
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Redemplo 25 mg Injektionslösung in einer Fertigspritze
Jede Einzeldosis-Fertigspritze enthält Plozasiran-Natrium entsprechend 25 mg Plozasiran in 0,5 mL Lösung.
Jeder Milliliter (mL) Lösung enthält 50 mg Plozasiran.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung (Injektion)
Klare, farblose bis gelbe Lösung mit einem pH-Wert von etwa 4,7–5,6 und einer Osmolalität von 320‑380 mOsm/kg.
Redemplo wird angewendet als Ergänzung zu Ernährungsmaßnahmen zur Senkung des Triglyceridspiegels bei erwachsenen Patienten mit familiärem Chylomikronämie-Syndrom (FCS) (siehe Abschnitt 4.2 bezüglich der Patientenauswahlkriterien).
Die Behandlung sollte von einem in der Behandlung von Patienten mit FCS erfahrenen Arzt eingeleitet und überwacht werden.
Patientenauswahl
Wenn die Anwendung von Redemplo in Betracht gezogen wird, muss die FCS-Diagnose des betroffenen Patienten entweder durch genetische Tests oder durch das Vorhandensein der folgenden klinischen Kriterien gesichert sein: Triglycerid(TG)-Nüchternwerte ≥ 10 mmol/L (≥ 880 mg/dL), die auf eine lipidsenkende Standardtherapie nicht ansprechen, sowie mindestens eines der folgenden: akute Pankreatitis in der Vorgeschichte, die nicht durch Alkohol oder Cholelithiasis verursacht wurde, wiederholte Krankenhausaufenthalte wegen starker Abdominalschmerzen ohne andere erklärbare Ursache in der Vorgeschichte, Pankreatitis im Kindesalter in der Vorgeschichte oder durch Hypertriglyceridämie verursachte Pankreatitis in der familiären Vorgeschichte.
Dosierung
Die empfohlene Plozasiran-Dosis beträgt 25 mg als einmalige subkutane Injektion alle 3 Monate.
Versäumte Dosis
Wenn eine Dosis versäumt wurde, sollte Plozasiran so schnell wie möglich verabreicht werden. Danach sollte die Dosierung alle 3 Monate ab der letzten verabreichten Dosis fortgesetzt werden.
Ältere Patienten
Bei älteren Patienten ≥ 65 Jahren ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Nierenfunktionsbeeinträchtigung
Bei Patienten mit leichter (geschätzte glomeruläre Filtrationsrate (eGFR) ≥ 60 bis ˂ 90 mL/min) oder mittelschwerer (eGFR ≥ 30 bis ˂ 60 mL/min) Nierenfunktionsbeeinträchtigung ist keine Dosisanpassung erforderlich. Plozasiran wurde bei Patienten mit schwerer Nierenfunktionsbeeinträchtigung oder terminaler Niereninsuffizienz (eGFR ˂ 30 mL/min) nicht untersucht und darf bei diesen Patienten nur angewendet werden, wenn der erwartete klinische Nutzen das potenzielle Risiko überwiegt (siehe Abschnitt 5.2).
Leberfunktionsbeeinträchtigung
Bei Patienten mit einer Erhöhung der Aspartataminotransferase (AST) über dem oberen Normwert (> ULN) und einem Gesamtbilirubin ≤ ULN oder einem Gesamtbilirubin > 1,0 bis 1,5 × ULN und einem beliebigen AST-Wert ist keine Dosisanpassung erforderlich. Plozasiran wurde bei Patienten mit mittelschwerer oder schwerer Leberfunktionsbeeinträchtigung nicht untersucht und darf bei diesen Patienten nur angewendet werden, wenn der erwartete klinische Nutzen das potenzielle Risiko überwiegt (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit dieses Arzneimittels bei Kindern und Jugendlichen im Alter von < 18 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Dieses Arzneimittel ist nur zur subkutanen Anwendung bestimmt. Es darf nicht intramuskulär oder intravenös angewendet werden.
Jede Fertigspritze ist nur zur einmaligen Anwendung bestimmt.
Die erste Injektion durch den Patienten selbst oder eine Pflegeperson ist unter Anleitung einer entsprechend qualifizierten medizinischen Fachperson zu erfolgen.
Als Injektionsstellen geeignet sind der Oberarm (bei Verabreichung durch eine Pflegeperson), Oberschenkel und Bauch (außer in einem Radius von 5 cm um den Nabel). Dieses Arzneimittel darf nicht in Bereiche injiziert werden, in denen die Haut empfindlich, gerötet, verhärtet oder verletzt ist oder Blutergüsse, Narben oder Dehnungsstreifen aufweist. Dieses Arzneimittel darf nicht in denselben Bereich injiziert werden, in den andere Arzneimittel injiziert werden.
Hinweise zur Handhabung des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
Detaillierte Anwendungshinweise sind am Ende der Packungsbeilage angegeben.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Hyperglykämie
Daten deuten darauf hin, dass Plozasiran bei einigen Patienten den Blutglukosewert erhöhen kann. In den placebokontrollierten Studien (siehe Abschnitt 4.8) traten Hyperglykämien unter Plozasiran bei mehr Patienten als unter Placebo auf. Bei einigen Patienten mit Diabetes oder einem erhöhten Diabetesrisiko kann eine Hyperglykämie auftreten, die eine diabetesspezifische Behandlung erfordert. Diese Patienten sollten gemäß den nationalen Leitlinien klinisch und biochemisch überwacht werden.
Natriumgehalt
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d. h. es ist nahezu „natriumfrei“.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Schwangerschaft
Es liegen keine Daten aus der Anwendung von Plozasiran bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Abschnitt 5.3).
Als Vorsichtsmaßnahme sollte die Anwendung von Plozasiran während der Schwangerschaft vorzugsweise vermieden werden.
Stillzeit
Es ist nicht bekannt, ob Plozasiran/Metaboliten in die Muttermilch ausgeschieden werden. Es liegen keine Informationen darüber vor, ob Plozasiran/Metaboliten beim Tier in die Milch ausgeschieden werden. Ein Risiko für das Neugeborene/den Säugling kann nicht ausgeschlossen werden.
Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Plozasiran verzichtet werden soll/die Behandlung mit Redemplo zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Es liegen keine klinischen Daten zur Auswirkung dieses Arzneimittels auf die menschliche Fertilität vor. Plozasiran hatte keinen Einfluss auf die Fertilität bei Ratten. Die bei Affen und Ratten gewonnenen Daten weisen insgesamt darauf hin, dass die klinische Relevanz der bei einer Untergruppe der männlichen Affen festgestellten geringeren Gewichte der Fortpflanzungsorgane unwahrscheinlich ist und dass das Risiko einer Beeinträchtigung der männlichen Fertilität und der Entwicklung der Fortpflanzungsorgane beim Menschen gering ist (siehe Abschnitt 5.3).
Plozasiran hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die häufigsten Nebenwirkungen sind Hyperglykämie (12,8 %), Kopfschmerzen (6,8 %), Übelkeit (4,7 %) und Reaktionen an der Injektionsstelle (4,7 %).
Unerwünschte Ereignisse, die zum Abbruch der Behandlung führten, waren Hyperglykämie (0,7 %) und Urtikaria (0,7 %).
Tabellarische Auflistung der Nebenwirkungen
Tabelle 1 zeigt die Nebenwirkungen, die bei Patienten unter Behandlung mit 25 mg Plozasiran in drei placebokontrollierten klinischen Studien (zwei Phase-II-Studien mit Patienten mit schwerer und mittelschwerer Hypertriglyceridämie sowie eine Phase-III-Studie mit Patienten mit FCS) berichtet wurden.
Die Nebenwirkungen sind nach MedDRA-Systemorganklasse und Häufigkeit aufgeführt. Die Häufigkeitskategorien sind wie folgt definiert: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 1. Nebenwirkungen
Systemorganklasse |
Nebenwirkung |
Häufigkeit |
Stoffwechsel- und Ernährungsstörungen |
Hyperglykämiea |
Sehr häufig |
Erkrankungen des Nervensystems |
Kopfschmerzen |
Häufig |
Erkrankungen des Gastrointestinaltrakts |
Übelkeit |
Häufig |
Leber und Gallenerkrankungen |
Lebererkrankung (ALT erhöht, AST erhöht) |
Gelegentlich |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Reaktion an der Injektionsstellea |
Häufig |
ALT = Alaninaminotransferase, AST = Aspartataminotransferase |
||
Beschreibung ausgewählter Nebenwirkungen
Hyperglykämie
Hyperglykämie trat in den placebokontrollierten Studien bei 12,8 % der mit Plozasiran behandelten Patienten und bei 9,8 % der Patienten in der Placebogruppe auf. Der Anteil der Patienten in jeder Gruppe, welche die Behandlung aufgrund von Hyperglykämie abbrachen, betrug 1,4 % bei den mit Plozasiran behandelten Patienten und 0 % bei den Patienten in der Placebogruppe. Zu den Hyperglykämie-Ereignissen bei mit Plozasiran behandelten Patienten zählten Glukose im Blut erhöht (1,4 %), Diabetes mellitus (1,4 %), glykosyliertes Hämoglobin erhöht (4,1 %), Hyperglykämie (1,4 %) und Diabetes mellitus Typ 2 (5,4 %) (siehe Abschnitt 4.4).
Reaktion an der Injektionsstelle
Reaktionen an der Injektionsstelle traten in den placebokontrollierten Studien bei 4,7 % der mit Plozasiran behandelten Patienten und bei 1,2 % der Patienten in der Placebogruppe auf. Alle diese Nebenwirkungen waren leicht. Kein Patient brach die Behandlung ab oder erforderte aufgrund von Reaktionen an der Injektionsstelle Änderungen oder zeitliche Verschiebungen der Dosierung. Zu den Reaktionen an der Injektionsstelle bei mit Plozasiran behandelten Patienten gehörten Erythem (0,7 %), Schmerzen an der Injektionsstelle (2,7 %) und Reaktionen an der Injektionsstelle (1,4 %). Die Häufigkeit von Reaktionen an der Injektionsstelle war nach der ersten Dosis am höchsten und nahm bei den folgenden Dosen ab.
Laboruntersuchungen
Erhöhte Lebertransaminasen
In klinischen Studien der Phase II und III traten bei Patienten unter Behandlung mit Plozasiran häufiger Erhöhungen der Lebertransaminasen im Serum > ULN auf als in der Placebogruppe. Asymptomatische, vorübergehende Erhöhungen der ALT- und AST-Werte > 3 × ULN traten bei 1,5 % bzw. 0,7 % der mit Plozasiran behandelten Teilnehmer auf. Diese Erhöhungen überschritten den Grenzwert von > 5 × ULN nicht und erforderten weder eine Dosisanpassung noch einen Behandlungsabbruch.
LDL-C-Werte
Die Behandlung mit Plozasiran kann zu einem Anstieg des Low-Density-Lipoprotein-Cholesterins (LDL-C) führen. In klinischen Studien stieg der mediane LDL-C-Wert von etwa 0,55 mmol/L zur Baseline auf 1,0–1,1 mmol/L nach 10 Monaten, wobei sich die Werte danach im Allgemeinen stabilisierten.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
In Phase-I-Studien wurden Dosen von bis zu 100 mg Plozasiran (das 4-Fache der empfohlenen Dosis) angewendet, ohne dass Sicherheitsbedenken aufkamen. Es gibt keine spezifische Behandlung bei einer Plozasiran-Überdosierung. Im Falle einer Überdosierung muss der Patient symptomatisch behandelt werden, und bei Bedarf sind unterstützende Maßnahmen einzuleiten.
Pharmakotherapeutische Gruppe: lipidmodifizierende Mittel, andere lipidmodifizierende Mittel, ATC-Code: noch nicht zugewiesen.
Wirkmechanismus
Plozasiran ist eine kleine interferierende RNA (small interfering RNA bzw. siRNA, doppelsträngiges Oligonukleotid), die mit N-Acetylgalactosamin konjugiert ist, um den Transport zu und die Aufnahme durch Hepatozyten zu erleichtern. In Hepatozyten baut Plozasiran die mRNA für Apolipoprotein C3 (APOC3) selektiv durch den RNA-Interferenzmechanismus ab, was zu einem reduzierten APOC3-Proteinspiegel in Leber und Serum führt. Dadurch wiederum erhöht sich die Aktivität der Lipoproteinlipase und die Aufnahme TG-reicher Lipoproteinreste durch Hepatozyten, was zu einer Senkung der TG-Werte im Serum führt.
Pharmakodynamische Wirkungen
In der Studie PALISADE senkte die Verabreichung von 25 mg Plozasiran alle 3 Monate bei Patienten mit FCS die Werte für APOC3, TG, Non-High-Density-Cholesterin (Non-HDL-C) und Very-Low-Density-Lipoprotein-Cholesterin (VLDL-C) (siehe auch nachstehend unter „Klinische Wirksamkeit“) und erhöhte das HDL-C und das LDL-C. Das LDL-C blieb bei den meisten Patienten im Normbereich. Die mediane Reduktion der Nüchternwerte des APOC3-Proteins und der Triglyceride im Serum in Monat 1 betrug 95 % bzw. 85 %, was darauf hindeutet, dass nach der ersten Dosis ein pharmakodynamischer Steady-State erreicht wird.
Kardiale Elektrophysiologie
Plozasiran in einer Dosis von 100 mg (dem 4-Fachen der empfohlenen Dosis) hatte keine Verlängerung des QT-Intervalls in klinisch relevantem Ausmaß zur Folge.
Klinische Wirksamkeit
Studie PALISADE bei Patienten mit FCS
PALISADE ist eine randomisierte, doppelblinde, placebokontrollierte klinische Studie mit 75 erwachsenen Patienten mit FCS, die eine fettarme Ernährung einhielten. Patienten ≥ 18 Jahren erhielten 4 einzelne subkutane Injektionen von 25 mg Plozasiran (N = 23), 50 mg Plozasiran (N = 22) oder Placebo (N = 19) im Abstand von 3 Monaten. An der Studie nahmen Patienten mit der Diagnose FCS und einem TG-Nüchternwert ≥ 10 mmol/L (≥ 880 mg/dL) teil, die auf eine lipidsenkende Standardtherapie nicht ansprachen.
Eine FCS-Diagnose war definiert als Patienten mit einem TG-Nüchternwert > 11,3 mmol/L (˃ 1 000 mg/dL) in der Vorgeschichte und entweder:
Einem bestätigenden Gentest (N = 41 [54,7 %]) oder nachgewiesener niedriger Lipoproteinlipase(LPL)-Aktivität; oder
Klinisch diagnostiziertem FCS (N = 34 [45,3 %]) mit entweder einer akuten Pankreatitis in der Vorgeschichte, die nicht durch Alkohol oder Cholelithiasis verursacht wurde, wiederholten Krankenhausaufenthalten wegen starker Abdominalschmerzen ohne andere erklärbare Ursache in der Vorgeschichte, Pankreatitis im Kindesalter in der Vorgeschichte oder durch Hypertriglyceridämie verursachte Pankreatitis in der familiären Vorgeschichte.
Das mittlere Alter betrug 46 Jahre, wobei in der mit 50 mg Plozasiran behandelten Gruppe mehr Patienten im Alter von < 50 Jahre waren (83,3 %) als in der mit 25 mg Plozasiran behandelten Gruppe oder in der Placebo-Gruppe (57,7 % bzw. 56,0 %). Die Zahl der Patienten ≥ 65 Jahre betrug 9 (12 %) und die der Patienten ≥ 75 Jahre betrug 2 (3 %). Ungefähr die Hälfte der Patienten in jeder Behandlungsgruppe war männlich. Die meisten Patienten waren weiß (73,3 %) oder asiatisch (21,3 %). Der mittlere Body-Mass-Index (BMI) betrug 25,5 kg/m2; 53,3 % der Teilnehmenden waren übergewichtig (BMI ≥ 25 kg/m2). Die Zahl der Patienten mit genetisch bestätigtem FCS betrug 41 und bei 34 Patienten lag keine genetische Bestätigung des FCS vor. Die Patienten, die Plozasiran erhielten, repräsentierten fünf Varianten: APOA5 – 2,3 %, APOC2 – 2,3 %, GPIHBP1 – 9,1 %, LMF1 – 6,8 %, LPL – 81,8 %. Bei insgesamt 89,3 % der Patienten war bereits eine Pankreatitis-Episode aufgetreten. Die Anteile der Patienten unter TG-senkender Therapie zur Baseline waren wie folgt: 66,7 % erhielten Fibrate, 29,3 % Icosapentethyl, Omega-3-Fettsäure oder Fischöl und 45,3 % Statine.
Die meisten Patienten erhielten alle 4 planmäßigen Dosen; 24 Patienten (92,3 %) in der mit 25 mg Plozasiran behandelten Gruppe, 22 Patienten (91,7 %) in der mit 50 mg Plozasiran behandelten Gruppe und 19 Patienten (76,0 %) in der Placebogruppe.
Der primäre Wirksamkeitsendpunkt war die mediane prozentuale Veränderung der Nüchtern-TGs gegenüber Baseline nach 10 Monaten. In Monat 10 war bei Anwendung von Plozasiran in der empfohlenen Dosis von 25 mg der mediane TG-Nüchternwert statistisch signifikant verringert (siehe Tabelle 2). Die TG-senkende Wirkung von 50 mg Plozasiran hatte keinen therapeutischen Nutzen gegenüber der empfohlenen Dosis von 25 mg.
In der Studie PALISADE führte die Verabreichung von 25 mg Plozasiran alle 3 Monate bei Patienten mit FCS zu einer signifikanten Senkung des medianen Nüchternspiegels von APOC3-Protein im Serum um 93 % (p < 0,0001).
Die bei den mit Plozasiran behandelten Patienten beobachteten Senkungen der TG-Werte waren bereits in Monat 1 (erste Messung nach Baseline) zu beobachten und blieben während der zwölfmonatigen Behandlungsdauer in der Studie PALISADE mit relativ geringen Schwankungen zwischen Spitzen- und Talspiegel konstant (siehe Abbildung 1). Die während des Behandlungszeitraums zu mehreren Zeitpunkten erreichten mittleren TG-Werte lagen unter dem anerkannten Grenzwert von 5,7 mmol/L (500 mg/dL) für ein erhöhtes Risiko einer akuten Pankreatitis (siehe Abbildung 1).
Tabelle 2: Mediane Unterschiede der prozentualen Veränderung der TG-Nüchternwerte und von APOC3 gegenüber Baseline bei Patienten mit FCS in Monat 10 in der Studie PALISADE
Behandlungsgruppe |
Placebo |
Plozasiran |
TG zur Baseline (mmol/L) | ||
N |
25 |
26 |
Median |
23,2 |
22,7 |
TG in Monat 10 (mmol/L) | ||
N |
19 |
24 |
Median |
18.2 |
5,0 |
Mediane prozentuale Veränderung des TG-Nüchternwerts in Monat 10 gegenüber Baseline |
-17 |
-80 |
Differenz zu Placebo |
-58,7 |
|
95%-KI |
-89,6; -27,9 |
|
p-Wert |
p < 0,0001 |
|
Mediane prozentuale Veränderung des APOC3-Nüchternwerts in Monat 10 gegenüber Baseline |
-1,3 |
-93,0 |
Differenz zu Placebo |
-90,5 |
|
95%-KI |
-108,3; -72,7 |
|
p-Wert |
p < 0,0001 |
|
APOC3 = Apolipoprotein C3; KI = Konfidenzintervall; FCS = familiäres Chylomikronämie-Syndrom; TG = Triglycerid.
Abbildung 1: Mediane absolute Triglycerid-Nüchternwerte bei Patienten mit FCS in der Studie PALISADE
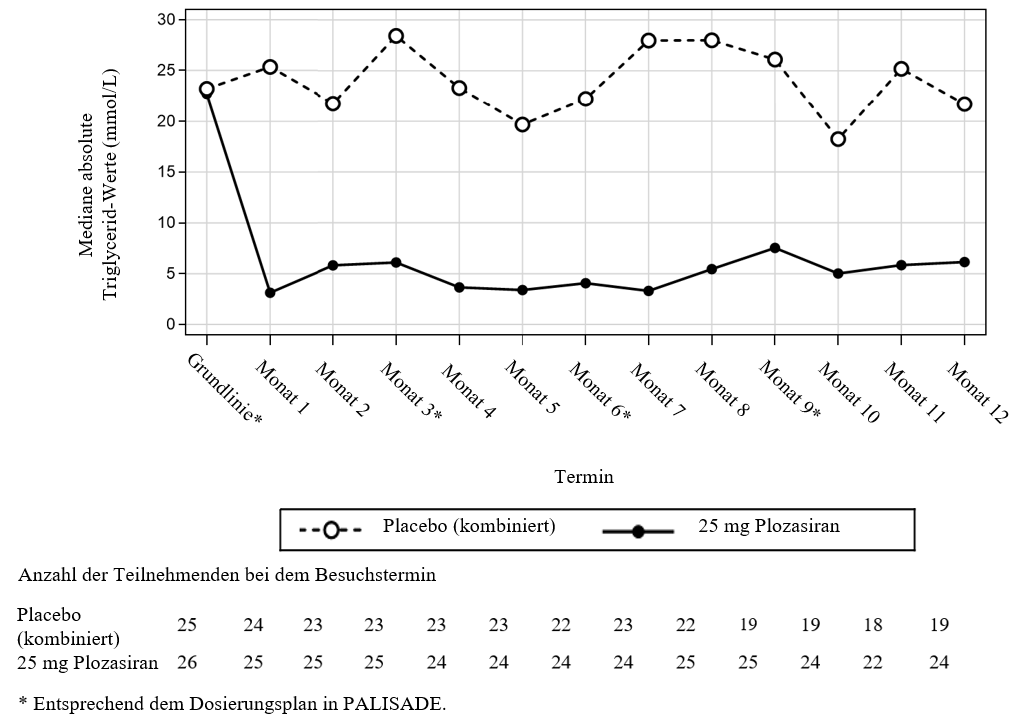
Eine vorab festgelegte Teilgruppenanalyse von Patienten mit genetisch bestätigtem versus klinisch diagnostiziertem FCS ergab, dass das TG-Ansprechen auf Plozasiran bei den Patienten, unabhängig von ihren bestätigten genetischen Merkmalen, ähnlich war.
Bei den Patienten mit Messung des TG-Nüchternwerts in Monat 10 war bei allen Patienten in der mit 25 mg Plozasiran behandelten Gruppe eine Reduzierung gegenüber Baseline und bei etwa 80 % der Patienten eine Reduzierung um mindestens > 50 % gegenüber Baseline zu verzeichnen. Darüber hinaus bewirkten die kombinierten Dosen von 25 mg und 50 mg Plozasiran im Vergleich zu Placebo eine signifikante Reduzierung der Inzidenz einer akuten Pankreatitis (Odds Ratio 0,169; p = 0,0292). Die Wahrscheinlichkeit einer akuten Pankreatitis war in den kombinierten Plozasiran-Gruppen im Vergleich zur Placebogruppe um 83 % niedriger, wobei 7 Pankreatitis-Ereignisse bei 5 Teilnehmenden (20 %) in der Placebogruppe auftraten und 2 Pankreatitis-Ereignisse bei 2 Teilnehmenden (4 %) in den kombinierten Plozasiran-Gruppen.
Offene Verlängerungsstudie (OLE) der Studie PALISADE bei Patienten mit FCS
Von den 64 Patienten, welche die 12-monatige randomisierte Studienbehandlung abgeschlossen hatten, wurden 62 (97 %) in den OLE-Zeitraum aufgenommen. Von diesen Patienten erhielten 18 (29 %) Placebo (Placebo-/Plozasiran-Gruppe) und 44 (71 %) Plozasiran (Plozasiran-/Plozasiran-Gruppe) während des randomisierten Zeitraums.
Erwartungsgemäß waren die medianen absoluten Triglycerid-Nüchternwerte bei OLE-Baseline (Monat 12) bei Patienten, die im randomisierten Zeitraum Placebo erhalten hatten (Placebo-/Plozasiran-Gruppe, 23,76 mmol/L [2 103 mg/dL]), höher als in der Plozasiran-/Plozasiran-Gruppe (6,31 mmol/L [558 mg/dL]). Es ist zu beachten, dass bei Patienten in der Placebo-/Plozasiran-Gruppe die medianen TG-Werte nach dem ersten Monat der Plozasiran-Behandlung bereits auf ein ähnliches Niveau wie in der Plozasiran-/Plozasiran-Gruppe gefallen waren (Monat 13; 3,67 mmol/L [325 mg/dL; -87,96 %] und 6,0 mmol/L [531 mg/dL; -75,23 %] in der Placebo-/Plozasiran- bzw. Plozasiran-/Plozasiran-Gruppe). Unter Berücksichtigung der erwarteten Variabilität der TG-Nüchternwerte und der Talwert-Messungen blieben diese Reduktionen bis Monat 18 des OLE-Zeitraums bestehen.
Immunogenität
In der PALISADE-Studie entwickelte keiner der 50 Patienten mit FCS, die über einen Zeitraum von 12 Monaten mit Plozasiran behandelt wurden, durch die Behandlung induzierte oder durch die Behandlung vermehrte Anti-Drug-Antikörper (ADA). Es gab keine Hinweise darauf, dass sich die Pharmakodynamik oder Wirksamkeit von Plozasiran im Laufe der Zeit nach mehrmaliger Gabe von Plozasiran veränderte. Bei den mit Plozasiran behandelten Patienten wurden keine Nebenwirkungen im Zusammenhang mit einer systemischen Immunreaktion festgestellt.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Plozasiran eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in der Behandlung des familiären Chylomikronämie-Syndroms gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Resorption
Nach einmaliger subkutaner Injektion von 25 mg Plozasiran betrug die maximale Plasmakonzentration (Cmax) 68,5 ng/mL. Die mediane Zeit bis zum Erreichen der Cmax (Tmax) betrug 6 Stunden.
Plozasiran wurde in den klinischen Studien nicht intravenös verabreicht, daher liegen keine Daten zur absoluten Bioverfügbarkeit beim Menschen vor. Nach subkutaner Verabreichung an Cynomolgus-Affen wurde die absolute Bioverfügbarkeit von Plozasiran auf 40 % geschätzt.
Verteilung
Nach mehrmaliger subkutaner Injektion von 25 mg Plozasiran wird das Arzneimittel in der terminalen Phase der Elimination mit einem scheinbaren Verteilungsvolumen (Vz/F) von 146 L im Plasma und extrazellulären Körperwasser verteilt. Im systemischen Kreislauf wird Plozasiran hauptsächlich in die Leber verteilt. Im Plasma hat Plozasiran einen ungebundenen Anteil von 22 %.
In-vitro-Studien lassen den Schluss zu, dass Plozasiran kein Substrat, Inhibitor oder Induktor von Transportern ist. Daher ist nicht zu erwarten, dass Wechselwirkungen zwischen Plozasiran und Transportern auftreten, die sich auf das Arzneimittel auswirken.
Biotransformation
Plozasiran wird hauptsächlich durch Nukleasen in der Leber zu kürzeren Oligonukleotiden unterschiedlicher Länge metabolisiert. In-vitro-Studien lassen den Schluss zu, dass Plozasiran kein Substrat von Cytochrom-P450(CYP450)-Enzymen ist.
In-vitro-Studien lassen den Schluss zu, dass Plozasiran kein Substrat, Inhibitor oder Induktor von CYP450-Enzymen ist. Daher ist nicht zu erwarten, dass Wechselwirkungen zwischen Plozasiran und CYP450-Enzymen auftreten, die sich auf das Arzneimittel auswirken.
Elimination
Die terminale Eliminationshalbwertszeit von Plozasiran im Plasma beträgt ungefähr 3 – 4 Stunden. Die mittlere scheinbare systemische Clearance beträgt 33,8 L/h. Etwa 16–19 % der Plozasiran-Dosis werden im Urin ausgeschieden.
Linearität/Nicht-Linearität
Plozasiran wies nach mehrmaliger subkutaner Injektion eine zeitlich invariante Pharmakokinetik auf. Nach Gabe mehrerer Dosen stiegen die Plasmaspiegel von Plozasiran (Cmax, AUC0-t und AUC0‑inf) im Dosisbereich von 10–50 mg proportional zur Dosis an.
Pharmakokinetische/pharmakodynamische Zusammenhänge
Plozasiran ist in Hepatozyten aktiv und weist eine verlängerte pharmakodynamische Aktivität auf, die von seinem pharmakokinetischen Profil im Plasmakompartiment unabhängig ist. Die lange Wirkdauer liegt über der Plasmaeliminationshalbwertszeit von 3 – 4 Stunden. Das pharmakodynamische Ansprechen ist bei der empfohlenen Dosis von 25 mg Plozasiran alle 3 Monate wahrscheinlich gesättigt.
Immunogenität
In der PALISADE-Studie entwickelte keiner der 50 Patienten mit FCS, die über einen Zeitraum von 12 Monaten mit Plozasiran behandelt wurden, durch die Behandlung induzierte oder durch die Behandlung vermehrte Anti-Drug-Antikörper (ADA). Es gab keine Hinweise darauf, dass sich die Pharmakokinetik von Plozasiran im Laufe der Zeit nach mehrmaliger Gabe von Plozasiran veränderte.
Besondere Patientengruppen
Ältere Patienten
In einer populationspharmakokinetischen Analyse mit Daten von gesunden erwachsenen Teilnehmenden und Patienten (N = 146); Alter 65–74 Jahre (N = 16); Alter 75–85 Jahre (N = 4), wurden keine klinisch signifikanten Unterschiede in der Pharmakokinetik von Plozasiran in Abhängigkeit vom Alter festgestellt (siehe Abschnitt 4.2).
Nierenfunktionsbeeinträchtigung
In einer populationspharmakokinetischen Analyse mit Daten von 23 Patienten mit leichter (eGFR ≥ 60 bis ˂ 90 mL/min) und 4 Patienten mit mittelschwerer (eGFR ≥ 30 bis ˂ 60 mL/min) Nierenfunktionsbeeinträchtigung wurden keine klinisch signifikanten Unterschiede in der Pharmakokinetik von Plozasiran festgestellt. Plozasiran wurde nicht bei Patienten mit schwerer Nierenfunktionsbeeinträchtigung oder terminaler Niereninsuffizienz (eGFR ≥ 30 mL/min) untersucht (siehe Abschnitt 4.2).
Leberfunktionsbeeinträchtigung
In einer populationspharmakokinetischen Analyse bei 4 Patienten mit einer AST-Erhöhung > ULN bei einem Gesamtbilirubin ≤ ULN oder einem Gesamtbilirubin-Wert > 1,0 bis 1,5 × ULN und jeglichem AST-Wert wurden keine klinisch signifikanten Unterschiede in der Pharmakokinetik von Plozasiran festgestellt. Plozasiran wurde nicht bei Patienten mit mittelschwerer oder schwerer Leberfunktionsbeeinträchtigung untersucht (siehe Abschnitt 4.2).
Körpergewicht, BMI
Die Plasmakonzentrationen von Plozasiran (Cmax und AUC) sind bei Patienten mit höherem Körpergewicht oder BMI in der Regel niedriger, ohne dass die Wirksamkeit der Behandlung reduziert ist. Daher wird keine Dosisanpassung bei schwereren Patienten empfohlen.
Geschlecht, Ethnie, ethnische Zugehörigkeit
In einer populationspharmakokinetischen Analyse, die Daten von 65 weiblichen (44,5 %) und 81 männlichen (55,5 %) Personen unterschiedlicher Ethnie (67,1 % Weiße, 11,0 % Schwarze, 9,6 % Asiaten, 2,1 % Ureinwohner Hawaiis oder der Pazifikinseln und 10,3 % multiethnisch oder nicht bekannt) umfasste, wurden keine klinisch signifikanten Unterschiede in der Plozasiran-Pharmakokinetik aufgrund von Geschlecht oder Ethnie bzw. ethnischer Zugehörigkeit festgestellt.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität, zum kanzerogenen Potenzial und zur Reproduktions- und Entwicklungstoxizität, lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
In einer Studie zur prä- und postnatalen Entwicklung wurde bei der hohen Dosis ein Anstieg der Zahl totgeborener Jungtiere und eine anschließende Verringerung des Lebendgeburtenindex festgestellt, wobei beim NOAEL (No-Observed-Adverse-Effect Level) vor der Entwöhnung und maternal/postnatal ein 3,1‑facher bzw. 31‑facher Sicherheitsabstand nach Anpassung auf die Körperoberfläche (BSA) bestand.
Es liegen keine Informationen darüber vor, ob Plozasiran oder seine Metaboliten beim Tier in die Milch ausgeschieden werden.
In einer 2-Jahres-Karzinogenitätsstudie an Ratten wurden bei der hohen Dosis benigne hepatozelluläre Adenome sowie in geringer Inzidenz Karzinome festgestellt. Die Sicherheitsabstände am NOAEL betragen das 10- bzw. 16-Fache, basierend auf der Körperoberfläche (BSA), und das 60- bzw. 53-Fache, basierend auf der AUC (area under the curve, Fläche unter der Kurve), für männliche bzw. weibliche Tiere. Obwohl die Relevanz für den Menschen nicht bekannt ist, ist das Risiko aufgrund der hohen Sicherheitsabstände wahrscheinlich niedrig.
Natriumchlorid
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
2 Jahre
Im Kühlschrank lagern (2 °C – 8 °C). Nicht einfrieren.
Kann das Arzneimittel einmalig für einen Zeitraum von bis zu 30 Tagen bei Raumtemperatur (15 °C – 25 °C) gelagert werden.
Das Entsorgungsdatum muss auf dem Umkarton vermerkt werden (d. h. bis zu 30 Tage nach dem Datum der Entnahme aus dem Kühlschrank).
Das Arzneimittel ist zu entsorgen, wenn es nicht innerhalb von 30 Tagen bei Aufbewahrung bei Raumtemperatur oder bis zum auf dem Umkarton aufgedruckten Verfallsdatum verwendet wird (je nachdem, welches Datum früher eintritt).
Einzeldosis-Fertigspritze aus Typ-I-Glas mit Brombutylstopfen und Nadel mit Nadelschutz. Jede Fertigspritze enthält 0,5 mL Injektionslösung.
Die Packungsgröße ist 1 Fertigspritze.
Das Arzneimittel sollte vor der Anwendung einer Sichtprüfung unterzogen werden. Die Lösung sollte klar und farblos bis gelb sein. Wenn die Lösung trüb ist oder sichtbare Partikel enthält, darf der Inhalt nicht injiziert werden und das Arzneimittel muss an die Apotheke zurückgegeben werden.
Die Fertigspritze sollte vor der Injektion Raumtemperatur (15 °C – 25 °C) annehmen. Sie sollte mindestens 30 Minuten vor der Anwendung aus dem Kühlschrank (2 °C – 8 °C) genommen werden. Es sollten keine anderen Erwärmungsmethoden (z. B. heißes Wasser oder Mikrowelle) verwendet werden.
Jede Fertigspritze darf nur einmal verwendet werden und muss anschließend gemäß den vor Ort geltenden Vorschriften in einem Kanülenabwurfbehälter entsorgt werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Arrowhead Pharmaceuticals Ireland Limited
One Spencer Dock
North Wall Quay
Dublin 1
D01 X9R7
Irland
EU/1/26/2041/001
Datum der Erteilung der Zulassung: 19. Juni 2026
Juni 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu. verfügbar.
Verschreibungspflichtig