
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Jascayd® 9 mg Filmtabletten
Jascayd® 18 mg Filmtabletten
Jascayd 9 mg Filmtabletten
Jede Filmtablette enthält 9 mg Nerandomilast.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 81,1 mg Lactose (als Monohydrat).
Jascayd 18 mg Filmtabletten
Jede Filmtablette enthält 18 mg Nerandomilast.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 162,2 mg Lactose (als Monohydrat).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette (Tablette)
Jascayd 9 mg Filmtabletten
Hellgelbe, ovale, bikonvexe Filmtablette mit der Prägung „F9“ auf der einen und dem Logo von Boehringer Ingelheim auf der anderen Seite (Tablettenlänge: 9,5 mm, Tablettenbreite: 4,6 mm)
Jascayd 18 mg Filmtabletten
Hellrote, ovale, bikonvexe Filmtablette mit der Prägung „F18“ auf der einen und dem Logo von Boehringer Ingelheim auf der anderen Seite (Tablettenlänge: 12 mm, Tablettenbreite: 5,9 mm)
Jascayd wird angewendet zur Behandlung von erwachsenen Patienten mit idiopathischer Lungenfibrose (Idiopathic Pulmonary Fibrosis, IPF).
Jascayd wird angewendet zur Behandlung von erwachsenen Patienten mit progredienter pulmonaler Fibrose (PPF).
Die Behandlung sollte von Ärzten eingeleitet werden, die in der Behandlung von Krankheiten, für die Jascayd zugelassen ist, erfahren sind.
Dosierung
Die empfohlene Dosis beträgt 18 mg zweimal täglich in einem ungefähren Abstand von 12 Stunden.
Abhängig von der individuellen Verträglichkeit des Patienten hinsichtlich der Nebenwirkungen Diarrhö oder Gewichtsverlust (siehe Abschnitt 4.4 und Abschnitt 4.8), und abhängig vom erwarteten Nutzen der Behandlung mit Nerandomilast (siehe Abschnitt 5.1), kann die Dosis auf 9 mg zweimal täglich reduziert werden (außer bei Patienten, die gleichzeitig Pirfenidon oder starke oder mäßige Induktoren von Cytochrom P450 3A (CYP3A) anwenden, siehe Abschnitt 4.5). Sobald die Nebenwirkung abgeklungen ist oder ausreichend unter Kontrolle gebracht wurde, kann die Behandlung mit der empfohlenen Dosis von zweimal täglich 18 mg fortgesetzt werden.
Gleichzeitige Anwendung mit starken CYP3A-Inhibitoren
Bei gleichzeitiger Anwendung mit starken CYP3A-Inhibitoren muss die Dosis auf 9 mg zweimal täglich reduziert werden (siehe Abschnitt 4.5).
Gleichzeitige Anwendung mit starken oder mäßigen CYP3A-Induktoren
Bei gleichzeitiger Anwendung mit starken oder mäßigen CYP3A-Induktoren beträgt die empfohlene Dosis 18 mg zweimal täglich und darf nicht auf 9 mg zweimal täglich reduziert werden (siehe Abschnitt 4.5).
Gleichzeitige Anwendung mit Pirfenidon
Die gleichzeitige Anwendung mit Pirfenidon reduzierte die Exposition gegenüber Nerandomilast um ca. 50 % (siehe Abschnitt 4.5). Bei Patienten, die Pirfenidon anwenden, beträgt die empfohlene Dosis 18 mg zweimal täglich; sie darf nicht auf 9 mg zweimal täglich reduziert werden.
Versäumte Dosis
Wird eine Dosis versäumt, ist die nächste Dosis zum nächsten planmäßigen Zeitpunkt einzunehmen.
Der Patient darf keine zusätzliche Dosis einnehmen. Die empfohlene maximale Dosis von 18 mg zweimal täglich darf nicht überschritten werden.
Besondere Patientengruppen
Ältere
Bei älteren Patienten ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Nierenfunktionsstörung
Bei Patienten mit leichter, mäßiger oder schwerer Niereninsuffizienz (eGFR ≥ 15 und < 90 ml/min, basierend auf der CKD‐EPI-Formel) ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Die Behandlung von Patienten mit terminaler Niereninsuffizienz (eGFR < 15 ml/min) wird nicht empfohlen. Die Pharmakokinetik, Sicherheit und Wirksamkeit von Nerandomilast wurden bei diesen Patienten nicht untersucht.
Leberfunktionsstörung
Bei Patienten mit leichter (Child-Pugh-Klasse A) oder mäßiger (Child-Pugh-Klasse B) Leberfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Die Behandlung von Patienten mit schwerer Leberfunktionsstörung (Child-Pugh-Klasse C) wird nicht empfohlen. Die Pharmakokinetik, Sicherheit und Wirksamkeit von Nerandomilast bei diesen Patienten wurden nicht untersucht.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit bei Kindern und Jugendlichen ist bisher noch nicht erwiesen.
Es liegen keine Daten vor.
Art der Anwendung
Jascayd ist zum Einnehmen. Die Tabletten sind unzerteilt mit Wasser zu schlucken.
Jascayd kann zu oder unabhängig von einer Mahlzeit eingenommen werden (siehe Abschnitt 5.2).
Alternative Art der Anwendung
Für Patienten, die Schwierigkeiten beim Schlucken der ganzen Tablette haben, kann die Tablette in einem Glas mit etwa 100 ml stillem Trinkwasser bei Raumtemperatur aufgelöst werden. Es dürfen keine anderen Flüssigkeiten verwendet werden. Die Tablette soll unzerkleinert in Wasser gegeben und etwa 15‑20 Minuten lang regelmäßig umgerührt werden, bis sie sich in sehr kleine Stücke aufgelöst hat (die Tablette löst sich nicht vollständig auf). Die entstandene Suspension soll innerhalb von 2 Stunden nach dem Anrühren getrunken werden. Wird die Suspension nicht sofort getrunken, muss sie vor dem Trinken erneut umgerührt werden. Das Glas sollte mit etwa 100 ml Wasser gespült werden; diese Flüssigkeit muss ebenfalls getrunken werden, um die vollständige Einnahme der Dosis sicherzustellen.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile
Diarrhö, Übelkeit und verminderter Appetit
In klinischen Studien wurden Diarrhö, Übelkeit und verminderter Appetit häufig als Nebenwirkungen berichtet (siehe Abschnitt 4.8), insbesondere bei Patienten, die Nerandomilast in Kombination mit Nintedanib oder Pirfenidon erhielten. Bei den meisten Patienten waren diese Nebenwirkungen von leichter bis mäßiger Ausprägung. Die Patienten sollten überwacht und Nebenwirkungen gegebenenfalls mit geeigneten symptomatischen Maßnahmen wie Flüssigkeitszufuhr sowie Antidiarrhoika oder Antiemetika behandelt werden. Eine Reduzierung der Hintergrundtherapie mit Nintedanib oder Pirfenidon sollte gemäß den Empfehlungen in den jeweiligen Fachinformationen erfolgen. Vorsicht ist geboten bei Patienten, die geschwächt sind, bereits gastrointestinale Symptome aufweisen oder zuvor unter Nintedanib oder Pirfenidon signifikante gastrointestinale Nebenwirkungen erlitten haben. Wenn Patienten mit schwerer oder anhaltender Diarrhö, Übelkeit oder vermindertem Appetit nicht auf eine symptomatische Behandlung, eine Dosisreduktion (siehe Abschnitt 4.2) oder eine Unterbrechung der Behandlung ansprechen, sollte die Behandlung mit Nerandomilast abgebrochen werden.
Gewichtsverlust
Die Behandlung mit Nerandomilast wurde mit einem dosisabhängigen Gewichtsverlust in Verbindung gebracht, insbesondere bei gleichzeitiger Anwendung von Nintedanib. Das Körpergewicht sollte während der Therapie regelmäßig kontrolliert werden. Wenn ein unerklärlicher Gewichtsverlust fortschreitet oder klinisch bedenklich wird, sollte die Behandlung neu bewertet und eine Dosisreduktion oder ein Abbruch der Behandlung in Betracht gezogen werden. Nerandomilast sollte bei Patienten, die untergewichtig sind oder an Erkrankungen leiden, bei denen ein weiterer Gewichtsverlust medizinisch unerwünscht sein könnte, mit Vorsicht angewendet werden (siehe Abschnitt 4.8).
Psychiatrische Erkrankungen
Psychiatrische Erkrankungen können bei Patienten mit IPF und PPF häufig als Komorbidität auftreten. In klinischen Studien mit Nerandomilast wurden über alle Behandlungsgruppen hinweg psychiatrische Ereignisse, darunter auch sehr seltene Fälle von Suizidgedanken und suizidalem Verhalten, berichtet. Es wird eine routinemäßige Überwachung auf psychiatrische Erkrankungen empfohlen.
Sonstige Bestandteile mit bekannter Wirkung
Lactose
Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht anwenden.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Filmtablette, d. h., es ist nahezu „natriumfrei“.
Basierend auf In-vitro-Bewertungen wird Nerandomilast hauptsächlich durch CYP3A metabolisiert, und es ist ein Substrat von P-Glykoprotein (P-gp).
Wirkungen anderer Arzneimittel auf Nerandomilast
Starke CYP3A-Inhibitoren
Klinische Auswirkungen: |
Unter der Gabe einer Einzeldosis von 6 mg Nerandomilast zusammen mit mehreren Dosen Itraconazol (einem starken dualen Inhibitor von CYP3A und P‑gp) an Probanden erhöhte sich die Exposition gegenüber Nerandomilast um das 2,2‑Fache hinsichtlich der AUC und um das 1,3‑Fache hinsichtlich der Cmax. |
Intervention: |
Bei gleichzeitiger Anwendung mit starken CYP3A‑Inhibitoren muss die Dosis auf 9 mg zweimal täglich reduziert werden (siehe Abschnitt 4.2). |
Beispiele: |
Clarithromycin, Itraconazol, Ritonavir |
Starke oder mäßige CYP3A-Induktoren
Klinische Auswirkungen: |
Bei der gleichzeitigen Verabreichung einer Einzeldosis von 18 mg Nerandomilast zusammen mit Mehrfachdosen von Carbamazepin (einem starken CYP3A-Induktor) an gesunde Probanden verringerte sich die Exposition von Nerandomilast um 51 % hinsichtlich der AUC und um 31 % hinsichtlich der Cmax. |
Intervention: |
Bei gleichzeitiger Anwendung mit starken oder mäßigen CYP3A-Induktoren beträgt die empfohlene Dosis 18 mg zweimal täglich und darf nicht auf 9 mg zweimal täglich reduziert werden (siehe Abschnitt 4.2). |
Beispiele: |
Carbamazepin, Johanniskraut, Rifampicin, Phenytoin, Bosentan, Metamizol |
Pirfenidon
Klinische Auswirkungen: |
Die gleichzeitige Anwendung mit Pirfenidon reduzierte die Exposition gegenüber Nerandomilast um ca. 50 %, basierend auf der Ctrough, ss; beobachtet wurde dies in der Phase III-Studie bei Patienten mit IPF. Basierend auf In-vitro-Daten kann Pirfenidon die CYP3A-Aktivität induzieren und zu einer verminderten Nerandomilast-Exposition führen. Bei der gleichzeitigen Anwendung von Nerandomilast mit Pirfenidon bei Patienten mit IPF in dieser Phase III‑Studie wurde unter der 9 mg-Dosis keine Wirksamkeit beobachtet, unter der 18 mg-Dosis wurde hingegen eine Reduktion der Abnahme der forcierten Vitalkapazität (FVC) beobachtet (siehe Abschnitt 5.1). |
Intervention: |
Bei Patienten, die Pirfenidon anwenden, beträgt die empfohlene Dosis 18 mg zweimal täglich; sie darf nicht auf 9 mg zweimal täglich reduziert werden (siehe Abschnitt 4.2). |
Nintedanib
Die gleichzeitige Anwendung mit Nintedanib veränderte die Exposition gegenüber Nerandomilast nicht – basierend auf den Talspiegelkonzentrationen im Steady State.
Wirkungen von Nerandomilast auf andere Arzneimittel
Bei gleichzeitiger Anwendung mit Nerandomilast wurden keine klinisch relevanten Unterschiede in der Pharmakokinetik der folgenden Wirkstoffe beobachtet: Pirfenidon, Nintedanib und orales Midazolam (CYP3A4‑Substrat). Basierend auf den Ergebnissen zur gleichzeitigen Anwendung mit Midazolam ist nicht zu erwarten, dass Nerandomilast die systemische Exposition anderer Arzneimittel, die hauptsächlich über CYP3A metabolisiert werden (z. B. orale Kontrazeptiva), verringert oder erhöht.
Empfängnisverhütung
Laut Erkenntnissen aus tierexperimentellen Studien kann Nerandomilast Fehlgeburten verursachen (siehe nachstehenden Abschnitt „Schwangerschaft“ sowie Abschnitt 5.3).
Frauen im gebärfähigen Alter sollten darauf hingewiesen werden, während der Behandlung eine Schwangerschaft zu vermeiden und zuverlässige Verhütungsmethoden anzuwenden.
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Nerandomilast bei Schwangeren vor. Laut Erkenntnissen aus tierexperimentellen Studien kann Nerandomilast Fehlgeburten verursachen (siehe Abschnitt 5.3).
Frauen im gebärfähigen Alter sollten darauf hingewiesen werden, während der Einnahme von Jascayd eine Schwangerschaft zu vermeiden. Die Anwendung von Jascayd während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen.
Patientinnen sollten darauf hingewiesen werden, ihren Arzt zu informieren, wenn sie während der Therapie mit Jascayd schwanger werden oder eine Schwangerschaft vermuten. Schwangere Frauen und Frauen im gebärfähigen Alter müssen über das mögliche Risiko einer Fehlgeburt aufgeklärt werden.
Stillzeit
Es liegen keine Daten zum Übergang von Nerandomilast in die Muttermilch oder zu dessen Auswirkungen auf das gestillte Kind oder die Milchproduktion vor. Studien an Ratten haben Hinweise auf eine Ausscheidung von Nerandomilast in die Milch gezeigt (siehe Abschnitt 5.3).
Ein Risiko für das gestillte Kind kann nicht ausgeschlossen werden. Das Stillen soll während der Behandlung mit Jascayd unterbrochen werden.
Fertilität
Zur Wirkung von Nerandomilast auf die menschliche Fertilität liegen keine Daten vor. Bei männlichen und weiblichen Ratten wurden unter der 4‑fachen Humanexposition keine negativen Auswirkungen auf die Fertilität beobachtet (siehe Abschnitt 5.3).
Jascayd hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die häufigsten Nebenwirkungen sind Diarrhö und Gewichtsverlust.
Tabellarische Auflistung von Nebenwirkungen
Tabelle 1 zeigt die Häufigkeit von Nebenwirkungen basierend auf 52‑Wochen-Daten aus zwei randomisierten, placebokontrollierten, doppelblinden klinischen Phase III-Studien (FIBRONEER‑IPF bei IPF und FIBRONEER‑ILD bei PPF) mit zweimal täglicher Gabe von 9 mg oder 18 mg Nerandomilast. Die Nebenwirkungen sind nach MedDRA-Systemorganklasse aufgeführt.
Die Häufigkeiten sind definiert als sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 1. Nebenwirkungen
Systemorganklasse |
Nebenwirkungen |
Häufigkeitskategorie |
|
IPFb |
PPF |
||
Stoffwechsel- und Ernährungsstörungen |
Verminderter Appetit |
Häufig |
Häufig |
Erkrankungen des Gastrointestinaltrakts |
Diarrhöa |
Sehr häufig |
Sehr häufig |
Übelkeit |
Häufig |
Häufig |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Rückenschmerzen |
Häufig |
Häufig |
Untersuchungen |
Gewichtsverlusta |
Häufig |
Häufig |
a siehe Abschnitt Beschreibung ausgewählter Nebenwirkungen
b Die Häufigkeiten wurden unter Ausschluss der Patienten in der 9 mg‑Gruppe berechnet, die gleichzeitig Pirfenidon erhielten, da diese Kombination aufgrund einer klinisch relevanten Arzneimittelwechselwirkung nicht empfohlen wird (siehe Abschnitt 4.2 und Abschnitt 4.5).
Beschreibung ausgewählter Nebenwirkungen
Diarrhö
Bei den meisten mit Nerandomilast behandelten Patienten war die Diarrhö von leichtem bis mäßigem Schweregrad und trat im Allgemeinen innerhalb der ersten 3 Monate der Behandlung auf.
Die meisten Fälle wurden durch eine symptomatische Therapie mit Antidiarrhoika und gegebenenfalls durch eine Reduzierung der Hintergrundtherapie mit Nintedanib oder Pirfenidon behandelt.
Über schwere Diarrhö wurde häufiger bei Patienten berichtet, die Nerandomilast in Kombination mit Nintedanib erhielten.
Die Häufigkeit von Diarrhö hing vom Vorliegen und der Art der Hintergrundtherapie ab (siehe Tabelle 2).
Tabelle 2. Häufigkeit von Diarrhö nach Untergruppen der Hintergrundtherapie über einen Zeitraum von 52 Wochen in der Studie FIBRONEER-IPF (Patienten mit IPF) und in der Studie FIBRONEER‑ILD (Patienten mit PPF)
IPF (FIBRONEER-IPF) |
PPF (FIBRONEER-ILD) |
|||||
Placebo |
Nerandomilast |
Nerandomilast |
Placebo |
Nerandomilast |
Nerandomilast |
|
Keine Hintergrundtherapie |
8 % |
17 % |
26 % |
16 % |
15 % |
27 % |
Nintedanib-Hintergrundtherapie |
27 % |
49 % |
62 % |
36 % |
48 % |
49 % |
Pirfenidon-Hintergrundtherapie |
8 % |
n.a.a |
23 % |
n.a.b |
n.a.b |
n.a.b |
a Nicht zutreffend, da Nerandomilast 9 mg zweimal täglich für Patienten, die Pirfenidon erhalten, nicht empfohlen wird.
b Nicht zutreffend, da Pirfenidon in FIBRONEER-ILD nicht als Hintergrundtherapie zugelassen war.
BID = zweimal täglich
IPF
Bei den Patienten, die im Rahmen der Studie FIBRONEER‑IPF ohne IPF‑Hintergrundtherapie behandelt wurden, trat keine schwere Diarrhö auf; mit Nintedanib-Hintergrundtherapie wurde sie bei 5 % der Patienten unter 18 mg, bei 2 % unter 9 mg und bei < 1 % unter Placebo berichtet.
Diarrhö war die häufigste Nebenwirkung, die mit einem Abbruch der Behandlung in Verbindung stand, und sie trat am häufigsten bei gleichzeitiger Hintergrundtherapie mit Nintedanib auf. Ohne Nintedanib‑Hintergrundtherapie traten Behandlungsabbrüche bei 1 % der Patienten unter 18 mg auf und wurden unter 9 mg oder Placebo nicht berichtet; bei Hintergrundtherapie mit Nintedanib traten sie bei 13 % unter 18 mg, bei 2 % unter 9 mg und bei 1 % unter Placebo auf; bei Hintergrundtherapie mit Pirfenidon wurden unter 18 mg oder Placebo keine Behandlungsabbrüche berichtet.
PPF
Bei den Patienten, die im Rahmen der Studie FIBRONEER‑ILD behandelt wurden, wurde ohne Hintergrundtherapie mit Nintedanib keine schwere Diarrhö berichtet; mit Nintedanib-Hintergrundtherapie wurde sie bei 2 % der Patienten unter 18 mg, bei 1 % unter 9 mg und bei < 1 % unter Placebo berichtet.
Diarrhö war die häufigste Nebenwirkung, die mit einem Abbruch der Behandlung in Verbindung stand, und sie trat am häufigsten bei gleichzeitiger Hintergrundtherapie mit Nintedanib auf. Ohne Nintedanib‑Hintergrundtherapie traten Behandlungsabbrüche bei 1 % der Patienten unter 18 mg auf und wurden unter 9 mg oder Placebo nicht berichtet; bei Hintergrundtherapie mit Nintedanib traten sie bei 4 % unter 18 mg, bei 3 % unter 9 mg und bei 1 % unter Placebo auf.
Gewichtsverlust
In klinischen Studien setzte der Gewichtsverlust nach der zweiten Behandlungswoche ein und stabilisierte sich nach 12 Wochen Behandlung.
IPF
In der Studie FIBRONEER‑IPF betrug die mittlere absolute Veränderung des Körpergewichts vom Ausgangswert bis Woche 52 ‑2,6 kg bei Patienten, die mit Nerandomilast 18 mg behandelt wurden, ‑2,4 kg bei Patienten, die mit Nerandomilast 9 mg behandelt wurden und ‑1,8 kg bei Patienten, die Placebo erhielten.
Die Häufigkeit von Gewichtsverlust hing vom Vorliegen und der Art der IPF-Hintergrundtherapie ab: ohne IPF-Hintergrundtherapie betrug sie 7 % unter 18 mg, 1 % unter 9 mg und 6 % unter Placebo; mit Nintedanib-Hintergrundtherapie betrug sie 16 % unter 18 mg, 13 % unter 9 mg und 11 % unter Placebo; mit Pirfenidon-Hintergrundtherapie betrug sie 5 % sowohl unter 18 mg als auch unter Placebo.
PPF
In der Studie FIBRONEER‑ILD betrug die mittlere absolute Veränderung des Körpergewichts vom Ausgangswert bis Woche 52 ‑3,2 kg bei Patienten, die mit Nerandomilast 18 mg behandelt wurden, ‑2,0 kg bei Patienten, die mit Nerandomilast 9 mg behandelt wurden, und ‑2,0 kg bei Patienten, die Placebo erhielten.
Die Häufigkeit von Gewichtsverlust hing vom Vorliegen der Nintedanib-Hintergrundtherapie ab: ohne Nintedanib-Hintergrundtherapie betrug sie 10 % unter 18 mg, 5 % unter 9 mg und 4 % unter Placebo; mit Nintedanib-Hintergrundtherapie betrug sie 11 % unter 18 mg, 9 % unter 9 mg und 8 % unter Placebo.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, 53175 Bonn, Website: https://www.bfarm.de anzuzeigen.
Symptome
In klinischen Studien wurden Einzeldosen von bis zu 48 mg Nerandomilast an Probanden untersucht, ohne dass dosislimitierende Toxizität auftrat. Fälle versehentlicher Überdosierung wurden bei Patienten berichtet, die Dosen von bis zu 72 mg/Tag über einen Zeitraum von bis zu 17 Tagen erhielten.
Die beobachteten Nebenwirkungen entsprachen dem bekannten Sicherheitsprofil von Nerandomilast.
Therapie
Im Falle einer Überdosierung wird empfohlen, den Patienten auf Anzeichen oder Symptome von Nebenwirkungen zu überwachen und eine angemessene symptomatische Behandlung einzuleiten.
Pharmakotherapeutische Gruppe: Selektive Immunsuppressiva, ATC‑Code: L04AA61
Wirkmechanismus
Nerandomilast ist ein selektiver Inhibitor der Phosphodiesterase 4 (PDE4) mit einer Präferenz für die Hemmung des Isoenzyms PDE4B gegenüber PDE4A, C und D. In‑vitro-Daten zeigen eine 9‑fache Selektivität gegenüber PDE4D, eine 24‑fache Selektivität gegenüber PDE4A und eine 870‑fache Selektivität gegenüber PDE4C. Bei den empfohlenen Dosen ist mit einer Hemmung von PDE4B und einer geringeren Hemmung von PDE4D zu rechnen. PDE4 hydrolysiert und inaktiviert zyklisches Adenosinmonophosphat (cAMP). Nerandomilast wirkt sowohl antifibrotisch als auch immunmodulatorisch, da die bevorzugte Inhibition von PDE4B den intrazellulären cAMP‑Spiegel erhöht und die Expression profibrotischer Wachstumsfaktoren und entzündungsfördernder Zytokine reduziert, die bei fibrotischen Lungenerkrankungen überexprimiert sind.
Pharmakodynamische Wirkungen
Kardiale Elektrophysiologie
Bei Einzeldosen von Nerandomilast von bis zu 48 mg (das 2,2‑Fache der geschätzten Cmax, ss unter der maximal empfohlenen Humandosis) wurde keine klinisch signifikante QTc‑Intervallverlängerung beobachtet.
Klinische Wirksamkeit und Sicherheit
Idiopathische Lungenfibrose (IPF)
In einer randomisierten, doppelblinden, placebokontrollierten Studie (FIBRONEER‑IPF) wurden die klinische Wirksamkeit und Sicherheit von Nerandomilast bei erwachsenen Patienten mit idiopathischer Lungenfibrose (IPF) mit und ohne Hintergrundtherapie mit Nintedanib oder Pirfenidon untersucht. Bei den Patienten mussten eine forcierte Vitalkapazität (Forced Vital Capacity, FVC) von mindestens 45 % des Sollwerts und eine Diffusionskapazität der Lunge für Kohlenmonoxid (Diffusing Capacity of the Lungs for Carbon Monoxide, DLCO) von mindestens 25 % des für Hämoglobin (Hb) korrigierten Sollwerts vorliegen. 1 177 Patienten wurden im Verhältnis 1:1:1 randomisiert und erhielten über mindestens 52 Wochen entweder zweimal täglich Nerandomilast 9 mg, zweimal täglich Nerandomilast 18 mg oder zweimal täglich Placebo. Die Randomisierung erfolgte stratifiziert nach dem Vorliegen bzw. Fehlen einer Hintergrundtherapie mit Nintedanib oder Pirfenidon zu Studienbeginn. Der primäre Endpunkt der Studie war die absolute Veränderung der FVC in ml nach 52 Wochen im Vergleich zu Placebo. Der wichtigste sekundäre Endpunkt war die Zeit bis zum ersten Auftreten einer der Komponenten des kombinierten Endpunkts: erste akute IPF‑Exazerbation, erste Hospitalisierung aufgrund einer respiratorischen Ursache oder Tod während der Studiendauer. Die Patienten wurden zum Zeitpunkt der Hauptanalyse im Median 14,6 Monate und zum Zeitpunkt der Analyse zum Studienende im Median 17,0 Monate nachbeobachtet.
Die Studienpopulation bestand zu 83 % aus Männern und zu 17 % aus Frauen mit einem durchschnittlichen Alter von 70 Jahren (Spanne: 42 bis 90 Jahre). 31 % der Patienten waren mindestens 75 Jahre alt. 68 % der Studienpopulation waren kaukasischer, 32 % asiatischer und 0,5 % afrikanischer/afroamerikanischer Herkunft. Bei 3 % der Patienten wurde zu Studienbeginn eine pulmonale Hypertonie festgestellt. 78 % der Patienten befanden sich in stabiler Behandlung mit Nintedanib (46 %) oder Pirfenidon (32 %), während 22 % keine dieser Therapien erhielten (15 % der Patienten waren therapienaiv und 8 % hatten die Behandlung mit Nintedanib oder Pirfenidon zuvor abgebrochen). Die mittlere FVC betrug zu Studienbeginn 2 843 ml und entsprach 78 % des Sollwerts. Patienten, die eine Hintergrundtherapie mit Nintedanib oder Pirfenidon erhielten, wiesen zu Studienbeginn ein fortgeschritteneres Krankheitsstadium auf als Patienten ohne Hintergrundtherapie, was sich in einem niedrigeren mittleren FVC-Prozentsatz des Sollwerts (77 % gegenüber 82 %), einem niedrigeren mittleren DLCO-Prozentsatz des Sollwerts (50 % gegenüber 55 %), einer längeren mittleren Zeit seit der Erstdiagnose (3,7 Jahre gegenüber 2,8 Jahre) und einer häufigeren Nutzung von Sauerstofftherapien (23 % gegenüber 16 %) äußerte.
Veränderung der FVC gegenüber dem Ausgangswert
Insgesamt zeigte sich im primären Endpunkt, der absoluten Veränderung der FVC (in ml) gegenüber dem Ausgangswert nach 52 Wochen, bei Patienten unter Nerandomilast im Vergleich zu Patienten unter Placebo eine statistisch signifikante Verbesserung. Der adjustierte mittlere Rückgang betrug bei Patienten, die zweimal täglich 18 mg bzw. 9 mg Nerandomilast erhielten, ‑115 ml bzw. ‑139 ml, während in der Placebogruppe ein adjustierter mittlerer Rückgang von ‑183 ml beobachtet wurde. Der jeweilige Behandlungsunterschied im Vergleich zur Placebogruppe betrug 69 ml (95 % KI: 30‑107; p = 0,0005) bzw. 45 ml (95 % KI: 6‑83; p = 0,0222).
Bei Patienten, die Pirfenidon als Hintergrundtherapie gegen IPF erhielten, wurde eine Arzneimittelwechselwirkung beobachtet (siehe Abschnitt 4.5). Bei diesen Patienten zeigte die zweimal tägliche Anwendung von 9 mg Nerandomilast keine Wirkung.
Die Ergebnisse für den primären Endpunkt in der Gesamtpopulation und nach IPF‑Hintergrundtherapie für die Nerandomilast-Dosen von 18 mg und 9 mg im Vergleich zu entsprechendem Placebo sind in Abbildung 1 bzw. Abbildung 2 dargestellt.
Abbildung 1. Absolute Veränderung der FVC (ml) gegenüber dem Ausgangswert nach 52 Wochen in der Studie FIBRONEER-IPF bei Patienten, die 18 mg Nerandomilast erhielten, im Vergleich zu Placebo

Bei Patienten, die vor Woche 52 verstarben, wurde die Veränderung im 10. Perzentil gegenüber dem Ausgangswert angenommen.
BID = zweimal täglich
Abbildung 2. Absolute Veränderung der FVC (ml) gegenüber dem Ausgangswert nach 52 Wochen in der Studie FIBRONEER‑IPF bei Patienten, die 9 mg Nerandomilast erhielten, im Vergleich zu Placebo
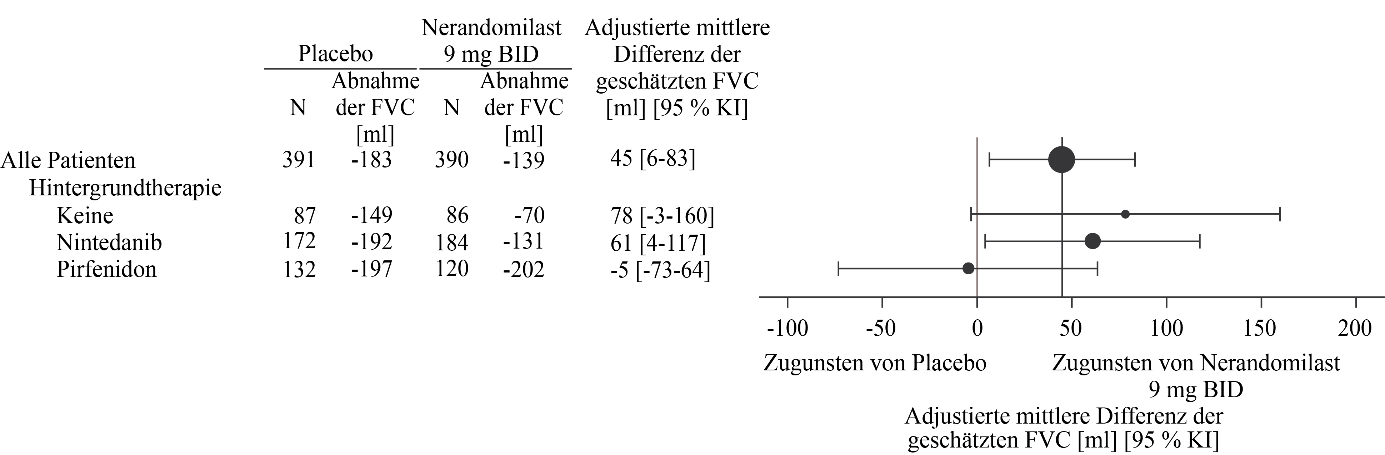
Bei Patienten, die vor Woche 52 verstarben, wurde die Veränderung im 10. Perzentil gegenüber dem Ausgangswert angenommen.
BID = zweimal täglich
Abbildung 3 zeigt die Veränderung der FVC gegenüber dem Ausgangswert im Zeitverlauf bei Patienten, die zweimal täglich Nerandomilast 18 mg erhielten, im Vergleich zu einem entsprechenden Placebo. Die Kurven für die mittlere adjustierte Veränderung der FVC gegenüber dem Ausgangswert trennten sich ab Woche 2 und divergierten bis Woche 52 und darüber hinaus weiter. Dieser Effekt wurde in allen Subgruppen mit unterschiedlichen IPF-Hintergrundtherapien konsistent beobachtet.
Abbildung 3. Veränderung der FVC (ml) gegenüber dem Ausgangswert im Zeitverlauf bei Patienten, die 18 mg Nerandomilast erhielten, im Vergleich zu Placebo in der Studie FIBRONEER‑IPF
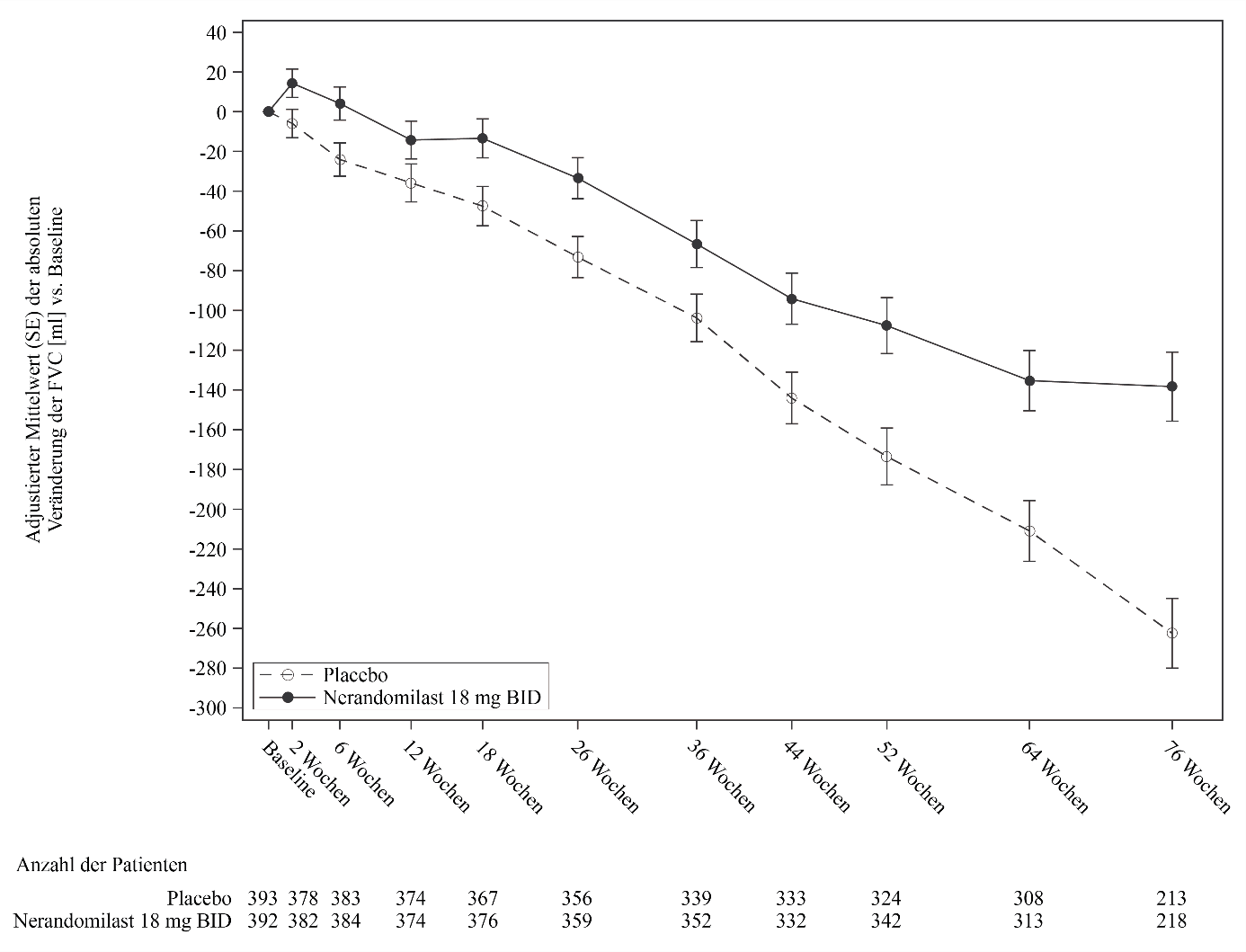
BID = zweimal täglich
Zeit bis zur ersten akuten IPF‑Exazerbation, ersten Hospitalisierung aufgrund einer respiratorischen Ursache oder Tod
Der wichtigste sekundäre kombinierte Endpunkt war die Zeit bis zum ersten Auftreten einer akuten IPF‑Exazerbation, einer Hospitalisierung aufgrund einer respiratorischen Ursache oder des Todes während der Studiendauer. Eine akute IPF‑Exazerbation war definiert als akute Verschlechterung oder Entwicklung von Dyspnoe, die typischerweise weniger als 1 Monat andauerte, neu aufgetretene beidseitige Milchglastrübung und/oder Konsolidierungen vor einem mit IPF vereinbaren Hintergrundmuster in der Computertomographie sowie eine Verschlechterung, die nicht vollständig durch Herzinsuffizienz oder Flüssigkeitsüberladung erklärt werden konnte. Weder akute IPF‑Exazerbationen noch Hospitalisierungen aufgrund einer respiratorischen Ursache wurden adjudiziert. Insgesamt ergab sich kein statistisch signifikanter Behandlungsunterschied bei den Gruppen mit Nerandomilast 18 mg bzw. 9 mg im Vergleich zu Placebo hinsichtlich des wichtigsten sekundären kombinierten Endpunkts. Zum Zeitpunkt der Hauptanalyse traten Ereignisse des wichtigsten sekundären Endpunkts bei 85 Patienten (22 %) in der 18 mg-Gruppe, bei 79 Patienten (20 %) in der 9 mg-Gruppe und bei 80 Patienten (20 %) in der Placebogruppe auf. Im Vergleich zu Placebo betrug die Hazard Ratio (HR) für die Zeit bis zum ersten Ereignis 1,17 (95 % KI: 0,86‑1,59; p = 0,3102) für die 18 mg-Dosis und 1,03 (95 % KI: 0,75‑1,41; p = 0,8568) für die 9 mg-Dosis.
Abbildung 4 und Abbildung 5 zeigen die Ergebnisse von Nerandomilast 18 mg und 9 mg im Vergleich zu Placebo für den wichtigsten sekundären Endpunkt und dessen Komponenten über die Dauer der FIBRONEER‑IPF-Studie zum Zeitpunkt der Analyse zum Studienende.
Abbildung 4. Akute IPF‑Exazerbation, Hospitalisierung aufgrund einer respiratorischen Ursache oder Tod bei Patienten, die 18 mg Nerandomilast erhielten, im Vergleich zu Placebo über die Dauer der Studie FIBRONEER‑IPF
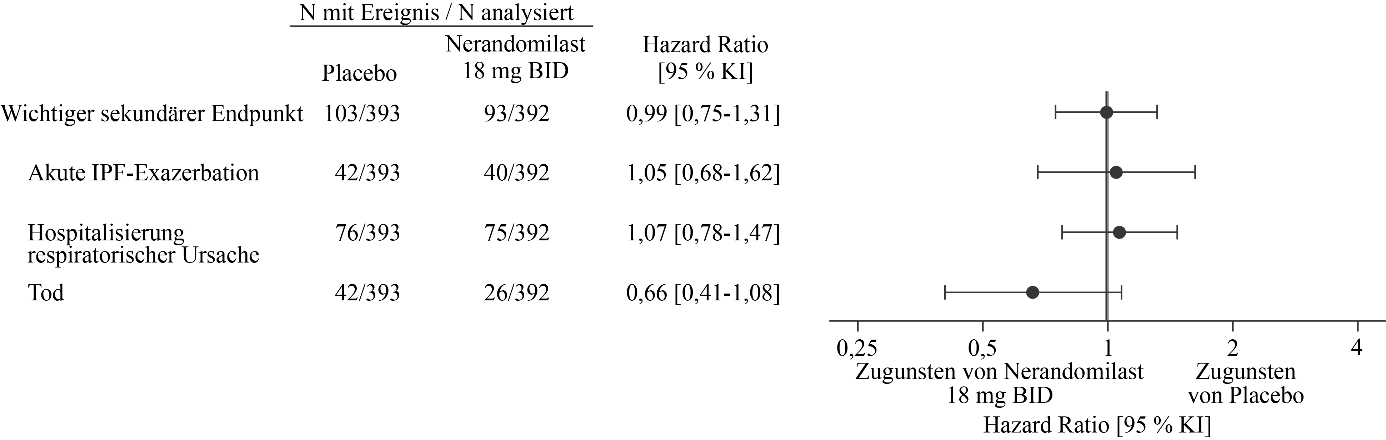
BID = zweimal täglich
Abbildung 5. Akute IPF‑Exazerbation, Hospitalisierung aufgrund einer respiratorischen Ursache oder Tod bei Patienten, die 9 mg Nerandomilast erhielten, im Vergleich zu Placebo über die Dauer der Studie FIBRONEER‑IPF
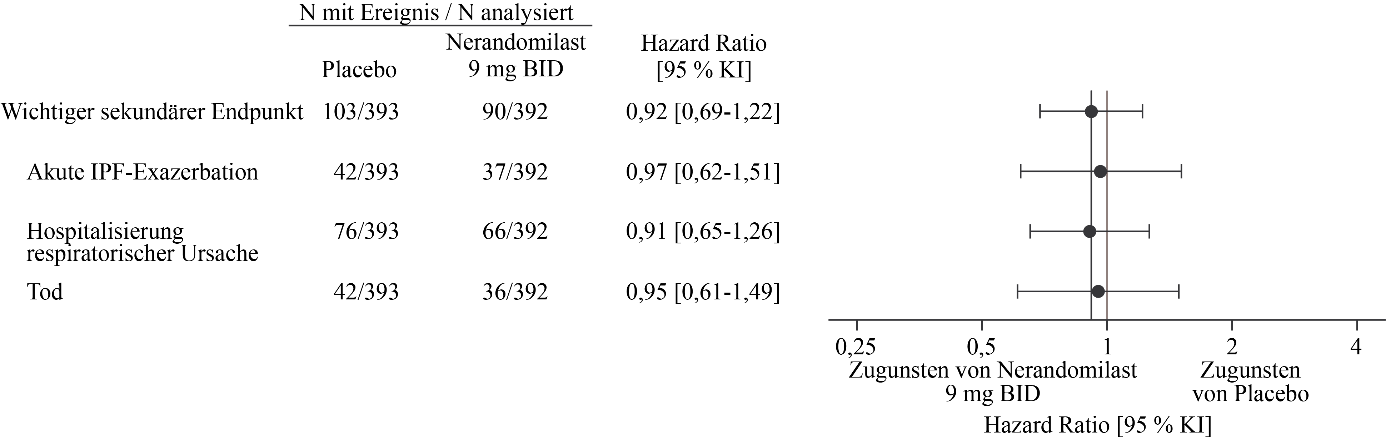
Es werden Ergebnisse für die Gesamtpopulation gezeigt, die zweimal täglich 9 mg Nerandomilast im Vergleich zu einem entsprechenden Placebo erhielt.
BID = zweimal täglich
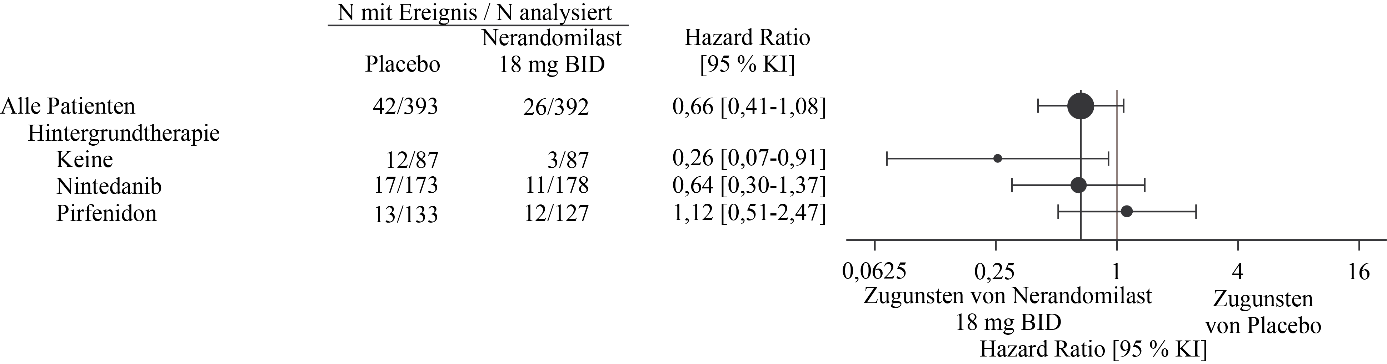
In der Analyse zum Studienende gab es 26 Todesfälle (7 %) in der 18 mg-Gruppe, 36 Todesfälle (9 %) in der 9 mg-Gruppe und 42 Todesfälle (11 %) in der Placebogruppe. Im Vergleich zu Placebo betrug die Hazard Ratio für die Zeit bis zum Tod 0,66 (95 % KI: 0,41‑1,08) für die 18 mg-Dosis und 0,95 (95 % KI: 0,61‑1,49) für die 9 mg-Dosis.
Abbildung 6 und Abbildung 7 zeigen die Analyse der Zeit bis zum Tod über die gesamte Studiendauer (Analyse zum Studienende) in den Nerandomilast-Dosisgruppen von 18 mg bzw. 9 mg im Vergleich zum entsprechenden Placebo für die nach Hintergrundtherapie unterteilten Subgruppen.
Abbildung 6. Zeit bis zum Tod unter Patienten, die 18 mg Nerandomilast erhielten, im Vergleich zu Placebo über die Dauer der Studie FIBRONEER‑IPF
BID = zweimal täglich
Abbildung 7. Zeit bis zum Tod unter Patienten, die 9 mg Nerandomilast erhielten, im Vergleich zu Placebo über die Dauer der Studie FIBRONEER‑IPF
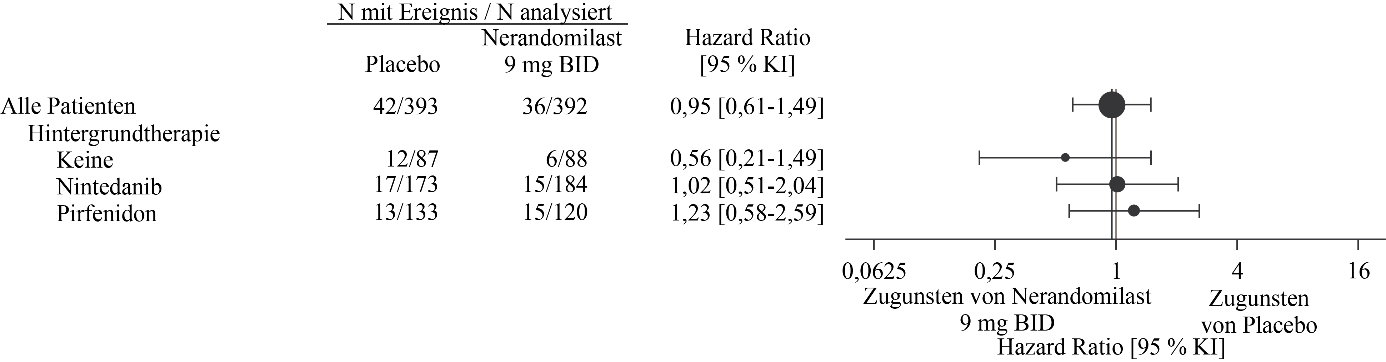
Es werden Ergebnisse für die Gesamtpopulation gezeigt, die zweimal täglich 9 mg Nerandomilast im Vergleich zu einem entsprechenden Placebo erhielt.
BID = zweimal täglich
Progrediente pulmonale Fibrose (PPF)
In einer randomisierten, doppelblinden, placebokontrollierten Studie (FIBRONEER‑ILD) wurden die klinische Wirksamkeit und Sicherheit von Nerandomilast bei erwachsenen Patienten mit PPF untersucht. Patienten mit PPF wurden ausgewählt, wenn sich bei ihnen eine relevante Fibrose (mehr als 10 % fibrotische Merkmale) in der hochauflösenden Computertomographie (High Resolution Computed Tomography, HRCT) darstellte und sie klinische Anzeichen einer Progression zeigten (definiert als Abnahme der forcierten Vitalkapazität [Forced Vital Capacity, FVC] von mindestens 10 %, FVC‑Abnahme von mindestens 5 % und weniger als 10 % mit Verschlechterung der Atemwegsbeschwerden oder der Bildgebung oder Verschlechterung der Atemwegsbeschwerden und der Bildgebung, alles innerhalb der 24 Monate vor dem Screening). Bei den Patienten mussten eine forcierte Vitalkapazität (Forced Vital Capacity, FVC) von mindestens 45 % des Sollwerts und eine Diffusionskapazität der Lunge für Kohlenmonoxid (Diffusing Capacity of the Lungs for Carbon Monoxide, DLCO) von mindestens 25 % des für Hämoglobin (Hb) korrigierten Sollwerts vorliegen. Geeignete Patienten erhielten eine stabile Hintergrundtherapie mit Nintedanib oder nicht. 1 178 Patienten wurden im Verhältnis 1:1:1 randomisiert und erhielten mindestens über einen Zeitraum von 52 Wochen zweimal täglich Nerandomilast 9 mg, zweimal täglich Nerandomilast 18 mg oder zweimal täglich Placebo. Die Randomisierung erfolgte stratifiziert nach Vorliegen oder Fehlen einer Hintergrundtherapie mit Nintedanib und nach dem Muster der hochauflösenden Computertomographie (HRCT) (usual interstitial pneumonia [UIP] oder UIP‑ähnliches fibrotisches Muster gegenüber anderen fibrotischen Mustern) unter Verwendung einer zentralen Überprüfung.
Der primäre Endpunkt der Studie war die absolute Veränderung der FVC in ml nach 52 Wochen gegenüber dem Ausgangswert im Vergleich zu Placebo. Der wichtigste sekundäre Endpunkt war die Zeit bis zum ersten Auftreten einer der Komponenten des kombinierten Endpunkts: die erste akute Exazerbation der interstitiellen Lungenerkrankung (Interstitial Lung Disease, ILD), die erste Hospitalisierung aufgrund einer respiratorischen Ursache oder der Tod während der Studiendauer. Die Patienten wurden zum Zeitpunkt der Hauptanalyse im Median 15,4 Monate und zum Zeitpunkt der Analyse zum Studienende im Median 17,2 Monate nachbeobachtet.
Die Studienpopulation bestand zu 56 % aus Männern und zu 44 % aus Frauen mit einem durchschnittlichen Alter von 66 Jahren (Spanne: 26 bis 88 Jahre). 20 % der Patienten waren mindestens 75 Jahre alt. 58 % der Studienpopulation waren kaukasischer, 39 % asiatischer und 1 % afrikanischer/afroamerikanischer Herkunft. Bei 5 % der Patienten wurde zu Studienbeginn eine pulmonale Hypertonie festgestellt.
44 % der Patienten befanden sich in stabiler Behandlung mit Nintedanib und 56 % waren nicht unter Nintedanib-Behandlung (44 % der Patienten waren therapienaiv und 12 % hatten die Behandlung mit Nintedanib zuvor abgebrochen). Bei der HRCT‑Untersuchung zu Studienbeginn wiesen 71 % der Patienten ein UIP- oder UIP‑ähnliches fibrotisches Muster auf, während bei 29 % der Patienten andere fibrotische Muster festgestellt wurden. Die zugrunde liegenden klinischen ILD‑Diagnosen waren autoimmune ILD (28 %), Hypersensitivitätspneumonitis (20 %), nicht klassifizierbare idiopathische interstitielle Pneumonie (20 %), idiopathische unspezifische interstitielle Pneumonie (19 %) und andere ILD (14 %). Die mittlere FVC betrug zu Studienbeginn 2 353 ml und entsprach 70 % des Sollwerts. Patienten, die eine Hintergrundtherapie mit Nintedanib erhielten, wiesen zu Studienbeginn ein fortgeschritteneres Krankheitsstadium auf als Patienten ohne Hintergrundtherapie, was sich in einem geringfügig niedrigeren mittleren FVC-Prozentsatz des Sollwerts (69 % gegenüber 71 %), einem niedrigeren mittleren DLCO-Prozentsatz des Sollwerts (45 % gegenüber 52 %), einer geringfügig längeren mittleren Zeit seit der ILD-Erstdiagnose (4,4 Jahre gegenüber 4,0 Jahre) und einer häufigeren Nutzung von Sauerstofftherapien (38 % gegenüber 19 %) äußerte.
Veränderung der FVC gegenüber dem Ausgangswert
Insgesamt zeigte sich im primären Endpunkt, der absoluten Veränderung der FVC (in ml) gegenüber dem Ausgangswert nach 52 Wochen, bei Patienten unter Nerandomilast im Vergleich zu Patienten unter Placebo eine statistisch signifikante Verbesserung.
Der adjustierte mittlere Rückgang betrug bei Patienten, die zweimal täglich 18 mg bzw. 9 mg Nerandomilast erhielten, ‑99 ml bzw. ‑85 ml, während in der Placebogruppe ein adjustierter mittlerer Rückgang von ‑166 ml beobachtet wurde. Der jeweilige Behandlungsunterschied im Vergleich zur Placebogruppe betrug 67 ml (95 % KI: 32‑102; p = 0,0002) bzw. 81 ml (95 % KI: 46‑116; p < 0,0001).
Die Primäranalyse war in allen vordefinierten Subgruppen hinsichtlich des Vorhandenseins oder Fehlens einer Nintedanib-Hintergrundtherapie, des HRCT‑Musters und der zugrunde liegenden klinischen ILD‑Diagnosen konsistent. Siehe Abbildung 8 und Abbildung 9.
Abbildung 8. Absolute Veränderung der FVC (ml) gegenüber dem Ausgangswert nach 52 Wochen in der Studie FIBRONEER‑ILD bei Patienten, die 18 mg Nerandomilast erhielten, im Vergleich zu Placebo
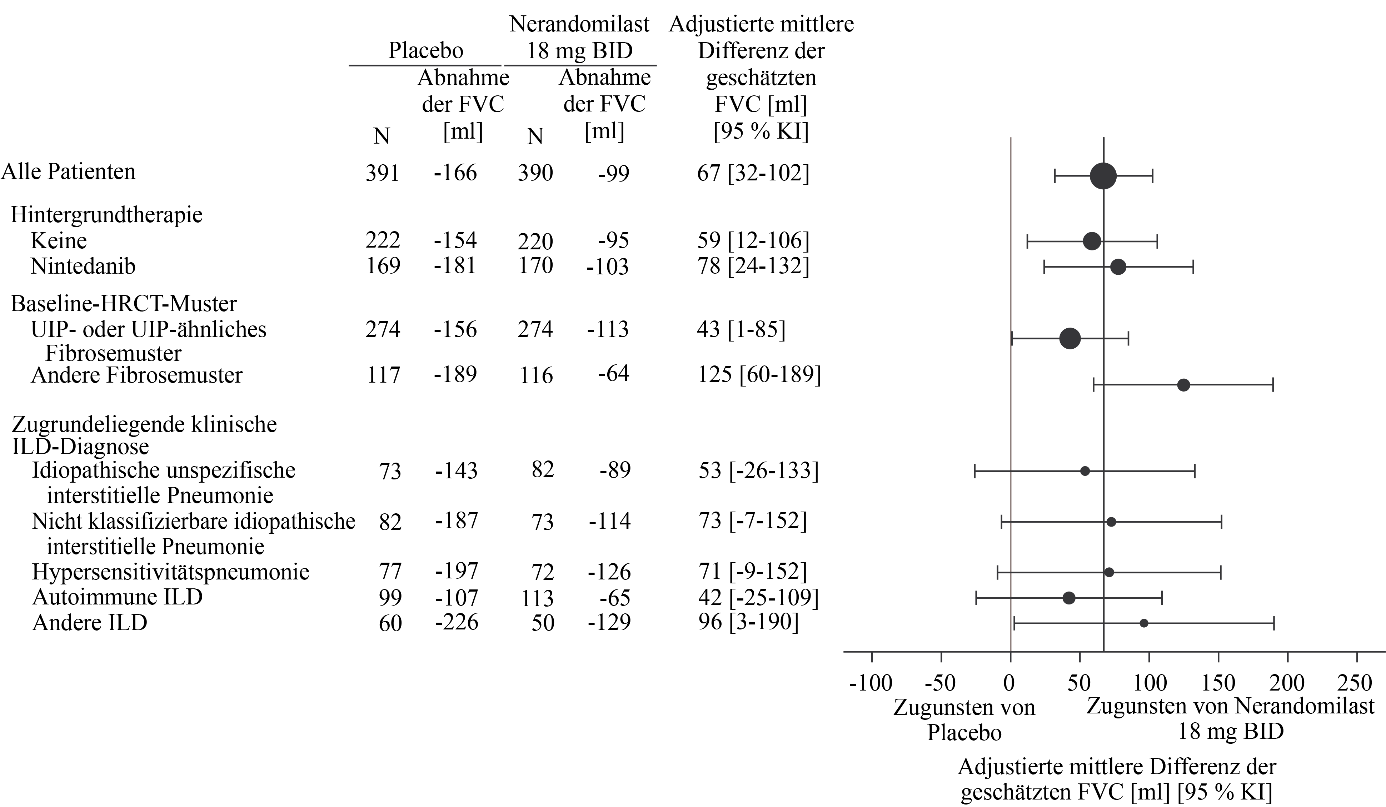
Bei Patienten, die vor Woche 52 verstarben, wurde die Veränderung im 10. Perzentil gegenüber dem Ausgangswert angenommen.
BID = zweimal täglich
Abbildung 9. Absolute Veränderung der FVC (ml) gegenüber dem Ausgangswert nach 52 Wochen in der Studie FIBRONEER‑ILD bei Patienten, die 9 mg Nerandomilast erhielten, im Vergleich zu Placebo
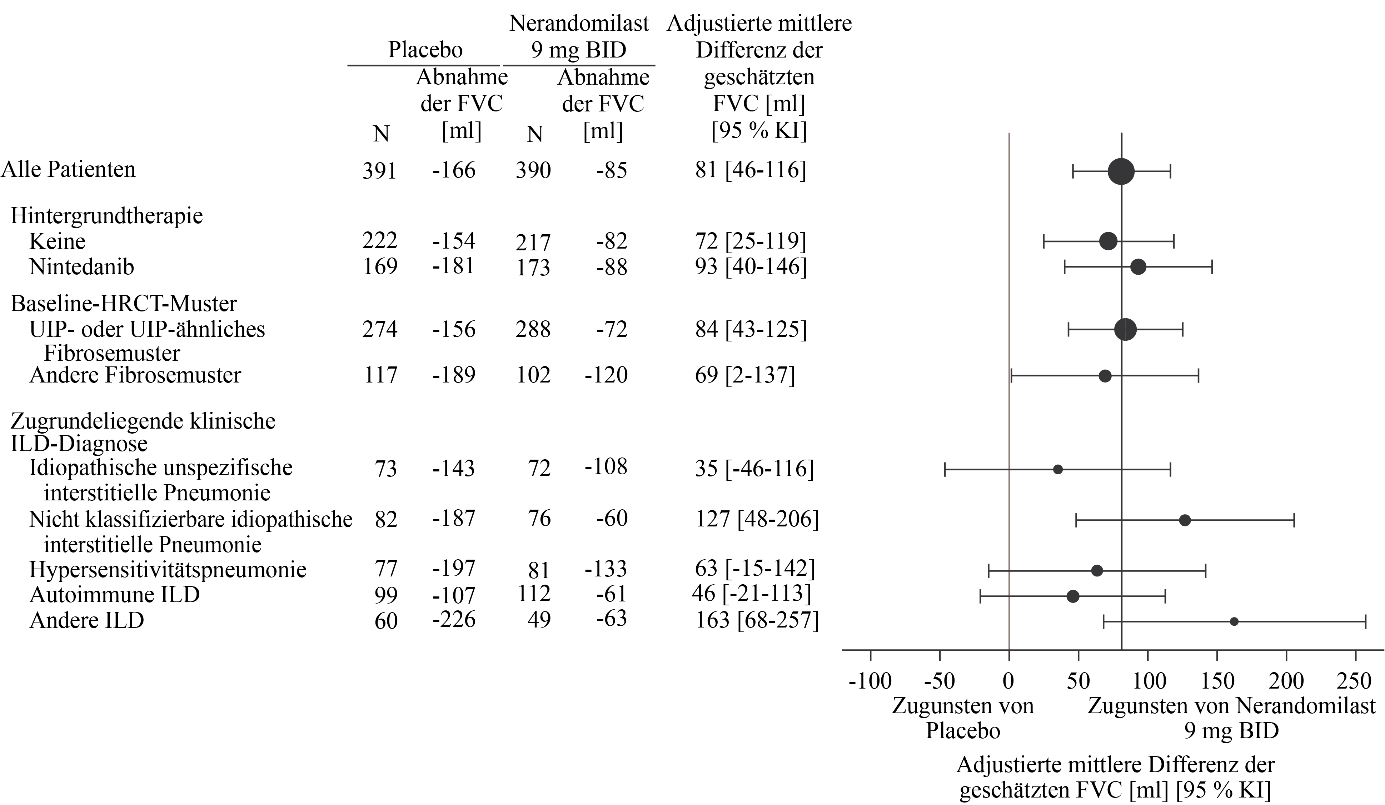
Bei Patienten, die vor Woche 52 verstarben, wurde die Veränderung im 10. Perzentil gegenüber dem Ausgangswert angenommen.
BID = zweimal täglich
Abbildung 10 zeigt die Veränderung der FVC gegenüber dem Ausgangswert im Zeitverlauf bei Patienten, die zweimal täglich Nerandomilast 18 mg erhielten, im Vergleich zu einem entsprechenden Placebo. Die Kurven für die mittlere adjustierte Veränderung der FVC gegenüber dem Ausgangswert trennten sich ab Woche 2 und divergierten bis Woche 52 und darüber hinaus weiter. Dieser Effekt war unabhängig von der Nintedanib-Hintergrundtherapie konsistent.
Abbildung 10. Veränderung der FVC (ml) gegenüber dem Ausgangswert bei Patienten, die 18 mg Nerandomilast erhielten, im Vergleich zu Placebo im Zeitverlauf in der Studie FIBRONEER‑ILD
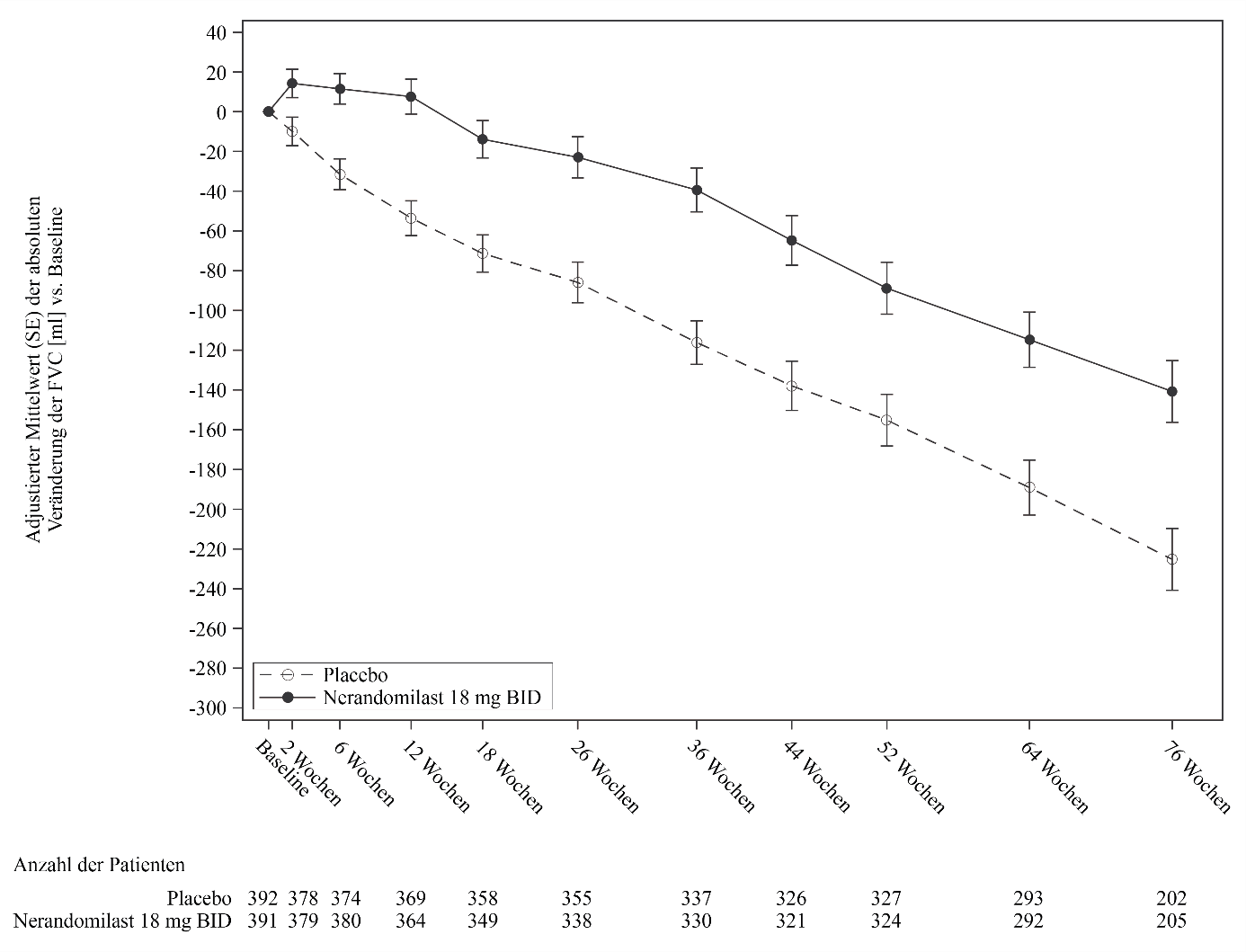
BID = zweimal täglich
Zeit bis zur ersten akuten ILD‑Exazerbation, ersten Hospitalisierung aufgrund einer respiratorischen Ursache oder Tod
Der wichtigste sekundäre kombinierte Endpunkt war die Zeit bis zum ersten Auftreten einer akuten ILD‑Exazerbation, einer Hospitalisierung aufgrund einer respiratorischen Ursache oder bis zum Tod während der Studiendauer. Eine akute ILD‑Exazerbation war definiert als akute Verschlechterung oder Entwicklung von Dyspnoe, die typischerweise weniger als 1 Monat andauerte, neu aufgetretene beidseitige Milchglastrübung und/oder Konsolidierungen vor einem mit fibrosierender ILD vereinbaren Hintergrundmuster in der Computertomographie sowie eine Verschlechterung, die nicht vollständig durch Herzinsuffizienz oder Flüssigkeitsüberladung erklärt werden konnte. Weder akute ILD‑Exazerbationen noch Hospitalisierungen aufgrund einer respiratorischen Ursache wurden adjudiziert.
Das Risiko für den wichtigsten sekundären Endpunkt war für beide Nerandomilast-Dosisgruppen im Vergleich zu Placebo numerisch geringer. Zum Zeitpunkt der Hauptanalyse traten Ereignisse des wichtigsten sekundären Endpunkts bei 95 Patienten (24 %) in der 18 mg-Gruppe, bei 110 Patienten (28 %) in der 9 mg-Gruppe und bei 122 Patienten (31 %) in der Placebogruppe auf. Im Vergleich zu Placebo betrug die Hazard Ratio für die Zeit bis zum ersten Ereignis 0,77 (95 % KI: 0,59‑1,01; p = 0,0602) für die 18 mg-Dosis und 0,88 (95 % KI: 0,68‑1,14; p = 0,3398) für die 9 mg-Dosis.
Abbildung 11 und Abbildung 12 zeigen die Ergebnisse von Nerandomilast 18 mg bzw. 9 mg im Vergleich zu Placebo für den wichtigsten sekundären Endpunkt und dessen Komponenten über die Dauer der FIBRONEER‑ILD-Studie zum Zeitpunkt der Analyse zum Studienende.
Abbildung 11. Akute ILD‑Exazerbation, Hospitalisierung aufgrund einer respiratorischen Ursache oder Tod bei Patienten, die 18 mg Nerandomilast erhielten, im Vergleich zu Placebo über die Dauer der Studie FIBRONEER‑ILD
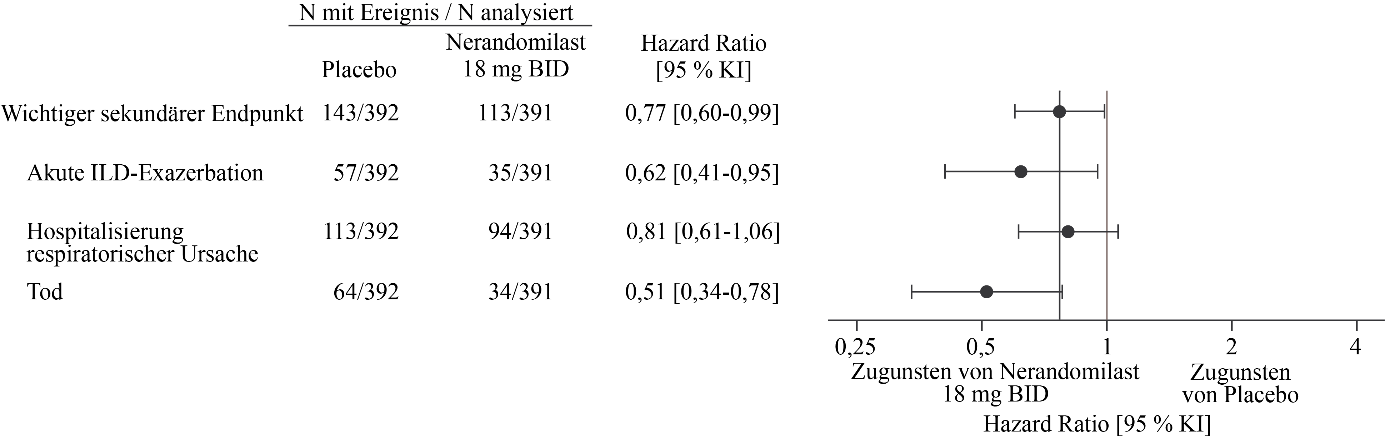
BID = zweimal täglich
Abbildung 12. Akute ILD‑Exazerbation, Hospitalisierung aufgrund einer respiratorischen Ursache oder Tod bei Patienten, die 9 mg Nerandomilast erhielten, im Vergleich zu Placebo über die Dauer der Studie FIBRONEER‑ILD
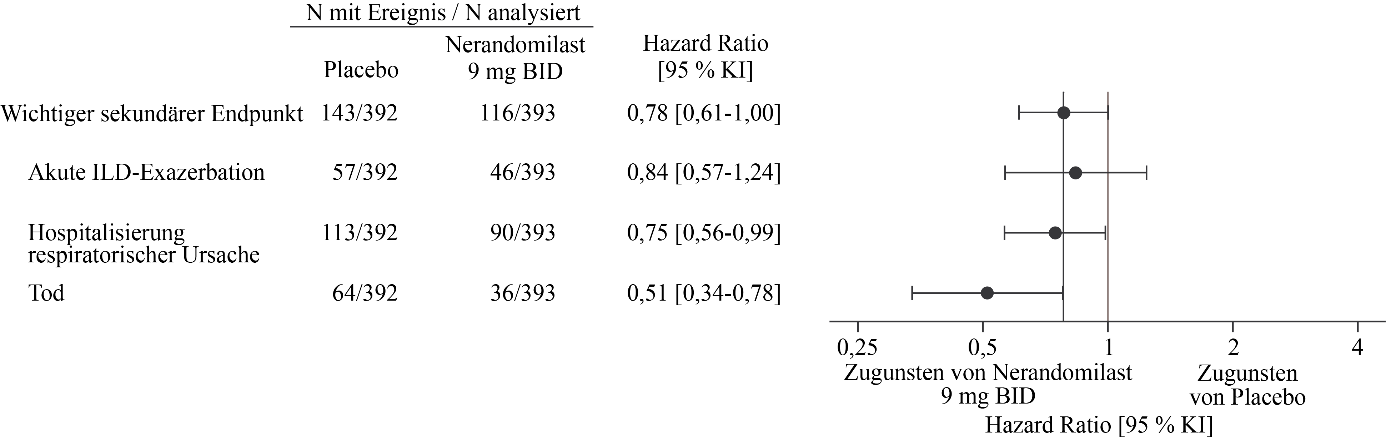
BID = zweimal täglich
Die Ergebnisse des wichtigsten sekundären Endpunkts waren unabhängig von einer Nintedanib-Hintergrundtherapie oder dem HRCT‑Muster im Allgemeinen konsistent.
In der Analyse zum Studienende gab es 34 Todesfälle (9 %) in der 18 mg-Gruppe, 36 Todesfälle (9 %) in der 9 mg-Gruppe und 64 Todesfälle (16 %) in der Placebogruppe. Im Vergleich zu Placebo betrug die Hazard Ratio für die Zeit bis zum Tod 0,51 (95 % KI: 0,34‑0,78) für die 18 mg-Dosis und 0,51 (95 % KI: 0,34‑0,78) für die 9 mg-Dosis.
Abbildung 13 und Abbildung 14 zeigen die Analyse der Zeit bis zum Tod über die gesamte Studiendauer (Analyse zum Studienende) in den Nerandomilast-Dosisgruppen von 18 mg bzw. 9 mg im Vergleich zum entsprechenden Placebo für die Subgruppen, die nach Hintergrundtherapie, HRCT-Muster sowie zugrundeliegender klinischer ILD-Diagnose unterteilt sind.
Abbildung 13. Zeit bis zum Tod unter Patienten, die 18 mg Nerandomilast erhielten, im Vergleich zu Placebo über die Dauer der Studie FIBRONEER‑ILD
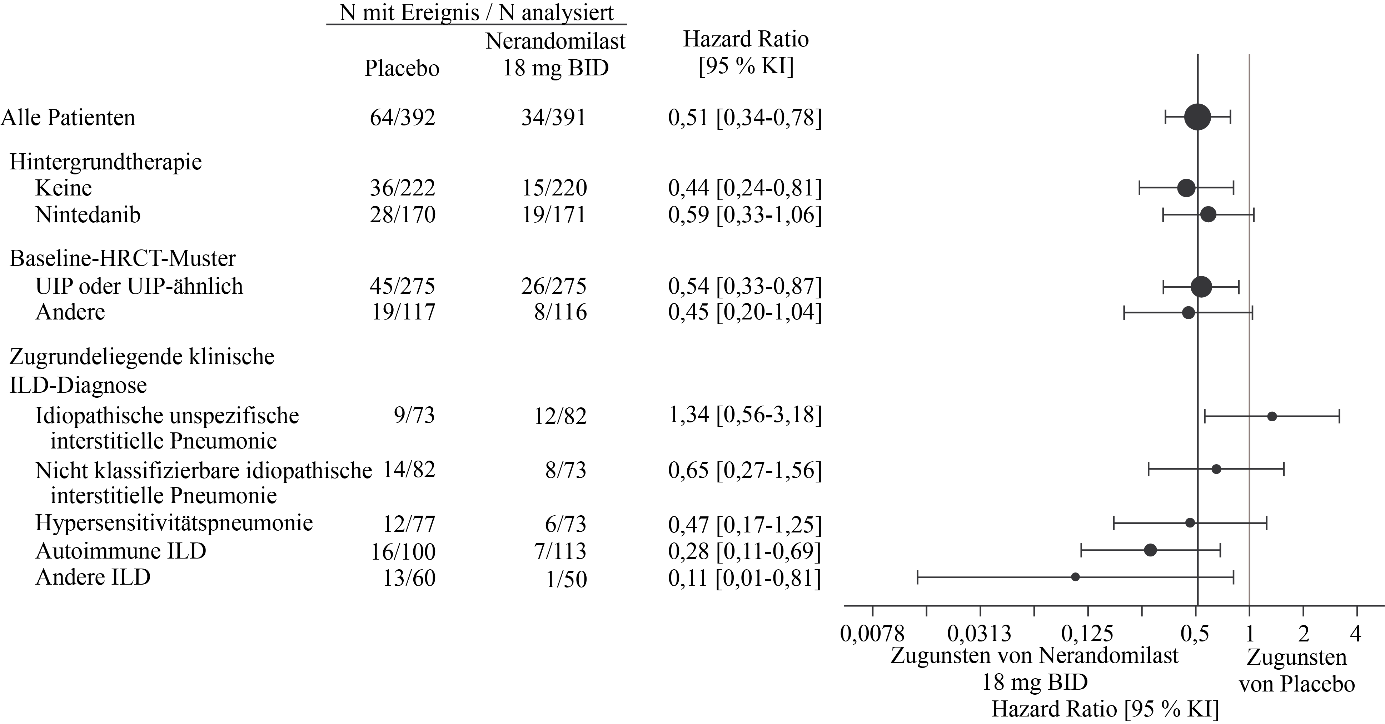
BID = zweimal täglich
Abbildung 14. Zeit bis zum Tod unter Patienten, die 9 mg Nerandomilast erhielten, im Vergleich zu Placebo über die Dauer der Studie FIBRONEER‑ILD
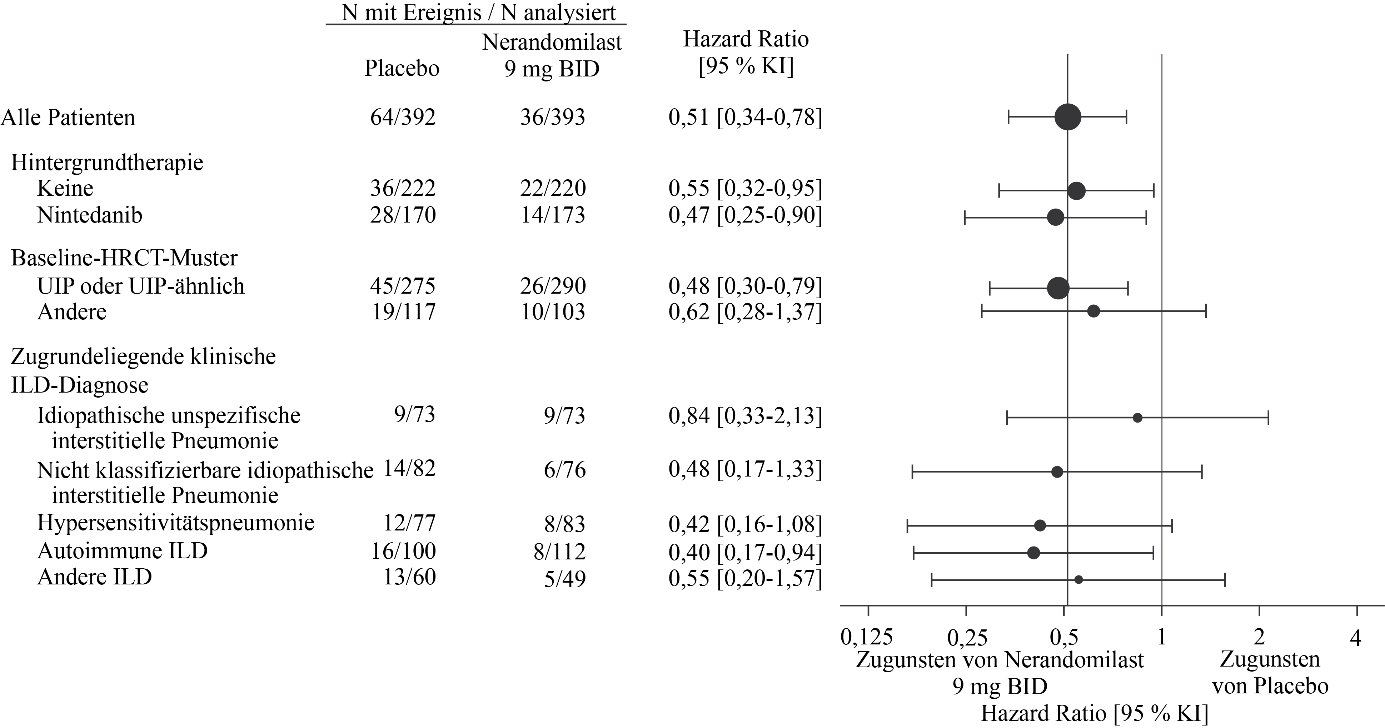
BID = zweimal täglich
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Jascayd eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in der fibrosierenden interstitiellen Lungenerkrankung gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Pharmakokinetik von Nerandomilast wurde an gesunden Probanden, Patienten mit IPF und Patienten mit PPF untersucht. Zwischen diesen Patientengruppen wurden keine klinisch relevanten Unterschiede in der Pharmakokinetik von Nerandomilast festgestellt.
Resorption
Nerandomilast erreichte nach oraler Anwendung von 9 mg- und 18 mg-Dosen nach im Median 1,00‑1,25 Stunden (Tmax) (Spanne: 0,5‑4 Stunden) maximale Plasmakonzentrationen (Cmax). Die absolute orale Bioverfügbarkeit von Nerandomilast betrug 73 % (90 % KI: 67‑79 %).
Die Anwendung von 18 mg Nerandomilast zu einer fett- und kalorienreichen Mahlzeit veränderte die Exposition gegenüber Nerandomilast nicht in klinisch relevantem Ausmaß (die AUC stieg um ca. 15 %, während die Cmax um ca. 14 % sank).
Verteilung
Nach einmaliger intravenöser Anwendung von Nerandomilast betrug das geometrische mittlere Verteilungsvolumen Vss ca. 94 l (gCV 32,0 %). In vitro war Nerandomilast ein Substrat des Transporterproteins P‑gp, jedoch nicht von BCRP, OATP1B1, OATP1B3, OAT1, OAT3 und OCT2.
Die humane Plasmaproteinbindung von Nerandomilast in vitro betrug 77 % und war nicht konzentrationsabhängig.
Bei Probanden verteilte sich Nerandomilast bevorzugt im Plasma mit einem Blut-Plasma-Verhältnis von 0,6‑0,8.
Biotransformation
Nerandomilast wird hauptsächlich durch Oxidation über CYP3A und Glucuronidierung über verschiedene Uridin-5′-diphosphat-Glucuronosyltransferase‑Enzyme metabolisiert.
Nach einmaliger oraler Anwendung war Nerandomilast der zirkulierende Hauptbestandteil und machte etwa 50 % der zirkulierenden Radioaktivität aus.
Nach mehrmaliger oraler Gabe von 12 mg Nerandomilast zweimal täglich war der einzige im Steady State im Plasma nachgewiesene Hauptmetabolit der dioxidative Metabolit BI 764333/M480(4). Dieser pharmakologisch inaktive Metabolit machte 12 % des gesamten Plasmamaterials im Steady State aus, das sowohl die Muttersubstanz als auch alle ihre Metaboliten umfasste.
Nerandomilast besitzt ein chirales Schwefelatom, und Jascayd enthält überwiegend chiral reines Nerandomilast (R‑Enantiomer). Nach oraler Anwendung von Nerandomilast erfolgt durch Metabolisierung eine chirale Inversion vom R- zum S‑Enantiomer. Das S‑Enantiomer wurde als ein unbedeutender (3 % der gesamten zirkulierenden Radioaktivität) Metabolit von Nerandomilast identifiziert und ist pharmakologisch inaktiv. Das pharmakologisch aktive R‑Enantiomer verblieb als das vorherrschende zirkulierende Enantiomer.
Elimination
Nach mehrmaliger oraler Anwendung von 18 mg Nerandomilast zweimal täglich betrug die terminale Halbwertszeit etwa 10 bis 17 Stunden und die geometrische mittlere scheinbare Plasma-Clearance im Steady State 274 ml/min mit einer interindividuellen Variabilität (gCV%) von 23,6 %. Nach oraler Anwendung einer Einzeldosis von 18 mg radioaktiv markiertem Nerandomilast wurden innerhalb von 9 Tagen nach der Dosisgabe etwa 95 % der Dosis wiedergefunden, davon 58 % in den Fäzes (13 % unverändert) und 36 % im Urin (12 % unverändert).
Linearität/Nicht-Linearität
Nerandomilast zeigte nach oraler Anwendung sowohl von Einzeldosen (0,06 bis 48 mg) als auch von Mehrfachdosen (1 bis 18 mg zweimal täglich) eine dosisproportionale Pharmakokinetik.
Nach zweimal täglicher oraler Anwendung von 18 mg Nerandomilast wurde innerhalb von 4 Tagen der Steady State erreicht, mit einem Akkumulationsverhältnis von bis zu 1,38, basierend auf AUC und Cmax.
PK bei bestimmten Patientengruppen
Alter, Geschlecht, ethnische Zugehörigkeit, Nieren- oder Leberfunktionsstörung
Es wurden keine klinisch relevanten Unterschiede in der Pharmakokinetik von Nerandomilast in Abhängigkeit von Alter (18‑90 Jahre), Geschlecht, ethnischer Zugehörigkeit (hispanisch/lateinamerikanisch oder nicht hispanisch/lateinamerikanisch), leichter, mittelschwerer und schwerer Nierenfunktionsstörung (eGFR ≥ 15 und < 90 ml/min, berechnet nach der CKD‑EPI‑Formel) oder leichter (Child-Pugh A) oder mittelschwerer (Child-Pugh B) Leberfunktionsstörung beobachtet. Patienten mit terminaler Niereninsuffizienz und Patienten mit schwerer Leberfunktionsstörung (Child-Pugh C) wurden nicht untersucht.
Ethnische Zugehörigkeit
Bei asiatischen Patienten ist die Talspiegelkonzentration von Nerandomilast um bis zu 47 % höher als bei Patienten kaukasischer Herkunft. Dieser Effekt dürfte klinisch nicht relevant sein.
Körpergewicht
Eine populationspharmakokinetische Analyse ergab eine höhere Exposition gegenüber Nerandomilast bei Patienten mit niedrigerem Körpergewicht und eine niedrigere Exposition bei Patienten mit höherem Körpergewicht. Der Einfluss des Körpergewichts auf die Plasmakonzentrationen von Nerandomilast dürfte klinisch nicht relevant sein.
Pharmakokinetische/pharmakodynamische Zusammenhänge
Ein Anstieg der Nerandomilast-Talspiegel im Steady State ging bei Patienten mit IPF und PPF mit einer besseren Wirksamkeit einher, was sich in einem geringeren Rückgang der absoluten forcierten Vitalkapazität (FVC) gegenüber dem Ausgangswert über einen Zeitraum von 52 Wochen zeigte.
Allgemeine Toxikologie
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Genotoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Es gibt keine Hinweise auf ein phototoxisches Potenzial.
Studien zur Toxizität bei wiederholter Gabe wurden an Ratten, Minischweinen und Affen durchgeführt. Eine Vaskulopathie (Entzündung, Hämorrhagie und Nekrose von Gefäßwänden) war der Hauptbefund bei Ratten und Minischweinen. Bei Affen, denen bis zu 30 mg/kg/Tag verabreicht wurden (10‑fache Humanexposition, basierend auf der AUC unter der maximal empfohlenen Humandosis [Maximum Recommended Human Dose, MRHD] [siehe Abschnitt 4.2]), wurden keine negativen Auswirkungen auf die Gefäße beobachtet. Die bei Ratten und Minischweinen beobachteten negativen Auswirkungen auf die Gefäße betrafen verschiedene Gewebe (z. B. Mesenterium und Gastrointestinaltrakt bei Ratten, Herz und Lunge bei Minischweinen). In Langzeitstudien wurden bei Ratten unter einer Dosierung von 2 mg/kg/Tag keine unerwünschten Gefäßveränderungen beobachtet, während bei einem Minischwein unter einer Dosierung von 3 mg/kg/Tag Gefäßveränderungen auftraten (beide Dosen entsprachen der Exposition beim Menschen, basierend auf der AUC unter der MRHD). Die Expositionsspanne bei Affen, die als die für den Menschen relevanteste Spezies gelten, deutet darauf hin, dass Primaten weniger empfindlich auf Gefäßeffekte reagieren als andere Spezies.
In toxikologischen Studien an Affen mit Expositionen, die mehr als das 10‑Fache der Humanexposition betrugen (basierend auf der AUC unter der MRHD), wurden Erbrechen und Herzveränderungen (fokale Degeneration oder Nekrose ohne begleitende Gefäßveränderungen) beobachtet.
Reproduktions- und Entwicklungstoxizität
Fertilität und frühe Embryonalentwicklung
In einer Fertilitätsstudie an männlichen Ratten zeigte die Anwendung von Nerandomilast in einer Dosis von 6 mg/kg/Tag (entspricht etwa der 4‑fachen Humanexposition, basierend auf der AUC unter der MRHD) keine Auswirkungen auf Paarung, Fertilität oder Spermienparameter.
Bei weiblichen Ratten wurden unter der höchsten getesteten Dosis von 9 mg/kg/Tag (entspricht etwa der 9‑fachen Humanexposition, basierend auf der AUC unter der MRHD) geringere Indizien hinsichtlich Paarung und Fertilität beobachtet, zurückzuführen auf eine allgemeine Toxizität. Unter der Dosis ohne beobachtbare schädliche Wirkungen (No Observed Adverse Effects Level, NOAEL) von 6 mg/kg/Tag (entspricht etwa der 4‑fachen Humanexposition unter der MRHD) wurden keine derartigen Effekte beobachtet. In den Studien zur Fertilität, zur frühen Embryonalentwicklung und zur embryo-fötalen Entwicklung wurde bei Ratten bei Dosen von ≥ 6 mg/kg/Tag eine Zunahme früher Resorption beobachtet. Unter der NOAEL-Dosis von 3 mg/kg/Tag (etwa die 3‑fache Humanexposition unter der MRHD) wurden keine derartigen Wirkungen beobachtet.
Bei geschlechtsreifen weiblichen Affen, denen Nerandomilast über einen Zeitraum von 39 Wochen verabreicht wurde, trat unter Dosierungen von 10 mg/kg/Tag und 30 mg/kg/Tag (entspricht etwa der 3- bzw. 10‑fachen Humanexposition, basierend auf der AUC unter der MRHD) eine sporadische Verlängerung des Menstruationszyklus auf. Bei einer Dosierung von 3 mg/kg/Tag (entspricht der Humanexposition, basierend auf der AUC unter der MRHD) wurden bei Affen keine Veränderungen des Menstruationszyklus beobachtet.
Bei Ratten wurden keine Veränderungen des Östruszyklus festgestellt.
Embryofetale Entwicklung
Nach Anwendung von radioaktiv markiertem Nerandomilast bei trächtigen Ratten wurde Radioaktivität in der Plazenta, im embryofetalen Blut und Gewebe nachgewiesen, was auf einen Übertritt durch die Plazentaschranke hindeutet.
Studien zur embryofetalen Entwicklung an Ratten und Kaninchen zeigten bis zu den höchsten getesteten Dosen von 9 mg/kg/Tag bzw. 15 mg/kg/Tag, entsprechend der 7- bzw. 4‑fachen Humanexposition, basierend auf der AUC unter der MRHD, keine Teratogenität, keine Skelettveränderungen und keine Fetotoxizität. Bei Ratten wurde eine embryofetale Letalität aufgrund von Verlusten nach der Implantation in Verbindung mit einer Zunahme früher Resorption bei einer Dosis von 6 mg/kg/Tag (die 5‑fache Humanexposition, basierend auf der AUC unter der MRHD) beobachtet, die vom 6. bis zum 17. Trächtigkeitstag verabreicht wurde. Bei Ratten und Kaninchen wurde unter der NOAEL‑Dosis von 3 mg/kg/Tag bzw. 15 mg/kg/Tag (etwa die 3- bzw. 4‑fache Humanexposition, basierend auf der AUC unter der MRHD) keine embryofetale Letalität beobachtet.
Prä- und postnatale Entwicklung
In einer Dosisfindungsstudie zur prä- und postnatalen Entwicklung an Ratten führte eine maternale Dosis von 6 mg/kg/Tag (entspricht etwa der 5- bis 6‑fachen Humanexposition unter der MRHD) vom 6. Trächtigkeitstag bis zum 6. Laktationstag zu einem geringeren Geburtsgewicht der Jungtiere. In der zulassungsrelevanten Studie zur prä- und postnatalen Entwicklung zeigte Nerandomilast, das trächtigen Ratten vom 6. Trächtigkeitstag bis zum 20. Laktationstag verabreicht wurde, bis zur höchsten getesteten Dosis von 3 mg/kg/Tag (entspricht etwa der 2‑fachen Humanexposition, basierend auf der AUC unter der MRHD) keine negativen Auswirkungen auf die Leistung der Muttertiere, keine Toxizität und keine Auswirkungen auf die Entwicklung, das Verhalten und die Reproduktionsleistung der F1‑Generation (Nachkommen). In dieser Studie war Nerandomilast während der Laktationsphase im Plasma der Rattenjungen nachweisbar.
In einer Studie zur Milchsekretion nach Einmalgabe bei säugenden Ratten, denen radioaktiv markiertes Nerandomilast oral verabreicht wurde, wurden in der Milch und im Plasma der laktierenden Weibchen ähnliche Konzentrationen der Gesamtradioaktivität beobachtet, wobei die maximale radioaktive Konzentration 1 Stunde nach der Gabe erreicht wurde und 24 Stunden nach der Gabe signifikant abgenommen hatte. Die Konzentration der Gesamtradioaktivität in Tiermilch lässt nicht zwangsläufig Rückschlüsse auf die Konzentration des Wirkstoffs in Muttermilch zu.
9 mg Filmtabletten
Lactose-Monohydrat
Mikrokristalline Cellulose
Hydroxypropylcellulose
Croscarmellose-Natrium
Magnesiumstearat
Hypromellose 2910
Talkum
Mannitol
Macrogol 8000
Eisen(III)-hydroxid-oxid × H2O (E 172)
18 mg Filmtabletten
Lactose-Monohydrat
Mikrokristalline Cellulose
Hydroxypropylcellulose
Croscarmellose-Natrium
Magnesiumstearat
Hypromellose 2910
Talkum
Mannitol
Macrogol 8000
Eisen(III)-oxid (E 172)
Eisen(II, III)-oxid (E 172)
Nicht zutreffend.
2 Jahre
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Für dieses Arzneimittel sind bezüglich der Temperatur keine besonderen Lagerungsbedingungen erforderlich.
Perforierte PVC/Aluminium-Einzeldosis-Blisterpackungen
Packungsgrößen: 10 × 1, 30 × 1 oder 60 × 1 Filmtablette
HDPE‑Flasche mit kindergesichertem Verschluss
Packungsgrößen: 60 oder 180 Filmtabletten
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Boehringer Ingelheim International GmbH
Binger Straße 173
55216 Ingelheim am Rhein
Deutschland
Jascayd 9 mg Filmtabletten
EU/1/26/2040/001
EU/1/26/2040/002
EU/1/26/2040/003
EU/1/26/2040/004
EU/1/26/2040/005
Jascayd 18 mg Filmtabletten
EU/1/26/2040/006
EU/1/26/2040/007
EU/1/26/2040/008
EU/1/26/2040/009
EU/1/26/2040/010
Datum der Erteilung der Zulassung: 15. Juli 2026
Juli 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.