
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Ekterly 300 mg Filmtabletten
Jede Filmtablette enthält 300 mg Sebetralstat.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette.
Gelbe, ovale (etwa 15 mm × 9 mm), bikonvexe Tabletten mit aufgeprägtem KalVista-Logo „K“ auf der einen und „300“ auf der anderen Seite.
Ekterly wird angewendet zur symptomatischen Behandlung von akuten Attacken eines hereditären Angioödems (HAE) bei Erwachsenen und Jugendlichen ab 12 Jahren.
Die Entscheidung, die Behandlung mit oralem Sebetralstat zu beginnen, soll von einem Arzt mit Erfahrung in der Behandlung von Patienten mit HAE getroffen werden.
Dosierung
Erwachsene und Jugendliche ab 12 Jahren
Die empfohlene Dosis beträgt eine 300-mg-Tablette Ekterly, anzuwenden bei den ersten Anzeichen einer bevorstehenden Attacke. Wenn kein ausreichendes Ansprechen erzielt wird oder die Symptome sich verschlimmern oder zurückkehren, kann 3 Stunden nach der ersten Dosis eine zweite Dosis eingenommen werden. Es dürfen nicht mehr als zwei Dosen in 24 Stunden eingenommen werden.
Patienten mit normalem C1-INH (nC1‑INH)
Bei HAE-Patienten mit einem normalem C1-INH (nC1‑INH) ist ein Absetzen der Behandlung in Erwägung zu ziehen, wenn kein klinisches Ansprechen beobachtet wird (siehe Abschnitte 4.4 und 5.1).
Ältere Patienten
Bei Patienten über 65 Jahren ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Nierenfunktionsstörungen
Bei Patienten mit Nierenfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Leberfunktionsstörungen
Bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse A oder B) ist keine Dosisanpassung erforderlich.
Die Anwendung bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh-Klasse C) wird nicht empfohlen (siehe Abschnitt 5.2).
Bei Patienten mit mittelschwerer Leberfunktionsstörung, die einen starken CYP3A4-Inhibitor einnehmen, wird zur Behandlung einer HAE-Attacke eine Einzeldosis von 300 mg empfohlen (siehe Abschnitt 4.5).
Patienten, die CYP3A4-Induktoren einnehmen
Bei der Einnahme von schwachen CYP3A4-Induktoren ist keine Dosisanpassung erforderlich.
Bei Patienten, die moderate oder starke CYP3A4-Induktoren einnehmen, wird eine Einzeldosis von 900 mg (3 × 300-mg-Tabletten) zur Behandlung einer HAE-Attacke empfohlen (siehe Abschnitt 4.5).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit bei Kindern unter 12 Jahren sind nicht erwiesen.
Es liegen keine Daten vor.
Art der Anwendung
Ekterly ist zum Einnehmen bestimmt.
Die Filmtabletten können unabhängig von den Mahlzeiten eingenommen werden (siehe Abschnitt 5.2).
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Larynxattacken
Nach der Behandlung von Larynxattacken müssen die Patienten sofort einen Arzt aufsuchen. Wenn sich die Symptome einer Larynxattacke nach der Behandlung verschlimmern, müssen die Patienten in einer geeigneten medizinischen Einrichtung behandelt werden.
Normaler C1-Esterase-Inhibitor (nC1-INH)
Es liegen keine Daten über die Anwendung von Ekterly bei HAE-Patienten mit nC1-INH vor.
Manche Unterkategorien des nC1-INH HAE sprechen unter Umständen nicht auf die Behandlung an, da es alternative Formen gibt, die keine Aktivierung des Plasmakallikreins beinhalten. Es wird empfohlen, einen Gentest, sofern verfügbar, gemäß den aktuellen HAE-Leitlinien durchzuführen und die Behandlung abzusetzen, wenn kein klinisches Ansprechen zu beobachten ist (siehe Abschnitte 4.2 und 5.1).
QT-Verlängerung
In einer klinischen Studie zur Beurteilung der Herzparameter bei gesunden Probanden wurde festgestellt, dass Sebetralstat das Potential zur Verlängerung des QT-Intervalls besitzt. Dies traf jedoch nur für hohe Konzentrationen zu, die mit der empfohlenen Dosis wahrscheinlich nicht erreicht werden. Es gibt keine Daten zur Anwendung von Sebetralstat bei Patienten mit unabhängigen Risikofaktoren für eine QT-Verlängerung, wie z. B. eine bekannte, bereits bestehende (erworbene oder angeborene) QT-Verlängerung, Elektrolytstörungen, Leberfunktionsstörungen, gleichzeitige Anwendung von Arzneimitteln, die mit dem Stoffwechsel von Sebetralstat interagieren, oder gleichzeitige Anwendung von anderen Arzneimittel, die bekanntlich das QT-Intervall verlängern. Bei solchen Patienten ist aufgrund des Risikos für eine QT-Verlängerung Vorsicht geboten. Dies gilt insbesondere für Patienten mit mehr als einem Risikofaktor (siehe Abschnitt 5.1).
Sonstige Bestandteile
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro Dosis, d. h. es ist nahezu „natriumfrei“.
Wirkung anderer Arzneimittel auf Sebetralstat
Sebetralstat ist ein Substrat von CYP3A4.
Itraconazol, ein starker CYP3A4-Inhibitor, erhöhte die Cmax von Sebetralstat um 135 % und die AUC um 420 %. Der moderate CYP3A4-Inhibitor Verapamil erhöhte die Cmax von Sebetralstat um 76 % und die AUC um 102 %. Die gleichzeitige Anwendung mit dem schwachen CYP3A4-Inhibitor Cimetidin verursachte keine Veränderung der Exposition gegenüber Sebetralstat. Bei der Einnahme von CYP3A4-Inhibitoren ist keine Dosisanpassung erforderlich.
Phenytoin, ein starker CYP3A4-Induktor, reduzierte die Cmax von Sebetralstat um 66 % und die AUC um 83 %. Der moderate CYP3A4-Induktor Efavirenz reduzierte die Cmax von Sebetralstat um 63 % und die AUC um 79 %. Die gleichzeitige Anwendung mit dem schwachen CYP3A4-Induktor Modafinil verursachte keine klinisch relevante Veränderung der Exposition gegenüber Sebetralstat. Bei der Einnahme von schwachen CYP3A4-Induktoren ist keine Dosisanpassung erforderlich.
Bei Patienten, die starke oder mäßige CYP3A4-Induktoren einnehmen (wie z. B. Rifampicin, Efavirenz, Carbamazepin, Phenytoin, Phenobarbital), wird empfohlen, eine HAE-Attacke mit einer Dosis von 900 mg (3 × 300-mg-Tabletten) zu behandeln.
Patienten mit mäßiger Leberfunktionsstörung, die einen starken CYP3A4-Inhibitor (z. B. Erythromycin, Clarithromycin, Itraconazol, Ketoconazol, Ritonavir) einnehmen, wird zur Behandlung einer HAE-Attacke eine Einzeldosis von 300 mg empfohlen.
Magensäurereduzierende Arzneimittel
Es wurde keine spezielle Studie zur Erfassung von Wechselwirkungen mit magensäurereduzierenden Arzneimitteln in vivo durchgeführt. Die Wirkung von magensäurereduzierenden Arzneimitteln auf die Pharmakokinetik von Sebetralstat ist somit nicht bekannt. Bei der gleichzeitigen Anwendung von Ekterly mit Arzneimitteln zur Veränderung des pH-Werts des Magens, wie z. B. Antazida, Protonenpumpenhemmer und Histamin-2-Rezeptorantagonisten, ist Vorsicht geboten.
Wirkung von Sebetralstat auf andere Arzneimittel
Es wurden keine klinischen Studien zur Erfassung von Wechselwirkungen bezüglich der Wirkung von Sebetralstat auf andere Arzneimittel durchgeführt.
Aus den In-vitro-Daten geht hervor, dass Sebetralstat die Enzyme CYP2C9, UGT1A4 und UGT1A9 sowie die Transporter OCT2, OATP1B3, MATE1 und MATE2‑K hemmen kann. Die klinische Bedeutung dieser Ergebnisse ist derzeit noch nicht geklärt. Die gleichzeitige Anwendung von Sebetralstat mit Substraten dieser Enzyme und Transporter, die eine geringe therapeutische Breite haben (wie z. B. Warfarin, Mycophenolsäure, Ciclosporin, Tacrolimus), ist angesichts des Risikos einer erhöhten pharmakokinetischen Exposition gegenüber diesen gleichzeitig verabreichten Arzneimitteln und somit eines Auftretens von Toxizität zu vermeiden, es sei denn, die Anwendung ist klinisch gerechtfertigt. Wenn sich eine gleichzeitige Anwendung nicht vermeiden lässt, wird eine engmaschige klinische Überwachung empfohlen, soweit dies möglich ist.
Kinder und Jugendliche
Studien zur Erfassung von Wechselwirkungen wurden nur bei Erwachsenen durchgeführt.
Gebärfähige Frauen
Gebärfähige Frauen müssen während der Behandlung mit Ekterly und für einen Zeitraum von 24 Stunden nach der letzten Dosis eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Ekterly bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3).
Ekterly darf während der Schwangerschaft nur dann angewendet werden, wenn der potenzielle Nutzen der Behandlung das potenzielle Risiko für den Fetus rechtfertigt (z. B. zur Behandlung von potenziell lebensbedrohlichen Larynxattacken).
Stillzeit
Es ist nicht bekannt, ob Sebetralstat oder seine Metaboliten in die Muttermilch übergehen. Die vorliegenden pharmakodynamischen/toxikologischen Daten bei Tieren zeigten, dass Sebetralstat und/oder seine Metaboliten in die Milch übergehen (Abschnitt 5.3).
Ein Risiko für das gestillte Kind kann nicht ausgeschlossen werden.
Es muss eine Entscheidung darüber getroffen werden, ob die Behandlung mit Ekterly unterbrochen oder ganz abgesetzt werden soll oder ob das Stillen nach der Einnahme von Ekterly für 24 Stunden unterbrochen werden soll, wobei der Nutzen des Stillens für das Kind und der Nutzen der Behandlung für die Mutter zu berücksichtigen sind.
Fertilität
Es liegen keine Daten zu den Auswirkungen von Ekterly auf die menschliche Fruchtbarkeit vor. In Tierstudien war keine Auswirkung auf die Fertilität zu beobachten (siehe Abschnitt 5.3).
Ekterly hat einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Nach der Anwendung von Ekterly wurde über das Auftreten von Schwindel berichtet. Dieses Symptom kann auch als Folge einer HAE-Attacke auftreten. Die Patienten sind anzuweisen, keine Fahrzeuge zu führen und keine Maschinen zu bedienen, wenn sie unter Schwindelgefühl leiden.
Zusammenfassung des Sicherheitsprofils
Ekterly wurde insgesamt 411 gesunden Probanden und 239 Patienten mit hereditärem Angioödem verabreicht. In den klinischen Zulassungsstudien wurden 1945 HAE-Attacken mit Ekterly behandelt.
Die häufigste Nebenwirkung bei den HAE-Patienten, die mit Ekterly behandelt wurden, war Kopfschmerzen (berichtet von 9,2 % der Patienten). Die berichteten Kopfschmerz-Ereignisse waren allgemein leicht bis mittelschwer, nicht ernsthaft und verschwanden ohne weitere Behandlung.
Tabellarische Auflistung der Nebenwirkungen
Die Häufigkeit der in der nachstehenden Tabelle aufgeführten Nebenwirkungen wird in den folgenden Kategorien angegeben:
Sehr häufig (≥ 1/10); häufig (≥ 1/100 bis < 1/10); gelegentlich (≥ 1/1 000 bis < 1/100); selten (≥ 1/10 000 bis < 1/1 000); sehr selten (< 1/10 000).
Tabelle 1. Zusammenfassung der Nebenwirkungen nach Systemorganklasse und Häufigkeit
Systemorganklasse |
Nebenwirkung |
Häufigkeit |
Erkrankungen des Nervensystems |
Kopfschmerzen |
Häufig |
Schwindelgefühl |
Häufig |
|
Erkrankungen des Gastrointestinaltrakts |
Erbrechen |
Häufig |
Übelkeit |
Häufig |
|
Abdominalschmerz* |
Häufig |
|
Diarrhoe |
Häufig |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Rückenschmerzen |
Häufig |
Gefäßerkrankungen |
Hitzewallung |
Häufig |
* Beinhaltet die Ereignisse Abdominalschmerz und Schmerzen Oberbauch. | ||
Kinder und Jugendliche
Bei 32 Jugendlichen im Alter von 12 bis < 18 Jahren wurden insgesamt 390 HAE-Attacken mit Sebetralstat behandelt. Das Sicherheitsprofil war dem bei Erwachsenen beobachteten ähnlich.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
In den klinischen Studien wurden keine Fälle von Überdosierung gemeldet. Es liegen keine Informationen darüber vor, wie potenzielle Anzeichen und Symptome einer Überdosierung zu erkennen sind. Falls Symptome auftreten sollten, wird eine symptomatische Behandlung empfohlen. Es gibt kein Antidot.
Pharmakotherapeutische Gruppe: Andere Hämatologika, Mittel zur Behandlung des hereditären Angioödems, ATC-Code: B06AC08.
Wirkmechanismus
Sebetralstat ist ein kompetitiver, reversibler Plasmakallikrein-Inhibitor (PKa). Durch die Hemmung von Pka verhindert Sebetralstat die Spaltung des hochmolekularen Kininogens (HK) und die anschließende Bildung von Bradykinin (BK) und hält so das Fortschreiten der HAE-Attacke auf, die mit einer erhöhten Gefäßpermeabilität und Ödembildung einhergeht. Sebetralstat hemmt auch die Aktivierung des positiven Rückkopplungsmechanismus des Kallikrein-Kinin-Systems (KKS) und verringert so die Bildung von Faktor XIIa (FXIIa) und weiterem PKa.
Klinische Wirksamkeit und Sicherheit
KONFIDENT-Studie
Die Wirksamkeit von Ekterly zur Behandlung von HAE-Attacken bei erwachsenen und jugendlichen Patienten ab 12 Jahren wurde in der KONFIDENT-Studie untersucht, einer randomisierten, doppelblinden, placebokontrollierten Studie im Dreifach-Crossover-Design.
Insgesamt 110 Patienten behandelten 264 Attacken. Das mediane Alter der Patienten betrug 39,5 Jahre und reichte von 13 bis 74 Jahren. Dies umfasste insbesondere 13 (11,8 %) Jugendliche und nur 4 (3,6 %) ältere Patienten. In der Studie waren Patienten mit HAE Typ 1 (101 Patienten [91,8 %]) und Typ 2 (9 Patienten [8,2 %]) vertreten. Bei Aufnahme in die Studie wendeten die Patienten entweder eine bedarfsabhängige konventionelle Behandlung an (86 [78,2 %]) oder eine Langzeitprophylaxe (24 [21,8 %]). Die Ausgangsmerkmale der behandelten Attacken schlossen alle Schweregrade von Attacken ein (113 [42,8 %] leicht, 102 [38,6 %] mittelschwer, 38 [14,4 %] schwer und 7 [2,7 %] sehr schwer) sowie alle anatomischen Lokalisationen (142 [53,8 %] subkutan, 120 [45,5 %] mukosal (8 [3 %] davon waren laryngeal)).
Die mediane Zeit bis zur Behandlung (IQR) betrug 41 (6 bis 140) Minuten.
Von den 264 behandelten Attacken wurden 87 mit 300 mg Sebetralstat behandelt, 93 wurden mit 600 mg Sebetralstat behandelt und 84 mit Placebo. Nach der Behandlung einer Attacke konnte nach frühestens 3 Stunden bei Bedarf eine weitere Dosis eingenommen werden, wenn der Patient dies aufgrund seiner Symptome für erforderlich hielt.
Der primäre Wirksamkeitsendpunkt war die Zeit bis zum Einsetzen einer Symptomlinderung, definiert als eine mindestens „leichte Besserung“ (bei zwei Zeitpunkten hintereinander) innerhalb von 12 Stunden nach Einnahme der ersten Dosis Sebetralstat, gemessen an der Gesamteinschätzung der Veränderung durch den Patienten (PGI-C; Patient Reported Global Impression of Change). Bei der PGI-C beurteilten die Patienten ihre Attackensymptome auf einer siebenstufigen Skala (von „sehr viel schlechter“ bis „sehr viel besser“). Damit der primäre Endpunkt als erreicht galt, musste innerhalb von 12 Stunden ein positives und anhaltendes Ansprechen in Form einer mindestens „leichten Besserung“ bei zwei Zeitpunkten hintereinander auf der PGI-C-Skala angegeben werden.
Die Zeit bis zum Einsetzen der Symptomlinderung war unter 300 mg Sebetralstat (Bonferroni-korrigiert p < 0,0001) und 600 mg Sebetratstat (Bonferroni-korrigiert p < 0,0013) statistisch signifikant kürzer als unter Placebo (Abbildung 1).
Abbildung 1. KONFIDENT-Studie – Kaplan-Meier-Kurve für die Zeit bis zum Einsetzen der Symptomlinderung innerhalb von 12 Stunden nach Einnahme der Dosis
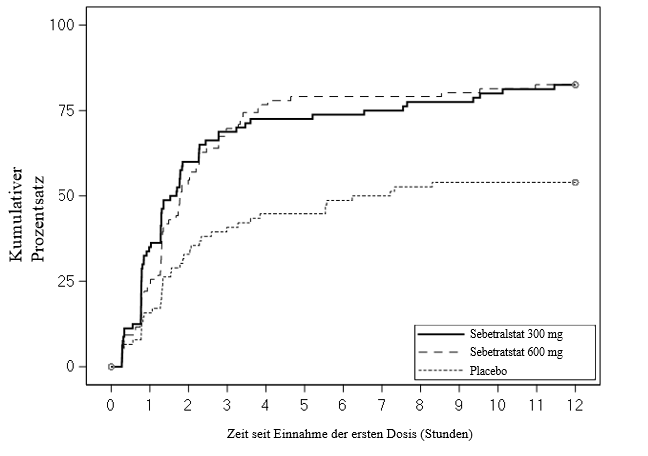
Der Anteil von Patienten, die innerhalb von 12 Stunden eine zweite Dosis einnahmen, betrug 29,9 % bei 300 mg Sebetralstat und 37,6 % bei 600 mg Sebetralstat und lag damit niedriger als unter Placebo mit 48,8 %. Der Anteil von Patienten, die den primären Endpunkt ohne Einnahme einer zweiten Dosis oder vor der zweiten Dosis von 300 mg oder 600 mg Sebetralstat erreichten, lag bei 93,9 % bzw. 95,8 %.
Der erste maßgebliche sekundäre Endpunkt war die Zeit bis zum Einsetzen einer Abnahme des Schweregrades gegenüber dem Ausgangswert (bei zwei Zeitpunkten hintereinander) innerhalb von 12 Stunden nach Einnahme der ersten Dosis Sebetralstat auf der PGI-S Skala (Patient Global Impression of Severity, Skala zur Beurteilung der Schwere der Erkrankung aus Sicht des Patienten). Bei der PGI-S beurteilten die Patienten ihre Attackensymptome auf einer fünfstufigen Skala (von „keine“ bis „sehr schwer“). Damit der erste maßgebliche sekundäre Endpunkt als erreicht galt, musste innerhalb von 12 Stunden eine positive und anhaltende Abnahme des Schweregrades um mindestens eine Stufe auf der PGI-S angegeben werden.
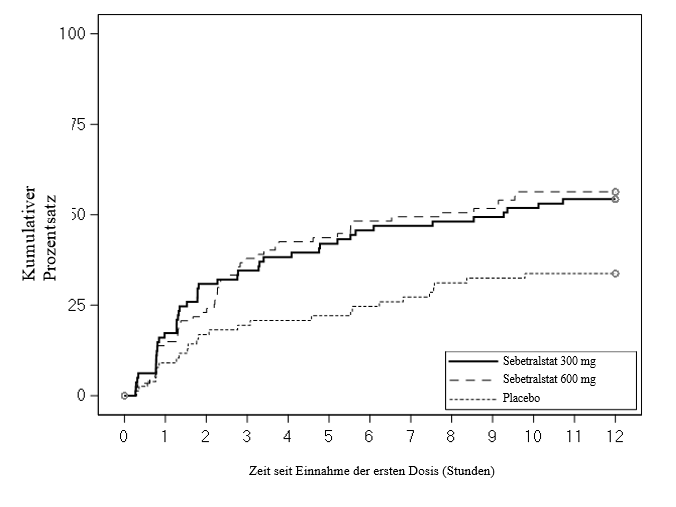
Die Zeit bis zur Abnahme des Schweregrades war unter 300 mg Sebetralstat (Bonferroni-korrigiert p = 0,0036) und 600 mg Sebetralstat (Bonferroni-korrigiert p = 0,0032) statistisch signifikant kürzer als unter Placebo (Abbildung 2).
Abbildung 2. KONFIDENT-Studie - Kaplan-Meier-Kurve der Zeit bis zur Abnahme des Schweregrades innerhalb von 12 Stunden nach Einnahme der Dosis
Der zweite maßgebliche sekundäre Endpunkt war die Zeit bis zum vollständigen Abklingen der Attacke, gemäß der Definition der PGI-S, innerhalb von 24 Stunden nach Einnahme der Dosis. Damit der zweite maßgebliche sekundäre Endpunkt als erreicht galt, musste auf der PGI‑S „keine“ innerhalb von 24 Stunden angegeben werden.
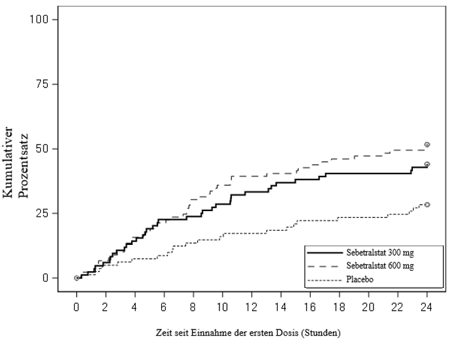
Die Zeit bis zum vollständigen Abklingen der Attacke war unter 300 mg Sebetralstat (Bonferroni-korrigiert p = 0,0022) und 600 mg Sebetralstat (Bonferroni-korrigiert p = 0,0001) statistisch signifikant kürzer als unter Placebo (Abbildung 3).
Abbildung 3. KONFIDENT-Studie - Kaplan-Meier-Kurve der Zeit bis zum vollständigen Abklingen der Attacke innerhalb von 24 Stunden nach Einnahme der Dosis
Die Auswertung der Ergebnisse des primären und der maßgeblichen sekundären Wirksamkeitsendpunkte in allen vordefinierten Untergruppen der KONFIDENT-Studie – einschließlich Anwendung einer nur bedarfsabhängigen Behandlung oder einer Langzeitprophylaxe – deckte sich mit den Ergebnissen der Gesamtpopulation.
KONFIDENT-S-Studie
In der offenen KONFIDENT-S-Studie behandelten Patienten bis zu 2 Jahre lang multiple Attacken mit 600 mg Sebetralstat. Insgesamt 134 Patienten (darunter 23 Jugendliche) behandelten 1706 Attacken. Die mediane Anzahl der behandelten Attacken pro Patient betrug 8 und die Spannweite reichte von 1 bis 61 Attacken. Die mediane Anzahl von behandelten Attacken lag zwischen 0 und 2 Attacken pro Monat. Die mediane Zeit vom Beginn der Attacke bis zur Behandlung betrug 10 Minuten. Bei den Jugendlichen betrug die mediane Zeit vom Beginn der Attacke bis zur Behandlung 4 Minuten. Die Wirksamkeitsergebnisse deckten sich mit denen der KONFIDENT-Studie. Die Wirksamkeit blieb auch bei wiederholter Anwendung erhalten.
Larynxattacken
In den klinischen Studien wurden insgesamt 36 Larynxattacken behandelt.
In der KONFIDENT-Studie wurden 4 Larynxattacken behandelt (2 mit 300 mg Sebetralstat, 2 mit 600 mg Sebetralstat). In der offenen KONFIDENT-S-Studie wurden 32 Larynxattacken mit 600 mg Sebetralstat behandelt. In Bezug auf die Zeit bis zum Einsetzen der Symptomlinderung deckten sich die Ergebnisse mit denen der Patienten mit nicht-laryngealen Attacken. Es wurden keine Schwierigkeiten beim Schlucken der Ekterly-Tabletten berichtet.
HAE-Population mit normalem C1-INH
Es liegen keine Daten über die Anwendung von Ekterly bei HAE-Patienten mit nC1-INH vor (siehe Abschnitte 4.2 und 4.4).
Kardiale Elektrophysiologie
In der klinischen Studie zur Untersuchung der kardialen Elektrophysiologie wurde festgestellt, dass Sebetralstat ein Potenzial zur Verlängerung des QT-Intervalls besitzt. Dies war jedoch nur unter hohen Konzentrationen der Fall, die mit der empfohlenen Dosis voraussichtlich nicht erreicht werden. Der größte mittlere Anstieg des QTc-Intervalls nach Gabe von Ekterly (dem 5-Fachen der empfohlenen Höchstdosis) an gesunde Probanden betrug 10,4 ms (Obergrenze des Konfidenzintervalls = 15,3 ms). Der Anstieg des QTc-Intervalls war konzentrationsabhängig (siehe Abschnitt 4.4).
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Ekterly eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen zur Behandlung des hereditären Angioödems gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Resorption
Nach der Einnahme einer Dosis von 300 mg wurde Sebetralstat rasch resorbiert, wobei die Spitzenkonzentrationen im Plasma nach etwa 1 Stunde erreicht wurden.
Einfluss von Nahrungsaufnahme
Bei einer Bewertung des Einflusses von Nahrungsaufnahme wurde nach der Einnahme einer Dosis von 600 mg zusammen mit einer fettreichen Mahlzeit kein Unterschied in der AUC von Sebetralstat beobachtet. Es kam zu einer Reduktion der Cmax um rund 29 % und einer Verzögerung der medianen Tmax um 2 Stunden. In klinischen Studien zur Sicherheit und Wirksamkeit wurde Ekterly ohne Berücksichtigung der Nahrungsaufnahme verabreicht und kann unabhängig von den Mahlzeiten eingenommen werden.
Verteilung
Die Plasmaproteinbindung beträgt beim Menschen rund 77 %. Nach einer Dosis von 600 mg radioaktiv markiertem Sebetralstat lag das Verhältnis der Radioaktivität zwischen Blut und Plasma bei etwa 0,65. Das geometrische Mittel des scheinbaren Verteilungsvolumens (Vz/F) nach einer Dosis von 300 mg betrug 208 l.
Elimination
Nach einer Dosis von 300 mg betrug das geometrische Mittel der Eliminationshalbwertszeit von Sebetralstat 3,7 Stunden. Das geometrische Mittel der scheinbaren Clearance (Cl/F) betrug 38,5 l/h.
Metabolismus
Sebetralstat wird in vitro hauptsächlich durch CYP3A4 verstoffwechselt. Sebetralstat ist ein In-vitro-Substrat von P‑Glycoprotein und BCRP. Nach einer Dosis von 600 mg radioaktiv markiertem Sebetralstat entfielen 64,1 % der gesamten Plasma-Radioaktivitäts-AUC0-24 auf Sebetralstat sowie auf 11 Metaboliten, die jeweils zwischen 0,39 % und 7,1 % der gesamten Radioaktivitäts-AUC0-24 ausmachten. Der Metabolit mit der höchsten Prävalenz im Plasma ist pharmakologisch inaktiv.
Sebetralstat ist ein In-vitro-Inhibitor von CYP3A4 und CYP2C9 sowie der Transporter OAT3, OCT2, MATE1, MATE2-K, OATP1B1 und OATP1B3.
Sebetralstat ist ein In-vitro-Induktor von CYP3A4. Angesichts seiner sporadischen Anwendung sowie seiner raschen Resorption und Elimination wird das Risiko einer Induktion von CYP3A4 nicht als klinisch relevant eingestuft.
Ausscheidung
Nach Verabreichung einer Dosis von 600 mg radioaktiv markiertem Sebetralstat an gesunde männliche Probanden wurden etwa 32 % der Radioaktivität im Urin ausgeschieden und 63 % im Stuhl. Dabei wurden rund 8,7 % der Dosis im Urin und 12,5 % der Dosis im Stuhl als unverändertes Sebetralstat vorgefunden. Sebetralstat wird primär durch hepatische Metabolisierung über den Stuhl eliminiert.
Linearität/Nicht-Linearität
Über einen Dosisbereich von 5 mg bis 600 mg war die Cmax von Sebetralstat dosisproportional, während die AUC stärker als dosisproportional anstieg, was wahrscheinlich auf eine längere terminale Eliminationsphase bei höheren Dosen zurückzuführen ist.
Spezielle Patientengruppen
Leberfunktionsstörungen
Die Pharmakokinetik von 600 mg Sebetralstat wurde bei Patienten mit leichter und mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse A oder B) untersucht. Im Vergleich zu Patienten mit normaler Leberfunktion waren die Cmax und die AUC bei Patienten mit leichter Leberfunktionsstörung um 7 % bzw. 16 % erhöht. Bei Patienten mit mittelschwerer Leberfunktionsstörung war die Cmax um 63 % und die AUC um 100 % erhöht. Bei Patienten mit mittelschwerer Leberfunktionsstörung, die einen starken CYP3A4-Inhibitor einnehmen, wird zur Behandlung einer HAE-Attacke eine Einzeldosis von 300 mg empfohlen (siehe Abschnitte 4.2 und 4.5).
Nierenfunktionsstörungen
Sebetralstat wird nicht primär über die Nieren eliminiert und wird nicht als Langzeittherapie angewendet.
Die Pharmakokinetik von Sebetralstat wurde nicht bei Patienten mit Nierenfunktionsstörung untersucht (siehe Abschnitt 4.2).
Ältere Patienten
Die KONFIDENT-Studie schloss keine ausreichende Zahl von Patienten ab 65 Jahren ein, um festzustellen, ob diese Patienten anders auf die Behandlung ansprechen als jüngere Erwachsene (siehe Abschnitt 4.2).
Pharmakokinetische/pharmakodynamische Zusammenhänge
Die konzentrationsabhängige Hemmung von Plasmakallikrein, gemessen als eine Reduktion der spezifischen Enzymaktivität gegenüber Baseline, trat nachweislich rasch ein und führte bereits 15 Minuten nach der Einnahme von 300 mg Ekterly zu einer nahezu vollständigen (≥ 95 %) Suppression des Plasmakallikreins bei Patienten mit HAE.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Das kanzerogene Potenzial von Sebetralstat wurde in einer 26-wöchigen Studie an rasH2-Tg-transgenen Mäusen sowie in einer 104-wöchigen Studie an Ratten untersucht. Es war kein vermehrtes Auftreten von malignen Tumoren zu verzeichnen und bei keiner Dosierung gab es Nachweise einer kanzerogenen Wirkung in einer der beiden Tierspezies. Die Expositionen nach den höchsten Dosen (bezogen auf die AUC von ungebundenem Sebetralstat im Plasma) bei männlichen und weiblichen Mäusen betrugen das 0,2- bzw. 0,4-Fache der für den Menschen empfohlenen Höchstdosis (MRHD; maximum recommended human dose) und bei Ratten das 5,7-Fache der MRHD.
In einer Fertilitätsstudie an Ratten wurden bei keiner Dosisstufe Auswirkungen auf das Paarungsverhalten oder die Fertilität festgestellt, aber es wurde eine Zunahme von Präimplantationsverlusten bei der höchsten Dosisstufe von 600 mg/kg/Tag (dem 7,7-Fachen der Exposition des Menschen bei der MRHD, bezogen auf die AUC von ungebundenem Sebetralstat im Plasma) beobachtet. Studien zur embryofetalen Entwicklung wurden an Ratten und Kaninchen durchgeführt. Bei Ratten wurde gezeigt, dass Sebetralstat und/oder seine Metaboliten in die Placenta übertreten. Unter 600 mg/kg/Tag (dem 12-Fachen der menschlichen Exposition bei der MRHD, bezogen auf die AUC von ungebundenem Sebetralstatim Plasma) wurden Missbildungen (Gaumenspalte, Ventrikelseptumdefekt) und embryofetale Letalität berichtet. Der Wert ohne beobachtete schädliche Wirkung (NOAEL) für die embryofetale Entwicklung lag bei 300 mg/kg/Tag (dem 3,0-Fachen der menschlichen Exposition bei der MRHD, bezogen auf die AUC von ungebundenem Sebetralstat). Bei Kaninchen wurden bei Dosen von bis zu 300 mg/kg/Tag (dem 6,8‑Fachen der menschlichen Exposition bei der MRHD, bezogen auf die AUC von ungebundenem Sebetralstat im Plasma) keine Missbildungen und keine embryofetale Letalität beobachtet. Potenzielle mit der PKa-Hemmung verbundene Entwicklungseffekte wurden bei Kaninchen aufgrund der Unterschiede in der pharmakologischen Aktivität von Sebetralstat zwischen den Tierspezies möglicherweise nicht vollständig erfasst. In einer Studie zur prä- und postnatalen Entwicklung an Ratten wurden bei Dosen von bis zu 450 mg/kg/Tag keine schädlichen Auswirkungen auf die Entwicklung festgestellt.
Die Verabreichung einer Einzeldosis von radioaktiv markiertem Sebetralstat an säugende Ratten führte zu vergleichbaren Konzentrationen an Gesamt-Radioaktivität in Milch und Plasma, wobei die Höchstkonzentration 1 Stunde nach Dosisgabe gemessen wurde. 24 Stunden nach der Dosisgabe war das mittlere Niveau der Radioaktivität sowohl in der Milch als auch im Plasma nah am Hintergrund.
Studien zur Umweltverträglichkeitsprüfung haben gezeigt, dass Sebetralstat das Potenzial hat, sich in einigen aquatischen Sedimentsystemen anzureichern und dort zu verbleiben.
Tablettenkern
Mikrokristalline Cellulose (E 460)
Croscarmellose-Natrium (E 468)
Povidon K30 (E 1201)
Magnesiumstearat (E 470b)
Filmüberzug
Macrogol-Poly(vinylalkohol)-Pfropfcopolymer (E 1209)
Talkum (E 553b)
Titandioxid (E 171)
Glycerol-Monocaprylocaprat (Typ 1) (E 471)
Poly(vinylalkohol) (E 1203)
Eisen(III)-hydroxid-oxid x H2O (E 172)
Eisen(II,III)-oxid (E 172)
Maltodextrin (E 1400)
Guargalactomannan (E 412)
Hypromellose (E 464)
Mittelkettige Triglyzeride
Nicht zutreffend.
3 Jahre
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
oPA/Al/PVC-Blisterpackung mit Aluminiumabdeckung (1 Filmtablette pro Blisterpackung).
Die Filmtabletten befinden sich in einer Blisterpackung, die in einer kindergesicherten Walletpackung aus Pappe verpackt ist. Die Walletpackungen sind in einer Schachtel verpackt.
Packungsgröße: 4 oder 6 Filmtabletten.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
KalVista Pharmaceuticals (Ireland) Ltd.
Magennis Place,
Block C,
Dublin 2,
D02 FK76,
Irland
EU/1/25/1975/001
EU/1/25/1975/002
Datum der Erteilung der Zulassung: 17. September 2025.
09/2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.