkt war Wirksamkeit. Von den 811 behandelten Patienten hatten 103 (13 %) einen ECOG‑Performance‑Status von 2, 686 (85 %) waren < 75 Jahre alt und 125 (15 %) waren ≥ 75 Jahre alt. Es wurden bei allen behandelten Patienten keine neuen Sicherheitssignale identifiziert und das Gesamtsicherheitsprofil von Nivolumab war über die Subgruppen hinweg vergleichbar. Die Wirksamkeitsergebnisse, basierend auf dem vom Prüfarzt bewerteten Gesamtansprechen, sind in Tabelle 24 dargestellt.
Tabelle 24: Gesamtansprechen (ORR) basierend auf den auswertbaren Patienten – Gesamtanzahl und Subgruppen (CA209171)
Ergebnisse |
Gesamtanzahl |
ECOG‑PS 2 |
< 75 Jahre |
≥ 75 Jahre |
N Responder/ N auswertbara |
66/671 |
1/64 |
55/568 |
11/103 |
a beinhaltet bestätigtes und unbestätigtes Ansprechen, Scans waren nur in Woche 8/9 und Woche 52 obligatorisch. | ||||
NSCLC mit nicht‑plattenepithelialer Histologie
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie vs. Docetaxel (CA209057)
Die Sicherheit und Wirksamkeit von Nivolumab 3 mg/kg als Monopräparat zur Behandlung des fortgeschrittenen oder metastatischen NSCLC mit nicht‑plattenepithelialer Histologie wurde in einer randomisierten, offenen Phase‑3‑Studie untersucht (CA209057). In die Studie wurden Patienten eingeschlossen, bei denen es während oder nach einer Vorbehandlung mit einer platinbasierten Kombinationschemotherapie, die eine Erhaltungstherapie beinhalten konnte, zu einer Progression kam. Die Patienten waren 18 Jahre oder älter und hatten einen ECOG‑Performance‑Status von 0 oder 1. Eine zusätzliche TKI‑Behandlungslinie für Patienten mit bekannter EGFR‑Mutation oder ALK‑Translokation war erlaubt. Der Einschluss von Patienten erfolgte unabhängig von ihrem Tumor-PD‑L1‑Status. Patienten mit aktiver Autoimmunerkrankung, symptomatischer interstitieller Lungenerkrankung oder aktiven Hirnmetastasen waren von der Studie ausgeschlossen. Patienten mit behandelten Hirnmetastasen konnten in die Studie eingeschlossen werden, wenn sich die neurologische Symptomatik mindestens 2 Wochen vor Einschluss in die Studie auf den Ausgangsbefund zurückgebildet hatte und die Patienten entweder Corticosteroide abgesetzt hatten oder eine stabile oder abnehmende Dosierung von < 10 mg Prednison‑Äquivalent pro Tag erhielten.
Insgesamt wurden 582 Patienten entweder für Nivolumab, das in einer Dosierung von 3 mg/kg alle 2 Wochen über 60 Minuten intravenös verabreicht wurde (n = 292), oder für Docetaxel, das zu 75 mg/m2 alle 3 Wochen verabreicht wurde (n = 290), randomisiert. Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen ersichtlich war oder bis die Behandlung nicht mehr vertragen wurde. Tumorbeurteilungen wurden gemäß RECIST, Version 1.1 durchgeführt. Der primäre Endpunkt für die Wirksamkeit war das Gesamtüberleben (Overall Survival = OS). Wichtige sekundäre Endpunkte für die Wirksamkeit waren die vom Prüfarzt beurteilte objektive Ansprechrate (Objective Response Rate = ORR) und das progressionsfreie Überleben (Progression‑Free Survival = PFS). Zusätzliche prädefinierte Subgruppen‑Analysen wurden durchgeführt, um die Wirksamkeit in Bezug auf eine Tumor‑PD‑L1‑Expression mit den prädefinierten Grenzwerten von 1 %, 5 % und 10 % zu bestimmen. Die Beurteilung der einzelnen PD‑L1‑Expressionsintervalle wurde aufgrund der kleinen Fallzahlen innerhalb der Intervalle nicht in die präspezifizierte Analyse einbezogen.
Tumorgewebeproben vor Studienbeginn wurden systematisch vor der Randomisierung gesammelt, um vorgeplante Wirksamkeitsanalysen in Abhängigkeit von der Tumor‑PD‑L1‑Expression durchzuführen. Die Tumor‑PD‑L1‑Expression wurde unter Verwendung des PD‑L1‑IHC‑28‑8‑PharmDx‑Assays bestimmt.
Das mediane Alter war 62 Jahre (Bereich: 21 bis 85) mit 34 % ≥65 Jahre und 7 % ≥75 Jahre. Die Mehrheit der Patienten war kaukasisch (92 %) und männlich (55 %). Der ECOG‑Performance‑Status zu Studienbeginn war 0 (31 %) oder 1 (69 %). 79 % der Patienten waren frühere/derzeitige Raucher.
Die Kaplan‑Meier‑Kurven des Gesamtüberlebens sind in Abbildung 16 dargestellt.
Abbildung 16: Kaplan‑Meier‑Kurven des Gesamtüberlebens (CA209057)
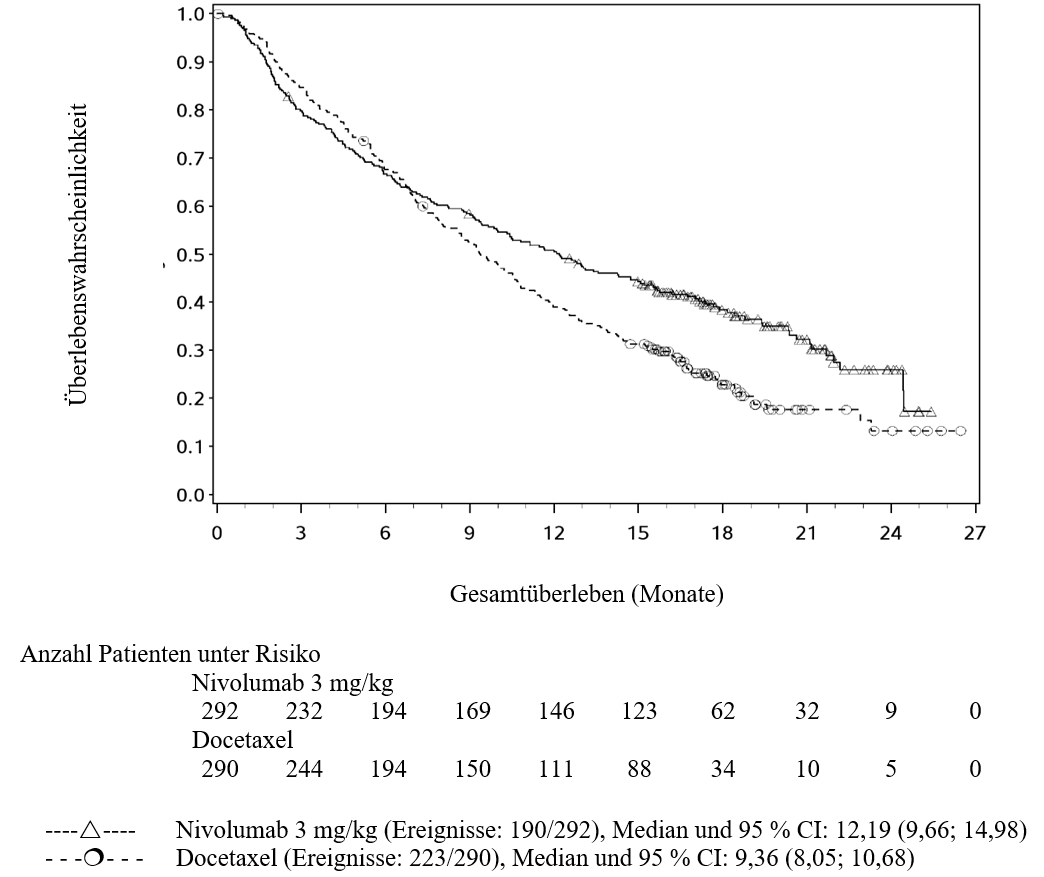
Die Studie zeigte bei der planmäßigen Interimsanalyse nach 413 Ereignissen (93 % der vorgesehenen Anzahl an Ereignissen für die Endauswertung) eine statistisch signifikante Verbesserung des Gesamtüberlebens bei den unter Nivolumab randomisierten Patienten verglichen mit denen unter Docetaxel. Die Wirksamkeitsergebnisse sind in Tabelle 25 dargestellt.
Tabelle 25: Wirksamkeitsergebnisse (CA209057)
Nivolumab |
Docetaxel |
|
Präspezifizierte Zwischenanalyse | ||
Gesamtüberleben |
||
Ereignisse |
190 (65,1 %) |
223 (76,9 %) |
Hazard Ratioa |
0,73 |
|
(95,92 % CI) |
(0,59; 0,89) |
|
p‑Wertb |
0,0015 |
|
Median (95 % CI) Monate |
12,19 (9,66; 14,98) |
9,36 (8,05; 10,68) |
Rate (95 % CI) nach 12 Monaten |
50,5 (44,6; 56,1) |
39,0 (33,3; 44,6) |
Bestätigtes objektives Ansprechen |
56 (19,2 %) |
36 (12,4 %) |
(95 % CI) |
(14,8; 24,2) |
(8,8; 16,8) |
Odds Ratio (95 % CI) |
1,68 (1,07; 2,64) |
|
p‑Wert |
0,0246 |
|
Vollständiges Ansprechen (Complete Response = CR) |
4 (1,4 %) |
1 (0,3 %) |
Teilweises Ansprechen (Partial Response = PR) |
52 (17,8 %) |
35 (12,1 %) |
Stabile Erkrankung (Stable Disease = SD) |
74 (25,3 %) |
122 (42,1 %) |
Mediane Ansprechdauer |
||
Monate (Spanne) |
17,15 (1,8 ‑ 22,6+) |
5,55 (1,2+ ‑ 15,2+) |
Mediane Zeit bis zum Ansprechen |
||
Monate (Spanne) |
2,10 (1,2 ‑ 8,6) |
2,61 (1,4 ‑ 6,3) |
Progressionsfreies Überleben |
||
Ereignisse |
234 (80,1 %) |
245 (84,5 %) |
Hazard Ratio |
0,92 |
|
95 % CI |
(0,77; 1,11) |
|
p‑Wert |
0,3932 |
|
Median (95 % CI) (Monate) |
2,33 (2,17; 3,32) |
4,21 (3,45; 4,86) |
Rate (95 % CI) nach 12 Monaten |
18,5 (14,1; 23,4) |
8,1 (5,1; 12,0) |
Aktualisierte Analyse | ||
Gesamtüberlebenc | ||
Ereignisse |
228 (78,1 %) |
247 (85,1 %) |
Hazard Ratioa |
0,75 |
|
(95 % CI) |
(0,63; 0,91) |
|
Rate (95 % CI) nach 24 Monaten |
28,7 (23,6; 34,0) |
15,8 (11,9; 20,3) |
Bestätigtes objektives Ansprechen |
19,2 % |
12,4 % |
(95 % CI) |
(14,8; 24,2) |
(8,8; 16,8) |
Mediane Ansprechdauer |
||
Monate (Spanne) |
17,2 (1,8 ‑ 33,7+) |
5,6 (1,2+ ‑ 16,8) |
Progressionsfreies Überleben |
||
Rate (95 % CI) nach 24 Monaten |
11,9 (8,3; 16,2) |
1,0 (0,2; 3,3) |
Aktualisierte Analyse | ||
Gesamtüberlebend |
||
Ereignisse |
250 (85,6 %) |
279 (96,2 %) |
Hazard Ratioa |
0,70 |
|
(95 % CI) |
(0,58; 0,83) |
|
Rate (95 % CI) nach 60 Monaten |
14,0 (10,2; 18,3) |
2,1 (0,9; 4,4) |
Bestätigtes objektives Ansprechen |
19,5 % |
12,4 % |
(95 % CI) |
(15,1; 24,5) |
(8,8; 16,8) |
Mediane Ansprechdauer |
||
Monate (Spanne) |
17,2 (1,8 ‑ 70,4+) |
5,6 (0,0+ ‑ 33,4) |
Progressionsfreies Überleben |
||
Rate (95 % CI) nach 60 Monaten |
7,5 (4,5; 11,4) |
Alle Patienten hatten entweder eine Progression, wurden zensiert oder konnten nicht mehr nachbeobachtet werden. |
a Mit einem stratifizierten Cox‑Modell für proportionale Hazards berechnet. |
Eine quantifizierbare Tumor‑PD‑L1‑Expression wurde bei 79 % der Patienten in der Nivolumab‑Gruppe und bei 77 % der Patienten der Docetaxel‑Gruppe gemessen. Der Grad der Tumor‑PD‑L1‑Expression war zwischen den beiden Behandlungsarmen (Nivolumab vs. Docetaxel) in allen vordefinierten Tumor‑PD‑L1‑Expressionsgraden von ≥ 1 % (53 % vs. 55 %), ≥ 5 % (41 % vs. 38 %) oder ≥ 10 % (37 % vs. 35 %) ausgeglichen.
In der Nivolumab‑Gruppe zeigten Patienten mit Tumor‑PD‑L1‑Expression bei allen vordefinierten Expressionsgraden eine größere Wahrscheinlichkeit für eine Verbesserung des Überlebens verglichen mit der Docetaxel‑Gruppe. Dahingegen war das Überleben für Patienten mit niedriger oder fehlender Tumor‑PD‑L1‑Expression ähnlich im Vergleich zu Docetaxel. In Bezug auf das objektive Ansprechen war eine höhere PD‑L1‑Expression mit einem höheren objektiven Ansprechen verbunden. Vergleichbar zur Gesamtpopulation war die mediane Dauer des Ansprechens bei Nivolumab vs. Docetaxel sowohl für Patienten ohne PD‑L1‑Expression erhöht (18,3 Monate vs. 5,6 Monate) als auch für Patienten mit PD‑L1‑Expression (16,0 Monate vs. 5,6 Monate).
Tabelle 26 fasst die Ergebnisse des objektiven Ansprechens und des Gesamtüberlebens anhand der Tumor‑PD‑L1‑Expression zusammen.
Tabelle 26: Objektive Ansprechrate (ORR) und Gesamtüberleben (OS) anhand der Tumor‑PD‑L1‑Expression (CA209057)
PD‑L1‑Expression |
Nivolumab |
Docetaxel |
|
ORR anhand der Tumor‑PD‑L1‑Expression | |||
Odds Ratio (95 % CI) |
|||
< 1 % |
10/108 (9,3 %) |
15/101 (14,9 %) |
0,59 (0,22; 1,48) |
≥ 1 % |
38/123 (30,9 %) |
15/123 (12,2 %) |
3,22 (1,60; 6,71) |
≥ 1 % bis < 10 %a |
6/37 (16,2 %) |
5/44 (11,4 %) |
1,51 (0,35; 6,85) |
≥ 10 % bis < 50 %a |
5/20 (25,0 %) |
7/33 (21,2 %) |
1,24 (0,26; 5,48) |
≥ 50 %a |
27/66 (40,9 %) |
3/46 (6,5 %) |
9,92 (2,68; 54,09) |
OS anhand der Tumor‑PD‑L1‑Expression | |||
Minimale Nachbeobachtungszeit: 13,2 Monate | |||
Anzahl der Ereignisse (Anzahl der Patienten) |
Unstratifiziertes Hazard Ratio (95 % CI) |
||
< 1 % |
77 (108) |
75 (101) |
0,90 (0,66; 1,24) |
≥ 1 % |
68 (123) |
93 (123) |
0,59 (0,43; 0,82) |
≥ 1 % bis < 10 %a |
27 (37) |
30 (44) |
1,33 (0,79; 2,24) |
≥ 10 % bis < 50 %a |
11 (20) |
26 (33) |
0,61 (0,30; 1,23) |
≥ 50 %a |
30 (66) |
37 (46) |
0,32 (0,20; 0,53) |
Aktualisierte Analyse | |||
< 1 % |
91 (108) |
86 (101) |
0,91 (0,67; 1,22) |
≥ 1 % |
87 (123) |
103 (123) |
0,62 (0,47; 0,83) |
Aktualisierte Analyse | |||
< 1 % |
100 (109) |
96 (101) |
0,87 (0,66; 1,16) |
≥ 1 % |
96 (122) |
119 (123) |
0,55 (0,42; 0,73) |
a Post‑hoc Analyse; aufgrund des geringen Stichprobenumfangs der Subgruppe sollten die Ergebnisse mit Vorsicht interpretiert werden, zudem war der PD‑L1‑IHC‑28‑8‑PharmDx‑Assay zum Zeitpunkt der Analyse nicht für die Expressionslevel 10 % oder 50 % analytisch validiert | |||
Im Nivolumab‑Arm verstarb eine größere Patientenanzahl (59/292; 20,2 %) innerhalb der ersten 3 Monate im Vergleich zum Docetaxel-Arm (44/290; 15,2 %). Die Ergebnisse einer explorativen, multivariaten Post‑hoc‑Analyse zeigten, dass mit Nivolumab behandelte Patienten, die prognostisch ungünstigere Faktoren und/oder einen aggressiven Krankheitsverlauf in Kombination mit einer niedrigen (z. B. < 50 %) oder fehlenden PD‑L1‑Expression aufweisen, ein höheres Risiko haben können innerhalb der ersten 3 Monate zu versterben.
In Subgruppen‑Analysen zeigte sich bei Patienten, die Nichtraucher waren oder deren Tumoren EGFR aktivierende Mutationen aufwiesen, kein Überlebensvorteil im Vergleich zu Docetaxel. Aufgrund der geringen Patientenanzahl können allerdings keine definitiven Schlussfolgerungen aus diesen Daten gezogen werden.
Nierenzellkarzinom (renal cell carcinoma, RCC)
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie mit Ipilimumab in Kombination mit Nivolumab vs. Sunitinib (CA209214)
Die Sicherheit und Wirksamkeit von Nivolumab 3 mg/kg in Kombination mit Ipilimumab 1 mg/kg zur Behandlung des fortgeschrittenen/metastasierten Nierenzellkarzinoms (RCC) wurden in einer randomisierten, offenen Phase‑III‑Studie (CA209214) untersucht. In die Studie wurden Patienten (ab 18 Jahren) mit nicht vorbehandeltem, fortgeschrittenem oder metastasiertem Nierenzellkarzinom mit klarzelliger Komponente eingeschlossen. Die primäre Population zur Untersuchung der Wirksamkeit bestand aus Patienten mit intermediärem/ungünstigem Risikoprofil mit mindestens 1 oder mehr von 6 prognostischen Risikofaktoren nach den International-Metastatic-RCC-Database-Consortium-(IMDC)‑Kriterien (weniger als ein Jahr seit dem Zeitpunkt der initialen Nierenzellkarzinom‑Diagnose bis zur Randomisierung, Karnofsky‑Performance‑Status < 80 %, Hämoglobin geringer als die untere Normgrenze, korrigierter Calciumwert größer als 10 mg/dl, Anzahl der Blutplättchen größer als die obere Normgrenze und absolute Anzahl an Neutrophilen größer als die obere Normgrenze). Diese Studie schloss Patienten unabhängig von ihrem Tumor‑PD‑L1‑Status ein. Patienten mit einem Karnofsky‑Performance‑Status < 70 %, aktiven Gehirnmetastasen oder Gehirnmetastasen in der Vorgeschichte, aktiver Autoimmunerkrankung oder Patienten mit einer Erkrankung, die eine Behandlung mit einer systemischen Immunsuppression erfordert, waren von der Studie ausgeschlossen. Die Patienten wurden nach IMDC‑Prognostic‑Score und Region stratifiziert.
Insgesamt wurden 1 096 Patienten in dieser Studie randomisiert, von denen 847 Patienten ein RCC mit intermediärem/ungünstigem Risikoprofil aufwiesen und entweder für 4 Dosiszyklen alle 3 Wochen 3 mg/kg Nivolumab (n = 425) intravenös über 60 Minuten in Kombination mit 1 mg/kg Ipilimumab intravenös über 30 Minuten verabreicht bekamen, gefolgt von einer Nivolumab‑Monotherapie mit 3 mg/kg alle 2 Wochen oder über 4 Wochen mit Sunitinib (n = 422) 50 mg täglich peroral behandelt wurden, gefolgt von einer 2‑wöchigen Einnahmepause in jedem Behandlungszyklus. Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen bestand oder bis die Behandlung nicht mehr vertragen wurde. Die erste Tumorbewertung fand 12 Wochen nach Randomisierung statt und wurde im ersten Jahr alle 6 Wochen und danach alle 12 Wochen bis zum Fortschreiten der Erkrankung oder dem Behandlungsende, je nachdem, was später eintrat, wiederholt. Eine Weiterbehandlung nach einer durch den Prüfarzt festgestellten Progression gemäß RECIST, Version 1.1 war erlaubt, wenn der Patient nach Einschätzung des Prüfarztes einen klinischen Nutzen hatte und die Studienmedikation tolerierte. Die primären Wirksamkeitsendpunkte waren OS, ORR und PFS bei Patienten mit intermediärem/ungünstigem Risikoprofil, welche durch ein Blinded Independent Central Review (BICR) bestimmt wurden.
Die Ausgangsmerkmale waren in beiden Gruppen etwa gleich verteilt. Das mediane Alter war 61 Jahre (Spanne: 21 ‑ 85) mit 38 % ≥ 65 Jahre und 8 % ≥ 75 Jahre. Die Mehrheit der Patienten war männlich (73 %) und kaukasisch (87 %) und 31 % bzw. 69 % der Patienten hatten einen Ausgangs‑KPS von 70 bis 80 % bzw. 90 bis 100 %. Die mediane Zeit von der initialen Diagnose bis zur Randomisierung betrug 0,4 Jahre sowohl in der Nivolumab-3 mg/kg-in-Kombination-mit-Ipilimumab-1 mg/kg- als auch in der Sunitinib‑Gruppe. Die mediane Behandlungszeit betrug 7,9 Monate (Spanne: 1 Tag ‑ 21,4+ Monate) bei mit Nivolumab mit Ipilimumab behandelten Patienten und 7,8 Monate (Spanne: 1 Tag ‑ 20,2+ Monate) bei mit Sunitinib behandelten Patienten. 29 % der Patienten wurden mit Nivolumab mit Ipilimumab über eine Progression hinaus weiterbehandelt.
Die Wirksamkeitsergebnisse für Patienten mit intermediärem/ungünstigem Risikoprofil sind in Tabelle 27 dargestellt (primäre Analyse mit minimaler Nachbeobachtungszeit von 17,5 Monaten und mit minimaler Nachbeobachtungszeit von 60 Monaten) und in Abbildung 17 (minimale Nachbeobachtungszeit von 60 Monaten).
Die OS‑Ergebnisse einer zusätzlichen deskriptiven Analyse, mit einer minimalen Nachbeobachtungszeit von 60 Monaten, zeigen Resultate, die mit der ursprünglichen primären Analyse übereinstimmen.
Tabelle 27: Wirksamkeitsergebnisse für Patienten mit intermediärem/ungünstigem Risikoprofil (CA209214)
Nivolumab + Ipilimumab |
Sunitinib |
||
Primäre Analyse | |||
Gesamtüberleben | |||
Ereignisse |
140 (33 %) |
188 (45 %) |
|
Hazard Ratioa |
0,63 |
||
99,8 % CI |
(0,44; 0,89) |
||
p‑Wertb, c |
< 0,0001 |
||
Median (95 % CI) |
NE (28,2; NE) |
25,9 (22,1; NE) |
|
Rate (95 % CI) |
|||
Nach 6 Monaten |
89,5 (86,1; 92,1) |
86,2 (82,4; 89,1) |
|
Nach 12 Monaten |
80,1 (75,9; 83,6) |
72,1 (67,4; 76,2) |
|
Progressionsfreies Überleben |
|||
Ereignisse |
228 (53,6 %) |
228 (54,0 %) |
|
Hazard Ratioa |
0,82 |
||
99,1 % CI |
(0,64; 1,05) |
||
p‑Wertb,h |
0,0331 |
||
Median (95 % CI) |
11,6 (8,71; 15,51) |
8,4 (7,03; 10,81) |
|
Bestätigtes objektives Ansprechen (BICR) |
177 (41,6 %) |
112 (26,5 %) |
|
(95 % CI) |
(36,9; 46,5) |
(22,4; 31,0) |
|
Differenz des ORR (95 % CI)d |
16,0 (9,8; 22,2) |
||
p‑Werte,f |
< 0,0001 |
||
Vollständiges Ansprechen (Complete Response = CR) |
40 (9,4 %) |
5 (1,2 %) |
|
Teilweises Ansprechen (Partial Response = PR) |
137 (32,2 %) |
107 (25,4 %) |
|
Stabile Erkrankung (Stable Disease = SD) |
133 (31,3 %) |
188 (44,5 %) |
|
Mediane Ansprechdauerg | |||
Monate (Spanne) |
NE (1,4+ ‑ 25,5+) |
18,17 (1,3+ ‑ 23,6+) |
|
Mediane Zeit bis zum Ansprechen |
|||
Monate (Spanne) |
2,8 (0,9 ‑ 11,3) |
3,0 (0,6 ‑ 15,0) |
|
Aktualisierte Analyse* | |||
Gesamtüberleben |
|||
Ereignisse |
242 (57 %) |
282 (67 %) |
|
Hazard Ratioa |
0,68 |
||
95 % CI |
(0,58; 0,81) |
||
Median (95 % CI) |
46,95 (35,35; 57,43) |
26,64 (22,08; 33,54) |
|
Rate (95 % CI) |
|||
Nach 24 Monaten |
66,3 (61,5; 70,6) |
52,4 (47,4; 57,1) |
|
Nach 36 Monaten |
54,6 (49,7; 59,3) |
43,7 (38,7; 48,5) |
|
Nach 48 Monaten |
49,9 (44,9; 54,6) |
35,8 (31,1; 40,5) |
|
Nach 60 Monaten |
43,0 (38,1; 47,7) |
31,3 (26,8; 35,9) |
|
Progressionsfreies Überleben |
|||
Ereignisse |
245 (57,6 %) |
253 (60,0 %) |
|
Hazard Ratioa |
0,73 |
||
95 % CI |
(0,61; 0,87) |
||
Median (95 % CI) |
11,6 (8,44; 16,63) |
8,3 (7,03; 10,41) |
|
Bestätigtes objektives Ansprechen (BICR) |
179 (42,1 %) |
113 (26,8 %) |
|
(95 % CI) |
(37,4; 47,0) |
(22,6; 31,3) |
|
Differenz des ORR (95 % CI)d,e |
16,2 (10,0; 22,5) |
||
Vollständiges Ansprechen (Complete Response = CR) |
48 (11,3 %) |
9 (2,1 %) |
|
Teilweises Ansprechen (Partial Response = PR) |
131 (30,8 %) |
104 (24,6 %) |
|
Stabile Erkrankung (Stable Disease = SD) |
131 (30,8 %) |
187 (44,3 %) |
|
Mediane Ansprechdauerg | |||
Monate (Spanne) |
NE (50,89 ‑ NE) |
19,38 (15,38 ‑ 25,10) |
|
Mediane Zeit bis zum Ansprechen |
|||
Monate (Spanne) |
2,8 (0,9 ‑ 35,0) |
3,1 (0,6 ‑ 23,6) |
|
a Basierend auf einem stratifizierten proportionalen Hazard‑Modell. | |||
Abbildung 17: Kaplan‑Meier‑Kurven des Gesamtüberlebens bei Patienten mit intermediärem/ungünstigem Risikoprofil (CA209214) – Minimale Nachbeobachtungszeit 60 Monate
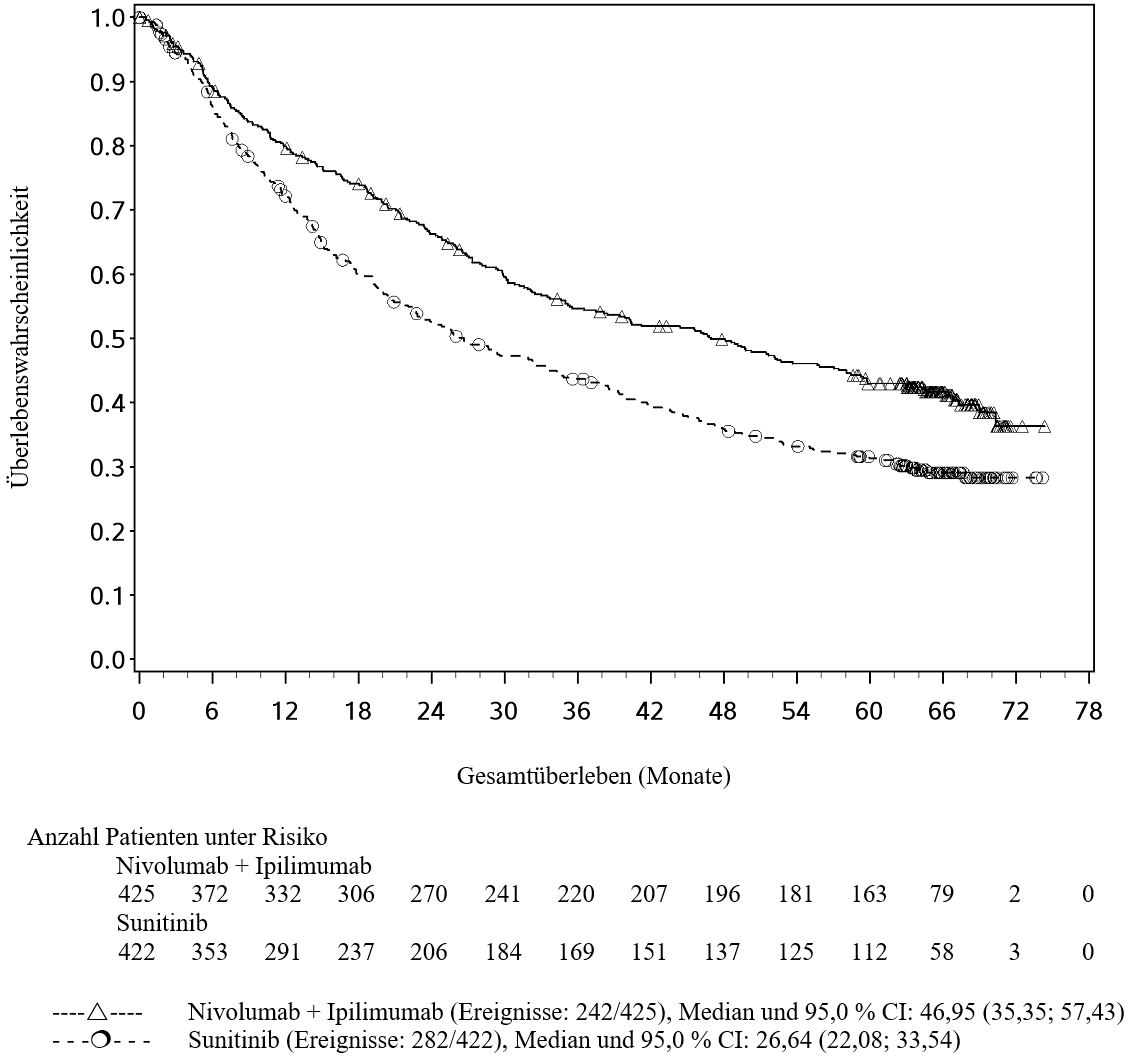
Eine deskriptive aktualisierte OS‑Analyse wurde durchgeführt, als alle Patienten eine minimale Nachbeobachtungszeit von 24 Monaten erreicht hatten. Zum Zeitpunkt der Analyse betrug das Hazard Ratio 0,66 (99,8 % CI 0,48‑0,91) mit 166/425 Ereignissen im Kombinations‑Arm und 209/422 Ereignissen im Sunitinib‑Arm. Bei Patienten mit intermediärem/ungünstigem Risikoprofil wurde ein Überlebensvorteil für den Nivolumab‑in‑Kombination‑mit‑Ipilimumab‑Arm im Vergleich zum Sunitinib‑Arm unabhängig von der Tumor‑PD‑L1‑Expression beobachtet. Das mediane OS bei Tumor‑PD‑L1‑Expression ≥ 1 % wurde für Nivolumab in Kombination mit Ipilimumab nicht erreicht und betrug im Sunitinib‑Arm 19,61 Monate (HR = 0,52; 95 % CI: 0,34; 0,78). Das mediane OS bei Tumor‑PD‑L1‑Expression < 1 % betrug 34,7 Monate für Nivolumab in Kombination mit Ipilimumab und betrug 32,2 Monate im Sunitinib‑Arm (HR = 0,70; 95 % CI: 0,54; 0,92).
In CA209214 wurden auch 249 Patienten mit günstigem Risikoprofil nach IMDC‑Kriterien im Nivolumab‑plus‑Ipilimumab‑Arm (n = 125) oder Sunitinib‑Arm (n = 124) eingeschlossen. Diese Patienten waren nicht Teil der ausgewerteten primären Population zur Untersuchung der Wirksamkeit. Patienten mit günstigem Risikoprofil zeigten bei minimaler Nachbeobachtungszeit von 24 Monaten unter Nivolumab plus Ipilimumab im Vergleich zu Sunitinib eine OS‑Hazard‑Ratio von 1,13 (95 % CI: 0,64; 1,99, p = 0,6710). Bei einer minimalen Nachbeobachtungszeit von 60 Monaten betrug die OS‑Hazard‑Ratio 0,94 (95 % CI: 0,65; 1,37).
Es liegen keine Daten zur Erstlinientherapie beim RCC mit Nivolumab in Kombination mit Ipilimumab bei Patienten mit ausschließlich nicht‑klarzelliger Histologie vor.
Der Anteil von Patienten ≥ 75 Jahre stellte 8 % der Patienten mit intermediärem/ungünstigem Risikoprofil in CA209214 dar. Nivolumab in Kombination mit Ipilimumab zeigte in dieser Subgruppe gegenüber der Gesamtpopulation numerisch einen geringeren Effekt auf OS (HR 0,97; 95 % CI: 0,48; 1,95) bei einer minimalen Nachbeobachtungszeit von 17,5 Monaten. Aufgrund der geringen Größe dieser Subgruppe, lassen sich daraus keine eindeutigen Schlussfolgerungen ziehen.
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie mit Nivolumab in Kombination mit Cabozantinib vs. Sunitinib (CA2099ER)
Die Sicherheit und Wirksamkeit von Nivolumab 240 mg in Kombination mit Cabozantinib 40 mg zur Erstlinientherapie des fortgeschrittenen/metastasierten RCC wurden in einer randomisierten, offenen Phase‑III‑Studie (CA2099ER) untersucht. In die Studie wurden Patienten (ab 18 Jahren) mit fortgeschrittenem oder metastasiertem RCC mit klarzelliger Komponente, Karnofsky‑Performance‑Status (KPS) ≥ 70 % und messbarer Erkrankung gemäß RECIST, Version 1.1 eingeschlossen, unabhängig von deren PD‑L1‑Status oder dem Risikoprofil nach IMDC‑Kriterien. Patienten mit einer Autoimmunerkrankung oder mit einer Erkrankung, die eine Behandlung mit einer systemischen Immunsuppression erfordert, Patienten, die mit einer Anti‑PD‑1‑, Anti‑PD‑L1‑, Anti‑PD‑L2‑, Anti‑CD137‑ oder Anti‑CTLA‑4‑Antikörper‑Therapie vortherapiert waren, mit einer schlecht kontrollierten Hypertonie trotz blutdrucksenkender Therapie, aktiven Hirnmetastasen oder unkontrollierter Nebenniereninsuffizienz waren von der Studie ausgeschlossen. Die Patienten wurden nach IMDC‑Prognostic‑Score, PD‑L1‑Tumorexoression und Region stratifiziert.
Insgesamt wurden 651 Patienten in dieser Studie randomisiert. Sie bekamen entweder Nivolumab 240 mg (n = 323) intravenös alle 2 Wochen in Kombination mit Cabozantinib 40 mg täglich peroral oder Sunitinib (n = 328) 50 mg täglich peroral für 4 Wochen, gefolgt von einer 2‑wöchigen Einnahmepause. Die Behandlung wurde bis zur Progression der Erkrankung oder nicht akzeptabler Toxizität fortgesetzt, wobei Nivolumab bis zu 24 Monate verabreicht wurde. Eine Weiterbehandlung nach einer durch den Prüfarzt festgestellten Progession gemäß RECIST, Version 1.1 war erlaubt, wenn der Patient nach Einschätzung des Prüfarztes einen klinischen Nutzen hatte und die Studienmedikation tolerierte. Die erste Tumorbewertung nach Baseline fand 12 Wochen (± 7 Tage) nach Randomisierung statt. Die nachfolgenden Tumorbewertungen wurden bis Woche 60 alle 6 Wochen (± 7 Tage) und dann alle 12 Wochen (± 14 Tage) bis zur radiologischen Progression, bestätigt durch BICR, durchgeführt. Der primäre Wirksamkeitsendpunkt war PFS, bestimmt durch ein BICR. Zusätzliche Wirksamkeitsmessungen umfassten OS und ORR als sekundäre Endpunkte.
Die Ausgangsmerkmale waren in beiden Gruppen etwa gleich verteilt. Das mediane Alter war 61 Jahre (Spanne: 28 ‑ 90) mit 38,4 % ≥ 65 Jahre und 9,5 % ≥ 75 Jahre. Die Mehrheit der Patienten war männlich (73,9 %) und kaukasisch (81,9 %). Der Anteil der asiatischen Patienten belief sich auf 8 %, und 23,2 % bzw. 76,5 % der Patienten hatten einen Ausgangs‑KPS von 70 bis 80 % bzw. 90 bis 100 %. Nach IMDC‑Kriterien hatten 22,6 % der Patienten ein günstiges, 57,6 % ein intermediäres und 19,7 % ein ungünstiges Risikoprofil. Nach der PD‑L1‑Tumorexpression hatten 72,5 % der Patienten eine PD‑L1‑Expression < 1 % oder unbestimmt und 24,9 % der Patienten eine PD‑L1‑Expression ≥ 1 %. Bei 11,5 % der Patienten hatten die Tumore sarkomatoide Merkmale. Die mediane Behandlungszeit betrug 14,26 Monate (Spanne: 0,2 ‑ 27,3) bei mit Nivolumab mit Cabozantinib behandelten Patienten und 9,23 Monate (Spanne: 0,8 ‑ 27,6 Monate) bei mit Sunitinib behandelten Patienten.
Die Studie zeigte eine statistisch signifikante Verbesserung des PFS, OS und ORR für Patienten, die in den Behandlungsarm Nivolumab in Kombination mit Cabozantinib gegenüber Sunitinib randomisiert worden waren. Die Wirksamkeitsergebnisse aus der primären Analyse (minimale Nachbeobachtungszeit 10,6 Monate; mediane Nachbeobachtungszeit 18,1 Monate) sind in Tabelle 28 dargestellt.
Tabelle 28: Wirksamkeitsergebnisse (CA2099ER)
Nivolumab + Cabozantinib |
Sunitinib |
|
Progressionsfreies Überleben | ||
Ereignisse |
144 (44,6 %) |
191 (58,2 %) |
Hazard Ratioa |
0,51 |
|
95 % CI |
(0,41; 0,64) |
|
p‑Wertb, c |
< 0,0001 |
|
Median (95 % CI) |
16,59 (12,45; 24,94) |
8,31 (6,97; 9,69) |
Gesamtüberleben |
||
Ereignisse |
67 (20,7 %) |
99 (30,2 %) |
Hazard Ratioa |
0,60 |
|
98,89 % CI |
(0,40; 0,89) |
|
p‑Wertb,c,e |
0,0010 |
|
Median (95 % CI) |
N.E. |
N.E. (22,6; N.E.) |
Rate (95 % CI) |
||
Nach 6 Monaten |
93,1 (89,7; 95,4) |
86,2 (81,9; 89,5) |
Bestätigtes objektives Ansprechen (BICR) |
180 (55,7 %) |
89 (27,1 %) |
(95 % CI)f |
(50,1; 61,2) |
(22,4; 32,3) |
Differenz des ORR (95 % CI)g |
28,6 (21,7; 35,6) |
|
p‑Werth |
< 0,0001 |
|
Vollständiges Ansprechen (Complete Response = CR) |
26 (8,0 %) |
15 (4,6 %) |
Teilweises Ansprechen (Partial Response = PR) |
154 (47,7 %) |
74 (22,6 %) |
Stabile Erkrankung (Stable Disease = SD) |
104 (32,2 %) |
138 (42,1 %) |
Mediane Ansprechdauerd | ||
Monate (Spanne) |
20,17 (17,31; N.E.) |
11,47 (8,31; 18,43) |
Mediane Zeit bis zum Ansprechen | ||
Monate (Spanne) |
2,83 (1,0 ‑ 19,4) |
4,17 (1,7 ‑ 12,3) |
a Mit einem stratifizierten Cox‑Modell für proportionale Hazards berechnet. Die Hazard Ratio ist Nivolumab und Cabozantinib gegenüber Sunitinib. | ||
Die primäre PFS‑Analyse beinhaltete eine Zensierung für neue Krebsbehandlungen (Tabelle 28). PFS‑Ergebnisse mit und ohne Zensierung für neue Krebsbehandlungen waren konsistent.
Es wurde ein PFS‑Vorteil, unabhängig von der IMDC‑Risikokategorie, im Nivolumab‑in‑Kombination‑mit‑Cabozantinib‑Arm gegenüber Sunitinib beobachtet. Für die Gruppe mit günstigem Risikoprofil wurde bei Nivolumab in Kombination mit Cabozantinib das mediane PFS nicht erreicht und betrug im Sunitinib‑Arm 12,81 Monate (HR = 0,60; 95 % CI: 0,37; 0,98). Für die Gruppe mit intermediärem Risikoprofil war das mediane PFS 17,71 Monate bei Nivolumab in Kombination mit Cabozantinib und betrug im Sunitinib‑Arm 8,38 Monate (HR = 0,54; 95 % CI: 0,41; 0,73). Für die Gruppe mit ungünstigem Risikoprofil war das mediane PFS 12,29 Monate bei Nivolumab in Kombination mit Cabozantinib und betrug im Sunitinib‑Arm 4,21 Monate (HR = 0,36; 95 % CI: 0,23; 0,58).
Ein PFS‑Vorteil wurde, unabhängig von der Tumor‑PD‑L1‑Expression, im Nivolumab‑in‑Kombination‑mit‑Cabozantinib‑Arm vs. Sunitinib beobachtet. Bei einer Tumor‑PD‑L1‑Expression ≥1 % lag das mediane PFS bei 13,08 Monaten im Nivolumab‑in‑Kombination‑mit‑Cabozantinib‑Arm und bei 4,67 Monaten im Sunitinib‑Arm (HR = 0,45; 95 % CI: 0,29; 0,68). Bei einer Tumor‑PD‑L1‑Expression < 1 % lag das mediane PFS bei 19,84 Monaten im Nivolumab‑in‑Kombination‑mit‑Cabozantinib‑Arm und bei 9,26 Monaten im Sunitinib‑Arm (HR = 0,50; 95 % CI: 0,38; 0,65).
Eine aktualisierte PFS‑ und OS‑Analyse wurde durchgeführt, als alle Patienten eine minimale Nachbeobachtungszeit von 16,0 Monaten und eine mediane Nachbeobachtungszeit von 23,5 Monaten erreicht hatten (siehe Abbildungen 18 und 19). Die PFS‑Hazard‑Ratio war 0,52 (95 % CI: 0,43, 0,64). Die OS‑Hazard‑Ratio war 0,66 (95 % CI: 0,50, 0,87). Aktualisierte Wirksamkeitsdaten (PFS und OS) in den Subgruppen entsprechend den IMDC‑Risikokategorien und PD‑L1‑Expressionslevel bestätigen die ursprünglichen Ergebnisse. Mit der aktualisierten Analyse wurde das mediane PFS für die Gruppe mit dem günstigen Risikoprofil erreicht.
Abbildung 18: Kaplan‑Meier‑Kurven des progressionsfreien Überlebens (CA2099ER)
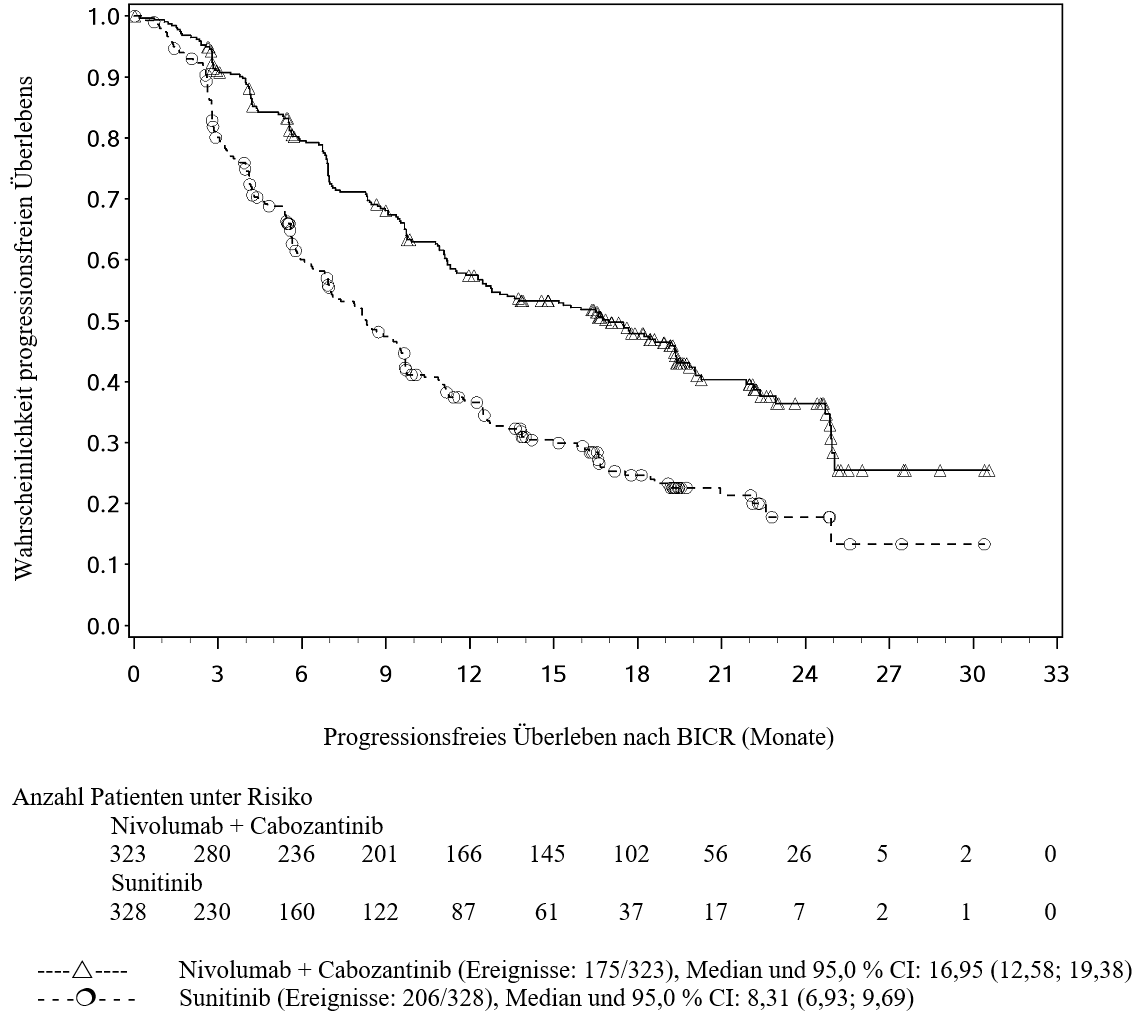
Abbildung 19: Kaplan‑Meier‑Kurven des Gesamtüberlebens (CA2099ER)
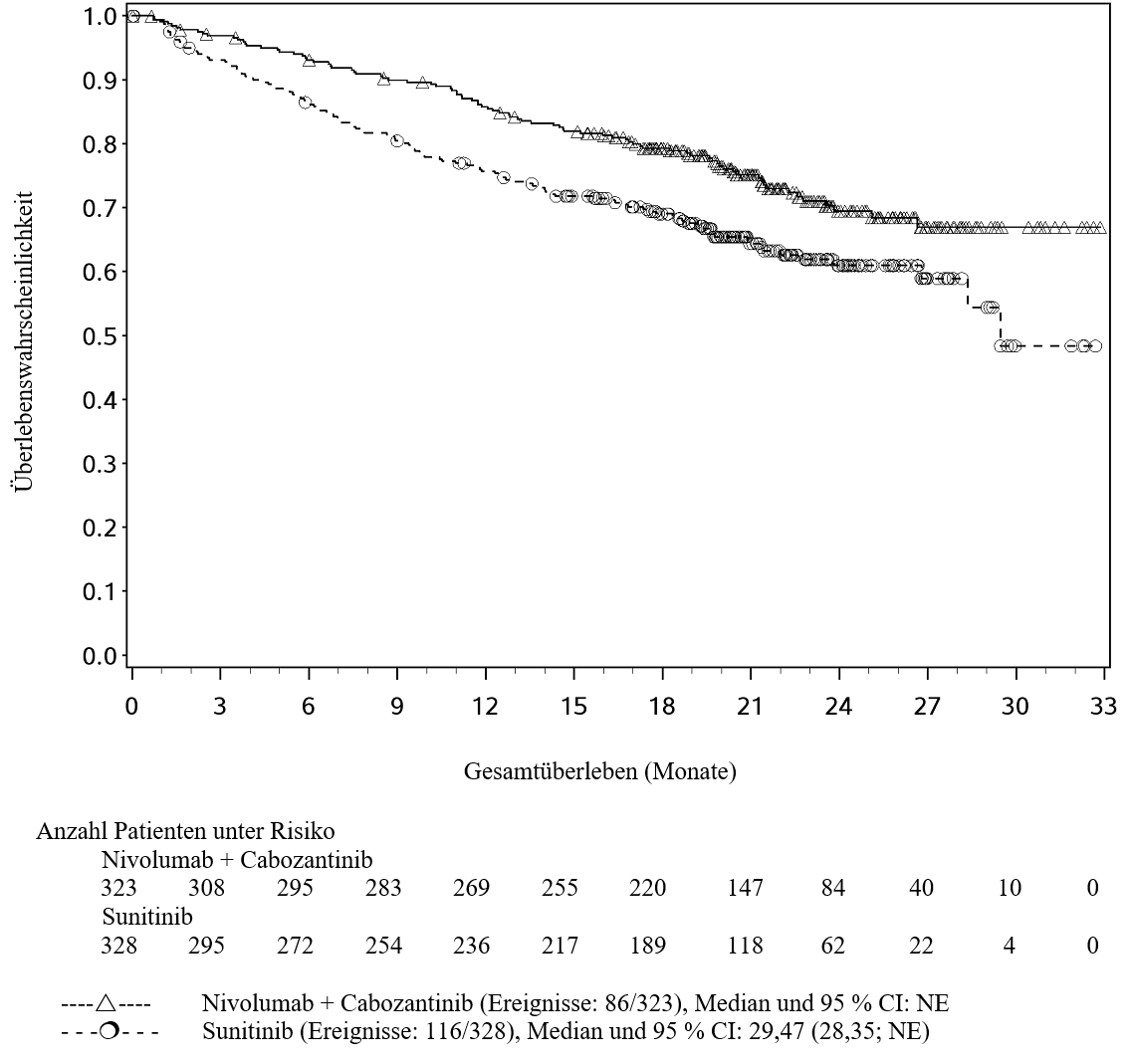
Subkutane Formulierung
Randomisierte, offene Phase‑III‑Studie vs. intravenös verabreichtes Nivolumab (CA20967T)
Die Sicherheit und Wirksamkeit der subkutanen Formulierung von Nivolumab wurden in einer multizentrischen, randomisierten, offenen Studie bei Patienten mit fortgeschrittenem oder metastasiertem klarzelligen RCC untersucht (CA20967T). Patienten ab 18 Jahren mit histologisch bestätigtem fortgeschrittenen oder metastasierten RCC mit klarzelliger Komponente, einschließlich Tumoren mit sarkomatoiden Merkmalen, die nicht mehr als 2 systemische Vortherapien erhalten hatten, wurden randomisiert Nivolumab 1 200 mg alle 4 Wochen subkutan oder Nivolumab 3 mg/kg alle 2 Wochen intravenös zu erhalten. Patienten mit unbehandelten, symptomatischen ZNS-Metastasen, leptomeningealen Metastasen, gleichzeitig aufgetretenen behandlungspflichtigen Malignomen oder einem Malignom innerhalb der letzten 2 Jahre in der Vorgeschichte, einer aktiven, bekannten oder vermuteten Autoimmunerkrankung oder Patienten, die eine vorherige Behandlung mit einem Checkpoint-Inhibitor erhalten hatten, waren von der Studie ausgeschlossen. Patienten mit asymptomatischen, stabilen ZNS-Metastasen, die keine sofortige Behandlung erforderten, konnten in die Studie eingeschlossen werden, wenn es innerhalb von 28 Tagen vor Verabreichung der ersten Dosis der Studienmedikation keine Nachweise für eine Progression gab.
Stratifikationsfaktoren für die Randomisierung waren das Körpergewicht (< 80 kg vs. ≥ 80 kg) und die Risikoklassifizierung gemäß dem International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) (günstiges vs. intermediäres vs. ungünstiges Risiko).
Das primäre Ziel der Studie war der Nachweis der Nichtunterlegenheit des Nivolumab‑Serumspiegels Cavgd28 und Cminss bei der subkutanen Verabreichung von Nivolumab im Vergleich zur intravenösen Verabreichung von Nivolumab (siehe Abschnitt 5.2). Das wichtigste sekundäre Ziel der Studie war der Nachweis der Nichtunterlegenheit der objektiven Ansprechrate bei der subkutanen Verabreichung von Nivolumab im Vergleich zur intravenösen Verabreichung von Nivolumab, bestimmt von einem unabhängigen zentralen Komitee (Blinded Independent Central Review, BICR). Weitere sekundäre Ziele beinhalteten die Bewertung der Ansprechdauer (duration of response, DOR), des progressionsfreien Überlebens (progression-free survival, PFS) und des Gesamtüberlebens (overall survival, OS).
Insgesamt wurden 495 Patienten randomisiert entweder subkutan verabreichtes Nivolumab (n = 248) oder intravenös verabreichtes Nivolumab (n = 247) zu erhalten. Das mediane Alter war 65 Jahre (Spanne: 20 ‑ 93), wobei 51 % ≥ 65 und 14 % ≥ 75 Jahre alt waren, 85 % waren weiß, 0,8 % waren asiatisch, 0,4 % waren schwarz und 68 % waren männlich. Siebenundfünfzig Prozent der Patienten wogen < 80 kg und 43 % ≥ 80 kg. Der Karnofsky‑Performance‑Status bei Einschluss in die Studie war 70 (7 %), 80 (20 %), 90 (34 %) oder 100 (39 %). Nach IMDC‑Risikokategorien hatten 21 % der Patienten ein günstiges, 62 % ein intermediäres und 17 % ein ungünstiges Risikoprofil.
Die Studie zeigte die Nichtunterlegenheit von subkutan verabreichtem Nivolumab 1 200 mg im Vergleich zu intravenös verabreichtem Nivolumab 3 mg/kg (siehe Abschnitt 5.2). Bei der primären Analyse (minimale Nachbeobachtungszeit 8 Monate) betrug die ORR 24,2 % (95 % CI: 19,0; 30,0) für subkutanes Nivolumab und 18,2 % (95 % CI: 13,6; 23,6) für intravenöses Nivolumab. Der Schätzer für das Relative Risiko des objektiven Ansprechens betrug 1,33 (95% CI: 0,94; 1,88). Zum Nachweis der Nichtunterlegenheit musste die untere Konfidenzgrenze des zweiseitigen 95% CI des relativen Risikos des objektiven Ansprechens ≥ 0,60 sein. Die aktualisierten Wirksamkeitsergebnisse mit einer minimalen Nachbeobachtungszeit von 14,9 Monaten (Datenschnitt 21. Februar 2024) sind in Tabelle 29 dargestellt.
Tabelle 29: Wirksamkeitsergebnisse ‑ CA20967T
Subkutan verabreichtes Nivolumab |
Intravenös verabreichtes Nivolumab |
|
ORRa nach BICR % (n/N) |
26,6 % (66/248) |
20,6 % (51/247) |
95 % CIb |
(21,2; 32,6) |
(15,8; 26,2) |
Schätzer für das relative Risiko des objektiven Ansprechens (95 % CI)c, d |
1,28 (0,93; 1,77) |
|
DORa nach BICR |
||
Median, Monate (95 % CI)e |
13,57 (8,57; NE) |
NR (15,7; NE) |
NR = nicht erreicht (not reached), NE = nicht abschätzbar (non‑estimable) | ||
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie von Nivolumab‑Monotherapie vs. Everolimus (CA209025)
Die Sicherheit und Wirksamkeit von Nivolumab 3 mg/kg als Monotherapie zur Behandlung von fortgeschrittenem Nierenzellkarzinom mit klarzelliger Histologie wurden in einer randomisierten, offenen Phase‑III‑Studie (CA209025) untersucht. Die Studie hat Patienten (ab 18 Jahren) eingeschlossen, bei denen es innerhalb oder nach 1 oder 2 vorangegangenen anti‑angiogenen Therapien und nicht mehr als insgesamt 3 systemischen Vortherapien, zu einer Krankheitsprogression kam. Die Patienten mussten einen Karnofsky‑Performance‑Status (KPS) von ≥ 70 % aufweisen. Der Einschluss von Patienten erfolgte unabhängig von ihrem Tumor‑PD‑L1‑Status. Von der Studie ausgeschlossen wurden Patienten mit aktuellen Hirnmetastasen oder Hirnmetastasen in der Vorgeschichte, Patienten mit vorangegangener Behandlung mit einem mTOR(mammalian target of rapamycin)-Inhibitor, aktiver Autoimmunerkrankung oder einer Erkrankung, die eine systemische Immunsuppression erfordert.
Insgesamt wurden 821 Patienten randomisiert, um entweder Nivolumab 3 mg/kg (n = 410) intravenös über 60 Minuten alle 2 Wochen oder Everolimus (n = 411) 10 mg täglich peroral zu erhalten. Die Behandlung wurde fortgesetzt, solange ein klinischer Nutzen beobachtet wurde oder die Behandlung nicht länger vom Patienten vertragen wurde. Die ersten Tumorbeurteilungen wurden 8 Wochen nach der Randomisierung durchgeführt. Anschließend wurde die Beurteilung im ersten Jahr alle 8 Wochen und danach alle 12 Wochen bis zum Progress oder bis zum Abbrechen der Behandlung durchgeführt, je nachdem, was später auftrat. Bei Patienten, die die Behandlung aus einem anderen Grund als Fortschreiten der Erkrankung abbrechen mussten, wurde die Tumorbeurteilung nach Abbrechen der Behandlung weiter fortgeführt. Eine Weiterbehandlung nach einer durch den Prüfarzt festgestellten Progression gemäß RECIST, Version 1.1 war erlaubt, wenn der Patient nach Einschätzung des Prüfarztes einen klinischen Nutzen hatte und die Studienmedikation tolerierte. Der primäre Endpunkt für die Wirksamkeit war das Gesamtüberleben (Overall Survival = OS). Die sekundären Endpunkte der Wirksamkeit beinhalteten die vom Prüfer beurteilte objektive Ansprechrate (Objective Response Rate = ORR) und das progressionsfreie Überleben (Progression‑Free Survival = PFS).
Die grundlegenden Patientenmerkmale waren zu Studienbeginn zwischen beiden Gruppen ausgeglichen. Das mediane Alter betrug 62 Jahre (Spanne: 18 – 88) mit 40 % ≥ 65 Jahre und 9 % ≥ 75 Jahre. Die Mehrzahl der Patienten war männlich (75 %) und kaukasisch (88 %), alle Memorial-Sloan-Kettering-Cancer-Center-Risikogruppen waren vertreten, 34 % der Patienten hatten einen Ausgangs‑Karnofsky‑Status (KPS) von 70 bis 80 % und 66 % der Patienten hatten einen Ausgangs‑KPS von 90 bis 100 %. Die Mehrheit der Patienten (72 %) hatte 1 Regime einer anti‑angiogenen Vortherapie erhalten. Die mediane Zeit von der initialen Diagnose bis zur Randomisierung betrug 2,6 Jahre in beiden Gruppen, Nivolumab und Everolimus. Die durchschnittliche Behandlungszeit betrug bei den mit Nivolumab behandelten Patienten 5,5 Monate (Spanne: 0 ‑ 29,6+ Monate) und 3,7 Monate (Spanne: 6 Tage ‑ 25,7+ Monate) bei den mit Everolimus behandelten Patienten.
44 % der Patienten wurden mit Nivolumab über eine Progression hinaus weiterbehandelt.
Die Kaplan‑Meier‑Kurven des Gesamtüberlebens sind in Abbildung 20 dargestellt.
Abbildung 20: Kaplan‑Meier‑Kurven des Gesamtüberlebens (CA209025)
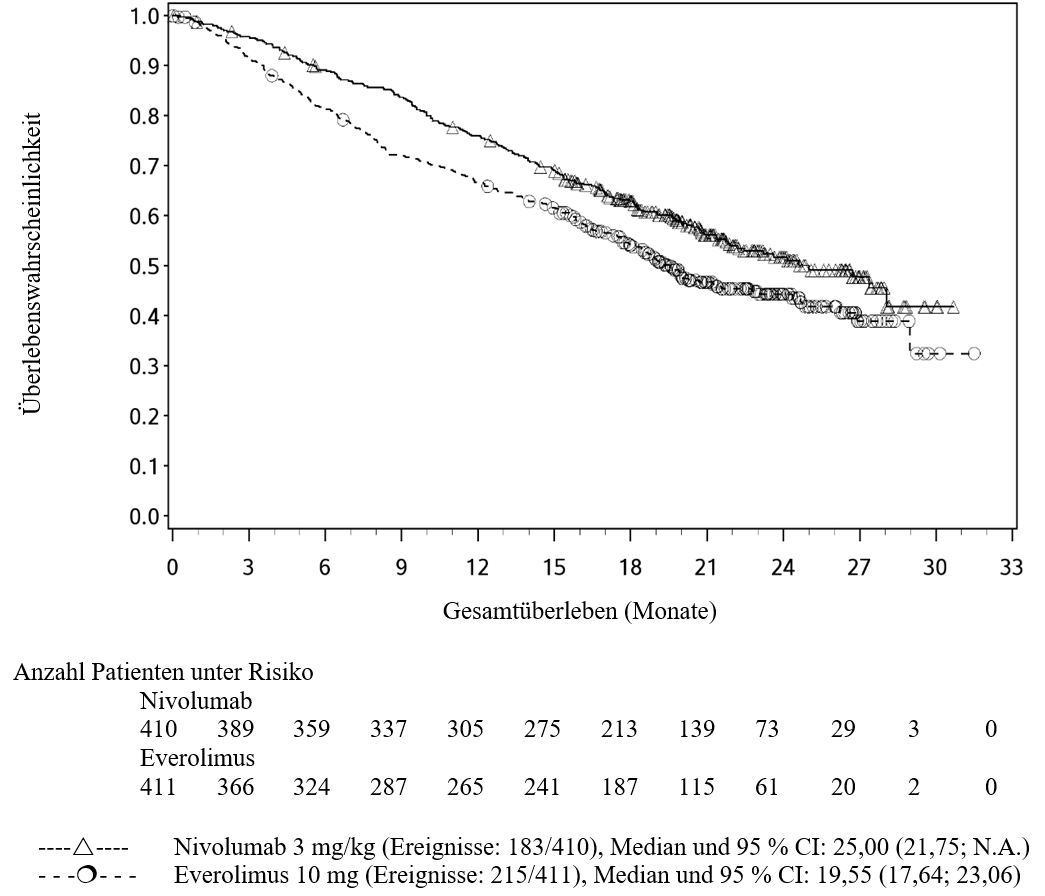
Die Studie zeigte bei der planmäßigen Interimsanalyse nach 398 Ereignissen (70 % der vorgesehenen Anzahl an Ereignissen für die Endauswertung) eine statistisch signifikante Verbesserung des Gesamtüberlebens bei den unter Nivolumab randomisierten Patienten verglichen mit denen unter Everolimus (Tabelle 30 und Abbildung 20). Die Verbesserung des Gesamtüberlebens wurde unabhängig vom PD‑L1‑Expressionsgrad beobachtet.
Die Wirksamkeitsergebnisse sind in Tabelle 30 dargestellt.
Tabelle 30: Wirksamkeitsergebnisse (CA209025)
Nivolumab |
Everolimus |
|
Gesamtüberleben |
||
Ereignisse |
183 (45 %) |
215 (52 %) |
Hazard-Ratio |
0,73 |
|
98,52 % CI |
(0,57; 0,93) |
|
p‑Wert |
0,0018 |
|
Median (95 % CI) |
25,0 (21,7; NE) |
19,6 (17,6; 23,1) |
Rate (95 % CI) |
||
Nach 6 Monaten |
89,2 (85,7; 91,8) |
81,2 (77,0; 84,7) |
Nach 12 Monaten |
76,0 (71,5; 79,9) |
66,7 (61,8; 71,0) |
Objektives Ansprechen |
103 (25,1 %) |
22 (5,4 %) |
(95 % CI) |
(21,0; 29,6) |
(3,4; 8,0) |
Odds Ratio (95 % CI) |
5,98 (3,68; 9,72) |
|
p‑Wert |
< 0,0001 |
|
Vollständiges Ansprechen (Complete Response = CR) |
4 (1,0 %) |
2 (0,5 %) |
Teilweises Ansprechen (Partial Response = PR) |
99 (24,1 %) |
20 (4,9 %) |
Stabile Erkrankung (Stable Disease = SD) |
141 (34,4 %) |
227 (55,2 %) |
Mediane Ansprechdauer |
||
Monate (Spanne) |
11,99 (0,0 ‑ 27.6+) |
11,99 (0,0+ ‑ 22,2+) |
Mediane Zeit bis zum Ansprechen |
||
Monate (Spanne) |
3,5 (1,4 ‑ 24,8) |
3,7 (1,5 ‑ 11,2) |
Progressionsfreies Überleben |
||
Ereignisse |
318 (77,6 %) |
322 (78,3 %) |
Hazard-Ratio |
0,88 |
|
95 % CI |
(0,75; 1,03) |
|
p‑Wert |
0,1135 |
|
Median (95 % CI) |
4,6 (3,71; 5,39) |
4,4 (3,71; 5,52) |
“+” kennzeichnet eine zensierte Beobachtung. | ||
Nach Beginn der Nivolumab‑Behandlung betrug die mediane Zeit bis zum Beginn des objektiven Ansprechens 3,5 Monate (Spanne: 1,4 ‑ 24,8 Monate). 49 Responder (47,6 %) zeigten ein anhaltendes Ansprechen über eine Dauer von 0,0‑27,6+ Monaten.
Das Gesamtüberleben konnte mit einer Verbesserung der krankheitsbedingten Symptome und nicht‑krankheitsspezifischer Lebensqualität (quality of life = QoL) im Verlauf in Verbindung gebracht werden. Diese wurden durch den Gebrauch der validierten und zuverlässigen Fragebögen des Functional Assessment of Cancer Therapy ‑ Kidney Symptom Index ‑ Disease Related Symptoms (FKSI‑DRS) und des EuroQoL EQ‑5D beurteilt. Die offensichtlich aussagekräftige Verbesserung der Symptome (MID = 2 Punkte Unterschied im FKSI‑DRS-Status; p < 0,001) und Zeit zur Verbesserung (HR = 1,66 (1,33; 2,08) p < 0,001) waren bei Patienten im Nivolumab‑Arm signifikant besser. Obwohl beide Studienarme eine aktive Therapie erhalten haben, sollten die QoL‑Daten im Kontext eines offenen Studiendesigns interpretiert und daher mit Vorsicht betrachtet werden.
Intravenöse Formulierung
Phase‑IIIb/IV‑Sicherheitsstudie (CA209374)
Zusätzliche Sicherheitsdaten und deskriptive Wirksamkeitsdaten sind aus der Studie CA209374 verfügbar. Dabei handelt es sich um eine offene Phase IIIb/IV Sicherheitsstudie mit Nivolumab‑Monotherapie (240 mg alle 2 Wochen) zur Behandlung von Patienten mit fortgeschrittenem oder metastasiertem RCC (n = 142), einschließlich 44 Patienten mit nicht‑klarzelliger Histologie.
Bei Patienten mit nicht‑klarzelliger Histologie betrugen die ORR und die mediane Dauer des Ansprechens 13,6 % bzw. 10,2 Monate nach einem minimalen Nachbeobachtungszeitraum von etwa 16,7 Monaten. Die klinische Aktivität wurde unabhängig vom PD‑L1‑Expressionsstatus des Tumors beobachtet.
Plattenepithelkarzinom des Kopf‑Hals‑Bereichs
Intravenöse Formulierung
Die Sicherheit und Wirksamkeit von Nivolumab 3 mg/kg als Monotherapie zur Behandlung des metastasierten oder rezidivierten SCCHN wurden in einer randomisierten, offenen Phase‑III‑Studie (CA209141) untersucht. In die Studie wurden Patienten (ab 18 Jahren) mit histologisch bestätigtem rezidiviertem oder metastasiertem SCCHN (Mundhöhle, Rachenraum, Kehlkopf) des Stadiums III/IV und nicht zugänglich für eine lokale kurative Behandlung (Operation oder Strahlentherapie mit oder ohne Chemotherapie) eingeschlossen, bei denen es während oder innerhalb von 6 Monaten nach Erhalt einer platinbasierten Therapie zu einer Progression kam, und die einen ECOG‑Performance‑Status von 0 oder 1 hatten. Die platinbasierte Vortherapie wurde in der adjuvanten, neo‑adjuvanten, primären, rezidivierten oder metastasierten Situation gegeben. Der Einschluss von Patienten erfolgte unabhängig von ihrem Tumor‑PD‑L1‑Status oder ihrem humanen Papilloma‑Virus (HPV)‑Status. Patienten mit aktiver Autoimmunerkrankung, mit Erkrankungen, die eine immunsuppressive Therapie erforderten, mit rezidivierten oder metastasierten Karzinomen des Nasopharynx, mit plattenepithelialen Karzinomen mit unbekanntem Primarius, mit Karzinomen der Speicheldrüse oder mit Karzinomen nicht‑plattenepithelialer Histologie (z. B. Schleimhautmelanomen) oder aktiven Hirnmetastasen oder leptomeningealen Metastasen waren von der Studie ausgeschlossen. Patienten mit behandelten Hirnmetastasen konnten in die Studie eingeschlossen werden, wenn sich die neurologische Symptomatik mindestens 2 Wochen vor Einschluss in die Studie auf den Ausgangsbefund zurückgebildet hatte und die Patienten entweder keine Corticosteroide erhielten oder eine stabile oder abnehmende Dosierung von < 10 mg Prednison‑Äquivalent pro Tag.
Insgesamt wurden 361 Patienten entweder auf Nivolumab 3 mg/kg, intravenös verabreicht alle 2 Wochen über 60 Minuten (n = 240), oder auf eine Behandlung nach Wahl des Prüfarztes randomisiert. Die Behandlung nach Wahl des Prüfarztes war entweder Cetuximab (n = 15) mit einer Anfangsdosis von 400 mg/m2 gefolgt von einer wöchentlichen Dosis von 250 mg/m2 oder Methotrexat (n = 52) in einer wöchentlichen Dosierung von 40 bis 60 mg/m2 oder Docetaxel (n = 54) in einer wöchentlichen Dosierung von 30 bis 40 mg/m2. Die Randomisierung wurde nach Vorbehandlung mit Cetuximab stratifiziert. Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen beobachtet werden konnte oder bis die Behandlung nicht mehr vertragen wurde. Tumorbeurteilungen wurden gemäß RECIST, Version 1.1, 9 Wochen nach Randomisierung und anschließend alle 6 Wochen durchgeführt. Eine Behandlung über die anfänglich vom Prüfarzt gemäß Definition nach RECIST, Version 1.1, festgestellten Progression hinaus war für Patienten, die Nivolumab erhielten, erlaubt, wenn der Patient nach Einschätzung des Prüfarztes einen klinischen Nutzen von der Behandlung hatte und die Therapie vertragen wurde. Der primäre Endpunkt für die Wirksamkeit war das Gesamtüberleben. Wichtige sekundäre Endpunkte der Wirksamkeit waren vom Prüfarzt beurteiltes progressionsfreies Überleben und Ansprechrate. Zusätzliche vorspezifizierte Untergruppen‑Analysen wurden durchgeführt, um die Wirksamkeit nach Tumor‑PD‑L1‑Expression bei den vordefinierten Grenzwerten von 1 %, 5 % und 10 % zu untersuchen.
Tumorgewebeproben vor Studienbeginn wurden systematisch vor Randomisierung gesammelt, um im Voraus geplante Wirksamkeitsanalysen nach Tumor‑PD‑L1‑Expression durchzuführen. Die Tumor‑PD‑L1‑Expression wurde mithilfe des PD‑L1‑IHC‑28‑8‑pharmDx‑Tests bestimmt.
Die Ausgangsmerkmale der beiden Gruppen waren im Allgemeinen ausgewogen. Das mediane Alter betrug 60 Jahre (Spanne: 28 ‑ 83), wobei 31 % ≥ 65 Jahre und 5 % ≥ 75 Jahre waren; 83 % waren Männer und 83 % waren weiß. Der Ausgangs‑ECOG‑Performance‑Status war 0 (20 %) oder 1 (78 %), 77 % waren früher oder derzeit Raucher, 90 % hatten eine Stadium‑IV‑Erkrankung, 66 % hatten zwei oder mehr Läsionen; 45 % hatten eine, 34 % hatten 2 und 20 % hatten 3 oder mehr Vortherapien erhalten; 25 % waren HPV‑16‑positiv.
Mit einer minimalen Nachbeobachtungszeit von 11,4 Monaten zeigte die Studie eine statistisch signifikante Verbesserung des Gesamtüberlebens für Patienten, die auf Nivolumab randomisiert waren, im Vergleich zu Patienten, die auf Therapie nach Wahl des Prüfarztes randomisiert waren. Die Kaplan‑Meier‑Kurven für das Gesamtüberleben sind in Abbildung 21 dargestellt. Die Wirksamkeitsergebnisse sind in Tabelle 31 dargestellt.
Abbildung 21: Kaplan‑Meier‑Kurven des Gesamtüberlebens (CA209141)
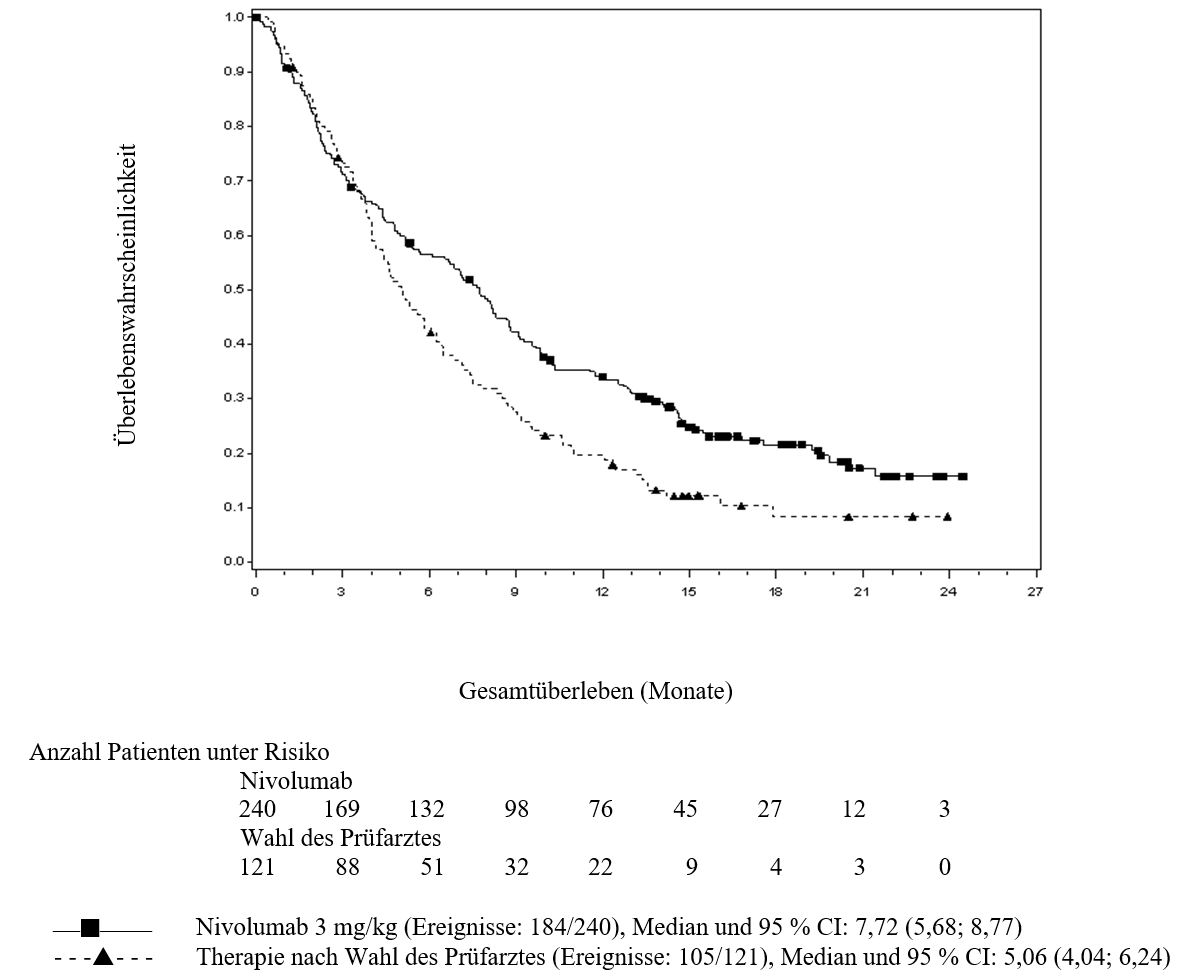
Tabelle 31: Wirksamkeitsergebnisse (CA209141)
Nivolumab |
Wahl des Prüfarztes |
||
Gesamtüberleben |
|||
Ereignisse |
184 (76,7 %) |
105 (86,8 %) |
|
Hazard Ratioa |
0,71 |
||
(95 % CI) |
(0,55; 0,90) |
||
p‑Wertb |
0,0048 |
||
Median (95 % CI) (Monate) |
7,72 (5,68; 8,77) |
5,06 (4,04; 6,24) |
|
Rate (95 % CI) nach 6 Monaten |
56,5 (49,9; 62,5) |
43,0 (34,0; 51,7) |
|
Rate (95 % CI) nach 12 Monaten |
34,0 (28,0; 40,1) |
19,7 (13,0; 27,3) |
|
Rate (95 % CI) nach 18 Monaten |
21,5 (16,2; 27,4) |
8,3 (3,6; 15,7) |
|
Progressionsfreies Überleben |
|||
Ereignisse |
204 (85,0 %) |
104 (86,0 %) |
|
Hazard Ratio |
0,87 |
||
95 % CI |
(0,69; 1,11) |
||
p‑Wert |
0,2597 |
||
Median (95 % CI) (Monate) |
2,04 (1,91; 2,14) |
2,33 (1,97; 3,12) |
|
Rate (95 % CI) nach 6 Monaten |
21,0 (15,9; 26,6) |
11,1 (5,9; 18,3) |
|
Rate (95 % CI) nach 12 Monaten |
9,5 (6,0; 13,9) |
2,5 (0,5; 7,8) |
|
Bestätigtes objektives Ansprechenc |
32 (13,3 %) |
7 (5,8 %) |
|
(95 % CI) |
(9,3; 18,3) |
(2,4; 11,6) |
|
Odds Ratio (95 % CI) |
2,49 (1,07; 5,82) |
||
Vollständiges Ansprechen (Complete Response = CR) |
6 (2,5 %) |
1 (0,8 %) |
|
Teilweises Ansprechen (Partial Response = PR) |
26 (10,8 %) |
6 (5,0 %) |
|
Stabile Erkrankung (Stable Disease = SD) |
55 (22,9 %) |
43 (35,5 %) |
|
Mediane Zeit bis zum Ansprechen |
|||
Monate (Spanne) |
2,1 (1,8 ‑ 7,4) |
2,0 (1,9 ‑ 4,6) |
|
Mediane Ansprechdauer |
|||
Monate (Spanne) |
9,7 (2,8 ‑ 20,3+) |
4,0 (1,5+ ‑ 8,5+) |
|
a Von einem stratifizierten Modell für proportionale Hazards berechnet. | |||
Eine quantifizierbare Tumor‑PD‑L1‑Expression wurde bei 67 % der Patienten in der Nivolumab‑Gruppe und bei 82 % der Patienten in der Gruppe Therapie nach Wahl des Prüfarztes gemessen. Der Grad der Tumor‑PD‑L1‑Expression war zwischen den beiden Behandlungsarmen (Nivolumab vs. Therapie nach Wahl des Prüfarztes) in allen vordefinierten Tumor‑PD‑L1‑Expressionsgraden von ≥ 1 % (55 % vs. 62 %), ≥ 5 % (34 % vs. 43 %), oder ≥ 10 % (27 % vs. 34 %) ausgeglichen.
In der Nivolumab‑Gruppe zeigten Patienten mit Tumor‑PD‑L1‑Expression bei allen vordefinierten Expressionsgraden eine größere Wahrscheinlichkeit für eine Verbesserung des Überlebens verglichen mit der Gruppe Therapie nach Wahl des Prüfarztes. Das Ausmaß der Verbesserung des Gesamtüberlebens war konsistent für die Tumor‑PD‑L1‑Expressionsgrade ≥ 1 %, ≥ 5 % oder ≥ 10 % (siehe Tabelle 32).
Tabelle 32: Gesamtüberleben in Abhängigkeit der Tumor‑PD‑L1‑Expression (CA209141)
PD‑L1 Expression |
Nivolumab |
Wahl des Prüfarztes |
|
Gesamtüberleben nach Tumor‑PD‑L1‑Expression | |||
Anzahl der Ereignisse (Patientenanzahl) |
Unstratifiziertes Hazard Ratio (95 % CI) |
||
< 1 % |
56 (73) |
32 (38) |
0,83 (0,54; 1,29) |
≥ 1 % |
66 (88) |
55 (61) |
0,53 (0,37; 0,77) |
≥ 5 % |
39 (54) |
40 (43) |
0,51 (0,32; 0,80) |
≥ 10 % |
30 (43) |
31 (34) |
0,57 (0,34; 0,95) |
In einer explorativen Post‑hoc‑Analyse wurde die PD‑L1‑Expression auf Tumorzellen und auf Tumor‑assoziierten Immunzellen (Tumor‑Associated Immune Cells = TAICs) mithilfe eines nicht‑validierten Tests in Bezug auf das Ausmaß des Behandlungseffektes von Nivolumab im Vergleich zur Therapie nach Wahl des Prüfarztes untersucht. Die Analyse zeigte, dass nicht nur die PD‑L1‑Expression auf Tumorzellen, sondern auch die PD‑L1-Expression auf TAICs mit einem Nutzen der Nivolumab‑Therapie im Vergleich zur Therapie nach Wahl des Prüfarztes verbunden zu sein scheint (siehe Tabelle 33). Aufgrund der geringen Patientenzahlen in den Subgruppen und des explorativen Charakters der Analyse können keine definitiven Schlussfolgerungen aus diesen Daten gezogen werden.
Tabelle 33: Wirksamkeit anhand der Tumorzell‑ und TAIC‑PD‑L1‑Expression (CA209141)
Medianes Gesamtüberlebena (Monate) |
Medianes progressionsfreies Überlebena (Monate) |
Gesamt-Ansprechrate (%) |
|||||
HRb (95 % CI) |
HRb (95 % CI) |
(95 % CI)c |
|||||
Nivolumab |
Therapie nach Wahl des Prüfarztes |
Nivolumab |
Therapie nach Wahl des Prüfarztes |
Nivolumab |
Therapie nach Wahl des Prüfarztes |
||
PD‑L1 ≥ 1 %, PD‑L1+ TAIC hochd |
9,10 |
4,60 |
3,19 |
1,97 |
19,7 |
0 |
|
0,43 (0,28; 0,67) |
0,48 (0,31; 0,75) |
(10,6; 31,8) |
(0; 7,5) |
||||
PD‑L1 ≥ 1 %, |
6,67 |
4,93 |
1,99 |
2,04 |
11,1 |
7,1 |
|
0,89 (0,44; 1,80) |
0,93 (0,46; 1,88) |
(2,4; 29,2) |
(0,2; 33,9) |
||||
PD‑L1 <1 %, PD‑L1+ TAIC hochd |
11,73 |
6,51 |
2,10 |
2,73 |
18,6 |
12,0 |
|
0,67 (0,38; 1,18) |
0,96 (0,55; 1,67) |
(8,4; 33,4) |
(2,5; 31,2) |
||||
PD‑L1 < 1 %, PD‑L1+ TAIC seltend |
3,71 |
4,85 |
1,84 |
2,12 |
3,7 |
10,0 |
|
1,09 (0,50; 2,36) |
1,91 (0,84; 4,36) |
(< 0,1; 19,0) |
(0,3; 44,5) |
||||
a Gesamtüberleben (OS) und progressionsfreies Überleben (PFS) wurden anhand der Kaplan‑Meier‑Methode bestimmt. | |||||||
Patienten, bei denen, nach Beurteilung des Prüfarztes, die primäre Lokalisation des Tumors im Bereich des Oropharynx lag, wurden auf HPV untersucht (bestimmt über p16‑Immunhistochemie [IHC]). Eine Verbesserung des Gesamtüberlebens wurde unabhängig vom HPV-Status beobachtet (HPV‑positiv: HR = 0,63; 95 % CI: 0,38, 1,04, HPV‑negativ: HR = 0,64; 95 % CI: 0,40; 1,03 und HPV‑unbekannt: HR = 0,78; 95 % CI: 0,55, 1,10).
Die Patienten‑bezogenen Endpunkte (Patient‑reported Outcomes [PROs]) wurden mithilfe des EORTC QLQ‑C30, EORTC QLQ‑H&N35 und des 3‑stufigen EQ‑5D beurteilt. Mit Nivolumab behandelte Patienten zeigten über einen Zeitraum von 15 Wochen stabile PROs, während Patienten, die mit einer Therapie nach Wahl des Prüfarztes behandelt wurden, deutliche Verschlechterungen in Bezug auf Funktionen (z. B. physische, soziale, Rollen‑Funktion) und Gesundheitsstatus zeigten sowie eine erhöhte Symptomatik (z. B. Ermüdung/Fatigue, Dyspnoe, Appetitverlust, Schmerz, sensorische Probleme, soziale Kontaktprobleme) aufwiesen. Die PRO‑Daten sollten im Kontext des offenen Studiendesigns gesehen und deswegen mit Vorsicht bewertet werden.
Urothelkarzinom
Adjuvante Behandlung des Urothelkarzinoms
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie von adjuvantem Nivolumab vs. Placebo (CA209274)
Die Sicherheit und Wirksamkeit der Nivolumab‑Monotherapie bei der adjuvanten Behandlung des Urothelkarzinoms wurde in einer multizentrischen, randomisierten, Placebo‑kontrollierten, doppelblinden Phase‑III‑Studie untersucht (CA209274). In die Studie wurden Patienten (ab 18 Jahren) eingeschlossen, die sich einer vollständigen Resektion eines muskelinvasiven Urothelkarzinoms (MIUC) unterzogen hatten, das in der Harnblase oder im oberen Urogenitaltrakt (Nierenbecken oder Harnleiter) angesiedelt war und bei denen ein hohes Rezidivrisiko bestand. Die pathologischen MIUC‑Einstufungskriterien zur Definition der Hochrisikopatienten waren ypT2‑ypT4a oder ypN+ bei erwachsenen Patienten, die neoadjuvante Cisplatin‑Chemotherapie erhalten hatten, und pT3‑pT4a oder pN+ bei erwachsenen Patienten, die keine neoadjuvante Cisplatin‑Chemotherapie erhalten hatten und für die eine adjuvante Cisplatin‑Chemotherapie nicht geeignet war oder die diese ablehnten. In die Studie wurden Patienten, unabhängig von ihrem Tumorzell‑PD‑L1‑Status, mit einem ECOG‑Performance‑Status 0 oder 1 eingeschlossen (ein ECOG‑Performance‑Status 2 war bei Patienten erlaubt, die für eine neoadjuvante Cisplatin‑Chemotherapie nicht geeignet waren). Die Tumorzell‑PD‑L1‑Expression wurde unter Verwendung des PD‑L1‑IHC‑28‑8‑pharmDx‑Assays bestimmt. Von der Studie ausgeschlossen waren Patienten mit aktiven, bekannten oder vermuteten Autoimmunerkrankungen und Patienten, die innerhalb von 28 Tagen vor Behandlungsbeginn mit der Studienmedikation eine Chemotherapie, Radiotherapie, biologische Krebstherapie, intravesikale Therapie oder investigative Behandlung erhalten hatten.
Insgesamt wurden 709 Patienten entweder für eine Behandlung mit Nivolumab 240 mg (n = 353) alle 2 Wochen oder Placebo (n = 356) alle 2 Wochen randomisiert. Die Behandlung wurde bis zum Wiederauftreten oder bis zu nicht akzeptabler Toxizität für eine Maximaldauer von 1 Jahr durchgeführt. Darunter befanden sich 282 Patienten mit Tumorzell‑PD‑L1‑Expression ≥ 1 %; 140 im Nivolumab‑Arm und 142 im Placebo‑Arm. Die Randomisierung wurde stratifiziert anhand des pathologischen Nodalstatus (N+ vs. N0/x mit < 10 Knoten entfernt vs. N0 mit ≥ 10 Knoten entfernt), des Tumorzell‑PD‑L1‑Expressionsstatus (≥ 1 % vs. < 1 %/unbestimmt) und der Anwendung von neoadjuvanter Cisplatin‑Chemotherapie. Tumorbeurteilungen anhand von bildgebenden Verfahren wurden nach Anwendung der ersten Dosis bis Woche 96 alle 12 Wochen durchgeführt, von Woche 96 bis Woche 160 alle 16 Wochen und danach alle 24 Wochen bis zum Wiederauftreten eines Rezidivs außerhalb des ableitenden Harntrakts oder Behandlungsabbruch (je nachdem, was später eintrat) für maximal 5 Jahre. Die primären Wirksamkeitsendpunkte waren das krankheitsfreie Überleben (disease‑free survival = DFS) bei allen randomisierten Patienten und DFS bei randomisierten Patienten mit Tumorzell‑PD‑L1‑Expression ≥ 1 %. DFS wurde definiert als der Zeitraum zwischen dem Datum der Randomisierung und dem Datum des ersten dokumentierten Wiederauftretens nach Einschätzung des Prüfarztes (Lokalrezidiv innerhalb des ableitenden Harntrakts, Rezidiv außerhalb des ableitenden Harntrakts oder Fernmetastase) oder des Todes (jeglicher Ursache), je nachdem, was zuerst eintrat. Sekundäre Wirksamkeitsendpunkte beinhalteten das Gesamtüberleben (overall survival = OS).
Die Ausgangsmerkmale waren im Allgemeinen über die Behandlungsgruppen hinweg ausgeglichen. Bei den Patienten mit Tumorzell‑PD‑L1‑Expression ≥ 1 % war das mediane Alter 66 Jahre (Spanne: 34 ‑ 92 Jahre), 76 % waren Männer und 76 % waren Weiße. 82 % hatten ein muskelinvasives Harnblasenkarzinom (MIBC), 18 % hatten ein Urothelkarzinom des oberen Harntrakts (upper tract urothelial carcinoma, UTUC) (Nierenbecken und Harnleiter), 42 % der Patienten hatten vorher Cisplatin im neoadjuvanten Setting erhalten, 45 % der Patienten hatten positive Lymphknoten (N+) nach vollständiger Tumorresektion. Der ECOG‑Performance‑Status der Patienten war 0 (61 %), 1 (37 %) oder 2 (2 %) und 7 % der Patienten hatten einen Hämoglobinwert < 10 g/dl.
Bei der primären präspezifizierten Zwischenauswertung von Patienten mit Tumorzell‑PD‑L1‑Expression ≥ 1 % (minimale Nachbeobachtungszeit 6,3 Monate und mediane Nachbeobachtungszeit 22,1 Monate im Nivolumab‑Arm), wurde eine signifikante Verbesserung des DFS bei den zu Nivolumab randomisierten Patienten im Vergleich zu Placebo gezeigt. Das mediane und vom Prüfarzt bestimmte DFS wurde für Nivolumab nicht erreicht (95 % CI: 21,19; NR) versus 8,41 Monate (95 % CI: 5,59; 21,19) für Placebo, HR 0,55 (98,72 % CI: 0,35; 0,85), p‑Wert = 0,0005. Die primäre Analyse des DFS beinhaltete die Zensierung für eine neue Krebstherapie. Die Ergebnisse für DFS mit und ohne Zensierung für eine neue Krebstherapie waren konsistent.
In einer aktualisierten deskriptiven DFS‑Analyse bei Patienten mit Tumorzell‑PD‑L1‑Expression ≥ 1 % (minimale Nachbeobachtungszeit 11,4 Monate und mediane Nachbeobachtungszeit 25,5 Monate im Nivolumab‑Arm) wurde die Verbesserung des DFS bestätigt.
Die Wirksamkeitsergebnisse dieser deskriptiven aktualisierten Analyse sind in Tabelle 34 und Abbildung 22 dargestellt.
Tabelle 34: Wirksamkeitsergebnisse bei Patienten mit Tumorzell‑PD‑L1 ≥ 1 % (CA209274)
Nivolumab |
Placebo |
||
Krankheitsfreies Überleben (DFS) |
Minimale Nachbeobachtungszeit 11,4 Monate |
||
Ergebnisse (%) |
56 (40,0) |
85 (59,9) |
|
Hazard Ratio (95 % CI)a |
0,53 (0,38; 0,75) |
||
Median (95 % CI) (Monate)b |
NR (22,11; NE) |
8,41 (5,59; 20,04) |
|
Rate (95 % CI) nach 6 Monaten |
74,5 (66,2; 81,1) |
55,7 (46,8; 63,6) |
|
Rate (95 % CI) nach 12 Monaten |
67,6 (59,0; 74,9) |
46,3 (37,6; 54,5) |
|
Rate (95 % CI) nach 24 Monaten |
58,6 (49,3; 66,9) |
37,4 (29,0; 45,8) |
|
NR = nicht erreicht (not reached), NE = nicht abschätzbar (non‑estimable). | |||
Abbildung 22: Kaplan‑Meier‑Kurven des DFS bei Patienten mit Tumor‑PD‑L1‑Expression ≥ 1 % (CA209274)
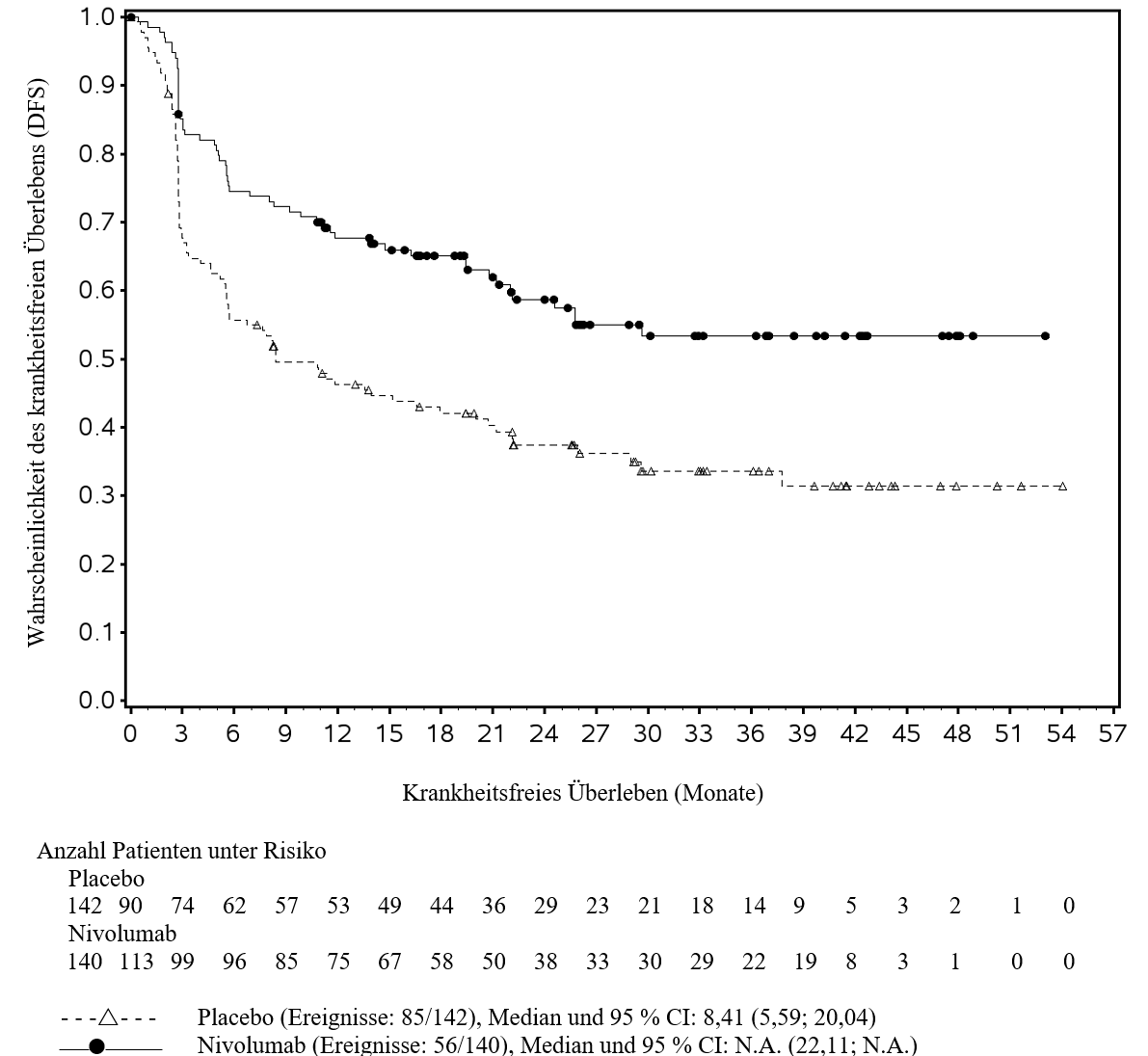
Minimale Nachbeobachtungszeit 11,4 Monate
Es wurden explorative, präspezifizierte, deskriptive Subgruppenanalysen durchgeführt, basierend auf Patienten, die im neoadjuvanten Setting zuvor mit Cisplatin behandelt wurden.
In der Subgruppe von Patienten mit Tumorzell‑PD‑L1‑Expression ≥ 1 %, die zuvor mit Cisplatin im neoadjuvanten Setting behandelt wurden (n = 118), betrug die DFS‑HR 0,37 (95 % CI: 0,22; 0,64) mit nicht erreichtem medianen DFS für den Nivolumab‑ und 8,41 Monate für den Placebo‑Arm. In der Patienten‑Subgruppe mit Tumorzell‑PD‑L1‑Expression ≥ 1 %, die zuvor kein Cisplatin im neoadjuvanten Setting erhalten hatten (n = 164), betrug die DFS‑HR 0,69 (95 % CI: 0,44; 1,08) mit medianem DFS 29,67 Monate für den Nivolumab‑ und 11,37 Monate für den Placebo‑Arm.
Behandlung des fortgeschrittenen Urothelkarzinoms
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie von Nivolumab in Kombination mit Chemotherapie vs. Chemotherapie (CA209901)
Die Sicherheit und Wirksamkeit von Nivolumab in Kombination mit Cisplatin und Gemcitabin, gefolgt von einer Nivolumab‑Monotherapie wurden in der randomisierten offenen Studie CA209901 mit Cisplatin‑geeigneten Patienten mit nicht resezierbarem oder metastasiertem Urothelkarzinom untersucht. In die Studie wurden Patienten (ab 18 Jahren) mit histologischem oder zytologischem Nachweis eines metastasierten oder chirurgisch nicht resezierbaren Urothelkarzinoms (transitional cell carcinoma, TCC) in Nierenbecken, Harnleiter, Harnblase oder Harnröhre eingeschlossen, die für Cisplatin und Gemcitabin geeignet sind. Geringe histologische Abweichungen (< 50 % insgesamt) waren akzeptabel (TCC musste die dominante Histologie sein). Alle Patienten mussten laut Computertomographie (CT) oder Magnetresonanztomographie (MRT) eine messbare Erkrankung gemäß RECIST-1.1-Kriterien haben. Eine vorherige systemische Therapie zur Behandlung eines metastasierten oder chirurgisch nicht resezierbaren Urothelkarzinoms war nicht erlaubt. Eine vorherige neoadjuvante Chemotherapie oder eine vorherige adjuvante platinbasierte Chemotherapie nach einer radikalen Zystektomie waren erlaubt, solange das Rezidiv der Erkrankung ≥ 12 Monate nach Abschluss der Therapie auftrat. Eine vorherige intravesikale Therapie war erlaubt, wenn sie mindestens 4 Wochen vor Beginn der Studienbehandlung abgeschlossen war. Eine kurative Strahlentherapie (mit oder ohne Chemotherapie) war erlaubt, wenn die Behandlung ≥ 12 Monate vor der Aufnahme abgeschlossen war. Eine palliative Strahlentherapie war erlaubt, solange sie mindestens 2 Wochen vor der Behandlung abgeschlossen war.
Insgesamt wurden 608 Patienten entweder für Nivolumab in Kombination mit Cisplatin und Gemcitabin (n = 304) oder für Cisplatin und Gemcitabin (n = 304) randomisiert. Die Randomisierung wurde nach Tumor‑PD‑L1‑Status (≥ 1 % vs. < 1 % oder unbestimmt) und Lebermetastasen (ja vs. nein) stratifiziert. Das mediane Alter betrug 65 Jahre (Spanne: 32 ‑ 86); 51 % der Patienten waren ≥ 65 Jahre und 12 % waren ≥ 75 Jahre, 23 % der Patienten waren Asiaten, 72 % waren Kaukasier, 0,3 % waren Schwarze; 77 % waren männlich, 23 % waren weiblich. Der ECOG‑Performance‑Status bei Einschluss in die Studie war 0 (53 %) oder 1 (46 %). Patienten im Behandlungsarm Nivolumab in Kombination mit Cisplatin und Gemcitabin wurden mit Nivolumab 360 mg alle drei Wochen, in Kombination mit Cisplatin und Gemcitabin für bis zu 6 Zyklen behandelt. Danach erhielten die Patienten eine Monotherapie mit Nivolumab 480 mg alle 4 Wochen für insgesamt bis zu 24 Monate. Die Patienten erhielten Gemcitabin in einer Dosis von 1 000 mg/m2 i.v. über 30 Minuten an den Tagen 1 und 8 des 3‑wöchigen Behandlungszyklus und Cisplatin in einer Dosis von 70 mg/m2 i.v. über 30 bis 120 Minuten an Tag 1 des 3‑wöchigen Behandlungszyklus. Insgesamt 92 Patienten (49 im Behandlungsarm Nivolumab in Kombination mit Cisplatin und Gemcitabin und 43 im Behandlungsarm Cisplatin und Gemcitabin) wurden nach mindestens einem Cisplatin‑Zyklus von Cisplatin auf Carboplatin umgestellt.
Die Studie zeigte eine statistisch signifikante Verbesserung des OS und PFS für Patienten, die in den Behandlungsarm Nivolumab in Kombination mit Cisplatin und Gemcitabin randomisiert worden waren im Vergleich zu Cisplatin und Gemcitabin allein. Die Wirksamkeitsergebnisse sind in Tabelle 35 und Abbildungen 23 und 24 dargestellt.
Tabelle 35: Wirksamkeitsergebnisse (CA209901)
Nivolumab und Cisplatin-Gemcitabin‑Chemotherapie |
Cisplatin‑Gemcitabin-Chemotherapie |
|
Gesamtüberlebena |
||
Ereignisse |
172 (56,6) |
193 (63,5) |
Median (Monate) |
21,7 |
18,9 |
Hazard Ratio (95 % CI)b |
0,78 |
|
p‑Wertc |
0,0171 |
|
Progressionsfreies Überlebena |
||
Ereignisse |
211 (69,4) |
191 (62,8) |
Median (Monate) |
7,92 |
7,56 |
Hazard Ratio (95 % CI)b |
0,72 |
|
p‑Wertc |
0,0012 |
|
Objektives Ansprechen |
||
Responder |
175 (57,6) |
131 (43,1) |
(95 % CI) |
(51,8; 63,2) |
(37,5; 48,9) |
a Basierend auf Kaplan‑Meier‑Schätzungen. | ||
Abbildung 23: Kaplan‑Meier‑Kurven des Gesamtüberlebens (CA209901)
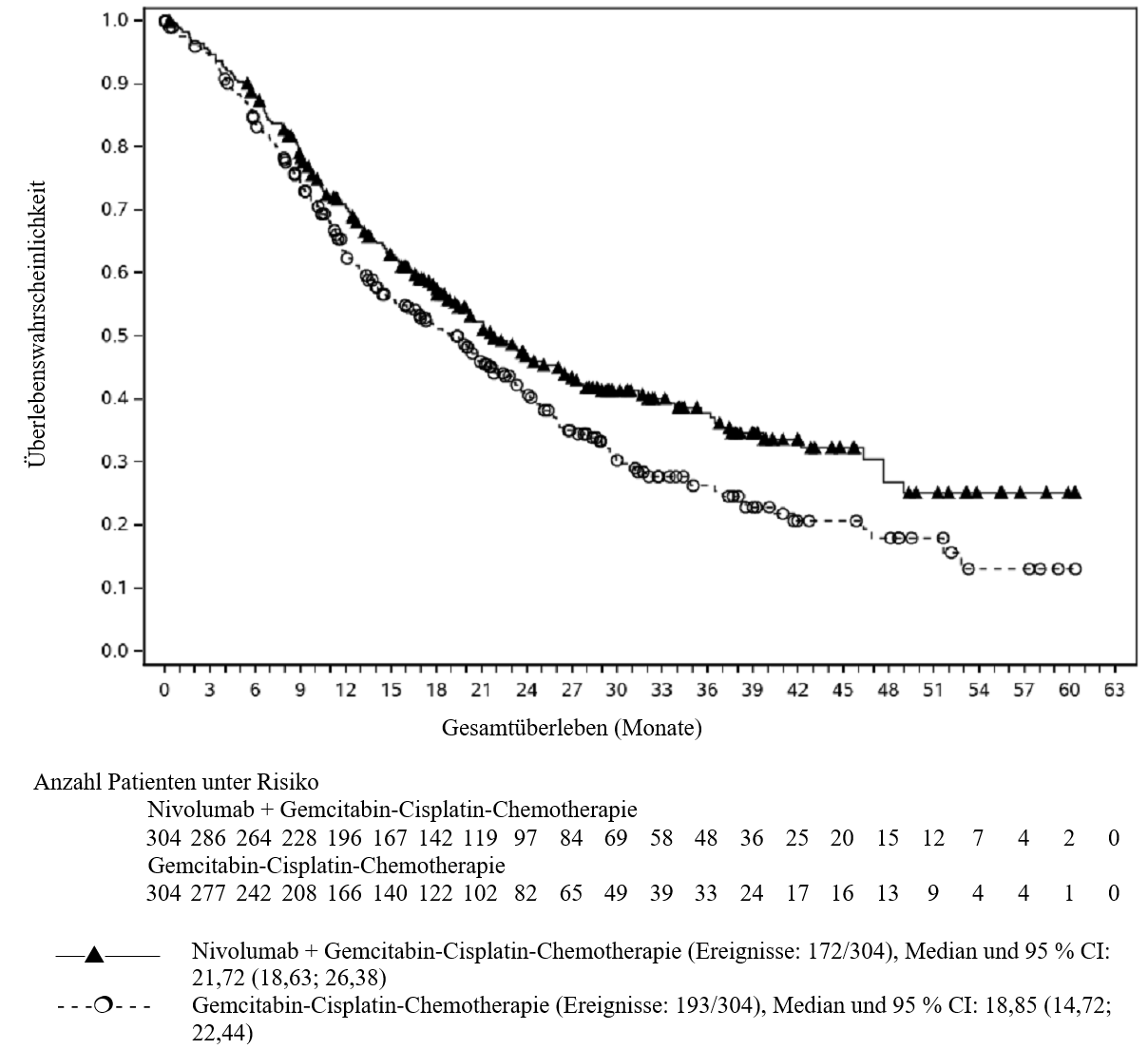
Basierend auf Datenschnitt: 09. Mai 2023, minimale Nachbeobachtungszeit von 7,4 Monaten
Abbildung 24: Kaplan‑Meier‑Kurven des progressionsfreien Überlebens (CA209901)
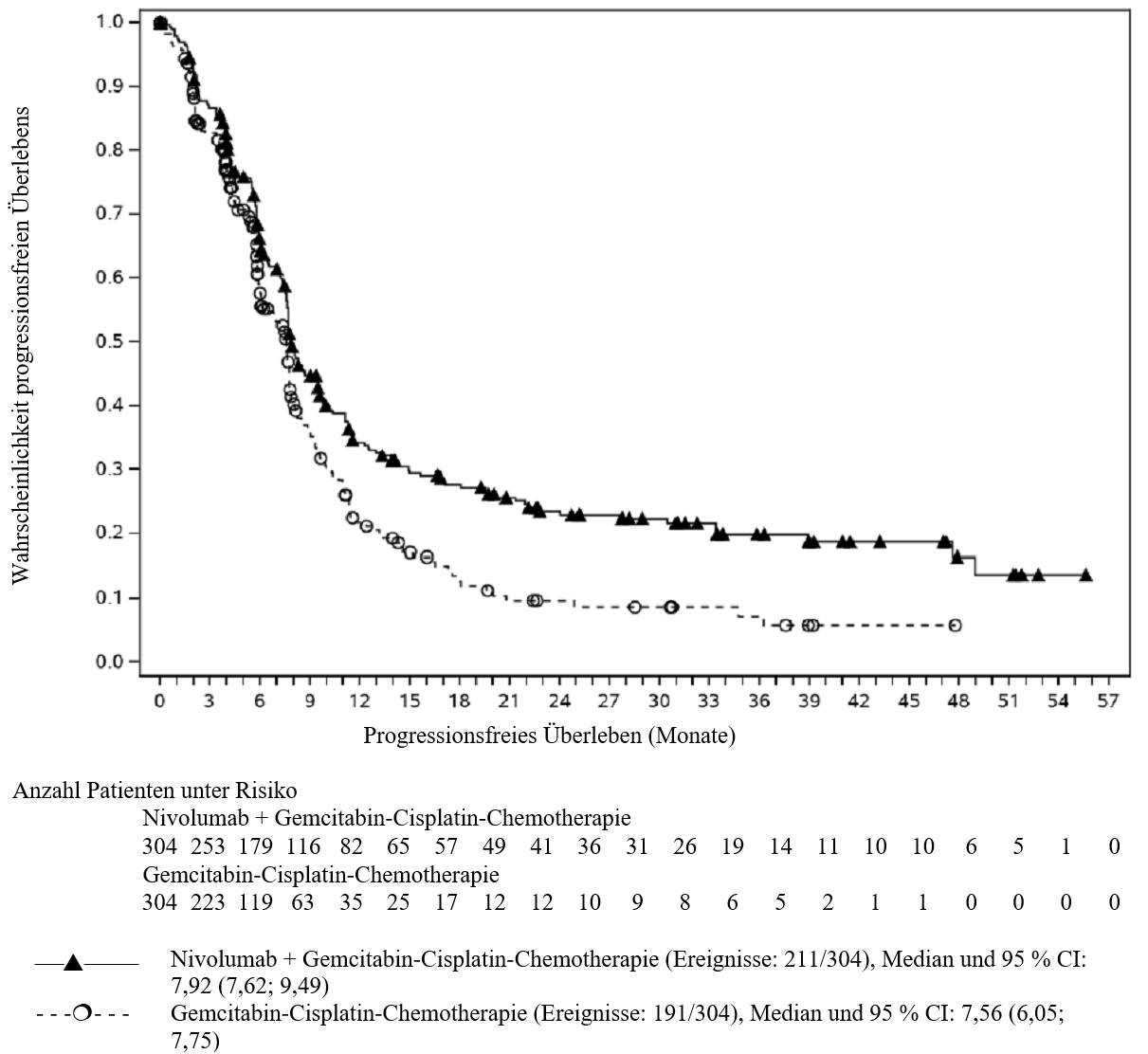
Basierend auf Datenschnitt: 09. Mai 2023, minimale Nachbeobachtungszeit von 7,4 Monaten
Die primäre PFS‑Analyse beinhaltete eine Zensierung für neue Krebsbehandlungen vor Krankheitsprogression (Tabelle 33). PFS‑Ergebnisse mit und ohne Zensierung für neue Krebsbehandlungen vor Krankheitsprogression waren konsistent.
Intravenöse Formulierung
Offene Phase‑II‑Studie (CA209275)
Die Sicherheit und Wirksamkeit von Nivolumab 3 mg/kg als Monotherapie zur Behandlung von Patienten mit lokal fortgeschrittenem oder metastasiertem Urothelkarzinom wurde in einer multizentrischen, offenen, einarmigen Phase‑II‑Studie untersucht (CA209275).
In die Studie wurden Patienten (ab 18 Jahren) eingeschlossen, die einen Progress während oder nach einer platinbasierten Chemotherapie bei fortgeschrittener oder metastasierter Erkrankung oder eine Progression innerhalb von 12 Monaten nach einer neoadjuvanten oder adjuvanten Behandlung mit einer platinbasierten Chemotherapie hatten. Die Patienten wiesen einen ECOG‑Performance‑Status von 0 oder 1 auf und wurden unabhängig vom Tumor‑PD‑L1‑Status eingeschlossen. Patienten mit aktiven Hirnmetastasen oder leptomeningealen Metastasen, mit aktiver Autoimmunerkrankung oder mit Erkrankungen, die eine immunsuppressive Therapie erforderten, waren von der Studie ausgeschlossen. Patienten mit Lebermetastasen, die mehr als 2 vorausgehende Chemotherapien erhalten hatten, waren ausgeschlossen.
Insgesamt waren 270 Patienten, die Nivolumab 3 mg/kg intravenös über 60 Minuten alle 2 Wochen erhielten, für die Beurteilung der Wirksamkeit mit einer minimalen Beobachtungszeit von 8,3 Monaten verfügbar. Die Behandlung wurde so lange fortgesetzt, wie ein klinischer Nutzen beobachtet wurde oder bis die Behandlung nicht mehr vertragen wurde. Die erste Beurteilung des Tumors wurde 8 Wochen nach Beginn der Behandlung und danach alle 8 Wochen für bis zu 48 Wochen durchgeführt, danach alle 12 Wochen bis zum Progress der Erkrankung oder der Beendigung der Therapie, je nachdem, was später erfolgte. Die Beurteilung des Tumors wurde bei Patienten, die die Therapie nicht aufgrund einer Progression beendet hatten, nach Beendigung der Therapie fortgeführt. Eine Behandlung über die anfänglich vom Prüfarzt gemäß Definition nach RECIST, Version 1.1, festgestellte Progression hinaus war erlaubt, wenn der Patient nach Einschätzung des Prüfarztes einen klinischen Nutzen von der Behandlung hatte, keine schnell fortschreitende Progression hatte und die Therapie vertrug. Der primäre Endpunkt für die Wirksamkeit war die objektive Ansprechrate (ORR), bestimmt von einem unabhängigen zentralen Komitee (Blinded Independent Central Review, BICR). Weitere Wirksamkeitsparameter waren die Dauer des Ansprechens, das progressionsfreie Überleben (PFS) und das Gesamtüberleben (OS) (siehe Tabelle 36).
Das mediane Alter war 66 Jahre (Spanne: 38 bis 90), wobei 55 % ≥65 Jahre und 14 % ≥75 Jahre alt waren. Die Mehrheit der Patienten war weiß (86 %) und männlich (78 %). Der ECOG-Performance-Status war zu Beginn 0 (54 %) oder 1 (46 %).
Tabelle 36: Wirksamkeitsergebnisse (CA209275)a
Nivolumab |
|||
Bestätigtes objektives Ansprechen |
54 (20,0 %) |
||
(95 % CI) |
(15,4; 25,3) |
||
Vollständiges Ansprechen (Complete Response = CR) |
8 (3,0 %) |
||
Teilweises Ansprechen (Partial Response = PR) |
46 (17,0 %) |
||
Stabile Erkrankung (Stable Disease = SD) |
60 (22,2 %) |
||
Mediane Dauer des Ansprechensb |
|||
Monate (Spanne) |
10,4 (1,9+ ‑ 12,0+) |
||
Mediane Zeit bis zum Ansprechen |
|||
Monate (Spanne) |
1,9 (1,6; 7,2) |
||
Progressionsfreies Überleben |
|||
Ereignisse (%) |
216 (80) |
||
Median (95 % CI) Monate |
2,0 (1,9; 2,6) |
||
Rate (95 % CI) nach 6 Monaten |
26,1 (20,9; 31,5) |
||
Gesamtüberlebenc |
|||
Ereignisse (%) |
154 (57) |
||
Median (95 % CI) Monate |
8,6 (6,05; 11,27) |
||
Rate (95 % CI) nach 12 Monaten |
41,0 (34,8; 47,1) |
||
Tumor‑PD‑L1‑Expressionslevel | |||
< 1 % |
≥ 1 % |
||
Bestätigtes objektives Ansprechen | |||
16 % (10,3; 22,7) |
25 % (17,7; 33,6) |
||
Mediane Dauer des Ansprechens | |||
10,4 (3,7; 12,0+) |
Nicht erreicht (1,9+; 12,0+) |
||
Progressionsfreies Überleben | |||
Median (95 % CI) Monate |
1,9 (1,8; 2,0) |
3,6 (1,9; 3,7) |
|
Rate (95 % CI) nach 6 Monaten |
22,0 (15,6; 29,2) |
30,8 (22,7; 39,3) |
|
Gesamtüberleben |
|||
Median (95 % CI) Monate |
5,9 (4,37; 8,08) |
11,6 (9,10; NE) |
|
Rate (95 % CI) nach 12 Monaten |
34,0 (26,1; 42,1) |
49,2 (39,6; 58,1) |
|
“+” stellt eine zensierte Beobachtung dar
a mediane Nachbeobachtung von 11,5 Monaten.
b Daten instabil wegen begrenzter Dauer des Ansprechens.
c beinhaltete 4 Todesfälle in Zusammenhang mit der Therapie: 1 Pneumonitis, 1 akuter Atemstillstand, 1 Atemstillstand und 1 Herzversagen.
NE: nicht abschätzbar (non‑estimable)
Ergebnisse einer explorativen Post‑hoc‑Analyse zeigen, dass bei Patienten mit einer niedrigen (z. B. < 1 %) oder fehlender Tumor‑PD‑L1‑Expression andere Patienten‑Charakteristika (z. B. Lebermetastasen, viszerale Metastasen, anfängliches Hämoglobin < 10 g/dl und ECOG‑Performance‑Status = 1) das klinische Ergebnis beeinflussen können.
Intravenöse Formulierung
Offene Phase‑I/II‑Studie (CA209032)
CA209032 war eine offene Phase‑I/II‑Multi‑Kohorten‑Studie mit einer Kohorte von 78 Patienten mit Urothelkarzinom, die, nach ähnlichen Einschlusskriterien wie in der Studie CA209275 mit Nivolumab‑Monotherapie 3 mg/kg behandelt wurden (18 Patienten wechselten entsprechend des Prüfplans zu einer Therapie mit Nivolumab 3 mg/kg in Kombination mit Ipilimumab 1 mg/kg über). Nach einer minimalen Nachbeobachtungszeit von 9 Monaten betrug die vom Prüfarzt festgestellte ORR 24,4 % (95 % CI: 15,3; 35,4). Die mediane Dauer des Ansprechens wurde nicht erreicht (Spanne: 4,4 ‑ 16,6+ Monate). Das mediane OS betrug 9,7 Monate (95 % CI: 7,26; 16,16) und die geschätzten OS‑Raten waren 69,2 % (CI: 57,7; 78,2) nach 6 Monaten und 45,6 % (CI: 34,2; 56,3) nach 12 Monaten.
dMMR‑ oder MSI‑H‑Kolorektalkarzinom
Intravenöse Formulierung
Offene Studie zu Nivolumab in Kombination mit Ipilimumab vs. Chemotherapie bei Patienten mit dMMR‑ oder MSI‑H‑Kolorektalkarzinom, die im metastasierten Setting nicht vorbehandelt worden sind
Die Sicherheit und Wirksamkeit von 240 mg Nivolumab in Kombination mit 1 mg/kg Ipilimumab alle 3 Wochen für maximal 4 Dosen, gefolgt von einer Nivolumab‑Monotherapie mit 480 mg alle 4 Wochen für die Erstlinienbehandlung des nicht resezierbaren oder metastasierten Kolorektalkarzinoms mit bekanntem MSI‑H‑ oder dMMR‑Status wurden in einer randomisierten, mehrarmigen, offenen Phase‑III‑Studie (CA2098HW) untersucht. Die Studienbehandlungsarme umfassten eine Nivolumab‑Monotherapie, Nivolumab in Kombination mit Ipilimumab oder eine Chemotherapie nach Wahl des Prüfarztes. Der MSI‑H‑ bzw. dMMR‑Tumorstatus wurde gemäß lokalem Behandlungsstandard mittels PCR, NGS bzw. IHC‑Assays bestimmt. Die zentrale Beurteilung des MSI‑H‑Status mittels PCR (Idylla MSI)‑Test und des dMMR‑Status mittels IHC (Omnis MMR)‑Test wurde retrospektiv an Tumorproben der Patienten durchgeführt, die für die Bestimmung des lokalen MSI‑H‑/dMMR‑Status verwendet worden waren. Die primäre Population zur Untersuchung der Wirksamkeit bestand aus Patienten mit durch zentrale Testung bestätigtem MSI‑H‑/dMMR‑Status. Patienten mit symptomatischen Hirnmetastasen, mit aktiver Autoimmunerkrankung, Patienten, die systemische Corticosteroide oder Immunsuppressiva einnahmen oder mit Checkpoint‑Inhibitoren vorbehandelt worden waren, waren von der Studie ausgeschlossen. Die Randomisierung wurde nach Lage des Tumors (rechts vs. links) stratifiziert. Die auf den Chemotherapie‑Arm randomisierten Patienten konnten bei Krankheitsprogression nach BICR-Beurteilung eine Kombinationstherapie mit Nivolumab plus Ipilimumab erhalten.
Insgesamt wurden 303 nicht vorbehandelte Patienten im metastasierten Setting in der Studie randomisiert, wobei 202 Patienten der Kombinationstherapie mit Nivolumab plus Ipilimumab und 101 Patienten der Chemotherapie‑Gruppe zugewiesen wurden. Hiervon hatten 255 Patienten einen durch zentrale Beurteilung bestätigten MSI‑H‑/dMMR‑Status, 171 im Nivolumab-plus-Ipilimumab-Kombinations-Arm und 84 im Chemotherapie-Arm. Die Patienten im Nivolumab-plus-Ipilimumab-Arm erhielten 240 mg Nivolumab alle 3 Wochen in Kombination mit 1 mg/kg Ipilimumab alle 3 Wochen für maximal 4 Dosen, gefolgt von einer Nivolumab-Monotherapie mit 480 mg alle 4 Wochen. Die Patienten im Chemotherapie‑Arm erhielten: mFOLFOX6 (Oxaliplatin, Leucovorin und Fluorouracil) mit oder ohne entweder Bevacizumab oder Cetuximab: Bolus-Regime mit Oxaliplatin 85 mg/m2, Leucovorin 400 mg/m2 und Fluorouracil 400 mg/m2, gefolgt von Fluorouracil 2 400 mg/m2 über 46 Stunden alle 2 Wochen. Bevacizumab 5 mg/kg oder Cetuximab 500 mg/m2, verabreicht vor mFOLFOX6 alle 2 Wochen; oder FOLFIRI (Irinotecan, Leucovorin und Fluorouracil) mit oder ohne entweder Bevacizumab oder Cetuximab: Bolus-Regime mit Irinotecan 180 mg/m2, Leucovorin 400 mg/m2 und Fluorouracil 400 mg/m2 und Fluorouracil 2 400 mg/m2 über 46 Stunden alle 2 Wochen. Bevacizumab 5 mg/kg oder Cetuximab 500 mg/m2, verabreicht vor FOLFIRI alle 2 Wochen. Die Behandlung wurde bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bei der Kombinationstherapie mit Nivolumab plus Ipilimumab bis zu 24 Monate fortgesetzt. Patienten, die die Kombinationstherapie aufgrund einer Nebenwirkung, die Ipilimumab zugeordnet wurde, abbrechen mussten, konnten mit Nivolumab als Monotherapie weiterbehandelt werden. Tumorbeurteilungen wurden gemäß RECIST, Version 1.1 während der ersten 24 Wochen alle 6 Wochen, dann alle 8 Wochen bis Woche 96, anschließend alle 16 Wochen bis Woche 146 und danach alle 24 Wochen durchgeführt.
Die Ausgangsmerkmale aller randomisierten, zuvor nicht aufgrund einer metastasierten Erkrankung behandelten Patienten waren wie folgt: Das mediane Alter betrug 63 Jahre (Spanne: 21 bis 87) mit 46 % ≥ 65 Jahre und 18 % ≥ 75 Jahre; 46 % der Patienten waren männlich und 86 % waren Kaukasier. Der ECOG‑Performance‑Status zu Studienbeginn war 0 (54 %) oder ≥ 1 (46 %); die Lage des Tumors war bei 68 % der Patienten rechts- und bei 32 % linksseitig; und 39 Patienten der 223 Patienten mit bekanntem Status hatten ein bestätigtes Lynch‑Syndrom. Die Ausgangsmerkmale der zuvor nicht aufgrund einer metastasierten Erkrankung behandelten Patienten mit zentral bestätigtem MSI‑H‑/dMMR‑Status waren konsistent mit denen aller randomisierten, nicht vorbehandelten Patienten. Von den 101 auf den Chemotherapie‑Arm randomisierten Patienten erhielten 88 eine Chemotherapie gemäß Protokoll, einschließlich Oxaliplatin‑basierter (58 %) und Irinotecan‑basierter (42 %) Behandlungsschemata. Darüber hinaus erhielten 66 Patienten entweder Bevacizumab (64 %) oder Cetuximab (11 %) als zielgerichtete Therapie.
Ein primärer Wirksamkeitsendpunkt der Studie war das durch ein BICR gemäß RECIST, Version 1.1‑Kriterien bestätigte progressionsfreie Überleben (PFS). Weitere Wirksamkeitsendpunkte umfassten objektive Ansprechrate (ORR) nach BICR, Gesamtüberleben (OS) und Dauer des Ansprechens.
Die Studie erreichte den primären Endpunkt bei der geplanten Zwischenanalyse und zeigte eine statistisch signifikante Verbesserung des PFS nach BICR bei Patienten mit zentral bestätigtem MSI-H-/dMMR-Status im Nivolumab-plus-Ipilimumab-Arm im Vergleich zum Chemotherapie-Arm. Die Ergebnisse bezüglich des progressionsfreien Überlebens, basierend auf der BICR‑Beurteilung, sind in Tabelle 37 und Abbildung 25 dargestellt. Zum Zeitpunkt der Zwischenanalyse waren die anderen Endpunkte, einschließlich der Daten aus dem Nivolumab‑Monotherapie‑Arm, aufgrund der Testhierarchie noch nicht getestet.
Tabelle 37: Wirksamkeitsergebnisse der Erstlinienbehandlung bei zentral bestätigtem MSI‑H‑/dMMR‑Kolorektalkarzinom (CA2098HW)a
Nivolumab + Ipilimumab |
Chemotherapie |
|
Progressionsfreies Überleben |
||
Ereignisse |
48 (28 %) |
52 (62 %) |
Hazard Ratio |
0,21 |
|
95 % CI |
(0,14; 0,32) |
|
p‑Wertb |
< 0,0001 |
|
Median (95 % CI) (Monate) |
NR (38,4; NR) |
5,9 (4,4; 7,8) |
a Mediane Nachbeobachtungsdauer 31,5 Monate (Spanne: 6,1 bis 48,4 Monate). | ||
Abbildung 25: Kaplan‑Meier‑Kurven des PFS in der Erstlinie bei Patienten mit zentral bestätigtem MSI‑H‑/dMMR‑Kolorektalkarzinom (CA2098HW)
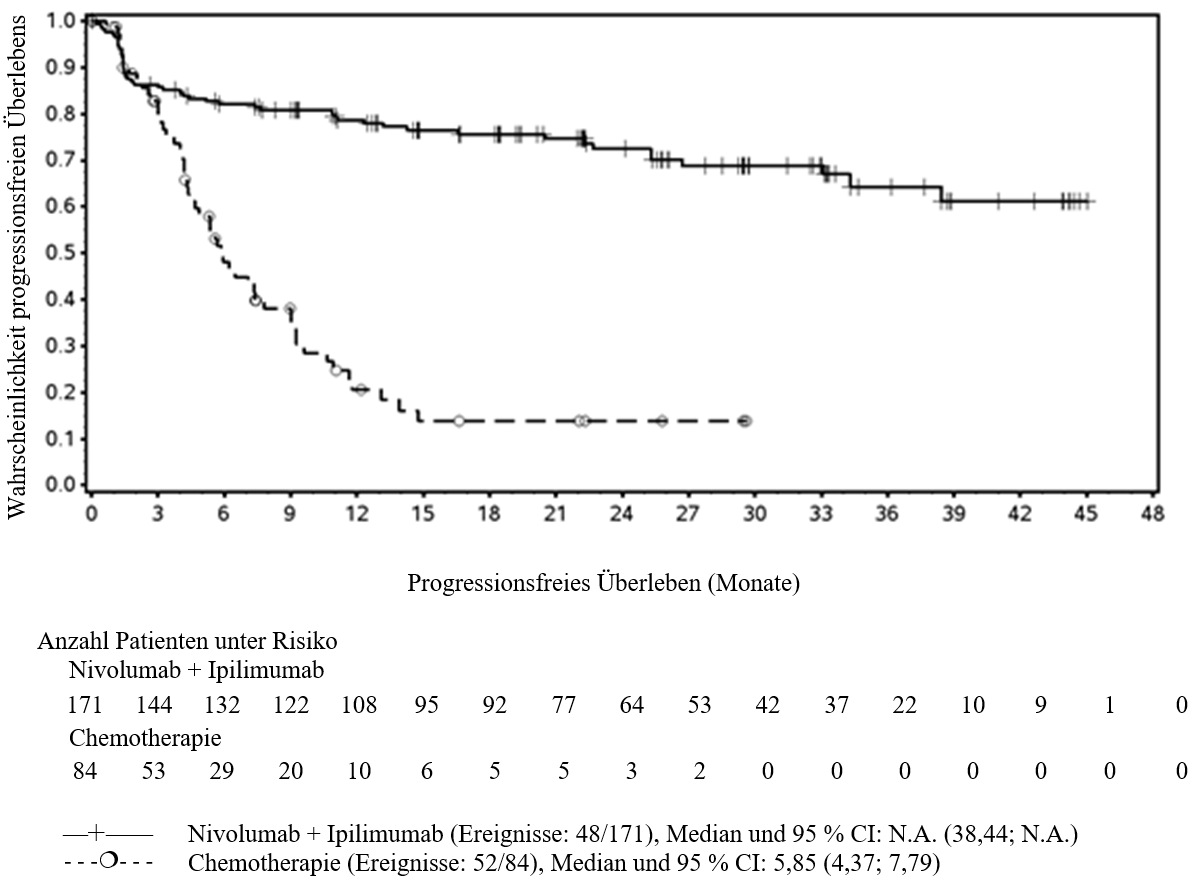
Offene Studie zu Nivolumab in Kombination mit Ipilimumab bei Patienten mit bestätigtem dMMR‑ oder MSI‑H‑Kolorektalkarzinom, die zuvor eine fluoropyrimidinbasierte Kombinationschemotherapie erhalten hatten
Die Sicherheit und Wirksamkeit von Nivolumab 3 mg/kg in Kombination mit Ipilimumab 1 mg/kg zur Behandlung des metastasierten Kolorektalkarzinoms mit Mismatch‑Reparatur‑Defizienz (dMMR) oder hoher Mikrosatelliteninstabilität (MSI‑H) wurden in einer multizentrischen, offenen, einarmigen Phase‑II‑Studie untersucht (CA209142).
In die Studie wurden Patienten (ab 18 Jahren) mit lokal bestimmtem dMMR‑ oder MSI‑H‑Status eingeschlossen, die einen Progress während oder nach einer Therapie mit Fluoropyrimidin und Oxaliplatin oder Irinotecan hatten, oder die gegen eine dieser Therapien intolerant waren. Patienten, deren letzte Vortherapie im adjuvanten Setting durchgeführt wurde, sollten mit oder innerhalb von 6 Monaten nach Abschluss der adjuvanten Chemotherapie progredient geworden sein. Die Patienten wiesen einen ECOG‑Performance‑Status von 0 oder 1 auf und wurden unabhängig vom Tumor‑PD‑L1‑Status eingeschlossen. Patienten mit aktiven Hirnmetastasen, mit aktiver Autoimmunerkrankung oder mit Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von der Studie ausgeschlossen.
Insgesamt wurden 119 Patienten behandelt und erhielten Nivolumab 3 mg/kg intravenös über 60 Minuten in Kombination mit Ipilimumab 1 mg/kg intravenös über 90 Minuten alle 3 Wochen für 4 Dosen gefolgt von einer Nivolumab‑Monotherapie mit 3 mg/kg alle 2 Wochen. Die Behandlung wurde fortgeführt, solange ein klinischer Nutzen bestand oder bis die Behandlung nicht mehr vertragen wurde. Untersuchungen des Tumors nach RECIST, Version 1.1 wurden alle 6 Wochen für die ersten 24 Wochen und dann alle 12 Wochen durchgeführt. Das primäre Wirksamkeitskriterium war das von den Prüfärzten bewertete ORR. Sekundäre Wirksamkeitskriterien waren ORR, bestätigt durch BICR, und die Krankheitskontrollrate. Die ORR‑Analyse beinhaltete die Ansprechdauer und die Zeit bis zum Ansprechen. Explorative Wirksamkeitskriterien schlossen PFS und OS mit ein.
Das mediane Alter war 58 Jahre (Spanne: 21 ‑ 88) wobei 32 % ≥ 65 und 9 % ≥ 75 Jahre alt waren, 59 % waren männlich und 92 % waren weiß. Der Ausgangs‑ECOG‑Performance‑Status betrug 0 (45 %) oder 1 (55 %), 25 % der Patienten hatten BRAF‑Mutationen, 37 % hatten KRAS‑Mutationen und bei 12 % war der Mutationsstatus unbekannt. Von den 119 behandelten Patienten, hatten 109 vorher eine fluoropyrimidinbasierte Chemotherapie im metastasierten und 9 im adjuvanten Setting erhalten. Vor Einschluss in die Studie hatten von den 119 behandelten Patienten bereits 118 (99 %) Fluorouracil, 111 (93 %) Oxaliplatin und 87 (73 %) Irinotecan als Bestandteil von vorherigen Therapieregimen erhalten; 82 Patienten (69 %) hatten eine Vortherapie mit Fluoropyrimidin, Oxaliplatin und Irinotecan erhalten. 23 %, 36 %, 24 % und 16 % haben 1, 2, 3 bzw. 4 oder mehr Vortherapien erhalten und 29 % der Patienten hatten einen EGFR‑Inhibitor erhalten.
Die Wirksamkeitsergebnisse (minimale Nachbeobachtungszeit 46,9 Monate; mediane Nachbeobachtungszeit 51,1 Monate) sind in Tabelle 38 dargestellt.
Tabelle 38: Wirksamkeitsergebnisse (CA209142)*
Nivolumab + Ipilimumab |
|
(n = 119) |
|
Bestätigtes objektives Ansprechen, n (%) |
77 (64,7) |
(95 % CI) |
(55,4; 73,2) |
Vollständiges Ansprechen |
15 (12,6) |
Teilweises Ansprechen |
62 (52,1) |
Stabile Erkrankung |
25 (21,0) |
Mediane Ansprechdauer |
|
Median (Spanne) Monate |
NR (1,4; 58,0+) |
Mediane Zeit bis zum Ansprechen |
|
Monate (Spanne) |
2,8 (1,1; 37,1) |
* nach Beurteilung des Prüfarztes | |
Das ORR, bestätigt durch BICR, war 61,3 % (95 % CI: 52,0; 70,1), einschließlich einer CR‑Rate von 20,2 % (95 % CI: 13,4; 28,5), PR‑Rate von 41,2 % (95 % CI: 32,2; 50,6) und mit einer stabilen Krankheit, die bei 22,7 % berichtet wurde. Die Beurteilung nach BICR war im Allgemeinen konsistent mit der Beurteilung durch die Prüfärzte. Ein bestätigtes Ansprechen wurde unabhängig vom BRAF‑ oder KRAS‑Mutationsstatus und des Tumor‑PD‑L1‑Expressionslevels beobachtet.
Von den 119 Patienten waren 11 (9,2 %) Patienten ≥ 75 Jahre. Das durch die Prüfärzte beurteilte ORR betrug bei den Patienten ≥ 75 Jahren 45,5 % (95 % CI: 16,7; 76,6).
Plattenepithelkarzinom des Ösophagus
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie mit Nivolumab in Kombination mit Ipilimumab vs. Chemotherapie und Nivolumab in Kombination mit Chemotherapie vs. Chemotherapie als Erstlinientherapie (CA209648)
Sicherheit und Wirksamkeit von Nivolumab in Kombination mit Ipilimumab und Nivolumab in Kombination mit Chemotherapie wurden in einer randomisierten, aktiv‑kontrollierten, offenen Studie untersucht (CA209648). In die Studie wurden erwachsene Patienten (ab 18 Jahren) mit nicht vorbehandeltem, nicht resezierbarem fortgeschrittenen, rezidivierten oder metastasierten Plattenepithelkarzinom des Ösophagus (ESCC) eingeschlossen. Patienten wurden unabhängig vom Tumor‑PD‑L1‑Status eingeschlossen und die Tumorzell‑PD‑L1‑Expression wurde mithilfe des PD‑L1‑IHC‑28‑8‑pharmDx‑Assays bestimmt. Die Patienten mussten ein Plattenepithelkarzinom oder ein adenosquamöses Karzinom des Ösophagus aufweisen und nicht geeignet sein für eine Chemo‑Strahlentherapie und/oder Operation. Vorangegangene adjuvante, neoadjuvante oder definitive Chemo‑, Strahlen‑ oder Chemoradiotherapie war zulässig, wenn sie als Teil einer kurativen Behandlung vor Beginn der Studie gegeben wurde. Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, mit symptomatischen Hirnmetastasen, mit aktiver Autoimmunerkrankung, Patienten, die systemische Corticosteroide oder Immunsuppressiva einnahmen, oder Patienten mit einem erhöhten Risiko für Blutungen und Fisteln aufgrund von offensichtlicher Tumorinvasion in angrenzende Organe des ösophagealen Tumors waren von der klinischen Studie ausgeschlossen. Die Randomisierung wurde nach Tumorzell‑PD‑L1‑Status (≥ 1 % vs. < 1 % oder unbestimmt), Region (Ostasien vs. übriges Asien vs. Rest der Welt), ECOG‑Performance‑Status (0 vs. 1) und Anzahl der Organe mit Metastasen (≤ 1 vs. ≥ 2) stratifiziert.
Insgesamt wurden 970 Patienten randomisiert und erhielten entweder Nivolumab in Kombination mit Ipilimumab (n = 325), Nivolumab in Kombination mit Chemotherapie (n = 321) oder Chemotherapie (n = 324). Davon hatten 473 Patienten eine Tumorzell‑PD‑L1‑Expression ≥ 1 %, 158 im Nivolumab‑plus‑Ipilimumab‑Arm, 158 im Nivolumab‑plus‑Chemotherapie‑Arm und 157 im Chemotherapie‑Arm. Patienten im Nivolumab‑plus‑Ipilimumab‑Arm erhielten 3 mg/kg Nivolumab alle 2 Wochen in Kombination mit Ipilimumab 1 mg/kg alle 6 Wochen und Patienten im Nivolumab‑plus‑Chemotherapie‑Arm erhielten 240 mg Nivolumab alle 2 Wochen an den Tagen 1 und 15, Fluorouracil 800 mg/m2/Tag intravenös von Tag 1 bis Tag 5 (für 5 Tage) und Cisplatin 80 mg/m2 intravenös an Tag 1 (eines vierwöchigen Zyklus). Patienten im Chemotherapie‑Arm erhielten Fluorouracil 800 mg/m2/Tag intravenös von Tag 1 bis Tag 5 (für 5 Tage) und Cisplatin 80 mg/m2 intravenös an Tag 1 (eines vierwöchigen Zyklus). Die Behandlung wurde bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate fortgesetzt. Patienten im Nivolumab‑plus‑Ipilimumab‑Arm, welche die Kombinationstherapie aufgrund einer Nebenwirkung, die Ipilimumab zugeordnet wurde, abbrechen mussten, konnten mit der Nivolumab‑Monotherapie weiter behandelt werden. Patienten im Nivolumab‑plus‑Chemotherapie‑Arm, bei welchen entweder die Fluorouracil‑ und/oder Cisplatinbehandlung abgebrochen wurde, konnten mit anderen Komponenten des Therapieregimes weiter behandelt werden.
Die Basischarakteristika der Behandlungsgruppen waren im Allgemeinen ausgewogen. Bei Patienten mit einer Tumorzell‑PD‑L1‑Expression ≥ 1 % betrug das mediane Alter 63 Jahre (Spanne: 26 ‑ 85), 8,2 % waren ≥ 75 Jahre, 81,8 % waren männlich, 73,1 % waren Asiaten und 23,3 % waren weiß. Patienten hatten histologisch bestätigte Plattenepithelkarzinome (98,9 %) oder adenosquamöse Karzinome (1,1 %) des Ösophagus. Der Ausgangs‑ECOG‑Performance‑Status war 0 (45,2 %) oder 1 (54,8 %).
Nivolumab in Kombination mit Chemotherapie vs. Chemotherapie
Die primären Wirksamkeitskriterien waren PFS (durch BICR) und OS bei Patienten mit Tumorzell‑PD‑L1‑Expression ≥ 1 %. Sekundäre Endpunkte gemäß der vordefinierten hierarchischen Testung enthielten OS, PFS (durch BICR) und ORR (durch BICR) bei allen randomisierten Patienten. Tumorbeurteilungen wurden gemäß RECIST, Version 1.1 alle 6 Wochen bis zu und einschließlich Woche 48, danach alle 12 Wochen durchgeführt.
Bei der primären, präspezifizierten Analyse mit einer minimalen Nachbeobachtungszeit von 12,9 Monaten zeigte die Studie eine statistisch signifikante Verbesserung des OS und PFS für Patienten mit einer Tumorzell‑PD‑L1‑Expression ≥ 1 %. Die Wirksamkeitsergebnisse sind in Tabelle 39 dargestellt.
Tabelle 39: Wirksamkeitsergebnisse bei Patienten mit Tumorzell‑PD‑L1 ≥ 1 % (CA209648)
Nivolumab + Chemotherapie |
Chemotherapiea |
|
Gesamtüberleben |
||
Ereignisse |
98 (62,0 %) |
121 (77,1 %) |
Hazard‑Ratio (99,5 % CI)b |
0,54 (0,37; 0,80) |
|
p‑Wertc |
< 0,0001 |
|
Median (95 % CI) (Monate)d |
15,44 (11,93; 19,52) |
9,07 (7,69; 9,95) |
Rate (95 % CI) nach 12 Monatend |
58,0 (49,8; 65,3) |
37,1 (29,2; 44,9) |
Progressionsfreies Überlebene |
||
Ereignisse |
117 (74,1 %) |
100 (63,7 %) |
Hazard‑Ratio (98,5 % CI)b |
0,65 (0,46; 0,92) |
|
p‑Wertc |
0,0023 |
|
Median (95 % CI) (Monate)d |
6,93 (5,68; 8,34) |
4,44 (2,89; 5,82) |
Rate (95 % CI) nach 12 Monatend |
25,4 (18,2; 33,2) |
10,5 (4,7; 18,8) |
Gesamtansprechen, n (%)e |
84 (53,2) |
31 (19,7) |
(95 % CI) |
(45,1; 61,1) |
(13,8; 26,8) |
Vollständiges Ansprechen |
26 (16,5) |
8 (5,1) |
Teilweises Ansprechen |
58 (36,7) |
23 (14,6) |
Ansprechdauere |
||
Median (95 % CI) (Monate)d |
8,38 (6,90; 12,35) |
5,68 (4,40; 8,67) |
Spanne |
1,4+; 34,6 |
1,4+; 31,8+ |
a Fluorouracil und Cisplatin. |
Bei einer aktualisierten deskriptiven Auswertung mit einer minimalen Nachbeobachtungszeit von 20 Monaten, waren die Verbesserungen des OS konsistent mit der primären Analyse. Das mediane OS betrug 15,05 Monate (95 % CI: 11,93; 18,63) für Nivolumab plus Chemotherapie vs. 9,07 Monate (95 % CI: 7,69; 10,02) für Chemotherapie (HR = 0,59; 95 % CI: 0,46; 0,76). Das mediane PFS betrug 6,93 Monate (95 % CI: 5,68; 8,35) für Nivolumab plus Chemotherapie vs. 4,44 Monate (95 % CI: 2,89; 5,82) für Chemotherapie (HR = 0,66; 95 % CI: 0,50; 0,87). Die ORR betrug 53,2 % (95 % CI: 45,1; 61,1) für Nivolumab plus Chemotherapie vs. 19,7 % (95 % CI: 13,8; 26,8) für Chemotherapie.
Die Kaplan‑Meier-Kurven für OS und PFS mit einer minimalen Nachbeobachtungszeit von 20 Monaten sind in den Abbildungen 26 und 27 dargestellt.
Abbildung 26: Kaplan‑Meier‑Kurven des OS bei Patienten mit Tumorzell‑PD‑L1 ≥ 1 % (CA209648)
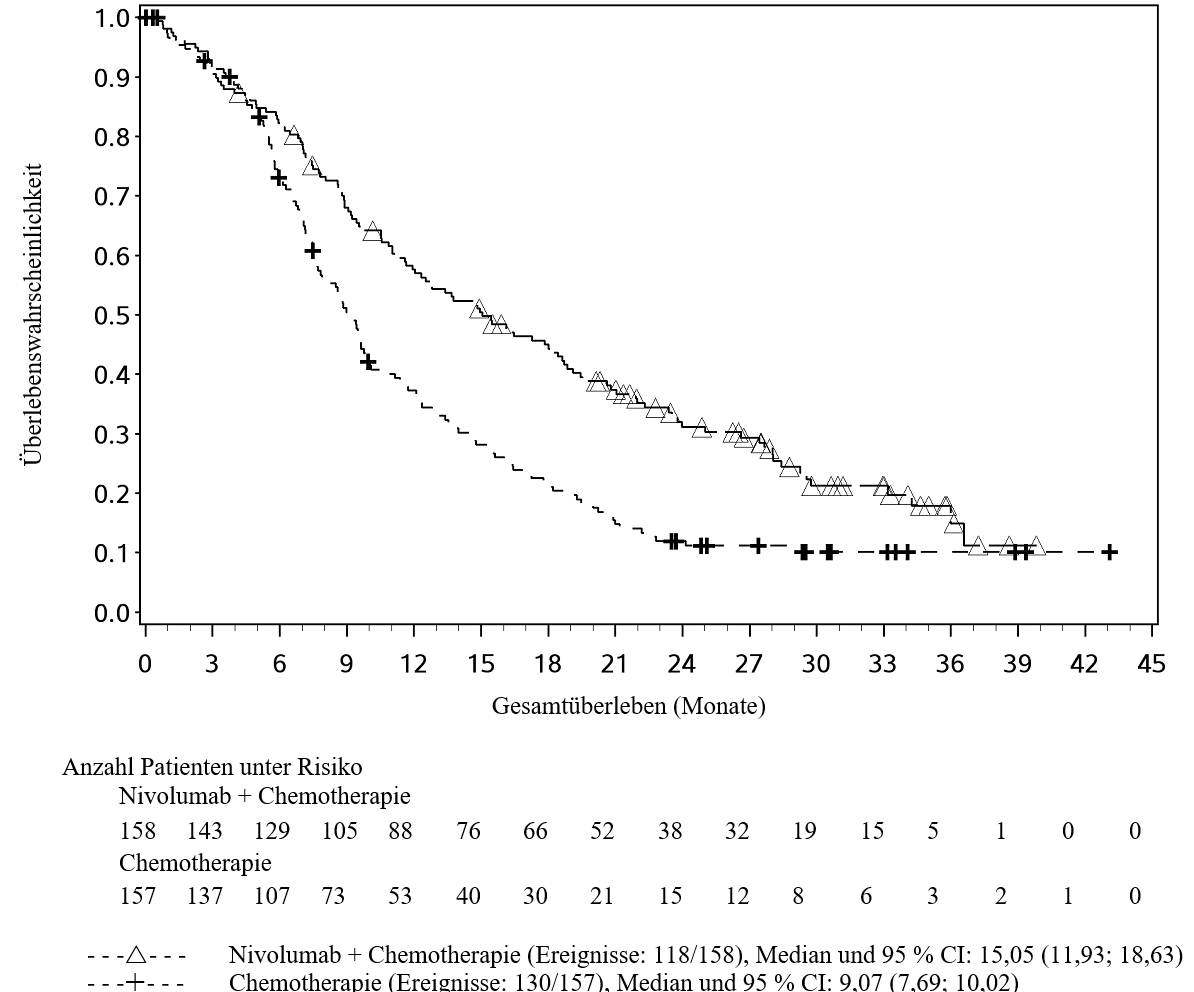
Basierend auf Datenschnitt: 23‑Aug‑2021, minimale Nachbeobachtungszeit von 20 Monaten
Abbildung 27: Kaplan‑Meier‑Kurven des PFS bei Patienten mit Tumorzell‑PD‑L1 ≥ 1 % (CA209648)
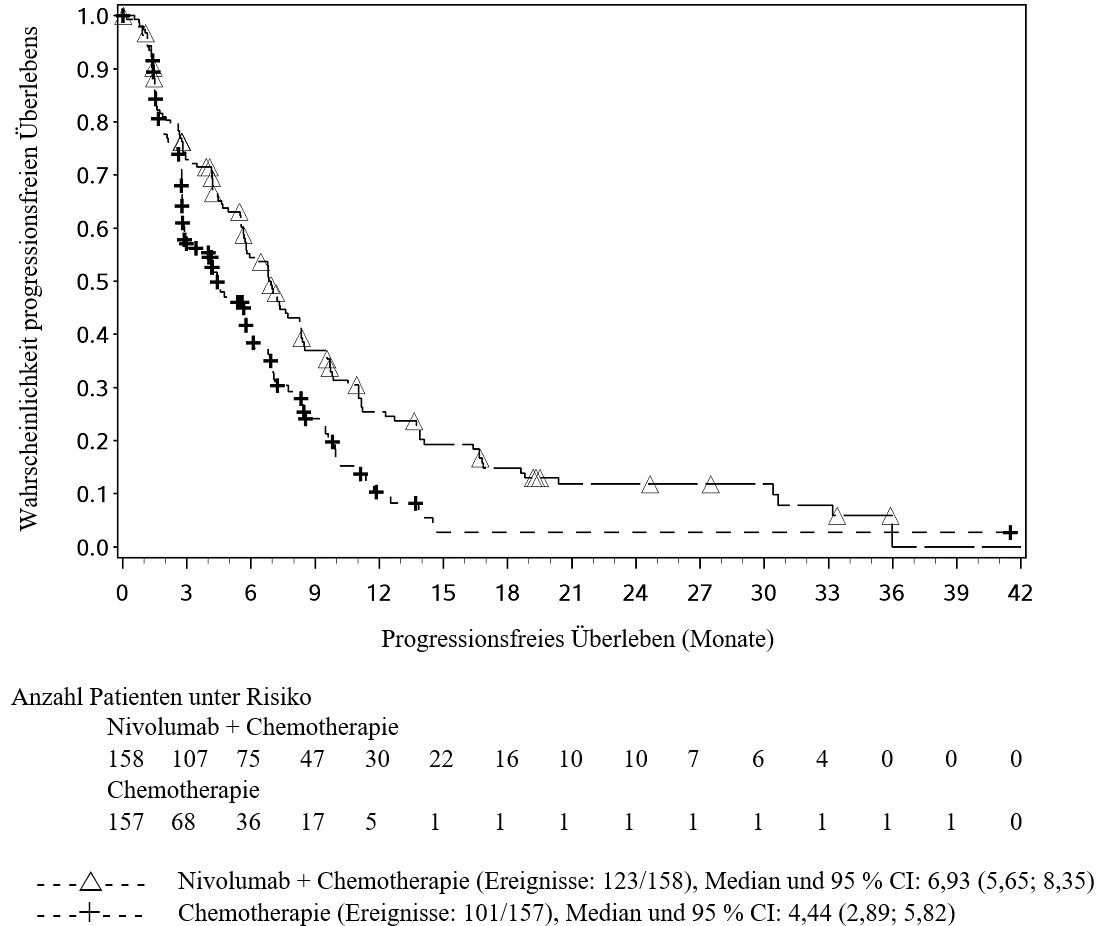
Basierend auf Datenschnitt: 23‑Aug‑2021, minimale Nachbeobachtungszeit von 20 Monaten
Intravenöse Formulierung
Randomisierte Phase‑III‑Studie mit Nivolumab‑Monotherapie in vorbehandelten Patienten (ONO‑4538‑24/ CA209473)
Die Sicherheit und Wirksamkeit von Nivolumab 240 mg als Monotherapie zur Behandlung des nicht‑resezierbaren fortgeschrittenen, rezidivierten oder metastasierten Plattenepithelkarzinoms des Ösophagus (ESCC) wurde in einer aktiv kontrollierten, offenen Phase‑III‑Studie untersucht (ONO‑4538‑24/CA209473). In die Studie wurden erwachsene Patienten (ab 20 Jahren) eingeschlossen, die gegenüber mindestens einem fluoropyrimidin‑ und platinbasierten Kombinationsregime refraktär oder intolerant waren. Patienten wurden unabhängig vom Tumor‑PD‑L1‑Expressionslevel eingeschlossen. Patienten, die gegenüber einer Taxan‑Therapie refraktär oder intolerant waren, Patienten mit symptomatischen oder behandlungsbedürftigen Hirnmetastasen, Patienten mit aktiver Autoimmunerkrankung oder mit Erkrankungen, die eine systemische immunsuppressive Therapie erforderten, und Patienten mit offensichtlicher Tumorinvasion in angrenzende Organe des Ösophagus (z. B. in die Aorta oder den Respirationstrakt) waren von der Studie ausgeschlossen.
Insgesamt wurden 419 Patienten 1:1 randomisiert und erhielten entweder Nivolumab 240 mg intravenös über 30 Minuten alle 2 Wochen (n = 210) oder eine Taxan‑haltige Chemotherapie nach Wahl des Prüfarztes: entweder Docetaxel (n = 65) 75 mg/m2 intravenös alle 3 Wochen oder Paclitaxel (n = 144) 100 mg/m2 intravenös einmal wöchentlich für 6 Wochen, gefolgt von 1 Woche Pause. Die Randomisierung wurde nach Region (Japan vs. Rest der Welt), Anzahl der Organe mit Metastasen (≤ 1 vs. ≥ 2) und Tumor‑PD‑L1‑Expression (≥ 1 % vs. < 1 % oder unbestimmt) stratifiziert. Die Behandlung wurde bis zum Progress der Erkrankung, beurteilt vom Prüfarzt gemäß Definition nach RECIST, Version 1.1, oder bis die Behandlung nicht mehr vertragen wurde fortgeführt. Untersuchungen des Tumors wurden alle 6 Wochen für 1 Jahr und danach alle 12 Wochen durchgeführt. Eine Behandlung über die anfänglich vom Prüfarzt festgestellte Progression hinaus war erlaubt, wenn der Patient, der Nivolumab bekam, keine schnell fortschreitende Progression aufwies, nach Einschätzung des Prüfarztes einen klinischen Nutzen von der Behandlung hatte, die Therapie vertrug, einen stabilen Performance‑Status aufwies und wenn die Behandlung über den Progress hinaus nicht zu einer Verzögerung einer dringenden Intervention zur Verhinderung von schweren Komplikationen im Zusammenhang mit dem Fortschreiten der Erkrankung führte (z. B. Hirnmetastasen). Der primäre Endpunkt für die Wirksamkeit war das Gesamtüberleben (OS). Wichtige sekundäre Endpunkte für die Wirksamkeit waren die objektive Ansprechrate (ORR) und das progressionsfreie Überleben (PFS), beurteilt vom Prüfarzt. Zusätzliche, präspezifizierte Subgruppen‑Analysen wurden durchgeführt, um die Wirksamkeit in Abhängigkeit der Tumor‑PD‑L1‑Expression mit der zuvor festgelegten Schwelle von 1 % zu evaluieren. Die Tumor‑PD‑L1‑Expression wurde mithilfe des PD‑L1‑IHC‑28‑8‑pharmDx‑Assays bestimmt.
Die Patientencharakteristika zu Beginn der Studie waren im Allgemeinen ausgewogen zwischen den zwei Gruppen. Das mediane Alter betrug 65 Jahre (Spanne: 33 ‑ 87), 53 % waren ≥ 65 Jahre, 10 % waren ≥ 75 Jahre, 87 % waren männlich, 96 % waren Asiaten und 4 % waren Kaukasier. Der ECOG‑Performance‑Status zu Studienbeginn war 0 (50 %) oder 1 (50 %).
Nach einer minimalen Nachbeobachtungszeit von 17,6 Monaten zeigte die Studie eine statistisch signifikante Verbesserung des Gesamtüberlebens für Patienten, die für Nivolumab randomisiert wurden, verglichen mit einer Taxan‑haltige Chemotherapie nach Wahl des Prüfarztes. Die Wirksamkeitsergebnisse sind in Tabelle 40 und Abbildung 28 dargestellt.
In den ersten 2,5 Monaten traten bei einem höheren Anteil von Patienten im Nivolumab‑Arm Todesfälle auf (32/210; 15,2 %) verglichen mit Patienten im Chemotherapie‑Arm (15/209; 7,2 %). Es konnten keine spezifischen Faktoren im Zusammenhang mit den frühen Todesfällen identifiziert werden.
Tabelle 40: Wirksamkeitsergebnisse (ONO‑4538‑24/CA209473)
Nivolumab |
Wahl des Prüfarztes |
|
Gesamtüberlebena |
||
Ereignisse |
160 (76 %) |
173 (83 %) |
Hazard Ratio (95 % CI)b |
0,77 (0,62; 0,96) |
|
p‑Wertc |
0,0189 |
|
Median (95 % CI) (Monate) |
10,9 (9,2; 13,3) |
8,4 (7,2; 9,9) |
Objektives Ansprechend;e |
33 (19,3 %) |
34 (21,5 %) |
(95 % CI) |
(13,7; 26,0) |
(15,4; 28,8) |
Vollständiges Ansprechen |
1 (0,6 %) |
2 (1,3 %) |
Teilweises Ansprechen |
32 (18,7 %) |
32 (20,3 %) |
Stabile Erkrankung |
31 (18,1 %) |
65 (41,1 %) |
Mediane Dauer des Ansprechens |
6,9 (5,4; 11,1) |
3,9 (2,8; 4,2) |
Progressionsfreies Überlebena |
||
Ereignisse |
187 (89 %) |
176 (84 %) |
Median (95 % CI) (Monate) |
1,7 (1,5; 2,7) |
3,4 (3,0; 4,2) |
Hazard Ratio (95 % CI)b |
1,1 (0,9; 1,3) |
|
a Basierend auf der ITT‑Analyse. | ||
Abbildung 28: Kaplan‑Meier‑Kurven des Gesamtüberlebens (ONO‑4538‑24/CA209473)
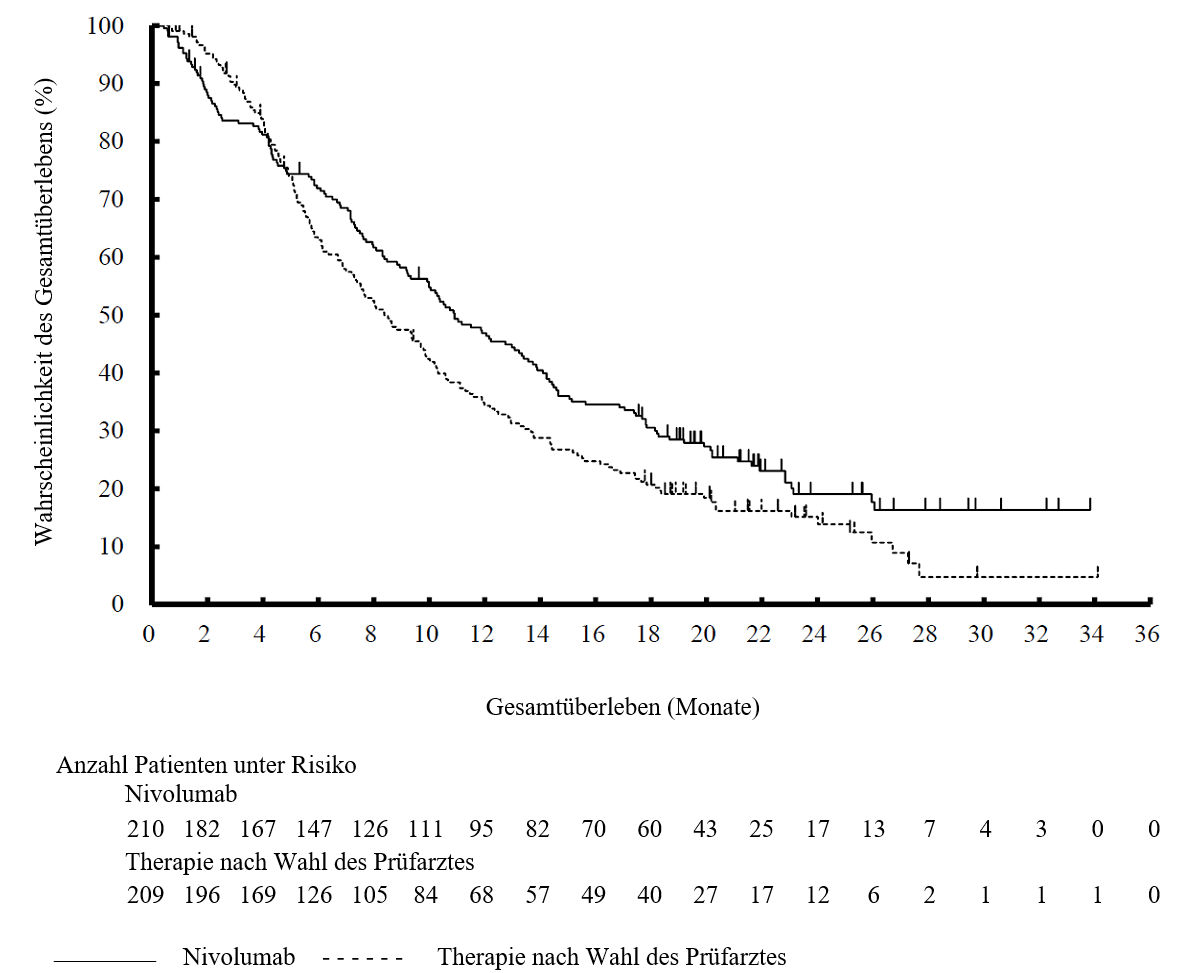
Eine Tumor‑PD‑L1‑Expression ≥ 1 % wurde bei 48 % der 419 Patienten gemessen. Die restlichen 52 % der Patienten wiesen eine Tumor‑PD‑L1‑Expression < 1 % auf. Die Hazard Ratio (HR) für OS in der Tumor‑PD‑L1‑positiven Subgruppe war 0,69 (95 % CI: 0,51; 0,94) mit einem medianen Gesamtüberleben von 10,9 bzw. 8,1 Monaten für Nivolumab bzw. Taxan‑haltige Chemotherapie nach Wahl des Prüfarztes. In der Tumor‑PD‑L1‑negativen ESCC‑Subgruppe lag die HR für OS bei 0,84 (95 % CI: 0,62; 1,14) mit einem medianen Gesamtüberleben von 10,9 bzw. 9,3 Monaten für Nivolumab bzw. Chemotherapie.
Adjuvante Behandlung der Karzinome des Ösophagus oder des gastroösophagealen Übergangs
Intravenöse Formulierung
Die Sicherheit und Wirksamkeit von Nivolumab als Monotherapie zur adjuvanten Behandlung der Karzinome des Ösophagus oder des gastroösophagealen Übergangs wurden in einer multizentrischen, randomisierten, Placebo‑kontrollierten, doppelblinden Phase‑III‑Studie untersucht (CA209577). In die Studie wurden erwachsene Patienten eingeschlossen, die eine CRT, gefolgt von einer operativen Komplettresektion des Karzinoms innerhalb von 16 Wochen vor Randomisierung erhalten hatten, und die, nach Beurteilung des Prüfarztes, eine pathologische Resterkrankung mit mindestens ypN1 oder ypT1 aufwiesen. Patienten mit einem anfänglichen ECOG‑Performance‑Status ≥ 2, Patienten, die keine gleichzeitige CRT vor der Operation erhalten hatten, Patienten mit Stadium IV resezierbarer Erkrankung, aktiver Autoimmunerkrankung oder Erkrankungen, die eine systemische immunsuppressive Therapie erfordern, waren von der Studie ausgeschlossen. Patienten wurden unabhängig vom Tumor‑PD‑L1‑Expressionslevel eingeschlossen.
Insgesamt wurden 794 Patienten 2:1 randomisiert und erhielten entweder Nivolumab 240 mg (n = 532) oder Placebo (n = 262). Die Patienten erhielten Nivolumab intravenös über 30 Minuten alle 2 Wochen für 16 Wochen, gefolgt von 480 mg infundiert über 30 Minuten alle 4 Wochen, beginnend in Woche 17. Patienten erhielten Placebo über 30 Minuten nach dem gleichen Dosierungsschema wie Nivolumab. Die Randomisierung wurde nach Tumor‑PD‑L1‑Expression (≥ 1 % vs. < 1 % oder unbestimmt oder nicht bewertbar), pathologischem Lymphknotenstatus (positiv ≥ ypN1 vs. negativ ypN0) und Histologie (Plattenepithel‑ vs. Adenokarzinom) stratifiziert. Die Behandlung wurde bis zum Wiederauftreten der Erkrankung, bis die Behandlung nicht mehr vertragen wurde oder bis zu einer Gesamtdauer von 1 Jahr fortgeführt. Der primäre Endpunkt für die Wirksamkeit war das krankheitsfreie Überleben (disease free survival, DFS), beurteilt vom Prüfarzt, definiert als die Zeit zwischen dem Zeitpunkt der Randomisierung und dem Zeitpunkt des ersten Wiederauftretens (lokal, regional oder entfernt von der primär resezierten Lokalisation) oder Tod jeglicher Ursache, je nachdem, was zuerst eintrat. Die behandelten Patienten wurden mithilfe von Bildgebung bezüglich eines Wiederauftretens des Tumors in den ersten 2 Jahren alle 12 Wochen und in den Jahren 3 bis 5 mindestens einmal alle 6 bis 12 Monate untersucht.
Die Patientencharakteristika zu Beginn der Studie waren im Allgemeinen ausgewogen zwischen den zwei Gruppen. Das mediane Alter betrug 62 Jahre (Spanne: 26 bis 86 Jahre), wobei 36 % ≥ 65 Jahre und 5 % ≥ 75 Jahre waren. Die Mehrheit der Patienten war weiß (82 %) und männlich (85 %). Der ECOG‑Performance‑Status zu Studienbeginn war 0 (58 %) oder 1 (42 %).
Bei der primären, präspezifizierten Interimsanalyse (minimale Nachbeobachtung 6,2 Monate und mediane Nachbeobachtung 24,4 Monate), zeigte die Studie eine statistisch signifikante Verbesserung des DFS für Patienten, die in den Nivolumab‑Arm randomisiert wurden im Vergleich zu Placebo. Das mediane DFS, beurteilt vom Prüfarzt, war 22,41 Monate (95 % CI: 16,62; 34,00) für Nivolumab versus 11,04 Monate (95 % CI: 8,34; 14,32) für Placebo, HR 0,69 (96,4 % CI: 0,56; 0,86), p‑Wert < 0,0003. Die primäre Analyse des DFS beinhaltete die Zensierung für eine neue Krebstherapie. Die Ergebnisse für DFS mit und ohne Zensierung für eine neue Krebstherapie waren konsistent. In einer aktualisierten, deskriptiven DFS‑Analyse mit einer minimalen Nachbeobachtungszeit von 14 Monaten und einer medianen Nachbeobachtungszeit von 32,2 Monaten wurde die Verbesserung des DFS bestätigt. Die Wirksamkeitsergebnisse dieser deskriptiven zweiten Analyse sind in Tabelle 41 und Abbildung 29 dargestellt.
Tabelle 41: Wirksamkeitsergebnisse (CA209577)
Nivolumab |
Placebo |
|
Krankheitsfreies Überleben (disease‑free survival, DFS)a mit minimaler Nachbeobachtung von 14 Monatenc | ||
Ereignisse (%) |
268 (50) |
171 (65) |
Hazard Ratio (95 % CI)b |
0,67 (0,55; 0,81) |
|
Median (95 % CI) (Monate) |
22,4 (17,0; 33,6) |
10,4 (8,3; 13,9) |
Rate (95 % CI) nach 6 Monaten |
72,6 (68,5; 76,3) |
61,5 (55,3; 67,1) |
Rate (95 % CI) nach 12 Monaten |
61,8 (57,4; 65,8) |
45,5 (39,3; 51,4) |
Rate (95 % CI) nach 24 Monaten |
48,3 (43,7; 52,8) |
36,0 (29,9; 42,0) |
a Basierend auf allen randomisierten Patienten. | ||
Abbildung 29: Kaplan‑Meier‑Kurven des krankheitsfreien Überlebens (DFS) (CA209577)
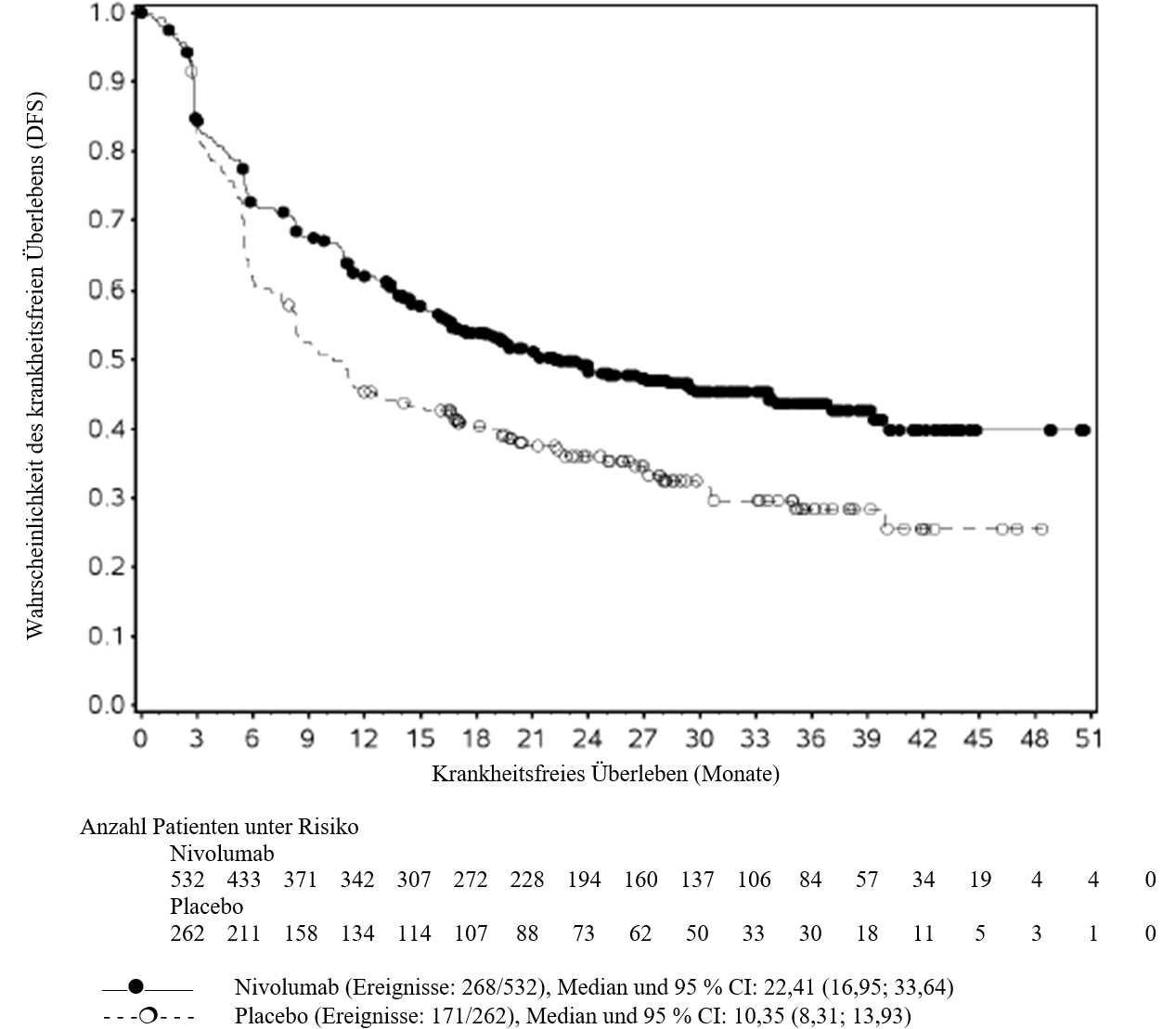
Basierend auf Datenschnitt: 18. Februar 2021, minimale Nachbeobachtungszeit von 14 Monaten
Bei der finalen OS‑Analyse mit einer minimalen Nachbeobachtungszeit von 60 Monaten betrug die HR für OS 0,85 (95,87 % CI: 0,70; 1,04), p‑Wert = 0,1064. Das mediane OS war 51,71 (95 % CI: 41,03; 61,63) Monate im Nivolumab‑Arm gegenüber 35,25 (95 % CI: 30,72; 48,76) Monate im Placebo‑Arm. Die Kaplan‑Meier‑Kurven für das OS mit einer minimalen Nachbeobachtungszeit von 60 Monaten sind in Abbildung 30 dargestellt.
Abbildung 30: Kaplan‑Meier‑Kurven des Gesamtüberlebens (OS) (CA209577)
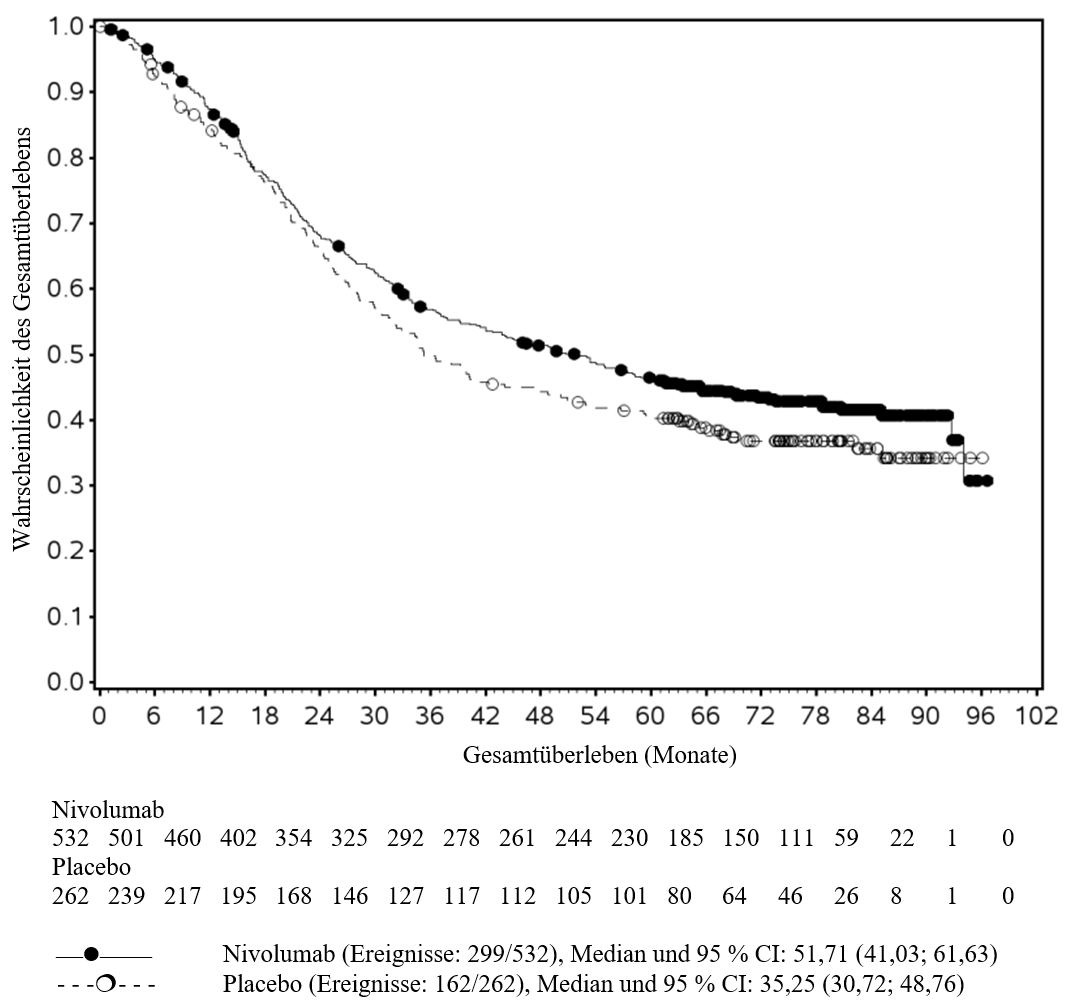
Basierend auf Datenschnitt: 17. Dez. 2024, minimale Nachbeobachtungszeit von 60 Monaten
Adenokarzinome des Magens, des gastroösophagealen Übergangs oder des Ösophagus
Intravenöse Formulierung
Sicherheit und Wirksamkeit von Nivolumab 240 mg alle 2 Wochen oder 360 mg alle 3 Wochen in Kombination mit Chemotherapie (Dosierung und Behandlungsschema von Nivolumab wurden in Abhängigkeit vom verwendeten Chemotherapieregime ausgewählt, siehe unten) wurden in einer randomisierten, offenen Phase‑III‑Studie (CA209649) untersucht. In die Studie wurden erwachsene Patienten (ab 18 Jahren) mit bis dahin unbehandelten fortgeschrittenen oder metastasierten Adenokarzinomen des Magens, des gastroösophagealen Übergangs (gastro‑esophageal junction, GEJ) oder des Ösophagus eingeschlossen, die keine vorherige systemische Behandlung (einschließlich HER2‑Inhibitoren) erhalten hatten und einen ECOG‑Performance‑Status von 0 oder 1 aufwiesen. Patienten wurden unabhängig ihres PD‑L1‑Expressions Status auf Tumorzellen eingeschlossen und die PD‑L1‑Expression der Tumorzellen wurde unter Verwendung des PD‑L1‑IHC‑28‑8‑pharmDx‑Assays bestimmt. Eine retrospektive Neubewertung des Tumor‑PD‑L1‑Status eines Patienten unter Verwendung des CPS‑Status wurde anhand der PD‑L1‑gefärbten Tumorproben durchgeführt, die zur Randomisierung verwendet worden waren. Patienten mit bekannten HER2‑positiven Tumoren, mit einem Ausgangs‑ECOG‑Performance‑Status ≥ 2, mit unbehandelten Metastasen im zentralen Nervensystem, oder mit einer aktiven, bekannten oder vermuteten Autoimmunerkrankung, oder deren Zustand eine systemische Immunsuppression erforderte, waren von der Studie ausgeschlossen. Insgesamt wurden 643 Patienten mit einem unbekannten HER2‑Status (40,3 % der Studienpopulation) in die Studie eingeschlossen. Die Randomisierung wurde anhand des PD‑L1‑Expressionsstatus der Tumorzellen stratifiziert (≥1 % vs. < 1 % oder unbestimmt), anhand der Region (Asien vs. US vs. Rest der Welt), des ECOG‑Performance‑Status (0 vs. 1) und des Chemotherapieregimes. Die Chemotherapie war FOLFOX (Fluorouracil, Leucovorin und Oxaliplatin) oder CapeOX (Capecitabin und Oxaliplatin).
Insgesamt wurden 1 581 Patienten randomisiert und erhielten entweder Nivolumab in Kombination mit Chemotherapie oder Chemotherapie. Darunter befanden sich 955 Patienten mit PD‑L1-CPS ≥ 5; 473 davon im Nivolumab‑plus‑Chemotherapie‑Arm und 482 im Chemotherapie‑Arm. Patienten im Nivolumab‑plus‑Chemotherapie‑Arm erhielten entweder Nivolumab 240 mg durch intravenöse Infusion über 30 Minuten in Kombination mit FOLFOX (Oxaliplatin 85 mg/m2, Leucovorin 400 mg/m2 und Fluorouracil 400 mg/m2 intravenös am Tag 1 und Fluorouracil 1 200 mg/m2 intravenös durch eine Dauerinfusion über 24 Stunden täglich oder per lokalem Standard am Tag 1 und 2) alle 2 Wochen, oder Nivolumab 360 mg durch intravenöse Infusion über 30 Minuten in Kombination mit CapeOX (Oxaliplatin 130 mg/m2 intravenös am Tag 1 und Capecitabin 1 000 mg/m2 oral zweimal täglich an den Tagen 1‑14) alle 3 Wochen. Die Behandlung wurde bis zur Progression der Krankheit, bis zum Auftreten nicht‑akzeptabler Toxizitäten oder, nur bei Nivolumab, 24 Monate lang fortgeführt. Den Patienten, die Nivolumab plus Chemotherapie erhielten und bei denen die Chemotherapie beendet wurde, war es erlaubt, Nivolumab‑Monotherapie mit 240 mg alle 2 Wochen, 360 mg alle 3 Wochen oder 480 mg alle 4 Wochen bis zu 24 Monate lang nach Behandlungsbeginn zu erhalten. Tumorbeurteilungen erfolgten alle 6 Wochen bis zur und einschließlich Woche 48, danach alle 12 Wochen.
Die bei Studieneintritt vorliegenden Merkmale der Patienten waren im Allgemeinen über die Behandlungsgruppen hinweg gleich verteilt. Bei Patienten mit PD‑L1-CPS ≥ 5 war das mediane Alter 62 Jahre (Bereich: 18‑90), 11 % waren ≥ 75 Jahre, 71 % waren Männer, 25 % waren Asiaten und 69 % waren Weiße. Der Ausgangs‑ECOG‑Performance‑Status war 0 (42 %) oder 1 (58 %). Die Verteilung der Lage des Tumors war Magen (70 %), GEJ (18 %) und Ösophagus (12 %).
Die primären Wirksamkeitskriterien waren PFS (durch BICR) und OS, beurteilt bei Patienten mit PD‑L1-CPS ≥ 5 basierend auf dem PD‑L1‑IHC‑28‑8‑pharmDX‑Test. Sekundäre Endpunkte gemäß der vordefinierten hierarchischen Testung waren OS bei Patienten mit PD‑L1-CPS ≥ 1 und bei allen randomisierten Patienten; weitere Endpunkte beinhalteten ORR (BICR) bei PD‑L1-CPS ≥ 5 und bei allen randomisierten Patienten. Bei der primären vordefinierten Auswertung mit minimaler Nachbeobachtungszeit von 12,1 Monaten zeigte die Studie eine statistisch signifikante Verbesserung von OS und PFS bei Patienten mit PD‑L1-CPS ≥ 5. Das mediane OS war 14,4 Monate (95 % CI: 13,1; 16,2) für Nivolumab in Kombination mit Chemotherapie vs. 11,1 Monate (95 % CI: 10,0; 12,1) für Chemotherapie (HR = 0,71; 98,4 % CI: 0,59; 0,86; p‑Wert < 0,0001). Das mediane PFS war 7,69 Monate (95 % CI: 7,03; 9,17) für Nivolumab in Kombination mit Chemotherapie vs. 6,05 Monate (95 % CI: 5,55; 6,90) für Chemotherapie (HR = 0,68; 98 % CI: 0,56; 0,81; p‑Wert < 0,0001). Das ORR war 60 % (95 % CI: 55; 65) für Nivolumab in Kombination mit Chemotherapie vs. 45 % (95 % CI: 40; 50) für Chemotherapie.
Bei der aktualisierten deskriptiven Auswertung mit einer minimalen Nachbeobachtungszeit von 19,4 Monaten, waren die Verbesserungen des OS konsistent mit der primären Auswertung. Die Wirksamkeitsergebnisse sind in Tabelle 42 und Abbildungen 31 und 32 dargestellt.
Tabelle 42: Wirksamkeitsergebnisse bei Patienten mit PD‑L1-CPS ≥ 5 (CA209649)
Nivolumab + Chemotherapie |
Chemotherapie |
|
Minimale Nachbeobachtungszeit 19,4 Monatea |
||
Gesamtüberleben |
||
Ereignisse |
344 (73 %) |
397 (82 %) |
Hazard-Ratio (95 % CI)b |
0,69 (0,60; 0,81) |
|
Median (95 % CI) (Monate)c |
14,4 (13,1; 16,3) |
11,1 (10,0; 12,1) |
Progressionsfreies Überlebend | ||
Ereignisse |
342 (72,3 %) |
366 (75,9 %) |
Hazard Ratio (95 % CI)b |
0,68 (0,59; 0,79) |
|
Median (95 % CI) (Monate)c |
8,31 (7,03; 9,26) |
6,05 (5,55; 6,90) |
Gesamtansprechen, nd,e |
227/378 (60 %) |
176/390 (45 %) |
(95 % CI) |
(54,9; 65,0) |
(40,1; 50,2) |
Vollständiges Ansprechen |
12,2 % |
6,7 % |
Teilweises Ansprechen |
47,9 % |
38,5 % |
Ansprechdauerd,e | ||
Median (95 % CI) (Monate)c |
9,69 (8,25; 12,22) |
6,97 (5,62; 7,85) |
a Deskriptive Analyse basierend auf Datenschnitt: 04. Januar 2021. | ||
Abbildung 31: Kaplan‑Meier‑Kurven des Gesamtüberlebens bei Patienten mit PD‑L1-CPS ≥ 5 (CA209649)
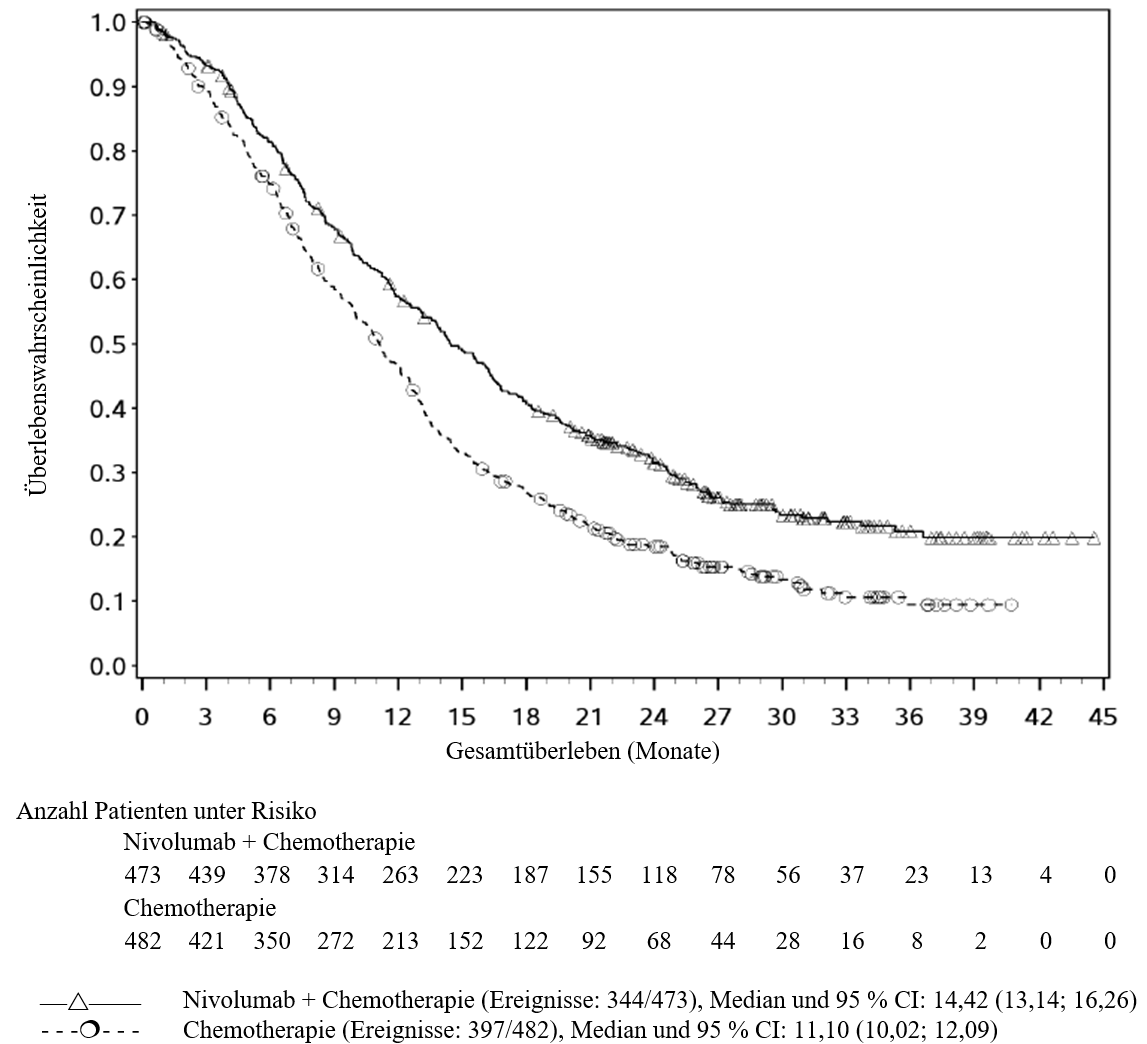
Minimale Nachbeobachtungszeit von 19,4 Monaten
Abbildung 32: Kaplan‑Meier‑Kurven des progressionsfreien Überlebens bei Patienten mit PD‑L1-CPS ≥ 5 (CA209649)
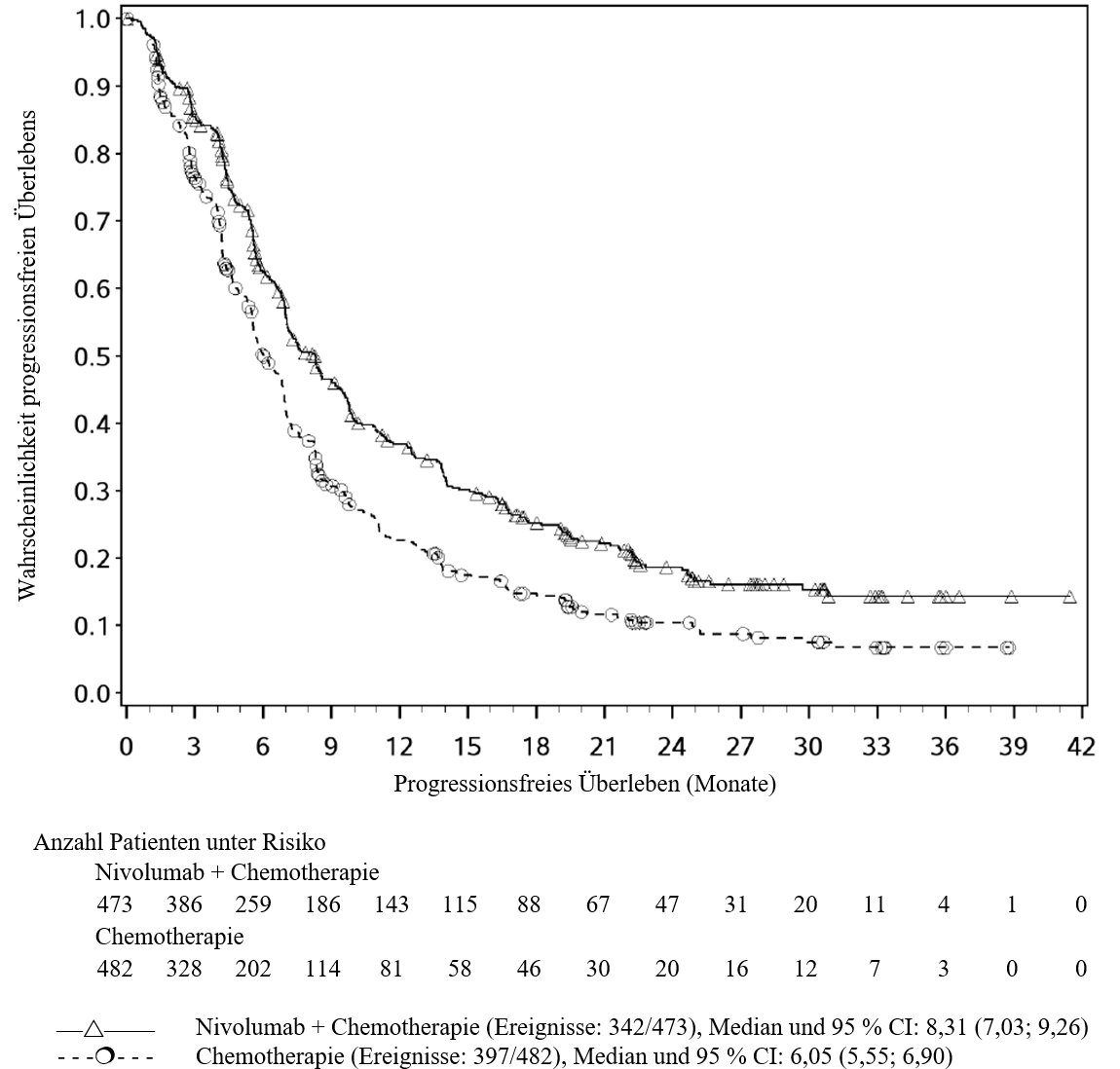
Minimale Nachbeobachtungszeit von 19,4 Monaten
Hepatozelluläres Karzinom
Intravenöse Formulierung
Die Sicherheit und Wirksamkeit von 1 mg/kg Nivolumab in Kombination mit 3 mg/kg Ipilimumab alle 3 Wochen für maximal 4 Dosen, gefolgt von einer Nivolumab‑Monotherapie mit 480 mg alle 4 Wochen für die Erstlinienbehandlung des nicht resezierbaren oder fortgeschrittenen hepatozellulären Karzinoms (HCC) wurden in einer randomisierten, aktiv kontrollierten, offenen Phase‑III‑Studie (CA2099DW) untersucht. In die Studie eingeschlossen wurden erwachsene Patienten (ab 18 Jahren) mit histologisch bestätigtem HCC, Child‑Pugh‑Klassifikation A, ECOG‑Performance‑Status 0 oder 1, die keine vorherige systemische Therapie für die fortgeschrittene Erkrankung erhalten hatten. Eine Ösophagogastroduodenoskopie war vor der Aufnahme in die Studie nicht vorgeschrieben. In die Studie wurden Erwachsene eingeschlossen, deren Erkrankung nicht durch einen operativen Eingriff und/oder lokoregionäre Therapien behandelt werden konnte oder danach fortgeschritten ist. Eine vorausgegangene neoadjuvante oder adjuvante systemische Therapie war zulässig. Patienten mit aktiver Autoimmunerkrankung, Hirnmetastasen oder leptomeningealen Metastasen, vorheriger Lebertransplantation, einer Vorgeschichte von hepatischer Enzephalopathie (innerhalb von 12 Monaten vor der Randomisierung), klinisch signifikantem Aszites, Erkrankungen, die eine systemische Immunsuppression erfordern, einer HIV-Infektion oder aktiver Koinfektion mit Hepatitis-B-Virus (HBV) und Hepatitis-C-Virus (HCV) oder HBV und Hepatitis-D-Virus (HDV) waren von der Studie ausgeschlossen. Die Randomisierung wurde nach Ätiologie (HBV vs. HCV vs. nicht‑viral), makrovaskulärer Invasion und/oder extrahepatischer Manifestation (vorhanden oder nicht vorhanden) und Alpha-Fetoprotein-Werten (≥ 400 oder < 400 ng/ml) stratifiziert.
Insgesamt wurden 668 Patienten randomisiert und erhielten Nivolumab in Kombination mit Ipilimumab (n = 335) oder eine Behandlung mit Lenvatinib oder Sorafenib nach Wahl des Prüfarztes (n = 333). In dem Arm, der eine durch den Prüfarzt ausgewählte Behandlung vorsah, erhielten 85 % der behandelten Patienten Lenvatinib und 15 % Sorafenib. Die Patienten im Nivolumab‑plus‑Ipilimumab‑Arm erhielten 1 mg/kg Nivolumab alle 3 Wochen in Kombination mit 3 mg/kg Ipilimumab alle 3 Wochen für bis zu maximal 4 Dosen, gefolgt von einer Nivolumab‑Monotherapie mit 480 mg alle 4 Wochen. Patienten in dem Arm, der eine durch den Prüfarzt ausgewählte Behandlung vorsah, erhielten entweder 8 mg Lenvatinib täglich peroral (bei einem Körpergewicht von < 60 kg) oder 12 mg täglich peroral (bei einem Körpergewicht von ≥ 60 kg) oder Sorafenib 400 mg zweimal täglich peroral. Die Behandlung wurde bis zur Progression der Erkrankung, nicht akzeptabler Toxizität oder bis zu 24 Monate fortgesetzt. Patienten, die die Kombinationstherapie aufgrund einer Nebenwirkung, die Ipilimumab zugeordnet wurde, abbrechen mussten, konnten mit Nivolumab als Monotherapie weiterbehandelt werden. Tumorbeurteilungen wurden bei Studienbeginn, nach Randomisierung in Woche 9 und Woche 16, dann alle 8 Wochen bis zu 48 Wochen und anschließend alle 12 Wochen bis zum Progress der Erkrankung, dem Abbruch der Behandlung oder Einleitung der nachfolgenden Therapie durchgeführt.
Die bei Studieneintritt vorliegenden Merkmale der Patienten waren im Allgemeinen über die Behandlungsgruppen hinweg gleichmäßig verteilt. Das mediane Alter betrug 66 Jahre (Spanne: 20 bis 89); 53 % der Patienten waren ≥ 65 Jahre und 16 % waren ≥ 75 Jahre, 53 % der Patienten waren Kaukasier, 44 % waren Asiaten, 2,2 % waren Schwarze und 82 % waren männlich. Der ECOG‑Performance‑Status bei Einschluss in die Studie war 0 (71 %) oder 1 (29 %). Vierunddreißig Prozent (34 %) der Patienten hatten eine HBV‑Infektion, 28 % eine HCV‑Infektion und bei 36 % gab es keine Nachweise für eine HBV- oder HCV‑Infektion. Neunzehn Prozent (19 %) der Patienten hatten eine alkoholbedingte Lebererkrankung und 11 % eine nichtalkoholische Fettlebererkrankung. Die Mehrheit der Patienten wies bei Studieneintritt BCLC‑Stadium C (73 %) auf, 19 % Stadium B und 6 % Stadium A. Der Anteil der Patienten mit Child‑Pugh‑Scores von 5, 6 und ≥ 7 betrug 77 %, 20 % bzw. 3 %. Bei insgesamt 54 % der Patienten lag eine extrahepatische Ausbreitung vor, 25 % hatten eine makrovaskuläre Invasion und 33 % wiesen AFP‑Werte ≥ 400 µg/l auf.
Die Studie zeigte eine statistisch signifikante Verbesserung des OS und der ORR für Patienten, die in den Behandlungsarm Nivolumab in Kombination mit Ipilimumab randomisiert worden waren, im Vergleich zu Lenvatinib oder Sorafenib nach Wahl des Prüfarztes. Die Wirksamkeitsergebnisse sind in Tabelle 43 und Abbildung 33 dargestellt.
Tabelle 43: Wirksamkeitsergebnisse bei HCC in der Erstlinie (CA2099DW)a
Nivolumab + Ipilimumab |
Lenvatinib oder Sorafenib |
|
Gesamtüberleben |
||
Ereignisse |
194 (58 %) |
228 (68 %) |
Median (Monate) |
23,7 |
20,6 |
Hazard Ratio (95 % CI)b |
0,79 (0,65; 0,96) |
|
p‑Wertc |
0,0180 |
|
Gesamtansprechen, n (%)d |
121 (36,1) |
44 (13,2) |
(95 % CI) |
(31,0; 41,5) |
(9,8; 17,3) |
p‑Werte |
< 0,0001 |
|
Vollständiges Ansprechen (%) |
23 (6,9) |
6 (1,8) |
Teilweises Ansprechen (%) |
98 (29,3) |
38 (11,4) |
Ansprechdauer (Monate)d |
||
Median |
30,4 |
12,9 |
a Minimale Nachbeobachtungszeit: 26,8 Monate. Mediane Nachbeobachtungszeit: 35,2 Monate. | ||
Abbildung 33: Kaplan‑Meier‑Kurven des OS in der Erstlinie bei Patienten mit HCC (CA2099DW)
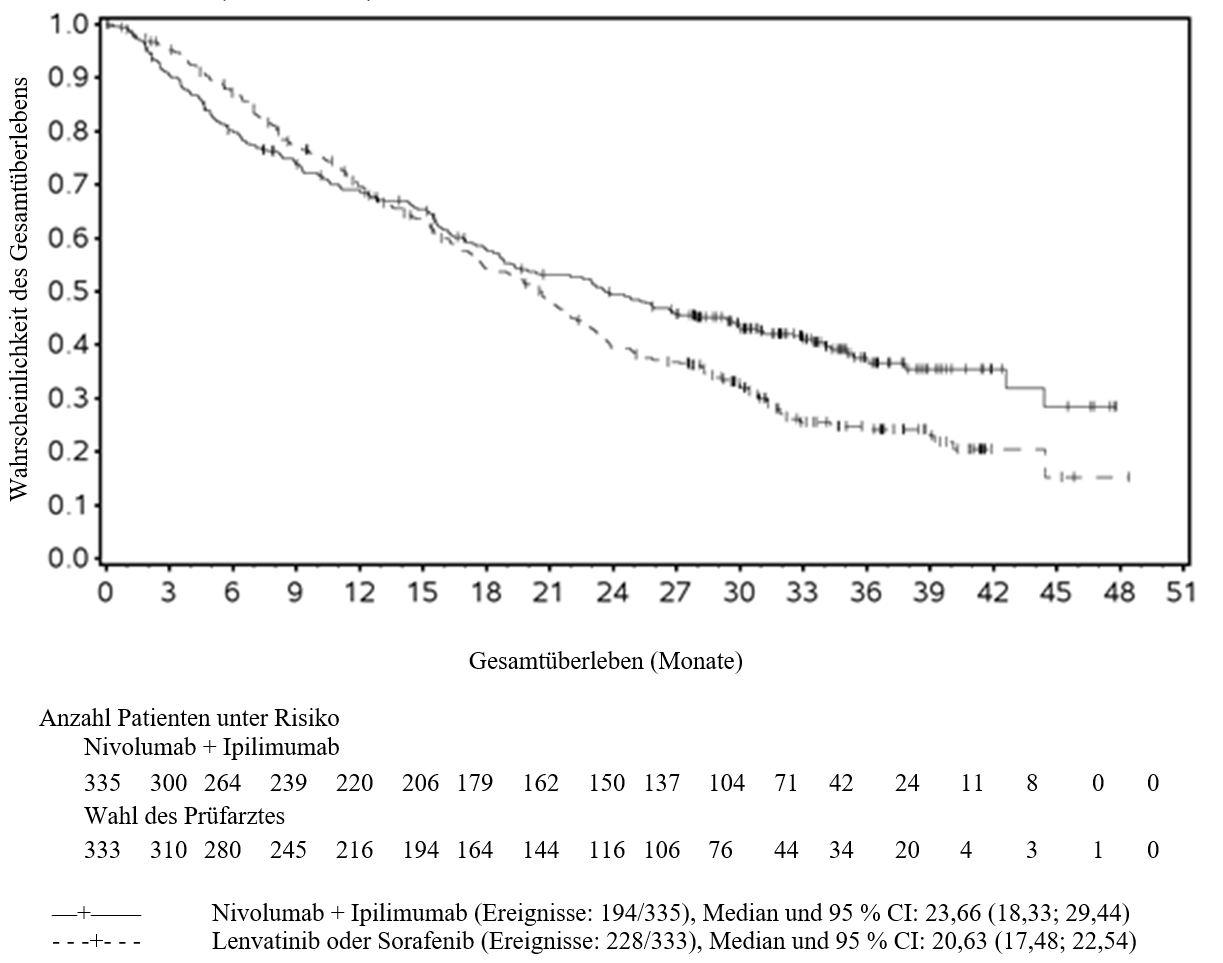
Kinder und Jugendliche
Subkutane Formulierung
Es wurden keine speziellen Studien mit OPDIVO‑Injektionslösung bei Kindern und Jugendlichen durchgeführt.
Die Europäische Arzneimittel‑Agentur hat für OPDIVO‑Injektionslösung zur subkutanen Anwendung eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in der Behandlung von bösartigen Neubildungen (außer Neubildungen des Zentralnervensystems sowie hämatopoetischen und lymphoiden Neubildungen mit Ausnahme des Hodgkin-Lymphoms) gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Sicherheit und Wirksamkeit bei älteren Patienten
Es wurden insgesamt keine Unterschiede bei Sicherheit oder Wirksamkeit zwischen älteren (≥ 65 Jahre) und jüngeren Patienten (< 65 Jahre) berichtet. Die Daten von Patienten mit SCCHN, adjuvanter Behandlung des Melanoms, und adjuvanter Behandlung der Karzinome des Ösophagus oder des gastroösophagealen Übergangs ab 75 Jahren sind begrenzt und lassen keine Schlussfolgerungen für diese Population zu.
Die Pharmakokinetik (PK) von Nivolumab‑Injektionslösung wurde mithilfe eines Populations-PK-Ansatzes bewertet. Die PK wurde bei einer Dosisstärke von 1 200 mg untersucht, die in mehreren Dosen alle 4 Wochen verabreicht wurde.
Die über 28 Tage zeitlich gemittelte Serumkonzentration von Nivolumab (Cavgd28) zeigte, dass subkutan verabreichtes Nivolumab (77,4 μg/ml) intravenös verabreichtem Nivolumab (36,9 μg/ml) nichtunterlegen ist, mit einem Quotienten des geometrischen Mittels von 2,098 (90 % CI: 2,001, 2,200). Die minimale Serumkonzentration von Nivolumab im Steady State (Cminss) zeigte, dass subkutan verabreichtes Nivolumab (122,2 μg/ml) intravenös verabreichtem Nivolumab (68,9 μg/ml) nichtunterlegen ist, mit einem Quotienten des geometrischen Mittels von 1,774 (90 % CI: 1,633, 1,927).
Resorption
Die mittlere Resorptionsgeschwindigkeitskonstante (Ka) von Nivolumab‑Injektionslösung beträgt 0,0123 h‑1 (oder 0,295 Tage‑1) und die Bioverfügbarkeit (F) 78,8 %. Die Spitzenkonzentrationen wurden nach etwa 6 Tagen erreicht.
Verteilung
Das geometrische Mittel (CV%) des Verteilungsvolumen im Steady State (Vss) beträgt 6,32 l (21,3 %).
Elimination
Die Clearance (CL) von Nivolumab‑Injektionslösung beim Menschen reduziert sich im Laufe der Zeit, mit einer mittleren maximalen Reduktion gegenüber den Ausgangswerten (CV%) von 24,6 % (15,8 %), was bei RCC‑Patienten zu einer geometrischen mittleren (CV%) Steady‑State‑Clearance (CLss) von 7,18 ml/h (52,3 %) führt. Die Reduktion der CLss wird als nicht klinisch relevant betrachtet.
Das geometrische Mittel (CV%) der Eliminationshalbwertszeit (t1/2) beträgt 26,5 Tage (32,1 %).
Spezielle Patientenpopulationen
Die folgenden Faktoren hatten keinen klinisch bedeutsamen Einfluss auf die Bioverfügbarkeit von Nivolumab‑Injektionslösung: Geschlecht und Performance‑Status. Die folgenden Faktoren hatten keinen klinisch bedeutsamen Einfluss auf die Clearance von Nivolumab‑Injektionslösung: Körpergewicht (35 bis 153 kg), Geschlecht, eGFR (24 bis 124 ml/min/1,73 m2) oder Performance‑Status.
Eingeschränkte Nierenfunktion
In Populations‑PK‑Analysen zu intravenös verabreichtem Nivolumab wurde die Auswirkung einer eingeschränkten Nierenfunktion auf die CL von Nivolumab bei Patienten mit leicht (GFR < 90 und ≥ 60 ml/min/1,73 m2; n = 379), mäßig (GFR < 60 und ≥30 ml/min/1,73 m2; n = 179) oder schwer (GFR < 30 und ≥ 15 ml/min/1,73 m2; n = 2) eingeschränkter Nierenfunktion im Vergleich zu Patienten mit normaler Nierenfunktion (GFR ≥ 90 ml/min/1,73 m2; n = 342) untersucht. Es wurden keine klinisch bedeutsamen Unterschiede der CL von Nivolumab zwischen Patienten mit leicht oder mäßig eingeschränkter und Patienten mit normaler Nierenfunktion festgestellt. Die Daten von Patienten mit schwer eingeschränkter Nierenfunktion sind zu begrenzt, als dass sich daraus Schlüsse für diese Population ableiten lassen (siehe Abschnitt 4.2).
Eingeschränkte Leberfunktion
In Populations‑PK‑Analysen zu intravenös verabreichtem Nivolumab wurde die Auswirkung einer eingeschränkten Leberfunktion auf die CL von Nivolumab bei Patienten mit verschiedenen Tumorarten (NSCLC, SCLC, Melanom, RCC, SCCHN, UC, GC und cHL) mit leicht eingeschränkter Leberfunktion (Gesamtbilirubin 1,0 × bis 1,5 × ULN oder AST > ULN gemäß der Definition der Kriterien des National Cancer Institute zur Leberfunktionsstörung; n = 351) und bei Patienten mit mäßig eingeschränkter Leberfunktion (Gesamtbilirubin > 1,5 × bis 3 × ULN und beliebige AST; n = 10) im Vergleich zu Patienten mit normaler Leberfunktion (Gesamtbilirubin und AST ≤ ULN; n = 3096) untersucht. Es wurden keine klinisch bedeutsamen Unterschiede der CL von Nivolumab zwischen Patienten mit leicht oder mäßig eingeschränkter und Patienten mit normaler Leberfunktion festgestellt. Ähnliche Ergebnisse wurden bei Patienten mit HCC beobachtet (leicht eingeschränkte Leberfunktion: n = 152; mäßig eingeschränkte Leberfunktion: n = 13). Nivolumab wurde bei Patienten mit schwer eingeschränkter Leberfunktion (Gesamtbilirubin > 3 × ULN und beliebige AST) nicht untersucht (siehe Abschnitt 4.2).
In Mausmodellen zur Schwangerschaft wurde gezeigt, dass durch eine Blockade des PD‑L1‑Signals die Toleranz gegenüber dem Fötus gestört wird und die Abortrate steigt. Die Wirkungen von Nivolumab auf die prä‑ und postnatale Entwicklung wurden in einer Studie an Affen untersucht, die Nivolumab nach Einsetzen der Organogenese im ersten Trimester bis zur Geburt zweimal wöchentlich mit Expositionen des 8‑ oder 35‑fachen derjenigen erhielten, die mit der klinischen Dosierung von 3 mg/kg Nivolumab beobachtet werden (basierend auf AUC). Mit Beginn des dritten Trimesters traten dosisabhängig eine höhere Abortrate und eine höhere Jungensterblichkeit auf.
Die anderen Nachkommen der mit Nivolumab behandelten Weibchen überlebten bis zur geplanten Termination ohne mit der Behandlung in Zusammenhang stehende klinische Symptome, Abweichungen von der normalen Entwicklung, Auswirkung auf das Organgewicht oder makro‑ oder mikroskopische pathologische Veränderungen. Die Ergebnisse für Wachstumsindizes sowie teratogene, immunologische und klinisch‑pathologische Parameter sowie neurologisch bedingtes Verhalten waren im gesamten postnatalen Zeitraum von 6 Monaten mit denen der Kontrollgruppe vergleichbar. Basierend auf dem Wirkmechanismus könnte eine Exposition des Fötus mit Nivolumab jedoch das Risiko für die Entwicklung einer immunvermittelten Erkrankung erhöhen oder die normale Immunantwort verändern und bei PD‑1‑Knockout‑Mäusen sind immunvermittelte Erkrankungen berichtet worden.
Fertilitätsstudien wurden für Nivolumab nicht durchgeführt.
Subkutane Formulierung
Hyaluronidase liegt in den meisten Geweben des menschlichen Körpers vor. Nichtklinische Daten zur rekombinanten humanen Hyaluronidase lassen auf der Grundlage konventioneller Studien zur Toxizität bei wiederholter Anwendung einschließlich sicherheitspharmakologischer Endpunkte keine besondere Gefährdung für den Menschen erkennen. Bei reproduktionstoxikologischen Studien mit rHuPH20 wurde bei Mäusen bei hoher systemischer Exposition embryofetale Toxizität, aber kein teratogenes Potenzial festgestellt.
Rekombinante humane Hyaluronidase (rHuPH20)
Histidin
Histidinhydrochlorid-Monohydrat
Saccharose
Pentetsäure
Polysorbat 80 (E 433)
Methionin
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Ungeöffnete Durchstechflasche
3 Jahre
Aufbewahrung in der Spritze
Aus mikrobiologischer Sicht soll das Arzneimittel nach dem Aufziehen von der Durchstechflasche in die Spritze sofort verwendet werden, da das Arzneimittel keine antimikrobiellen Konservierungsmittel und keine Bakteriostatika enthält. Wenn die OPDIVO‑Injektionslösung nicht sofort verwendet wird, kann sie in eine Spritze aufgezogen im Kühlschrank bei 2 °C bis 8 °C, vor Licht geschützt bis zu 7 Tage und/oder bei einer Raumtemperatur von 20 °C bis 25 °C und Umgebungslicht bis zu 8 Stunden aufbewahrt werden. Die Injektionslösung ist zu entsorgen, wenn die Aufbewahrungsdauer diese Grenzen überschreitet. Bei der Vorbereitung der Spritze für die Injektion ist auf eine aseptische Handhabung zu achten.
Im Kühlschrank lagern (2 °C ‑ 8 °C).
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Vorbereitung der Spritze, siehe Abschnitt 6.3.
OPDIVO 300 mg Injektionslösung: Durchstechflasche (Glas Typ I) mit Butylkautschuk‑Stopfen und Aluminiumverschluss mit einer violettfarbenen Flip-Off-Kunststoffkappe, die 2,5 ml Injektionslösung enthält.
OPDIVO 600 mg Injektionslösung: Durchstechflasche (Glas Typ I) mit Butylkautschuk‑Stopfen und Aluminiumverschluss mit einer orangefarbenen Flip-Off-Kunststoffkappe, die 5 ml Injektionslösung enthält.
Packung mit einer Durchstechflasche.
Die Zubereitung soll, besonders im Hinblick auf die Asepsis, durch geschultes Personal im Einklang mit den Richtlinien zur guten Herstellungspraxis durchgeführt werden.
Vorbereitung der Spritze
OPDIVO‑Injektionslösung ist nur zur Einmalanwendung bestimmt und ist gebrauchsfertig.
OPDIVO‑Injektionslösung darf NICHT verdünnt und nicht mit anderen Arzneimitteln vermischt werden.
OPDIVO‑Injektionslösung ist kompatibel mit Polypropylen, Polycarbonat, Polyethylen, Polyurethan, Polyvinylchlorid, Fluorethylenpropylen und Edelstahl.
OPDIVO‑Injektionslösung sollte eine klare bis opaleszierende, farblose bis gelbe Lösung sein. Vor der Anwendung ist die Lösung einer Sichtprüfung zu unterziehen und zu verwerfen, falls sie eine Verfärbung aufweist oder mehr als nur wenige transluzente bis weiße Schwebstoffe enthält.
Durchstechflasche nicht schütteln.
Die korrekte Anzahl an benötigten Durchstechflaschen mit OPDIVO-Injektionslösung basierend auf der verordneten Dosis berechnen.
Die Durchstechflasche(n) vor der Entnahme der Dosis Raumtemperatur annehmen lassen.
Für die Entnahme des Arzneimittels aus der Durchstechflasche werden eine Spritze und eine Transferkanüle benötigt. OPDIVO‑Injektionslösung kann subkutan mit einer hypodermischen Injektionsnadel mit 23 G ‑ 25 G oder einem subkutanen Verabreichungsbesteck (z. B. Flügel‑/Butterfly‑Kanüle) verabreicht werden.
Die benötigte Menge entsprechend der empfohlenen Dosis entnehmen: entweder 5 ml (600 ml), 7,5 ml (900 mg) oder 10 ml (1 200 mg).
Die hypodermische Injektionsnadel darf erst unmittelbar vor der Verabreichung auf die Spritze aufgesetzt werden, um ein Verstopfen zu vermeiden.
Es wird empfohlen, die zubereitete Dosis sofort zu verwenden.
Falls eine Aufbewahrung erforderlich ist (siehe Abschnitt 6.3), ist die Spritze vor der Aufbewahrung mit einer Spritzenkappe zu verschließen.
Bei einer Aufbewahrung im Kühlschrank muss die Lösung vor der Verabreichung auf Raumtemperatur gebracht werden.
Entsorgung
In der Durchstechflasche verbliebene Restmengen der Lösung sind zu entsorgen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Bristol‑Myers Squibb Pharma EEIG
Plaza 254
Blanchardstown Corporate Park 2
Dublin 15, D15 T867
Irland
EU/1/15/1014/005
EU/1/15/1014/006
Datum der Erteilung der Zulassung: 19. Juni 2015
Datum der letzten Verlängerung der Zulassung: 23. April 2020
Mai 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel‑Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig
Bristol-Myers Squibb GmbH & Co. KGaA
Arnulfstraße 29
80636 München
Medizinische Information
Telefon: 0800 0752002
E-Mail: medwiss.info@bms.com
www.bmsmedinfo.de