
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Prufibry 1 g Pulver und Lösungsmittel zur Herstellung einer Injektions-/Infusionslösung
Humanes Fibrinogen
Eine Durchstechflasche enthält nominal 1 g humanes Fibrinogen.
Nach Rekonstitution mit 50 ml Lösungsmittel (Wasser für Injektionszwecke) enthält Prufibry etwa 20 mg/ml humanes Fibrinogen.
Der Gehalt an gerinnungsfähigem Protein wird entsprechend der Monografie des Europäischen Arzneibuchs für humanes Fibrinogen bestimmt.
Hergestellt aus Plasma menschlicher Spender.
Sonstige Bestandteile mit bekannter Wirkung
Eine Durchstechflasche enthält etwa 132,2 mg Natrium (5,8 mmol) und 25,5 mg Polysorbat 80.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Pulver und Lösungsmittel zur Herstellung einer Injektions-/Infusionslösung
Weißes Pulver und klares, farbloses Lösungsmittel.
Prufibry hat einen pH-Wert von 6,5–7,5 und eine Osmolalität von ≥240 mOsmol/kg.
Prufibry wird angewendet:
zur Behandlung und perioperativen Prophylaxe von Blutungen bei Patienten mit kongenitaler Hypo- oder Afibrinogenämie mit Blutungsneigung.
als Komplementärtherapie zur Behandlung unkontrollierter schwerwiegender Blutungen bei erworbener Hypofibrinogenämie aufgrund von Operationen oder Traumata.
Prufibry wird bei Erwachsenen, Kindern und Jugendlichen (0–18 Jahre) angewendet.
Die Therapie sollte unter Überwachung eines Arztes eingeleitet werden, der Erfahrung in der Behandlung von Gerinnungsstörungen hat.
Dosierung
Die Dosierung und Dauer der Substitutionstherapie richten sich nach der Schwere der Erkrankung, dem Ort und Ausmaß der Blutungen sowie dem klinischen Zustand des Patienten.
Die individuelle Dosierung sollte auf Grundlage des (funktionalen) Fibrinogenspiegels berechnet werden. Die Dosierung und Häufigkeit der Anwendung sollten für jeden Patienten individuell durch regelmäßige Messung des Fibrinogenspiegels im Plasma sowie durch kontinuierliche Überwachung des klinischen Zustands des Patienten und anderer angewandter Substitutionstherapien bestimmt werden.
Der normale Fibrinogenspiegel im Plasma beträgt 1,5–4,5 g/l. Bei kongenitaler Hypo- oder Afibrinogenämie liegt der kritische Fibrinogenspiegel im Plasma, unterhalb dessen es zu Blutungen kommen kann, bei etwa 0,5–1,0 g/l.
Bei größeren chirurgischen Eingriffen ist eine präzise Überwachung der Substitutionstherapie durch Gerinnungstests unbedingt erforderlich.
1. Perioperative Prophylaxe und Behandlung von Blutungen bei Patienten mit kongenitaler Hypo- oder Afibrinogenämie mit Blutungsneigung
Zur Vorbeugung übermäßiger Blutungen bei chirurgischen Eingriffen wird eine prophylaktische Behandlung empfohlen, um den Fibrinogenspiegel auf 1 g/l anzuheben und auf diesem Wert zu halten, bis die Hämostase unter Kontrolle ist, und auf einem Wert über 0,5 g/l zu halten, bis die Wundheilung abgeschlossen ist.
Im Fall von Blutungsepisoden wird ein Ziel-Fibrinogenspiegel im Plasma von 1 g/l empfohlen.
In beiden Fällen, chirurgischer Eingriff oder Behandlung einer Blutungsepisode, ist die Dosis wie folgt zu berechnen:
Erwachsene, Kinder und Jugendliche (6–18 Jahre):
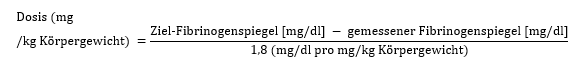
Kinder <6 Jahren:
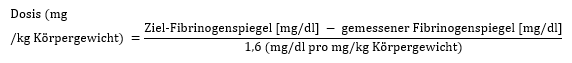
Wenn der Fibrinogenspiegel nicht bekannt ist, wird die folgende Anfangsdosis empfohlen:
Erwachsene, Kinder und Jugendliche (0–18 Jahre):
70 mg/kg Körpergewicht.
Die nachfolgende Dosierung (Injektionsdosen und -häufigkeit) sollte auf Grundlage des klinischen Zustands des Patienten und der Laborergebnisse angepasst werden.
Die biologische Halbwertszeit von Fibrinogen beträgt 3–4 Tage. Daher ist bei fehlendem Verbrauch eine wiederholte Behandlung mit humanem Fibrinogen in der Regel nicht erforderlich. Aufgrund der Akkumulation bei wiederholter Verabreichung zur Prophylaxe sollten die Dosis und die Häufigkeit entsprechend den therapeutischen Zielen des Arztes für den jeweiligen Patienten bestimmt werden.
2. Behandlung von Blutungen bei Patienten mit erworbener Hypofibrinogenämie
Erwachsene:
Im Allgemeinen werden initial 1–2 g verabreicht, mit nachfolgenden Infusionen nach Bedarf. Bei schwerwiegenden Blutungen, z. B. bei größeren Operationen, können größere Mengen an Fibrinogen (4–8 g) erforderlich sein.
Kinder und Jugendliche (0–18 Jahre):
Die Dosierung sollte nach dem Körpergewicht und der klinischen Notwendigkeit bestimmt werden, beträgt jedoch in der Regel 20–30 mg/kg.
Art der Anwendung
Intravenöse Infusion oder Injektion.
Prufibry sollte langsam intravenös verabreicht werden. Die empfohlene maximale Geschwindigkeit beträgt:
Patientengruppe |
Maximale Infusionsgeschwindigkeit |
Erwachsene, Kinder und Jugendliche (6–18 Jahre) mit kongenitaler Hypo- oder Afibrinogenämie |
5 ml/min (100 mg/min) |
Kinder (<6 Jahre) mit kongenitaler Hypo- oder Afibrinogenämie |
<5 ml/min (<100 mg/min) |
Erwachsene, Kinder und Jugendliche (0–18 Jahre) mit erworbenem Fibrinogenmangel |
250 ml/min (5 g/min) |
Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung siehe Abschnitte 6.2 und 6.6.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Thromboembolie
Es besteht ein Thromboserisiko, wenn Patienten mit angeborenem oder erworbenem Mangel mit humanem Fibrinogen behandelt werden, insbesondere bei hohen Dosen oder wiederholter Gabe. Patienten, die humanes Fibrinogen erhalten, sollten engmaschig auf Anzeichen oder Symptome einer Thrombose überwacht werden.
Bei Patienten mit koronarer Herzkrankheit oder Myokardinfarkt in der Anamnese, Patienten mit Lebererkrankung, peri- oder postoperativen Patienten, Neugeborenen oder Patienten mit einem Risiko thromboembolischer Ereignisse oder disseminierter intravaskulärer Gerinnung muss der potenzielle Nutzen einer Behandlung mit Fibrinogen aus humanem Blutplasma gegen das Risiko thromboembolischer Komplikationen abgewogen werden. Vorsicht und eine engmaschige Überwachung sind ebenfalls geboten.
Erworbene Hypofibrinogenämie ist mit niedrigen Plasmakonzentrationen aller Gerinnungsfaktoren (nicht nur Fibrinogen) und Gerinnungshemmern assoziiert, daher sollte eine Behandlung mit Blutprodukten, die Gerinnungsfaktoren enthalten, erwogen werden. Eine sorgfältige Überwachung des Gerinnungssystems ist erforderlich.
Allergische oder anaphylaktische Reaktionen
Wenn allergische oder anaphylaktische Reaktionen auftreten, muss die Injektion/Infusion sofort gestoppt werden. Beim Auftreten eines anaphylaktischen Schocks ist die medizinische Standardtherapie bei Schock durchzuführen.
Übertragbare Erreger
Standardmaßnahmen zur Verhütung von Infektionen durch die Verabreichung von Arzneimitteln, die
aus menschlichem Blut oder Plasma hergestellt wurden, beinhalten Spenderauswahl, Testung jeder Einzelspende und jedes Plasmapools auf spezifische Infektionsmarker und Einschluss effektiver
Herstellungsschritte zur Inaktivierung/Eliminierung von Viren. Dennoch kann die Möglichkeit der Übertragung von Erregern bei der Verabreichung von Arzneimitteln, die aus menschlichem Blut oder
Plasma hergestellt worden sind, nicht völlig ausgeschlossen werden. Dies trifft auch für bisher unbekannte oder neu auftretende Viren und andere Erreger zu.
Die ergriffenen Maßnahmen werden als wirksam gegenüber umhüllten Viren wie dem Humanen Immundefizienz-Virus (HIV), dem Hepatitis-B-Virus (HBV) und dem Hepatitis-C-Virus (HCV),
sowie dem nicht umhüllten Hepatitis-A-Virus (HAV) angesehen.
Gegen nicht umhüllte Viren wie Parvovirus B19 sind die getroffenen Maßnahmen möglicherweise von begrenztem Wert. Parvovirus-B19-Infektionen können bei schwangeren Frauen (Infektion des Fötus) und bei Personen mit Immundefekt oder verstärkter Erythropoese (z. B. hämolytische Anämie) schwerwiegende Folgen haben.
Bei Patienten, die regelmäßig/wiederholt mit aus humanem Plasma gewonnenen Arzneimitteln behandelt werden, sollte eine entsprechende Impfung (Hepatitis A und B) erwogen werden.
Immunogenität
Bei Substitutionstherapien mit Gerinnungsfaktoren von anderen kongenitalen Mangelkrankheiten wurden Antikörperreaktionen beobachtet. Zu Fibrinogen liegen jedoch bisher keine Daten vor.
Sonstige Bestandteile mit bekannter Wirkung
Natriumgehalt
Dieses Arzneimittel enthält etwa 132,2 mg (5,8 mmol) Natrium pro Durchstechflasche, entsprechend 6,6 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
Polysorbat 80 Gehalt
Dieses Arzneimittel enthält etwa 25,5 mg Polysorbat 80 pro Durchstechflasche. Polysorbate können allergische Reaktionen hervorrufen.
Kinder und Jugendliche
Die für Erwachsene genannten besonderen Warnhinweise und Vorsichtsmaßnahmen sind auch für
Kinder und Jugendliche zu beachten.
Es sind keine Wechselwirkungen zwischen Produkten mit humanem Fibrinogen und anderen Arzneimitteln bekannt.
Schwangerschaft
Die Sicherheit von Prufibry bei der Anwendung in der Schwangerschaft wurde nicht in kontrollierten klinischen Studien untersucht. Die klinische Erfahrung mit der Anwendung von Fibrinogenprodukten bei geburtshilflichen Komplikationen lässt keine schädlichen Auswirkungen auf den Verlauf der Schwangerschaft oder auf die Gesundheit des Fötus oder Neugeborenen erwarten.
Stillzeit
Die Sicherheit von Prufibry bei der Anwendung in der Stillzeit wurde bisher nicht in kontrollierten klinischen Studien untersucht.
Fertilität
Es liegen keine Daten zur Fertilität vor.
Prufibry hat keinen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Tabellarische Zusammenfassung der Nebenwirkungen
Die nachstehende Tabelle entspricht der MedDRA-Systemorganklassifizierung (SOC und bevorzugter Begriff).
Die Häufigkeiten wurden gemäß der folgenden Konvention bestimmt: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1 000, <1/100); selten (≥1/10 000, <1/1 000); sehr selten (<1/10 000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Nachstehend sind die Nebenwirkungen aufgeführt, die in zwei kontrollierten klinischen Studien zu Prufibry beobachtet wurden bzw. von anderen Fibrinogenprodukten bekannt sind:
Systemorganklasse nach MedDRA-Klassifikation |
Nebenwirkung |
Häufigkeit |
Erkrankungen des Blutes und des Lymphsystems |
Thrombozytopenie |
Gelegentlich |
Erkrankungen des Immunsystems |
Allergische/anaphylaktische Reaktionen |
Nicht bekannt |
Gefäßerkrankungen |
Thromboembolische Ereignisse (einschließlich Myokardinfarkt und Lungenembolie (siehe Abschnitt 4.4)) |
Häufig* |
Erkrankungen der Haut und des Unterhautgewebes |
Urtikaria, Pruritus |
Gelegentlich |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber |
Gelegentlich |
* Häufigkeitsangaben aus einer aggregierten Analyse einer einarmigen und einer aktiv kontrollierten klinischen Studie (Inzidenz: 1,3 % der Teilnehmer). In der aktiv kontrollierten Studie war die Häufigkeit in der Vergleichspräparat-Gruppe höher (0,9 % vs. 3,6 % der Teilnehmer).
Hinweise zur Sicherheit hinsichtlich übertragbarer Erreger siehe Abschnitt 4.4.
Kinder und Jugendliche
Vierundzwanzig Patienten im Alter von 1 bis <18 Jahren wurden in einer klinischen Studie der Phase I/ III bei kongenitalem Fibrinogenmangel behandelt (siehe Abschnitt 5.1). Das allgemeine Sicherheitsprofil unterscheidet sich bei Erwachsenen, Jugendlichen und Kindern nicht. Es liegen keine Daten zur Anwendung von Prufibry bei Kindern und Jugendlichen mit erworbenem Fibrinogenmangel vor.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung anzuzeigen über:
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51-59
63225 Langen
Tel.: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
Zur Vermeidung von Überdosierungen sind während der Therapie regelmäßige Kontrollen des Fibrinogenspiegels im Plasma angezeigt (siehe Abschnitt 4.2).
Im Falle einer Überdosierung besteht ein erhöhtes Risiko für thromboembolische Komplikationen.
Pharmakotherapeutische Gruppe: Antihämorrhagika, humanes Fibrinogen, ATC-Code: B02BB01
Humanes Fibrinogen (Gerinnungsfaktor I) wird in Anwesenheit von Thrombin, aktiviertem Gerinnungsfaktor XIII (FXIIIa) und Calciumionen in ein stabiles und elastisches dreidimensionales hämostatisches Fibringerinnsel umgebaut.
Wirkmechanismus
Die Gabe von humanem Fibrinogen erhöht den Fibrinogenspiegel im Plasma und kann den Blutgerinnungsdefekt bei Patienten mit Fibrinogenmangel vorübergehend korrigieren.
Pharmakodynamische Wirkungen
In einer prospektiven, offenen, multizentrischen klinischen Studie der Phase I wurden die pharmakodynamischen Eigenschaften von Prufibry bei 27 Patienten aller Altersgruppen mit Afibrinogenämie oder schwerer Hypofibrinogenämie untersucht. Die Beurteilung erfolgte anhand der Fibrinogen-Aktivität (FiAc) im Plasma nach einmaliger i.v. Gabe von 70 mg/kg Körpergewicht (KG). Die maximale Gerinnselfestigkeit (maximum clot firmness; MCF) als Surrogatmarker für die klinische Wirksamkeit ergab für Prufibry bei Patienten aller Altersgruppen eine starke positive Korrelation mit der FiAc (Korrelationskoeffizient nach Pearson >0,7).
Klinische Wirksamkeit und Sicherheit
Kongenitale Hypo- oder Afibrinogenämie
In einer prospektiven, offenen, multizentrischen klinischen Phase-III-Studie wurden die Wirksamkeit und Sicherheit der einmaligen oder wiederholten Gabe von Prufibry zur Behandlung bei Bedarf (on-demand treatment; ODT) bzw. Prophylaxe bei Bedarf (on-demand prophylaxis; ODP) bei 175 Blutungsereignissen bei 36 Patienten aller Altersgruppen mit kongenitalem Fibrinogenmangel untersucht. Als Wirksamkeitsendpunkt wurde die hämostatische Gesamtwirkung (overall haemostatic response; OHR) bei jedem der chirurgischen Verfahren und jedem behandelten Blutungsereignis durch den Prüfarzt auf einer vorgegebenen Skala bewertet. Die im Hinblick auf operationsbedingte Blutungsereignisse präoperativ verabreichten Dosen Prufibry betrugen etwa 58 mg/kg KG bei Erwachsenen und 71–83 mg/kg KG in bei Kindern und Jugendlichen. Die OHR in Bezug auf alle spontanen Blutungen sowie chirurgisch oder traumatisch bedingten Blutungen zusammengenommen belegte einen Behandlungserfolg bei 173 von 175 Blutungsereignissen (98,9 %; zusammengesetzt aus hervorragender OHR [85,7 %] und guter OHR [13,1 %]). Ein signifikanter Anstieg des mittleren MCF-Werts nach i.v. Gabe von Prufibry im Zeitverlauf war in allen Altersgruppen zu verzeichnen.
Bei allen Wirksamkeitsendpunkten wiesen die Ergebnisse weder erhebliche Unterschiede zwischen den Altersgruppen noch Unterschiede je nach Art der Behandlung (ODT vs. ODP) oder der Schwere der Blutung auf.
Die ein- oder mehrmalige Infusion von Prufibry erwies sich als sicher und gut verträglich für Patienten aller Altersgruppen mit kongenitalem Fibrinogenmangel. Das Sicherheitsprofil steht im Einklang mit der Art der zugrunde liegenden Erkrankung, dem pharmakologischen Wirkmechanismus und dem erwarteten Sicherheitsprofil von humanen Fibrinogenprodukten.
Erworbene Hypofibrinogenämie
In einer prospektiven, randomisierten, aktiv kontrollierten, multizentrischen, partiell verblindeten klinischen Phase-III-Studie bei Erwachsenen mit erworbener Hypofibrinogenämie, die sich einer größeren spinalen oder abdominalen (zytoreduktiven Pseudomyxoma-peritonei [PMP]) Operation unterzogen, wurden die Wirksamkeit und Sicherheit der intraoperativen i.v. Gabe von Prufibry als Komplementärtherapie zur Behandlung von unkontrollierten schwerwiegenden Blutungen untersucht. In den zwei chirurgischen Populationen wurden aufgrund von Unterschieden in den Operationsverfahren unterschiedliche Dosierungsschemata angewendet (spinale Operation: individualisierte, FIBTEM-gesteuerte Dosierung; PMP-Operation: präemptive feste Dosierung). Die Ergebnisse aus beiden chirurgischen Populationen bestätigten die Dosierungsangaben für Prufibry bei erworbener Hypofibrinogenämie in Abschnitt 4.2. Der primäre Wirksamkeitsendpunkt war der Nachweis der Nichtunterlegenheit von Prufibry gegenüber der Standardtherapie (gefrorenes Frischplasma/Kryopräzipitat [FFP/Cryo]) mit einer Nichtunterlegenheitsmarge von 150 ml hinsichtlich der Reduktion des intraoperativen Blutverlusts.
Die Nichtunterlegenheit von Prufibry gegenüber FFP/Cryo wurde nachgewiesen (van-Elteren-p-Wert < 0,001). Prufibry ist in unterschiedlichen Untergruppen durchgängig hochwirksam, wie der signifikant geringere mittlere Blutverlust in der Prufibry-Gruppe im Vergleich zur FFP/Cryo-Gruppe belegt und Sensitivitätsanalysen bestätigen. Auf alle Patienten bezogen betrug der mittlere (SD) Blutverlust nach der Entscheidung für die Behandlung mit Prufibry bzw. FFP/Cryo bis zum Ende der Operation 1444,4 ml (992,77 ml) in der Prufibry-Gruppe und 1735,1 ml (1029,17 ml) in der FFP/Cryo-Gruppe. Die Differenz der adjustierten Mittelwerte (Least Squares Means) zwischen beiden Behandlungsgruppen betrug -279,43 ml, und die obere Grenze des zweiseitigen 95%-KI für die Differenz betrug -6,48 ml, was nicht größer war als die vordefinierte Nichtunterlegenheitsgrenze von 150 ml.
Die verabreichte Gesamtdosis Prufibry reichte von 1 g bis 9 g, wobei die Mehrheit der Patienten zwischen 1 g und 4 g erhielt. Die Behandlung führte in der Prufibry-Gruppe zu einem geringeren Gesamt-Blutverlust im Vergleich zur FFP/Cryo-Gruppe.
Die Daten bestätigten das günstige Sicherheitsprofil von Prufibry. Die beobachteten Nebenwirkungen und klinischen Parameter stehen im Einklang mit den Erwartungen für diese Art von chirurgischen Eingriffen und für humane Fibrinogenprodukte.
Kinder und Jugendliche
Prufibry wurde in einer klinischen Studie der Phase I/III bei insgesamt 28 Kindern und Jugendlichen mit kongenitalem Fibrinogenmangel angewendet (12 Patienten in Phase I und 16 Patienten in Phase III; 4 Patienten wurden im Rahmen beider Phasen mit Prufibry behandelt). In der klinischen Phase-I-Studie wurden die PK- und PD-Eigenschaften bei 6 Kindern im Alter von <6 Jahren, 3 Kindern im Alter von 6 bis <12 Jahren und 3 Jugendlichen im Alter von 12 bis <18 Jahren untersucht. Zu den in der klinischen Phase-III-Studie behandelten Kindern und Jugendlichen gehörten 3 Kinder im Alter von <6 Jahren, 9 Kinder im Alter von 6 bis <12 Jahren und 4 Jugendliche im Alter von 12 bis <18 Jahren.
Die Daten zeigen keine bemerkenswerten Unterschiede in der Exposition gegenüber Prufibry oder der Erholung des Fibrinogenspiegels zwischen Erwachsenen, Kindern und Jugendlichen.
Die Europäische Arzneimittel-Agentur hat für Prufibry eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in der Behandlung des erworbenen Fibrinogenmangels gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Humanes Fibrinogen ist ein normaler Bestandteil des menschlichen Plasmas und verhält sich wie körpereigenes Fibrinogen.
Prufibry wird intravenös verabreicht und ist sofort in einer der verabreichten Dosis entsprechenden Plasmakonzentration verfügbar. Die biologische Halbwertszeit von Fibrinogen im Plasma beträgt 3–4 Tage.
Die pharmakokinetischen Eigenschaften von Prufibry wurden anhand der FiAg- und FiAc-Werte im Plasma nach einmaliger i.v. Gabe von 70 mg/kg KG an Patienten aller Altersgruppen mit kongenitaler Afibrinogenämie oder schwerer Hypofibrinogenämie untersucht.
Die PK-Eigenschaften des FiAg nach der i.v. Gabe von Prufibry wurden mit einem Zwei-Kompartiment-Dispositionsmodell mit Elimination erster Ordnung gut beschrieben. Die getroffenen Vorhersagen für die nicht-kompartimentelle Analyse (NCA) des FiAg-Plasmaspiegels deckten sich gut mit den individuellen FiAg-Messungen in der Studie. In der folgenden Tabelle sind die PK-Parameter für FiAg und FiAc zusammengefasst, basierend auf individuellen NCA-Werten aus einem Populations-PK/PD-Modell.
Tabelle 1: Zusammenfassung der pharmakokinetischen Parameter (n = 27) von Prufibry
Parameter |
Mittelwert (SD) |
Bereich |
Fibrinogen-Antigen | ||
t1/2 [h] |
67,9 (15,3) |
45,7–96,4 |
Cmax [g/l] |
1,81 (0,42) |
0,713–2,81 |
AUC0-∞ [g × h/l)] |
173 (45,4) |
104–289 |
MRT0-∞ [h] |
133 (17,4) |
99,4–166 |
Vdss pro kg [ml/kg] |
57,8 (19,1) |
32,9–112 |
CL pro kg (ml/[h*kg]) |
0,43 (0,1) |
0,24–0,67 |
IR (mg/dl pro mg/kg-Dosis) |
2,63 (0,652) |
(1–4,42) |
Fibrinogen-Aktivität | ||
t1/2 [h] |
60,3 (13,3) |
41,2–90,3 |
Cmax [g/l] |
1,26 (0,4) |
0,403–2,26 |
AUC0-∞ [g × h/l)] |
104 (33,5) |
55,5–200 |
MRT0-∞ [h] |
124 (16,2) |
95,8–159 |
Vdss pro kg [ml/kg] |
92,4 (34,7) |
43–201 |
CL pro kg (ml/[h*kg]) |
0,733 (0,2) |
0,35–1,26 |
IR (mg/dl pro mg/kg Dosis) |
1,88 (0,61) |
0,56–3,17 |
Abkürzungen: AUC0-∞: Fläche unter der Kurve von 0 bis unendlich; Cmax: Höchstkonzentration; CL: Clearance; IR: inkrementelle Wiederfindung laut Beobachtung; MRT0-∞: mittlere Verweilzeit, extrapoliert bis unendlich; n: Anzahl Patienten; SD: Standardabweichung; t1/2: Halbwertszeit; Vdss: Verteilungsvolumen im Gleichgewichtszustand.
Die gewonnenen Daten belegen, dass die Gabe von Prufibry zu einem Anstieg der systemischen Fibrinogenkonzentration (FiAg) der Patienten führt (von 0 g/l vor der Verabreichung auf bis zu 1,86 g/l am Ende der Infusion), verbunden mit einem Anstieg der Fibrinogen-Aktivität (FiAc) (bis zu 1,28 g/l bei Kindern und Jugendlichen und 1,34 g/l bei Erwachsenen). Dies erbrachte den Nachweis für Prufibry als geeignete Substitutionstherapie für Patienten mit Fibrinogenmangel.
Kinder und Jugendliche
Die mittlere AUC0-∞ sowohl des FiAg als auch der FiAc waren in allen 3 pädiatrischen Gruppen niedriger als bei den Erwachsenen. Die verminderte AUC0-∞ bei den Kindern und Jugendlichen war eine erwartete Auswirkung der KG-basierten Dosierung, da bekannt ist, dass die metabolische Kapazität pro Kilogramm zunimmt, je niedriger das KG ist.
Andere PK-Parameter zeigen keine klinisch bedeutsamen Unterschiede zwischen den Altersgruppen.
Darüber hinaus belegen die PK/PD-Daten die Angemessenheit der Anwendung von Prufibry in allen Altersgruppen mit einer körpergewichtsbasierten Dosierung.
Präklinische Daten, basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei einmaliger Gabe sowie Studien zur Thrombogenität (Wessler-Test), lassen keine besonderen Gefahren für den Menschen erkennen.
Präklinische Studien mit wiederholten Dosisgaben (chronische oder Reproduktionstoxizität, Kanzerogenität und Mutagenität) können in herkömmlichen Tiermodellen nicht sinnvoll durchgeführt werden, da infolge der Verabreichung heterologer humaner Proteine Antikörper gebildet werden.
Pulver: Argininhydrochlorid, Polysorbat 80, Natriumchlorid, Natriumcitrat-Dihydrat, Trehalose-Dihydrat (Ph. Eur).
Lösungsmittel: Wasser für Injektionszwecke.
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden und muss über eine separate Injektions-/Infusionsleitung verabreicht werden.
Es wird empfohlen, die intravenöse Gabe der rekonstituierten Lösung bei Raumtemperatur mit einem Standard-Infusionsbesteck durchzuführen.
2 Jahre
Die chemische und physikalische Stabilität nach Rekonstitution ist für 24 Stunden bei Raumtemperatur (max. 25 °C) nachgewiesen.
Aus mikrobiologischer Sicht sollte das Arzneimittel sofort nach der Rekonstitution verwendet werden. Wenn es nicht sofort verwendet wird, liegen die Aufbewahrungszeit und -bedingungen vor der Anwendung in der Verantwortung des Anwenders. Die rekonstituierte Lösung darf nicht eingefroren oder im Kühlschrank gelagert werden.
Nicht über 30 °C lagern. Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
1 Packung Prufibry enthält:
1 g humanes Fibrinogen in einer farblosen 100 ml Durchstechflasche (Typ-I-Glas), verschlossen mit einem Brombutyl-Gummistopfen und einem Aluminium-Kunststoff Flip-Off-Kappe.
50 ml Lösungsmittel (Wasser für Injektionszwecke) in einer farblosen 50 ml Durchstechflasche (Typ-I-Glas), verschlossen mit einem Chlorbutyl-Gummistopfen und einem Aluminium-Kunststoff Flip-Off-Kappe.
1 Transfersystem mit integriertem Filter
Hinweise zum Gebrauch und zur Handhabung:
Bei allen Verfahrensschritten ist auf aseptische Bedingungen zu achten!
Abb. 1 |
Abb. 2 |
Abb. 3 |
Abb. 4 |
Abb. 5 |
 |
 |
 |
 |
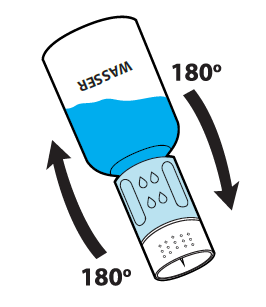 |
Abb. 6 |
Abb. 7 |
Abb. 8 |
Abb. 9 |
|
 |
 |
 |
 |
Auflösung des Konzentrats:
Bringen Sie die ungeöffneten Durchstechflaschen mit Lösungsmittel (Wasser für Injektionszwecke) und Pulver auf Raumtemperatur.
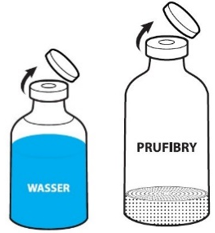
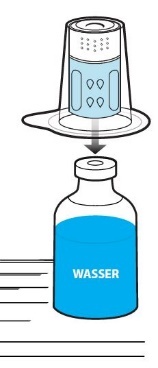
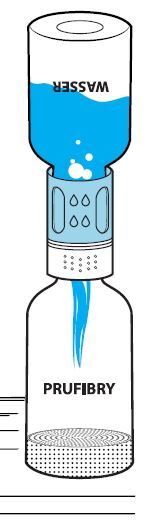
Entfernen Sie die Kappen der Durchstechflaschen mit Lösungsmittel und Pulver, um den mittleren Teil des Gummistopfens freizulegen (Abb. 1). Reinigen Sie die Gummistopfen der Durchstechflaschen mit Pulver und Lösungsmittel mit einem Desinfektionsmittel.
Entfernen Sie die Oberseite der Verpackung des Transfersystems vollständig (Abb. 2). Nehmen Sie das Transfersystem nicht aus der durchsichtigen Blisterpackung, um die Sterilität aufrechtzuerhalten. Berühren Sie den Dorn nicht.
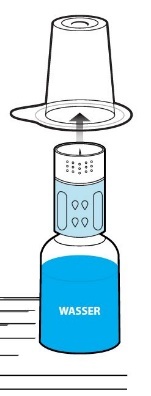
Stellen Sie die Durchstechflasche mit Lösungsmittel auf eine ebene Fläche. Setzen Sie den blauen Teil des Transfersystems mitsamt der Blisterpackung gerade auf die aufrecht stehende Durchstechflasche mit Lösungsmittel (Abb. 3), bis es einrastet. Das Transfersystem nicht drehen!
Entfernen Sie den verbleibenden Teil der Blisterpackung vom Transfersystem. Jetzt erscheint der weiße Teil des Transfersystems (Abb. 4).
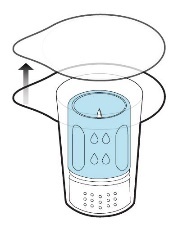
Stellen Sie die Durchstechflasche mit dem Pulver auf eine ebene Fläche.
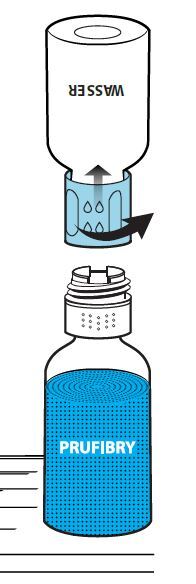
Drehen Sie die Einheit aus dem Transfersystem und der Durchstechflasche mit dem Lösungsmittel auf den Kopf (Abb. 5). Stechen Sie den Adapter mit dem Dorn seines weißen Teils senkrecht in den Stopfen der aufrecht stehenden Durchstechflasche mit dem Pulver (Abb. 6), bis er einrastet. Durch das in der Durchstechflasche mit dem Pulver vorhandene Vakuum fließt das Lösungsmittel in diese Durchstechflasche (Abb. 7).
Vorsichtiges Schwenken der Einheit aus Transfersystem, Pulver- und Lösungsmittel-Durchstechflasche hilft beim Auflösen des Pulvers. Nicht schütteln, um Schaumbildung zu vermeiden. Das Pulver sollte innerhalb von etwa 3 Minuten vollständig aufgelöst sein. Es sollte nicht länger als 30 Minuten dauern, das Pulver aufzulösen. Wenn das Pulver nicht innerhalb von 30 Minuten aufgelöst ist, sollte das Arzneimittel verworfen werden. Die Lösung ist klar oder leicht opalisierend.
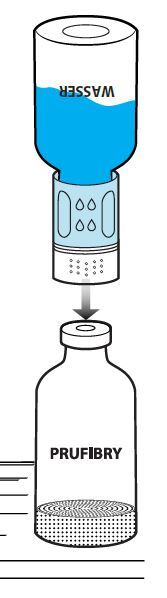
Drehen Sie danach den blauen Teil des Transfersystems zusammen mit der Durchstechflasche mit dem Lösungsmittel gegen den Uhrzeigersinn ab (Abb. 8). Entsorgen Sie die Durchstechflasche mit dem Lösungsmittel und den daran befestigten blauen Teil des Transfersystems. Der Luer-Lock-Anschluss ist jetzt sichtbar. Um die Sterilität aufrechtzuerhalten, darf der Luer-Lock-Anschluss nicht berührt werden.
Die gebrauchsfertige Lösung sollte unmittelbar nach der Auflösung verabreicht werden. Das rekonstituierte Arzneimittel sollte vor der Verabreichung visuell auf Partikel und Verfärbungen untersucht werden.
Die Lösung soll nahezu farblos sein. Verwenden Sie keine Lösungen, die trüb sind oder sichtbare Partikel enthalten.
Injektion:
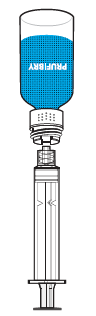
Nachdem Sie das Pulver wie oben beschrieben aufgelöst haben, schrauben Sie eine Spritze auf den Luer-Lock-Anschluss der Arzneimittelflasche mit dem weißen Teil des Transfersystems (Abb. 9). So können Sie das gelöste Arzneimittel leicht in die Spritze aufziehen. Dies muss mit gleichmäßigem Krafteinsatz geschehen, um Schaumbildung zu vermeiden. Ein separater Filter ist nicht erforderlich, da das Transfersystem über einen eigenen integrierten Filter verfügt.
Schrauben Sie die Durchstechflasche mit dem weißen Teil des Transfersystems vorsichtig von der Spritze ab. Für die intravenöse Verabreichung der rekonstituierten Lösung wird ein Standard-Infusionsset empfohlen. Prufibry darf nicht mit anderen Arzneimitteln gemischt werden und sollte über eine separate Injektions-/Infusionsleitung verabreicht werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Biotest Pharma GmbH
Landsteinerstraße 5
63303 Dreieich
Deutschland
Tel.: +49 6103 801 0
Fax: +49 6103 801 150
E-Mail: mail@biotest.com
Zul.-Nr. PEI.H.12230.01.1
Datum der Erteilung der Zulassung: 07.11.2025
01/2026
Verschreibungspflichtig
Deutschland, Kanada, Österreich, Slowakei, Tschechische Republik, Ungarn und USA