
Cofact 500 I.E. Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
Cofact liegt als Pulver und Lösungsmittel zur Herstellung einer Injektionslösung mit humanem Prothrombinkomplex vor. Das Arzneimittel enthält nominal folgende Internationale Einheiten (I.E.) der in der Tabelle unten genannten humanen Gerinnungsfaktoren:
Cofact 500 I.E. |
Nach Rekonstitution* (I.E./ml) |
|
Arzneilich wirksame Bestandteile |
||
Gerinnungsfaktor II |
280 – 700 |
14 – 35 |
Gerinnungsfaktor VII |
140 – 400 |
7 – 20 |
Gerinnungsfaktor IX |
500 |
25 |
Gerinnungsfaktor X |
280 – 700 |
14 – 35 |
Weitere arzneilich wirksame Bestandteile |
||
Protein C |
222 - 780 |
11 - 39 |
Protein S |
20 - 160 |
1 – 8 |
* Nach Rekonstitution mit 20 ml Wasser für Injektionszwecke | ||
Der Gesamtproteingehalt je Durchstechflasche mit 500 I.E. beträgt 260 – 700 mg. Die spezifische Produktaktivität beträgt ≥ 0,6 I.E./mg, ausgedrückt als Aktivität von Faktor IX.
Die Aktivitäten aller Gerinnungsfaktoren sowie Protein C und S (Antigen) wurden nach den aktuellen Standards der WHO oder des Europäischen Arzneibuchs getestet.
Sonstige Bestandteil(e) mit bekannter Wirkung
Nach Rekonstitution enthält dieses Arzneimittel 125 – 195 mmol Natrium/l, bis zu 89,6 mg Natrium pro Durchstechflasche mit 500 I.E..
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
Das Pulver ist von bläulicher Farbe. Das Lösungsmittel ist eine klare, farblose Flüssigkeit ohne sichtbare Partikel.
Behandlung von Blutungen und perioperative Vorbeugung von Blutungen bei erworbenem Mangel an Gerinnungsfaktoren des Prothrombinkomplexes, wie beispielsweise bei einem Mangel infolge einer Behandlung mit Vitamin-K-Antagonisten oder im Falle einer Überdosis Vitamin-K-Antagonisten, wenn eine schnelle Korrektur des Mangels erforderlich ist.
Behandlung von Blutungen und perioperative Vorbeugung bei erblichem Mangel an einem der Vitamin-K-abhängigen Gerinnungsfaktoren, wenn kein gereinigtes spezifisches Gerinnungsprodukt zur Verfügung steht.
Dosierung
Bei den nachfolgenden Dosierungsangaben handelt es sich lediglich um Richtwerte. Die Behandlung ist unter Aufsicht eines Arztes einzuleiten, der in der Behandlung von Gerinnungsstörungen erfahren ist. Dosierung und Dauer der Ersatztherapie richten sich nach der Schwere der Störung, nach dem Ort und der Stärke der Blutung sowie nach dem klinischen Zustand des Patienten.
Die Verabreichungsmenge und die Häufigkeit der Verabreichung sind bei jedem Patienten individuell festzulegen. Dosisintervalle sind den unterschiedlichen Halbwertszeiten der verschiedenen Gerinnungsfaktoren im Prothrombinkomplex im Blutkreislauf anzupassen (vgl. Abschnitt 5.2). Individuelle Dosierungsmaßgaben lassen sich nur auf Basis regelmäßiger Bestimmungen der individuellen Plasmaspiegel der relevanten Gerinnungsfaktoren oder durch allgemeine Tests des Prothrombinkomplex-Spiegels (Thromboplastinzeit, INR) und durch kontinuierliche Überwachung des klinischen Zustandes des Patienten identifizieren.
Bei größeren chirurgischen Eingriffen ist eine präzise Überwachung der Ersatztherapie mittels Gerinnungstests essentiell (spezifische Gerinnungsfaktortests und/oder allgemeine Tests des Prothrombinkomplex-Spiegels).
Blutungen und perioperative Vorbeugung gegen Blutungen bei Behandlung mit Vitamin-K-Antagonisten:
Die Dosis richtet sich nach dem INR-Ausgangswert vor der Behandlung, dem INR-Zielwert und dem Körpergewicht. In den folgenden Tabellen sind ungefähre Dosismengen aufgeführt, die für eine Normalisierung des INR –Wertes bei verschiedenen INR-Ausgangswerten erforderlich sind.
Die Dosistabelle dient nur als allgemeine Dosierungsrichtlinie. Sie ist nicht als Ersatz für die individuelle Abstimmung der Dosis bei jedem einzelnen Patienten und für eine engmaschige Überwachung der INR und anderer Gerinnungsparameter während der Therapie bestimmt.
Empfohlene Dosierungen von Cofact in ml zum Erreichen eines INR-Zielwertes ≤ 2,1
|
INR-Ausgangswert Körpergewicht |
7,5 |
5,9 |
4,8 |
4,2 |
3,6 |
3,3 |
50 kg |
40 |
40 |
40 |
30 |
30 |
30 |
60 kg |
50 |
50 |
40 |
40 |
30 |
30 |
70 kg |
60 |
50 |
50 |
50 |
40 |
40 |
80 kg |
60 |
60 |
60 |
50 |
50 |
40 |
90 kg |
60 |
60 |
60 |
60 |
50 |
50 |
100 kg |
60 |
60 |
60 |
60 |
60 |
50 |
|
INR-Ausgangswert Körpergewicht |
3,0 |
2,8 |
2,6 |
2,5 |
2,3 |
2,2 |
50 kg |
20 |
20 |
X |
X |
X |
X |
60 kg |
30 |
20 |
X |
X |
X |
X |
70 kg |
30 |
30 |
X |
X |
X |
X |
80 kg |
40 |
30 |
X |
X |
X |
X |
90 kg |
40 |
30 |
X |
X |
X |
X |
100 kg |
40 |
40 |
X |
X |
X |
X |
Empfohlene Dosierungen von Cofact in ml zum Erreichen eines INR-Zielwertes ≤ 1,5
|
INR- Körpergewicht |
7,5 |
5,9 |
4,8 |
4,2 |
3,6 |
3,3 |
50 kg |
60 |
60 |
60 |
50 |
50 |
50 |
60 kg |
80 |
70 |
70 |
60 |
60 |
60 |
70 kg |
90 |
80 |
80 |
70 |
70 |
70 |
80 kg |
100 |
100 |
90 |
90 |
90 |
80 |
90 kg |
100 |
100 |
100 |
90 |
90 |
90 |
100 kg |
100 |
100 |
100 |
100 |
100 |
90 |
|
INR- Körpergewicht |
3,0 |
2,8 |
2,6 |
2,5 |
2,3 |
2,2 |
50 kg |
40 |
40 |
30 |
30 |
30 |
30 |
60 kg |
50 |
50 |
40 |
40 |
40 |
30 |
70 kg |
60 |
60 |
50 |
40 |
40 |
40 |
80 kg |
80 |
70 |
60 |
50 |
50 |
40 |
90 kg |
80 |
80 |
70 |
60 |
50 |
40 |
100 kg |
90 |
80 |
70 |
70 |
60 |
50 |
Die Dosierungen werden, aufgrund seiner relativ kurzen Halbwertszeit und der niedrigen Ausbeute nach der Infusion im Vergleich zu den anderen Gerinnungsfaktoren in Cofact, auf Basis der Faktor-IX-Konzentration in Cofact berechnet, Man geht davon aus, dass eine mittlere Plasmakonzentration von Faktor IX von ≥ 30% für einen INR-Wert von ≤ 2,1 bzw. ≥ 60% für einen INR-Wert von ≤ 1,5 ausreichend ist. Die berechneten Mengen werden auf ein Vielfaches von 10 ml abgerundet, und es wurde ein oberer Grenzwert von 60 bzw. 100 ml insgesamt festgesetzt (vgl. Tabellen oben). Die INR-Zielwerte entsprechen den Empfehlungen der Federation of Dutch Thrombosis Services und bewegen sich in der gleichen Größenordnung wie die Empfehlungen der englischen und deutschen Fachgesellschaften.
Die Korrektur der durch den Vitamin-K-Antagonisten induzierten Verschlechterung der Hämostase bleibt für rund 6-8 Stunden bestehen. Die Wirkung von Vitamin K wird dagegen bei gleichzeitiger Verabreichung üblicherweise innerhalb von 4-6 Stunden erreicht. Eine wiederholte Behandlung mit humanem Prothrombinkomplex ist daher in der Regel nicht erforderlich, wenn Vitamin K gegeben wurde.
Da diese Empfehlungen rein empirischer Art sind und Wiederfindung und Wirkungsdauer variieren können, ist eine Überwachung der INR während der Behandlung zwingend erforderlich.
Blutungen und perioperative Vorbeugung bei erblichem Mangel an einem der Vitamin-K-abhängigen Gerinnungsfaktoren, wenn kein spezifisches Gerinnungsprodukt zur Verfügung steht:
Die für eine Behandlung erforderliche berechnete Dosierung beruht auf dem empirischen Befund, dass etwa 1 I.E. Faktor VII oder Faktor IX je kg Körpergewicht die Plasmaaktivität von Faktor VII bzw. Faktor IX um 0,01 I.E./ml erhöht, und dass 1 I.E. von Faktor II oder X je kg Körpergewicht die Plasmaaktivität von Faktor II bzw. X um 0,02 bzw. 0,017 I.E./ml erhöht.
Die Dosis eines bestimmten verabreichten Faktors wird in Internationalen Einheiten (I.E.) ausgedrückt, die zum aktuellen WHO-Standard für jeden Faktor in Bezug stehen. Die Aktivität eines bestimmten Gerinnungsfaktors im Plasma wird entweder prozentual (relativ zu Normalplasma) oder in Internationalen Einheiten (relativ zum internationalen Standard für den jeweiligen Gerinnungsfaktor) ausgedrückt.
Eine Internationale Einheit (I.E.) einer Gerinnungsfaktoraktivität ist äquivalent zu der Menge in einem ml normalem Humanplasma.
Beispielsweise beruht die Berechnung der erforderlichen Dosierung von Faktor X auf dem empirischen Befund, dass 1 Internationale Einheit (I.E.) Faktor X je kg Körpergewicht die Plasmaaktivität von Faktor X um 0,017 I.E./ml erhöht. Die erforderliche Dosis wird daher anhand der folgenden Formel ermittelt:
Erforderliche Einheiten = Körpergewicht (kg) x gewünschter Anstieg von Faktor X (I.E./ml) x 60
Dabei ist 60 (ml/kg) der Kehrwert der geschätzten Wiederfindungsrate.
Wenn die individuelle Wiederfindungsrate bekannt ist, sollte für die Berechnung der betreffende Wert verwendet werden.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit der Anwendung von Cofact bei pädiatrischen Patienten wurden nicht untersucht.
Art und Dauer der Anwendung
Das Arzneimittel wie unter Abschnitt 6.6 beschrieben auflösen. Cofact ist intravenös zu verabreichen.
Es wird empfohlen, das aufgelöste Arzneimittel mit einer Geschwindigkeit von etwa 2 ml pro Minute zu verabreichen.
Überempfindlichkeit gegen die arzneilich wirksamen Bestandteile oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Es sollte ein Spezialist mit Erfahrung bei der Behandlung von Gerinnungsstörungen hinzugezogen werden.
Bei Patienten mit erworbenem Mangel an Vitamin-K-abhängigen Gerinnungsfaktoren (z. B. induziert durch Behandlung mit Vitamin-K-Antagonisten) sollte Cofact nur verwendet werden, wenn eine schnelle Korrektur des Prothrombinkomplex-Spiegels angezeigt ist, beispielsweise bei starken Blutungen oder einer Notoperation. Ansonsten ist eine Reduktion der Dosis des Vitamin-K-Antagonisten und/oder die Gabe von Vitamin K für gewöhnlich ausreichend.
Patienten, die mit Vitamin-K-Antagonisten behandelt werden, können eine grundlegende Hyperkoagulationsbereitschaft aufweisen, die durch die Infusion von humanem Prothrombinkomplex ggf. verstärkt wird.
Bei einem angeborenen Mangel an einem der Vitamin-K-abhängigen Gerinnungsfaktoren sollte, falls verfügbar, ein spezifisches Gerinnungsfaktorprodukt verwendet werden.
Bei Auftreten von allergischen oder anaphylaktischen Reaktionen ist die Injektion/Infusion sofort abzubrechen. Bei Schock ist die medizinische Standardbehandlung von Schockzuständen anzuwenden.
Zu den Standardmaßnahmen zur Prävention von Infektionen infolge der Verwendung von Arzneimitteln, die aus menschlichem Blut oder Humanplasma hergestellt sind, gehören die Auswahl der Spender, die Untersuchung der einzelnen Blutspenden und der Plasmapools hinsichtlich spezifischer Infektionsmarker und die Durchführung wirksamer Schritte während der Herstellung zur Inaktivierung/Entfernung von Viren. Dessen ungeachtet kann die Möglichkeit der Übertragung infektiöser Erreger bei Verabreichung von Arzneimitteln, die aus menschlichem Blut oder Plasma hergestellt sind, nicht gänzlich ausgeschlossen werden. Dies gilt auch für unbekannte oder neu auftretende Viren und andere Pathogene.
Die getroffenen Maßnahmen werden als wirksam erachtet für umhüllte Viren wie das humane Immunschwächevirus (HIV), das Hepatitis-B-Virus (HBV) und das Hepatitis-C-Virus (HCV)sowie für das nicht umhüllte Hepatitis-A-Virus (HAV). Die getroffenen Maßnahmen sind unter Umständen von eingeschränkter Wirksamkeit bei anderen nicht umhüllten Viren wie Parvovirus B19. Eine Infektion mit Parvovirus B19 kann bei Schwangeren (Infektion des Fetus) und bei Personen mit Immunschwäche oder erhöhter Erythropoese (z. B. bei hämolytischer Anämie) schwerwiegend sein.
Bei Patienten, die regelmäßig oder wiederholt Prothrombinkomplex-Produkte erhalten, die aus Humanplasma gewonnen werden, sollte eine geeignete Impfung (Hepatitis A und B) in Betracht gezogen werden.
Es besteht ein Risiko einer Thrombose oder einer disseminierten intravasalen Gerinnung, wenn Patienten mit angeborenem oder erworbenem Mangel mit humanem Prothrombinkomplex behandelt werden, vor allem bei wiederholter Gabe. Das Risiko ist unter Umständen höher bei Behandlung einer isolierten Faktor-VII-Defizienz, da die anderen Vitamin-K-abhängigen Gerinnungsfaktoren mit längerer Halbwertszeit auf wesentlich höhere Mengen als im Normalzustand akkumulieren können.
Patienten, die einen humanen Prothrombinkomplex erhalten, sind engmaschig auf Anzeichen oder Symptome einer intravasalen Gerinnung oder Thrombose zu beobachten. Aufgrund des Risikos thromboembolischer Komplikationen sind Patienten mit der Anamnese einer koronaren Herzerkrankung, Patienten mit Lebererkrankung, peri- oder postoperative Patienten, Neugeborene oder Patienten mit einem Risiko für thromboembolische Ereignisse oder eine disseminierte intravasale Gerinnung bei Verabreichung von humanem Prothrombinkomplex engmaschig zu überwachen. In all diesen Situationen ist der mögliche Nutzen der Behandlung gegen die Risiken dieser Komplikationen abzuwägen.
Hinsichtlich der Verwendung von Cofact bei perinatalen Blutungen infolge eines Vitamin-K-Mangels bei Neugeborenen liegen keine Daten vor.
Sonstige Bestandteile
Cofact enthält bis zu 448 mg Natrium pro 100 ml. Dies entspricht bis zu 22 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Aufnahme von 2 g Natrium. Dies ist von Patienten, die eine natriumarme Diät einhalten, zu berücksichtigen.
Kinder und Jugendliche
Es liegen keine ausreichenden Daten für die Empfehlung zur Anwendung von Cofact bei Kindern und Jugendlichen vor.
Humane Prothrombinkomplexprodukte neutralisieren die Wirkung einer Behandlung mit Vitamin-K-Antagonisten, aber es sind keine Wechselwirkungen mit anderen Arzneimitteln bekannt.
Die Sicherheit von humanem Prothrombinkomplex für die Verwendung während der Schwangerschaft und Stillzeit beim Menschen wurde nicht untersucht.
Tierexperimentelle Studien sind nicht geeignet, um die Sicherheit im Hinblick auf Schwangerschaft, embryonale/fetale Entwicklung, Entbindung oder postnatale Entwicklung zu beurteilen. Humaner Prothrombinkomplex sollte daher während der Schwangerschaft und Stillzeit nur verwendet werden, wenn eine eindeutige Indikation vorliegt. Siehe Abschnitt 4.4 bezüglich Hinweisen zum Risiko einer Parvovirus-B19-Infektion bei Schwangeren.
Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und der Fähigkeit zum Bedienen von Maschinen durchgeführt.
Tabellarische Zusammenfassung von Nebenwirkungen von Cofact.
Die aufgeführten Nebenwirkungen wurden während klinischer Studien und während der Anwendung von Cofact nach Markteinführung berichtet. Die nachstehende Tabelle entspricht der MedDRA-Systemorganklassifikation (Systemorganklasse und bevorzugter Begriff). Die Häufigkeit der Nebenwirkungen wird definiert gemäß der folgenden Konvention: sehr häufig (≥ 1/10); häufig (≥ 1/100 bis < 1/10); gelegentlich (≥ 1/1 000 bis < 1/100); selten (≥ 1/10 000 bis < 1/1 000); sehr selten (< 1/10 000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Häufigkeit von Nebenwirkungen
MedDRA |
Nebenwirkung |
Häufigkeit |
Erkrankungen des Immunsystems |
Anaphylaktische Reaktion, Überempfindlichkeit |
Nicht bekannt |
Erkrankungen des Nervensystems |
Apoplektischer Insult, Schwindelgefühl |
Nicht bekannt |
Herzerkrankungen |
Akuter Myokardinfarkt |
Nicht bekannt |
Gefäßerkrankungen |
Thromboembolische Ereignisse (Embolie, tiefe Venenthrombose); siehe Abschnitt 4.4 |
Häufig |
Hypotonie |
Gelegentlich |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Lungenembolie, respiratorische Insuffizienz |
Nicht bekannt |
Erkrankungen des Gastrointestinaltrakts |
Übelkeit, Erbrechen |
Nicht bekannt |
Erkrankungen der Haut und des Unterhautgewebes |
Hyperhidrosis, Pruritus, Urtikaria |
Nicht bekannt |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Rötung an der Infusionsstelle, Reizung an der Infusionsstelle, Schwellung an der Infusionsstelle Unwohlsein |
Nicht bekannt |
Untersuchungen |
Anomale Leberfunktion |
Nicht bekannt |
Eine Ersatztherapie kann zur Bildung von Antikörpern im Blut führen, die einen oder mehrere der Faktoren des humanen Prothrombinkomplexes hemmen. Wenn eine solche Hemmung stattfindet, zeigt sich dieser Zustand als eingeschränkte klinische Wirksamkeit, zum Beispiel anhaltende Blutungen.
Angaben zur Sicherheit im Hinblick auf übertragbare Krankheitserreger siehe Abschnitt 4.4.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Telefon: +49 6103 77 0, Telefax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
Die Verwendung hoher Dosen humaner Prothrombinkomplexprodukte wurde mit dem Auftreten von Herzinfarkt, disseminierter intravasaler Gerinnung, Venenthrombose und Lungenembolie in Verbindung gebracht. Bei einer Überdosis ist daher das Risiko der Entwicklung thromboembolischer Komplikationen oder einer disseminierten intravasalen Gerinnung erhöht.
Pharmakotherapeutische Gruppe: Antihämorrhagika, Blutgerinnungsfaktoren IX, II, VII und X in Kombination, ATC-Code: B02BD01.
Die in der Leber unter Beteiligung von Vitamin K synthetisierten Gerinnungsfaktoren II, VII, IX und X werden allgemein als Prothrombinkomplex bezeichnet. Zusätzlich zu den Gerinnungsfaktoren enthält Cofact die Vitamin K abhängigen Gerinnungshemmer Protein C und Protein S.
Faktor VII ist das Zymogen der aktiven Serinprotease VIIa, die am Anfang des extrinsischen Pfades der Blutgerinnung steht. Der Komplex aus Gewebefaktor und Faktor VIIa aktiviert die Gerinnungsfaktoren X und IX, wodurch sich Faktor IXa und Xa bilden. Im Verlauf der weiteren Aktivierung der Gerinnungskaskade wird Prothrombin (Faktor II) aktiviert und in Thrombin umgewandelt. Durch Thrombin wird aus Fibrinogen zu Fibrin gebildet, was zur Bildung eines Gerinnsels führt. Die normale Entstehung von Thrombin ist demnach für die Thrombozytenfunktion als Teil der primären Hämostase von ausschlaggebender Bedeutung.
Ein isolierter schwerer Mangel an Faktor VII bewirkt eine reduzierte Thrombinbildung und eine Neigung zu Blutungen infolge einer beeinträchtigten Fibrinbildung und einer verschlechterten primären Hämostase. Ein isolierter Mangel an Faktor IX ist eine der klassischen Hämophilien (Hämophilie B). Ein isolierter Mangel an Faktor II oder Faktor X ist sehr selten, verursacht aber in schwerer Ausprägung eine Blutungsneigung ähnlich der bei klassischen Hämophilien.
Die weiteren arzneilich wirksamen Bestandteile, die Gerinnungshemmer Protein C und Protein S, werden ebenfalls in der Leber synthetisiert. Die biologische Aktivität von Protein C wird durch den Cofaktor Protein S verstärkt.
Aktiviertes Protein C hemmt die Gerinnung durch Inaktivierung der Gerinnungsfaktoren Va und VIIIa. Protein S als Cofaktor von Protein C unterstützt die Inaktivierung der Gerinnung. Ein Protein C-Mangel wird mit einem erhöhten Thrombose-Risiko assoziiert.
Ein erworbener Mangel der Vitamin-K-abhängigen Gerinnungsfaktoren ist die Folge einer Behandlung mit Vitamin-K-Antagonisten. Verstärkt sich der Mangelzustand, kommt es zu einer starken Blutungsneigung, die sich als retroperitoneale oder zerebrale Blutung und weniger als Muskel- und Gelenkblutung äußert. Auch eine schwere Leberinsuffizienz führt zu deutlich verringerten Spiegeln der Vitamin-K-abhängigen Gerinnungsfaktoren und einer klinischen Blutungsneigung. Diese ist jedoch wegen einer anhaltenden intravaskulären Koagulation geringen Grades, niedriger Thrombozytenzahlen, eines Mangels an Gerinnungsinhibitoren und einer gestörten Fibrinolyse vergleichsweise komplex.
Die Gabe von humanem Prothrombinkomplex bewirkt einen Anstieg der Plasmaspiegel der Vitamin-K-abhängigen Gerinnungsfaktoren und kann den Gerinnungsdefekt bei Patienten mit einem Mangel an einem oder mehreren dieser Faktoren vorübergehend korrigieren.
Das Spektrum der Plasma-Halbwertszeiten der vier in Cofact vorhandenen Gerinnungsfaktoren ist in der Literatur beschrieben:
Gerinnungsfaktor |
Halbwertszeit |
Faktor II |
40 - 60 Stunden |
Faktor VII |
4 -6 Stunden |
Faktor IX |
18 -25 Stunden |
Faktor X |
30 - 60 Stunden |
Abgesehen von einer Studie bei Ratten hinsichtlich eines möglichen hypotensiven Effektes (der sich als nicht vorhanden erwies), wurden keine tierexperimentellen Studien mit Cofact durchgeführt.
Es sind tierexperimentelle toxikologische Untersuchungen mit TNBP und Tween 80 durchgeführt worden. Cofact enthält nicht mehr als 0,4 µg TNBP je I.E. Faktor IX und nicht mehr als 4 µg Tween 80 je I.E. Faktor IX. Wenn Cofact in der empfohlenen Dosis verwendet wird, bleiben die Mengen an TNBP und Tween 80, die ein Patient erhält, deutlich unter den Mengen, die sich bei Versuchstieren als schädlich erwiesen haben.
Pulver: Trinatriumcitratdihydrat, Natriumchlorid, Antithrombin ≤0,6 I.E./ml.
Lösungsmittel: Wasser für Injektionszwecke.
Dieses Arzneimittel darf nicht mit anderen Arzneimitteln gemischt werden.
Cofact ist kompatibel mit Polypropylen-Material. Die Behandlung könnte infolge einer Adsorption von Gerinnungsfaktor an die Innenfläche von Injektions-/Infusionsbestecken anderen Materials versagen.
3 Jahre.
Die chemische und physikalische Stabilität der gebrauchsfertigen Lösung wurde für 3 Stunden bei 15 °C – 25 °C belegt. Aus mikrobiologischer Sicht sollte Cofact nach der Rekonstitution unmittelbar verwendet werden. Wird es nicht sofort verwendet, ist der Anwender für Lagerdauer und Lagerbedingungen verantwortlich.
Im Kühlschrank bei 2 °C – 8 °C lagern. Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Innerhalb der Haltbarkeitsdauer kann das Arzneimittel über einen begrenzten Zeitraum von bis zu 6 Monaten bei bis zu 25°C aufbewahrt werden. Sobald das Arzneimittel aus dem Kühlschrank entnommen wurde, darf es nicht in den Kühlschrank zurückgestellt werden. Wenn das Arzneimittel während dieses Zeitraums nicht benutzt wird, muss es entsorgt werden. Das Datum des Beginns der Raumtemperaturlagerung (bei maximal 25 °C) muss auf dem Umkarton vermerkt werden.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
Eine Packung enthält:
500 I.E. eines Pulvers in einer Durchstechflasche (Typ-I-Glas), verschlossen mit einem Gummistopfen (Brombutyl mit einer Beschichtung aus einem fluorierten Polymer) und einem Aluminiumverschluss mit Flip-Off-Schutzkappe aus Kunststoff
20 ml Lösungsmittel in einer Durchstechflasche (Typ-I-Glas), verschlossen mit einem Gummistopfen (Brombutyl mit einer Beschichtung aus einem fluorierten Polymer) und einem Aluminiumverschluss mit Flip-Off-Schutzkappe aus Kunststoff
1 Transfersystem nextaro v (15-µm-Filter nominal)
Allgemeine Hinweise bei Verwendung eines nextaro v Transfersystems
Der getrocknete Proteinanteil wird in 20 ml Wasser für Injektionszwecke aufgelöst. Wenn das Arzneimittel bei 2 °C bis 8 °C gelagert wird, müssen die verschlossenen Durchstechflaschen mit dem Pulver bzw. dem Lösungsmittel (Wasser für Injektionszwecke) vor dem Auflösen des Präparates auf Raumtemperatur (15 °C – 25 °C) gebracht werden. Diese Temperatur ist während der Rekonstitution beizubehalten. Wird zum Erwärmen ein Wasserbad verwendet, muss darauf geachtet werden, dass das Wasser nicht mit den Gummistopfen oder den Flip-Off-Schutzkappen der Durchstechflaschen in Berührung kommt. Die Temperatur des Wasserbads darf 37 °C nicht überschreiten.
Während des unten beschriebenen Verfahrens ist eine aseptische Arbeitstechnik erforderlich. Stellen Sie sicher, dass die Flip-Off-Schutzkappen der Durchstechflaschen mit Pulver bzw. Lösungsmittel entfernt wurden und dass der Bördelrand und der Gummistopfen mit einer antiseptischen Lösung desinfiziert wurden und vor dem Öffnen der Verpackung des Transfersystems getrocknet sind. Berühren Sie nicht die Gummistopfen der Durchstechflaschen mit dem Lösungsmittel bzw. dem Pulver.
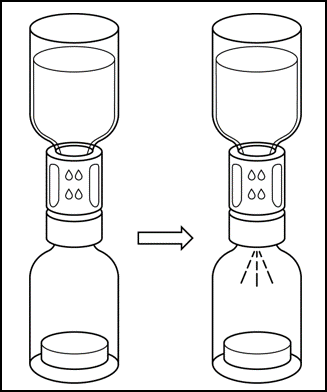
Durch das Vakuum in der Durchstechflasche mit dem Pulver wird das Lösungsmittel automatisch in die Durchstechflasche mit dem Pulver überführt.
In der Regel sollte sich das Pulver innerhalb von 10 Minuten vollständig gelöst haben und eine blau gefärbte Lösung bilden. Die Lösung sollte klar oder leicht opaleszent sein. Verwenden Sie keine Lösungen, die trüb sind oder Ablagerungen aufweisen. Die Lösung muss vor der Verabreichung visuell auf Partikel und Verfärbungen überprüft werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Verfahren unter Verwendung einer nextaro v Transfersystems
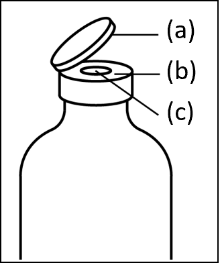
1. Entfernen Sie die Flip-Off-Schutzkappen (a) sowohl von der Durchstechflasche mit Lösungsmittel als auch von der Durchstechflasche mit Pulver. Desinfizieren Sie den Bördelrand (b) einschließlich des Gummistopfens (c) sowohl der Durchstechflasche mit Lösungsmittel als auch der Durchstechflasche mit Pulver mit einer antiseptischen Lösung. |
 |
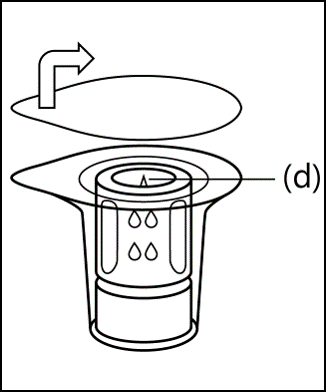
2. Öffnen Sie die Verpackung des Transfersystems, indem Sie den Deckel öffnen und vollständig abziehen. Um die Sterilität aufrechtzuerhalten, nehmen Sie das Einweg-Transfersystem nicht aus der Verpackung und berühren Sie nicht den Dorn (d) des Transfersystems. |
 |
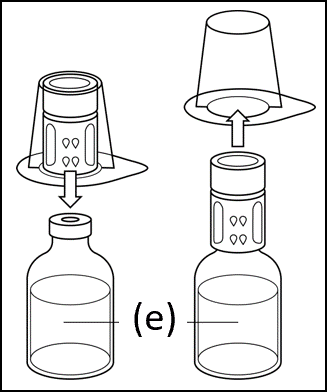
3. Stellen Sie die Durchstechflasche mit dem Lösungsmittel auf eine ebene und saubere Fläche und halten Sie diese mit einer Hand fest. Ohne die Verpackung des Transfersystems zu entfernen, setzen Sie den blauen Teil des Transfersystems oben auf die Lösungsmittelflasche (e) und drücken Sie das Verbindungsstück gerade und fest nach unten, bis es einrastet. Die Umverpackung beim Anbringen nicht drehen. |
 |
4. Halten Sie die Lösungsmittelflasche fest und entfernen Sie vorsichtig die Umverpackung vom Transfersystem. Die Umverpackung nicht drehen. Achten Sie darauf, dass das Transfersystem weiterhin fest mit der Lösungsmittelflasche verbunden bleibt. |
|
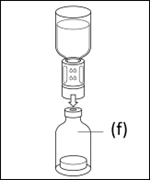
5. Stellen Sie die Durchstechflasche mit dem Pulver (f) auf eine ebene Fläche und halten Sie diese fest. Nehmen Sie die Lösungsmittelflasche samt aufgesetztem Transfersystem und drehen Sie diese auf den Kopf. Setzen Sie den weißen Teil des Transfersystems oben auf die Durchstechflasche mit dem Pulver und drücken Sie das Verbindungsstück fest nach unten, bis es einrastet. Beim Anbringen nicht drehen. |
 |
6. Das Lösungsmittel fließt automatisch in die Durchstechflasche mit dem Pulver. |
 |
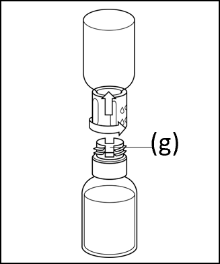
7. Nach Abschluss des Transfervorgangs und der vollständigen Auflösung des Präparats, halten Sie den weißen Teil fest und drehen den damit verbundenen blauen Teil gegen den Uhrzeigersinn, um das Transfersystem in zwei Teile zu trennen. Entfernen und entsorgen Sie den blauen Teil zusammen mit der leeren Durchstechflasche. Den Luer-Lock-Adapter (g) nicht berühren. |
 |
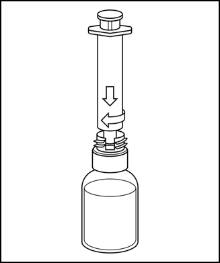
8. Halten Sie die Durchstechflasche mit der rekonstituierten Lösung fest und schrauben Sie eine Spritze (mindestens 20 ml) in den Luer-Lock-Adapter (g) am weißen Teil des Transfersystems. |
 |
|
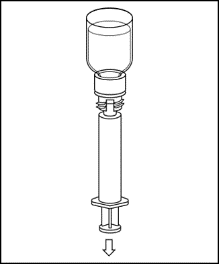
9. Drehen Sie die Durchstechflasche auf den Kopf und ziehen Sie die Lösung in die Spritze auf. 10. Sobald die Lösung aufgezogen wurde, halten Sie den Spritzenzylinder fest (wobei der Spritzenkolben nach unten zeigt) und entfernen Sie die Spritze vom weißen Teil des Transfersystems. Entsorgen Sie den weißen Teil zusammen mit der leeren Durchstechflasche. |
 |
Prothya Biosolutions Netherlands B.V.
Plesmanlaan 125
NL-1066 CX Amsterdam
Niederlande
PEI.H.03453.02.1
Niederlande: 1. Oktober 1997/ 24. September 2011
August 2024
Deutschland, Niederlande, Ungarn und Vereinigte Staaten von Amerika.