
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Itvisma® 1,2 × 1014 Vektorgenome Injektionslösung
Onasemnogen abeparvovec ist ein Gentherapeutikum, das das humane Survival-Motoneuron-(SMN‑)Protein exprimiert. Es handelt sich um einen nicht replizierenden rekombinanten adeno-assoziierten Vektor auf der Basis des Virus-Serotyps 9 (AAV9), der die cDNA des humanen SMN-Gens unter der Kontrolle des Cytomegalievirus-Enhancers/Hühner-β-Aktin-Hybrid-Promotors enthält.
Onasemnogen abeparvovec wird durch DNA-Rekombinationstechnologie in menschlichen embryonalen Nierenzellen gebildet.
Jede Einzeldosis-Durchstechflasche enthält 1,2 × 1014 Vektorgenome (vg) von Onasemnogen abeparvovec in 3 ml Lösung.
Onasemnogen abeparvovec intrathekale Injektion hat eine nominale Konzentration von 4 × 1013 vg/ml und jede Durchstechflasche enthält ein extrahierbares Volumen von mindestens 3 ml.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung.
Eine klare bis leicht opake, farblose bis leicht weiße Lösung mit einem pH-Wert von 7,7 bis 8,3 und einer Osmolalität von 390 bis 430 mOsm/kg.
Itvisma wird angewendet zur Behandlung von Patienten ab 2 Jahren und älter mit 5q‑assoziierter spinaler Muskelatrophie (SMA) mit einer biallelischen Mutation im SMN1‑Gen.
Eine Behandlung sollte in klinischen Zentren erfolgen und von einem Arzt überwacht werden, der über Erfahrung in der Therapie von Patienten mit SMA verfügt.
Vor der Verabreichung von Itvisma sind Ausgangs-Laboruntersuchungen erforderlich, einschließlich, aber nicht beschränkt auf:
AAV9‑Antikörpertestung mit einem entsprechend validierten Test (siehe Abschnitt 4.4),
Leberfunktion: Alaninaminotransferase (ALT), Aspartataminotransferase (AST) und Gesamtbilirubin (siehe Abschnitt 4.4),
Kreatinin und
Großes Blutbild (einschließlich Hämoglobin und Thrombozytenzahl) (siehe Abschnitt 4.4).
Es wird empfohlen, dass die Patienten vor der Injektion einen klinisch stabilen allgemeinen Gesundheitszustand aufweisen (siehe Abschnitt 4.4). Das Nutzen-Risiko-Profil von Itvisma bei Patienten mit respiratorischer Insuffizienz, die dauerhaft beatmet werden müssen und/oder nicht schlucken können, ist nicht erwiesen.
Da der Effekt der Behandlung in sich nicht teilenden Zellen andauert, sollten Patienten, die zuvor mit Onasemnogen abeparvovec (unabhängig von der Art der Anwendung) behandelt wurden, nicht mit Itvisma behandelt werden (siehe Abschnitt 5.1).
Dosierung
Itvisma wird als Einzeldosis von 1,2 × 1014 vg verabreicht.
Immunmodulatorisches Therapieregime
Zur Abschwächung der Immunantwort wird eine Immunmodulation mit Corticosteroiden empfohlen. Nach der Behandlung kann eine Erhöhung der Leberaminotransferasen oder eine Verringerung der Thrombozytenzahl auftreten (siehe Abschnitte 4.4 und 4.8). Wenn möglich, sollte der Impfplan des Patienten an die begleitende Corticosteroid-Gabe vor und nach der Injektion von Itvisma angepasst werden (siehe Abschnitt 4.5).
Tabelle 1 enthält das empfohlene immunmodulatorische Therapieregime vor und nach der Injektion.
Tabelle 1 Empfohlenes immunmodulatorisches Therapieregime vor und nach der Injektion
Vor der Injektion |
24 Stunden vor der Itvisma-Injektion |
Prednisolon p.o. 1 mg/kg/Tag (oder Äquivalent) |
Nach der Injektion |
30 Tage (einschließlich Tag der Verabreichung von Itvisma) |
Prednisolon p.o. 1 mg/kg/Tag (oder Äquivalent) |
Anschließend 28 Tage: |
Systemische Corticosteroide sollten schrittweise ausgeschlichen werden. |
|
Bei Patienten mit unauffälligem Befund (unauffälliger klinischer Untersuchungsbefund, Gesamtbilirubin, ALT- und AST-Werte jeweils unter dem 2‑Fachen der Normobergrenze [upper limit of normal, ULN]) am Ende des 30‑Tages-Zeitraums: |
Ausschleichende Reduktion von Prednisolon (oder äquivalent, falls ein anderes Corticosteroid verwendet wird), z. B. durch wöchentliche Reduktionen um 0,20 mg/kg/Tag über einen Zeitraum von mindestens 4 Wochen bei oralem Prednisolon. |
|
oder |
||
Bei Patienten mit abweichenden Leberfunktionswerten am Ende des 30‑Tages-Zeitraums: Fortführung, bis die AST- und ALT-Werte unter 2‑facher ULN liegen und alle anderen Untersuchungsergebnisse (z. B. Gesamtbilirubin) wieder in den Normalbereich zurückgegangen sind, anschließend ausschleichende Reduktion über 28 Tage oder länger, falls notwendig. |
Systemische Corticosteroide (Äquivalent zu Prednisolon p.o. 1 mg/kg/Tag) Systemische Corticosteroide sollten schrittweise ausgeschlichen werden. |
Wenn Patienten zu irgendeinem Zeitpunkt nicht ausreichend auf das Äquivalent von 1 mg/kg/Tag oralem Prednisolon ansprechen, sollte basierend auf dem klinischen Verlauf des Patienten eine sofortige Konsultation mit einem Gastroenterologen oder Hepatologen und eine Anpassung an das empfohlene immunmodulatorische Therapieregime, einschließlich einer Dosiserhöhung, längerer Anwendungsdauer oder Verlängerung der Corticosteroid-Ausschleichung, in Betracht gezogen werden (siehe Abschnitt 4.4). Wenn eine orale Corticosteroidtherapie nicht vertragen wird oder nicht wirksam ist, kann ein intravenöses Corticosteroid als klinisch indiziert in Erwägung gezogen werden.
Verwendet der behandelnde Arzt ein anderes Corticosteroid anstelle von Prednisolon, sollten gegebenenfalls ähnliche Überlegungen und Ansätze zur ausschleichenden Corticosteroid-Dosierung nach 30 Tagen angewandt werden.
Besondere Patientengruppen
Eingeschränkte Nierenfunktion
Die Sicherheit und Wirksamkeit von Itvisma bei Patienten mit eingeschränkter Nierenfunktion ist nicht erwiesen. Eine Dosisanpassung sollte nicht in Betracht gezogen werden.
Eingeschränkte Leberfunktion
Bei Patienten mit eingeschränkter Leberfunktion sollte die Behandlung mit Itvisma sorgfältig überlegt werden (siehe Abschnitt 4.4). Eine Dosisanpassung sollte nicht in Betracht gezogen werden.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Itvisma bei Kindern unter 2 Jahren ist nicht erwiesen. Zurzeit vorliegende Daten werden in den Abschnitten 4.8 und 5.1 beschrieben; eine Dosierungsempfehlung kann jedoch nicht gegeben werden bei Kindern zwischen 6 Monaten und < 2 Jahren. Es liegen keine Daten bei Kindern < 6 Monate vor.
Erwachsene
Es liegen keine klinischen Studiendaten bei Patienten ab 18 Jahren und älter vor.
Art der Anwendung
Zur intrathekalen Anwendung. Die Behandlung sollte intrathekal mittels Lumbalpunktion durch medizinisches Fachpersonal erfolgen, das Erfahrung in der Durchführung von Lumbalpunktionen hat.
Unmittelbar vor der Verabreichung den Inhalt aus der Durchstechflasche in die Spritze aufziehen, Luft aus der Spritze entfernen, das Dosisvolumen von 3 ml in der Spritze bestätigen, die Spritze verschließen und an den Ort der Injektion des Patienten bringen.
Es sollte eine Sedierung in Erwägung gezogen werden, wenn dies durch den klinischen Zustand des Patienten angezeigt ist.
Es sollten bildgebende Verfahren zur Unterstützung der intrathekalen Injektion in Betracht gezogen werden.
Den Patienten vor und nach der intrathekalen Injektion auf mögliche Anomalien im Zusammenhang mit einer Lumbalpunktion untersuchen, um schwerwiegende verfahrensbedingte Komplikationen zu vermeiden.
Vor der Verabreichung sind 3 ml Zerebrospinalflüssigkeit (CSF) mithilfe einer Lumbalpunktionsnadel zu entnehmen.
Itvisma wird als Einzeldosis mittels intrathekaler Bolusinjektion über ungefähr 1 bis 2 Minuten verabreicht.
Nach der intrathekalen Injektion wird die Trendelenburg-Lagerung (je nach klinischem Zustand des Patienten) empfohlen.
Für detaillierte Hinweise zur Zubereitung, Handhabung, versehentlichen Exposition und Entsorgung des Arzneimittels siehe Abschnitt 6.6.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Gründe für einen Aufschub der Behandlung
Aufgrund des erhöhten Risikos einer schwerwiegenden systemischen Immunantwort wird empfohlen, dass die Patienten vor der Injektion einen klinisch stabilen allgemeinen Gesundheitszustand (z. B. Hydratations- und Ernährungszustand, keine Infektion, respiratorischer Status) aufweisen (siehe Abschnitt 4.4). Liegt eine akute (z. B. respiratorische) oder chronische, unkontrollierte Infektion vor, sollte die Behandlung mit Itvisma so lange aufgeschoben werden, bis die Infektion abgeklungen und der Patient klinisch stabil ist. Zum Zeitpunkt der Injektion dürfen keine klinischen Anzeichen oder Symptome einer Infektion vorhanden sein.
Vorbestehende Immunität gegenüber AAV9
In klinischen Studien mit Itvisma mussten die Patienten zu Beginn der Studie einen Anti-AAV9-Antikörpertiter im Serum von ≤ 1:50 aufweisen. Die Sicherheit und Wirksamkeit von Itvisma bei Patienten mit erhöhten Anti-AAV9-Antikörpertitern wurden beim Menschen nicht untersucht.
Lebertoxizität
Nach der Anwendung von Onasemnogen abeparvovec als intrathekale Injektion wurde über Lebertoxizität berichtet, die sich im Allgemeinen in erhöhten ALT- und/oder AST-Werten manifestierte (siehe Abschnitt 4.8). Um mögliche Aminotransferase-Erhöhungen zu mildern, sollte jedem Patienten vor und nach der intrathekalen Injektion ein systemisches Corticosteroid verabreicht werden. Die immunvermittelte Lebertoxizität kann die Anpassung des immunmodulatorischen Regimes einschließlich einer längeren Anwendungsdauer, einer Dosiserhöhung oder einer Verlängerung der Corticosteroid-Ausschleichung erfordern (siehe Abschnitt 4.2).
Patienten mit einer vorbestehenden Leberfunktionsstörung oder einer akuten Virusinfektion der Leber haben möglicherweise ein erhöhtes Risiko für eine Leberschädigung. Patienten mit erhöhten Leberfunktionswerten wurden in klinischen Studien zur intrathekalen Injektion von Onasemnogen abeparvovec nicht untersucht.
Vor der intrathekalen Injektion von Onasemnogen abeparvovec sollte die Leberfunktion aller Patienten durch klinische Untersuchung und Labortests beurteilt werden. Die Leberfunktion sollte nach der intrathekalen Injektion von Onasemnogen abeparvovec mindestens 3 Monate und zu anderen Zeitpunkten, sofern dies klinisch indiziert ist, überwacht werden. Nach der intrathekalen Injektion von Onasemnogen abeparvovec sollten AST, ALT und Gesamtbilirubin im ersten Monat und während des gesamten Zeitraums der Corticosteroid-Ausschleichung wöchentlich beurteilt werden. Wenn der Patient am Ende der Ausschleichphase klinisch stabil ist und unauffällige Befunde aufweist, sollte die Leberfunktion einen weiteren Monat alle zwei Wochen überwacht werden. Das Ausschleichen von systemischen Corticosteroiden sollte nicht in Betracht gezogen werden, bis die AST/ALT-Spiegel weniger als 2 × ULN betragen (siehe Abschnitt 4.2).
Patienten mit sich verschlechternden Leberfunktionstestergebnissen und/oder Anzeichen oder Symptomen einer akuten Erkrankung sollten sofort klinisch untersucht und engmaschig überwacht werden. Bei Verdacht auf eine Leberschädigung wird eine sofortige Konsultation mit einem Gastroenterologen oder Hepatologen und weitere Tests empfohlen (z. B. Albumin, Prothrombinzeit, PTT und INR).
Thrombozytopenie
Vorübergehend verminderte Thrombozytenzahlen wurden typischerweise innerhalb der ersten Woche nach der intrathekalen Injektion von Onasemnogen abeparvovec beobachtet. In den meisten Fällen betrug die Thrombozytenzahl ˃ 75 × 109/l und stieg in den zwei Wochen nach der intrathekalen Injektion von Onasemnogen abeparvovec wieder bis auf den Ausgangswert an.
Die Thrombozytenzahl sollte vor der intrathekalen Injektion von Onasemnogen abeparvovec ermittelt und danach weiterhin regelmäßig überwacht werden, im ersten Monat mindestens wöchentlich und wenn klinisch angezeigt, bis die Thrombozytenzahl wieder auf den Ausgangswert zurückgeht.
Thrombotische Mikroangiopathie
Es kann eine thrombotische Mikroangiopathie (TMA) auftreten. Die TMA ist durch Thrombozytopenie, mikroangiopathische hämolytische Anämie und akute Nierenschädigung gekennzeichnet. Eine simultane Aktivierung des Immunsystems (z. B. Infektionen, Impfungen) kann dazu beitragen.
Anzeichen und Symptome einer TMA sollten unverzüglich untersucht werden, da TMA einen lebensbedrohlichen oder tödlichen Ausgang haben kann.
Die Thrombozytopenie ist ein Hauptmerkmal der TMA, daher sollte die Thrombozytenzahl nach der intrathekalen Injektion von Onasemnogen abeparvovec regelmäßig überwacht werden und die Patienten sollten auf Anzeichen und Symptome einer TMA wie Hypertonie, eine erhöhte Zahl von Blutergüssen, Krampfanfälle oder verminderte Urinausscheidung überwacht werden. Im Falle, dass diese Anzeichen und Symptome begleitend zu einer Thrombozytopenie auftreten, sollten weitere diagnostische Abklärungen auf hämolytische Anämie und Nierenfunktionsstörung unverzüglich vorgenommen werden. Bei klinischen Anzeichen, Symptomen und/oder Laborbefunden, die auf eine TMA hinweisen, sollte sofort ein Hämatologe und/oder Nephrologe konsultiert werden, um die TMA wie klinisch angezeigt zu behandeln. Patienten und Betreuungspersonen sollten über die Anzeichen und Symptome einer TMA informiert und angewiesen werden, sich beim Auftreten dieser Symptome unverzüglich in ärztliche Notfallbehandlung zu begeben.
Periphere sensorische Neuropathie
Bei der intrathekalen Injektion von Onasemnogen abeparvovec ist eine periphere sensorische Neuropathie aufgetreten. Die intrathekale Injektion von Onasemnogen abeparvovec kann zu sensorischen Symptomen führen (z. B. Taubheitsgefühl, Kribbeln, Prickeln oder Schmerzen in Armen, Händen, Beinen und/oder Füßen), wobei der Symptombeginn in klinischen Studien ungefähr drei Wochen nach der Injektion auftrat. Symptome, die eine Behandlung mit zusätzlichen Therapien erfordern, können fortbestehen, sich jedoch im Laufe der Zeit auch schrittweise verbessern (siehe Abschnitt 4.8). Sensorische Symptome, die auf eine periphere sensorische Neuropathie hindeuten, können auch im Rahmen des natürlichen Verlaufs einer SMA auftreten.
Eine vollständige neurologische Abklärung und andere Untersuchungen und/oder eine symptomatische Behandlung sollten in Abhängigkeit vom klinischen Bild des Patienten in Erwägung gezogen werden. Patienten und Betreuungspersonen sollten über die Anzeichen und Symptome einer peripheren sensorischen Neuropathie informiert und angewiesen werden, den behandelnden Arzt unverzüglich zu informieren, wenn diese Symptome auftreten.
Risiko der Tumorigenität infolge der Vektorintegration
Es besteht ein theoretisches Risiko der Tumorigenität aufgrund der potenziellen Integration der AAV-Vektor-DNA von Onasemnogen abeparvovec in das Wirtsgenom.
Onasemnogen abeparvovec intrathekale Injektion besteht aus einem nicht replizierenden AAV9-Vektor, dessen DNA größtenteils in episomaler Form persistiert. Bei AAV-Gentherapien wurde über eine zufällige Integration rekombinanter AAV-Vektor-DNA in die menschliche DNA berichtet. Die klinische Relevanz einzelner Integrationsereignisse ist nicht bekannt, aber es wird anerkannt, dass einzelne Integrationsereignisse potenziell zu einem Risiko der Tumorigenität beitragen könnten.
Bisher wurden keine Fälle von Malignitäten im Zusammenhang mit einer Onasemnogen abeparvovec-Behandlung mittels intrathekaler Injektion berichtet. Im Falle eines Tumors sollte der Inhaber der Zulassung kontaktiert werden, um eine Anleitung zur Entnahme von Patientenproben für Testzwecke zu erhalten.
Blut-, Organ-, Gewebe- und Zellspende
Patienten, die mit Onasemnogen abeparvovec mittels intrathekaler Injektion behandelt wurden, sollten kein Blut, keine Organe, keine Gewebe oder Zellen für Transplantationen spenden.
Natriumgehalt
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro 3‑ml‑Dosis, d. h., es ist nahezu „natriumfrei“.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Impfungen
Wenn möglich, sollte der Impfplan des Patienten an die begleitende Corticosteroid-Gabe vor und nach der intrathekalen Injektion von Onasemnogen abeparvovec angepasst werden. Eine saisonale Prophylaxe gegen das Respiratorische Synzytialvirus (RSV) wird empfohlen. Lebendimpfstoffe wie Masern, Mumps und Röteln (MMR) und Varizellen sollten bei Patienten unter einer immunsuppressiven Steroiddosis (d. h. ≥ 2 Wochen täglicher Gabe von 20 mg oder 2 mg/kg Körpergewicht Prednisolon oder Äquivalent) nicht angewendet werden.
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Onasemnogen abeparvovec bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Abschnitt 5.3). Es ist nicht bekannt, ob Onasemnogen abeparvovec auf den Fötus übertragen werden könnte. Daher sollten Frauen, die schwanger sind oder schwanger werden möchten, nur nach einer gründlichen Nutzen-Risiko-Bewertung mit Itvisma behandelt werden.
Stillzeit
Es liegen keine Daten über das Vorhandensein von Onasemnogen abeparvovec in der Muttermilch und über die Auswirkungen auf den gestillten Säugling oder auf die Milchproduktion vor. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Es liegen keine Daten über den Einfluss von Onasemnogen abeparvovec auf die Fertilität beim Menschen vor. In tierexperimentellen Fertilitätsstudien hatte Onasemnogen abeparvovec in einer Dosis von 1,1 × 1014 vg/kg intravenös verabreicht keine Auswirkungen auf die Fertilität bei männlichen und weiblichen Mäusen (siehe Abschnitt 5.3).
Die intrathekale Injektion von Onasemnogen abeparvovec hat einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Patienten, bei denen Schwindel auftritt (siehe Abschnitt 4.8), sollten das Führen von Fahrzeugen und das Bedienen von Maschinen vermeiden.
Zusammenfassung des Sicherheitsprofils
Die in diesem Abschnitt beschriebenen Sicherheitsdaten stammen von 127 Patienten aus den Studien COAV101B12301, COAV101B12302 und COAV101A12102 über einen Nachbeobachtungszeitraum von 52 Wochen (siehe Abschnitt 5.1). Die am häufigsten berichteten Nebenwirkungen nach der Anwendung von Itvisma waren Infektion der oberen Atemwege (41,7 %), Fieber (36,2 %), Erbrechen (28,3 %), Kopfschmerzen (13,4 %) und erhöhte Leberenzyme (9,4 %). Die am häufigsten gemeldeten schwerwiegenden unerwünschten Nebenwirkungen waren Erbrechen (2,4 %), erhöhte Leberenzymwerte (1,6 %), Kopfschmerzen (1,6 %) und Fieber (1,6 %). Die gemeldeten Nebenwirkungen waren in allen Studien ähnlich.
Tabellarische Liste der Nebenwirkungen
Die mit Itvisma aufgetretenen Nebenwirkungen bei Patienten, die eine intrathekale Injektion der empfohlenen Dosierung erhalten haben, sind in Tabelle 2 dargestellt. Die Nebenwirkungen sind gemäß MedDRA-Systemorganklassifikation und nach Häufigkeit geordnet angegeben. Die Häufigkeitskategorien basieren auf folgender Konvention: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1 000, < 1/100); selten (≥ 1/10 000, < 1/1 000); sehr selten (< 1/10 000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 2 Liste der Nebenwirkungen von Itvisma
Nebenwirkungen nach MedDRA-SOC/PT und Häufigkeit | |
Infektionen und parasitäre Erkrankungen | |
Sehr häufig |
Infektion der oberen Atemwegea) |
Erkrankungen des Blutes und des Lymphsystems | |
Häufig |
Thrombozytopenieb) |
Erkrankungen des Nervensystems | |
Sehr häufig |
Kopfschmerzenc) |
Häufig |
Schwindelgefühlc) |
Häufig |
Periphere sensorische Neuropathied) |
Häufig |
Hypoästhesie |
Häufig |
Parästhesie |
Erkrankungen des Gastrointestinaltrakts | |
Sehr häufig |
Erbrechen |
Leber- und Gallenerkrankungen | |
Häufig |
Leberenzyme erhöhte) |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Sehr häufig |
Fieber |
| |
Beschreibung ausgewählter Nebenwirkungen
Periphere sensorische Neuropathie
In klinischen Studien wurden innerhalb von etwa drei Wochen nach Verabreichung von Itvisma Fälle von peripherer sensorischer Neuropathie (1,6 %) beobachtet. Die Patienten wiesen Hypoästhesie und Parästhesie auf. Bei diesen Fällen war eine längerfristige symptomatische Behandlung erforderlich, und einige Symptome waren zum Studienende nicht vollständig abgeklungen (siehe Abschnitt 4.4).
Hepatische Laboranomalien
In klinischen Studien erhielten alle Patienten eine prophylaktische Corticosteroid-Behandlung. Die meisten Patienten erhielten das empfohlene immunmodulatorische Regime über eine mediane Dauer von ca. 60 Tagen (siehe Abschnitt 4.2). Es wurden AST- oder ALT-Erhöhungen von > 3 × ULN bei 4,7 % der Patienten nach Verabreichung von Itvisma beobachtet. Die Erhöhungen der Serum-Aminotransferasen bildeten sich unter Prednisolon-Behandlung zurück und die Patienten erholten sich ohne klinische Folgen.
Immunogenität
In den klinischen Studien COAV101B12301 und COAV101B12302 kam es bei allen Patienten nach einer einmaligen Itvisma-Injektion zu Erhöhungen der Anti-AAV9-Antikörpertiter im Serum gegenüber dem Ausgangswert. Die medianen AAV9-Antikörpertiter im Serum 12 Monate nach der Itvisma-Injektion betrugen in beiden Studien > 1:800 000. In der untersuchten Population zeigte sich über den Nachbeobachtungszeitraum von 12 Monaten nach der Dosierung kein klarer Zusammenhang zwischen dem Anti‑AAV9‑Antikörperspiegel nach der Anwendung und Sicherheitsbefunden oder einem Wirksamkeitsverlust.
Während des 12‑monatigen Zeitraums nach der Itvisma-Injektion wurden in den Studien COAV101B12301 und COAV101B12302 bei 5/75 (6,7 %) bzw. 2/27 (7,4 %) der mit Itvisma behandelten Patienten positive Anti-SMN-Antikörper beobachtet. Es kann kein eindeutiger Zusammenhang zwischen einer positiven Anti-SMN-Antikörperreaktion und der Sicherheit oder Wirksamkeit von Itvisma während des Nachbeobachtungszeitraums von 12 Monaten nach Dosisgabe hergestellt werden.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel.: + 49 6103 770, Fax: + 49 6103 77 1234, Website: www.pei.de, anzuzeigen.
In Bezug auf eine Überdosierung von Itvisma liegen keine Daten aus klinischen Studien vor. Die Dosis von Itvisma ist eine einzelne, fixe Dosis und wird nur einmal verabreicht, daher wird eine Überdosierung als unwahrscheinlich erachtet.
Pharmakotherapeutische Gruppe: andere Mittel gegen Störungen des Muskel- und Skelettsystems, ATC-Code: M09AX09
Wirkmechanismus
Onasemnogen abeparvovec ist eine Gentherapie, die darauf abzielt, eine funktionsfähige Kopie des Survival-Motoneuron-Gens (SMN1) in die transduzierten Zellen einzubringen, um die monogenetische Grundursache der spinalen Muskelatrophie (SMA) zu behandeln. Durch das Bereitstellen einer alternativen Quelle der SMN-Proteinexpression in Motoneuronen wird erwartet, dass das Überleben und die Funktion der transduzierten Motoneuronen gefördert wird.
Onasemnogen abeparvovec ist ein nicht replizierender rekombinanter AAV-Vektor, der ein AAV9‑Kapsid verwendet, um ein stabiles, voll funktionsfähiges menschliches SMN-Transgen bereitzustellen. Das in Onasemnogen abeparvovec vorhandene SMN1‑Gen ist so konzipiert, dass es als episomale DNA im Kern der transduzierten Zellen liegt und in postmitotischen Zellen stabil exprimiert wird. Das Transgen wird als selbst-komplementäres doppelsträngiges Molekül in die Zielzellen eingebracht. Die Expression des Transgens wird durch einen konstitutiven Promotor (Hybrid aus Cytomegalievirus-Enhancer und Hühner-β-Aktin-Promotor) ermöglicht, der zu einer kontinuierlichen, anhaltenden SMN‑Proteinexpression führt. Der Nachweis des Wirkmechanismus wird hauptsächlich durch präklinische Studien gestützt.
Klinische Wirksamkeit und Sicherheit
Phase‑III‑Studie COAV101B12301 (STEER) bei Patienten mit SMA
Dies ist eine 52‑wöchige, randomisierte, doppelblinde, Sham-kontrollierte multizentrische Studie.
Die Wirksamkeit wurde bei 126 nicht vorbehandelten Patienten mit SMA im Alter von 2 bis < 18 Jahren untersucht, die in der Lage waren, selbstständig zu sitzen, aber nie ohne Hilfe gehen konnten. Die Patienten wurden im Verhältnis 3:2 randomisiert und erhielten entweder Itvisma (1,2 × 1014 vg) durch intrathekale Lumbalinjektion (n = 75) oder ein Sham-Verfahren (n = 51). Die Randomisierung war nach Alter und Score auf der Hammersmith Functional Motor Scale – Expanded (HFMSE) beim Screening vor der Behandlung stratifiziert. Patienten mit einem erhöhten (Referenzwert > 1:50) Anti-AAV9-Antikörpertiter im Serum zu Studienbeginn waren ausgeschlossen.
Der primäre Endpunkt war die Veränderung des HFMSE-Gesamtscores am Ende der Nachbeobachtung, definiert als der Durchschnitt der Beurteilungen in Woche 48 und Woche 52, gegenüber dem Ausgangswert in der Gesamtpopulation der Studie (Altersgruppe 2 bis < 18 Jahre) mit Itvisma im Vergleich zur Sham-Behandlung. Ein sekundärer Endpunkt war der Anteil der Patienten mit Verbesserung des HFMSE-Gesamtscores um mindestens 3 Punkte gegenüber dem Ausgangswert am Ende der Nachbeobachtung. Mit der HFMSE wird die motorische Funktion bei Patienten mit SMA mit eingeschränkter Gehfähigkeit beurteilt. Die Skala besteht aus 33 bewerteten Items zur Beurteilung von Bewegungen vom Sitzen bis hin zur Benutzung einer Treppe. Jedes Item wird von 0 bis 2 bewertet, mit einem maximalen Gesamtscore von 66. Höhere Werte bedeuten eine bessere motorische Funktion. Ein weiterer sekundärer Endpunkt war die Veränderung im Revised Upper Limb Module (RULM) gegenüber dem Ausgangswert am Ende der Nachbeobachtung. Der RULM ist ein SMA-spezifisches Bewertungsinstrument der motorischen Funktion der oberen Gliedmaßen (proximal und distal). Der RULM enthält 19 bewertete Items (18 von 0 bis 2 und 1 von 0 bis 1), und es gibt eine maximal erreichbare Punktzahl von 37, wobei höhere Werte auf eine bessere motorische Funktion hinweisen.
Das mediane Alter beim Screening betrug 4,54 Jahre (Bereich: 2,0 bis 16,5 Jahre), und das mediane berichtete Alter beim ersten Auftreten klinischer Anzeichen und Symptome von SMA betrug 11 Monate (Bereich: 6 bis 33 Monate). Zu den höchsten motorischen Meilensteinen, die die Patienten je erreicht haben, gehörten Sitzen, Stehen mit oder ohne Unterstützung oder Gehen mit Unterstützung. Zu Studienbeginn betrug der mittlere HFMSE-Score 17,97 und 18,17 in der mit Itvisma behandelten Gruppe bzw. in der Sham-behandelten Kontrollgruppe. Der mittlere RULM-Ausgangswert betrug 16,52 in der mit Itvisma behandelten Gruppe und 17,42 in der Sham-behandelten Kontrollgruppe. Die demografischen Merkmale waren zur Baseline zwischen dem Itvisma-Arm und dem Sham-Arm ausgewogen.
Die Analyse des primären Endpunkts zeigte eine statistisch signifikante und klinisch bedeutsame Verbesserung der HFMSE-Scores gegenüber dem Ausgangswert am Ende der Nachbeobachtung in der mit Itvisma behandelten Gruppe im Vergleich zur Sham-Kontrollgruppe (Tabelle 3, Abbildung 1).
Tabelle 3 Primärer Endpunkt der Studie COAV101B12301
Endpunkt |
Mit Itvisma behandelte Patienten (N = 75) |
Patienten in der Sham-Kontrollgruppe (N = 51) |
Mittlerer HFMSE-Gesamtwert bei Studienbeginn (SD) |
17,97 (10,110) |
18,17 (9,756) |
Mittlerer HFMSE-Gesamtwert am Ende der Nachbeobachtung1 (SD) |
20,49 (11,356) |
19,05 (10,132) |
Veränderung des HFMSE-Gesamtscores2 am Ende der Nachbeobachtung gegenüber dem Ausgangswert in der Gesamtpopulation der Studie (Altersgruppe 2 bis < 18 Jahre)1, 2, 3 |
2,39 (0,439) |
0,51 (0,532) |
Unterschied zur Sham-Kontrollgruppe |
1,88 |
|
SD = Standardabweichung | ||
Abbildung 1 Veränderung des HFMSE-Scores gegenüber dem Ausgangswert bei Patienten im Alter von 2 bis zu < 18 Jahren in der Studie COAV101B12301
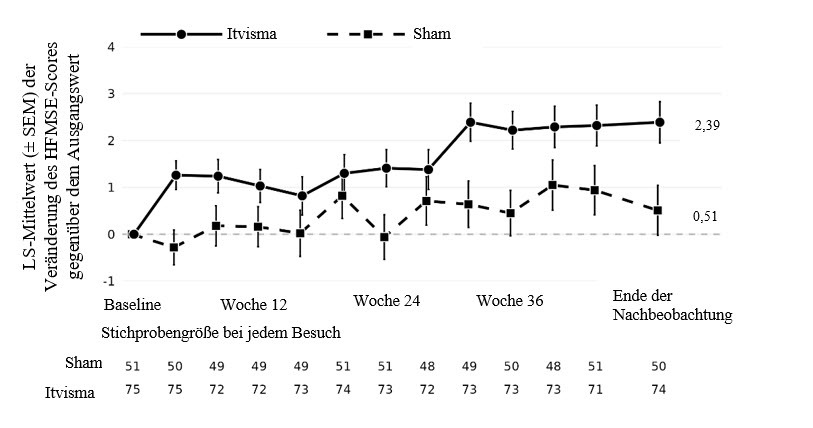
Hinweis: Die Daten stellen den LS‑Mittelwert ± SEM dar; die Zwischenzeitpunkte sind rein deskriptiv und wurden nicht auf Mehrfachvergleiche korrigiert.
Die Veränderung im RULM gegenüber dem Ausgangswert am Ende der Nachbeobachtung wurde in der mit Itvisma behandelten Gruppe und in der Sham-Kontrollgruppe beurteilt. Der Anstieg der LS‑Mittelwerte der RULM-Gesamtscores von Baseline bis zum Ende der Nachbeobachtung betrug 2,44 Punkte in der mit Itvisma behandelten Gruppe und 0,92 Punkte in der Sham-Kontrollgruppe. Die Behandlungsdifferenz von 1,52 Punkten zugunsten der mit Itvisma behandelten Gruppe erreichte keine statistische Signifikanz (95%‑KI: 0,34; 2,71) bei einem Alpha-Level von 0,0025 gemäß der vorab geplanten multiplen Teststrategie.
Der Prozentsatz der Patienten mit einer Verbesserung des HFMSE-Gesamtscores von Baseline bis zum Ende der Nachbeobachtung um mindestens 3 Punkte war in der mit Itvisma behandelten Gruppe zahlenmäßig höher (39,2 %) als in der Sham-Kontrollgruppe (26,0 %), obwohl der Unterschied keine statistische Signifikanz erreichte (Odds Ratio [OR]: 2,03; 95%‑KI: 0,9; 4,57).
Siebenundsechzig von 75 Patienten aus der Studie COAV101B12301, die Itvisma erhalten hatten, wurden nach dem Ende der Nachbeobachtungszeit noch weitere 3 Monate beobachtet. Während des gesamten Nachbeobachtungszeitraums von bis zu 15 Monaten nach der Verabreichung von Itvisma stiegen die durchschnittlichen HFMSE-Werte weiter an, was auf eine verbesserte und anhaltende motorische Funktion hinweist.
Phase‑III‑Studie COAV101B12302 (STRENGTH) bei SMA-Patienten nach Vorbehandlung mit anderen SMA-modifizierenden Therapien
Dies ist eine unverblindete, einarmige, multizentrische Phase‑III‑Studie zur intrathekalen Verabreichung von Itvisma (1,2 × 1014 vg) bei 27 Patienten mit SMA im Alter von 2 bis < 18 Jahren (medianes Alter beim Screening: 7,0 Jahre; Bereich: 2,3 bis 17,6 Jahre), die eine vorhergehende SMA-Therapie abgebrochen hatten (Nusinersen n = 21, Risdiplam n = 4, Nusinersen und Risdiplam [nicht gleichzeitig] n = 2). Vor der Behandlung mit Itvisma hatten die Patienten Nusinersen im Mittel 4,3 Jahre (Bereich: 1,86 bis 6,18 Jahre) und Risdiplam im Mittel 3,0 Jahre (Bereich: 0,39 bis 6,28 Jahre) erhalten. Alle Patienten konnten sitzen, aber nie selbstständig gehen. Die motorische Funktion der Patienten zur Baseline umfasste Sitzen, Stehen mit oder ohne Unterstützung und/oder Gehen mit Unterstützung. Patienten mit einem erhöhten (Referenzwert > 1:50) Anti-AAV9-Antikörpertiter im Serum zu Studienbeginn waren ausgeschlossen.
In Woche 52 zeigte sich bei den Patienten eine allgemeine Stabilisierung der motorischen Funktion, gemessen mittels HFMSE (n = 21) mit einer mittleren Veränderung (SD) gegenüber dem Ausgangswert von 0,17 (2,88) und mittels RULM (n = 21) mit einer mittleren Veränderung (SD) gegenüber dem Ausgangswert von 0,29 (2,85). Nach Adjustierung für die Altersstratifizierung zur Baseline betrug die bereinigte mittlere (LS‑Mittelwert) Veränderung beim HFMSE- und beim RULM-Gesamtscore zwischen Baseline und Woche 52 1,05 bzw. 0,59. Bei den meisten Patienten war in Woche 52 eine Aufrechterhaltung der motorischen Baseline-Meilensteine oder höhere motorische Meilensteine festzustellen.
Phase‑I/II‑Studie COAV101A12102 (STRONG) bei Patienten mit SMA
Dies ist eine unverblindete Studie der Phase I/II, in der 25 Patienten im Alter von 6 Monaten bis < 5 Jahren zum Zeitpunkt der Behandlung eine einmalige intrathekale Injektion von Itvisma (1,2 × 1014 vg) erhielten. Alle Patienten waren nicht vorbehandelt und konnten zur Baseline sitzen, aber nie selbstständig stehen oder gehen. Patienten mit einem erhöhten (Referenzwert > 1:50) Anti-AAV9-Antikörpertiter im Serum zu Studienbeginn waren ausgeschlossen. Die Patienten waren nach ihrem Alter bei Dosisgabe in zwei Gruppen stratifiziert (6 Monate bis < 2 Jahre [N = 13]; 2 bis < 5 Jahre [N = 12]). Das mediane Alter bei Dosisgabe betrug 17,7 Monate (Bereich: 7 bis 23 Monate) in der Altersgruppe 6 Monate bis < 2 Jahre und 33,7 Monate (Bereich: 26 bis 55 Monate) in der Altersgruppe 2 bis < 5 Jahre. Das mediane Alter bei Auftreten klinischer Anzeichen und Symptome der SMA betrug 8,0 Monate in der Altersgruppe 6 Monate bis < 2 Jahre und 8,5 Monate in der Altersgruppe 2 bis < 5 Jahre.
Der primäre Wirksamkeitsendpunkt bei Patienten im Alter von 2 bis < 5 Jahren war die Veränderung des HFMSE-Scores 12 Monate nach der Itvisma-Injektion gegenüber dem Ausgangswert. Der mittlere HFMSE-Score (SD) betrug 14,8 (9,98) zur Baseline und 21,3 (11,94) bei Monat 12. Die primäre Analyse in dieser Altersgruppe zeigte eine Veränderung der LS‑Mittelwerte (95%‑KI) der HFMSE-Scores zwischen Baseline und Monat 12 von 6,0 (3,7; 8,3) Punkten.
Der primäre Wirksamkeitsendpunkt für Patienten im Alter von 6 Monaten bis < 2 Jahren war die Fähigkeit, bei jedem Besuchstermin nach Baseline bis 12 Monate nach der Itvisma-Injektion für mindestens 3 Sekunden selbständig zu stehen (Bayley Scales of Infant and Toddler Development [Bayley‑III] – Gross Motor [GM] Subtest Item 40). Der primäre Endpunkt wurde nicht erreicht; Einer der 13 Patienten erreichte diesen Meilenstein in Monat 12.
Exploratorische Analysen bei Patienten im Alter von 6 Monaten bis < 2 Jahren zeigen Verbesserungen der Grob- und der Feinmotorik gegenüber Baseline, gemessen anhand der Bayley-III-Subtests für Grob- und Feinmotorik. Alle 13 Patienten zeigten bei den Gesamt-Scores in den Subtests für Grob- und Feinmotorik eine Verbesserung von Baseline bis Monat 12, mit einer mittleren Veränderung (SD) von 6,7 (6,46) bzw. 12,7 (3,71) gegenüber Baseline. Darüber hinaus zeigten in einer Teilgruppe der Patienten, die während der Studie das Alter von 2 Jahren erreichten und für die HFMSE-Daten über mindestens sieben Monate nach Baseline vorlagen (n = 6), alle Patienten einen Anstieg des HFMSE-Scores um 1 bis 14 Punkte (mittlere Veränderung ± SD: 6,7 ± 4,72) von der Erstbeurteilung mittels HFMSE bis zum Ende von Monat 7. Die Wirksamkeit in der Altersgruppe von 6 Monaten bis ˂ 2 Jahren wurde nicht nachgewiesen.
Zwölf von 25 Patienten aus der Studie COAV101A12102 wurden für bis zu 7,2 Jahre in eine Langzeitstudie aufgenommen. Bis zum 30. Juni 2025 konnte die Mehrheit der Patienten ihre motorischen Funktionen beibehalten oder weiter verbessern. Neun von 12 Patienten erhielten zu einem beliebigen Zeitpunkt während der Langzeitstudie eine gleichzeitige Behandlung mit Nusinersen oder Risdiplam. Der Grund für die gleichzeitige Gabe von Nusinersen oder Risdiplam ist nicht bekannt, und es kann keine Aussage über die Wirkung der Behandlung bei diesen Patienten getroffen werden.
Es wurden Studien zur Freisetzung des Onasemnogen abeparvovec-Vektors durchgeführt, die die Menge der durch Speichel, Urin, Fäzes und Nasensekret aus dem Körper ausgeschiedenen Vektor‑DNA nach intrathekaler Verabreichung ermitteln.
Onasemnogen abeparvovec-Vektor‑DNA war nach intrathekaler Injektion in Freisetzungsproben nachweisbar. Die Freisetzung (Ausscheidung) von Onasemnogen abeparvovec erfolgte vorwiegend über die Fäzes. Der Großteil der Vektor‑DNA (> 90 %) wird innerhalb von 2 Wochen nach der Verabreichung der Dosis ausgeschieden. Die höchste Freisetzung über die Fäzes wurde zwischen 0,005 % bis 0,03 % der Gesamtdosis geschätzt. Die maximale Ausscheidungskonzentration in den Fäzes war nach intrathekaler Verabreichung von 1,2 × 1014 vg in COAV101B12301 im Vergleich zur intravenösen Verabreichung von 1,1 × 1014 vg/kg (Studien AVXS-101-CL-302 und AVXS-101-CL-303) um das ~70-Fache niedriger.
Bei Patienten im Alter von 6 Monaten bis < 2 Jahren erhöht sich das CSF‑Volumen um das 1,0‑ bis 1,6‑Fache. Vor diesem Hintergrund kann die Exposition von Onasemnogen abeparvovec im CSF bei pädiatrischen Patienten im Alter von 6 Monaten bis < 2 Jahren im Vergleich zu Patienten im Alter von ≥ 2 Jahren um das bis zu 1,6‑Fache höher sein.
Bioverteilung
Nach intrathekaler Verabreichung bei nicht-menschlichen Primaten wurde der Vektor mit anschließender Expression von Transgen-mRNA weit verteilt. Die höchste Vektor-DNA-Konzentration wurde in der Leber nachgewiesen, gefolgt von den Dorsalwurzelganglien (DRG) und dem Rückenmark, die niedrigste Konzentration befand sich in den Keimdrüsen. Die höchsten Konzentrationen an Transgen-mRNA traten tendenziell in Herz, Leber und Muskel auf.
Bei Mäusen, die am 1. postnatalen Tag mit Onasemnogen abeparvovec intravenös oder intrazerebroventrikulär dosiert wurden, war der Virusvektor in den Keimzellen von Männchen und Weibchen in der 3., 8. oder 24. Woche nach der Verabreichung nicht nachweisbar. Bei juvenilen nicht-menschlichen Primaten, die mit einem scAAV9‑Virusvektor behandelt wurden, der in Onasemnogen abeparvovec verwendet wird, hier aber das grün fluoreszierende Protein (GFP) oder mCherry als Transgen trägt, konnte gezeigt werden, dass dieser nach intrathekaler Verabreichung, Verabreichung in die Cisterna magna und intravenöser Verabreichung Oozyten von zyklischen Weibchen im Alter von 13–17 Monaten transduzierte, nicht aber die Samenkanälchen oder Keimzellen von geschlechtsreifen männlichen Tieren.
Bei nicht-menschlichen Primaten zeigten sich nach intrathekaler Verabreichung keine Auswirkungen auf die Verteilung der DNA des scAAV9-Vektors (verwendet in Onasemnogen abeparvovec) im Rückenmark durch hohe vorbestehende Anti-AAV9-Antikörpertiter im Serum (entsprechend Titerwerten von bis zu etwa 1:25 000 beim Menschen).
Allgemeine Toxizität
In einer 12‑monatigen Toxizitätsstudie an juvenilen nicht-menschlichen Primaten führte die intrathekale Verabreichung einer Einzeldosis Onasemnogen abeparvovec in Dosen von 1,20 × 1013, 3,0 × 1013 oder 6,0 × 1013 vg/Tier 6 Wochen nach Dosisgabe zu akuter, minimaler bis mäßiger Entzündung der mononukleären Zellen und neuronaler Degeneration in den Dorsalwurzelganglien (DRG) und im Ganglion trigeminale (TG) sowie zu axonaler Degeneration und/oder Gliose im Rückenmark. Nach 12 Monaten hatten sich diese nicht-progredienten Befunde teilweise bis vollständig zurückgebildet. Für diese Befunde bei nicht-menschlichen Primaten gab es keine entsprechenden klinischen Beobachtungen, daher ist die klinische Relevanz beim Menschen nicht bekannt. Leberbefunde bei juvenilen nicht-menschlichen Primaten, einschließlich erhöhter Aminotransferasewerte und Einzelzellnekrose von Hepatozyten, hatten sich nach 12 Monaten vollständig zurückgebildet. Eine Dosis ohne beobachtbare schädliche Wirkung (no observed adverse effect level, NOAEL) konnte aufgrund der bereits bei der niedrigsten Dosis auftretenden Entzündung und Degeneration in ZNS/DRG/TG nicht ermittelt werden.
Genotoxizität und Karzinogenität
Studien zur Genotoxizität und Karzinogenität wurden mit Onasemnogen abeparvovec nicht durchgeführt.
Reproduktions- und Entwicklungstoxizität
In Studien zur Fertilität und frühen embryonalen Entwicklung (FEED) an Mäusen wurden nach intravenöser Verabreichung von Onasemnogen abeparvovec in Dosen von 1,1 × 1013 vg/kg oder 1,1 × 1014 vg/kg keine unerwünschten Wirkungen auf die Fertilität von männlichen oder weiblichen Tieren festgestellt.
In einer Studie zur embryofetalen Entwicklung (EFD) an Mäusen erhielten trächtige Tiere eine intravenöse Dosis Onasemnogen abeparvovec von entweder 1,1 × 1013 vg/kg oder 1,1 × 1014 vg/kg am 6. Trächtigkeitstag. Es gab keine Hinweise auf maternale Toxizität, embryofetale Toxizität, Teratogenität oder verminderte Lebensfähigkeit. Trotz des Vorhandenseins von Vektor-DNA in Plazenta, Ovarien und Uterus konnte in keinem fetalen Gewebe am 18. Trächtigkeitstag Onasemnogen-abeparvovec-DNA nachgewiesen werden.
Die NOAEL für die EFD-Studie und die FEED-Studien liegt bei 1,1 × 1014 vg/kg, was mindestens dem 7‑Fachen der empfohlenen klinischen intrathekalen Dosis, basierend auf dem Körpergewicht, entspricht.
Tromethamin
Magnesiumchlorid
Natriumchlorid
Poloxamer 188
Salzsäure (zur pH‑Wert-Einstellung)
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Der Kontakt des Arzneimittels mit Medizinprodukten aus Polyvinylchlorid (PVC), Bisphenol-A (BPA), Bis(2-ethylhexyl)phthalat (DEHP) oder Latex muss vermieden werden.
2 Jahre
Nach dem Auftauen
Nach dem Auftauen darf das Arzneimittel nicht wieder eingefroren werden.
Es kann 14 Tage lang bei 2 °C bis 8 °C gekühlt in Originalverpackung gelagert werden. Bevor das Produkt im Kühlschrank gelagert wird, ist das Empfangsdatum auf der Originalverpackung zu vermerken.
Nach dem Aufziehen der Dosis in die Spritze kann sie bis zu 24 Stunden bei 2 °C bis 8 °C aufbewahrt werden. Innerhalb dieses 24‑Stunden-Zeitraums ist ein maximaler Zeitraum von 5 Stunden ohne Kühlung zulässig. Wenn die Spritze mit dem Vektor nicht innerhalb dieses Zeitraums verwendet wird, muss sie entsorgt werden.
Tiefgekühlt lagern und transportieren (≤ -60 °C).
Sofort nach Erhalt im Kühlschrank lagern (2 °C–8 °C).
In der Originalverpackung aufbewahren.
Aufbewahrungsbedingungen nach Auftauen des Arzneimittels, siehe Abschnitt 6.3.
Itvisma ist in einer durchsichtigen Einzeldosis-Durchstechflasche (5 ml, Cyclo-Olefin-Polymer) mit Stopfen (20 mm Chlorbutylkautschuk) und Dichtung (Aluminium, Flip-off) mit farbiger Verschlusskappe (Kunststoff) erhältlich. Jede Durchstechflasche hat ein Füllvolumen von 3 ml.
Jeder Umkarton enthält 1 Durchstechflasche.
Auftauen
Itvisma im Kühlschrank (2 °C bis 8 °C) für ca. 4 Stunden oder bei Raumtemperatur (20 °C bis 25 °C) für ca. 1 Stunde auftauen.
Vor der intrathekalen Injektion sollte Itvisma auf Raumtemperatur gebracht werden.
Itvisma ist eine klare bis leicht opake, farblose bis leicht weiße Lösung. Sie dürfen dieses Arzneimittel nicht verwenden, wenn Sie nach dem Auftauen des gefrorenen Produkts und vor der Anwendung Partikel, Trübungen oder Verfärbungen feststellen.
Nicht schütteln.
Nach dem Auftauen das Arzneimittel nicht wieder einfrieren.
Nach dem Auftauen sollte Itvisma so schnell wie möglich verabreicht werden. Nach dem Aufziehen des Dosisvolumens in die Spritze, kann sie bis zu 24 Stunden im Kühlschrank (2 °C bis 8 °C) aufbewahrt werden, wobei ein maximaler Zeitraum von 5 Stunden ohne Kühlung innerhalb des 24‑Stunden-Zeitraums zulässig ist. Wenn die Spritze mit dem Vektor nicht innerhalb dieses Zeitraums verwendet wird, muss sie entsorgt werden.
Vorsichtsmaßnahmen vor der Handhabung bzw. vor der Anwendung des Arzneimittels
Die Handhabung von Itvisma muss aseptisch unter sterilen Bedingungen erfolgen.
Dieses Arzneimittel enthält genetisch veränderte Organismen (GVO).
Bei der Handhabung oder Verabreichung von Itvisma sollte persönliche Schutzausrüstung (einschließlich Handschuhe, Schutzbrille, Laborkittel und Ärmelschoner) getragen werden.
Die Durchstechflasche muss vor Gebrauch aufgetaut werden. Itvisma ist nur im aufgetauten Zustand zu verwenden.
Unmittelbar vor der Verabreichung den Inhalt aus der Durchstechflasche in die Spritze aufziehen, Luft aus der Spritze entfernen, das Dosisvolumen von 3 ml in der Spritze bestätigen, die Spritze verschließen und an den Ort der Injektion des Patienten bringen.
Bei der Zusammensetzung der Injektion muss sichergestellt werden, dass die mit der Itvisma-Lösung in Kontakt kommenden Oberflächen der Komponenten aus den in Tabelle 4 aufgeführten kompatiblen Materialien bestehen. Die verwendeten Komponenten müssen für die intrathekale oder neuroaxiale Anwendung indiziert sein.
Tabelle 4 Mit Itvisma kompatibles Komponentenmaterial
Komponente |
Konstruktionsmaterial |
Kanüle 18 G bis 19 G zum Aufziehen, maximal 40 mm lang |
Edelstahl |
5‑ml- bis 10‑ml-Spritzea |
Polypropylen |
Spritzenkappea |
Polypropylen, Polyethylen oder Methacrylat-Acrylnitril-Butadien-Styrol |
Spinalkanüle 22 G bis 27 G, maximal 156 mm lang |
Edelstahl |
a Darf nicht aus Polyvinylchlorid (PVC), Bisphenol-A (BPA), Bis(2‑ethylhexyl)phthalat (DEHP) oder Latex bestehen | |
Vorsichtsmaßnahmen bei der Entsorgung und versehentlichen Exposition gegenüber dem Arzneimittel
Verschüttetes Itvisma muss mit einem saugfähigen Gaze-Pad aufgewischt werden und der betroffene Bereich muss mit einer Bleichlösung und anschließend mit Alkoholtüchern desinfiziert werden. Alle Reinigungsmaterialien müssen doppelt verpackt und gemäß den nationalen Anforderungen zum Umgang mit biologischen Abfällen entsorgt werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zum Umgang mit biologischen Abfällen zu beseitigen.
Alle Materialien, die mit Itvisma in Berührung gekommen sein könnten (z. B. Durchstechflasche, alle für die Injektion verwendeten Materialien, einschließlich steriler Abdecktücher und Nadeln), müssen in Übereinstimmung mit den nationalen Anforderungen zum Umgang mit biologischen Abfällen entsorgt werden.
Ein versehentlicher Kontakt mit Itvisma muss vermieden werden. Bei versehentlicher Hautexposition muss der betroffene Bereich mindestens 15 Minuten gründlich mit Wasser und Seife gereinigt werden. Bei versehentlichem Kontakt mit den Augen muss der betroffene Bereich mindestens 15 Minuten gründlich mit Wasser gespült werden.
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Irland
EU/1/26/2038/001
30. Juni 2026
Juni 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur
https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig
Novartis Pharma GmbH
Sophie-Germain-Straße 10
90443 Nürnberg
Telefon: (09 11) 273-0
Medizinischer InfoService
Telefon: (09 11) 273-12 100
Telefax: (09 11) 273-12 160
E-Mail: infoservice.novartis@novartis.com

V001