
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Ogsiveo 50 mg Filmtabletten
Ogsiveo 100 mg Filmtabletten
Ogsiveo 150 mg Filmtabletten
Ogsiveo 50 mg Filmtabletten
Jede Filmtablette enthält 50 mg Nirogacestat (als Nirogacestat-Dihydrobromid).
Sonstige Bestandteile mit bekannter Wirkung
Jede Filmtablette enthält 57,8 mg Lactose-Monohydrat.
Jede Filmtablette enthält Gelborange S (E110).
Ogsiveo 100 mg Filmtabletten
Jede Filmtablette enthält 100 mg Nirogacestat (als Nirogacestat-Dihydrobromid).
Sonstige Bestandteile mit bekannter Wirkung
Jede Filmtablette enthält 115,7 mg Lactose-Monohydrat.
Jede Filmtablette enthält Gelborange S (E110).
Ogsiveo 150 mg Filmtabletten
Jede Filmtablette enthält 150 mg Nirogacestat (als Nirogacestat-Dihydrobromid).
Sonstige Bestandteile mit bekannter Wirkung
Jede Filmtablette enthält 173,5 mg Lactose-Monohydrat.
Jede Filmtablette enthält Gelborange S (E110).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette (Tablette).
Ogsiveo 50 mg Filmtabletten
Runde, bikonvexe, orangefarbene Filmtabletten mit einem Durchmesser von 8 mm und der Prägung „50“ auf einer Seite.
Ogsiveo 100 mg Filmtabletten
Runde, bikonvexe, hellorangefarbene Filmtabletten mit einem Durchmesser von 10 mm und der Prägung „100“ auf einer Seite.
Ogsiveo 150 mg Filmtabletten
Ovale, gelb-orangefarbene Filmtabletten mit einer Breite von 8,5 mm und einer Länge von 17,5 mm und der Prägung „150“ auf einer Seite.
Ogsiveo als Monotherapie wird angewendet für die Behandlung erwachsener Patienten mit fortschreitenden Desmoidtumoren, die eine systemische Behandlung erfordern.
Die Behandlung mit Ogsiveo sollte von einem in der Anwendung von Krebstherapien erfahrenen Arzt eingeleitet und überwacht werden.
Dosierung
Die empfohlene Dosis beträgt 150 mg Ogsiveo zweimal täglich: morgens eine Dosis und abends eine Dosis. Diese Dosis darf nicht überschritten werden.
Dauer der Behandlung
Die Behandlung mit Ogsiveo sollte bis zum Fortschreiten der Erkrankung oder bis zu einer inakzeptablen Toxizität fortgeführt werden.
Auslassen einer Dosis
Wenn eine Dosis Ogsiveo ausgelassen wird, sollten die Patienten keine zusätzliche Dosis einnehmen, sondern die nächste verordnete Dosis einnehmen.
Dosisanpassung bei Nebenwirkungen
Tabelle 1 zeigt die empfohlenen Dosismodifikationen bei ausgewählten Nebenwirkungen.
Bei anderen schweren Nebenwirkungen oder im Falle lebensbedrohlicher Nebenwirkungen ist Ogsiveo auszusetzen, bis die Reaktion auf Grad ≤ 1 oder den Ausgangszustand abgeklungen ist. Die Behandlung mit Ogsiveo sollte nur mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden, nachdem der potenzielle Nutzen und die Wahrscheinlichkeit des Wiederauftretens der Nebenwirkung sorgfältig abgewogen wurde. Ogsiveo muss dauerhaft abgesetzt werden, wenn eine schwere oder lebensbedrohliche Nebenwirkung bei erneuter Verabreichung der verringerten Dosis wiederkehrt.
Dosismodifikationen sollten erfolgen, wenn bei Patienten folgende Nebenwirkungen auftreten (die Grade beziehen sich auf die Common Terminology Criteria for Adverse Events):
Tabelle 1: Empfohlene Dosismodifikationen bei Nebenwirkungen bei mit Ogsiveo behandelten Patienten
Nebenwirkung |
Empfohlene Maßnahme |
Diarrhö | |
Diarrhö Grad 3, die trotz maximaler medizinischer Therapie ≥ 3 Tage anhält |
Ogsiveo sollte ausgesetzt werden, bis die Reaktion auf Grad ≤ 1 oder den Ausgangszustand abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden. |
Hautreaktionen | |
Follikulitis Grad 3 |
Ogsiveo sollte ausgesetzt werden, bis die Reaktion auf Grad ≤ 1 oder den Ausgangszustand abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden. |
Makulo-papulöser Ausschlag Grad 3 |
Ogsiveo sollte ausgesetzt werden, bis die Reaktion auf Grad ≤ 1 oder den Ausgangszustand abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden. |
Hidradenitis Grad 3 |
Ogsiveo sollte ausgesetzt werden, bis die Reaktion auf Grad ≤ 1 oder den Ausgangszustand abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden. |
Elektrolytanomalien | |
Hypophosphatämie Grad 3, die trotz maximaler Ersatztherapie ≥ 7 Tage anhält |
Ogsiveo sollte ausgesetzt werden, bis die Reaktion auf Grad ≤ 1 oder den Ausgangszustand abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden. |
Hypokaliämie Grad 3 trotz maximaler Ersatztherapie |
Ogsiveo sollte ausgesetzt werden, bis die Reaktion auf Grad ≤ 1 oder den Ausgangszustand abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden. |
Anomalien der Leberwerte | |
Alanin-Aminotransferase (ALT) oder Aspartat-Aminotransferase (AST) ≥ 3 bis 5 x ULN |
Ogsiveo sollte ausgesetzt werden, bis die ALT-, AST- oder beide Werte auf < 3 x ULN oder den Ausgangswert zurückgegangen sind, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden. |
ALT oder AST > 5 x ULN |
Ogsiveo muss dauerhaft abgesetzt werden. |
Andere Nebenwirkungen | |
Anaphylaxie oder andere schwere Überempfindlichkeitsreaktion |
Ogsiveo muss dauerhaft abgesetzt werden. |
Besondere Patientengruppen
Ältere Patienten
Für über 65-jährige Patienten wird keine Dosisanpassung empfohlen.
Klinische Daten über Patienten im Alter von über 65 Jahren sind begrenzt.
Nierenfunktionsstörung
Bei Patienten mit leichter bis mäßiger Nierenfunktionsstörung wird eine Dosisanpassung nicht empfohlen. Bei Patienten mit schwerer Nierenfunktionsstörung wird die Verabreichung nicht empfohlen (siehe Abschnitt 5.2).
Leberfunktionsstörung
Bei Patienten mit leichter bis mäßiger Leberfunktionsstörung wird eine Dosisanpassung nicht empfohlen.
Bei Patienten mit schwerer Leberfunktionsstörung wird die Verabreichung nicht empfohlen (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Ogsiveo bei Kindern und Jugendlichen im Alter von 2 bis 18 Jahren ist nicht erwiesen. Bei Kindern von der Geburt bis zum Alter von 2 Jahren darf Ogsiveo aufgrund von potenziellen Sicherheitsbedenken bezüglich des strukturellen und funktionellen Wachstums nicht angewendet werden. Zurzeit vorliegende Daten werden in Abschnitt 4.8 und 5.1 beschrieben; eine Dosierungsempfehlung kann jedoch nicht gegeben werden.
Art der Anwendung
Ogsiveo ist zur oralen Einnahme vorgesehen.
Die Tabletten können mit oder unabhängig von den Mahlzeiten eingenommen werden. Die Tabletten dürfen nicht zerteilt, zerstoßen oder gekaut werden, weil derzeit keine Daten vorliegen, die andere Arten der Anwendung unterstützen.
Die Patienten sollten während der Einnahme von Ogsiveo auf den Verzehr von Grapefruit und Grapefruitsaft verzichten (siehe Abschnitt 4.5).
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Schwangerschaft (siehe Abschnitt 4.4 und 4.6)
Gebärfähige Frauen, die keine hochwirksame Empfängnisverhütung anwenden (siehe Abschnitt 4.4 und 4.6)
Stillzeit (siehe Abschnitt 4.6)
Diarrhö
Diarrhö wurde bei mit Nirogacestat behandelten Patienten berichtet (siehe Abschnitt 4.8). Patienten, bei denen während der Behandlung mit Nirogacestat Diarrhö auftritt, sollten überwacht und mit Antidiarrhoika behandelt werden. Bei Diarrhö Grad 3, die trotz maximaler medizinischer Therapie ≥ 3 Tage anhält, sollte Nirogacestat ausgesetzt werden, bis die Diarrhö auf Grad ≤ 1 oder den Ausgangszustand abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden (siehe Abschnitt 4.2).
Erkrankungen der Haut und des Unterhautgewebes
Dermatologische Reaktionen, einschließlich makulo-papulöser Ausschlag, Follikulitis und Hidradenitis, wurden bei mit Nirogacestat behandelten Patienten berichtet (siehe Abschnitt 4.8). Die Patienten sollten während des gesamten Behandlungsverlaufs auf dermatologische Reaktionen überwacht und je nach klinischer Indikation behandelt werden. Bei dermatologischen Reaktionen Grad 3 sollte Nirogacestat ausgesetzt werden, bis die Reaktion auf Grad ≤ 1 oder den Ausgangszustand abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden (siehe Abschnitt 4.2).
Funktionsstörung der Eierstöcke
Funktionsstörungen der Eierstöcke wurden bei mit Nirogacestat behandelten gebärfähigen Patientinnen berichtet (siehe Abschnitt 4.8). Bei 75 % der in der DeFi-Studie mit Nirogacestat behandelten gebärfähigen Frauen wurden Funktionsstörungen der Eierstöcke berichtet; diese wurde aufgrund von anomalen Fortpflanzungshormonspiegeln oder peri-menopausalen Symptomen festgestellt. Es wurde berichtet, dass die Funktionsstörungen der Eierstöcke bei 79 % der gebärfähigen Frauen während der Behandlung abgeklungen waren. Nachbeobachtungsinformationen liegen mit Ausnahme von zwei für alle 27 Patientinnen vor. Es wurde berichtet, dass die Funktionsstörungen der Eierstöcke bei allen gebärfähigen Frauen, für die Daten vorliegen, abgeklungen waren (siehe Abschnitt 4.8). Die Wirkungen von Nirogacestat auf die menschliche Fertilität sind nicht bekannt. Auf Grundlage von Erkenntnissen aus tierexperimentellen Studien kann die weibliche Fertilität beeinträchtigt werden. Gebärfähige Frauen sollten vor Beginn der Behandlung mit Nirogacestat über das Risiko von Funktionsstörungen der Eierstöcke aufgeklärt werden. Patientinnen sollten auf eine veränderte Regelmäßigkeit des Menstruationszyklus und auftretende Symptome von Östrogenmangel überwacht werden, darunter Hitzeanfälle, nächtliche Schweißausbrüche und vaginale Trockenheit.
Elektrolytanomalien
Elektrolytanomalien, einschließlich Hypophosphatämie und Hypokaliämie, wurden bei mit Nirogacestat behandelten Patienten berichtet (siehe Abschnitt 4.8). Der Phosphat- und Kaliumspiegel sollte regelmäßig überwacht werden, und Phosphat bzw. Kalium sollten bei Bedarf supplementiert werden. Bei Hypophosphatämie Grad 3, die trotz maximaler Ersatztherapie ≥ 7 Tage anhält, sollte Nirogacestat ausgesetzt werden, bis diese auf Grad ≤ 1 oder den Ausgangswert abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden (siehe Abschnitt 4.2). Bei Hypokaliämie Grad 3, die trotz maximaler Ersatztherapie beliebig lang anhält, sollte Nirogacestat ausgesetzt werden, bis diese auf Grad ≤ 1 oder den Ausgangswert abgeklungen ist, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden (siehe Abschnitt 4.2).
Anomalien der Leberwerte
Anstiege der ALT- oder AST-Werte wurden bei mit Nirogacestat behandelten Patienten berichtet (siehe Abschnitt 4.8). Leberfunktionstests sollten regelmäßig überwacht werden. Bei ALT- oder AST-Werten ≥ 3 bis 5 x ULN sollte Nirogacestat ausgesetzt werden, bis die ALT-, AST- oder beide Werte auf < 3 x ULN oder den Ausgangswert abgeklungen sind, dann sollte die Behandlung mit einer Dosis von 100 mg zweimal täglich wieder aufgenommen werden. Bei ALT- oder AST-Werten > 5 x ULN sollte Nirogacestat dauerhaft abgesetzt werden (siehe Abschnitt 4.2).
Nichtmelanozytärer Hautkrebs
Nichtmelanozytärer Hautkrebs (Basalzellkarzinom und Plattenepithelkarzinom) wurden bei mit Nirogacestat behandelten Patienten berichtet (siehe Abschnitt 4.8). Hautuntersuchungen sollten vor der Einleitung von Nirogacestat und routinemäßig während der Behandlung mit Nirogacestat durchgeführt werden. Fälle sollten gemäß klinischen Praktiken behandelt werden, und die Patienten können die Nirogacestat-Behandlung ohne Dosisanpassung fortführen.
Embryofetale Toxizität – Empfängnisverhütung bei Männern und Frauen
Nirogacestat kann bei Verabreichung an eine Schwangere den Fetus schädigen (siehe Abschnitt 4.6 und 5.3). Die Patienten sollten über das potenzielle Risiko für einen Fetus aufgeklärt werden. Bei Frauen im gebärfähigen Alter muss vor Einleitung der Nirogacestat-Behandlung ein negativer Schwangerschaftstest vorliegen. Schwangerschaftstests während der Behandlung mit Nirogacestat sollten für Frauen im gebärfähigen Alter, bei denen Amenorrhö vorliegt, in Betracht gezogen werden. Mit Nirogacestat behandelte Frauen im gebärfähigen Alter müssen während der Behandlung mit Nirogacestat und für eine Woche nach der letzten Dosis Nirogacestat hochwirksame Empfängnisverhütungsmethoden anwenden (siehe Abschnitt 4.6). Frauen im gebärfähigen Alter sollten angewiesen werden, ihren Arzt sofort bei Bekanntwerden einer oder Verdacht auf eine Schwangerschaft zu informieren, und müssen die Einnahme von Nirogacestat bei Eintreten einer Schwangerschaft beenden.
Männliche Patienten mit Partnerinnen im gebärfähigen Alter sollten angewiesen werden, während der Behandlung mit Nirogacestat und für eine Woche nach der letzten Nirogacestat-Dosis hochwirksame Empfängnisverhütungsmethoden anzuwenden (siehe Abschnitt 4.6).
Sonstige Bestandteile
Dieses Arzneimittel enthält Lactose (siehe Abschnitt 2 und 6.1). Patienten mit den seltenen hereditären Störungen Galaktoseunverträglichkeit, Laktasemangel oder Glukose-Galaktose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
Dieses Arzneimittel enthält den Bestandteil Gelborange S (E110) (siehe Abschnitt 2 und 6.1), der allergische Reaktionen verursachen kann.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Filmtablette, d. h. es ist nahezu „natriumfrei“ (siehe Abschnitt 6.1).
Studien zur Erfassung von Wechselwirkungen wurden nur bei Erwachsenen durchgeführt.
Nirogacestat wird vorwiegend durch CYP3A4 metabolisiert und ist ein Substrat von P‑Glykoprotein (P-gp).
Substanzen, die die Serumkonzentrationen von Nirogacestat erhöhen können
Wirkung von mittelstarken und starken CYP3A4-Inhibitoren
In einer klinischen Studie erhöhte sich bei gleichzeitiger Gabe von Itraconazol (ein starker CYP3A4‑Inhibitor und P‑gp-Inhibitor) die Cmax von Nirogacestat um das 2,5-Fache und die AUC um das 8,2-Fache. Es wird erwartet, dass die gleichzeitige Gabe mit mittelstarken CYP3A4-Inhibitoren auch zu klinisch relevanten Expositionsanstiegen führt.
Die gleichzeitige Anwendung mit starken Inhibitoren von CYP3A4 (z. B. Clarithromycin, orales Ketoconazol, Itraconazol) und mittelstarken Inhibitoren von CYP3A4 (z. B. Erythromycin und Fluconazol) ist daher zu vermeiden.
Alternative begleitende Arzneimittel ohne oder mit minimaler CYP3A4-Inhibition sollten erwogen werden. Falls keine therapeutischen Alternativen verfügbar sind, muss die Behandlung mit Ogsiveo sofort unterbrochen werden, und zwar für den Zeitraum der Anwendung eines starken oder mittelstarken CYP3A4-Inhibitors.
Die Patienten sollten während der Einnahme von Ogsiveo auf den Verzehr von Grapefruit und Grapefruitsaft verzichten, da diese Inhibitoren von CYP3A4 enthalten (siehe Abschnitt 4.2).
Substanzen, die die Serumkonzentrationen von Nirogacestat herabsetzen können
Wirkung von starken und mittelstarken CYP3A-Induktoren
Die Wirkungen von CYP3A4-Induktoren auf die Nirogacestat-Exposition sind in einer klinischen Studie nicht untersucht worden. Es wird erwartet, dass mittelstarke und starke Induktoren klinisch relevante Verringerungen der Nirogacestat-Exposition zur Folge haben, die zu einer verminderten Wirksamkeit führen könnten. Eine Begleittherapie mit starken Induktoren von CYP3A4 (z. B. Carbamazepin, Phenytoin, Rifampicin, Phenobarbital und Johanniskraut) und mittelstarken CYP3A‑Induktoren (z. B. Efavirenz und Etravirin) ist daher zu vermeiden. Bei Patienten, für die CYP3A4-Induktoren indiziert sind, sollten alternative Wirkstoffe mit einem geringeren Enzyminduktionspotenzial ausgewählt werden.
Wirkung von säuresenkenden Wirkstoffen
Nirogacestat verfügt über eine pH‑abhängige Löslichkeit, wobei die Löslichkeit bei pH-Werten über 6.0 erheblich verringert ist. Die Wirkungen von säuresenkenden Wirkstoffen (d. h. H2‑Rezeptor-Antagonisten, Protonenpumpenhemmer und Antazida) auf die Nirogacestat-Expositionen sind nicht in einer klinischen Studie untersucht worden, jedoch kann die gleichzeitige Gabe dieser Arzneimittel die Bioverfügbarkeit von Nirogacestat reduzieren. Die gleichzeitige Anwendung von Ogsiveo mit Protonenpumpenhemmern und H2-Blockern wird nicht empfohlen. Falls jedoch die gleichzeitige Anwendung mit säuresenkenden Wirkstoffen nicht vermeidbar ist, können Ogsiveo und Antazida zeitlich gestaffelt werden, indem Ogsiveo 2 Stunden vor oder 2 Stunden nach dem Antazidum eingenommen wird.
Wirkungen von Nirogacestat auf die Pharmakokinetik anderer Arzneimittel
CYP-Substrate
Eine Studie zu Arzneimittelwechselwirkungen mit gesunden Probanden, in der die Wirkungen von mehreren Gaben Nirogacestat bei einer Dosis von 95 mg einmal täglich auf die Exposition des sensitiven CYP3A4-Substrats Midazolam untersucht wurden, ergab einen 1,3‑fachen Anstieg der Cmax von Midazolam und einen 1,6‑fachen Anstieg der AUC von Midazolam. Die Wirkung der klinischen Dosis Nirogacestat (150 mg zweimal täglich) auf die Midazolam-Exposition ist nicht untersucht worden und kann davon abweichen. Ogsiveo sollte nicht gleichzeitig mit CYP3A4-Substraten angewendet werden, die enge therapeutische Indizes aufweisen (z. B. Cyclosporin, Tacrolimus, Digitoxin, Warfarin, Carbamazepin).
Da keine Studie zur Untersuchung der Wirkung von Nirogacestat auf die systemische Exposition von steroidalen Kontrazeptiva durchgeführt wurde, ist nicht bekannt, ob Nirogacestat die Wirksamkeit von systemisch wirkenden hormonellen Kontrazeptiva verringert. Gebärfähige Frauen müssen hochwirksame Empfängnisverhütungsmethoden anwenden (siehe Abschnitt 4.6).
In-vitro-Studien haben gezeigt, dass Nirogacestat ein Induktor von CYP2C8, CYP2C9, CYP2C19 und CYP2B6 sein kann und insofern das Risiko besteht, dass Nirogacestat eine verringerte Exposition von Substraten dieser Enzyme verursachen kann. Wenn Substrate von CYP2C8, CYP2C9, CYP2C19 und CYP2B6 zusammen mit Ogsiveo angewendet werden, sollte beurteilt werden, ob die Wirksamkeit des Substrats verringert ist; zur Aufrechterhaltung optimaler Plasmakonzentrationen ist möglicherweise eine Dosisanpassung des Substrats erforderlich.
Arzneistofftransportersysteme
Eine Studie zu Arzneimittelwechselwirkungen mit einer Einzeldosis bewies, dass Nirogacestat die Exposition des P-gp-Substrats Dabigatran nicht beeinflusste, was belegt, dass keine klinisch relevante P-gp-Inhibition durch Nirogacestat stattfindet.
Frauen im gebärfähigen Alter/Empfängnisverhütung bei Männern und Frauen
Frauen im gebärfähigen Alter und Männer mit Partnerinnen im gebärfähigen Alter sind anzuweisen, während der Behandlung mit Ogsiveo eine Schwangerschaft zu vermeiden (siehe Abschnitt 4.4).
Frauen im gebärfähigen Alter müssen während der Behandlung mit Ogsiveo und für eine Woche nach der letzten Dosis Ogsiveo hochwirksame Empfängnisverhütungsmethoden anwenden (siehe Abschnitt 4.4). Es ist nicht bekannt, ob Nirogacestat die Wirksamkeit von systemisch wirkenden hormonellen Kontrazeptiva verringert. Die Patienten sollten angewiesen werden, während der Behandlung mit Ogsiveo und für eine Woche nach der letzten Dosis Ogsiveo mindestens eine hochwirksame Empfängnisverhütungsmethode (z. B. ein nicht-hormonelles Intrauterinpessar) oder zwei zusätzliche Verhütungsmethoden, einschließlich einer Barrieremethode, anzuwenden. Frauen im gebärfähigen Alter sollten angewiesen werden, ihren Arzt sofort bei Bekanntwerden einer oder bei Verdacht auf eine Schwangerschaft zu informieren, und müssen die Einnahme von Ogsiveo bei Eintreten einer Schwangerschaft beenden. Frauen im gebärfähigen Alter dürfen während der Behandlung mit Ogsiveo und für eine Woche nach der letzten Ogsiveo-Dosis keine Eizellen (Oozyten) spenden.
Männliche Patienten mit Partnerinnen im gebärfähigen Alter müssen während der Behandlung mit Ogsiveo und für eine Woche nach der letzten Ogsiveo-Dosis hochwirksame Empfängnisverhütungsmethoden anwenden (siehe Abschnitt 4.4). Männliche Patienten sollten während der Behandlung mit Ogsiveo und für eine Woche nach der letzten Ogsiveo-Dosis kein Sperma spenden.
Schwangerschaft
Auf Grundlage von tierexperimentellen Studien und des Wirkmechanismus von Ogsiveo kann das Arzneimittel bei Verabreichung an eine schwangere Frau den Fetus schädigen. Ogsiveo ist bei schwangeren Frauen kontraindiziert (siehe Abschnitt 4.3 und 5.3). Bei Frauen im gebärfähigen Alter muss vor Einleitung der Ogsiveo-Behandlung ein negativer Schwangerschaftstest vorliegen. Schwangerschaftstests während der Behandlung mit Ogsiveo sollten für Frauen im gebärfähigen Alter, bei denen Amenorrhö vorliegt, in Betracht gezogen werden. Die Patienten sollten über das potenzielle Risiko für einen Fetus aufgeklärt werden. Wenn eine Patientin während der Einnahme von Ogsiveo schwanger wird, muss die Behandlung abgesetzt werden. In der DeFi-Studie wurde über einen Spontanabort von einer Frau berichtet, die während der Behandlung mit Nirogacestat schwanger wurde.
Stillzeit
Es liegen keine Daten darüber vor, ob Nirogacestat oder die zugehörigen Metaboliten in die Milch von Menschen oder Tieren übergeht oder sich auf gestillte Kinder oder auf die Milchproduktion auswirken. Wegen der Möglichkeit schwerwiegender Nebenwirkungen bei gestillten Kindern dürfen Frauen während der Behandlung mit Ogsiveo und für eine Woche nach der letzten Dosis Ogsiveo nicht stillen (siehe Abschnitt 4.3).
Fertilität
An Menschen wurden keine Fertilitätsstudien durchgeführt. Die Wirkung von Ogsiveo auf die Fertilität bei Menschen ist nicht bekannt. Auf Grundlage von Erkenntnissen aus tierexperimentellen Studien kann die männliche und weibliche Fertilität beeinträchtigt werden (siehe Abschnitt 5.3).
Ogsiveo hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen. Da bei Patienten, die Nirogacestat einnehmen, Ermüdung und Schwindelgefühl auftreten können (siehe Abschnitt 4.8), sollten Patienten mit diesen Nebenwirkungen beim Führen eines Fahrzeugs oder Bedienen von Maschinen Vorsicht walten lassen.
Zusammenfassung des Sicherheitsprofils
Die häufigsten Nebenwirkungen sind Diarrhö (85 %), Ausschlag (65 %), Funktionsstörungen der Eierstöcke bei gebärfähigen Frauen (60 %), Übelkeit (59 %), Ermüdung (50 %), Hypophosphatämie (50 %), Kopfschmerz (40 %) und Stomatitis (40 %).
Die am häufigsten gemeldete schwerwiegende Nebenwirkung war eine Funktionsstörung der Eierstöcke (vorzeitige Menopause, 3 %). Die häufigsten schweren Nebenwirkungen waren Diarrhö (16 %) und Hypophosphatämie (13 %).
Bei 19 % der Patienten wurde Nirogacestat aufgrund einer Nebenwirkung dauerhaft abgesetzt. Die häufigsten Nebenwirkungen, die zum Absetzen führten, waren Diarrhö (5 %), Funktionsstörungen der Eierstöcke (5 %) und erhöhte ALT (3 %).
Die Häufigkeit einer Unterbrechung der Nirogacestat-Behandlung aufgrund von Nebenwirkungen betrug 59 %. Die häufigsten Nebenwirkungen, die zur Behandlungsunterbrechung führten, waren Diarrhö (11 %), makulo-papulöser Ausschlag (10 %), Hypophosphatämie (6 %) und Übelkeit (5 %).
Die Häufigkeit einer Nirogacestat-Dosisreduktion aufgrund von Nebenwirkungen betrug 44 %. Die häufigsten Nebenwirkungen, die zur Dosisreduktion führten, waren Diarrhö (9 %), makulo-papulöser Ausschlag (6 %), Stomatitis (3 %) und Hypophosphatämie (3 %).
Tabellarische Auflistung von Nebenwirkungen
Sofern nicht anders angegeben, beruhen die Häufigkeiten der Nebenwirkungen auf Gesamthäufigkeiten von Nebenwirkungen jeglicher Ursache, festgestellt bei 88 Patienten in klinischen Studien, die über eine mediane Dauer von 21,5 Monaten Nirogacestat 150 mg zweimal täglich erhielten.
Die Nebenwirkungen sind nach Häufigkeit gemäß folgender Konvention geordnet: sehr häufig (>1/10), häufig (>1/100, <1/10), gelegentlich (>1/1 000, <1/100), selten (>1/10 000, <1/1 000), sehr selten (<1/10 000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 2: Gemeldete Nebenwirkungen
Systemorganklasse |
Nebenwirkung |
Alle Grade |
Grad 3–4 |
Erkrankungen des Gastrointestinaltrakts |
Diarrhö |
Sehr häufig |
Sehr häufig |
Übelkeit |
Sehr häufig |
Häufig |
|
Stomatitisa |
Sehr häufig |
Häufig |
|
Mundtrockenheit |
Sehr häufig |
-- |
|
Erkrankungen der Haut und des Unterhautgewebes |
Ausschlagb |
Sehr häufig |
Häufig |
Alopezie |
Sehr häufig |
-- |
|
Follikulitis |
Sehr häufig |
Häufig |
|
Hidradenitis |
Häufig |
Häufig |
|
Trockene Haut |
Sehr häufig |
-- |
|
Pruritis |
Sehr häufig |
-- |
|
Gutartige, bösartige und nicht spezifizierte Neubildungen |
Basalzellkarzinom |
Häufig |
-- |
Plattenepithelkarzinom |
Häufig |
-- |
|
Stoffwechsel- und Ernährungsstörungen |
Hypophosphatämie |
Sehr häufig |
Sehr häufig |
Hypokaliämie |
Sehr häufig |
Häufig |
|
Erkrankungen des Nervensystems |
Kopfschmerzen |
Sehr häufig |
-- |
Schwindelgefühl |
Sehr häufig |
-- |
|
Untersuchungen |
Proteinurie |
Sehr häufig |
-- |
Glykosurie |
Sehr häufig |
-- |
|
Erkrankungen des Blutes und des Lymphsystems |
Eosinophilie |
Sehr häufig |
-- |
Erkrankungen der Nieren und Harnwege |
Nierentubuluserkrankung |
Häufig |
-- |
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen |
Knochenfrakturd |
Häufig |
-- |
Leber- und Gallenerkrankungen |
ALT erhöht |
Sehr häufig |
Häufig |
AST erhöht |
Sehr häufig |
Häufig |
|
Erkrankungen der Geschlechtsorgane und der Brustdrüse |
Funktionsstörung der Eierstöckee |
Sehr häufig |
-- |
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Husten |
Sehr häufig |
-- |
Infektion der oberen Atemwegef |
Sehr häufig |
-- |
|
Dyspnoe |
Sehr häufig |
-- |
|
Epistaxis |
Sehr häufig |
-- |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Ermüdung |
Sehr häufig |
Häufig |
Grippeähnliche Erkrankung |
Sehr häufig |
-- |
|
Augenerkrankungen |
Photopsie |
Häufig |
-- |
Mouches volantes |
Häufig |
-- |
|
Trockenes Auge |
Häufig |
-- |
|
Erkrankungen des Immunsystems |
Überempfindlichkeit* |
Nicht bekannt |
a Stomatitis umfasst Stomatitis, Mundulzeration, Mundschmerzen und Schmerzen im Oropharynx.
b Ausschlag umfasst makulo-papulären Ausschlag, Dermatitis akneiform, Ausschlag, erythematöser Hautausschlag, Ausschlag mit Juckreiz und papulöser Ausschlag.
c Plattenepithelkarzinom umfasst Plattenepithelkarzinom der Haut und Plattenepithelkarzinom.
d Knochenfraktur umfasst Fraktur, Fraktur des Fußes, Fraktur der Hand, Radiusfraktur, Fraktur der Hüfte und Rippenfraktur.
e Funktionsstörung der Eierstöcke umfasst Nachlassen der ovariellen Funktion, vorzeitige Menopause, Amenorrhö, Oligomenorrhö, menstruelle Unregelmäßigkeit, Dysmenorrhö, heftige Menstruationsblutung, vulvovaginale Trockenheit, Hitzewallung, erniedrigter Anti-Müller-Hormonspiegel (AMH) und erhöhtes follikelstimulierendes Hormon (FSH).
f Infektion der oberen Atemwege umfasst Infektion der oberen Atemwege, Virusinfektion der oberen Atemwege, akute Sinusitis und Sinusitis.
-- bedeutet, dass keine Fälle gemeldet wurden.
* Fälle aus der Zeit nach der Markteinführung.
Beschreibung ausgewählter Nebenwirkungen
Die nachstehend beschriebenen Daten zeigen die Ergebnisse der randomisierten, doppelt verblindeten Phase-III-DeFi-Studie bei Patienten mit Desmoidtumoren, die 150 mg BID Nirogacestat (N=69) oder Placebo (N=72) zweimal täglich erhielten.
Diarrhö
In dem doppelt verblindeten Zeitraum der DeFi-Studie wurde Diarrhö bei 84 % der mit Nirogacestat behandelten Patienten und bei 35 % der Patienten, die das Placebo erhielten, berichtet. Grad-3-Ereignisse traten bei 16 % bzw. 1 % der Patienten auf (siehe Abschnitt 4.4). Diarrhö Grad ≤ 2 klang bei 74 % der Patienten ab, die weiterhin mit Nirogacestat behandelt wurden. Die mediane Dauer bis zum ersten Einsetzen von Diarrhö bei den mit Nirogacestat behandelten Patienten betrug 9 Tage (Bereich 2 bis 434 Tage). Unter den Patienten, die Nirogacestat erhielten, führte Diarrhö bei 10 % zu einer Dosisreduktion und bei 7 % zum Behandlungsabbruch.
Erkrankungen der Haut und des Unterhautgewebes
Es wurde berichtet, dass in dem doppelt verblindeten Zeitraum der DeFi-Studie dermatologische Reaktionen eine höhere Inzidenz bei mit Nirogacestat behandelten Patienten hatten als bei Patienten, die das Placebo erhielten; dazu gehörten makulo-papulärer Ausschlag (32 % bzw. 6 %), Hidradenitis (9 % bzw. 0 %) und Follikulitis (13 % bzw. 0 %) (siehe Abschnitt 4.4). Die mediane Dauer bis zum Auftreten von Ausschlag-Fällen betrug 22 Tage (Bereich 2 bis 603 Tage). Erkrankungen der Haut und des Unterhautgewebes führten bei 9 % der mit Nirogacestat behandelten Patienten zu einer Dosisreduktion, darunter makulo-papulärer Ausschlag bei 4 % und Hidradenitis bei 3 %. Makulo-papulärer Ausschlag führte bei 1 % zum Behandlungsabbruch.
Funktionsstörung der Eierstöcke
In dem doppelt verblindeten Zeitraum der DeFi-Studie wurde bei 75 % der gebärfähigen Frauen, die Nirogacestat erhielten, über Funktionsstörungen der Eierstöcke (definiert als Nachlassen der ovariellen Funktion, vorzeitige Menopause, Amenorrhö, Oligomenorrhö und Menopause) berichtet, während bei den Patientinnen, die das Placebo erhielten, keine Fälle gemeldet wurden. Es gab drei schwerwiegende Reaktionen von Funktionsstörungen der Eierstöcke; bei allen handelte es sich um vorzeitige Menopause, was 11 % aller Teilnehmerinnen, die Funktionsstörungen der Eierstöcke meldeten, entspricht. Die mediane Dauer bis zum ersten Einsetzen von Funktionsstörungen der Eierstöcke betrug 8,9 Wochen (Bereich 1 Tag bis 54 Wochen), und die gesamte mediane Dauer betrug 18,9 Wochen (Bereich 11 Tage bis 215 Wochen). Es wurde berichtet, dass die Funktionsstörungen der Eierstöcke bei 79 % der gebärfähigen Frauen während der Behandlung abgeklungen waren. Nachbeobachtungsinformationen liegen mit Ausnahme von zwei für alle 27 Patientinnen vor. Es wurde berichtet, dass die Funktionsstörungen der Eierstöcke bei allen gebärfähigen Frauen, für die Daten vorliegen, abgeklungen waren. Die mediane Dauer bis zum Abklingen nach dem Absetzen von Nirogacestat betrug 10,9 Wochen (Bereich 4 bis 18 Wochen). Die Wirkungen von Nirogacestat auf die Fertilität sind nicht bekannt (siehe Abschnitt 4.4). Eine Dosis-Wirkungs-Beziehung wurde zwischen Nirogacestat und dem Serumspiegel von follikelstimulierendem Hormon (FSH), wobei sich der FSH-Spiegel linear zu ansteigenden Serumkonzentrationen von Nirogacestat erhöhte.
Elektrolytanomalien
Elektrolytanomalien wurden in dem doppelt verblindeten Zeitraum der DeFi-Studie bei mit Nirogacestat behandelten Patienten berichtet, darunter Hypophosphatämie (43 %) und Hypokaliämie (12 %), und bei 7 % bzw. 1 % der Patienten, die das Placebo erhielten. Die mediane Dauer bis zum ersten Einsetzen von Hypophosphatämie und Hypokaliämie betrug 15 Tage (Bereich 1 Tag bis 833 Tage) bzw. 15 Tage (Bereich 1 Tag bis 57 Tage). Ereignisse einer Hypophosphatämie und Hypokaliämie Grad 3 traten bei 3 % der mit Nirogacestat behandelten Patienten und bei keinen der Patienten, die das Placebo erhielten, auf (siehe Abschnitt 4.4). Hypophosphatämie und Hypokaliämie führten zu einer Dosisreduktion bei 4 % bzw. 1 % der mit Nirogacestat behandelten Patienten. Unter den Patienten, die Nirogacestat erhielten, führte Hypophosphatämie bei 1% zum Behandlungsabbruch.
Anomalien der Leberwerte
Anstiege der ALT- und AST-Werte wurden bei 19 % bzw. 17 % der mit Nirogacestat behandelten Patienten in dem doppelt verblindeten Zeitraum der DeFi-Studie gemeldet, gegenüber 8 % bzw. 11 % bei den Patienten, die das Placebo erhielten. Die mediane Dauer bis zum ersten Einsetzen von ALT- und AST-Anstiegen betrug 22 Tage (ALT: Bereich 8 bis 924 Tage; AST: Bereich 1 Tag bis 1 023 Tage). Anstiege der ALT und AST von Grad 3 (> 5 x ULN) traten bei 3 % der mit Nirogacestat behandelten Patienten und bei 1 % im Placebo-Arm auf (siehe Abschnitt 4.4). Unter den Patienten, die Nirogacestat erhielten, führten ALT- und AST-Anstiege bei 1% zu einer Dosisreduktion. Anstiege der ALT- und AST-Werte führten zum Behandlungsabbruch bei 4 % bzw. 3% der mit Nirogacestat behandelten Patienten.
Nichtmelanozytärer Hautkrebs
Nichtmelanozytäre Hautkrebserkrankungen wurden in dem doppelt verblindeten Zeitraum der DeFi-Studie bei den mit Nirogacestat behandelten Patienten mit höherer Inzidenz als bei den Patienten, die das Placebo erhielten, berichtet, darunter Plattenepithelkarzinom (3 % bzw. 0 %) und Basalzellkarzinom (1 % bzw. 0 %), wobei ein Patient beide Arten von nichtmelanozytärem Hautkrebs meldete (siehe Abschnitt 4.4). Weitere zwei Fälle von nichtmelanozytärem Hautkrebs wurden außerhalb des doppelt verblindeten Zeitraums der DeFi-Studie berichtet.
Wirkung auf den proximalen Nierentubulus
Glykosurie und Proteinurie wurden in dem doppelt verblindeten Zeitraum der DeFi-Studie bei 52 % bzw. 46 % der mit Nirogacestat behandelten Patienten beobachtet, gegenüber 1 % bzw. 39 % bei den Patienten, die das Placebo erhielten. Die mediane Dauer bis zum Einsetzen von Glykosurie und Proteinurie betrug 85 Tage (Bereich 55 bis 600 Tage) bzw. 72 Tage (Bereich 38 bis 937 Tage). Ein Patient in der der DeFi-Studie meldete eine Erkrankung des Nierentubulus mit erhöhter Ausscheidung von Harnsäure, Glucose und Phosphat im Urin, jedoch ohne eine übermäßige Ausscheidung von Proteinen mit niedrigem Molekulargewicht (Beta2‑Mikroglobulin) oder eine veränderte Nierenfunktion. Das Ereignis wurde mittels Dosisreduktion behandelt.
Knochenfraktur
In dem doppelt verblindeten Zeitraum der DeFi‑ Studie wurden Knochenfrakturen bei 6 % der mit Nirogacestat behandelten Patienten berichtet, während bei Patienten, die das Placebo erhielten, keine Knochenfrakturen gemeldet wurden. Alle gemeldeten Knochenfrakturen waren nicht schwerwiegend und entsprachen Grad 1 oder 2. Die mediane Dauer bis zum ersten Einsetzen von Knochenfrakturereignissen bei den mit Nirogacestat behandelten Patienten betrug 125 Tage (Bereich 1 Tag bis 739 Tage). Knochenfrakturereignisse führten bei keinem der mit Nirogacestat behandelten Patienten zu einer Dosisreduktion oder zum Behandlungsabbruch.
Kinder und Jugendliche
Epiphysenerkrankungen, die sich als Erweiterung der Epiphysenfuge äußerten, wurden bei 4 von 26 (15 %) der Kinder und Jugendlichen mit offenen Wachstumsfugen berichtet, die außerhalb der DeFi-Studie mit Nirogacestat behandelt wurden. Die Ereignisse umfassten Epiphyseolyse, Fraktur der Hüfte, Epiphysenerkrankung und Osteonekrose. Alle vier pädiatrischen Patienten waren im Alter zwischen 11 und 12 Jahren. Informationen zur Anwendung bei Kindern und Jugendlichen, siehe Abschnitt 4.2.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
Anzeichen und Symptome
Es wird erwartet, dass die Symptome einer Ogsiveo-Überdosierung einer Verstärkung seiner pharmakologischen Wirkungen entsprechen, darunter möglicherweise Diarrhö, Übelkeit, Erbrechen, Hypophosphatämie, erhöhte Transaminasen und Epistaxis.
Behandlung von Überdosierung
Aufgrund des hohen Maßes der Proteinbindung wird nicht davon ausgegangen, dass Ogsiveo bei Patienten mit normalen Serum-Proteinwerten dialysierbar ist. Im Fall einer Überdosierung sollte die Behandlung mit Ogsiveo ausgesetzt werden und allgemeine unterstützende Maßnahmen sollten eingeleitet werden.
Pharmakotherapeutische Gruppe: Antineoplastisches Mittel, andere antineoplastische Mittel, ATC-Code: L01XX81
Wirkmechanismus
Nirogacestat ist ein reversibler und nicht-kompetitiver Inhibitor der Gamma-Sekretase, der die proteolytische Aktivierung des Notch-Rezeptors blockiert.
Kardiale Elektrophysiologie
Die Wirkungen der Nirogacestat-Konzentration auf die Verlängerung des QTc-Intervalls wurden mit einer modell-basierten Analyse vorhergesagt. Die 90-%-Konfidenzintervalle für die vorhergesagte mittlere Veränderung von QTcF lagen bei dem erwarteten Cmax bei supratherapeutischen Dosen unter 10 ms. Daher ist keine klinisch signifikante Verlängerung des QTcF-Intervalls mit der therapeutischen Dosierung von Ogsiveo assoziiert.
Klinische Wirksamkeit und Sicherheit
Die DeFi-Studie war eine internationale, multizentrische, randomisierte (1:1), doppelt verblindete, placebokontrollierte Phase-III-Studie bei erwachsenen Patienten mit fortschreitenden Desmoidtumoren. Patienten mit histologisch bestätigten Desmoidtumoren, die innerhalb der 12 Monate vor dem Screening um ≥ 20 % (gemessen nach RECIST v1.1) fortgeschritten waren und bei denen die fortgesetzte Progression kein unmittelbares signifikantes Risiko für den Patienten darstellte, kamen für die Studienteilnahme infrage. Die Randomisierung wurde nach Lokalisation(en) der Zieltumoren (intraabdominell bzw. extraabdominell) stratifiziert. Patienten mit mehreren Zieltumoren sowohl an der intraabdominellen als auch der extraabdominellen Lokalisation wurden als intraabdominell klassifiziert. Die Patienten erhielten in 28-Tages-Zyklen zweimal täglich 150 mg orales Nirogacestat oder Placebo bis zum Eintreten von Progression, Tod oder inakzeptabler Toxizität. Die primäre Wirksamkeitsmessgröße war das progressionsfreie Überleben (progression-free survival, PFS). Die Progression wurde radiografisch mittels RECIST v1.1 durch eine verblindete, unabhängige zentrale Bildgebungsprüfung bestimmt oder vom Prüfarzt klinisch bewertet und durch eine verblindete, unabhängige zentrale Prüfung oder Tod aus beliebigem Grund qualifiziert. Weitere Wirksamkeitsmessgrößen umfassten die objektive Ansprechrate (objective response rate, ORR), Veränderung der Schmerzen gegenüber Baseline in Zyklus 10, Veränderung des Desmoidtumor-spezifischen Symptomschweregrads gegenüber Baseline in Zyklus 10, Veränderung der Rollenfunktion und körperlichen Funktionsfähigkeit gegenüber Baseline in Zyklus 10 und die Veränderung der Gesamtlebensqualität gegenüber Baseline in Zyklus 10. Die Schmerzen wurden anhand des 7-Tages-Durchschnitts von Frage 3 (d. h. stärkste Schmerzen) aus dem Brief Pain Inventory (BPI) in der Kurzform (Short Form, SF). Der Desmoidtumor-spezifische Symptomschweregrad und die körperliche Funktionsfähigkeit wurden mit der GOunder/DTRF DEsmoid Symptom/Impact Scale (GODDESS) gemessen.
Insgesamt wurden 142 Patienten randomisiert: 70 zu Nirogacestat und 72 zum Placebo. Insgesamt betrug das mediane Alter 34 Jahre (Bereich: 18 bis 76); 4 % waren 65 Jahre oder älter; 65 % waren weiblich; die ethnische Herkunft: 83 % hellhäutig, 6 % schwarz, 3 % asiatisch und 8 % andere; 73 % hatten einen ECOG-Leistungsstatus (performance status, PS) von 0, 27 % hatten einen ECOG-PS von 1 und < 1 % hatten einen ECOG-PS von 2. 23 % der Patienten hatten eine intraabdominelle Erkrankung oder eine sowohl intra- als auch extraabdominelle Erkrankung, und 77 % hatten nur eine extraabdominelle Erkrankung. Bei 41 % der Patienten lag eine Erkrankung mit mehreren Herden und bei 59 % eine Erkrankung mit einem Herd vor. Von 105 Patienten mit bekanntem somatischen Tumor-Mutationsstatus wiesen 81 % eine CTNNB1-Mutation und 21 % eine APC-Mutation auf. Bei 17 % der Patienten lag eine Familienanamnese von familiärer adenomatöser Polypose (FAP) vor. 23 % der Patienten hatten keine vorherige Therapie erhalten und 44 % hatten ≥ 3 vorherige Therapielinien erhalten. Die vorherige Therapie umfasste systemische Therapie (61 %), Operation (53 %) und Strahlentherapie (23 %). 36 % der Patienten waren zuvor mit Chemotherapie behandelt und 33 % wurden mit einem Tyrosinkinasehemmer vorbehandelt worden. 55 % hatten bei Baseline bei Frage 3 (stärkste Schmerzen) im BPI‑SF einen Score von ≥ 2.
Die Wirksamkeitsergebnisse bei der ITT-Population, die alle randomisierten Patienten umfasste, sind unten dargestellt. Die Verbesserungen von PFS und ORR waren zugunsten von Nirogacestat, und zwar unabhängig von den Baseline-Charakteristika, einschließlich Tumorlokalisation und Art der vorherigen Therapien.
Tabelle 3: Wirksamkeitsergebnisse bei Patienten mit fortschreitenden Desmoidtumoren gemäß RECIST 1.1
Nirogacestat |
Placebo |
|
Progressionsfreies Überleben | ||
Anzahl (%) der Patienten mit dem Ereignis |
12 (17) |
37 (51) |
Radiografische Progression a |
11 (16) |
30 (42) |
Klinische Progression a |
1 (1) |
6 (8) |
Tod |
0 |
1 (1) |
Median (Monate) (95%-KI) b |
NE (NE, NE) |
15,1 (8,4; NE) |
Hazard-Ratio (95%-KI) |
0,29 (0,15; 0,55) |
|
P-Wert c |
< 0,001 |
|
Objektive Ansprechrate (objective response rate, ORR) a | ||
ORR, n (%) |
29 (41) |
6 (8) |
CR |
5 (7) |
0 |
PR |
24 (34) |
6 (8) |
P-Wert e |
< 0,001 |
|
Abkürzungen: KI: Konfidenzintervall; CR: vollständiges Ansprechen (complete response); ORR: objektive Ansprechrate (objective response rate); PR: partielles Ansprechen (partial response); NE: nicht erreicht
a Durch verblindete, unabhängige zentrale Prüfung beurteilt.
b Mit der Kaplan-Meier-Methode bestimmt.
c Der P-Wert stammte aus einem einseitigen stratifizierten Log-Rank-Test.
d Bestimmt mit der exakten Methode auf Basis von Binomialverteilung.
e Der P-Wert stammte aus einem zweiseitigen Cochran-Mantel-Haenszel-Test.
Abbildung 1: Kaplan-Meier-Kurve des PFS
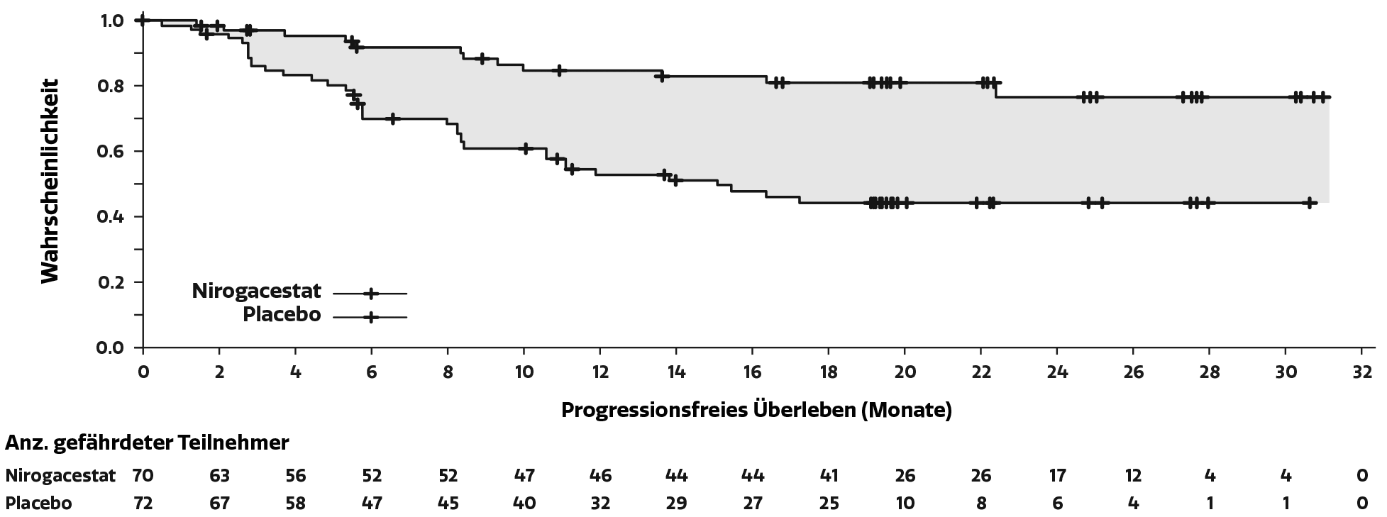
Hinweis: Medianwert und 95 %-Konfidenzintervalle wurden anhand der Kaplan-Meier-Methode bestimmt. Aufgrund der geringen Anzahl an Ereignissen im Nirogacestat-Arm konnte die Kaplan-Meier-Schätzung der medianen Dauer bis zur Progression nicht bewertet werden.
Behandlungsergebnisse aus Patientenperspektive
Die PFS-Ergebnisse wurden durch die Veränderung der vom Patienten berichteten stärksten Schmerzen gegenüber Baseline unterstützt und waren in Zyklus 10 zugunsten des Nirogacestat-Arms (‑1,6 gegenüber ‑0,2; mittlere LS-Differenz: ‑1,3; 95%-Konfidenzintervall: ‑2,1 bis ‑0,6; p < 0,001).
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Ogsiveo eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen für die Behandlung von Weichgewebssarkom gewährt. Informationen zur Anwendung bei Kindern und Jugendlichen, siehe Abschnitt 4.2.
Resorption
Die Spitzenkonzentrationen von Nirogacestat werden etwa 1,5 Stunden nach oraler Anwendung erreicht. Die absolute Bioverfügbarkeit von Nirogacestat nach der oralen Gabe beträgt ungefähr 19,2 % (Bereich: 16,2 % bis 24,3 %).
Verteilung
Der Blut-Plasma-Quotient wird beim Menschen auf ungefähr 0,5 geschätzt. Die Serum-Proteinbindung beträgt in vitro ungefähr 99,6 %. Nirogacestat wird stark sowohl an humanes Serumalbumin als auch an saures α-1-Glykoprotein gebunden, wobei die Affinität bei saurem α-1-Glykoprotein größer ist. Auf Basis der Analyse der populationsbezogenen Pharmakokinetik wurde das scheinbare orale Verteilungsvolumen von Nirogacestat bei Patienten mit Desmoidtumoren auf 1430 Liter geschätzt.
Biotransformation
Nirogacestat wird weitgehend hauptsächlich durch CYP3A4 metabolisiert. Das Wissen über in vivo vorkommende wichtige oder aktive Metaboliten ist aufgrund der Einschränkungen bei der Erkennung von nicht radiomarkierten Metaboliten begrenzt. Zahlreiche Metaboliten mit geringerem Anteil wurden in der Zirkulation und in Exkreten festgestellt.
Elimination
Nach einer oralen Einzeldosis von radiomarkiertem Nirogacestat bei gesunden Probanden fanden sich ungefähr 65 % der Dosis innerhalb von 13 Tagen nach der Gabe wieder; 38 % werden im Stuhl eliminiert, 17 % werden im Urin eliminiert, und 10 % der Markierung fanden sich in der Ausatemluft wieder. Von einer verabreichten Dosis Nirogacestat in unveränderter Form fanden sich im Urin weniger als 0,01 % und im Stuhl weniger als 0,5 % wieder.
Gemäß der Analyse der populationsbezogenen Pharmakokinetik in der Population mit Desmoidtumoren wird die scheinbare terminale Eliminationshalbwertszeit auf ungefähr 23 Stunden geschätzt. Die scheinbare orale systemische Clearance beträgt ungefähr 45 Liter/Stunde.
Linearität/Nicht‑Linearität
Die Exposition von Nirogacestat steigt mit eskalierenden Einzel- und Wiederholungsdosen, wobei proportionelle Anstiege im Dosisbereich 50–150 mg liegen.
Steady-State-Bedingungen werden nach wiederholter Gabe nach ungefähr 7 Tagen erreicht. Gemäß der Analyse der populationsbezogenen Pharmakokinetik wird das Akkumulationsverhältnis bei Patienten mit Desmoidtumor auf ungefähr 1,5 geschätzt.
Besondere Patientengruppen
Auswirkungen von Leberfunktionsstörungen
Die Pharmakokinetik von Nirogacestat wurde bei Patienten mit mäßiger Leberfunktionsstörung nach der Child-Pugh-Klassifikation untersucht. Die Nirogacestat-Gesamtexposition (AUC) wurde durch mäßige Leberfunktionsstörung nicht beeinflusst, aber die Spitzenexposition (Cmax) wurde um 28 % verringert; das Verteilungsvolumen war höher und die Halbwertszeit länger.
Auswirkungen von Nierenfunktionsstörungen
Die Wirkungen von Nierenfunktionsstörungen auf die Pharmakokinetik von Nirogacestat sind nicht in einer speziellen klinischen Studie untersucht worden. In einem PopPK-Modell wurde kein klinisch aussagefähiger Zusammenhang zwischen Nierenfunktionstests und der Pharmakokinetik von Nirogacestat beobachtet. Unter den 335 in der PopPK-Analyse eingeschlossenen Studienteilnehmern hatten zwei Patienten eine leichte und mäßige Nierenfunktionsstörung. In die PopPK-Analyse wurden keine Patienten mit schwerer Nierenfunktionsstörung eingeschlossen.
In Studien zur Toxizität bei wiederholter Gabe an Ratten und Hunde wurden die meisten Toxizitäten mit Gamma-Sekretase-Inhibition assoziiert. Zu den Auswirkungen gehörten Atrophie der Eierstöcke, Veränderungen des Östruszyklus, verringerte Zellularität in darmassoziiertem lymphatischem Gewebe und verringerte Zellularität der mesenterialen Lymphknoten. In der Studie an Ratten wurde eine Verdickung der Wachstumsfuge beobachtet. Darüber hinaus zeigte sich bei allen in der Studie an Ratten untersuchten Dosisstufen dosisabhängig chronische progressive Nephropathie, pulmonale Phospholipidose und Nekrose der Speicheldrüse. In der Studie an Hunden lagen behandlungsbedingte Auswirkungen im Darm, in der Milz, Gallenblase, Leber, Niere, in den Hoden und im Eierstock vor. Die Darm- und Leberbefunde gingen bei den meisten Hunden mit einer generalisierten Entzündung und assoziierten klinischen pathologischen Veränderungen einher. In den 3‑monatigen oralen Toxizitätsstudien mit Ratten und Hunden wurde keine höchste Dosis ohne beobachtete schädliche Auswirkung (No Oberserved Adverse Effect Level, NOAEL) identifiziert. Die niedrigste Dosis in der Studie an Ratten betrug 5 mg/kg/Tage (humanäquivalente Dosis 50 mg/Tag) und in der Studie mit Hunden 2 mg/kg/Tag (humanäquivalente Dosis 70 mg/Tag). Die systemischen Expositionen lagen auch unter den verabreichten humanen systemischen Expositionen (AUC): 150 mg Nirogacestat BID.
Kanzerogenität
Der Notch-Signalweg scheint sowohl eine onkogene als auch eine tumorsuppressive Funktion zu haben. Das karzinogene Potenzial von Nirogacestat wurde in einer 6-monatigen Studie mit transgenen rasH2-Mäusen untersucht. Bei Dosen bis zu 100 mg/kg/Tag wurde eine erhöhte Inzidenz von Hämangiosarkomen beobachtet. Bei 100 mg/kg/Tag lagen die systemischen Expositionen (AUC) unter (0,2‑fach) denjenigen bei Menschen, denen 150 mg Nirogacestat BID verabreicht wurde. Das karzinogene Potenzial bei Ratten wurde nicht untersucht.
Reproduktions- und Entwicklungstoxizität
Nirogacestat verringerte die Fertilitätsindizes sowohl bei männlichen als auch weiblichen Ratten, was mit Atrophie der Eierstöcke, geringerem Hodengewicht und erniedrigter Spermienbeweglichkeit sowie Auswirkungen auf die Spermienmorphologie korrelierte. Darüber hinaus kam es in Fertilitätsstudien zu frühzeitigem Embryonenverlust. In einer vorläufigen Studie zur embryonalen/fetalen Entwicklung induzierte Nirogacestat signifikanten und dosisabhängigen Embryonenverlust, Fälle von frühzeitiger Resorption und vermindertes Gewicht bei überlebenden Embryonen. Diese Auswirkungen traten bei 20 mg/kg/Tag auf, was zu systemischen Expositionen unter (ungefähr 0,45‑fach) humanen Expositionen nach Gabe von 150 mg Nirogacestat BID (siehe Abschnitt 4.4) führte.
Tablettenkern
Mikrokristalline Cellulose
Lactose-Monohydrat
Carboxymethylstärke-Natrium (Ph.Eur.)
Magnesiumstearat
Tablettenüberzug
Macrogol-Polyvinylalkohol-graft-Copolymer (E1209)
Talkum
Titandioxid (E171)
Glycerolmonocaprylocaprat (Ph.Eur.) Typ 1/Mono-/Diglyceride (E471)
Poly(vinylalkohol) – teilweise hydrolysiert (E1203)
Gelborange S (E110)
Eisen(III)-hydroxid-oxid x H2O (E172)
Nicht zutreffend.
2 Jahre.
Unter 25 °C lagern.
Ogsiveo 50 mg Filmtabletten
HDPE-Flasche mit kindersicherem Verschluss und Schutzfolie mit 120 oder 180 Tabletten.
Ogsiveo 100 mg Filmtabletten
Ogsiveo 150 mg Filmtabletten
Transparente PVC/PVDC-Blister mit Aluminium-Rückwand mit 14 Tabletten. Eine Packung enthält 56 Tabletten in 4 Blistern.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Merck Europe B.V.
Gustav Mahlerplein 102
1082 MA Amsterdam
Niederlande
EU/1/25/1932/001
EU/1/25/1932/002
EU/1/25/1932/003
EU/1/25/1932/004
Datum der Erteilung der Zulassung: 14. August 2025
Juli 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel‑Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig