
SOMAVERT® 10 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
SOMAVERT® 15 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
SOMAVERT® 20 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
SOMAVERT® 25 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
SOMAVERT® 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
SOMAVERT 10 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
1 Durchstechflasche enthält 10 mg Pegvisomant.
Nach Rekonstitution enthält 1 ml Lösung 10 mg Pegvisomant.*
Sonstiger Bestandteil mit bekannter Wirkung
Die 10-mg-Stärke des Arzneimittels enthält 0,4 mg Natrium pro Durchstechflasche.
SOMAVERT 15 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
1 Durchstechflasche enthält 15 mg Pegvisomant.
Nach Rekonstitution enthält 1 ml Lösung 15 mg Pegvisomant.*
Sonstiger Bestandteil mit bekannter Wirkung
Die 15-mg-Stärke des Arzneimittels enthält 0,4 mg Natrium pro Durchstechflasche.
SOMAVERT 20 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
1 Durchstechflasche enthält 20 mg Pegvisomant.
Nach Rekonstitution enthält 1 ml Lösung 20 mg Pegvisomant.*
Sonstiger Bestandteil mit bekannter Wirkung
Die 20-mg-Stärke des Arzneimittels enthält 0,4 mg Natrium pro Durchstechflasche.
SOMAVERT 25 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
1 Durchstechflasche enthält 25 mg Pegvisomant.
Nach Rekonstitution enthält 1 ml Lösung 25 mg Pegvisomant.*
Sonstiger Bestandteil mit bekannter Wirkung
Die 25-mg-Stärke des Arzneimittels enthält 0,5 mg Natrium pro Durchstechflasche.
SOMAVERT 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
1 Durchstechflasche enthält 30 mg Pegvisomant.
Nach Rekonstitution enthält 1 ml Lösung 30 mg Pegvisomant.*
Sonstiger Bestandteil mit bekannter Wirkung
Die 30-mg-Stärke des Arzneimittels enthält 0,6 mg Natrium pro Durchstechflasche.
*in Escherichia-coli-Zellen durch rekombinante DNA-Technologie hergestellt.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung (Pulver zur Herstellung einer Injektionszubereitung)
Das Pulver ist weiß bis leicht gelblich.
Behandlung der Akromegalie bei erwachsenen Patienten, bei denen Operation und/oder Strahlentherapie nicht den gewünschten Behandlungserfolg erzielten und bei denen eine adäquate medikamentöse Behandlung mit Somatostatinanaloga die IGF-I-Konzentration nicht normalisierte bzw. nicht vertragen wurde.
Die Behandlung muss unter Aufsicht eines Arztes eingeleitet werden, der in der Behandlung der Akromegalie erfahren ist.
Dosierung
Eine Startdosis von 80 mg Pegvisomant ist subkutan unter ärztlicher Aufsicht zu verabreichen.
Nachfolgend sind täglich SOMAVERT 10 mg gelöst in 1 ml Lösungsmittel als subkutane Injektion zu geben.
Dosisanpassungen müssen auf Serumspiegeln von Insulin-like Growth Factor I (IGF‑I) beruhen. Alle 4 bis 6 Wochen sind die Serumkonzentrationen von IGF‑I zu bestimmen und es müssen geeignete Dosisanpassungen in Schritten von 5 mg/Tag erfolgen, um die IGF‑I-Serumkonzentrationen im altersgemäßen Normbereich zu halten und einen optimalen Therapieerfolg zu erzielen.
Beurteilung der Ausgangs-Leberenzymwerte vor Einleitung der Behandlung mit SOMAVERT
Vor Beginn der Behandlung mit SOMAVERT sollte eine Beurteilung der Ausgangs‑Leberwerte (LW) der Patienten erfolgen [Alanin‑Aminotransferase (ALT) im Serum, Aspartat‑Aminotransferase (AST), Gesamtbilirubin im Serum (Serum Total Bilirubin, TBIL) und alkalische Phosphatase (ALP)]. Empfehlungen zur Einleitung der Behandlung mit SOMAVERT basierend auf den Ausgangs‑LW und Empfehlungen zur Überwachung der LW während der Behandlung mit SOMAVERT, siehe Tabelle A im Abschnitt Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung (4.4).
Die Höchstdosis sollte 30 mg/Tag nicht überschreiten.
Für die verschiedenen Dosierungen stehen folgende Stärken zur Verfügung: SOMAVERT 10 mg, SOMAVERT 15 mg, SOMAVERT 20 mg, SOMAVERT 25 mg und SOMAVERT 30 mg.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von SOMAVERT bei Kindern im Alter von 0 bis 17 Jahren sind nicht erwiesen. Es liegen keine Daten vor.
Ältere Menschen
Es ist keine Dosisanpassung erforderlich.
Eingeschränkte Leber- oder Nierenfunktion
Die Sicherheit und Wirksamkeit von SOMAVERT wurden bei Patienten mit einer eingeschränkten Nieren- oder Leberfunktion nicht untersucht.
Art der Anwendung
Pegvisomant sollte als subkutane Injektion gegeben werden.
Die Injektionsstelle ist täglich zu wechseln, um einer Lipohypertrophie vorzubeugen.
Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Wachstumshormon-sezernierende Tumore
Da sich Wachstumshormon-sezernierende Hypophysentumore manchmal vergrößern und schwerwiegende Komplikationen verursachen können (z. B. Gesichtsfeldausfälle), müssen alle Patienten sorgfältig überwacht werden. Wird ein Tumorwachstum nachgewiesen, sind möglicherweise alternative Verfahren angezeigt.
Überwachung der IGF-1-Konzentration im Serum
Pegvisomant ist ein potenter Antagonist der Wirkung des Wachstumshormons. Aus der Anwendung dieses Arzneimittels kann daher ein Wachstumshormon-Mangel-Status resultieren, obwohl erhöhte Serumspiegel an Wachstumshormon vorliegen. Die Konzentration von IGF-I im Serum muss überwacht werden und durch Anpassung der Pegvisomant-Dosis innerhalb des altersgemäßen Normalbereiches gehalten werden.
ALT- oder AST-Erhöhungen
Vor Beginn der Behandlung mit SOMAVERT sollte eine Beurteilung der Ausgangs-Leberwerte der Patienten erfolgen [Alanin‑Aminotransferase (ALT) im Serum, Aspartat‑Aminotransferase (AST), Gesamtbilirubin im Serum (Serum Total Bilirubin, TBIL) und alkalische Phosphatase (ALP)].
Bei Patienten mit ALT- und AST‑Erhöhungen oder bei Patienten mit einer Behandlung mit einem Somatostatinanalogon in der Vorgeschichte müssen Anzeichen einer obstruktiven Gallengangserkrankung ausgeschlossen sein. Wenn Anzeichen für eine Lebererkrankung bestehen bleiben, ist die Behandlung mit Pegvisomant abzubrechen.
Empfehlungen zur Einleitung der Behandlung mit SOMAVERT basierend auf den Ausgangs-Leberwerten (LW) und Empfehlungen zur Überwachung der Leberwerte während der Behandlung mit SOMAVERT, siehe Tabelle A.
Tabelle A: Empfehlungen für die Einleitung der Behandlung mit SOMAVERT basierend auf den Ausgangs‑LW und für die regelmäßige Überwachung der LW während der Behandlung mit SOMAVERT
Ausgangs-LW |
Empfehlungen |
Normal |
|
Erhöht, aber unterhalb oder gleich dem 3‑fachen der Obergrenze des Normbereichs (ULN) |
|
Über dem 3‑fachen der Obergrenze des Normbereichs (ULN) |
|
Abkürzungen: ALT = Alanin-Aminotransferase; AST = Aspartat-Aminotransferase; LW = Leberwerte; ULN = upper limit of normal (Obergrenze des Normbereichs).
Falls ein Patient während der Behandlung mit SOMAVERT eine Erhöhung der LW oder andere Anzeichen oder Symptome einer Leberfunktionsstörung entwickelt, wird die folgende Behandlung des Patienten empfohlen (Tabelle B).
Tabelle B. Klinische Empfehlungen basierend auf anormalen Ergebnissen von Leberfunktionstests während der Anwendung von SOMAVERT
Leberwerte und klinische Anzeichen/Symptome |
Empfehlungen |
|
Erhöht, aber unterhalb oder gleich dem 3‑fachen der Obergrenze des Normbereichs (ULN) |
|
|
Höher als das 3‑fache, aber niedriger als das 5‑fache der Obergrenze des Normbereichs (ULN) (ohne Anzeichen/Symptome einer Hepatitis oder einer anderen Leberschädigung bzw. einer Erhöhung des TBIL im Serum) |
|
|
Mindestens das 5‑fache der Obergrenze des Normbereichs (ULN) oder Transaminase-Erhöhungen, die mindestens dem 3‑fachen der Obergrenze des Normbereichs (ULN) entsprechen und mit einer Erhöhung des TBIL im Serum in Zusammenhang stehen (mit oder ohne Anzeichen/Symptome einer Hepatitis oder einer anderen Leberschädigung) |
|
|
Anzeichen oder Symptome, die auf Hepatitis oder eine andere Leberschädigung hinweisen (z. B. Gelbsucht, Bilirubinurie, Müdigkeit, Übelkeit, Erbrechen, Schmerzen im rechten oberen Quadranten, Aszites, Ödem mit ungeklärter Ursache, leichtes Entstehen von Blutergüssen) |
|
|
Hypoglykämie
Eine klinische Prüfung mit Pegvisomant an Diabetes-Patienten, die entweder mit Insulin oder oralen Antidiabetika behandelt wurden, zeigte ein Hypoglykämierisiko in dieser Gruppe. Daher kann es bei Akromegalie-Patienten mit Diabetes mellitus erforderlich sein, die Dosis für Insulin oder orale Antidiabetika zu senken (siehe Abschnitt 4.5).
Verbesserte Fertilität
Die therapeutischen Vorteile einer Reduktion der IGF-I-Konzentration, die zu einer Verbesserung des klinischen Zustandes der Patienten führt, können möglicherweise auch die Fertilität bei weiblichen Patienten verbessern (siehe Abschnitt 4.6).
Schwangerschaft
Die Kontrolle der Akromegalie kann sich während der Schwangerschaft verbessern. Die Gabe von Pegvisomant während der Schwangerschaft wird nicht empfohlen (siehe Abschnitt 4.6). Falls Pegvisomant während der Schwangerschaft angewendet wird, sollten die IGF-I-Werte engmaschig überwacht und die Pegvisomant-Dosis gegebenenfalls basierend auf den IGF-I-Werten angepasst werden (siehe Abschnitt 4.2).
Natriumgehalt
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosiervolumen. Patienten unter einer natriumarmen Diät können informiert werden, dass dieses Arzneimittel nahezu „natriumfrei“ ist.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt. Die Fortsetzung der Behandlung mit Somatostatinanaloga ist kritisch abzuwägen. Die Anwendung dieses Arzneimittels in Kombination mit anderen Arzneimitteln zur Behandlung der Akromegalie wurde nicht gesondert untersucht.
Bei Patienten, die Insulin oder orale Antidiabetika erhalten, kann wegen der Auswirkung von Pegvisomant auf die Insulinsensitivität eine Dosisreduktion dieser Wirkstoffe erforderlich sein (siehe Abschnitt 4.4).
Pegvisomant besitzt eine ausgeprägte strukturelle Ähnlichkeit mit Wachstumshormon, was zu Kreuzreaktionen mit kommerziell verfügbaren Wachstumshormon-Assays führt. Da bei Akromegalie-Patienten die Serumkonzentrationen dieses Arzneimittels in therapeutisch wirksamen Dosen im Allgemeinen 100- bis 1.000‑fach höher liegen als die tatsächlichen Serumkonzentrationen von Wachstumshormon, werden bei Anwendung kommerziell verfügbarer Wachstumshormon-Assays falsche Werte für Wachstumshormon-Serumkonzentrationen berichtet. Daher darf eine Pegvisomant-Behandlung nicht auf der Grundlage von Wachstumshormonkonzentrationen im Serum, die mit solchen Assays bestimmt werden, überwacht oder angepasst werden.
Schwangerschaft
Es liegen begrenzte Daten zur Anwendung von Pegvisomant bei schwangeren Frauen vor. Es liegen keine ausreichenden tierexperimentellen Studien in Bezug auf die Reproduktionstoxizität vor (siehe Abschnitt 5.3).
SOMAVERT wird während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die keine Empfängnisverhütung anwenden, nicht empfohlen.
Falls Pegvisomant während der Schwangerschaft angewendet wird, sollten die IGF-I-Werte engmaschig überwacht werden, insbesondere im ersten Trimenon. Möglicherweise muss die Pegvisomant-Dosis während der Schwangerschaft angepasst werden (siehe Abschnitt 4.4).
Stillzeit
Der Übergang von Pegvisomant in die Muttermilch wurde in tierexperimentellen Studien nicht untersucht. Klinische Daten liegen begrenzt vor (ein Fall wurde berichtet) und erlauben keine Rückschlüsse auf den Übergang von Pegvisomant in die Muttermilch beim Menschen. Deshalb sollte Pegvisomant bei stillenden Frauen nicht angewendet werden. Bei Absetzen dieses Arzneimittels kann jedoch weiter gestillt werden: Der Nutzen der Pegvisomant-Behandlung für die Mutter und der Nutzen des Stillens für das Kind sind bei dieser Entscheidung abzuwägen.
Fertilität
Für Pegvisomant liegen keine Daten zur Fertilität vor.
Der therapeutische Nutzen durch die Verringerung der IGF-I-Konzentration, der zu einer Verbesserung des klinischen Zustands der Patienten führt, könnte bei Patientinnen auch die Fertilität verbessern.
Studien zu Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen wurden nicht durchgeführt.
Zusammenfassung des Sicherheitsprofils
Die folgende Liste enthält Nebenwirkungen, die in klinischen Prüfungen mit SOMAVERT beobachtet wurden.
In klinischen Studien an mit Pegvisomant behandelten Patienten (n = 550) war die Mehrzahl der unerwünschten Wirkungen von Pegvisomant gering bis mäßig ausgeprägt, von begrenzter Dauer und erforderte kein Absetzen der Behandlung.
Die am häufigsten berichteten unerwünschten Reaktionen, die bei mehr als 10 % der mit Pegvisomant behandelten Akromegalie-Patienten in klinischen Prüfungen auftraten, waren Kopfschmerzen 25 %, Arthralgie 16 % und Diarrhö 13 %.
Tabellarische Auflistung der Nebenwirkungen
Die nachfolgend aufgeführten Nebenwirkungen wurden in klinischen Studien beobachtet oder stammen aus Spontanberichten und sind nach Organsystemen und entsprechend ihrer Häufigkeiten in Kategorien eingeteilt.
Die Nebenwirkungen sind mit folgenden Häufigkeiten aufgelistet:
Sehr häufig: |
≥ 1/10 |
Systemorganklasse |
Sehr häufig |
Häufig |
Gelegentlich |
Nicht bekannt |
Erkrankungen des Blutes und des Lymphsystems |
Thrombozytopenie, Leukopenie, Leukozytose, hämorrhagische Diathese |
|||
Erkrankungen des Immunsystems |
Hypersensitivitätsreaktionenb |
anaphylaktische Reaktionb, anaphylaktoide Reaktionb |
||
Stoffwechsel- und Ernährungsstörungen |
Hypercholesterinämie, Hyperglykämie, Hypoglykämie, Gewichtszunahme |
Hypertriglyzeridämie |
||
Psychiatrische Erkrankungen |
anormale Träume |
Panikattacke, Ausfall des Kurzzeitgedächtnisses, Apathie, Verwirrung, Schlafstörung, gesteigerte Libido |
Zornesausbruch |
|
Erkrankungen des Nervensystems |
Kopfschmerzen |
Somnolenz, Tremor, Schwindel, Hypästhesie |
Narkolepsie, Migräne, Geschmacksstörungen |
|
Augenerkrankungen |
Schmerzen am Auge |
Asthenopie |
||
Erkrankungen des Ohrs und des Labyrinths |
Ménière-Krankheit |
|||
Herzerkrankungen |
periphere Ödeme |
|||
Gefäßerkrankungen |
Hypertonie |
|||
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Dyspnoe |
Laryngospasmusb |
||
Erkrankungen des Gastrointestinaltrakts |
Diarrhö |
Erbrechen, Konstipation, Übelkeit, aufgetriebenes Abdomen, Dyspepsie, Flatulenz |
Hämorrhoiden, verstärkter Speichelfluss, trockener Mund, Störungen an den Zähnen |
|
Leber- und Gallenerkrankungen |
anormale Leberfunktionstests (z. B. Erhöhung der Transaminase) (siehe Abschnitt 4.4) |
|||
Erkrankungen der Haut und des Unterhautzellgewebes |
Hyperhidrose, Quetschung, Pruritusb, Hautausschlagb |
Gesichtsödem, trockene Haut, Hämatomneigung, nächtliches Schwitzen, Erythemb, Urtikariab |
Angioödemb |
|
Skelettmuskulatur und Bindegewebserkrankungen |
Arthralgie |
Myalgie, Arthritis |
||
Erkrankungen der Nieren und Harnwege |
Hämaturie |
Proteinurie, Polyurie, Nierenfunktionsstörungen |
||
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Reaktionen (einschließlich Hypersensitivität) an der Injektionsstelle, Hämatom oder Blutung an der Injektionsstelle, Hypertrophie (z. B. Lipohypertrophie) an der Injektionsstellea, grippeähnliche Erkrankung, Müdigkeit, Asthenie, Pyrexie |
abnormes Befinden, Wundheilungsstörungen, Hunger |
a siehe unten Beschreibung spezieller Nebenwirkungen.
b Nebenwirkungen im Zusammenhang mit Hypersensitivitätsreaktionen
Beschreibung ausgewählter Nebenwirkungen
Die meisten Reaktionen an der Injektionsstelle, beschrieben als lokalisierte Erytheme und Wundsein, verbesserten sich spontan durch eine lokale symptomatische Behandlung, während die Behandlung mit Pegvisomant fortgesetzt wurde. Das Auftreten von Hypertrophie an der Injektionsstelle, einschließlich Lipohypertrophie, wurde beobachtet.
Bei 16,9 % der mit Pegvisomant behandelten Patienten wurde die Entwicklung von isolierten niedrigtitrigen Antikörpern gegenüber Wachstumshormon beobachtet. Die klinische Signifikanz dieser Antikörper ist nicht bekannt.
Es wurde über systemische Hypersensitivitätsreaktionen, einschließlich Anaphylaxie/ anaphylaktoide Reaktionen, Laryngospasmus, Angioödeme und generalisierte Hautreaktionen (Hautausschlag, Erythem, Pruritus, Urtikaria) während der Anwendung nach Markteinführung berichtet. Bei einigen Patienten war eine Krankenhauseinweisung erforderlich. Bei erneuter Verabreichung traten die Symptome nicht bei allen Patienten wieder auf.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das aufgeführte nationale Meldesystem anzuzeigen.
Deutschland |
Österreich |
Zur Überdosierung mit Pegvisomant liegen begrenzte Erfahrungen vor. In dem einen berichteten Fall einer akuten Überdosierung, bei dem 80 mg/Tag über 7 Tage gegeben wurden, entwickelte der Patient eine geringfügig verstärkte Müdigkeit und Mundtrockenheit. In der Woche nach Absetzen der Behandlung wurden folgende Nebenwirkungen beobachtet: Insomnie, verstärkte Müdigkeit, periphere Ödeme, Tremor und Gewichtszunahme. Zwei Wochen nach Behandlungsende wurden Leukozytose und mäßige Blutungen an den Injektions- und Venenpunktionsstellen beobachtet, welche als möglicherweise mit Pegvisomant in Zusammenhang stehend angesehen wurden.
Im Falle einer Überdosierung sollte die Anwendung dieses Arzneimittels unterbrochen und nicht wieder aufgenommen werden, bis die IGF-I-Spiegel in den oder über den Normalbereich zurückgekehrt sind.
Pharmakotherapeutische Gruppe: Andere Hypophysenvorderlappenhormone und Analoga, ATC‑Code: H01AX01.
Wirkmechanismus
Pegvisomant ist ein Analogon des menschlichen Wachstumshormons und wurde gentechnologisch zu einem Wachstumshormon-Rezeptor-Antagonisten verändert. Pegvisomant bindet an Wachstumshormonrezeptoren der Zelloberfläche, wo es die Bindung von Wachstumshormon blockiert und hierdurch mit der intrazellulären Signaltransduktion von Wachstumshormon interferiert. Pegvisomant ist hochselektiv für den Wachstumshormon-Rezeptor, weshalb keine Kreuzreaktionen mit anderen Zytokinrezeptoren, einschließlich des Prolaktinrezeptors auftreten.
Pharmakodynamische Wirkungen
Eine Hemmung der Wachstumshormon-Wirkung durch Pegvisomant führt zu einer Abnahme der Serumkonzentration von IGF-I und anderen Wachstumshormon-abhängigen Serumproteinen wie dem freien IGF‑I, der säurelabilen Untereinheit von IGF-I (ALS) und dem IGF-Bindungsprotein-3 (IGFBP-3).
Klinische Wirksamkeit und Sicherheit
In einer 12-wöchigen randomisierten, doppelblinden Multizenterstudie zum Vergleich von Placebo und Pegvisomant, wurden Patienten mit Akromegalie (n = 112) behandelt. Zu allen Untersuchungszeitpunkten nach Behandlungsbeginn wurden in den mit Pegvisomant behandelten Gruppen dosisabhängige, statistisch signifikante Reduktionen der Mittelwerte für IGF-I (p < 0,0001), freies IGF-I (p < 0,05), IGFBP-3 (p < 0,05) und ALS (p < 0,05) beobachtet. Am Ende dieser Studie (Woche 12) normalisierte sich das Serum-IGF-I bei 9,7 %, 38,5 %, 75 % und 82 % der Personen, die mit Placebo, Pegvisomant 10 mg/Tag, 15 mg/Tag bzw. 20 mg/Tag, behandelt wurden.
Statistisch signifikante (p < 0,05) Verbesserungen des Gesamt-Symptomen-Scores wurden für alle Dosisgruppen gegenüber Placebo beobachtet.
Eine Gruppe von 38 Akromegalie-Patienten wurde über mindestens 12 aufeinander folgende Monate (Mittelwert = 55 Wochen) im Rahmen einer offenen Dosis-Titrations-Langzeitstudie mit täglicher Pegvisomant-Gabe beobachtet. In dieser Gruppe sank die durchschnittliche IGF-I-Konzentration von 917 ng/ml auf 299 ng/ml nach Pegvisomant-Gabe, wobei 92 % eine normale (altersgemäße) IGF‑I‑Konzentration erreichten.
In verschiedenen Studien, so auch in der Studie Acrostudy, normalisierte sich unter Pegvisomant die IGF-1-Konzentration bei einem hohen Prozentsatz von Patienten (> 70 %). Des Weiteren wurde eine signifikante Verringerung der Konzentration von Nüchternplasmaglukose (fasting plasma glucose, FPG) und Nüchternplasmainsulin (fasting plasma insulin, FPI) festgestellt.
Pegvisomant verbessert zudem die Insulinsensitivität. Dies ist wahrscheinlich auf eine Blockade der GH-Rezeptoren im Gewebe zurückzuführen, vor allem in der Leber, aber auch im Fettgewebe, in den Nieren und in der Skelettmuskulatur, wodurch die nachteilige Wirkung von GH auf Insulin-Signalwege, Lipolyse und Gluconeogenese aufgehoben wird. Der Wirkmechanismus aller genannten Effekte ist jedoch nicht abschließend bekannt. Bei Akromegalie-Patienten mit Diabetes mellitus kann eine Verringerung der Insulin- oder Antidiabetika-Dosis erforderlich sein (siehe Abschnitt 4.4 und 4.5).
Resorption
Nach subkutaner Gabe ist die Resorption von Pegvisomant langsam und verzögert, und Serum-Spitzenspiegel von Pegvisomant werden im Allgemeinen erst 33 bis 77 Stunden nach der Gabe erreicht. Der mittlere Resorptionsgrad einer subkutanen Dosis lag, verglichen mit einer intravenösen Dosis, bei 57 %.
Verteilung
Das scheinbare Verteilungsvolumen von Pegvisomant ist relativ gering (7 bis 12 l).
Biotransformation
Der Metabolismus von Pegvisomant wurde nicht untersucht.
Elimination
Die mittlere Gesamtkörperclearance von Pegvisomant nach Mehrfachdosierung liegt schätzungsweise bei 28 ml/Stunde für subkutane Dosierungen und im Bereich von 10 bis 20 mg/Tag. Die renale Clearance von Pegvisomant ist vernachlässigbar, sie beträgt weniger als 1 % der Gesamtkörperclearance. Pegvisomant wird langsam aus dem Serum eliminiert. Die geschätzte mittlere Halbwertszeit nach Einzel- oder Mehrfachdosierungen liegt im Allgemeinen im Bereich von 74 bis 172 Stunden.
Linearität/Nicht-Linearität
Nach einer subkutanen Einzeldosis von Pegvisomant wurde mit steigenden Dosen von 10, 15 oder 20 mg keine Linearität beobachtet. Im Steady State wurde bei pharmakokinetischen Populationsstudien eine annähernd lineare Pharmakokinetik beobachtet. Die Daten von 145 Patienten, die in 2 Langzeitstudien täglich 10, 15 oder 20 mg erhielten, zeigen mittlere Pegvisomant-Serumkonzentrationen (± SD) von ungefähr 8.800 ± 6.300, 13.200 ± 8.000 und 15.600 ± 10.300 ng/ml.
Die Pharmakokinetik von Pegvisomant ist bei normalen gesunden Probanden und Akromegalie-Patienten vergleichbar, obwohl Individuen mit höherem Körpergewicht zu einer höheren Gesamtkörperclearance von Pegvisomant tendieren als Individuen mit geringerem Körpergewicht und daher höhere Dosen an Pegvisomant benötigen können.
Basierend auf den Studien zur Toxizität bei wiederholter Gabe an Ratten und Affen lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Wegen der ausgeprägten pharmakologischen Wirkung am Affen wurde jedoch keine systemische Exposition untersucht, welche die bei Patienten mit therapeutischer Dosierung übersteigt.
Maligne fibröse Histiozytome mit Fibrose und histiozytärer Entzündung wurden an den Injektionsstellen bei den Männchen in einer Kanzerogenitätsstudie an Ratten beobachtet. Hierbei wurde von einer Exposition ausgegangen, die der 3‑fachen Exposition beim Menschen basierend auf den mittleren Plasmakonzentrationen in 2 Langzeitstudien mit einer täglichen Dosis von 30 mg entspricht. Die Bedeutung dieser Reaktion für den Menschen ist derzeit nicht bekannt. Die erhöhte Inzidenz von Tumoren an der Injektionsstelle war am wahrscheinlichsten auf eine Irritation und die hohe Empfindlichkeit der Ratte für wiederholte subkutane Injektionen zurückzuführen.
Studien zur frühen embryonalen und embryofetalen Entwicklung wurden an trächtigen Kaninchen mit Pegvisomant in subkutan verabreichten Dosen von 1 mg/kg/Tag, 3 mg/kg/Tag und 10 mg/kg/Tag durchgeführt. Es gab keine Anzeichen für teratogene Wirkungen in Verbindung mit der Verabreichung von Pegvisomant während der Organogenese. Bei 10 mg/kg/Tag (dem 6-Fachen der therapeutischen Höchstdosis beim Menschen basierend auf der Körperoberfläche) wurde in beiden Studien ein erhöhter Postimplantationsverlust beobachtet. Eine Studie zur Fertilität wurde nicht durchgeführt.
Pulver
Glycin
Mannitol (E 421)
Dinatriumhydrogenphosphat
Natriumdihydrogenphosphat 1 H2O
Lösungsmittel
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
3 Jahre
Nach Rekonstitution ist das Arzneimittel sofort zu verwenden.
Die Durchstechflasche(n) im Kühlschrank lagern (2 ºC - 8 ºC). Nicht einfrieren. Die Durchstechflasche(n) in der/den Faltschachtel(n) aufbewahren, um den Inhalt vor Licht zu schützen.
Die Faltschachtel(n) mit der/den Durchstechflasche(n) mit SOMAVERT-Pulver kann/können einmalig bis zu 30 Tage lang bei einer Raumtemperatur von maximal 25 °C aufbewahrt werden. Das Verbrauchsdatum ist auf der Faltschachtel zu notieren (bis zu 30 Tage ab dem Datum der Entnahme aus dem Kühlschrank). Die Durchstechflasche(n) muss/müssen vor Licht geschützt werden und darf/dürfen nicht wieder in den Kühlschrank gestellt werden. Die Durchstechflasche(n) mit SOMAVERT-Pulver muss/müssen entsorgt werden, wenn sie nicht innerhalb von 30 Tagen bei Lagerung bei Raumtemperatur verwendet wird/werden oder das auf dem Umkarton aufgedruckte Verfalldatum erreicht ist, je nachdem, was früher eintritt.
Die Fertigspritze(n) nicht über 30 °C lagern oder im Kühlschrank lagern (2 °C ‑ 8 °C). Nicht einfrieren.
Nach Rekonstitution
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
Durchstechflasche (Typ-I-Flintglas) mit einem Stopfen (Chlorobutylkautschuk), die 10 mg oder 15 mg oder 20 mg oder 25 mg oder 30 mg Pegvisomant als Pulver enthält, und Fertigspritze (Typ-I-Borosilikatglas) mit einem Kolbenstopfen (Bromobutylkautschuk) und einer Schutzkappe (Bromobutylkautschuk), die 1 ml Lösungsmittel (Wasser für Injektionszwecke) enthält. Die Farbe der Kunststoffschutzkappe ist spezifisch für die Stärke des Arzneimittels.
SOMAVERT 10 mg und 15 mg
Packungsgröße zu 30 Durchstechflaschen, Fertigspritzen und Sicherheitsnadeln.
SOMAVERT 20 mg, 25 mg und 30 mg
Packungsgröße zu 1 und 30 Durchstechflasche(n), Fertigspritze(n) und Sicherheitsnadel(n).
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Die Spritze und die Sicherheitsnadel zur Gabe der Injektion werden mit dem Arzneimittel mitgeliefert.
Bevor die mitgelieferte Sicherheitsnadel auf die Spritze aufgesteckt werden kann, muss die Kappe von der Fertigspritze entfernt werden. Diese kann abgeknickt werden. Die Spritze sollte aufrecht gehalten werden, um ein Auslaufen zu verhindern. Das Ende der Spritze darf mit nichts in Berührung kommen.

Das Pulver ist mit 1 ml Lösungsmittel zu rekonstituieren. Beim Hinzufügen des Lösungsmittels aus der Spritze sollten Durchstechflasche und Spritze in einem bestimmten Winkel gehalten werden, wie unten im Bild dargestellt.
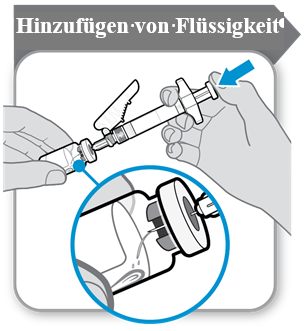
Das Lösungsmittel wird in die Durchstechflasche mit dem Pulver gegeben. Das Lösungsmittel ist langsam in die Durchstechflasche zu entleeren, um eine mögliche Schaumbildung zu verhindern. Dies würde das Arzneimittel unbrauchbar machen. Zum Auflösen das Pulver vorsichtig mit leichter, kreisender Bewegung schwenken. Nicht heftig schütteln, da dies zur Denaturierung des Wirkstoffs führen kann.
Vor Verabreichung ist die rekonstituierte Lösung visuell auf Fremdstoffe (oder -körper) oder irgendwelche Veränderungen der physischen Erscheinung zu überprüfen. Im Falle einer entsprechenden Beobachtung muss das Arzneimittel entsorgt werden.
Bevor das aufgelöste SOMAVERT in die Spritze aufgezogen wird, wird die Durchstechflasche mit der immer noch darin steckenden Spritze umgedreht und sichergestellt, dass die Öffnung im Stopfen sichtbar ist, wie unten im Bild dargestellt.
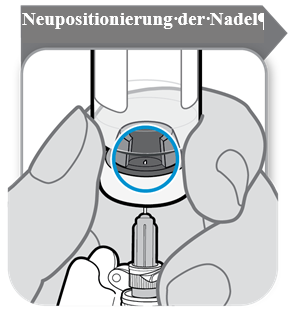
Die Nadel wird so weit herunter gezogen, dass sich die Nadelspitze am niedrigsten Punkt der Flüssigkeit befindet. Der Kolben wird in der Spritze langsam zurückgezogen, um das Arzneimittel aus der Durchstechflasche zu entnehmen. Sollte Luft in der Spritze zu sehen sein, auf den Spritzenzylinder klopfen, damit die Luftblasen nach oben steigen, und danach die Luftblasen vorsichtig in die Durchstechflasche drücken.
Vor der Entsorgung der Spritze und Nadel wird der Nadelschutz über die Nadel gelegt und sichergestellt, dass er einrastet. Spritze und Nadel sollten niemals wiederverwendet werden.
Nur zum einmaligen Gebrauch. Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Brüssel
Belgien
EU/1/02/240/001 10 mg 30 Durchstechflaschen
EU/1/02/240/002 15 mg 30 Durchstechflaschen
EU/1/02/240/004 20 mg 1 Durchstechflasche
EU/1/02/240/003 20 mg 30 Durchstechflaschen
EU/1/02/240/009 25 mg 1 Durchstechflasche
EU/1/02/240/010 25 mg 30 Durchstechflaschen
EU/1/02/240/011 30 mg 1 Durchstechflasche
EU/1/02/240/012 30 mg 30 Durchstechflaschen
Datum der Erteilung der Zulassung: 13. November 2002
Datum der letzten Verlängerung der Zulassung: 20. September 2007
Februar 2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
__________________________________________________________________________
VERKAUFSABGRENZUNG IN DEUTSCHLAND
Verschreibungspflichtig
REZEPTPFLICHT/APOTHEKENPFLICHT IN ÖSTERREICH
Rezept- und apothekenpflichtig, wiederholte Abgabe verboten
PACKUNGSGRÖSSEN IN DEUTSCHLAND
SOMAVERT 10 mg:
Packung mit je 30 Durchstechflaschen (N3)
SOMAVERT 15 mg:
Packung mit je 30 Durchstechflaschen (N3)
SOMAVERT 20 mg:
Packung mit je 1 Durchstechflasche (N1)
Packung mit je 30 Durchstechflaschen (N3)
SOMAVERT 25 mg:
Packung mit je 30 Durchstechflaschen (N3)
SOMAVERT 30 mg:
Packung mit je 30 Durchstechflaschen (N3)
PACKUNGSGRÖSSEN IN ÖSTERREICH
SOMAVERT 10 mg:
Packung mit je 30 Durchstechflaschen
SOMAVERT 15 mg:
Packung mit je 30 Durchstechflaschen
SOMAVERT 20 mg:
Packung mit je 30 Durchstechflaschen
SOMAVERT 25 mg:
Packung mit je 30 Durchstechflaschen
SOMAVERT 30 mg:
Packung mit je 30 Durchstechflaschen
REPRÄSENTANT IN DEUTSCHLAND
PFIZER PHARMA GmbH
Friedrichstr. 110
10117 Berlin
Tel.: 030 550055-51000
Fax: 030 550054-10000
REPRÄSENTANT IN ÖSTERREICH
Pfizer Corporation Austria Ges.m.b.H.
Floridsdorfer Hauptstraße 1
A-1210 Wien
Tel.: +43 (0)1 521 15-0
spcde-v19sv-pv-0