
Kisplyx 4 mg Hartkapseln
Kisplyx 10 mg Hartkapseln
Kisplyx 4 mg Hartkapseln
Eine Hartkapsel enthält 4 mg Lenvatinib (als Mesilat).
Kisplyx 10 mg Hartkapseln
Eine Hartkapsel enthält 10 mg Lenvatinib (als Mesilat).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Hartkapsel.
Kisplyx 4 mg Hartkapseln
Gelblich-rotes Unterteil und gelblich-rotes Oberteil, Länge ca. 14,3 mm; das Oberteil ist mit „Є” in schwarzer Farbe und das Unterteil mit „LENV 4 mg” gekennzeichnet.
Kisplyx 10 mg Hartkapseln
Gelbes Unterteil und gelblich-rotes Oberteil, Länge ca. 14,3 mm; das Oberteil ist mit „Є” in schwarzer Farbe und das Unterteil mit „LENV 10 mg” gekennzeichnet.
Kisplyx ist indiziert zur Behandlung von Erwachsenen mit fortgeschrittenem Nierenzellkarzinom (renal cell carcinoma, RCC):
in Kombination mit Pembrolizumab als Erstlinientherapie (siehe Abschnitt 5.1).
in Kombination mit Everolimus nach einer gegen den vaskulären endothelialen Wachstumsfaktor (VEGF) gerichteten vorangegangenen Behandlung (siehe Abschnitt 5.1).
Die Behandlung sollte von einem qualifizierten Arzt eingeleitet und überwacht werden, der Erfahrung in der Tumorbehandlung besitzt.
Dosierung
Kisplyx in Kombination mit Pembrolizumab als Erstlinientherapie
Die empfohlene Dosis von Lenvatinib beträgt 20 mg (zwei 10 mg Kapseln) oral einmal täglich in Kombination mit Pembrolizumab entweder 200 mg alle 3 Wochen oder 400 mg alle 6 Wochen, verabreicht als intravenöse Infusion über einen Zeitraum von 30 Minuten. Die Tagesdosis von Lenvatinib ist dem Bedarf entsprechend gemäß dem Dosis-/Toxizitäts-Managementplan anzupassen. Die Behandlung mit Lenvatinib ist fortzusetzen, bis eine Krankheitsprogression oder eine inakzeptable Toxizität auftritt. Die Behandlung mit Pembrolizumab ist fortzusetzen, bis eine Krankheitsprogression oder eine inakzeptable Toxizität auftritt oder die für Pembrolizumab festgelegte maximale Behandlungsdauer erreicht ist.
Für vollständige Informationen zur Dosierung von Pembrolizumab siehe Fachinformation zu Pembrolizumab.
Kisplyx in Kombination mit Everolimus als Zweitlinientherapie
Die empfohlene Tagesdosis von Lenvatinib beträgt 18 mg (eine 10 mg Kapsel und zwei 4 mg Kapseln) oral einmal täglich in Kombination mit 5 mg Everolimus einmal täglich. Die Tagesdosis von Lenvatinib und, falls erforderlich, von Everolimus ist dem Bedarf entsprechend gemäß dem Dosis-/Toxizitäts-Managementplan anzupassen.
Für vollständige Informationen zur Dosierung von Everolimus siehe Fachinformation zu Everolimus.
Wenn ein Patient eine Dosis von Lenvatinib vergisst und diese nicht innerhalb von 12 Stunden eingenommen werden kann, sollte diese Dosis ausgelassen und die nächste Dosis dann zum üblichen Einnahmezeitpunkt eingenommen werden.
Die Behandlung sollte so lange fortgesetzt werden, wie ein klinischer Nutzen zu beobachten ist oder bis eine inakzeptable Toxizität auftritt.
Dosisanpassung und Absetzen der Lenvatinib-Therapie
Die Behandlung von Nebenwirkungen kann eine Therapieunterbrechung, eine Dosisanpassung oder ein Absetzen der Lenvatinib-Therapie erforderlich machen (siehe Abschnitt 4.4). Leichte bis mittelschwere Nebenwirkungen (z. B. Grad 1 oder 2) erfordern im Allgemeinen keine Unterbrechung der Lenvatinib-Therapie, es sei denn, sie sind für den Patienten trotz eines optimalen Behandlungsmanagements nicht tolerierbar. Schwere (z. B. Grad 3) oder nicht tolerierbare Nebenwirkungen erfordern eine Unterbrechung der Lenvatinib-Therapie bis zur Besserung der Nebenwirkung auf Grad 0 - 1 oder bis zur Rückkehr zum Ausgangszustand.
Bevor die Lenvatinib-Therapie unterbrochen oder Dosisreduktionen vorgenommen werden, ist ein optimales medizinisches Management (d. h. Behandlung oder Therapie) von Übelkeit, Erbrechen und Diarrhoe einzuleiten; gastrointestinale Toxizitäten sind aktiv zu behandeln, um das Risiko für das Auftreten von Nierenfunktionsstörungen oder Nierenversagen zu reduzieren (siehe Abschnitt 4.4).
Bei Toxizitäten, bei denen man davon ausgeht, dass sie in Zusammenhang mit Lenvatinib stehen (siehe Tabelle 2), muss nach Abklingen/Besserung einer Nebenwirkung auf Grad 0 bis 1 oder bis zur Rückkehr zum Ausgangszustand die Behandlung mit einer reduzierten Lenvatinib-Dosis gemäß den Empfehlungen in Tabelle 1 fortgesetzt werden.
Tabelle 1 Dosisanpassungen der empfohlenen Lenvatinib-Tagesdosisa
Lenvatinib-Dosis in Kombination mit Pembrolizumab |
Lenvatinib-Dosis in Kombination mit Everolimus |
|
Empfohlene Tagesdosis |
20 mg oral einmal täglich |
18 mg oral einmal täglich |
Erste Dosisreduktion |
14 mg oral einmal täglich |
14 mg oral einmal täglich |
Zweite Dosisreduktion |
10 mg oral einmal täglich |
10 mg oral einmal täglich |
Dritte Dosisreduktion |
8 mg oral einmal täglich |
8 mg oral einmal täglich (zwei 4 mg Kapseln) |
a: Es liegen nur wenige Daten für Dosen unter 8 mg vor. | ||
Bei Anwendung in Kombination mit Pembrolizumab muss die Einnahme eines oder beider Arzneimittel gegebenenfalls unterbrochen werden. Gegebenenfalls ist die Behandlung mit Lenvatinib zu unterbrechen, die Dosis zu reduzieren oder die Behandlung abzusetzen. Die Unterbrechung oder das Absetzen der Pembrolizumab-Behandlung müssen gemäß den Anweisungen in der Fachinformation zu Pembrolizumab erfolgen. Für Pembrolizumab werden keine Dosisreduktionen empfohlen.
Bei Toxizitäten, bei denen man davon ausgeht, dass sie in Zusammenhang mit Everolimus stehen, muss die Behandlung unterbrochen werden, auf jeden zweiten Tag reduziert oder beendet werden (siehe Fachinformation zu Everolimus für Empfehlungen zu Dosisanpassungen im Hinblick auf spezifische Nebenwirkungen).
Bei Toxizitäten, bei denen man davon ausgeht, dass sie in Zusammenhang mit Lenvatinib und Everolimus stehen, muss zuerst die Lenvatinib-Dosis reduziert werden (siehe Tabelle 1), bevor die Everolimus-Dosis reduziert wird.
Bei Auftreten von lebensbedrohlichen Reaktionen (z. B. Grad 4) müssen alle Behandlungen abgesetzt werden, außer bei Laborwertabweichungen, die als nicht lebensbedrohlich eingestuft werden. In diesem Fall sollten die Reaktionen wie eine schwere Nebenwirkung (z. B. Grad 3) eingestuft und behandelt werden.
Die Schweregrade basieren auf den Common Terminology Criteria for Adverse Events (CTCAE) des National Cancer Institute (NCI).
Tabelle 2 Nebenwirkungen, die eine Anpassung der Lenvatinib-Dosis erfordern
Nebenwirkung |
Schweregrad |
Maßnahme |
Dosisreduktion und Fortsetzung der Lenvatinib-Behandlung |
Hypertonie |
Grad 3 |
Unterbrechung der Behandlung |
Abklingen auf Grad 0, 1 oder 2. |
Grad 4 |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
|
Proteinurie |
≥ 2 g / 24 Stunden |
Unterbrechung der Behandlung |
Abklingen auf weniger als 2 g / 24 Stunden. |
Nephrotisches Syndrom |
------- |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
Nierenfunktionsstörungen oder Niereninsuffizienz |
Grad 3 |
Unterbrechung der Behandlung |
Abklingen auf Grad 0 - 1 oder Rückbildung zum Ausgangszustand. |
Grad 4* |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
|
Herzinsuffizienz |
Grad 3 |
Unterbrechung der Behandlung |
Abklingen auf Grad 0 - 1 oder Rückbildung zum Ausgangszustand |
Grad 4 |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
|
PRES/RPLS |
Jeder Grad |
Unterbrechung der Behandlung |
Bei Abklingen auf Grad 0 - 1 ist eine Fortsetzung der Behandlung mit reduzierter Dosis zu erwägen. |
Hepatotoxizität |
Grad 3 |
Unterbrechung der Behandlung |
Abklingen auf Grad 0 - 1 oder Rückbildung zum Ausgangszustand |
Grad 4* |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
|
Arterielle Thromboembolien |
Jeder Grad |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
Blutungen |
Grad 3 |
Unterbrechung der Behandlung |
Abklingen auf Grad 0 - 1 |
Grad 4 |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
|
Gastrointestinale Perforation oder Fistel |
Grad 3 |
Unterbrechung der Behandlung |
Abklingen auf Grad 0 - 1 oder Rückbildung zum Ausgangszustand. |
Grad 4 |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
|
Nichtgastrointestinale Fistel |
Grad 4 |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
QT-Zeit-Verlängerung |
> 500 ms |
Unterbrechung der Behandlung |
Abklingen auf <480 ms oder Rückbildung zum Ausgangszustand |
Diarrhoe |
Grad 3 |
Unterbrechung der Behandlung |
Abklingen auf Grad 0 - 1 oder Rückbildung zum Ausgangszustand. |
Grad 4 (trotz medikamentöser Behandlung) |
Beenden der Behandlung |
Keine Fortsetzung der Behandlung |
|
*: Laborwertabweichungen (Grad 4), die als nicht lebensbedrohlich eingestuft werden, können wie schwere Nebenwirkungen (z. B. Grad 3) behandelt werden. | |||
Spezielle Patientengruppen
Für Informationen zur klinischen Erfahrung mit der Kombinationsbehandlung von Lenvatinib und Pembrolizumab, siehe Abschnitt 4.8.
Patienten im Alter von ≥ 65 Jahren mit Hypertonie zum Behandlungsbeginn oder Patienten mit Nierenfunktionsstörungen scheinen eine geringere Verträglichkeit gegenüber Lenvatinib aufzuweisen (siehe Abschnitt 4.8).
Für die meisten speziellen Patientengruppen liegen keine Daten für die Kombination von Lenvatinib und Everolimus vor. Die folgenden Angaben leiten sich aus den klinischen Erfahrungen mit Lenvatinib als Einzelwirkstoff bei Patienten mit differenziertem Schilddrüsenkarzinom (DTC; siehe Fachinformation zu Lenvima) ab.
Außer Patienten mit schweren Leber- und/oder Nierenfunktionsstörungen (siehe unten) sollten alle Patienten die Behandlung mit der empfohlenen Dosis von 20 mg Lenvatinib täglich mit Pembrolizumab oder 18 mg Lenvatinib mit 5 mg Everolimus einmal täglich gemäß Indikation beginnen. Die Dosis sollte auf Basis der individuellen Verträglichkeit weiter angepasst werden.
Patienten mit Hypertonie
Der Blutdruck sollte vor der Behandlung mit Lenvatinib gut eingestellt sein und während der Behandlung regelmäßig überwacht werden (siehe Abschnitte 4.4 und 4.8).
Patienten mit Leberfunktionsstörungen
Es liegen nur begrenzte Daten zur Kombination von Lenvatinib mit Pembrolizumab bei Patienten mit Leberfunktionsstörungen vor. Bei Patienten mit leichter (Child-Pugh A) oder mittelschwerer (Child-Pugh B) Leberfunktionsstörung ist keine Anpassung der Anfangsdosis der Kombination basierend auf der Leberfunktion erforderlich. Bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh C) beträgt die empfohlene Lenvatinib-Anfangsdosis 10 mg einmal täglich. Informationen zur Dosierung bei Patienten mit Leberfunktionsstörungen sind der Fachinformation zu Pembrolizumab zu entnehmen. Je nach individueller Verträglichkeit können weitere Dosisanpassungen erforderlich sein. Die Kombination sollte bei Patienten mit schwerer Leberfunktionsstörung nur angewendet werden, wenn der erwartete Nutzen das Risiko überwiegt (siehe Abschnitt 4.8).
Für Patienten mit Leberfunktionsstörungen liegen keine Daten für die Kombination von Lenvatinib mit Everolimus vor. Bei Patienten mit leichter (Child-Pugh A) oder mittelschwerer (Child-Pugh B) Leberfunktionsstörung ist keine Anpassung der Anfangsdosis der Kombination erforderlich. Bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh C) beträgt die empfohlene Lenvatinib-Anfangsdosis 10 mg einmal täglich in Kombination mit der in der Fachinformation zu Everolimus empfohlenen Everolimus-Dosis für Patienten mit schwerer Leberfunktionsstörung. Je nach individueller Verträglichkeit können weitere Dosisanpassungen erforderlich sein. Die Kombination sollte bei Patienten mit schwerer Leberfunktionsstörung nur angewendet werden, wenn der erwartete Nutzen das Risiko überwiegt (siehe Abschnitt 4.8).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter oder mittelschwerer Nierenfunktionsstörung ist keine Anpassung der Anfangsdosis erforderlich. Bei Patienten mit schwerer Nierenfunktionsstörung beträgt die empfohlene Anfangsdosis 10 mg Lenvatinib einmal täglich. Informationen zur Dosierung bei Patienten mit Nierenfunktionsstörungen sind den Fachinformationen zu Pembrolizumab oder Everolimus zu entnehmen. Je nach individueller Verträglichkeit können weitere Dosisanpassungen erforderlich sein. Patienten mit einer terminalen Niereninsuffizienz wurden nicht untersucht, sodass die Anwendung von Lenvatinib bei diesen Patienten nicht empfohlen wird (siehe Abschnitt 4.8).
Ältere Patienten
Es ist keine Anpassung der Anfangsdosis auf Grund des Alters erforderlich. Über die Anwendung bei Patienten ≥ 75 Jahre liegen nur begrenzte Daten vor (siehe Abschnitt 4.8).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Lenvatinib bei Kindern im Alter von 2 bis < 18 Jahren ist nicht erwiesen. Zurzeit vorliegende Daten werden in den Abschnitten 4.8, 5.1 und 5.2 beschrieben; eine Dosierungsempfehlung kann jedoch nicht gegeben werden. Lenvatinib darf bei Kindern im Alter unter 2 Jahren nicht angewendet werden, da Bedenken hinsichtlich der Sicherheit aus Tierstudien abzuleiten sind (siehe Abschnitt 5.3).
Ethnische Abstammung
Es ist keine Anpassung der Anfangsdosis aufgrund der ethnischen Abstammung erforderlich (siehe Abschnitt 5.2). Die aktuell verfügbaren Daten sind in Abschnitt 4.8 beschrieben.
Körpergewicht unter 60 kg
Eine Anpassung der Anfangsdosis auf der Grundlage des Körpergewichts ist nicht erforderlich. Zur Behandlung mit Lenvatinib in Kombination mit Everolimus bei Patienten mit einem Körpergewicht unter 60 kg und RCC liegen bisher nur begrenzte Daten vor (siehe Abschnitt 4.8).
Leistungsstatus
Patienten mit einem ECOG (Eastern Cooperative Oncology Group) Leistungsstatus von 2 oder höher waren von RCC-Studie 205 ausgeschlossen (siehe Abschnitt 5.1). Patienten mit einem KPS (Karnofsky Performance Status [Karnofsky-Leitungsstatus]) von < 70 waren von Studie 307 (CLEAR) ausgeschlossen. Das Nutzen-Risiko-Verhältnis dieser Patienten wurde nicht bewertet.
Art der Anwendung
Lenvatinib ist zum Einnehmen. Die Kapseln sollen jeden Tag etwa zur gleichen Tageszeit, mit einer Mahlzeit oder unabhängig von den Mahlzeiten, eingenommen werden (siehe Abschnitt 5.2). Pflegepersonen dürfen die Kapseln nicht öffnen, um den wiederholten Kontakt mit dem Kapselinhalt zu vermeiden.
Die Lenvatinib‑Kapseln können unzerkaut mit Wasser geschluckt oder als Suspension verabreicht werden, indem zu deren Herstellung die ganze(n) Kapsel(n) in Wasser, Apfelsaft oder Milch aufgelöst wird bzw. werden. Die Suspension kann oral oder über eine Ernährungssonde verabreicht werden. Bei Verabreichung über eine Ernährungssonde sollte die Suspension mit Hilfe von Wasser zubereitet werden (siehe Abschnitt 6.6 zur Zubereitung und Verabreichung der Suspension).
Sollte die Lenvatinib‑Suspension nicht zum Zeitpunkt der Zubereitung verbraucht werden, kann sie in einem abgedeckten Behältnis im Kühlschrank bei einer Temperatur von 2 ºC bis 8 ºC für eine maximale Dauer von 24 Stunden aufbewahrt werden. Nach der Entnahme aus dem Kühlschrank muss die Suspension vor der Anwendung etwa 30 Sekunden lang geschüttelt werden. Erfolgt die Verabreichung nicht innerhalb von 24 Stunden, muss die Suspension entsorgt werden.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Stillzeit (siehe Abschnitt 4.6).
Hypertonie
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über eine in der Regel früh im Behandlungsverlauf auftretende Hypertonie berichtet (siehe Abschnitt 4.8). Der Blutdruck sollte vor der Behandlung mit Lenvatinib gut eingestellt werden und Patienten mit bekannter Hypertonie sollten vor Beginn der Lenvatinib-Behandlung für mindestens 1 Woche eine antihypertensive Behandlung in stabiler Dosierung erhalten haben. Es wurde über schwere Komplikationen einer schlecht eingestellten Hypertonie, darunter Aortendissektion, berichtet. Die frühzeitige Erkennung und wirksame Behandlung der Hypertonie sind wichtig, um Behandlungsunterbrechungen oder Dosisreduktionen von Lenvatinib möglichst zu vermeiden. Die Behandlung mit Antihypertensiva sollte begonnen werden, sobald das Vorliegen einer Hypertonie bestätigt ist. Der Blutdruck sollte nach der ersten Behandlungswoche mit Lenvatinib kontrolliert werden, anschließend in den ersten 2 Monaten alle 2 Wochen und dann sollte die Kontrolle monatlich erfolgen. Die Wahl der antihypertensiven Behandlung sollte individuell auf die klinische Situation des Patienten abgestimmt werden und sich an dem medizinischen Standard orientieren. Bei bisher normotensiven Patienten sollte eine Monotherapie mit einem Standard-Antihypertensivum begonnen werden, sobald eine Hypertonie festgestellt wird. Bei denjenigen Patienten, die bereits ein Antihypertensivum erhalten, kann die Dosis des angewendeten Arzneimittels erhöht werden, wenn es angemessen ist, oder es können zusätzlich ein oder mehrere Arzneimittel einer anderen Klasse von Antihypertensiva gegeben werden. Sofern erforderlich, sollte die Behandlung der Hypertonie nach den Empfehlungen in Tabelle 3 durchgeführt werden.
Tabelle 3 Empfohlene Hypertonie-Behandlung
Blutdruckwerte (BD) |
Empfohlene Maßnahme |
Systolischer BD ≥ 140 mmHg bis < 160 mmHg oder diastolischer BD ≥ 90 mmHg bis < 100 mmHg |
Weiterbehandlung mit Lenvatinib und Beginn einer antihypertensiven Therapie, sofern diese nicht bereits erfolgt |
Systolischer BD ≥ 160 mmHg oder |
1. Vorübergehendes Absetzen von Lenvatinib |
Lebensbedrohliche Folgen |
Eine Notfallbehandlung ist indiziert. Lenvatinib absetzen und eine adäquate medizinische Behandlung durchführen. |
Aneurysmen und Arteriendissektionen
Die Verwendung von VEGF-Signalweg-Hemmern bei Patienten mit oder ohne Hypertonie kann die Entstehung von Aneurysmen und/oder Arteriendissektionen begünstigen. Vor Beginn der Behandlung mit Lenvatinib sollte dieses Risiko bei Patienten mit Risikofaktoren wie Hypertonie oder Aneurysmen in der Vorgeschichte sorgfältig abgewogen werden.
Gebärfähige Frauen
Gebärfähige Frauen müssen während der Einnahme von Lenvatinib sowie einen Monat lang nach Therapieende eine hochwirksame Methode der Empfängnisverhütung anwenden (siehe Abschnitt 4.6). Es ist bisher nicht bekannt, ob Lenvatinib das Risiko für thromboembolische Ereignisse erhöht, wenn es in Kombination mit oralen Kontrazeptiva angewendet wird.
Proteinurie
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über eine in der Regel früh im Behandlungsverlauf auftretende Proteinurie berichtet (siehe Abschnitt 4.8). Der Urin sollte regelmäßig auf Proteine kontrolliert werden. Wenn mit dem Urin-Teststreifen eine Proteinurie von ≥ 2+ festgestellt wird, ist möglicherweise eine Unterbrechung der Behandlung, eine Dosisanpassung oder ein Absetzen der Behandlung erforderlich (siehe Abschnitt 4.2). Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über Fälle von nephrotischem Syndrom berichtet. Bei Auftreten eines nephrotischen Syndroms muss Lenvatinib abgesetzt werden.
Nierenversagen und Nierenfunktionsstörung
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über Nierenfunktionsstörungen und Nierenversagen berichtet (siehe Abschnitt 4.8). Als Hauptrisikofaktor wurde eine Dehydrierung und/oder Hypovolämie aufgrund von gastrointestinaler Toxizität ermittelt. Die gastrointestinale Toxizität muss aktiv behandelt werden, um das Risiko einer Nierenfunktionsstörung oder eines Nierenversagens zu reduzieren. Bei Patienten, die mit Arzneimitteln behandelt werden, welche auf das Renin-Angiotensin-Aldosteron-System wirken, ist Vorsicht geboten, da die Kombinationsbehandlung möglicherweise mit einem erhöhten Risiko für akutes Nierenversagen verbunden ist. Möglicherweise ist eine Unterbrechung der Behandlung, eine Dosisanpassung oder ein Absetzen der Behandlung erforderlich (siehe Abschnitt 4.2).
Bei schwerer Nierenfunktionsstörung muss die Anfangsdosis von Lenvatinib angepasst werden (siehe Abschnitt 4.2 und 5.2).
Herzinsuffizienz
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über eine Herzinsuffizienz (< 1 %) und eine reduzierte linksventrikuläre Ejektionsfraktion berichtet (siehe Abschnitt 4.8). Die Patienten sollten hinsichtlich klinischer Symptome und Anzeichen für eine kardiale Dekompensation überwacht werden, da eine Unterbrechung der Behandlung, eine Dosisanpassung oder ein Absetzen der Behandlung erforderlich sein könnte (siehe Abschnitt 4.2).
Posteriores reversibles Enzephalopathie-Syndrom (PRES)/reversibles posteriores Leukenzephalopathie-Syndrom (RPLS)
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über das Auftreten eines PRES, auch bekannt als RPLS, berichtet (< 1 %; siehe Abschnitt 4.8). PRES ist eine neurologische Störung, die mit Kopfschmerzen, Krampfanfällen, Lethargie, Verwirrtheit, veränderter mentaler Funktion, Blindheit und anderen Sehstörungen oder neurologischen Störungen einhergehen kann. Es kann eine leichte bis schwere Hypertonie vorliegen. Die Diagnose eines PRES muss durch eine Magnetresonanztomographie bestätigt werden. Es sollten geeignete Maßnahmen zur Blutdruckeinstellung getroffen werden (siehe Abschnitt 4.4, Hypertonie). Bei Patienten mit Anzeichen oder Symptomen eines PRES ist möglicherweise eine Unterbrechung der Behandlung, eine Dosisanpassung oder ein Absetzen der Behandlung erforderlich (siehe Abschnitt 4.2).
Hepatotoxizität
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde am häufigsten über einen Anstieg von Alaninaminotransferase, Aspartataminotransferase und Bilirubin im Blut als die Leber betreffende Nebenwirkungen berichtet. Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über Fälle von Leberversagen und akuter Hepatitis berichtet (< 1 %; siehe Abschnitt 4.8). Die Fälle von Leberversagen wurden im Allgemeinen bei Patienten mit fortgeschrittenen Lebermetastasen berichtet. Die Leberfunktionswerte sollten vor Beginn der Behandlung kontrolliert werden, anschließend sollte die Kontrolle in den ersten 2 Monaten alle 2 Wochen und danach monatlich während der Behandlung erfolgen. Bei einer Hepatotoxizität ist möglicherweise eine Unterbrechung der Behandlung, eine Dosisanpassung oder ein Absetzen der Behandlung erforderlich (siehe Abschnitt 4.2).
Bei Patienten mit schwerer Leberinsuffizienz muss die Anfangsdosis von Lenvatinib angepasst werden (siehe Abschnitte 4.2 und 5.2).
Arterielle Thromboembolien
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über Fälle von arteriellen Thromboembolien (Schlaganfall, transitorische ischämische Attacke und Myokardinfarkt) berichtet (siehe Abschnitt 4.8). Lenvatinib wurde bei Patienten, bei denen in den vergangenen 6 Monaten eine arterielle Thromboembolie aufgetreten war, nicht untersucht. Lenvatinib sollte daher bei diesen Patienten mit Vorsicht angewendet werden. Die Behandlungsentscheidung sollte auf Basis des individuellen Nutzen-Risiko-Verhältnisses für den jeweiligen Patienten getroffen werden. Nach dem Auftreten einer arteriellen Thromboembolie muss Lenvatinib abgesetzt werden.
Blutungen
In klinischen Studien sind schwerwiegende tumorbedingte Blutungen, einschließlich tödlich verlaufener Blutungen, aufgetreten und wurden auch aus Erfahrungen nach dem Inverkehrbringen berichtet (siehe Abschnitt 4.8). Im Rahmen der Marktüberwachung wurden schwerwiegende und tödlich verlaufene Karotis-Blutungen bei Patienten mit anaplastischem Schilddrüsenkarzinom (ATC) häufiger beobachtet als bei Patienten mit DTC oder anderen Tumorarten. Der Grad der Tumorinvasion/-infiltration von wichtigen Blutgefäßen (wie z. B. der Arteria carotis) sollte berücksichtigt werden, weil durch Schrumpfen/Nekrose des Tumors infolge der Lenvatinib-Behandlung ein Risiko für schwere Blutungen bestehen kann. Infolge des Schrumpfens des Tumors und Fistelbildung, wie z. B. Ösophagotrachealfisteln, kam es zu einigen Blutungsfällen. Fälle von tödlich verlaufenen intrakranialen Blutungen wurden bei einigen Patienten mit oder ohne Hirnmetastasen gemeldet. Es liegen auch Berichte über Blutungen in anderen Körperregionen außer dem Gehirn vor (z. B. in der Trachea, innerhalb des Abdomens oder in der Lunge).
Bei Auftreten von Blutungen kann eine Behandlungsunterbrechung, eine Dosisanpassung oder ein Absetzen der Behandlung erforderlich sein (siehe Abschnitt 4.2, Tabelle 2).
Gastrointestinale Perforation oder Fistelbildung
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über Fälle von gastrointestinalen Perforationen oder Fisteln berichtet (siehe Abschnitt 4.8). In den meisten Fällen traten gastrointestinale Perforation oder Fisteln bei Patienten mit Risikofaktoren wie einer vorausgegangenen Operation oder einer Strahlentherapie auf. Bei einer gastrointestinalen Perforation oder Fistel ist möglicherweise eine Unterbrechung der Behandlung, eine Dosisanpassung oder ein Absetzen der Behandlung erforderlich (siehe Abschnitt 4.2).
Nichtgastrointestinale Fisteln
Die Patienten können während der Behandlung mit Lenvatinib einem erhöhten Risiko für die Bildung von Fisteln ausgesetzt sein. In klinischen Studien und gemäß Erfahrungen nach dem Inverkehrbringen wurden Fälle von Fistelbildung oder Fistelvergrößerung in anderen Körperregionen außer dem Magen oder Darm beobachtet (z. B. Trachealfisteln, Ösophagotrachealfisteln, Ösophagusfisteln, Hautfisteln, Fisteln im weiblichen Genitaltrakt). Außerdem wurde über Pneumothorax mit und ohne eindeutigen Nachweis einer Bronchopleuralfistel berichtet. Einige berichtete Fälle von Fisteln und Pneumothorax traten im Zusammenhang mit einer Tumorregression oder -nekrose auf. Frühere Operationen oder Radiotherapien können Risikofaktoren sein, die hierzu beitragen. Lungenmetastasen können ebenfalls das Risiko eines Pneumothorax erhöhen. Bei Patienten mit Fisteln sollte keine Behandlung mit Lenvatinib begonnen werden, um eine Verschlimmerung der Fisteln zu vermeiden; bei Patienten mit Beteiligung der Speiseröhre oder des Tracheobronchialtrakts und Fisteln jeglicher Art von Grad 4 (siehe Abschnitt 4.2) muss die Behandlung mit Lenvatinib dauerhaft abgesetzt werden. Über den Nutzen einer Behandlungsunterbrechung oder Dosisreduktion zur Kontrolle von anderen Ereignissen stehen nur begrenzte Informationen zur Verfügung, aber in manchen Fällen wurde eine Zustandsverschlechterung beobachtet und es ist daher Vorsicht geboten. Wie andere Wirkstoffe der gleichen Klasse, kann auch Lenvatinib die Wundheilung ungünstig beeinflussen.
QT-Zeit-Verlängerung
Eine Verlängerung der QT-/QTc-Zeit wurde häufiger bei Patienten berichtet, die mit Lenvatinib behandelt wurden, als bei Patienten, die mit Placebo behandelt wurden (siehe Abschnitt 4.8). Bei allen Patienten, unter besonderer Berücksichtigung derjenigen mit kongenitalem Long-QT-Syndrom, Myokardinsuffizienz und Bradyarrhythmien, und bei Patienten, die Arzneimittel einnehmen, von denen bekannt ist, dass sie die QT-Zeit verlängern (z. B. Antiarrhythmika der Klasse Ia und III), sollten regelmäßig Elektrokardiogramme durchgeführt werden. Lenvatinib sollte vorübergehend abgesetzt werden, wenn sich QT-Zeit-Verlängerungen von über 500 ms entwickeln. Nach Rückbildung der QTc-Zeit-Verlängerung auf < 480 ms oder zum Ausgangswert kann die Lenvatinib-Behandlung mit einer reduzierten Dosis fortgesetzt werden.
Elektrolytstörungen wie Hypokaliämie, Hypokalzämie oder Hypomagnesiämie können das Risiko für eine QT-Zeit-Verlängerung erhöhen und daher sollten Elektrolytabweichungen bei allen Patienten vor dem Beginn der Behandlung überwacht und korrigiert werden. Ferner sollten während der Behandlung regelmäßige EKG-Kontrollen und Untersuchungen der Elektrolyte (Magnesium, Kalium und Kalzium) erwogen werden. Die Kalziumspiegel im Blut sollten mindestens einmal monatlich kontrolliert werden und bei Bedarf sollte während der Lenvatinib-Behandlung eine Kalziumsupplementierung erfolgen. Je nach Schwere der Elektrolytstörungen und bei EKG-Veränderungen oder persistierender Hypokalzämie sollte die Lenvatinib-Behandlung unterbrochen oder die Dosis gegebenenfalls angepasst werden.
Störung der Suppression des Thyreoidea-stimulierenden Hormons/Schilddrüsenfehlfunktion
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über Hypothyreose berichtet (siehe Abschnitt 4.8). Die Schilddrüsenfunktion muss vor Einleitung und in regelmäßigen Abständen während der Behandlung mit Lenvatinib überwacht werden. Eine Hypothyreose ist entsprechend der gängigen medizinischen Praxis zu behandeln, um den euthyreoten Zustand aufrecht zu erhalten.
Lenvatinib stört die exogene Schilddrüsensuppression (siehe Abschnitt 4.8). Die Spiegel des Thyreoidea-stimulierenden Hormons (TSH) sollten regelmäßig kontrolliert werden und die Schilddrüsenhormontherapie sollte angepasst werden, um angemessene TSH-Spiegel entsprechend dem therapeutischen Ziel des Patienten zu erzielen.
Diarrhoe
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde häufig über Diarrhoe berichtet, die in der Regel bereits zu einem frühen Zeitpunkt während der Behandlung auftrat (siehe Abschnitt 4.8). Zur Vermeidung einer Dehydrierung sollte umgehend eine medizinische Behandlung der Diarrhoe eingeleitet werden. Bei Fortbestehen einer Diarrhoe von Grad 4 trotz medizinischer Behandlung muss Lenvatinib abgesetzt werden.
Wundheilungsstörungen
Zur Wirkung von Lenvatinib auf die Wundheilung wurden keine formellen Studien durchgeführt. Es wurde über verzögerte Wundheilung bei Patienten unter Lenvatinib berichtet. Bei größeren operativen Eingriffen an Patienten, die Lenvatinib erhalten, sollte in Erwägung gezogen werden, Lenvatinib vorübergehend zu pausieren. Es liegen nur begrenzte klinische Erfahrungen mit dem Zeitpunkt der Wiederaufnahme der Behandlung mit Lenvatinib nach einem größeren operativen Eingriff vor. Die Entscheidung zur Wiederaufnahme der Lenvatinib-Behandlung nach einem größeren operativen Eingriff sollte daher nach klinischem Ermessen angesichts eines angemessenen Wundheilungsverlaufs erfolgen.
Kieferosteonekrose
Bei Patienten, die mit Lenvatinib behandelt wurden, wurde über Fälle von Kieferosteonekrose berichtet. In manchen berichteten Fällen handelte es sich um Patienten, die eine vorherige oder gleichzeitige antiresorptive Knochentherapie und/oder andere Angiogenese-Hemmer wie z. B. Bevacizumab, TKI oder mTOR-Inhibitoren erhalten hatten. Daher ist Vorsicht geboten, wenn Lenvatinib entweder gleichzeitig mit oder im Anschluss an antiresorptive Medikamente und/oder Angiogenese-Hemmer verabreicht wird.
Invasive Dentaleingriffe stellen einen bekannten Risikofaktor dar. Vor der Behandlung mit Lenvatinib sollten eine zahnärztliche Untersuchung und eine angemessene Zahnvorsorge in Betracht gezogen werden. Bei Patienten, die zuvor intravenöse Bisphosphonate erhalten haben oder diese derzeit erhalten, sollten invasive Dentaleingriffe nach Möglichkeit vermieden werden (siehe Abschnitt 4.8).
Tumorlysesyndrom (TLS)
Lenvatinib kann ein TLS verursachen, das tödlich verlaufen kann. Zu den TLS-Risikofaktoren gehören unter anderem eine hohe Tumorlast, eine vorbestehende Nierenfunktionsstörung und Dehydrierung. Diese Patienten sollten engmaschig überwacht und bei klinischer Indikation behandelt werden, und eine prophylaktische Hydratation sollte in Erwägung gezogen werden.
Spezielle Patientengruppen
Über die Anwendung bei Patienten anderer ethnischer Abstammung als der kaukasischen oder asiatischen sowie bei Patienten ≥ 75 Jahre liegen bisher nur begrenzte Daten vor. Lenvatinib sollte angesichts der herabgesetzten Verträglichkeit bei Asiaten und älteren Patienten in dieser Patientengruppe mit Vorsicht angewendet werden (siehe Abschnitt 4.8).
Über die Anwendung von Lenvatinib unmittelbar nach einer Behandlung mit Sorafenib oder anderen Krebsmitteln liegen keine Daten vor und es kann ein potenzielles Risiko für additive Toxizitäten bestehen, wenn zwischen diesen Behandlungen kein ausreichend langer Auswaschzeitraum eingehalten wird. In klinischen Prüfungen betrug der Auswaschzeitraum mindestens 4 Wochen.
Auswirkung anderer Arzneimittel auf Lenvatinib
Chemotherapeutika
Die gleichzeitige Anwendung von Lenvatinib, Carboplatin und Paclitaxel hat keine signifikante Auswirkung auf die Pharmakokinetik dieser 3 Wirkstoffe. Darüber hinaus wurde die Pharmakokinetik von Lenvatinib bei Patienten mit RCC durch die gleichzeitige Anwendung von Everolimus nicht wesentlich beeinträchtigt.
Auswirkung von Lenvatinib auf andere Arzneimittel
CYP3A4-Substrate
Eine klinische Studie zu Arzneimittelwechselwirkungen (DDI-Studie) mit Krebspatienten hat ergeben, dass die Plasmakonzentrationen von Midazolam (ein empfindliches CYP3A- und Pgp-Substrat) durch den Einfluss von Lenvatinib nicht verändert wurden. Darüber hinaus wurde die Pharmakokinetik von Everolimus bei Patienten mit RCC durch die gleichzeitige Anwendung von Lenvatinib nicht wesentlich beeinträchtigt. Es wird daher keine signifikante Arzneimittelwechselwirkung zwischen Lenvatinib und anderen CYP3A4/Pgp-Substraten erwartet.
Orale Kontrazeptiva
Es ist bisher nicht bekannt, ob Lenvatinib die Wirksamkeit von hormonalen Kontrazeptiva herabsetzen kann, und deshalb müssen Frauen, die orale hormonale Kontrazeptiva anwenden, zusätzlich eine wirksame Methode zur Empfängnisverhütung anwenden (siehe Abschnitt 4.6).
Gebärfähige Frauen/Empfängnisverhütung bei Frauen
Gebärfähige Frauen sollten während der Behandlung mit Lenvatinib sowie mindestens bis zu einem Monat nach Therapieende nicht schwanger werden und eine hochwirksame Verhütungsmethode anwenden. Es ist bisher nicht bekannt, ob Lenvatinib die Wirksamkeit von hormonalen Kontrazeptiva herabsetzen kann, und deshalb sollen Frauen, die orale hormonale Kontrazeptiva anwenden, zusätzlich eine Barrieremethode verwenden.
Schwangerschaft
Es liegen keine Erfahrungen zur Anwendung von Lenvatinib bei Schwangeren vor. Bei der Anwendung an Ratten und Kaninchen zeigte Lenvatinib eine embryotoxische und teratogene Wirkung (siehe Abschnitt 5.3).
Während der Schwangerschaft darf Lenvatinib nicht angewendet werden, es sei denn dies ist eindeutig erforderlich. Dabei ist der Nutzen für die Mutter gegen das Risiko für den Fetus sorgfältig abzuwägen.
Stillzeit
Es ist nicht bekannt, ob Lenvatinib beim Menschen in die Muttermilch übergeht. Bei Ratten werden Lenvatinib und seine Metaboliten in die Muttermilch ausgeschieden (siehe Abschnitt 5.3).
Da ein Risiko für Neugeborene oder Säuglinge nicht ausgeschlossen werden kann, ist Lenvatinib während der Stillzeit kontraindiziert (siehe Abschnitt 4.3).
Fertilität
Es sind keine humanen Daten bekannt. Jedoch wurde bei Ratten, Hunden und Affen eine Toxizität an Hoden und Eierstöcken beobachtet (siehe Abschnitt 5.3).
Lenvatinib hat geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen, da es Nebenwirkungen wie Müdigkeit und Schwindel hervorrufen kann. Patienten, bei denen diese Symptome auftreten, sollten beim Fahren oder Bedienen von Maschinen vorsichtig sein.
Zusammenfassung des Sicherheitsprofils
Das Sicherheitsprofil von Lenvatinib basiert auf zusammengefassten Daten zu 497 RCC-Patienten, die mit Lenvatinib in Kombination mit Pembrolizumab behandelt wurden, einschließlich Studie 307 (CLEAR), zusammengefassten Daten zu 623 RCC-Patienten, die mit Lenvatinib in Kombination mit Everolimus behandelt wurden, sowie 458 DTC-Patienten und 496 HCC-Patienten, die mit Lenvatinib als Monotherapie behandelt wurden.
Lenvatinib in Kombination mit Pembrolizumab bei RCC
Das Sicherheitsprofil von Lenvatinib in Kombination mit Pembrolizumab basiert auf Daten von 497 RCC-Patienten. Die am häufigsten berichteten Nebenwirkungen (bei ≥ 30 % der Patienten) waren Diarrhoe (61,8 %), Hypertonie (51,5 %), Fatigue (47,1 %), Hypothyreose (45,1 %), verminderter Appetit (42,1 %), Übelkeit (39,6 %), Stomatitis (36,6 %), Proteinurie (33,0 %), Dysphonie (32,8 %) und Arthralgie (32,4 %).
Die häufigsten schweren (Grad ≥ 3) Nebenwirkungen (≥ 5 %) waren Hypertonie (26,2 %), erhöhte Lipase (12,9 %), Diarrhoe (9,5 %), Proteinurie (8,0 %), erhöhte Amylase (7,6 %), Gewichtsverlust (7,2 %) und Fatigue (5,2 %).
Bei 33,4 % der Patienten wurden Lenvatinib, Pembrolizumab oder beide Arzneimittel aufgrund einer Nebenwirkung abgesetzt. Bei 23,7 % wurde Lenvatinib und bei 12,9 % wurden beide Arzneimittel abgesetzt. Die häufigsten Nebenwirkungen (≥ 1 %), die zum Absetzen von Lenvatinib, Pembrolizumab oder beiden Arzneimitteln führten, waren Myokardinfarkt (2,4 %), Diarrhoe (2,0 %), Proteinurie (1,8 %) und Hautausschlag (1,4 %). Die Nebenwirkungen, die am häufigsten zum Absetzen von Lenvatinib führten (≥ 1 %), waren Myokardinfarkt (2,2 %), Proteinurie (1,8 %) und Diarrhoe (1,0 %).
Behandlungsunterbrechungen von Lenvatinib, Pembrolizumab oder beiden Arzneimitteln aufgrund einer Nebenwirkung erfolgten bei 80,1 % der Patienten; Lenvatinib wurde bei 75,3 % und beide Arzneimittel bei 38,6 % der Patienten unterbrochen. Bei 68,4 % der Patienten wurde die Lenvatinib-Dosis reduziert. Die häufigsten Nebenwirkungen (≥ 5 %), die zu einer Dosisreduktion oder zu einer Unterbrechung von Lenvatinib führten, waren Diarrhoe (25,6 %), Hypertonie (16,1 %), Proteinurie (13,7 %), Fatigue (13,1 %), verminderter Appetit (10,9 %), palmar-plantares Erythrodysästhesie-Syndrom (PPE) (10,7 %), Übelkeit (9,7 %), Asthenie (6,6 %), Stomatitis (6,2 %), erhöhte Lipase (5,6 %) und Erbrechen (5,6 %).
Lenvatinib in Kombination mit Everolimus bei RCC
Das Sicherheitsprofil von Lenvatinib in Kombination mit Everolimus basiert auf Daten von 623 Patienten.
Die am häufigsten berichteten Nebenwirkungen (bei ≥ 30 % der Patienten) waren Diarrhoe (69,0 %), Fatigue (41,9 %), Hypertonie (41,7 %), verminderter Appetit (41,6 %), Stomatitis (40,6 %), Übelkeit (38,8 %), Proteinurie (34,2 %), Erbrechen (32,7 %) und Gewichtsverlust (31,3 %).
Die häufigsten schweren (≥ Grad 3) Nebenwirkungen (≥ 5 %) waren Hypertonie (19,3 %), Diarrhoe (13,8 %), Proteinurie (8,8 %), Fatigue (7,1 %), verminderter Appetit (6,3 %) und Gewichtsverlust (5,8 %).
Bei 27,0 % der Patienten wurden Lenvatinib, Everolimus oder beide Arzneimittel aufgrund einer Nebenwirkung abgesetzt; Lenvatinib wurde bei 21,7 % und beide Arzneimittel bei 18,7 % der Patienten abgesetzt. Die häufigsten Nebenwirkungen (≥ 1 %), die zum Absetzen von Lenvatinib, Everolimus oder beiden Arzneimitteln führten, waren Proteinurie (2,7 %), Diarrhoe (1,0 %) und verminderter Appetit (1,0 %). Die Nebenwirkung, die am häufigsten zum Absetzen von Lenvatinib führte (≥ 1 %), war Proteinurie (2,1 %).
Dosisunterbrechungen von Lenvatinib, Everolimus oder beiden Arzneimitteln aufgrund einer Nebenwirkung erfolgten bei 82,2 % der Patienten. Bei 74,3 % der Patienten, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurde Lenvatinib pausiert. Beide Arzneimittel wurden bei 71,9 % dieser Patienten pausiert. Die häufigsten Nebenwirkungen (≥ 5 %), die zu einer Dosisreduktion oder Dosisunterbrechung von Lenvatinib führten, waren Diarrhoe (30,4 %), Fatigue (15,3 %), Proteinurie (14,7 %), verminderter Appetit (13,4 %), Stomatitis (13,2 %), Übelkeit (10,9 %), Erbrechen (10,2 %), Hypertonie (9,2 %), Asthenie (7,9 %), verminderte Thrombozytenzahl (5,7 %) und Gewichtsverlust (5,1 %).
Tabellarische Auflistung der Nebenwirkungen
Die in klinischen Studien sowie nach der Markteinführung von Lenvatinib beobachteten Nebenwirkungen sind in Tabelle 4 aufgeführt. Nebenwirkungen, die bekanntermaßen im Zusammenhang mit Lenvatinib oder mit allein verabreichten Komponenten der Kombinationstherapie auftreten, können während der Kombinationsbehandlung mit diesen Arzneimitteln auftreten, selbst wenn diese Nebenwirkungen in klinischen Studien mit der Kombinationstherapie nicht berichtet wurden.
Weitere Sicherheitsinformationen bei Anwendung von Lenvatinib im Rahmen einer Kombinationstherapie sind den Fachinformationen zu den jeweiligen Komponenten der Kombinationstherapie zu entnehmen.
Die Häufigkeiten sind wie folgt definiert:
Sehr häufig (≥1/10)
Häufig (≥1/100, <1/10)
Gelegentlich (≥1/1.000, <1/100)
Selten (≥ 1/10.000, < 1/1.000)
Sehr selten (< 1/10.000)
Nicht bekannt (Häufigkeit auf der Grundlage der verfügbaren Daten nicht abschätzbar)
In jeder Häufigkeitskategorie werden die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 4 Berichtete Nebenwirkungen bei Patienten, die mit Lenvatinib behandelt wurden§
Systemorganklasse |
Lenvatinib-Monotherapie |
Kombination mit Everolimus |
Kombination mit Pembrolizumab |
Infektionen und parasitäre Erkrankungen | |||
Sehr häufig |
Harnwegsinfektion |
||
Häufig |
Harnwegsinfektion |
Harnwegsinfektion |
|
Gelegentlich |
Perinealabszess |
Perinealabszess |
Perinealabszess |
Erkrankungen des Blutes und des Lymphsystems | |||
Sehr häufig |
Thrombozytopenie‡ |
Thrombozytopenie‡ |
Thrombozytopenie‡ |
Gelegentlich |
Milzinfarkt |
||
Endokrine Erkrankungen | |||
Sehr häufig |
Hypothyreose* |
Hypothyreose* |
Hypothyreose* |
Häufig |
Nebenniereninsuffizienz |
||
Gelegentlich |
Nebenniereninsuffizienz |
Nebenniereninsuffizienz |
|
Stoffwechsel- und Ernährungsstörungen | |||
Sehr häufig |
Hypokalzämie*, ‡ |
Hypokalzämie ‡ |
Hypokalzämie ‡ |
Häufig |
Dehydrierung |
Dehydrierung |
Dehydrierung |
Selten |
Tumorlysesyndrom† |
Tumorlysesyndrom† |
Tumorlysesyndrom† |
Psychiatrische Erkrankungen | |||
Sehr häufig |
Insomnie |
Insomnie |
Insomnie |
Erkrankungen des Nervensystems | |||
Sehr häufig |
Schwindel |
Kopfschmerzen |
Schwindel |
Häufig |
Apoplektischer Insult† |
Schwindel |
|
Gelegentlich |
Posteriores reversibles Enzephalopathiesyndrom |
Apoplektischer Insult† |
Apoplektischer Insult |
Herzerkrankungen | |||
Häufig |
Myokardinfarkta, † |
Myokardinfarkta, † |
Myokardinfarkta |
Gelegentlich |
Reduzierte Ejektionsfraktion |
Herzinsuffizienz† |
|
Gefäßerkrankungen | |||
Sehr häufig |
Blutungb, *, † |
Blutungb, *, † |
Blutungb, *, † |
Häufig |
Hypotonie |
Hypotonie |
|
Nicht bekannt |
Aneurysmen und Arteriendissektionen |
Aneurysmen und Arteriendissektionen |
Aneurysmen und Arteriendissektionen |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |||
Sehr häufig |
Dysphonie |
Dysphonie |
Dysphonie |
Häufig |
Lungenembolie† |
Lungenembolie |
Lungenembolie |
Gelegentlich |
Pneumothorax |
Pneumothorax |
|
Erkrankungen des Gastrointestinaltrakts | |||
Sehr häufig |
Diarrhoe* |
Diarrhoe* |
Diarrhoe* |
Häufig |
Analfistel |
Mundtrockenheit |
Pankreatitisg |
Gelegentlich |
Pankreatitisg |
Pankreatitisg |
Analfistel |
Leber- und Gallenerkrankungen | |||
Sehr häufig |
Bilirubin im Blut erhöht*, ‡ |
Hypoalbuminämie*, ‡ |
Bilirubin im Blut |
Häufig |
Leberversagenh, † |
Cholezystitis |
Cholezystitis |
Gelegentlich |
Hepatozelluläre Schädigung/Hepatitisj |
Leberversagenh, † |
Leberversagenh, † |
Erkrankungen der Haut und des Unterhautgewebes | |||
Sehr häufig |
Palmar-plantares Erythrodysästhesie-Syndrom |
Palmar-plantares Erythrodysästhesie-Syndrom |
Palmar-plantares Erythrodysästhesie-Syndrom |
Häufig |
Hyperkeratose |
Alopezie |
Hyperkeratose |
Gelegentlich |
Hyperkeratose |
||
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |||
Sehr häufig |
Rückenschmerzen |
Rückenschmerzen |
Rückenschmerzen |
Häufig |
Myalgie |
||
Gelegentlich |
Kieferosteonekrose |
Kieferosteonekrose |
|
Erkrankungen der Nieren und Harnwege | |||
Sehr häufig |
Proteinurie* |
Proteinurie* |
Proteinurie* |
Häufig |
Nierenversagenk, *, † |
Nierenversagenk, *, † |
Nierenversagenk, * |
Gelegentlich |
Nephrotisches Syndrom |
Nephrotisches Syndrom |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |||
Sehr häufig |
Fatigue |
Fatigue |
Fatigue |
Häufig |
Unwohlsein |
Unwohlsein |
Unwohlsein |
Gelegentlich |
Verzögerte Heilung |
Verzögerte Heilung |
Verzögerte Heilung |
Nicht bekannt |
Nichtgastrointestinale Fistelnl |
||
§: Die in Tabelle 4 aufgeführten Häufigkeiten von Nebenwirkungen sind möglicherweise nicht vollständig Lenvatinib allein zuzuschreiben, sondern können auch Faktoren enthalten, die aus Grunderkrankungen oder von in Kombination angewendeten Arzneimitteln einfließen.
*: Siehe Abschnitt 4.8, Beschreibung ausgewählter Nebenwirkungen für weitere Beschreibungen.
†: Beinhaltet Fälle mit tödlichem Ausgang.
‡: Häufigkeit basierend auf Labordaten.
Die folgenden Begriffe wurden zusammengefasst:
a: Myokardinfarkt umfasst Myokardinfarkt und akuten Myokardinfarkt.
b: Umfasst alle Blutungen betreffenden Begriffe:
Blutungen betreffende Begriffe, die bei mindestens 5 Patienten mit RCC in den mit Lenvatinib und Pembrolizumab behandelten Gruppen auftraten, waren: Epistaxis, Hämaturie, Kontusion, Zahnfleischbluten, Rektalblutung, Hämoptyse, Ekchymose und Hämatochezie.
c: Hypertonie umfasst: Hypertonie, hypertensive Krise, erhöhter diastolischer Blutdruck, orthostatische Hypertonie und erhöhten Blutdruck.
d: Gastrointestinale und abdominale Schmerzen umfassen: abdominale Beschwerden, Abdominalschmerz, Schmerzen im Unterbauch, Schmerzen im Oberbauch, abdominaler Druckschmerz, epigastrische Beschwerden und gastrointestinale Schmerzen.
e: Orale Entzündung umfasst: Stomatitis aphthosa, aphthöses Ulkus, Zahnfleischerosion, Zahnfleischulkus, Mundschleimhautbläschen, Stomatitis, Glossitis, Mundulzeration und Schleimhautentzündung.
f: Schmerzen im Mundbereich umfasst: Mundschmerzen, Glossodynie, Zahnfleischschmerzen, oropharyngeale Beschwerden, oropharyngeale Schmerzen und Zungenbeschwerden.
g: Pankreatitis umfasst: Pankreatitis und akute Pankreatitis.
h: Leberversagen umfasst: Leberversagen, akutes Leberversagen und chronisches Leberversagen.
i: Hepatische Enzephalopathie umfasst: hepatische Enzephalopathie, hepatisches Koma, metabolische Enzephalopathie und Enzephalopathie.
j: Hepatozelluläre Schädigung und Hepatitis umfassen: arzneimittelinduzierte Leberschädigung, hepatische Steatose und cholestatische Leberschädigung.
k: Nierenversagen umfasst: akute prärenale Insuffizienz, Nierenversagen, akutes Nierenversagen, akute Nierenschädigung und Nierentubulusnekrose.
l: Nichtgastrointestinale Fisteln umfasst Fälle von Fisteln, die außerhalb des Magens und des Darms auftreten, wie z. B. Trachealfisteln, Ösophagotrachealfisteln, Ösophagusfisteln, Hautfisteln und Fisteln im weiblichen Genitaltrakt.
Beschreibung ausgewählter Nebenwirkungen
Hypertonie (siehe Abschnitt 4.4)
In der CLEAR-Studie (siehe Abschnitt 5.1) wurde Hypertonie bei 56,3 % der Patienten in der mit Lenvatinib und Pembrolizumab behandelten Gruppe und bei 42,6 % der Patienten in der mit Sunitinib behandelten Gruppe berichtet. Die expositionsbereinigte Häufigkeit von Hypertonie betrug 0,65 Episoden pro Patientenjahr in der mit Lenvatinib und Pembrolizumab behandelten Gruppe und 0,73 Episoden pro Patientenjahr in der mit Sunitinib behandelten Gruppe. Die mediane Zeit bis zum Eintritt der Hypertonie bei mit Lenvatinib und Pembrolizumab behandelten Patienten betrug 0,7 Monate. Reaktionen von Grad 3 oder höher traten bei 28,7 % der Patienten in der mit Lenvatinib und Pembrolizumab behandelten Gruppe auf, verglichen mit 19,4 % der Patienten in der mit Sunitinib behandelten Gruppe. Bei 16,8 % der Patienten mit Hypertonie wurden Dosisanpassungen von Lenvatinib (9,1 % Dosisunterbrechung und 11,9 % Dosisreduktion) vorgenommen. Bei 0,9 % der Patienten führte Hypertonie zu einem dauerhaften Absetzen der Behandlung mit Lenvatinib.
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde bei 42,5 % der Patienten über Hypertonie berichtet (die Inzidenz von Hypertonien von Grad 3 oder Grad 4 war 19,7 %). Bei 9,8 % der Patienten mit Hypertonie, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (5,3 % Dosisreduktion und 6,2 % Dosisunterbrechung) vorgenommen. Bei 0,9 % dieser Patienten führte Hypertonie zu einem dauerhaften Absetzen der Behandlung mit Lenvatinib. Die mediane Zeit bis zum Auftreten der Hypertonie bei mit Lenvatinib und Everolimus behandelten Patienten betrug 0,5 Monate.
Proteinurie (siehe Abschnitt 4.4)
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde bei 34,8 % der Patienten (9,0 % waren ≥ Grad 3) über Proteinurie berichtet. Bei 15,1 % der Patienten mit Proteinurie, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (9,6 % Dosisreduktion und 9,8 % Dosisunterbrechung) vorgenommen. Proteinurie führte bei 2,1 % dieser Patienten zum dauerhaften Absetzen der Behandlung. Die mediane Zeit bis zum Auftreten der Proteinurie bei mit Lenvatinib und Everolimus behandelten Patienten betrug 1,4 Monate.
Nierenversagen und Nierenfunktionsstörung (siehe Abschnitt 4.4)
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population kam es bei 1,3 % der Patienten zu Nierenversagen (0,6 % waren ≥ Grad 3) und 5,3 % entwickelten eine akute Nierenschädigung (2,7 % waren ≥ Grad 3). Die Nieren betreffende Nebenwirkungen wurden bei 17,2 % der Patienten berichtet (4,3 % waren ≥ Grad 3). Bei 5,5 % der Patienten mit die Nieren betreffenden Nebenwirkungen, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (2,3 % Dosisreduktion und 4,0 % Dosisunterbrechung) vorgenommen. Die Nieren betreffende Nebenwirkungen führten bei 1,9 % dieser Patienten zum dauerhaften Absetzen der Behandlung. Die mediane Zeit bis zum Auftreten der die Nieren betreffenden Nebenwirkungen bei mit Lenvatinib und Everolimus behandelten Patienten betrug 3,5 Monate.
Herzinsuffizienz (siehe Abschnitt 4.4)
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde bei 3,5 % der Patienten über Herzinsuffizienz (1,8 % waren ≥ Grad 3) berichtet. Bei 0,9 % der Patienten mit Herzinsuffizienz, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (0,4 % Dosisreduktion und 0,8 % Dosisunterbrechung) vorgenommen. Herzinsuffizienz führte bei 0,6 % dieser Patienten zum dauerhaften Absetzen der Behandlung. Die mediane Zeit bis zum Auftreten der Herzinsuffizienz bei mit Lenvatinib und Everolimus behandelten Patienten betrug 3,6 Monate.
Posteriores reversibles Enzephalopathie-Syndrom (PRES)/reversibles posteriores Leukenzephalopathie-Syndrom (RPLS) (siehe Abschnitt 4.4)
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde 1 Fall von PRES berichtet (Grad 2), welches nach einer Behandlungsdauer von 1,3 Monaten auftrat und weder Dosisanpassungen noch ein Absetzen der Behandlung erforderte.
Hepatotoxizität (siehe Abschnitt 4.4)
In der CLEAR-Studie (siehe Abschnitt 5.1) waren in der mit Lenvatinib und Pembrolizumab behandelten Gruppe die am häufigsten gemeldeten die Leber betreffenden Nebenwirkungen Anstiege der Leberenzymwerte, darunter Anstiege der Alaninaminotransferase (11,9 %), der Aspartataminotransferase (11,1 %) und des Bilirubins im Blut (4,0 %). Ähnliche Ereignisse traten in der mit Sunitinib behandelten Gruppe mit Häufigkeiten von 10,3 %, 10,9 % bzw. 4,4 % auf. Die mediane Zeit bis zum Auftreten der die Leber betreffenden Nebenwirkungen betrug bei der mit Lenvatinib und Pembrolizumab behandelten Gruppe 3,0 Monate (alle Grade) und bei der mit Sunitinib behandelten Gruppe 0,7 Monate. Die expositionsbereinigte Häufigkeit von Hepatotoxizitäts-Ereignissen betrug in der mit Lenvatinib und Pembrolizumab behandelten Gruppe 0,39 Episoden pro Patientenjahr und in der mit Sunitinib behandelten Gruppe 0,46 Episoden pro Patientenjahr. Die die Leber betreffenden Nebenwirkungen von Grad 3 traten bei 9,9 % der mit Lenvatinib und Pembrolizumab behandelten Patienten und bei 5,3 % der mit Sunitinib behandelten Patienten auf. Die die Leber betreffenden Nebenwirkungen führten zu Behandlungsunterbrechungen und Dosisreduktionen von Lenvatinib bei 8,5 % bzw. 4,3 % der Patienten und zum dauerhaften Absetzen der Behandlung mit Lenvatinib bei 1,1 % der Patienten.
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population waren die am häufigsten gemeldeten die Leber betreffenden Nebenwirkungen Anstiege der Leberenzymwerte, einschließlich Anstiege der Alaninaminotransferase (11,9 %), der Aspartataminotransferase (11,4 %) und der Gamma-Glutamyltransferase (2,7 %). Die Leber betreffende Nebenwirkungen von Grad 3 traten bei 6,1 % der mit Lenvatinib und Everolimus behandelten Patienten auf. Bei 6,0 % der Patienten mit Hepatotoxizität, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (2,8 % Dosisreduktion und 4,2 % Dosisunterbrechung) vorgenommen. Die Leber betreffende Nebenwirkungen führten bei 0,9 % dieser Patienten zum dauerhaften Absetzen der Behandlung. Die mediane Zeit bis zum Auftreten der die Leber betreffenden Nebenwirkungen bei den mit Lenvatinib und Everolimus behandelten Patienten betrug 1,8 Monate.
Arterielle Thromboembolien (siehe Abschnitt 4.4)
In der CLEAR-Studie (siehe Abschnitt 5.1) wurde bei 5,4 % der Patienten in der mit Lenvatinib und Pembrolizumab behandelten Gruppe über arterielle thromboembolische Ereignisse (von denen 3,7 % Grad 3 oder höher waren) berichtet, verglichen mit 2,1 % der Patienten in der mit Sunitinib behandelten Gruppe (wobei 0,6 % Grad 3 oder höher waren). Keines der Ereignisse verlief tödlich. Die expositionsbereinigte Häufigkeit von Episoden arterieller thromboembolischer Ereignisse betrug in der mit Lenvatinib und Pembrolizumab behandelten Gruppe 0,04 Episoden pro Patientenjahr und in der mit Sunitinib behandelten Gruppe 0,02 Episoden pro Patientenjahr. Das am häufigsten berichtete arterielle thromboembolische Ereignis in der mit Lenvatinib und Pembrolizumab behandelten Gruppe war Myokardinfarkt (3,4 %). In der mit Sunitinib behandelten Gruppe trat ein Myokardinfarkt-Ereignis (0,3 %) auf. Die mediane Zeit bis zum Auftreten arterieller thromboembolischer Ereignisse betrug in der mit Lenvatinib und Pembrolizumab behandelten Gruppe 10,4 Monate.
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde bei 2,7 % der Patienten über arterielle thromboembolische Ereignisse (2,2 % waren ≥ Grad 3) berichtet. Bei 0,6 % der Patienten mit arteriellen thromboembolischen Ereignissen, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (0,6 % Dosisunterbrechung) vorgenommen. Arterielle thromboembolische Ereignisse führten bei 1,5 % dieser Patienten zum dauerhaften Absetzen der Behandlung. Das am häufigsten berichtete arterielle thrombolische Ereignis in der mit Lenvatinib und Everolimus behandelten Gruppe war Myokardinfarkt (1,3 %). Die mediane Zeit bis zum Auftreten der arteriellen thromboembolischen Ereignisse bei mit Lenvatinib und Everolimus behandelten Patienten betrug 6,8 Monate.
Blutungen (siehe Abschnitt 4.4)
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde bei 28,6 % der Patienten über Blutungen (3,2 % waren ≥ Grad 3) berichtet. Bei 4,9 % der Patienten mit Blutungen, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (4,2 % Dosisunterbrechung und 0,8 % Dosisreduktion) vorgenommen. Blutungen führten bei 0,6 % dieser Patienten zum dauerhaften Absetzen der Behandlung. Die am häufigsten berichteten Blutungsereignisse in der mit Lenvatinib und Everolimus behandelten Gruppe waren Epistaxis (19,4 %) und Hämaturie (4,2 %). Die mediane Zeit bis zum Auftreten der Blutungen bei mit Lenvatinib und Everolimus behandelten Patienten betrug 1,9 Monate.
Hypokalzämie (siehe Abschnitt 4.4, QT-Zeit-Verlängerung)
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde bei 4,8 % der Patienten über Hypokalzämie (1,1 % waren ≥ Grad 3) berichtet. Bei 0,8 % der Patienten mit Hypokalzämie, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (0,6 % Dosisunterbrechung und 0,4 % Dosisreduktion) vorgenommen. Keiner dieser Patienten brach die Behandlung aufgrund von Hypokalzämie dauerhaft ab. Die mediane Zeit bis zum Auftreten der Hypokalzämie bei mit Lenvatinib und Everolimus behandelten Patienten betrug 2,9 Monate.
Gastrointestinale Perforation oder Fistelbildung (siehe Abschnitt 4.4)
In der mit Lenvatinib und Everolimus behandelten zusamengefassten RCC-Population wurde bei 3,7 % der Patienten über gastrointestinale Perforation (2,9 % waren ≥ Grad 3) berichtet. Bei 2,1 % der Patienten mit gastrointestinalen Perforationen, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (1,5 % Dosisunterbrechung und 0,6 % Dosisreduktion) vorgenommen. Gastrointestinale Perforationen führten bei 1,1 % dieser Patienten zum dauerhaften Absetzen der Behandlung. Die mediane Zeit bis zum Auftreten der gastrointestinalen Perforation bei mit Lenvatinib und Everolimus behandelten Patienten betrug 3,6 Monate.
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde bei 1,0 % der Patienten über Fistelbildung (0,5 % waren ≥ Grad 3) berichtet. Bei 0,8 % der Patienten mit gastrointestinalen Perforationen, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (0,8 % Dosisunterbrechung) vorgenommen. Fistelbildung führte bei 0,4 % dieser Patienten zum dauerhaften Absetzen der Behandlung. Die mediane Zeit bis zum Auftreten der Fistelbildung bei mit Lenvatinib und Everolimus behandelten Patienten betrug 3,7 Monate.
Nichtgastrointestinale Fisteln (siehe Abschnitt 4.4)
Die Anwendung von Lenvatinib war mit Fällen von Fistelbildung, einschließlich zum Tod führender Reaktionen, verbunden. Fälle von Fistelbildung in anderen Körperregionen außer dem Magen oder Darm wurden bei verschiedenen Indikationen beobachtet. Die Reaktionen wurden zu unterschiedlichen Zeitpunkten während der Behandlung gemeldet, angefangen von zwei Wochen bis zu über 1 Jahr nach Therapiebeginn. Die mediane Latenzzeit lag bei ca. 3 Monaten.
QT-Zeit-Verlängerung (siehe Abschnitt 4.4)
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde bei 9,8 % der Patienten in der mit Lenvatinib und Everolimus behandelten Gruppe über QTc-Zeit-Verlängerungen von über 60 ms berichtet. Die Inzidenz von QTc-Zeit-Verlängerungen von mehr als 500 ms betrug in der mit Lenvatinib und Everolimus behandelten Gruppe 3,3 %. Die mediane Zeit bis zum Auftreten von QTc-Zeit-Verlängerungen bei mit Lenvatinib und Everolimus behandelten Patienten betrug 3,0 Monate.
Erhöhte Blutwerte von Thyreoidea-stimulierendem Hormon (TSH)/Hypothyreose (siehe Abschnitt 4.4)
In der Studie CLEAR (siehe Abschnitt 5.1) trat bei 47,2 % der Patienten in der mit Lenvatinib und Pembrolizumab behandelten Gruppe und bei 26,5 % der Patienten in der mit Sunitinib behandelten Gruppe Hypothyreose auf. Die expositionsbereinigte Häufigkeit von Hypothyreose betrug in der mit Lenvatinib und Pembrolizumab behandelten Gruppe 0,39 Episoden pro Patientenjahr und in der mit Sunitinib behandelten Gruppe 0,33 Episoden pro Patientenjahr. Im Allgemeinen war der Großteil der Hypothyreose-Ereignisse in der mit Lenvatinib und Pembrolizumab behandelten Gruppe vom Grad 1 oder 2. Hypothyreose vom Grad 3 wurde bei 1,4 % der Patienten in der mit Lenvatinib und Pembrolizumab behandelten Gruppe berichtet, verglichen mit keinen Patienten in der mit Sunitinib behandelten Gruppe. Zum Studienbeginn hatten 90,0 % der Patienten in der mit Lenvatinib und Pembrolizumab behandelten Gruppe und 93,1 % der Patienten in der mit Sunitinib behandelten Gruppe Ausgangs-TSH-Spiegel ≤ obere Normgrenze. Erhöhungen der TSH-Spiegel > obere Normgrenze wurden nach Studienbeginn bei 85,0 % der mit Lenvatinib und Pembrolizumab behandelten Patienten beobachtet, verglichen mit 65,6 % der mit Sunitinib behandelten Patienten. Bei mit Lenvatinib und Pembrolizumab behandelten Patienten führten Hypothyreose-Ereignisse bei 2,6 % der Patienten zu einer Dosisanpassung von Lenvatinib (Reduktion oder Unterbrechung) und bei 1 Patienten zum Absetzen von Lenvatinib.
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population trat Hypothyreose bei 24,1 % der Patienten auf. Im Allgemeinen war die Mehrzahl der Hypothyreose-Ereignisse von Schweregrad 1 oder 2. Hypothyreose vom Schweregrad 3 wurde bei 0,3 % der mit Lenvatinib und Everolimus behandelten Patienten berichtet. Die mediane Zeit bis zum Auftreten von Hypothyreose-Ereignissen bei mit Lenvatinib und Everolimus behandelten Patienten betrug 2,7 Monate. Zu Studienbeginn wiesen 83,0 % der Patienten in der mit Lenvatinib und Everolimus behandelten Gruppe TSH-Spiegel ≤ der oberen Normgrenze auf. Erhöhungen der TSH-Spiegel > der oberen Normgrenze wurden nach Studienbeginn bei 71,3 % der mit Lenvatinib und Everolimus behandelten Patienten berichtet. Bei 1,3 % der Patienten mit Hypothyreose-Ereignissen, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (0,4 % Dosisreduktion und 0,9 % Dosisunterbrechung) vorgenommen. Es wurden keine Behandlungsabbrüche berichtet.
Diarrhoe (siehe Abschnitt 4.4)
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population wurde bei 69,0 % der Patienten über Diarrhoe (13,8 % waren Grad ≥ 3) berichtet. Bei 30,4 % der Patienten mit Diarrhoe, bei denen Daten zu individuellen Medikamentenanpassungen erhoben wurden, wurden Dosisanpassungen von Lenvatinib (17,7 % Dosisunterbrechung und 19,6 % Dosisreduktion) vorgenommen. Diarrhoe führte bei 0,6 % dieser Patienten zum dauerhaften Absetzen der Behandlung.
Kinder und Jugendliche
In den Studien 216 und 231 an Kindern und Jugendlichen (siehe Abschnitt 5.1) stimmte das Gesamtsicherheitsprofil von Lenvatinib als Monotherapie oder in Kombination mit Everolimus mit demjenigen überein, das bei Erwachsenen unter Behandlung mit Lenvatinib beobachtet wurde.
In Studie 216 wurde bei 3 Patienten (4,7 %) mit Ewing-Sarkom, Rhabdomyosarkom (RMS) und Wilms-Tumor ein Pneumothorax berichtet; bei allen 3 Patienten lagen zu Studienbeginn Lungenmetastasen vor. In Studie 231 wurde bei 8 Patienten (6,3 %) mit Spindelzellsarkom, undifferenziertem Sarkom, RMS, bösartigem peripherem Nervenscheidentumor, Synovialsarkom, Spindelzellkarzinom und bösartigem ossifizierenden fibromyxoiden Tumor ein Pneumothorax berichtet; bei allen 8 Patienten lagen zu Studienbeginn Lungenmetastasen oder eine Primärerkrankung in der Brustwand oder Pleurahöhle vor. In Studie 216 wurde bei keinem Patienten die Studienbehandlung aufgrund eines Pneumothorax abgebrochen; in Studie 231 wurde bei einem Patienten die Studienbehandlung aufgrund eines Pneumothorax abgebrochen (weitere Informationen zu Kindern und Jugendlichen finden Sie auch in der Fachinformation zu Lenvima, Abschnitt 4.8.).
In Phase 1 (Kombinationstherapie-Dosisfindungskohorte) der Studie 216 waren die am häufigsten berichteten Nebenwirkungen (≥ 40 %) Hypertonie, Hypothyreose, Hypertriglyzeridämie, abdominale Schmerzen und Diarrhoe; und in Phase 2 (Expansionskohorte mit der Kombinationstherapie) waren die am häufigsten berichteten Nebenwirkungen (≥ 35 %) Hypertriglyzeridämie, Proteinurie, Diarrhoe, verminderte Lymphozytenzahl, verminderte Anzahl weißer Blutkörperchen, erhöhtes Cholesterin im Blut, Fatigue und verminderte Thrombozytenzahl.
In Studie 231 waren die am häufigsten berichteten Nebenwirkungen (≥ 15 %) Hypothyreose, Hypertonie, Proteinurie, verminderter Appetit, Diarrhoe und verminderte Thrombozytenzahl.
Andere spezielle Patientengruppen
Ältere Patienten
In der CLEAR-Studie hatten ältere Patienten (≥ 75 Jahre) eine höhere (Differenz ≥ 10 %) Inzidenz von Proteinurie als jüngere Patienten (< 65 Jahre).
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population hatten ältere Patienten (≥ 75 Jahre) eine höhere (Differenz ≥ 10 %) Inzidenz von verminderter Thrombozytenzahl, Gewichtsverlust, Proteinurie und Hypertonie als jüngere Patienten (< 65 Jahre).
Geschlecht
In der CLEAR-Studie hatten Männer eine höhere (Differenz ≥ 10 %) Inzidenz von Diarrhoe als Frauen.
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population hatten Frauen eine höhere (Differenz ≥ 10 %) Inzidenz von Übelkeit, Erbrechen, Asthenie und Hypertonie als Männer.
Ethnische Abstammung
In der CLEAR-Studie hatten Patienten mit asiatischer Abstammung eine höhere (Differenz ≥ 10 %) Inzidenz von palmar-plantarem Erythrodysästhesie-Syndrom, Proteinurie und Hypothyreose (einschließlich erhöhter Schilddrüsenhormone im Blut) als Patienten kaukasischer Abstammung, während Patienten kaukasischer Abstammung eine höhere Inzidenz von Fatigue, Übelkeit, Arthralgie, Erbrechen und Asthenie hatten.
In der mit Lenvatinib und Everolimus behandelten zusamengefassten RCC-Population hatten Patienten mit asiatischer Abstammung eine höhere (Differenz ≥ 10 %) Inzidenz von Hypothyreose, Stomatitis, verminderter Thrombozytenzahl, Proteinurie, PPE und Hypertonie als Patienten kaukasischer Abstammung, während Patienten kaukasischer Abstammung eine höhere Inzidenz von Übelkeit, Asthenie, Fatigue und Hypercholesterinämie hatten.
Hypertonie bei Studienbeginn
In der CLEAR-Studie hatten Patienten mit Hypertonie bei Studienbeginn eine höhere Inzidenz von Proteinurie als Patienten ohne Hypertonie bei Studienbeginn.
Diabetes bei Studienbeginn
In der mit Lenvatinib und Everolimus behandelten zusammengefassten RCC-Population hatten Patienten mit Diabetes bei Studienbeginn eine höhere (Differenz ≥ 10 %) Inzidenz von Proteinurie als Patienten ohne Diabetes bei Studienbeginn.
Leberfunktionsstörung
Bei RCC liegen für Patienten mit Leberfunktionsstörungen nur begrenzte Daten vor.
Nierenfunktionsstörung
Bei RCC hatten mit Lenvatinib und Everolimus behandelte Patienten mit einer Nierenfunktionsstörung bei Studienbeginn eine höhere Inzidenz von Thrombozytopenie oder verminderter Thrombozytenzahl als Patienten mit normaler Nierenfunktion.
Patienten mit einem Körpergewicht < 60 kg
Bei RCC hatten mit Lenvatinib und Everolimus behandelte Patienten mit einem niedrigen Körpergewicht (< 60 kg) eine höhere (Differenz ≥ 10 %) Inzidenz von verminderter Thrombozytenzahl und Hypertonie.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
Die höchsten in klinischen Studien untersuchten Dosen von Lenvatinib waren 32 mg und 40 mg pro Tag. In klinischen Prüfungen traten auch Medikationsfehler auf, die zu Einzeldosen von 40 bis 48 mg führten. Die unter diesen Dosen am häufigsten beobachteten Nebenwirkungen waren Hypertonie, Übelkeit, Diarrhoe, Fatigue, Stomatitis, Proteinurie, Kopfschmerz sowie eine Verschlimmerung des PPE. Es gibt auch Berichte über Überdosierungen von Lenvatinib, bei denen Einzelgaben mit dem 6- bis 10-Fachen der empfohlenen Tagesdosis eingesetzt wurden. Diese Fälle gingen mit Nebenwirkungen einher, die dem bekannten Sicherheitsprofil von Lenvatinib entsprechen (d. h. Nieren- und Herzinsuffizienz) oder es traten keine Nebenwirkungen auf.
Es gibt kein spezifisches Antidot bei einer Überdosierung mit Lenvatinib. Bei dem Verdacht auf eine Überdosierung sollte die Behandlung mit Lenvatinib unterbrochen und bei Bedarf eine geeignete unterstützende Behandlung erfolgen.
Pharmakotherapeutische Gruppe: antineoplastische Mittel, Proteinkinase-Inhibitoren, ATC-Code: L01EX08
Wirkmechanismus
Lenvatinib ist ein Rezeptor-Tyrosinkinase (RTK)-Inhibitor, der selektiv die Kinaseaktivitäten der Rezeptoren VEGFR1 (FLT1), VEGFR2 (KDR) und VEGFR3 (FLT4) des vaskulären Endothelwachstumsfaktors (VEGF) sowie andere, mit dem proangiogenen und onkogenen Signalweg in Zusammenhang stehende RTK, einschließlich der Rezeptoren FGFR1, 2, 3 und 4 des Fibroblasten-Wachstumsfaktors (FGF) und den Rezeptor PDGFRα, des Blutplättchen-Wachstumsfaktors (PDGF), sowie die Rezeptoren KIT und RET, hemmt. In syngenen Maustumormodellen führte Lenvatinib zu einer Verminderung der tumorassoziierten Makrophagen sowie zu einer Erhöhung der aktivierten zytotoxischen T-Zellen und zeigte in Kombination mit einem monoklonalen Anti-PD-1-Antikörper eine stärkere Antitumoraktivität als eine der beiden Behandlungen allein.
Die Kombination von Lenvatinib und Everolimus wies eine im Vergleich zu jedem der beiden Wirkstoffe allein erhöhte antiangiogene und Antitumoraktivität auf, belegt anhand einer Abnahme der humanen Endothelzellproliferation, der Kapillarröhrchenbildung und der VEGF-Signalgebung in vitro sowie des Tumorvolumens in Xenograft-Mausmodellen des humanen Nierenzellkarzinoms.
Obwohl nicht direkt untersucht, wird angenommen, dass die hypertensive Wirkung von Lenvatinib durch die Hemmung von VEGFR2 in den Endothelzellen der Blutgefäße vermittelt wird. Ebenso wurde die Ursache der Lenvatinib-induzierten Proteinurie nicht direkt untersucht, diese wird aber vermutlich über eine Herunterregulierung von VEGFR1 und VEGFR2 in den Podozyten des Glomerulus vermittelt.
Der Wirkungsmechanismus bei Hypothyreose ist bisher nicht vollständig geklärt.
Der Wirkungsmechanismus, der bei der Kombination von Lenvatinib und Everolimus zu einer Verschlimmerung der Hypercholesterinämie führt, wurde nicht direkt untersucht und ist nicht vollständig geklärt.
Obwohl auch der Wirkungsmechanismus für die Verschlimmerung der Diarrhoe bei der Kombination von Lenvatinib und Everolimus nicht direkt untersucht wurde, geht man davon aus, dass diese Wirkung durch eine Störung der Darmfunktion vermittelt wird, die mit den Wirkungsmechanismen der einzelnen Wirkstoffe zusammenhängt – Hemmung von VEGF/VEGFR und c-KIT durch Lenvatinib in Verbindung mit der Hemmung von mTOR/NHE3 durch Everolimus.
Klinische Wirksamkeit und Sicherheit
Erstlinientherapie von Patienten mit RCC (in Kombination mit Pembrolizumab)
Die Wirksamkeit von Lenvatinib in Kombination mit Pembrolizumab wurde in Studie 307 (CLEAR), einer multizentrischen, offenen, randomisierten Studie untersucht, in die 1.069 Patienten mit fortgeschrittenem RCC mit klarzelliger Komponente, einschließlich anderer histologischer Merkmale wie sarkomatoider oder papillärer Merkmale, als Erstlinien-Behandlung aufgenommen wurden. Die Patienten wurden unabhängig vom PD-L1-Expressionsstatus des Tumors aufgenommen. Patienten mit einer aktiven Autoimmunerkrankung oder einer Erkrankung, die eine Immunsuppression erforderte, waren für die Studie nicht geeignet. Die Randomisierung wurde nach geographischer Region (Nordamerika und Westeuropa vs. „Rest der Welt“) und nach Memorial Sloan Kettering Cancer Center (MSKCC)-Prognosegruppen (günstig, intermediär und ungünstig) stratifiziert.
Die Patienten wurden randomisiert Lenvatinib 20 mg oral einmal täglich in Kombination mit Pembrolizumab 200 mg intravenös alle 3 Wochen (n = 355) oder Lenvatinib 18 mg oral einmal täglich in Kombination mit Everolimus 5 mg oral einmal täglich (n = 357) oder Sunitinib 50 mg oral einmal täglich über einen Zeitraum von 4 Wochen mit anschließender Behandlungspause von 2 Wochen (n = 357) zugeteilt. Alle Patienten im mit Lenvatinib und Pembrolizumab behandelten Arm begannen die Behandlung mit Lenvatinib 20 mg oral einmal täglich. Die mediane Zeit bis zur ersten Dosisreduktion von Lenvatinib betrug 1,9 Monate. Die mediane durchschnittliche Tagesdosis für Lenvatinib betrug 14 mg. Die Behandlung wurde so lange fortgesetzt, bis eine inakzeptable Toxizität oder eine Krankheitsprogression nach Einschätzung des Prüfarztes und gemäß Bestätigung durch eine unabhängige radiologische Prüfungskommission (independent radiologic review comittee, IRC) anhand der Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1)-Kriterien eintrat. Die Verabreichung von Lenvatinib zusammen mit Pembrolizumab war über eine Krankheitsprogression gemäß RECIST-Definition hinaus zulässig, wenn der Patient klinisch stabil war und nach Ansicht des Prüfarztes einen klinischen Nutzen aus der Behandlung zog. Die Behandlung mit Pembrolizumab wurde maximal 24 Monate lang fortgesetzt; die Behandlung mit Lenvatinib konnte hingegen über 24 Monate hinaus fortgesetzt werden. Eine Beurteilung des Tumorstatus erfolgte zu Studienbeginn und danach alle 8 Wochen.
Die Merkmale der Studienpopulation (355 Patienten in dem mit Lenvatinib und Pembrolizumab behandelten Arm und 357 Patienten in dem mit Sunitinib behandelten Arm) lauteten wie folgt: medianes Alter von 62 Jahren (Bereich: 29 bis 88 Jahre); 41 % im Alter von 65 Jahren oder älter, 74 % männlich; 75 % Weiße, 21 % Asiaten, 1 % Schwarze und 2 % anderer ethnischer Abstammung; 17 % bzw. 83 % hatten zu Studienbeginn einen KPS von 70 bis 80 bzw. 90 bis 100. Die Patienten waren gemäß Risikokategorien des IMDC (International Metastatic RCC Database Consortium) wie folgt verteilt: 33 % günstig, 56 % intermediär und 10 % ungünstig. Die Verteilung gemäß den MSKCC-Prognosegruppen lautete 27 % günstig, 64 % intermediär und 9 % ungünstig. Bei 99 % war eine metastasierte Erkrankung vorhanden, und bei 1 % der Patienten war eine lokal fortgeschrittene Erkrankung vorhanden. Häufige von Metastasen betroffene Stellen bei den Patienten waren Lunge (69 %), Lymphknoten (45 %) und Knochen (26 %).
Der primäre Wirksamkeitsendpunkt war das progressionsfreie Überleben (progression-free survival, PFS) basierend auf RECIST 1.1 gemäß IRC. Sekundäre Wirksamkeitsendpunkte waren Gesamtüberleben (overall survival, OS) und objektive Ansprechrate (objective response rate, ORR). Für Lenvatinib in Kombination mit Pembrolizumab waren bei der vorher festgelegten Zwischenanalyse (finale Analyse des PFS) im Vergleich zu Sunitinib statistisch signifikante Verbesserungen von PFS, OS und ORR zu verzeichnen. Das mediane PFS für Lenvatinib in Kombination mit Pembrolizumab betrug 23,9 Monate (95%-KI: 20,8; 27,7) im Vergleich zu 9,2 Monaten (95%-KI: 6,0; 11,0) für Sunitinib, HR 0,39 (95%-KI: 0,32; 0,49; p-Wert < 0,0001). Für das OS betrug die HR 0,66 (95%-KI: 0,49; 0,88; p-Wert 0,0049) mit einer medianen OS-Nachbeobachtungszeit von 26,5 Monaten und einer medianen Behandlungsdauer von 17,0 Monaten für Lenvatinib plus Pembrolizumab. Die ORR für Lenvatinib in Kombination mit Pembrolizumab betrug 71 % (95%-KI: 66; 76) vs. 36 % (95%-KI: 31; 41) p-Wert < 0,0001 für Sunitinib. Die Wirksamkeitsergebnisse für PFS, OS und ORR bei der im Prüfplan spezifizierten finalen Analyse (mediane Nachbeobachtungszeit von 49,4 Monaten) sind in Tabelle 5, Abbildung 1 und Abbildung 2 zusammengefasst. Die PFS-Ergebnisse waren über die vorab festgelegten Subgruppen, über die MSKCC-Prognosegruppen und über die verschiedenen PD-L1-Tumorexpressionsstatus hinweg konsistent. Die Wirksamkeitsergebnisse nach MSKCC-Prognosegruppe sind in Tabelle 6 zusammengefasst.
Die finale OS-Analyse wurde nicht bereinigt, um nachfolgende Therapien zu berücksichtigen: 195/357 (54,6 %) Patienten im Sunitinib-Arm und 56/355 (15,8 %) Patienten im Lenvatinib- plus Pembrolizumab-Arm erhielten eine anschließende Anti-PD-1/PD-L1-Therapie.
Tabelle 5 Wirksamkeitsergebnisse beim Nierenzellkarzinom gemäß IRC in der CLEAR-Studie
Lenvatinib 20 mg mit Pembrolizumab 200 mg |
Sunitinib 50 mg |
|
Progressionsfreies Überleben (PFS)* | ||
Anzahl der Ereignisse, n (%) |
207 (58 %) |
214 (60 %) |
Medianes PFS in Monaten (95 %-KI)a |
23,9 (20,8; 27,7) |
9,2 (6,0; 11,0) |
Hazard Ratio (95 %-KI)b, c |
0,47 (0,38; 0,57) |
|
p-Wertc |
< 0,0001 |
|
Gesamtüberleben (OS) | ||
Anzahl der Todesfälle, n (%) |
149 (42 %) |
159 (45 %) |
Medianes OS in Monaten (95 %-KI) |
53,7 (48,7; NE) |
54,3 (40,9; NE) |
Hazard Ratio (95 %-KI)b, c |
0,79 (0,63; 0,99) |
|
p-Wert |
0,0424 |
|
Objektive Ansprechrate (bestätigt) | ||
Objektive Ansprechrate, n (%) |
253 (71,3 %) |
131 (36,7 %) |
(95%-KI) |
(66,6, 76,0) |
(31,7, 41,7) |
Anzahl der Fälle mit vollständigem Ansprechen (CR), n (%) |
65 (18,3 %) |
17 (4,8 %) |
Anzahl der Fälle mit partiellem Ansprechen (PR), n (%) |
188 (53,0 %) |
114 (32 %) |
p-Wertd |
< 0,0001 |
|
Dauer des Ansprechensa | ||
Median in Monaten (Bereich) |
26,7 (1,64+, 55,92+) |
14,7 (1,64+, 54,08+) |
Die Tumorbeurteilung erfolgte anhand der RECIST 1.1-Kriterien; in die ORR flossen nur Fälle mit bestätigtem Ansprechen ein | ||
Abbildung 1 Kaplan-Meier-Kurven für das progressionsfreie Überleben in der CLEAR-Studie*
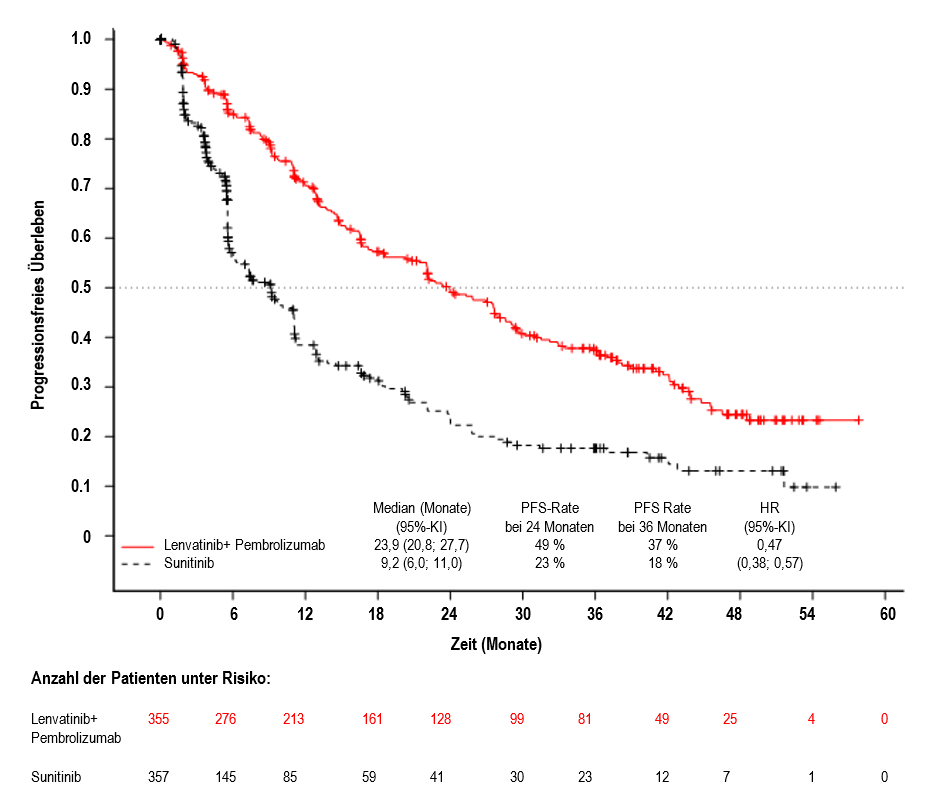
Datenstichtag: 31. Juli 2022
*: Basierend auf der aktualisierten PFS-Analyse, die zum Zeitpunkt der im Prüfplan spezifizierten finalen OS-Analyse durchgeführt wurde.
Abbildung 2 Kaplan-Meier-Kurven für das Gesamtüberleben in der CLEAR-Studie*
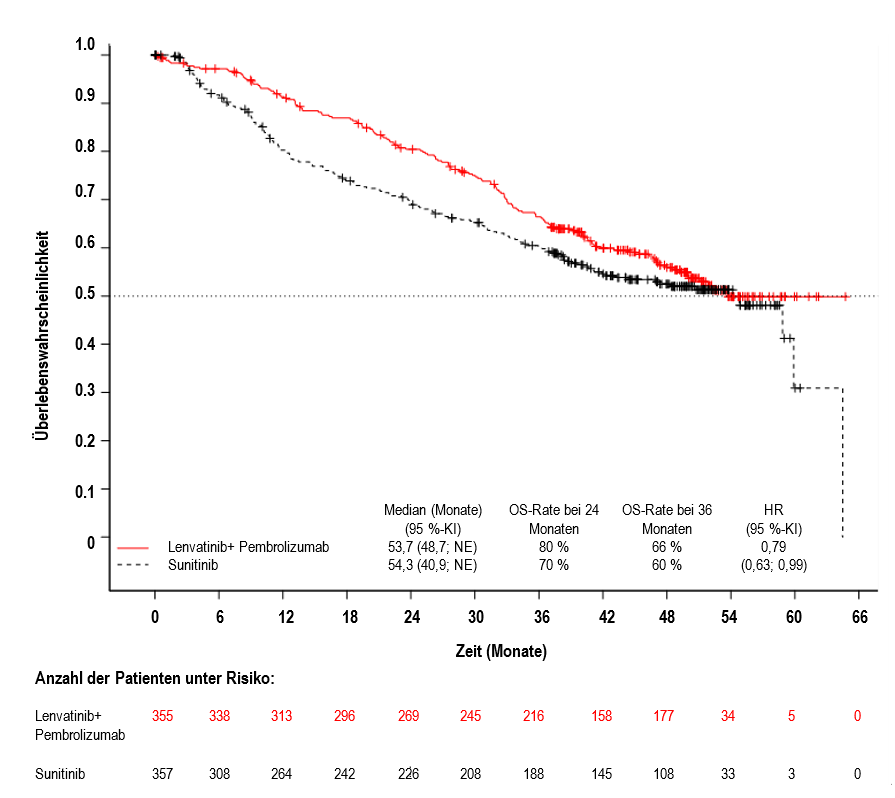
NE = nicht schätzbar
Datenstichtag: 31. Juli 2022
*: Basierend auf der im Prüfplan spezifizierten finalen OS-Analyse
Die CLEAR-Studie lieferte keine ausreichende Teststärke, um die Wirksamkeit bei individuellen Subgruppen zu bewerten. Tabelle 6 fasst die Wirksamkeitsergebnisse nach MSKCC-Prognosegruppe basierend auf der finalen OS-Analyse bei einer medianen Nachbeobachtungszeit von 49,4 Monaten zusammen.
Tabelle 6 Wirksamkeitsergebnisse in der CLEAR-Studie nach MSKCC-Prognosegruppe
|
Lenvatinib + Pembrolizumab |
Sunitinib |
Lenvatinib + Pembrolizumab vs. Sunitinib |
||
|
Anzahl der Patienten |
Anzahl der Ereignisse |
Anzahl der Patienten |
Anzahl der Ereignisse |
|
Progressionsfreies Überleben (PFS) gemäß IRCa |
PFS-HR (95 %-KI) |
||||
Günstig |
96 |
56 |
97 |
65 |
0,46 (0,32; 0,67) |
Intermediär |
227 |
129 |
228 |
130 |
0,51 (0,40; 0,65) |
Ungünstig |
32 |
22 |
32 |
19 |
0,18 (0,08; 0,42) |
Gesamtüberleben (OS)a |
OS-HR (95 %-KI) |
||||
Günstig |
96 |
27 |
97 |
31 |
0,89 (0,53; 1,50) |
Intermediär |
227 |
104 |
228 |
108 |
0,81 (0,62; 1,06) |
Ungünstig |
32 |
18 |
32 |
20 |
0,59 (0,31; 1,12) |
a: Mediane Nachbeobachtungszeit 49,4 Monate (Datenstichtag – 31. Juli 2022) | |||||
Offene, einarmige Phase 2-Studie
Es stehen zusätzliche Daten aus der offenen, einarmigen Phase 2-Studie KEYNOTE-B61 zu Lenvatinib (20 mg einmal täglich) in Kombination mit Pembrolizumab (400 mg alle 6 Wochen) als Erstlinientherapie von Patienten mit fortgeschrittenem oder metastasiertem RCC mit einer nicht-klarzelligen Histologie (n=158) zur Verfügung, darunter 59 % papillär, 18 % chromophob, 4 % Translokation, 1 % medullär, 13 % nicht klassifiziert und 6 % sonstige. Die ORR betrug 50,6 % (95%-KI (42,6; 58,7)) und die mediane Dauer des Ansprechens betrug 19,5 Monate (95%-KI 15,3; NR).
Zweitlinientherapie von Patienten mit RCC (in Kombination mit Everolimus)
Studie 205, eine multizentrische, randomisierte, offene Studie, wurde durchgeführt, um die Sicherheit und Wirksamkeit der Gabe von Lenvatinib allein oder in Kombination mit Everolimus bei Patienten mit inoperablen fortgeschrittenen oder metastasierten RCC zu bestimmen. Die Studie bestand aus einem Phase 1b-Teil zur Dosisfindung und einem Phase 2-Teil. Im Phase 1b-Teil wurden 11 Patienten eingeschlossen, die die Kombination von Lenvatinib 18 mg und Everolimus 5 mg erhielten. Im Phase 2-Teil wurden insgesamt 153 Patienten mit inoperablem fortgeschrittenem oder metastasiertem RCC nach einer vorhergehenden gegen VEGF gerichteten Therapie eingeschlossen. Die Kombination von Lenvatinib und Everolimus in der empfohlenen Dosierung erhielten insgesamt 62 Patienten. Die Patienten mussten unter anderem ein histologisch gesichertes, überwiegend klarzelliges RCC sowie den radiologischen Nachweis der Krankheitsprogression, ermittelt unter Verwendung von RECIST 1.1 aufweisen, sowie eine gegen VEGF gerichtete Therapie erhalten haben und einen ECOG (Eastern Cooperative Oncology Group)-Leistungsstatus von 0 oder 1 aufweisen.
Die Patienten wurden nach dem Zufallsprinzip einem von 3 Armen zugeordnet: 18 mg Lenvatinib plus 5 mg Everolimus, 24 mg Lenvatinib oder 10 mg Everolimus im Verhältnis 1:1:1. Die Patienten wurden nach dem Hämoglobinwert (≤ 13 g/dl vs. > 13 g/dl für Männer bzw. ≤ 11,5 g/dl vs. > 11,5 g/dl für Frauen) und dem korrigierten Serum-Kalzium (≥ 10 mg/dl vs. < 10 mg/dl) stratifiziert. Der Median der durchschnittlichen Tagesdosis pro Patient betrug im Kombinationsarm 13,5 mg Lenvatinib (75,0 % der vorgesehenen Dosis von 18 mg) und 4,7 mg Everolimus (93,6 % der vorgesehenen Dosis von 5 mg). Die abschließende Dosisstärke im Kombinationsarm betrug bei 29 % der Patienten 18 mg, bei 31 % der Patienten 14 mg, bei 23 % der Patienten 10 mg, bei 16 % der Patienten 8 mg und bei 2 % der Patienten 4 mg.
Von den nach dem Zufallsprinzip zugeteilten 153 Patienten waren 73 % männlich, das mediane Alter betrug 61 Jahre, 37 % waren 65 Jahre oder älter, 7 % waren 75 Jahre oder älter, und 97 % waren kaukasischer Abstammung. 95 % der Patienten hatten Metastasen und 5 % eine inoperable fortgeschrittene Erkrankung. Alle Patienten hatten bei Studienbeginn einen ECOG-Leistungsstatus von 0 (55 %) oder 1 (45 %) mit ähnlicher Verteilung über die 3 Therapiearme. Eine schlechte Prognose, definiert durch die MSKCC-Kriterien (Memorial Sloan-Kettering Cancer Center Criteria), wurde bei 39 % der Patienten im Studienarm mit Lenvatinib und Everolimus, bei 44 % im Lenvatinib-Studienarm und bei 38 % im Everolimus-Studienarm beobachtet. Eine schlechte Prognose gemäß IMDC (International mRCC Database Consortium) wurde bei 20 % der Patienten im Lenvatinib- und Everolimus-Arm, bei 23 % der Patienten im Lenvatinib-Arm und bei 24 % der Patienten im Everolimus-Arm beobachtet. Die mediane Zeit von der Diagnose bis zur ersten Dosis betrug 32 Monate für den Lenvatinib- und Everolimus-Behandlungsarm, 33 Monate für den Lenvatinib-Arm und 26 Monate für den Everolimus-Arm. Alle Patienten hatten zuvor eine Behandlung mit 1 VEGF-Hemmer erhalten, davon 65 % mit Sunitinib, 23 % mit Pazopanib, 4 % mit Tivozanib, 3 % mit Bevacizumab und jeweils 2 % mit Sorafenib oder Axitinib.
Der primäre Wirksamkeitsendpunkt, basierend auf dem vom Prüfarzt bestimmten Tumoransprechen, war das PFS im Lenvatinib- plus Everolimus-Arm im Vergleich zum Everolimus-Arm und im Lenvatinib-Arm im Vergleich zum Everolimus-Arm. Weitere Wirksamkeitsendpunkte umfassten das OS und die vom Prüfarzt beurteilte ORR. Die Tumorbeurteilung erfolgte nach RECIST 1.1.
Der Lenvatinib- plus Everolimus-Arm zeigte eine statistisch signifikante und klinisch bedeutsame Verbesserung des PFS gegenüber dem Everolimus-Arm (siehe Tabelle 7 und Abbildung 3). Auf der Grundlage der Ergebnisse einer nachträglich durchgeführten explorativen Analyse mit einer begrenzten Anzahl von Patienten pro Subgruppe wurde eine positive Wirkung auf das PFS festgestellt, unabhängig davon, welche frühere gegen VEGF-gerichtete Therapie angewendet worden war: Sunitinib (Hazard Ratio [HR] = 0,356 [95 %-KI: 0,188; 0,674]) oder andere Therapien (HR = 0,350 [95 %-KI: 0,148; 0,828]). Der Lenvatinib-Arm zeigte ebenfalls eine Verbesserung des PFS im Vergleich mit dem Everolimus-Arm. Das Gesamtüberleben war höher im Lenvatinib- plus Everolimus-Arm (siehe Tabelle 7 und Abbildung 4). Die Studie besaß keine ausreichende statistische Power für eine Analyse des OS.
Der Behandlungseffekt der Kombination auf das PFS und die ORR wurde auch durch eine nachträgliche retrospektive unabhängige verblindete Beurteilung der Scans unterstützt. Der Lenvatinib- plus Everolimus-Arm zeigte eine statistisch signifikante und klinisch bedeutsame Verbesserung des PFS im Vergleich zum Everolimus-Arm. Die Ergebnisse für ORR entsprachen den Ergebnissen der Prüfarzt-Beurteilung, 35,3 % im Lenvatinib- plus Everolimus-Arm, mit vollständigem Ansprechen und 17 Fällen von teilweisem Ansprechen; im Everolimus-Arm hatte kein Patient ein objektives Ansprechen (p < 0,0001) zu Gunsten des Lenvatinib- plus Everolimus-Arms.
Tabelle 7 Wirksamkeitsergebnisse nach einer vorherigen gegen VEGF gerichteten Therapie in RCC-Studie 205
Lenvatinib 18 mg + Everolimus 5 mg |
Lenvatinib 24 mg |
Everolimus 10 mg |
|
Progressionsfreies Überleben (PFS)a nach der Beurteilung durch den Prüfarzt | |||
Medianes PFS in Monaten (95 %-KI) |
14,6 (5,9; 20,1) |
7,4 (5,6; 10,2) |
5,5 (3,5; 7,1) |
Hazard-Ratio (95 %-KI)b |
0,40 (0,24; 0,67) |
- |
- |
p-Wert |
0,0005 |
- |
- |
Progressionsfreies Überleben (PFS)a durch eine nachträgliche unabhängige retrospektive Beurteilung | |||
Medianes PFS in Monaten (95 %-KI) |
12,8 (7,4; 17,5) |
9,0 (5,6; 10,2) |
5,6 (3,6; 9,3) |
Hazard Ratio (95 %-KI)b |
0,45 (0,26; 0,79) |
- |
- |
p-Wert |
0,003 |
- |
- |
Gesamtüberlebenc | |||
Anzahl der Todesfälle, n (%) |
32 (63) |
34 (65) |
37 (74) |
Medianes OS in Monaten (95 %-KI) |
25,5 (16,4; 32,1) |
19,1 (13,6; 26,2) |
15,4 (11,8; 20,6) |
Hazard-Ratio (95 %-KI)b |
0,59 (0,36; 0,97) |
- |
- |
Objektive Ansprechrate, n (%) nach der Beurteilung durch den Prüfarzt | |||
Vollständiges Ansprechen |
1 (2) |
0 |
0 |
Partielles Ansprechen |
21 (41) |
14 (27) |
3 (6) |
Objektive Ansprechrate |
22 (43) |
14 (27) |
3 (6) |
Stabile Erkrankung |
21 (41) |
27 (52) |
31 (62) |
Dauer des Ansprechens, Monate, Median (95 %-KI) |
13,0 (3,7; NE) |
7,5 (3,8; NE) |
8,5 (7,5; 9,4) |
Die Tumorbeurteilung erfolgte anhand der RECIST 1.1-Kriterien. Datenstichtag = 13. Juni 2014 | |||
Abbildung 3 Kaplan-Meier-Kurve des progressionsfreien Überlebens
(Einschätzung des Prüfarztes)
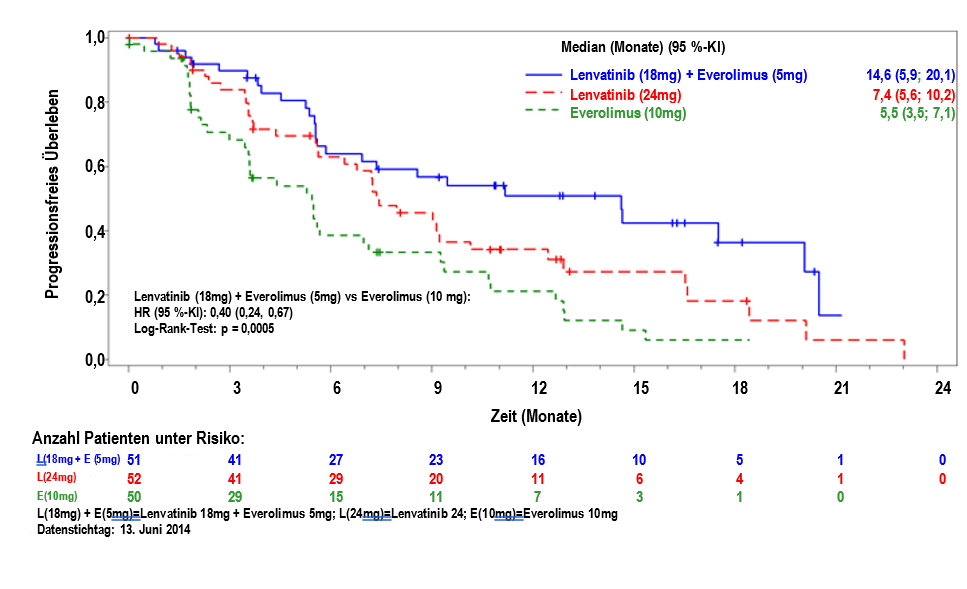
Abbildung 4 Kaplan-Meier-Kurve des Gesamtüberlebens
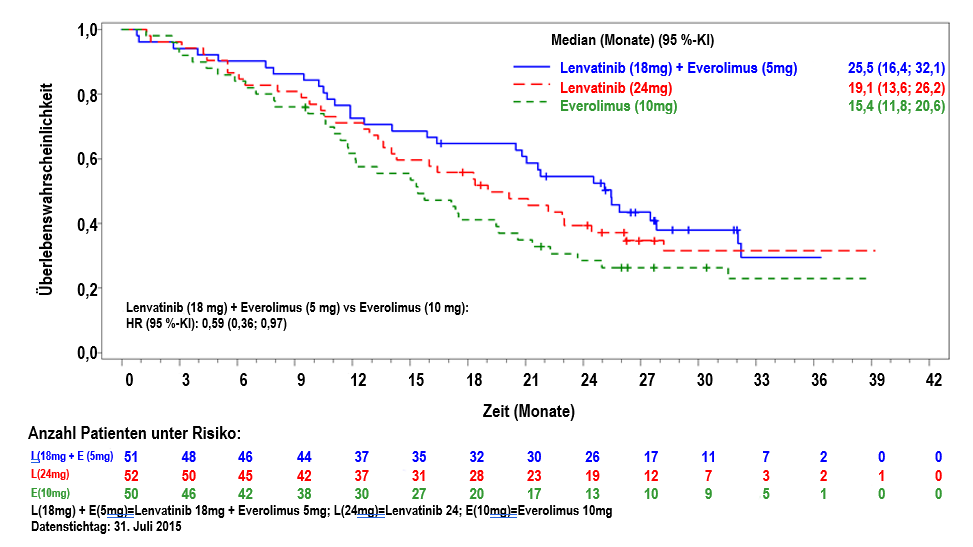
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Lenvatinib eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in der Behandlung des Nierenzellkarzinoms (RCC, renal cell carcinoma) gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen)
Studien an Kindern und Jugendlichen
Die Wirksamkeit von Lenvatinib wurde in zwei offenen Studien untersucht, jedoch nicht bestätigt (weitere Informationen zu Kindern und Jugendlichen finden Sie auch in der Fachinformation zu Lenvima, Abschnitt 5.1):
Bei Studie 216 handelte es sich um eine multizentrische, offene, einarmige Phase 1-/2-Studie zur Beurteilung der Sicherheit, Verträglichkeit und Antitumoraktivität von Lenvatinib, das Kindern und Jugendlichen (sowie jungen Erwachsenen im Alter von ≤ 21 Jahren) mit rezidivierenden oder refraktären soliden Malignitäten, darunter Tumoren des ZNS) in Kombination mit Everolimus verabreicht wurde. Es wurden insgesamt 64 Patienten in die Studie eingeschlossen und behandelt. In Phase 1 (Kombinationstherapie-Dosisfindung) wurden 23 Patienten eingeschlossen und behandelt: 5 mit Dosierungsstufe –1 (Lenvatinib 8 mg/m² und Everolimus 3 mg/m²) und 18 mit Dosierungsstufe 1 (Lenvatinib 11 mg/m² und Everolimus 3 mg/m²). Die für die Kombination empfohlene Dosis betrug Lenvatinib 11 mg/m² und Everolimus 3 mg/m² einmal täglich. In Phase 2 (Kombinationstherapie-Expansion) wurden 41 Patienten eingeschlossen und in den folgenden Kohorten mit der empfohlenen Dosis behandelt: Ewing-Sarkom (EWS, n=10), Rhabdomyosarkom (RMS, n=20) und hochmaligne Gliome (HGG, n=11). Der primäre Wirksamkeitsendpunkt war die objektive Ansprechrate (ORR; Objective Response Rate) in Woche 16 bei auswertbaren Patienten, basierend auf der Beurteilung durch den Prüfarzt anhand von RECIST v1.1 oder RANO (bei Patienten mit HGG). In den Kohorten mit EWS und HGG wurde kein objektives Ansprechen beobachtet; 2 Fälle mit partiellem Ansprechen (PR) wurden in der RMS-Kohorte beobachtet, woraus sich eine ORR in Woche 16 von 10 % (95%-KI: 1,2; 31,7) ergibt.
Bei Studie 231 handelt es sich um eine multizentrische, offene Phase-2-Basket-Studie zur Beurteilung der Antitumoraktivität und Sicherheit von Lenvatinib bei Kindern, Jugendlichen und jungen Erwachsenen im Alter von 2 bis ≤ 21 Jahren mit rezidivierenden oder refraktären soliden Malignitäten, darunter EWS, RMS und HGG. Insgesamt 127 Patienten wurden in die Studie eingeschlossen und in den folgenden Kohorten mit der empfohlenen Dosis Lenvatinib (14 mg/m²) behandelt: EWS (n=9), RMS (n=17), HGG (n=8) und sonstige solide Tumore (n=9 jeweils für diffuses Mittelliniengliom, Medulloblastom und Ependymom; alle sonstigen soliden Tumore n=66). Der primäre Wirksamkeitsendpunkt war die ORR in Woche 16 bei auswertbaren Patienten, basierend auf der Beurteilung durch den Prüfarzt anhand von RECIST v1.1 oder RANO (bei Patienten mit HGG). Bei Patienten mit HGG, diffusem Mittelliniengliom, Medulloblastom oder Ependymom wurde kein objektives Ansprechen beobachtet. Zwei Fälle mit partiellem Ansprechen wurden in der EWS- und in der RMS-Kohorte beobachtet, woraus sich jeweils eine ORR in Woche 16 von 22,2 % (95%-KI: 2,8; 60,0) bzw. 11,8 % (95%-KI: 1,5; 36,4) ergibt. In der Gruppe mit sonstigen soliden Tumoren wurden fünf Fälle mit partiellem Ansprechen (bei Patienten mit Synovialsarkom [n=2], kaposiformem Hämangioendotheliom [n=1], Wilms-Tumor/Nephroblastom [n=1] und Klarzellkarzinom [n=1]) beobachtet, woraus sich eine ORR in Woche 16 von 7,7 % (95%-KI: 2,5; 17,0) ergibt.
Die pharmakokinetischen Parameter von Lenvatinib wurden an gesunden erwachsenen Probanden, erwachsenen Patienten mit Leberinsuffizienz, Niereninsuffizienz und soliden Tumoren untersucht.
Resorption
Lenvatinib wird nach oraler Einnahme rasch resorbiert. Die tmax ist in der Regel 1 bis 4 Stunden nach der Einnahme erreicht. Nahrungsmittel beeinflussen das Ausmaß der Resorption nicht, verlangsamen jedoch die Resorption. Bei Einnahme zu den Mahlzeiten verzögern sich bei gesunden Probanden die Spitzenkonzentrationen im Plasma um 2 Stunden. Die absolute Bioverfügbarkeit wurde nicht am Menschen untersucht. Die Daten einer Massenbilanz-Studie lassen jedoch darauf schließen, dass sie sich in einer Größenordnung von 85 % bewegt.
Verteilung
In vitro ist die Bindung von Lenvatinib an menschliche Plasmaproteine hoch. Sie reichte von 98 % bis 99 % (0,3 – 30 μg/ml, Mesilat). Die Bindung erfolgte hauptsächlich an Albumin und in geringerem Ausmaß an das saure α1-Glykoprotein und γ‑Globulin. Bei Patienten mit Leberfunktionsstörung, Patienten mit Nierenfunktionsstörung sowie entsprechenden gesunden Probanden wurde eine ähnliche Plasmaproteinbindung (97 % bis 99 %) ohne Abhängigkeiten von den Lenvatinib-Konzentrationen (0,2 μg/ml bis 1,2 μg/ml) beobachtet.
In vitro betrug das Verhältnis der Blut-Plasma-Konzentration von Lenvatinib 0,589 bis 0,608 (0,1 ‑ 10 μg/ml, Mesilat).
In-vitro-Studien legen nahe, dass Lenvatinib ein Substrat für P-gp und BCRP ist. Lenvatinib zeigt nur eine minimale oder gar keine Wirkung gegen P-gp-vermittelte und BCRP-vermittelte Transportaktivitäten. Auch wurde keine Induktion der P-gp mRNA-Expression beobachtet. Lenvatinib ist kein Substrat für OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2 oder BSEP. In menschlichem Leberzytosol führte Lenvatinib zu keiner Hemmung der Aldehydoxidase-Aktivität.
Bei Patienten lag das mediane Verteilungsvolumen (Vz/F) der ersten Dosis zwischen 50,5 l und 92 l und war im Allgemeinen konsistent in den Dosisgruppen von 3,2 mg bis 32 mg. Das analoge mediane Verteilungsvolumen im Steady-State (Vz/Fss) war ebenfalls im Allgemeinen konsistent und bewegte sich zwischen 43,2 l und 121 l.
Biotransformation
In vitro wurde Lenvatinib im Hinblick auf die P450-vermittelte Metabolisierung nachweislich vor allem (> 80 %) über Cytochrom P450 3A4 metabolisiert. In-vivo-Daten deuteten darauf hin, dass auch nicht P450-vermittelte Stoffwechselwege einen erheblichen Anteil am Gesamtstoffwechsel von Lenvatinib hatten. Folglich wirkten sich Induktoren und Inhibitoren von CYP 3A4 in vivo nur minimal auf die Exposition gegenüber Lenvatinib aus (siehe Abschnitt 4.5).
In menschlichen Lebermikrosomen wurde die demethylierte Form von Lenvatinib (M2) als Hauptmetabolit identifiziert. M2’ und M3’, die Hauptmetaboliten in menschlichen Fäzes, wurden durch die Aldehydoxidase aus M2 bzw. Lenvatinib gebildet.
In Plasmaproben, die bis zu 24 Stunden nach der Gabe entnommen wurden, bildete Lenvatinib 97 % der Radioaktivität in Plasma-Radiochromatogrammen, während weitere 2,5 % auf den M2-Metaboliten entfielen. Auf der Basis der AUC0- inf entfielen 60 % der Gesamtradioaktivität im Plasma und 64 % im Blut auf Lenvatinib.
Daten einer Massenbilanz-/Ausscheidungsstudie beim Menschen zeigen, dass Lenvatinib beim Menschen weitgehend verstoffwechselt wird. Als Hauptstoffwechselwege beim Menschen wurden die Oxidation durch die Aldehydoxidase, die Demethylierung über CYP3A4, die Glutathion-Konjugation mit Elimination der O‑Arylgruppe (funktionelle Chlorphenylgruppe) und Kombinationen dieser Wege mit anschließender weiterer Biotransformation identifiziert (z. B. Glukuronidierung, Hydrolyse der funktionellen Glutathiongruppe, Abbau der funktionellen Cysteingruppe und intramolekulare Umstellung der Cysteinylglycin- und Cystein-Konjugate mit anschließender Dimerisierung). Diese Stoffwechselwege in vivo entsprechen den Daten der In-vitro-Studien mit menschlichem Biomaterial.
In-vitro-Studien zu Transportern
Siehe Abschnitt Verteilung.
Elimination
Plasmakonzentrationen fallen nach Cmax biexponentiell ab. Die mittlere terminale exponentielle Halbwertszeit von Lenvatinib beträgt ca. 28 Stunden.
Nach Verabreichung von radioaktiv markiertem Lenvatinib an 6 Patienten mit soliden Tumoren wurden etwa zwei Drittel der radioaktiven Markierung mit den Fäzes und ein Viertel mit dem Urin ausgeschieden. Der M3-Metabolit war der Hauptanalyt der Exkrete (ca. 17 % der Dosis), gefolgt von M2’ (ca. 11 % der Dosis) und M2 (ca. 4,4 % der Dosis).
Linearität/Nicht-Linearität
Dosisproportionalität und Akkumulation
Bei Patienten mit soliden Tumoren, die eine oder mehrere Dosen Lenvatinib einmal täglich erhielten, stieg die Exposition gegenüber Lenvatinib (Cmax und AUC) im Bereich von 3,2 bis 32 mg einmal täglich direkt proportional zur verabreichten Dosis an.
Lenvatinib weist im Steady State nur eine minimale Akkumulation auf. Im genannten Bereich betrug der mediane Akkumulationsindex (Rac) 0,96 (20 mg) bis 1,54 (6,4 mg).
Spezielle Patientengruppen
Leberfunktionsstörung
Bei 6 Patienten mit leichter und mittelgradiger Leberinsuffizienz (Child-Pugh A bzw. Child-Pugh B) wurde die Pharmakokinetik von Lenvatinib nach einer Einzeldosis von 10 mg untersucht. Eine Dosis von 5 mg wurde bei 6 Patienten mit hochgradiger Leberinsuffizienz (Child-Pugh C) untersucht. Acht gesunde Probanden mit übereinstimmenden demographischen Daten dienten als Kontrollen und erhielten eine Dosis von 10 mg. Die mediane Halbwertszeit von Patienten mit leichter, mittelgradiger und hochgradiger Leberinsuffizienz war untereinander sowie mit der von Patienten mit normaler Leberfunktion vergleichbar und betrug 26 bis 31 Stunden. Der prozentuale Anteil der Lenvatinib-Dosis, die mit dem Urin ausgeschieden wurde, war bei allen Kohorten niedrig (< 2,16 % bei allen Behandlungskohorten).
Die Exposition gegenüber Lenvatinib auf der Basis der dosiskorrigierten AUC(0-t) und AUC(0-inf)-Daten betrug 119 %, 107 % bzw. 180 % des Normalwerts bei Patienten mit leichter, mittelgradiger bzw. hochgradiger Leberinsuffizienz. Es wurde festgestellt, dass die Plasmaproteinbindung im Plasma von leberinsuffizienten Patienten der Plasmaproteinbindung bei den entsprechenden übereinstimmenden gesunden Probanden ähnlich ist, und es wurde keine Konzentrationsabhängigkeit beobachtet. Dosierungsempfehlungen siehe Abschnitt 4.2.
Nierenfunktionsstörung
Bei 6 Patienten mit leichter, mittelschwerer und schwerer Nierenfunktionsstörung wurde die Pharmakokinetik von Lenvatinib nach einer Einzeldosis von 24 mg untersucht und mit der von 8 gesunden Probanden mit übereinstimmenden demographischen Daten verglichen. Patienten mit einer terminalen Niereninsuffizienz wurden nicht untersucht.
Die auf AUC(0-inf)-Daten beruhende Lenvatinib-Exposition betrug für Patienten mit leichter, mittelschwerer bzw. schwerer Nierenfunktionsstörung 101 %, 90 % bzw. 122 % des Normalwerts. Es wurde festgestellt, dass die Plasmaproteinbindung im Plasma von niereninsuffizienten Patienten der Plasmaproteinbindung bei den entsprechenden übereinstimmenden gesunden Probanden ähnlich ist, und es wurde keine Konzentrationsabhängigkeit beobachtet. Dosierungsempfehlungen siehe Abschnitt 4.2.
Alter, Geschlecht, Gewicht, ethnische Abstammung
Auf der Grundlage einer pharmakokinetischen Populationsanalyse von Patienten, die bis zu 24 mg Lenvatinib einmal täglich erhielten, hatten Alter, Geschlecht, Gewicht und ethnische Abstammung (Japaner vs. Andere, Kaukasier vs. Andere) keine signifikanten Auswirkungen auf die Ausscheidung (siehe Abschnitt 4.2).
Kinder und Jugendliche
Basierend auf einer populationspharmakokinetischen Analyse bei Kindern im Alter von 2 bis 12 Jahren, darunter Daten von 3 pädiatrischen Patienten im Alter von 2 bis < 3 Jahren, 28 pädiatrischen Patienten im Alter von ≥ 3 bis < 6 Jahren und 89 pädiatrischen Patienten im Alter von 6 bis ≤ 12 Jahren, die auf alle Gruppen des pädiatrischen Programms zu Lenvatinib verteilt waren, war die orale Clearance (CL/F) von Lenvatinib vom Körpergewicht abhängig, jedoch nicht vom Alter. Die erwarteten Konzentrationen, gemessen als Fläche unter der Konzentrations-Zeit-Kurve im Steady-State (AUCss) bei pädiatrischen Patienten, die 14 mg/m2 erhielten, waren mit denen erwachsener Patienten, die eine feststehende Dosis von 24 mg erhielten, vergleichbar. In diesen Studien wurden bei Kindern (2 bis 12 Jahre), Jugendlichen und jungen erwachsenen Patienten, welche die untersuchten Tumorarten aufwiesen, keine auffallenden Unterschiede in der Pharmakokinetik des Wirkstoffs Lenvatinib beobachtet. Für Kinder liegen jedoch nur relativ begrenzte Daten vor, um daraus eine eindeutige Schlussfolgerung ziehen zu können (siehe Abschnitt 4.2).
Die in den chronischen Toxizitätsstudien (bis zu 39 Wochen) aufgetretenen Befunde in verschiedenen Organen und Geweben entsprachen den pharmakologischen Wirkungen von Lenvatinib. Hierzu zählen Glomerulopathie, testikuläre Atrophie, ovarielle Follikelatresie, gastrointestinale Veränderungen, Knochenveränderungen, Veränderungen der Nebennieren (Ratten und Hunde) und arterielle Schädigungen (arterielle fibrinoide Nekrose, Mediadegeneration oder Blutung) bei Ratten, Hunden und Cynomolgus-Affen. Bei Ratten, Hunden und Affen wurden ferner erhöhte Transaminase-Spiegel verbunden mit Zeichen für eine Hepatotoxizität beobachtet. Am Ende einer vierwöchigen Erholungszeit wurde bei allen untersuchten Tierarten eine Reversibilität der toxikologischen Veränderungen festgestellt.
Genotoxizität
Lenvatinib war nicht genotoxisch.
Mit Lenvatinib wurden keine Karzinogenitätsstudien durchgeführt.
Reproduktions- und Entwicklungstoxizität
Es wurden mit Lenvatinib keine speziellen Tierstudien zur Beeinflussung der Fertilität durchgeführt. Es wurden jedoch in den chronischen Toxizitätsstudien bei Tieren Veränderungen von Hoden (Hypozellularität des samenbildenden Epithels) und Ovarien (Follikelatresie) bei einer Exposition gegenüber dem 11- bis 15-Fachen (Ratte) bzw. dem 0,6- bis 7-Fachen (Affe) der zu erwartenden klinischen Exposition (auf der Basis der AUC) bei der maximal verträglichen humanen Dosis festgestellt. Diese Befunde waren, am Ende einer 4-wöchigen Erholungsphase, reversibel.
Die Gabe von Lenvatinib während der Organogenese führte bei Dosierungen unterhalb der klinischen Exposition (auf der Basis der AUC) bei der maximal verträglichen humanen Dosis zur Embryoletalität und Teratogenität bei Ratten (äußere und skelettale Anomalien des Fetus) und ebenso bei Kaninchen (äußere, viszerale oder skelettale Anomalien des Fetus) bei Dosierungen unterhalb der klinischen Exposition (auf Basis der Körperoberfläche mg/m²) bei der maximal verträglichen humanen Dosis. Diese Ergebnisse zeigen ein teratogenes Potenzial von Lenvatinib, das wahrscheinlich durch die pharmakologische Aktivität von Lenvatinib als antiangiogener Wirkstoff bedingt ist.
Lenvatinib und seine Metaboliten gehen bei Ratten in die Muttermilch über.
Juvenile tierexperimentelle Toxizitätsstudien
Bei juvenilen Ratten, bei denen die Behandlung am 7. postnatalen Tag (PND) oder am PND21 begonnen wurde, war die Mortalität die dosislimitierende Toxizität. Sie wurde bei Expositionen beobachtet, die um das 125-Fache bzw. 12-Fache niedriger waren als die Exposition, nach der es bei adulten Ratten zur Mortalität kam, was auf eine im jüngeren Alter ausgeprägtere Sensitivität gegenüber der Toxizität schließen lässt. Die Mortalität kann daher Komplikationen zugeschrieben werden, die mit Primärläsionen des Duodenums einhergehen und bei denen möglicherweise auch zusätzliche toxische Wirkungen auf nicht vollständig ausgebildete Zielorgane eine Rolle spielen.
Die Toxizität von Lenvatinib war bei jüngeren Ratten ausgeprägter (die Gabe begann am PND7) als bei Ratten, bei denen die Gabe am PND21 begann, und eine Mortalität sowie bestimmte Toxizitätserscheinungen wurden bei juvenilen Ratten unter 10 mg/kg früher beobachtet als bei adulten Ratten, die die gleiche Dosierung erhielten. Bei juvenilen Ratten wurden zudem eine Wachstumsretardierung und daraufhin eine verzögerte körperliche Entwicklung sowie auf pharmakologische Wirkungen zurückzuführende Läsionen (Nagezähne, Femur [epiphysäre Wachstumsfuge], Nieren, Nebennieren und Duodenum) festgestellt.
Kapselinhalt
Calciumcarbonat
Mannitol
Mikrokristalline Cellulose
Hyprolose
Hyprolose (niedrig substituiert)
Talkum
Kapselhülse
Hypromellose
Titandioxid (E 171)
Eisen(III)-hydroxid-oxid x H2O (E 172)
Eisen(III)-oxid (E 172)
Druckfarbe
Schellack
Eisen(II,III)-oxid (E 172)
Kaliumhydroxid
Propylenglycol
Nicht zutreffend.
4 Jahre.
Nicht über 25 °C lagern.
In der Original-Blisterpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Polyamid/Aluminium/PVC/Aluminium-Blisterpackungen mit 10 Kapseln. Jeder Umkarton enthält 30, 60 oder 90 Hartkapseln.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Pflegepersonen dürfen die Kapseln nicht öffnen, um wiederholten Kontakt mit dem Kapselinhalt zu vermeiden.
Zubereitung und Verabreichung einer Suspension
Die Suspension kann mit Hilfe von Wasser, Apfelsaft oder Milch zubereitet werden. Bei Verabreichung über eine Ernährungssonde sollte die Suspension mit Hilfe von Wasser zubereitet werden.
Geben Sie die Kapsel(n) entsprechend der verschriebenen Dosis (bis zu 5 Kapseln) in ein kleines Behältnis (mit einer Kapazität von etwa 20 ml (4 Teelöffeln)) oder in eine orale Spritze (20 ml); die Kapseln nicht zerbrechen oder zerkleinern.
Geben Sie in das Behältnis oder in die orale Spritze 3 ml Flüssigkeit hinzu. Warten Sie 10 Minuten, bis sich die Kapselhülle (äußere Oberfläche) auflöst, rühren oder schütteln Sie die Mixtur dann 3 Minuten lang, bis sich die Kapseln vollständig aufgelöst haben.
Bei Verwendung einer oralen Spritze: Verschließen Sie die Spritze, entfernen Sie den Kolben und geben Sie mit einer zweiten Spritze oder kalibrierten Pipette die Flüssigkeit zu der ersten Spritze hinzu; setzen Sie vor dem Mischen den Kolben wieder ein.
Verabreichen Sie den gesamten Inhalt des Behältnisses oder der oralen Spritze. Die Suspension kann dabei direkt aus dem Behältnis getrunken oder von der oralen Spritze direkt in den Mund oder über eine Ernährungssonde verabreicht werden.
Fügen Sie im nächsten Schritt mit Hilfe einer zweiten Spritze oder einer Pipette weitere 2 ml Flüssigkeit zu dem Behältnis oder der oralen Spritze hinzu; rühren oder schütteln und verabreichen Sie den Inhalt. Wiederholen Sie diesen Vorgang mindestens zweimal, bis keine Rückstände mehr sichtbar sind, sodass sichergestellt ist, dass die gesamte Medikamentenmenge eingenommen wurde.
Hinweis: Die Kompatibilität wurde für Spritzen aus Polypropylen und für Ernährungssonden mit einem Mindestdurchmesser von 5 French/Charrière (für Schläuche aus Polyvinylchlorid oder Polyurethan), mit einem Mindestdurchmesser von 6 French/Charrière (für Schläuche aus Silikon) und mit einem Durchmesser von bis zu 16 French/Charrière für Schläuche aus Polyvinylchlorid, Polyurethan oder Silikon bestätigt.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Eisai GmbH
Edmund-Rumpler-Straße 3
60549 Frankfurt am Main
Deutschland
E-Mail: medinfo_de@eisai.net
Kisplyx 4 mg Hartkapseln
EU/1/16/1128/001
EU/1/16/1128/003
EU/1/16/1128/004
Kisplyx 10 mg Hartkapseln
EU/1/16/1128/002
EU/1/16/1128/005
EU/1/16/1128/006
Datum der Erteilung der Zulassung: 25. August 2016
Datum der letzten Verlängerung der Zulassung: 17. Juni 2021
03.2026
Verschreibungspflichtig
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu verfügbar.