
RINVOQ® 15 mg Retardtabletten
RINVOQ® 30 mg Retardtabletten
RINVOQ® 45 mg Retardtabletten
RINVOQ 15 mg Retardtabletten
Eine Retardtablette enthält Upadacitinib 0,5 H2O, entsprechend 15 mg Upadacitinib.
RINVOQ 30 mg Retardtabletten
Eine Retardtablette enthält Upadacitinib 0,5 H2O, entsprechend 30 mg Upadacitinib.
RINVOQ 45 mg Retardtabletten
Eine Retardtablette enthält Upadacitinib 0,5 H2O, entsprechend 45 mg Upadacitinib.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Retardtablette
RINVOQ 15 mg Retardtabletten
Purpurfarbene längliche, bikonvexe Retardtabletten mit den Abmessungen 14 x 8 mm und der Prägung ‚a15‘ auf einer Seite.
RINVOQ 30 mg Retardtabletten
Rote längliche, bikonvexe Retardtabletten mit den Abmessungen 14 x 8 mm und der Prägung ‚a30‘ auf einer Seite.
RINVOQ 45 mg Retardtabletten
Gelbe bis gelb gesprenkelte, längliche, bikonvexe Retardtabletten mit den Abmessungen 14 x 8 mm und der Prägung ‚a45‘ auf einer Seite.
Rheumatoide Arthritis
RINVOQ wird angewendet zur Behandlung der mittelschweren bis schweren aktiven rheumatoiden Arthritis bei erwachsenen Patienten, die auf ein oder mehrere krankheitsmodifizierende Antirheumatika (DMARDs) unzureichend angesprochen oder diese nicht vertragen haben. RINVOQ kann als Monotherapie oder in Kombination mit Methotrexat angewendet werden.
Psoriasis-Arthritis
RINVOQ wird angewendet zur Behandlung der aktiven Psoriasis-Arthritis bei erwachsenen Patienten, die auf ein oder mehrere DMARDs unzureichend angesprochen oder diese nicht vertragen haben. RINVOQ kann als Monotherapie oder in Kombination mit Methotrexat angewendet werden.
Axiale Spondyloarthritis
Nicht röntgenologische axiale Spondyloarthritis (nr‑axSpA)
RINVOQ wird angewendet zur Behandlung der aktiven nicht röntgenologischen axialen Spondyloarthritis bei erwachsenen Patienten mit objektiven Anzeichen einer Entzündung, angezeigt durch erhöhtes C‑reaktives Protein (CRP) und/oder Nachweis durch Magnetresonanztomografie (MRT), die unzureichend auf nicht steroidale Antirheumatika (NSAR) angesprochen haben.
Ankylosierende Spondylitis (AS, röntgenologische axiale Spondyloarthritis)
RINVOQ wird angewendet zur Behandlung der aktiven ankylosierenden Spondylitis bei erwachsenen Patienten, die auf eine konventionelle Therapie unzureichend angesprochen haben.
Riesenzellarteriitis
RINVOQ wird angewendet zur Behandlung der Riesenzellarteriitis bei erwachsenen Patienten.
Atopische Dermatitis
RINVOQ wird angewendet zur Behandlung der mittelschweren bis schweren atopischen Dermatitis bei Erwachsenen und Jugendlichen ab 12 Jahren, die für eine systemische Therapie infrage kommen.
Colitis ulcerosa
RINVOQ wird angewendet zur Behandlung der mittelschweren bis schweren aktiven Colitis ulcerosa bei erwachsenen Patienten, die auf eine konventionelle Therapie oder ein Biologikum unzureichend angesprochen haben, nicht mehr darauf ansprechen oder diese nicht vertragen haben.
Morbus Crohn
RINVOQ wird angewendet zur Behandlung des mittelschweren bis schweren aktiven Morbus Crohn bei erwachsenen Patienten, die auf eine konventionelle Therapie oder ein Biologikum unzureichend angesprochen haben, nicht mehr darauf ansprechen oder diese nicht vertragen haben.
Die Behandlung mit Upadacitinib sollte von einem Arzt eingeleitet und überwacht werden, der über Erfahrung in der Diagnose und Behandlung von Erkrankungen verfügt, für die Upadacitinib indiziert ist.
Dosierung
Rheumatoide Arthritis, Psoriasis-Arthritis und axiale Spondyloarthritis
Die empfohlene Dosis von Upadacitinib beträgt 15 mg einmal täglich.
Bei Patienten mit axialer Spondyloarthritis, die nach 16 Wochen der Behandlung kein klinisches Ansprechen zeigen, ist ein Absetzen der Behandlung in Erwägung zu ziehen. Bei einigen Patienten mit anfänglich partiellem Ansprechen kann es im Verlauf der Weiterbehandlung über 16 Wochen hinaus zu Verbesserungen kommen.
Riesenzellarteriitis
Die empfohlene Dosis von Upadacitinib beträgt 15 mg einmal täglich in Kombination mit einer Ausschleichtherapie der Kortikosteroide. Upadacitinib-Monotherapie sollte nicht zur Behandlung akuter Rezidive angewendet werden (siehe Abschnitt 4.4).
Aufgrund der chronischen Natur der Riesenzellarteriitis kann Upadacitinib 15 mg einmal täglich nach Absetzen der Kortikosteroide als Monotherapie fortgesetzt werden.
Atopische Dermatitis
Die empfohlene Dosis von Upadacitinib beträgt 15 mg oder 30 mg einmal täglich, je nach individuellem Krankheitsbild.
Eine Dosis von 15 mg wird für Patienten mit einem erhöhten Risiko für venöse Thromboembolien (VTE), schwerwiegende unerwünschte kardiovaskuläre Ereignisse (MACE) und maligne Erkrankungen empfohlen (siehe Abschnitt 4.4).
Eine Dosis von 30 mg einmal täglich kann bei Patienten mit hoher Krankheitslast, die kein erhöhtes Risiko für VTE, MACE und maligne Erkrankungen aufweisen (siehe Abschnitt 4.4) oder bei Patienten mit unzureichendem Ansprechen auf 15 mg einmal täglich angemessen sein.
Bei Jugendlichen (12 bis 17 Jahren) mit einem Körpergewicht von mindestens 30 kg beträgt die empfohlene Dosis 15 mg. Wenn der Patient nicht ausreichend auf 15 mg einmal täglich anspricht, kann die Dosis auf 30 mg einmal täglich erhöht werden.
Es sollte die niedrigste wirksame Dosis verwendet werden, um das Ansprechen aufrechtzuerhalten.
Bei Patienten im Alter von 65 Jahren und älter beträgt die empfohlene Dosis 15 mg einmal täglich (siehe Abschnitt 4.4).
Begleitende topische Therapien
Upadacitinib kann mit oder ohne topische Kortikosteroide angewendet werden. Topische Calcineurininhibitoren können für empfindliche Bereiche wie Gesicht, Hals, intertriginöse und Genitalbereiche verwendet werden.
Bei Patienten, die nach 12 Behandlungswochen keine Anzeichen für einen therapeutischen Nutzen zeigen, ist ein Absetzen von Upadacitinib in Erwägung zu ziehen.
Colitis ulcerosa
Einleitung der Therapie
Die empfohlene Anfangsdosis von Upadacitinib beträgt 45 mg einmal täglich über 8 Wochen. Bei Patienten, die bis Woche 8 keinen ausreichenden therapeutischen Nutzen erzielen, können 45 mg Upadacitinib einmal täglich für einen weiteren 8-wöchigen Zeitraum angewendet werden (siehe Abschnitt 5.1). Bei Patienten, die bis Woche 16 keine Anzeichen für einen therapeutischen Nutzen zeigen, sollte Upadacitinib abgesetzt werden.
Erhaltungstherapie
Die empfohlene Erhaltungsdosis von Upadacitinib beträgt 15 mg oder 30 mg einmal täglich, je nach individuellem Krankheitsbild des Patienten:
Eine Dosis von 15 mg wird für Patienten mit einem erhöhten Risiko für VTE, MACE und maligne Erkrankungen empfohlen (siehe Abschnitt 4.4).
Eine Dosis von 30 mg einmal täglich kann bei einigen Patienten, z. B. bei Patienten mit hoher Krankheitslast oder denjenigen, die eine 16‑wöchige Einleitungstherapie benötigen, und bei denen kein erhöhtes Risiko für VTE, MACE und maligne Erkrankungen besteht (siehe Abschnitt 4.4) oder die mit einer Dosis von 15 mg einmal täglich keinen ausreichenden therapeutischen Nutzen erzielen, angemessen sein.
Es sollte die niedrigste wirksame Dosis verwendet werden, um das Ansprechen aufrechtzuerhalten.
Bei Patienten im Alter von 65 Jahren und älter beträgt die empfohlene Dosis 15 mg einmal täglich (siehe Abschnitt 4.4).
Bei Patienten, die auf die Behandlung mit Upadacitinib angesprochen haben, können Kortikosteroide entsprechend dem Versorgungsstandard reduziert und/oder abgesetzt werden.
Morbus Crohn
Einleitung der Therapie
Die empfohlene Anfangsdosis von Upadacitinib beträgt 45 mg einmal täglich über 12 Wochen. Bei Patienten, die nach der anfänglichen 12-wöchigen Induktion keinen ausreichenden therapeutischen Nutzen erzielt haben, kann eine verlängerte Induktion über weitere 12 Wochen mit einer Dosis von 30 mg einmal täglich in Betracht gezogen werden. Bei diesen Patienten sollte Upadacitinib abgesetzt werden, wenn nach 24 Behandlungswochen kein therapeutischer Nutzen nachweisbar ist.
Erhaltungstherapie
Die empfohlene Erhaltungsdosis von Upadacitinib beträgt 15 mg oder 30 mg einmal täglich, je nach individuellem Krankheitsbild des Patienten:
Eine Dosis von 15 mg wird bei Patienten mit einem erhöhten Risiko für VTE, MACE und maligne Erkrankungen empfohlen (siehe Abschnitt 4.4).
Eine Dosis von 30 mg einmal täglich kann bei Patienten mit hoher Krankheitslast, bei denen kein erhöhtes Risiko für VTE, MACE und maligne Erkrankungen besteht (siehe Abschnitt 4.4) oder die mit einer Dosis von 15 mg einmal täglich keinen ausreichenden therapeutischen Nutzen erzielen, angemessen sein.
Es sollte die niedrigste wirksame Dosis verwendet werden, um das Ansprechen aufrechtzuerhalten.
Bei Patienten im Alter von 65 Jahren und älter beträgt die empfohlene Erhaltungsdosis 15 mg einmal täglich (siehe Abschnitt 4.4).
Bei Patienten, die auf die Behandlung mit Upadacitinib angesprochen haben, können Kortikosteroide entsprechend dem Versorgungsstandard reduziert und/oder abgesetzt werden.
Wechselwirkungen
Bei Patienten mit Colitis ulcerosa und Morbus Crohn, die starke Inhibitoren des Cytochroms P450 (CYP) 3A4 erhalten (z. B. Ketoconazol, Clarithromycin), beträgt die empfohlene Anfangsdosis 30 mg einmal täglich und die empfohlene Erhaltungsdosis 15 mg einmal täglich (siehe Abschnitt 4.5).
Behandlungsbeginn
Die Behandlung sollte bei Patienten mit einer absoluten Lymphozytenzahl (ALC) von weniger als 0,5 x 109 Zellen/l, einer absoluten Neutrophilenzahl (ANC) von weniger als 1 x 109 Zellen/l oder einem Hämoglobinwert (Hb) von weniger als 8 g/dl nicht begonnen werden (siehe Abschnitte 4.4 und 4.8).
Behandlungsunterbrechung
Wenn bei einem Patienten eine schwere Infektion auftritt, sollte die Behandlung unterbrochen werden, bis die Infektion unter Kontrolle ist.
Eine Unterbrechung der Behandlung kann erforderlich sein, bis die in Tabelle 1 beschriebenen Laborwertabweichungen entsprechend normalisiert sind.
Tabelle 1 Überwachung der Laborparameter
Laborparameter |
Maßnahme |
Überwachung |
Absolute Neutrophilenzahl (ANC) |
Die Behandlung sollte bei einer ANC von < 1 x 109 Zellen/l unterbrochen werden und nach Anstieg der ANC über diesen Wert wieder begonnen werden. |
Bestimmung der Werte vor Beginn und dann spätestens zwölf Wochen nach Beginn der Behandlung. Anschließend entsprechend der individuellen Behandlung des Patienten. |
Absolute Lymphozytenzahl (ALC) |
Die Behandlung sollte bei einer ALC von < 0,5 x 109 Zellen/l unterbrochen werden und nach Anstieg der ALC über diesen Wert wieder begonnen werden. |
|
Hämoglobin (Hb) |
Die Behandlung sollte bei einem Hb-Wert von < 8 g/dl unterbrochen werden und darf erst nach Anstieg des Hb über diesen Wert wieder begonnen werden. |
|
Lebertransaminasen |
Bei Verdacht auf arzneimittelinduzierte Leberschäden sollte die Behandlung vorübergehend unterbrochen werden. |
Bestimmung der Werte vor Beginn und während der Behandlung entsprechend der routinemäßigen Behandlung des Patienten. |
Lipide |
Die Patienten sollten entsprechend den internationalen klinischen Leitlinien für Hyperlipidämie behandelt werden. |
Bestimmung der Werte zwölf Wochen nach Beginn der Behandlung, danach entsprechend den internationalen klinischen Leitlinien für Hyperlipidämie. |
Besondere Patientengruppen
Ältere Patienten
Rheumatoide Arthritis, Psoriasis-Arthritis und axiale Spondyloarthritis
Bisher liegen nur begrenzte Erfahrungen bei Patienten ab 75 Jahren vor (siehe Abschnitt 4.4).
Atopische Dermatitis
Bei atopischer Dermatitis werden Dosen über 15 mg einmal täglich bei Patienten ab 65 Jahren nicht empfohlen (siehe Abschnitte 4.4 und 4.8).
Colitis ulcerosa und Morbus Crohn
Bei Colitis ulcerosa und Morbus Crohn werden Dosen über 15 mg einmal täglich als Erhaltungstherapie bei Patienten ab 65 Jahren nicht empfohlen (siehe Abschnitte 4.4 und 4.8). Die Sicherheit und Wirksamkeit von Upadacitinib bei Patienten ab 75 Jahren sind bisher noch nicht erwiesen.
Niereninsuffizienz
Bei Patienten mit leichter oder mittelschwerer Niereninsuffizienz ist keine Dosisanpassung erforderlich. Bisher liegen nur begrenzte Erfahrungen mit der Anwendung von Upadacitinib bei Patienten mit schwerer Niereninsuffizienz vor (siehe Abschnitt 5.2). Upadacitinib ist bei Patienten mit schwerer Niereninsuffizienz mit Vorsicht anzuwenden (siehe Tabelle 2). Upadacitinib wurde bei Patienten mit terminaler Niereninsuffizienz nicht untersucht und wird daher zur Behandlung dieser Patienten nicht empfohlen.
Tabelle 2 Empfohlene Dosis bei schwerer Niereninsuffizienza
Anwendungsgebiet |
Empfohlene Dosis (einmal täglich) |
Rheumatoide Arthritis, Psoriasis-Arthritis, axiale Spondyloarthritis, Riesenzellarteriitis, atopische Dermatitis |
15 mg |
Colitis ulcerosa, Morbus Crohn |
Anfangsdosis: 30 mg |
Erhaltungsdosis: 15 mg | |
a Geschätzte glomeruläre Filtrationsrate (eGFR) 15 bis < 30 ml/min/1,73 m2 | |
Leberinsuffizienz
Bei Patienten mit leichter (Child-Pugh A) oder mittelschwerer (Child-Pugh B) Leberinsuffizienz ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Upadacitinib darf bei Patienten mit schwerer Leberinsuffizienz (Child-Pugh C) nicht angewendet werden (siehe Abschnitt 4.3).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von RINVOQ bei Kindern unter 12 Jahren mit atopischer Dermatitis sind nicht erwiesen. Es liegen keine Daten vor.
Die Sicherheit und Wirksamkeit von RINVOQ bei Kindern und Jugendlichen im Alter von 0 bis unter 18 Jahren mit rheumatoider Arthritis, Psoriasis-Arthritis, axialer Spondyloarthritis, Colitis ulcerosa und Morbus Crohn sind bisher noch nicht erwiesen. Es liegen keine Daten vor.
Es gibt in der Indikation Riesenzellarteriitis keinen relevanten Nutzen von RINVOQ bei Kindern und Jugendlichen.
Art der Anwendung
RINVOQ ist einmal täglich mit oder unabhängig von einer Mahlzeit zu einer beliebigen Uhrzeit einzunehmen. Die Tabletten sind im Ganzen zu schlucken und dürfen nicht geteilt, zerdrückt oder zerkaut werden, damit sichergestellt ist, dass die gesamte Dosis korrekt eingenommen wird.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Aktive Tuberkulose (TB) oder aktive schwerwiegende Infektionen (siehe Abschnitt 4.4).
Schwere Leberinsuffizienz (siehe Abschnitt 4.2).
Schwangerschaft (siehe Abschnitt 4.6).
|
Upadacitinib sollte nur angewendet werden, wenn keine geeigneteren Behandlungsoptionen zur Verfügung stehen:
|
Anwendung bei Patienten im Alter von 65 Jahren und älter
In Anbetracht des in einer großen randomisierten Studie zu Tofacitinib (ein weiterer Januskinase(JAK)-Inhibitor) beobachteten erhöhten Risikos für MACE, maligne Erkrankungen, schwere Infektionen und Gesamtmortalität bei Patienten im Alter von 65 Jahren und älter sollte Upadacitinib bei diesen Patienten nur angewendet werden, wenn keine geeigneteren Behandlungsoptionen zur Verfügung stehen.
Bei Patienten im Alter von 65 Jahren und älter besteht bei 30 mg Upadacitinib einmal täglich ein erhöhtes Risiko für Nebenwirkungen. Daher beträgt die empfohlene Dosis für die Langzeitanwendung bei dieser Patientengruppe 15 mg einmal täglich (siehe Abschnitte 4.2 und 4.8).
Immunsuppressiva
Die Kombination mit anderen potenten Immunsuppressiva wie Azathioprin, 6-Mercaptopurin, Ciclosporin, Tacrolimus und biologischen DMARDs oder anderen JAK-Inhibitoren wurde in klinischen Studien nicht untersucht und wird nicht empfohlen, da das Risiko einer zusätzlichen immunsuppressiven Wirkung nicht ausgeschlossen werden kann.
Schwerwiegende Infektionen
Bei Patienten, die Upadacitinib erhielten, wurden schwere Infektionen, darunter auch solche mit tödlichem Ausgang, berichtet. Die am häufigsten berichteten schwerwiegenden Infektionen, die unter Upadacitinib berichtet wurden, umfassten Pneumonie (siehe Abschnitt 4.8) und Cellulitis. Bei Patienten, die Upadacitinib erhalten haben, wurden Fälle von bakterieller Meningitis und Sepsis berichtet. Als opportunistische Infektionen wurden Tuberkulose, multidermatomaler Herpes zoster, orale/ösophageale Candidose und Kryptokokkose unter Upadacitinib-Behandlung berichtet.
Bei Patienten mit aktiver, schwerwiegender Infektion, einschließlich lokaler Infektionen, darf mit der Behandlung mit Upadacitinib nicht begonnen werden (siehe Abschnitt 4.3).
Bei folgenden Patienten sollte Upadacitinib nur nach Nutzen-Risiko-Abwägung angewendet werden:
bei Patienten mit chronischen oder wiederkehrenden Infektionen
bei Patienten mit Exposition gegenüber Tuberkulose
bei Patienten mit einer schweren oder opportunistischen Infektion in der Anamnese
bei Patienten, die in Gebieten mit endemischer Tuberkulose oder endemischen Mykosen gelebt oder solche Gebiete bereist haben, oder
bei Patienten mit Grunderkrankungen, aufgrund derer sie anfällig für Infektionen sind.
Patienten sollten während und nach Behandlung mit Upadacitinib engmaschig auf Anzeichen und Symptome einer Infektion überwacht werden. Entwickelt ein Patient eine schwerwiegende oder opportunistische Infektion, muss die Behandlung mit Upadacitinib unterbrochen werden. Patienten, bei denen unter Behandlung mit Upadacitinib eine neue Infektion auftritt, sind unverzüglich einer für immungeschwächte Patienten angemessenen, vollständigen diagnostischen Abklärung zu unterziehen; eine entsprechende Antibiotikatherapie ist einzuleiten, die Patienten sind engmaschig zu überwachen und die Behandlung mit Upadacitinib ist zu unterbrechen, falls der Patient nicht auf die Antibiotikatherapie anspricht. Sobald die Infektion unter Kontrolle ist, kann die Behandlung mit Upadacitinib fortgesetzt werden.
Bei 30 mg Upadacitinib wurde im Vergleich zu 15 mg Upadacitinib eine höhere Rate an schwerwiegenden Infektionen beobachtet.
Da Infektionen bei älteren Patienten und bei Diabetikern häufiger auftreten, ist bei der Behandlung dieser Patienten Vorsicht geboten. Bei Patienten im Alter von 65 Jahren und älter sollte Upadacitinib nur angewendet werden, wenn keine geeigneteren Behandlungsoptionen zur Verfügung stehen (siehe Abschnitt 4.2).
Tuberkulose
Vor Therapiebeginn mit Upadacitinib ist ein Tuberkulose(TB)-Screening durchzuführen. Upadacitinib darf nicht bei Patienten mit aktiver TB angewendet werden (siehe Abschnitt 4.3). Bei Patienten mit unbehandelter latenter TB oder bei Patienten mit Risikofaktoren für eine TB-Infektion ist vor Einleitung der Behandlung mit Upadacitinib eine Anti-TB-Therapie in Erwägung zu ziehen.
Die Konsultation eines in der Tuberkulosebehandlung erfahrenen Arztes ist empfehlenswert, wenn entschieden werden soll, ob eine Anti-TB-Therapie im Einzelfall geeignet ist.
Die Patienten müssen auf die Entwicklung von Anzeichen und Symptomen einer TB überwacht werden; dies gilt auch für Patienten mit negativem Befund auf eine latente TB-Infektion vor Therapiebeginn.
Virusreaktivierung
In klinischen Studien wurden Virusreaktivierungen, einschließlich Fällen der Reaktivierung von Herpesviren (z. B. Herpes zoster), berichtet (siehe Abschnitt 4.8). Das Risiko für Herpes zoster scheint bei Patienten japanischer Herkunft, die mit Upadacitinib behandelt werden, erhöht zu sein. Tritt bei einem Patienten Herpes zoster auf, sollte eine Unterbrechung der Behandlung mit Upadacitinib bis zum Abklingen der Infektion in Erwägung gezogen werden.
Vor Beginn und während einer Therapie mit Upadacitinib sollte ein Screening auf eine virale Hepatitis und die Überwachung einer möglichen Reaktivierung durchgeführt werden. Patienten mit positivem Ergebnis beim Test auf Hepatitis-C-Antikörper und Hepatitis-C-Virus-RNA waren von den klinischen Studien ausgeschlossen. Patienten mit positivem Ergebnis beim Test auf Hepatitis-B-Oberflächen-Antigen oder Hepatitis-B-Virus-DNA waren von den klinischen Studien ausgeschlossen. Falls während der Behandlung mit Upadacitinib Hepatitis-B-Virus-DNA festgestellt wird, ist ein Hepatologe zu konsultieren.
Impfung
Es liegen keine Daten zum Ansprechen auf Impfungen mit Lebendimpfstoffen bei Patienten unter Upadacitinib-Behandlung vor. Die Anwendung von attenuierten Lebendimpfstoffen während oder unmittelbar vor einer Behandlung mit Upadacitinib wird nicht empfohlen. Vor Einleitung der Therapie mit Upadacitinib wird empfohlen, den Impfstatus der Patienten entsprechend den aktuellen Impfleitlinien zu überprüfen und alle erforderlichen Impfungen nachzuholen; dazu zählt auch die prophylaktische Impfung gegen Herpes zoster (siehe Abschnitt 5.1).
Maligne Erkrankungen
Lymphome und andere maligne Erkrankungen wurden bei Patienten, die JAK-Inhibitoren einschließlich Upadacitinib erhielten, berichtet.
In einer großen randomisierten, aktiv kontrollierten Studie zu Tofacitinib (ein weiterer JAK-Inhibitor) bei Patienten mit rheumatoider Arthritis ab 50 Jahren mit mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurde unter Tofacitinib im Vergleich zu Tumornekrosefaktor-Inhibitoren (TNF-Inhibitoren) eine höhere Rate an malignen Erkrankungen, insbesondere Lungenkrebs, Lymphom und nichtmelanozytärer Hautkrebs (NMSC) beobachtet.
Bei 30 mg Upadacitinib wurde im Vergleich zu 15 mg Upadacitinib eine höhere Rate an malignen Erkrankungen beobachtet.
Bei Patienten im Alter von 65 Jahren und älter, bei Patienten, die Raucher oder ehemalige Langzeitraucher sind, oder bei Patienten mit anderen Risikofaktoren für maligne Erkrankungen (z. B. aktuelle oder zurückliegende maligne Erkrankungen) sollte Upadacitinib nur dann angewendet werden, wenn keine geeigneteren Behandlungsoptionen zur Verfügung stehen.
Nichtmelanozytärer Hautkrebs (NMSC)
NMSCs wurden bei Patienten berichtet, die mit Upadacitinib behandelt wurden (siehe Abschnitt 4.8). Unter 30 mg Upadacitinib wurde eine höhere Rate an NMSC beobachtet als unter 15 mg Upadacitinib. Eine regelmäßige Hautuntersuchung wird bei allen Patienten empfohlen, insbesondere bei denjenigen mit Risikofaktoren für Hautkrebs.
Hämatologische Anomalien
In klinischen Studien wurde bei ≤ 1 % der Patienten eine absolute Neutrophilenzahl (ANC) von < 1 x 109 Zellen/l, eine absolute Lymphozytenzahl (ALC) von < 0,5 x 109 Zellen/l und ein Hämoglobinwert von < 8 g/dl berichtet (siehe Abschnitt 4.8). Bei Patienten, bei denen im Rahmen routinemäßiger Untersuchungen eine ANC von < 1 x 109 Zellen/l, eine ALC von < 0,5 x 109 Zellen/l oder ein Hämoglobinwert von < 8 g/dl beobachtet wird, sollte mit der Behandlung nicht begonnen werden bzw. sollte die Behandlung vorübergehend unterbrochen werden (siehe Abschnitt 4.2).
Gastrointestinale Perforationen
Fälle von Divertikulitis und gastrointestinalen Perforationen wurden in klinischen Studien sowie nach der Markteinführung berichtet (siehe Abschnitt 4.8).
Upadacitinib sollte bei Patienten mit Risiko für eine gastrointestinale Perforation mit Vorsicht angewendet werden (z. B. Patienten mit divertikulärer Erkrankung, mit Divertikulitis in der Anamnese oder Patienten, die nicht steroidale Antirheumatika [NSAR], Kortikosteroide oder Opioide anwenden). Patienten mit aktivem Morbus Crohn haben ein erhöhtes Risiko, eine intestinale Perforation zu entwickeln. Patienten mit neu auftretenden abdominalen Anzeichen und Symptomen sind zur frühzeitigen Erkennung einer Divertikulitis oder einer gastrointestinalen Perforation umgehend zu untersuchen.
Schwerwiegende unerwünschte kardiovaskuläre Ereignisse
In klinischen Studien zu Upadacitinib wurden Fälle von MACE beobachtet.
In einer großen randomisierten, aktiv kontrollierten Studie zu Tofacitinib (ein weiterer JAK-Inhibitor) bei Patienten mit rheumatoider Arthritis ab 50 Jahren mit mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurde unter Tofacitinib im Vergleich zu TNF-Inhibitoren eine höhere Rate an MACE, definiert als kardiovaskulärer Tod, nicht tödlicher Myokardinfarkt (MI) und nicht tödlicher Schlaganfall, beobachtet.
Daher sollte Upadacitinib bei Patienten im Alter von 65 Jahren und älter, bei Patienten, die Raucher oder ehemalige Langzeitraucher sind, und bei Patienten mit atherosklerotischen kardiovaskulären Erkrankungen in der Vorgeschichte oder anderen kardiovaskulären Risikofaktoren nur dann angewendet werden, wenn keine geeigneteren Behandlungsoptionen zur Verfügung stehen.
Lipide
Die Behandlung mit Upadacitinib war mit einem dosisabhängigen Anstieg der Lipidwerte verbunden. Es wurde ein Anstieg des Gesamtcholesterins, des Low-Density-Lipoproteins (LDL) und des High-Density-Lipoproteins (HDL) beobachtet (siehe Abschnitt 4.8). Erhöhungen des LDL-Cholesterins ließen sich mit einer Statintherapie wieder auf die Werte vor Behandlungsbeginn senken, wobei die Evidenzlage begrenzt ist. Die Auswirkungen dieser Lipidwerterhöhungen auf die kardiovaskuläre Morbidität und Mortalität wurden nicht untersucht (zur Überwachung siehe Abschnitt 4.2).
Anstieg der Lebertransaminasen
Die Behandlung mit Upadacitinib war im Vergleich zu Placebo mit einer höheren Inzidenz für erhöhte Leberwerte verbunden (siehe Abschnitt 4.8).
Lebertransaminasen müssen vor Beginn und während der Behandlung entsprechend der routinemäßigen Untersuchung des Patienten bestimmt werden. Es wird empfohlen, unverzüglich die Ursache der Leberwerterhöhungen zu ermitteln, um eine mögliche arzneimittelinduzierte Leberschädigung zu erkennen.
Falls im Rahmen von routinemäßigen Untersuchungen des Patienten ein ALT- oder AST-Anstieg beobachtet und eine arzneimittelinduzierte Leberschädigung vermutet wird, sollte Upadacitinib abgesetzt werden, bis eine solche Diagnose ausgeschlossen werden kann.
Venöse Thromboembolie
In klinischen Studien zu Upadacitinib wurden Fälle von tiefer Venenthrombose (TVT) und Lungenembolie (LE) beobachtet.
In einer großen randomisierten, aktiv kontrollierten Studie zu Tofacitinib (ein weiterer JAK-Inhibitor) bei Patienten mit rheumatoider Arthritis ab 50 Jahren mit mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurde unter Tofacitinib im Vergleich zu TNF-Inhibitoren eine dosisabhängig höhere Rate an VTE, einschließlich TVT und LE, beobachtet.
Bei Patienten mit kardiovaskulären oder malignen Risikofaktoren (siehe auch Abschnitt 4.4 „Schwerwiegende unerwünschte kardiovaskuläre Ereignisse“ und „Maligne Erkrankungen“) sollte Upadacitinib nur angewendet werden, wenn keine geeigneteren Behandlungsoptionen zur Verfügung stehen.
Bei Patienten mit bekannten VTE-Risikofaktoren, die keine kardiovaskulären oder malignen Risikofaktoren sind, sollte Upadacitinib mit Vorsicht angewendet werden. Zu den VTE-Risikofaktoren, die keine kardiovaskulären oder malignen Risikofaktoren sind, gehören frühere VTE, Patienten, die sich einer größeren Operation unterziehen müssen, Immobilisierung, die Anwendung kombinierter hormoneller Kontrazeptiva oder eine Hormonersatztherapie sowie vererbte Gerinnungsstörungen. Die Patienten sollten während der Behandlung mit Upadacitinib in regelmäßigen Abständen erneut untersucht werden, um Veränderungen des VTE-Risikos festzustellen. Patienten mit Anzeichen und Symptomen einer VTE sollten umgehend untersucht werden, und die Behandlung sollte bei Patienten mit Verdacht auf VTE unabhängig von der Dosis abgesetzt werden.
Netzhautvenenverschluss
Netzhautvenenverschluss wurde bei Patienten berichtet, die mit JAK-Inhibitoren, einschließlich Upadacitinib, behandelt wurden. Den Patienten sollte geraten werden, umgehend einen Arzt aufzusuchen, falls bei ihnen Symptome auftreten, die auf einen Netzhautvenenverschluss hindeuten.
Überempfindlichkeitsreaktionen
Schwerwiegende Überempfindlichkeitsreaktionen wie Anaphylaxie und Angioödem wurden bei Patienten, die Upadacitinib erhielten, berichtet. Wenn eine klinisch signifikante Überempfindlichkeitsreaktion auftritt, muss die Behandlung mit Upadacitinib abgesetzt und eine geeignete Therapie eingeleitet werden (siehe Abschnitte 4.3 und 4.8).
Hypoglykämie bei Patienten, die wegen Diabetes behandelt werden
Es liegen Berichte über Hypoglykämie nach Beginn der Gabe von JAK-Inhibitoren, einschließlich Upadacitinib, bei Patienten vor, die eine Behandlung gegen Diabetes erhielten. Falls eine Hypoglykämie auftritt, kann eine Dosisanpassung der Antidiabetika erforderlich sein.
Medikamentenrückstände im Stuhl
Bei Patienten, die Upadacitinib einnahmen, wurde über Arzneimittelrückstände im Stuhl oder über Stomaausgänge berichtet. In den meisten Berichten wurden anatomische (z. B. Ileostomie, Kolostomie, Darmresektion) oder funktionelle gastrointestinale Erkrankungen mit verkürzten gastrointestinalen Transitzeiten beschrieben. Patienten sollten angewiesen werden, ihren Arzt zu kontaktieren, wenn wiederholt Arzneimittelrückstände beobachtet werden. Die Patienten sollten klinisch überwacht werden und eine alternative Behandlung sollte in Betracht gezogen werden, wenn das therapeutische Ansprechen unzureichend ist.
Riesenzellarteriitis
Upadacitinib-Monotherapie sollte nicht zur Behandlung akuter Rezidive angewendet werden, da die Wirksamkeit in diesem Setting nicht untersucht wurde. Kortikosteroide sollen gemäß medizinischer Beurteilung und Behandlungsleitlinien gegeben werden.
Potenzielle Auswirkungen anderer Arzneimittel auf die Pharmakokinetik von Upadacitinib
Upadacitinib wird hauptsächlich durch CYP3A4 metabolisiert. Daher kann die Plasmaexposition von Upadacitinib durch Arzneimittel beeinflusst werden, die CYP3A4 stark hemmen oder induzieren.
Gleichzeitige Anwendung mit CYP3A4-Inhibitoren
Die Upadacitinib-Exposition ist bei gleichzeitiger Anwendung mit starken CYP3A4-Inhibitoren (z. B. Ketoconazol, Itraconazol, Posaconazol, Voriconazol, Clarithromycin und Grapefruit) erhöht. In einer klinischen Studie führte die gleichzeitige Anwendung von Upadacitinib und Ketoconazol zu einem Anstieg der Cmax von Upadacitinib um 70 % und der AUC um 75 %. 15 mg Upadacitinib einmal täglich ist bei Patienten unter Langzeitbehandlung mit starken CYP3A4-Inhibitoren mit Vorsicht anzuwenden. 30 mg Upadacitinib wird bei Patienten mit atopischer Dermatitis, die eine dauerhafte Behandlung mit starken CYP3A4‑Inhibitoren erhalten, nicht empfohlen. Bei Patienten mit Colitis ulcerosa oder Morbus Crohn, die starke CYP3A4-Inhibitoren anwenden, beträgt die empfohlene Anfangsdosis 30 mg einmal täglich und die empfohlene Erhaltungsdosis 15 mg einmal täglich (siehe Abschnitt 4.2). Bei langfristiger Anwendung sind Alternativen zu starken CYP3A4-Inhibitoren in Betracht zu ziehen. Nahrungsmittel oder Getränke, die Grapefruit enthalten, müssen während der Behandlung mit Upadacitinib vermieden werden.
Gleichzeitige Anwendung mit CYP3A4-Induktoren
Die Upadacitinib-Exposition ist bei gleichzeitiger Anwendung mit starken CYP3A4-Induktoren (z. B. Rifampicin und Phenytoin) verringert, was zu einer abgeschwächten Wirkung von Upadacitinib führen kann. In einer klinischen Prüfung führte die Anwendung von Upadacitinib zusammen mit mehrfacher Gabe von Rifampicin (einem starken CYP3A-Induktor) zu einer Abnahme der Cmax von Upadacitinib um ca. 50 % und der AUC um ca. 60 %. Veränderungen der Krankheitsaktivität von Patienten sollten überwacht werden, wenn Upadacitinib gleichzeitig mit starken CYP3A4-Induktoren angewendet wird.
Methotrexat und den pH-Wert modifizierende Arzneimittel (z. B. Antacida oder Protonenpumpeninhibitoren) haben keinen Einfluss auf die Plasmaexposition von Upadacitinib.
Potenzielle Auswirkungen von Upadacitinib auf die Pharmakokinetik anderer Arzneimittel
Die mehrfache Anwendung von 30 mg oder 45 mg Upadacitinib einmal täglich bei gesunden Probanden hatte eine eingeschränkte Auswirkung auf die Plasmaexposition von Midazolam (sensitives CYP3A-Substrat) (Abnahme der AUC und Cmax von Midazolam um 24–26 %), was darauf hindeutet, dass 30 mg oder 45 mg Upadacitinib einmal täglich einen schwachen induzierenden Effekt auf CYP3A haben kann. In einer klinischen Studie führte die mehrfache Anwendung von 30 mg Upadacitinib einmal täglich bei gesunden Probanden zu einer Abnahme der AUC von Rosuvastatin um 33 % und von Atorvastatin um 23 % sowie einer Abnahme der Cmax von Rosuvastatin um 23 %. Upadacitinib hatte keine relevante Auswirkung auf die Cmax von Atorvastatin oder auf die Plasmaexposition von ortho-Hydroxy-Atorvastatin (hauptsächlicher aktiver Metabolit von Atorvastatin). Die mehrfache Anwendung von 45 mg Upadacitinib einmal täglich bei gesunden Probanden hatte eine begrenzte Zunahme der AUC und Cmax von Dextromethorphan (sensitives CYP2D6-Substrat) um 30 % bzw. 35 % zur Folge, was darauf hindeutet, dass 45 mg Upadacitinib einmal täglich einen schwachen inhibitorischen Effekt auf CYP2D6 hat. Bei gleichzeitiger Anwendung von Upadacitinib wird keine Dosisanpassung von CYP3A-Substraten, CYP2D6-Substraten, Rosuvastatin oder Atorvastatin empfohlen.
Upadacitinib hat keine relevanten Auswirkungen auf die Plasmaexposition von Ethinylestradiol, Levonorgestrel, Methotrexat oder Arzneimitteln, die als Substrate von CYP1A2, CYP2B6, CYP2C9 oder CYP2C19 metabolisiert werden.
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während der Behandlung und für 4 Wochen nach der letzten Dosis von Upadacitinib eine zuverlässige Verhütungsmethode anwenden. Mädchen und/oder ihre Eltern/betreuenden Personen sind zu informieren, dass sie sich mit dem behandelnden Arzt in Verbindung setzen müssen, sobald bei der Patientin unter der Behandlung mit Upadacitinib die Menarche stattfindet.
Schwangerschaft
Bisher liegen keine oder nur begrenzte Erfahrungen mit der Anwendung von Upadacitinib bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Upadacitinib war bei Ratten und Kaninchen teratogen und hatte bei Exposition in utero bei Rattenföten Auswirkungen auf die Knochen und bei Kaninchenföten Auswirkungen auf das Herz.
Upadacitinib ist während der Schwangerschaft kontraindiziert (siehe Abschnitt 4.3).
Falls eine Patientin während der Behandlung mit Upadacitinib schwanger wird, sollten die Eltern über das potenzielle Risiko für den Fötus informiert werden.
Stillzeit
Es ist nicht bekannt, ob Upadacitinib oder dessen Metaboliten in die menschliche Muttermilch übergehen. Die zur Verfügung stehenden pharmakodynamischen/toxikologischen Daten aus tierexperimentellen Studien haben gezeigt, dass Upadacitinib in die Milch übergeht (siehe Abschnitt 5.3).
Ein Risiko für das Neugeborene/Kind kann nicht ausgeschlossen werden.
Upadacitinib sollte während der Stillzeit nicht angewendet werden.
Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Upadacitinib verzichtet werden soll / die Behandlung mit Upadacitinib zu unterbrechen ist. Dabei soll sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau berücksichtigt werden.
Fertilität
Die Wirkung von Upadacitinib auf die Fertilität des Menschen wurde nicht untersucht. Tierexperimentelle Studien ergaben keine Hinweise auf Auswirkungen auf die Fertilität (siehe Abschnitt 5.3).
Upadacitinib kann einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen haben, weil ein Schwindelgefühl und Drehschwindel bei der Behandlung mit RINVOQ auftreten kann (siehe Abschnitt 4.8).
Zusammenfassung des Sicherheitsprofils
In den placebokontrollierten klinischen Studien zu rheumatoider Arthritis, Psoriasis-Arthritis und axialer Spondyloarthritis waren die am häufigsten berichteten Nebenwirkungen unter 15 mg Upadacitinib (≥ 2 % der Patienten in mindestens einer der Indikationen mit der höchsten Rate der vorgestellten Indikationen) Infektionen der oberen Atemwege (19,5 %), Erhöhungen der Kreatinphosphokinase (CPK) im Blut (8,6 %), erhöhte Alanintransaminase (4,3 %), Bronchitis (3,9 %), Übelkeit (3,5 %), Neutropenie (2,8 %), Husten (2,2 %), erhöhte Aspartattransaminase (2,2 %) und Hypercholesterinämie (2,2 %).
In den placebokontrollierten klinischen Studien zur atopischen Dermatitis waren die am häufigsten berichteten Nebenwirkungen (≥ 2 % der Patienten) unter 15 mg oder 30 mg Upadacitinib Infektionen der oberen Atemwege (25,4 %), Akne (15,1 %), Herpes simplex (8,4 %), Kopfschmerzen (6,3 %), eine Erhöhung der CPK im Blut (5,5 %), Husten (3,2 %), Follikulitis (3,2 %), Bauchschmerzen (2,9 %), Übelkeit (2,7 %), Neutropenie (2,3 %), Fieber (2,1 %) und Influenza (2,1 %).
In den placebokontrollierten klinischen Studien zur Einleitungs‑ und Erhaltungstherapie bei Colitis ulcerosa und Morbus Crohn waren die am häufigsten berichteten Nebenwirkungen (≥ 3 % der Patienten) unter 45 mg, 30 mg oder 15 mg Upadacitinib Infektionen der oberen Atemwege (19,9 %), Fieber (8,7 %), eine Erhöhung der CPK im Blut (7,6 %), Anämie (7,4 %), Kopfschmerzen (6,6 %), Akne (6,3 %), Herpes zoster (6,1 %), Neutropenie (6,0 %), Ausschlag (5,2 %), Pneumonie (4,1 %), Hypercholesterinämie (4,0 %), Bronchitis (3,9 %), erhöhte Aspartattransaminase (3,9 %), Fatigue (3,9 %), Follikulitis (3,6 %), erhöhte Alanintransaminase (3,5 %), Herpes simplex (3,2 %) und Influenza (3,2 %).
Die häufigsten schwerwiegenden Nebenwirkungen waren schwerwiegende Infektionen (siehe Abschnitt 4.4).
Das Sicherheitsprofil von Upadacitinib bei langfristiger Behandlung war bei allen Indikationen im Allgemeinen vergleichbar mit dem Sicherheitsprofil in der placebokontrollierten Phase.
Tabellarische Auflistung der Nebenwirkungen
Die folgende Auflistung der Nebenwirkungen basiert auf Erfahrungen aus klinischen Studien und Erfahrungen nach der Markteinführung. Die Häufigkeit der unten aufgeführten Nebenwirkungen ist wie folgt definiert:
sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000). Die Häufigkeiten in Tabelle 3 basieren auf der jeweils höheren Rate der Nebenwirkungen, die in klinischen Studien mit RINVOQ zu rheumatischen Erkrankungen (15 mg), zu atopischer Dermatitis (15 mg und 30 mg), zu Colitis ulcerosa (15 mg, 30 mg und 45 mg) oder zu Morbus Crohn (15 mg, 30 mg und 45 mg) berichtet wurden. Wenn zwischen den Indikationen deutliche Unterschiede in der Häufigkeit beobachtet wurden, sind diese in den Fußnoten unter der Tabelle aufgeführt.
Tabelle 3 Nebenwirkungen
Systemorganklasse |
Sehr häufig |
Häufig |
Gelegentlich |
Selten |
Infektionen und parasitäre Erkrankungen |
Infektionen der oberen Atemwegea |
Bronchitisa,b |
Orale Candidose |
|
Gutartige, bösartige und nicht spezifizierte Neubildungen (einschl. Zysten und Polypen) |
Nichtmelanozytärer Hautkrebsf |
|||
Erkrankungen des Blutes und des Lymphsystems |
Anämiea |
|||
Erkrankungen des Immunsystems |
Urtikariac,g |
Schwerwiegende Überempfindlichkeitsreaktionena,e |
||
Stoffwechsel- und Ernährungsstörungen |
Hypercholesterinämiea,b |
Hypertriglyzeridämie |
||
Erkrankungen des Nervensystems |
Kopfschmerzena,j |
|||
Erkrankungen des Ohrs und des Labyrinths |
Drehschwindela |
|||
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Husten |
|||
Erkrankungen des Gastrointestinaltrakts |
Bauchschmerzena |
Gastrointestinale Perforationi |
||
Erkrankungen der Haut und des Unterhautgewebes |
Aknea,c,d,g |
Ausschlaga |
||
Erkrankungen der Geschlechtsorgane und der Brustdrüse |
Samenverfärbungl |
|||
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fatigue |
|||
Untersuchungen |
CPK im Blut erhöht |
|||
a Dargestellt als gruppierter Begriff | ||||
Beschreibung ausgewählter Nebenwirkungen
Rheumatoide Arthritis
Infektionen
In placebokontrollierten klinischen Studien, in denen Upadacitinib mit csDMARDs kombiniert wurde, betrug die Infektionsrate über einen Zeitraum von 12/14 Wochen im Upadacitinib-15-mg-Behandlungsarm 27,4 % gegenüber 20,9 % im Placeboarm. In Methotrexat(MTX)-kontrollierten Studien betrug die Infektionsrate über einen Zeitraum von 12/14 Wochen im Behandlungsarm mit 15 mg Upadacitinib als Monotherapie 19,5 % gegenüber 24,0 % unter MTX. Insgesamt betrug die Langzeit-Infektionsrate unter 15 mg Upadacitinib in allen fünf klinischen Phase-III-Studien (2 630 Patienten) 93,7 Ereignisse pro 100 Patientenjahre.
In placebokontrollierten klinischen Studien, in denen Upadacitinib mit csDMARDs kombiniert wurde, betrug die Rate schwerwiegender Infektionen über einen Zeitraum von 12/14 Wochen im Upadacitinib-15-mg-Behandlungsarm 1,2 % gegenüber 0,6 % im Placeboarm. In MTX-kontrollierten Studien betrug die Rate schwerwiegender Infektionen über einen Zeitraum von 12/14 Wochen im Behandlungsarm mit 15 mg Upadacitinib als Monotherapie 0,6 % gegenüber 0,4 % unter MTX. Insgesamt betrug die Langzeitrate schwerwiegender Infektionen unter 15 mg Upadacitinib in allen fünf klinischen Phase-III-Studien 3,8 Ereignisse pro 100 Patientenjahre. Die häufigste schwerwiegende Infektion war Pneumonie. Die Rate an schwerwiegenden Infektionen blieb unter Langzeitanwendung stabil.
Opportunistische Infektionen (außer Tuberkulose)
In placebokontrollierten klinischen Studien, in denen Upadacitinib mit csDMARDs kombiniert wurde, betrug die Rate opportunistischer Infektionen über einen Zeitraum von 12/14 Wochen im Upadacitinib-15-mg-Behandlungsarm 0,5 % gegenüber 0,3 % im Placeboarm. In MTX-kontrollierten Studien gab es über einen Zeitraum von 12/14 Wochen unter 15 mg Upadacitinib als Monotherapie keine Fälle opportunistischer Infektionen; im MTX-Kontrollarm lag die Rate opportunistischer Infektionen bei 0,2 %. Insgesamt betrug die Langzeitrate opportunistischer Infektionen unter 15 mg Upadacitinib in allen fünf klinischen Phase-III-Studien 0,6 Ereignisse pro 100 Patientenjahre.
Die Langzeitrate von Herpes zoster unter 15 mg Upadacitinib betrug in allen fünf klinischen Phase‑III-Studien 3,7 Ereignisse pro 100 Patientenjahre. Die meisten Herpes-zoster-Ereignisse betrafen ein einzelnes Dermatom und waren nicht schwerwiegend.
Anstieg der Lebertransaminasen
In placebokontrollierten Studien, in denen Upadacitinib mit csDMARDs kombiniert wurde, wurden über einen Zeitraum von bis zu 12/14 Wochen bei 2,1 % bzw. 1,5 % der mit 15 mg Upadacitinib behandelten Patienten bei mindestens einer Bestimmung ein Anstieg der Alanintransaminase (ALT) und Aspartattransaminase (AST) von ≥ 3 x des oberen normalen Grenzwerts (upper limit of normal, ULN) beobachtet, verglichen mit 1,5 % bzw. 0,7 % bei den mit Placebo behandelten Patienten. Von den 22 Fällen eines Anstiegs der Lebertransaminasen waren die meisten asymptomatisch und vorübergehend.
In MTX‑kontrollierten Studien wurden über einen Zeitraum von bis zu 12/14 Wochen bei 0,8 % bzw. 0,4 % der mit 15 mg Upadacitinib behandelten Patienten bei mindestens einer Bestimmung ein ALT- und AST-Anstieg von ≥ 3 x des ULN beobachtet, verglichen mit 1,9 % bzw. 0,9 % bei den mit MTX behandelten Patienten.
Das Muster und die Inzidenz der ALT-/AST-Anstiege blieben im zeitlichen Verlauf stabil; dies gilt auch für die Langzeit-Fortsetzungsstudien.
Anstiege der Lipidwerte
Die Behandlung mit 15 mg Upadacitinib war mit einem Anstieg der Lipidwerte assoziiert, einschließlich eines Anstiegs des Gesamtcholesterins, der Triglyzeride, des LDL- und HDL-Cholesterins. Es gab keine Veränderung beim LDL/HDL-Quotienten. Der Anstieg wurde nach zwei bis vier Wochen Behandlung beobachtet und blieb unter Langzeittherapie stabil. Bei den Patienten in den kontrollierten Studien, deren Baselinewerte unterhalb der festgelegten Grenzen lagen, wurde in folgender Häufigkeit beobachtet, dass die Werte mindestens einmal innerhalb von 12/14 Wochen über den festgelegten Grenzen lagen (einschließlich Patienten mit einem einzelnen erhöhten Wert):
Gesamtcholesterin ≥ 5,17 mmol/l (200 mg/dl): 62 % unter 15 mg Upadacitinib vs. 31 % unter Placebo
LDL-Cholesterin ≥ 3,36 mmol/l (130 mg/dl): 42 % unter 15 mg Upadacitinib vs. 19 % unter Placebo
HDL-Cholesterin ≥ 1,03 mmol/l (40 mg/dl): 89 % unter 15 mg Upadacitinib vs. 61 % unter Placebo
Triglyzeride > 2,26 mmol/l (200 mg/dl): 25 % unter 15 mg Upadacitinib vs. 15 % unter Placebo
Kreatinphosphokinase
In placebokontrollierten Studien, in denen Upadacitinib mit csDMARDs kombiniert wurde, wurden über einen Zeitraum von bis zu 12/14 Wochen Anstiege der CPK-Werte beobachtet. CPK-Anstiege von > 5 x des ULN wurden über einen Zeitraum von 12/14 Wochen bei 1,0 % der Patienten im Upadacitinib-15-mg-Behandlungsarm und bei 0,3 % im Placeboarm beobachtet. Die meisten Anstiege von > 5 x des ULN waren vorübergehend und erforderten keinen Behandlungsabbruch. Die mittleren CPK‑Werte stiegen bis Woche 4 an, der mittlere Anstieg lag in Woche 12 bei 60 U/l und die Werte blieben danach, auch unter Langzeittherapie, auf dem höheren Wert stabil.
Neutropenie
In placebokontrollierten Studien, in denen Upadacitinib mit csDMARDs kombiniert wurde, kam es über einen Zeitraum von bis zu 12/14 Wochen bei 1,1 % der Patienten im Upadacitinib-15-mg-Behandlungsarm und bei < 0,1 % im Placeboarm bei mindestens einer Bestimmung zu einem Rückgang der Neutrophilenzahl unter 1 x 109 Zellen/l. In klinischen Studien wurde die Behandlung bei einer ANC von < 1 x 109 Zellen/l unterbrochen (siehe Abschnitt 4.2). Die mittlere Neutrophilenzahl nahm über 4 bis 8 Wochen ab. Der Rückgang der Neutrophilenzahl blieb im Zeitverlauf auch unter Langzeittherapie stabil auf einem niedrigeren Wert als bei Baseline.
Psoriasis-Arthritis
Insgesamt entsprach das Sicherheitsprofil bei Patienten mit aktiver Psoriasis-Arthritis unter 15 mg Upadacitinib dem Sicherheitsprofil bei Patienten mit rheumatoider Arthritis. Bei Patienten, die mit Upadacitinib in Kombination mit MTX behandelt wurden, wurde im Vergleich zu Patienten unter Monotherapie eine höhere Rate an schwerwiegenden Infektionen (2,6 Ereignisse pro 100 Patientenjahre bzw. 1,3 Ereignisse pro 100 Patientenjahre) und an erhöhten Lebertransaminasewerten (ALT-Anstieg Grad 3 und höher bei 1,4 % bzw. 0,4 %) beobachtet.
Axiale Spondyloarthritis
Insgesamt entsprach das Sicherheitsprofil bei Patienten mit aktiver axialer Spondyloarthritis unter 15 mg Upadacitinib dem Sicherheitsprofil bei Patienten mit rheumatoider Arthritis. Es wurden keine neuen Ergebnisse zur Sicherheit beobachtet.
Riesenzellarteriitis
Insgesamt entsprach das Sicherheitsprofil bei Patienten mit Riesenzellarteriitis unter 15 mg Upadacitinib dem allgemein bekannten Sicherheitsprofil von Upadacitinib.
Schwerwiegende Infektionen
In der placebokontrollierten klinischen Studie betrug die Häufigkeit schwerwiegender Infektionen über einen Zeitraum von 52 Wochen in dem Behandlungsarm mit 15 mg Upadacitinib 5,7 % gegenüber 10,7 % unter Placebo. Die Langzeitrate von schwerwiegenden Infektionen betrug 5,9 Ereignisse pro 100 Patientenjahre im Behandlungsarm mit 15 mg Upadacitinib und 10,5 Ereignisse pro 100 Patientenjahre im Placeboarm.
Opportunistische Infektionen (außer Tuberkulose)
In der placebokontrollierten klinischen Studie betrug die Häufigkeit der opportunistischen Infektionen (außer Tuberkulose und Herpes zoster) über einen Zeitraum von 52 Wochen 1,9 % im Behandlungsarm mit 15 mg Upadacitinib gegenüber 0,9 % unter Placebo. Die Langzeitrate der opportunistischen Infektionen (außer Tuberkulose und Herpes zoster) betrug 1,8 Ereignisse pro 100 Patientenjahre im Behandlungsarm mit 15 mg Upadacitinib und 1,5 Ereignisse pro 100 Patientenjahre im Placeboarm.
In der placebokontrollierten klinischen Studie betrug die Häufigkeit von Herpes zoster über einen Zeitraum von 52 Wochen 5,3 % im Behandlungsarm mit 15 mg Upadacitinib gegenüber 2,7 % unter Placebo. Die Langzeitrate von Herpes zoster betrug 5,9 Ereignisse pro 100 Patientenjahre im Behandlungsarm mit 15 mg Upadacitinib und 3,0 Ereignisse pro 100 Patientenjahre im Placeboarm.
Atopische Dermatitis
Infektionen
In der placebokontrollierten Phase der klinischen Studien betrug die Infektionsrate über einen Zeitraum von 16 Wochen in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 39 % bzw. 43 % gegenüber 30 % unter Placebo. Die Langzeitrate von Infektionen betrug in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 98,5 bzw. 109,6 Ereignisse pro 100 Patientenjahre.
In placebokontrollierten klinischen Studien betrug die Häufigkeit schwerwiegender Infektionen über einen Zeitraum von 16 Wochen in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 0,8 % bzw. 0,4 % gegenüber 0,6 % unter Placebo. Die Langzeitrate schwerwiegender Infektionen betrug in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 2,3 bzw. 2,8 Ereignisse pro 100 Patientenjahre.
Opportunistische Infektionen (außer Tuberkulose)
In der placebokontrollierten Phase der klinischen Studien handelte es sich bei allen opportunistischen Infektionen (mit Ausnahme von TB und Herpes zoster) um ein Ekzema herpeticatum. Die Häufigkeit von Ekzema herpeticatum über 16 Wochen betrug in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 0,7 % bzw. 0,8 % gegenüber 0,4 % unter Placebo. Die Langzeitrate von Ekzema herpeticatum betrug in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 1,6 bzw. 1,8 Ereignisse pro 100 Patientenjahre. Unter 30 mg Upadacitinib wurde ein Fall einer ösophagealen Candidiasis berichtet.
Die Langzeitrate von Herpes zoster betrug in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 3,5 bzw. 5,2 Ereignisse pro 100 Patientenjahre. Die meisten Herpes-zoster-Ereignisse betrafen ein einzelnes Dermatom und waren nicht schwerwiegend.
Auffällige Laborwerte
Die dosisabhängigen Erhöhungen der ALT‑ und/oder AST‑Werte (≥ 3 x ULN), Veränderungen der Lipidparameter, der CPK‑Werte (> 5 x ULN) und der Neutropenie (ANC < 1 x 109 Zellen/l) im Zusammenhang mit der Upadacitinib-Behandlung waren vergleichbar mit den Beobachtungen in den klinischen Studien zu rheumatischen Erkrankungen.
In Studien zu atopischer Dermatitis wurde nach Woche 16 ein geringer Anstieg des LDL‑Cholesterins beobachtet. Der Anstieg (Mittelwert) des LDL-Cholesterins im Vergleich zur Baseline war zu Woche 52 0,41 mmol/l für 15 mg Upadacitinib und 0,56 mmol/l für 30 mg Upadacitinib.
Colitis ulcerosa
Das allgemeine Sicherheitsprofil bei Patienten mit Colitis ulcerosa entsprach im Allgemeinen dem Sicherheitsprofil von Patienten mit rheumatoider Arthritis.
Im Vergleich zur 8-wöchigen Einleitungsphase wurde bei einer 16-wöchigen Induktionsphase eine höhere Rate an Herpes zoster beobachtet.
Infektionen
In den placebokontrollierten Einleitungsstudien betrug die Infektionsrate über einen Zeitraum von 8 Wochen im Behandlungsarm mit 45 mg Upadacitinib 20,7 % gegenüber 17,5 % unter Placebo. In der placebokontrollierten Erhaltungsstudie betrug die Infektionsrate über einen Zeitraum von 52 Wochen in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 40,4 % bzw. 44,2 % gegenüber 38,8 % unter Placebo. Die Langzeitrate von Infektionen betrug unter 15 mg und 30 mg Upadacitinib 64,5 bzw. 77,8 Ereignisse pro 100 Patientenjahre.
In den placebokontrollierten Einleitungsstudien betrug die Häufigkeit schwerwiegender Infektionen über einen Zeitraum von 8 Wochen sowohl im Behandlungsarm mit 45 mg Upadacitinib als auch unter Placebo 1,3 %. In einer um 8 Wochen verlängerten Behandlung mit 45 mg Upadacitinib wurden keine weiteren schwerwiegenden Infektionen beobachtet. In der placebokontrollierten Erhaltungsstudie betrug die Häufigkeit schwerwiegender Infektionen über einen Zeitraum von 52 Wochen in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 3,6 % bzw. 3,2 % gegenüber 3,3 % unter Placebo. Die Langzeitrate schwerwiegender Infektionen betrug in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 3,0 bzw. 4,6 Ereignisse pro 100 Patientenjahre. Die am häufigsten berichtete schwerwiegende Infektion in den Einleitungs‑ und Erhaltungsphasen war eine COVID‑19-Pneumonie.
Opportunistische Infektionen (außer Tuberkulose)
In den placebokontrollierten Einleitungsstudien über einen Zeitraum von 8 Wochen betrug die Häufigkeit der opportunistischen Infektionen (außer Tuberkulose und Herpes zoster) im Behandlungsarm mit 45 mg Upadacitinib 0,4 % gegenüber 0,3 % unter Placebo. In einer um 8 Wochen verlängerten Behandlung mit 45 mg Upadacitinib wurden keine weiteren opportunistischen Infektionen (außer Tuberkulose und Herpes zoster) beobachtet. In der placebokontrollierten Erhaltungsstudie über einen Zeitraum von 52 Wochen betrug die Häufigkeit opportunistischer Infektionen (außer Tuberkulose und Herpes zoster) in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 0,8 % bzw. 0,8 % gegenüber 0,8 % unter Placebo. Die Langzeitrate der opportunistischen Infektionen (außer Tuberkulose und Herpes zoster) betrug in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 0,3 bzw. 0,6 Ereignisse pro 100 Patientenjahre.
In den placebokontrollierten Einleitungsstudien über einen Zeitraum von 8 Wochen betrug die Häufigkeit von Herpes zoster im Behandlungsarm mit 45 mg Upadacitinib 0,6 % gegenüber 0 % unter Placebo. Die Rate von Herpes zoster betrug über einen 16‑wöchigen Behandlungszeitraum mit 45 mg Upadacitinib 3,9 %. In der placebokontrollierten Erhaltungsstudie über einen Zeitraum von 52 Wochen betrug die Häufigkeit von Herpes zoster in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 4,8 % bzw. 5,6 % gegenüber 0 % unter Placebo. Die Langzeitrate von Herpes zoster betrug in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 4,5 bzw. 7,2 Ereignisse pro 100 Patientenjahre.
Gastrointestinale Perforationen
In der placebokontrollierten Erhaltungsphase wurde eine gastrointestinale Perforation bei 1 Patienten, der mit Placebo (1,5 pro 100 Patientenjahre) und bei keinen Patienten, die mit 15 mg oder 30 mg Upadacitinib behandelt wurden, berichtet. In der Langzeit-Fortsetzungsstudie berichteten 1 Patient, der mit 15 mg Upadacitinib (0,1 pro 100 Patientenjahre) behandelt wurde, und 1 Patient, der mit 30 mg Upadacitinib (< 0,1 pro 100 Patientenjahre) behandelt wurde, Ereignisse.
Auffällige Laborwerte
In den klinischen Einleitungs‑ und Erhaltungsstudien waren Laborwertveränderungen in Form einer Erhöhung der ALT‑ und/oder AST‑Werte (≥ 3 x ULN), Veränderungen der CPK‑Werte (> 5 x ULN) und Neutropenie (ANC < 1 x 109 Zellen/l) im Zusammenhang mit der Upadacitinib-Behandlung generell vergleichbar mit den Beobachtungen in den klinischen Studien zu rheumatischen Erkrankungen und atopischer Dermatitis. Dosisabhängige Veränderungen dieser Laborparameter im Zusammenhang mit einer Behandlung mit 15 mg und 30 mg Upadacitinib wurden beobachtet.
In den placebokontrollierten Einleitungsstudien über einen Zeitraum von bis zu 8 Wochen kam es bei 2,0 % der Patienten im Behandlungsarm mit 45 mg Upadacitinib und bei 0,8 % unter Placebo bei mindestens einer Messung zu einem Rückgang der Lymphozytenzahl unter 0,5 x 109 Zellen/l. In der placebokontrollierten Erhaltungsstudie über einen Zeitraum von bis zu 52 Wochen kam es bei 1,6 % bzw. 1,2 % der Patienten im Behandlungsarm mit 15 mg bzw. 30 mg Upadacitinib und bei 0,8 % unter Placebo bei mindestens einer Messung zu einem Rückgang der Lymphozytenzahl unter 0,5 x 109 Zellen/l. In klinischen Studien wurde die Behandlung bei einer ALC < 0,5 x 109 Zellen/l unterbrochen (siehe Abschnitt 4.2). Während der Behandlung mit Upadacitinib wurden keine nennenswerten mittleren Veränderungen der Lymphozytenzahl beobachtet.
Nach 8 Wochen unter 45 mg Upadacitinib wurden Anstiege der Lipidwerte beobachtet, die unter der Langzeittherapie mit 15 mg und 30 mg Upadacitinib im Allgemeinen stabil blieben. Bei den Patienten in den placebokontrollierten Einleitungsstudien, deren Baselinewerte unterhalb der festgelegten Grenzen lagen, wurde in folgender Häufigkeit beobachtet, dass die Werte mindestens einmal innerhalb von 8 Wochen über den festgelegten Grenzen lagen (einschließlich Patienten mit einem einzelnen erhöhten Wert):
Gesamtcholesterin ≥ 5,17 mmol/l (200 mg/dl): 49 % unter 45 mg Upadacitinib vs. 11 % unter Placebo
LDL-Cholesterin ≥ 3,36 mmol/l (130 mg/dl): 27 % unter 45 mg Upadacitinib vs. 9 % unter Placebo
HDL-Cholesterin ≥ 1,03 mmol/l (40 mg/dl): 79 % unter 45 mg Upadacitinib vs. 36 % unter Placebo
Triglyzeride ≥ 2,26 mmol/l (200 mg/dl): 6 % unter 45 mg Upadacitinib vs. 4 % unter Placebo
Morbus Crohn
Insgesamt entsprach das Sicherheitsprofil bei Patienten mit Morbus Crohn unter Upadacitinib dem bekannten Sicherheitsprofil von Upadacitinib.
Schwerwiegende Infektionen
In den placebokontrollierten Einleitungsstudien betrug die Häufigkeit schwerwiegender Infektionen über einen Zeitraum von 12 Wochen im Behandlungsarm mit 45 mg Upadacitinib 1,9 % gegenüber 1,7 % unter Placebo. In der placebokontrollierten Erhaltungsstudie betrug die Häufigkeit schwerwiegender Infektionen über einen Zeitraum von 52 Wochen in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib 3,2 % bzw. 5,7 % gegenüber 4,5 % unter Placebo. Die Langzeitrate schwerwiegender Infektionen betrug in den Behandlungsarmen mit 15 mg und 30 mg Upadacitinib bei Patienten, die auf 45 mg Upadacitinib als Einleitungstherapie ansprachen, 5,1 bzw. 7,3 Ereignisse pro 100 Patientenjahre. Die am häufigsten berichtete schwerwiegende Infektion in den Einleitungs‑ und Erhaltungsstudien waren gastrointestinale Infektionen.
Gastrointestinale Perforationen
Während des placebokontrollierten Zeitraums der klinischen Phase-III-Einleitungsstudien über 12 Wochen wurde eine gastrointestinale Perforation bei 1 Patienten (0,1 %), der mit 45 mg Upadacitinib behandelt wurde, und bei keinem Patienten, der Placebo erhielt, berichtet. Unter allen Patienten, die während der Einleitungsstudien mit 45 mg Upadacitinib (n = 938) behandelt wurden, wurde bei 4 Patienten (0,4 %) eine gastrointestinale Perforation berichtet.
In der placebokontrollierten Langzeit-Fortsetzungsstudie wurde eine gastrointestinale Perforation bei jeweils 1 Patienten, der mit Placebo (0,7 Ereignisse pro 100 Patientenjahre), 15 mg Upadacitinib (0,4 Ereignisse pro 100 Patientenjahre) und 30 mg Upadacitinib (0,4 Ereignisse pro 100 Patientenjahre) behandelt wurde, berichtet. Unter allen Patienten, die eine Notfallbehandlung mit 30 mg Upadacitinib (n = 336) erhielten, wurde während der Langzeitbehandlung bei 3 Patienten (0,8 Ereignisse pro 100 Patientenjahre) eine gastrointestinale Perforation berichtet.
Auffällige Laborwerte
In den Einleitungs‑ und Erhaltungsstudien waren Laborwertveränderungen in Form einer Erhöhung der ALT‑ und/oder AST‑Werte (≥ 3 x ULN), Veränderungen der CPK‑Werte (> 5 x ULN), der Neutropenie (ANC < 1 x 109 Zellen/l) und der Lipidparameter im Zusammenhang mit der Behandlung mit Upadacitinib generell vergleichbar mit den Beobachtungen in den klinischen Studien zu rheumatischen Erkrankungen, atopischer Dermatitis und Colitis ulcerosa. Dosisabhängige Veränderungen dieser Laborparameter im Zusammenhang mit einer Behandlung mit 15 mg und 30 mg Upadacitinib wurden beobachtet.
In den placebokontrollierten Einleitungsstudien über einen Zeitraum von bis zu 12 Wochen kam es bei 2,2 % der Patienten im Behandlungsarm mit 45 mg Upadacitinib und bei 2,0 % unter Placebo bei mindestens einer Messung zu einem Rückgang der Lymphozytenzahl unter 0,5 x 109 Zellen/l. In der placebokontrollierten Erhaltungsstudie über einen Zeitraum von bis zu 52 Wochen kam es bei 4,6 % bzw. 5,2 % der Patienten im Behandlungsarm mit 15 mg bzw. 30 mg Upadacitinib und bei 1,8 % unter Placebo bei mindestens einer Messung zu einem Rückgang der Lymphozytenzahl unter 0,5 x 109 Zellen/l. In klinischen Studien wurde die Behandlung bei einer ALC < 0,5 x 109 Zellen/l unterbrochen (siehe Abschnitt 4.2). Während der Behandlung mit Upadacitinib wurden keine nennenswerten mittleren Veränderungen der Lymphozytenzahl beobachtet.
In den placebokontrollierten Einleitungsstudien über einen Zeitraum von bis zu 12 Wochen kam es bei 2,7 % der Patienten im Behandlungsarm mit 45 mg Upadacitinib und bei 1,4 % unter Placebo bei mindestens einer Messung zu einem Rückgang der Hämoglobinkonzentration unter 8 g/dl. In der placebokontrollierten Erhaltungsstudie über einen Zeitraum von bis zu 52 Wochen kam es bei 1,4 % bzw. 4,4 % der Patienten im Behandlungsarm mit 15 mg bzw. 30 mg Upadacitinib und bei 2,8 % unter Placebo bei mindestens einer Messung zu einem Rückgang der Hämoglobinkonzentration unter 8 g/dl. In klinischen Studien wurde die Behandlung bei einer Hämoglobinkonzentration von < 8 g/dl unterbrochen (siehe Abschnitt 4.2). Während der Behandlung mit Upadacitinib wurden keine nennenswerten mittleren Veränderungen der Hämoglobinkonzentration beobachtet.
Ältere Patienten
Basierend auf den begrenzten Daten bei Patienten im Alter ab 65 Jahren mit atopischer Dermatitis, Colitis ulcerosa und Morbus Crohn war die Rate der Nebenwirkungen insgesamt unter der 30‑mg-Dosis von Upadacitinib höher als unter der 15‑mg-Dosis (siehe Abschnitt 4.4).
Kinder und Jugendliche
In den globalen Phase‑III-Studien (n = 343) und den ergänzenden Teilstudien zu Jugendlichen (n = 198) wurden insgesamt 541 Jugendliche im Alter von 12 bis 17 Jahren mit atopischer Dermatitis behandelt; davon erhielten 264 die 15‑mg-Dosis und 265 erhielten die 30-mg-Dosis. Das Sicherheitsprofil von 15 mg und 30 mg Upadacitinib bei Jugendlichen war vergleichbar mit dem bei Erwachsenen. Bei Langzeitanwendung von 15 mg bzw. 30 mg Upadacitinib wurde Hautpapillom als Nebenwirkung bei 3,4 % bzw. 6,8 % der jugendlichen Patienten mit atopischer Dermatitis beobachtet.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem anzuzeigen:
Deutschland
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: http://www.bfarm.de
Upadacitinib wurde in klinischen Studien in Dosierungen eingesetzt, die in Bezug auf die AUC Dosis äquivalent zu einer Dosierung von bis zu 60 mg einmal täglich als Retardtablette sind. Die Nebenwirkungen waren vergleichbar mit solchen bei niedrigerer Dosierung, und es wurden keine spezifischen Toxizitäten erkannt. Etwa 90 % von Upadacitinib im systemischen Kreislauf werden innerhalb von 24 Stunden nach Anwendung ausgeschieden (innerhalb des in klinischen Studien untersuchten Dosisbereichs). Im Falle einer Überdosierung wird empfohlen, Patienten auf Anzeichen und Symptome von Nebenwirkungen zu überwachen. Bei Patienten, bei denen es zu Nebenwirkungen kommt, muss eine adäquate Behandlung eingeleitet werden.
Pharmakotherapeutische Gruppe: Immunsuppressiva, Janus-assoziierte Kinase(JAK)-Inhibitoren, ATC-Code: L04AF03
Wirkmechanismus
Upadacitinib ist ein selektiver und reversibler Januskinase(JAK)-Inhibitor. JAKs sind intrazelluläre Enzyme, die Signale von Zytokinen und Wachstumsfaktoren weiterleiten, die an einer Vielzahl von zellulären Prozessen, wie Entzündungsreaktionen, Hämatopoese und Immunüberwachung, beteiligt sind. Die JAK-Enzymfamilie umfasst vier Mitglieder – JAK1, JAK2, JAK3 und TYK2 –, die paarweise Signaltransduktoren und Aktivatoren der Transkription (signal transducers and activators of transcription, STATs) phosphorylieren und dadurch aktivieren. Diese Phosphorylierung moduliert wiederum die Genexpression und Zellfunktion. JAK1 ist für Signalwege von inflammatorischen Zytokinen von Bedeutung, während JAK2 für die Reifung von Erythrozyten wichtig ist und JAK3-Signale eine Rolle im Rahmen der Immunüberwachung und Lymphozytenfunktion spielen.
In humanzellbasierten Assays inhibiert Upadacitinib bevorzugt JAK1- oder JAK1/3-Signalwege im Vergleich zu anderen Zytokin-Signalwegen, die über JAK2-Paare vermittelt werden. Atopische Dermatitis wird durch proinflammatorische Zytokine (darunter IL‑4, IL‑13, IL‑22, TSLP, IL‑31 und IFN‑γ) gesteuert, die Signale über den JAK1‑Signalweg übertragen. Die Hemmung von JAK1 durch Upadacitinib verringert die Signalübertragung vieler Mediatoren, die die Anzeichen und Symptome der atopischen Dermatitis, beispielsweise ekzematöse Hautläsionen und Pruritus, auslösen.
Proinflammatorische Zytokine (hauptsächlich IL‑6, IL‑7, IL‑15 und IFN-γ) übertragen Signale über den JAK1‑Signalweg und sind an der Pathologie chronisch-entzündlicher Darmerkrankungen beteiligt. Die JAK1‑Hemmung durch Upadacitinib moduliert die Signalgebung der JAK‑abhängigen Zytokine, die der Entzündungslast und den Anzeichen und Symptomen von chronisch-entzündlichen Darmerkrankungen zugrunde liegen.
Pharmakodynamische Wirkungen
Hemmung der durch IL-6 induzierten STAT3‑Phosphorylierung und durch IL-7 induzierten STAT5-Phosphorylierung
Bei gesunden Probanden führte die Anwendung von Upadacitinib (Formulierung mit sofortiger Freisetzung) zu einer dosis- und konzentrationsabhängigen Hemmung der durch IL-6 (JAK1/JAK2) induzierten STAT3-Phosphorylierung und der durch IL-7 (JAK1/JAK3) induzierten STAT5-Phosphorylierung im Vollblut. Die maximale Hemmung wurde 1 Stunde nach Anwendung beobachtet und fiel bis zum Ende des Anwendungsintervalls wieder nahezu auf Baselinewerte ab.
Lymphozyten
Bei Patienten mit rheumatoider Arthritis ging die Behandlung mit Upadacitinib mit einem geringen, vorübergehenden Anstieg der mittleren ALC-Werte von Baseline bis zu Woche 36 einher, der unter weiterer Behandlung allmählich auf Baseline- oder nahezu Baselinewerte zurückging.
hsCRP
Bei Patienten mit rheumatoider Arthritis war die Behandlung mit Upadacitinib bereits nach Woche 1 mit einer Abnahme des mittleren hsCRP-Spiegels gegenüber Baseline verbunden und diese Abnahme hielt unter weiterer Behandlung an.
Impfstoffstudien
Der Einfluss von Upadacitinib auf die humorale Antwort nach Verabreichung eines adjuvantierten rekombinanten Herpes-zoster-Glykoprotein-E-Impfstoffs wurde bei 93 Patienten mit rheumatoider Arthritis unter stabiler Behandlung mit 15 mg Upadacitinib untersucht. 98 % der Patienten erhielten gleichzeitig Methotrexat. 49 % der Patienten erhielten orale Kortikosteroide bei Baseline. Der primäre Endpunkt war der Anteil der Patienten mit zufriedenstellendem humoralen Ansprechen, definiert als ≥ 4‑facher Anstieg der Konzentration des Anti-Glykoprotein-E-Titers vor der Impfung in Woche 16 (4 Wochen nach der zweiten Impfung). Die Impfung der mit 15 mg Upadacitinib behandelten Patienten führte bei 79 von 90 (88 % [95 %-CI: 81,0; 94,5]) der Patienten in Woche 16 zu einem zufriedenstellenden humoralen Ansprechen.
Der Einfluss von Upadacitinib auf die humorale Antwort nach Verabreichung eines inaktivierten Pneumokokkenpolysaccharid-Konjugatimpfstoffs (13-valent, adsorbiert) wurde bei 111 Patienten mit rheumatoider Arthritis unter stabiler Behandlung mit 15 mg Upadacitinib (n = 87) oder 30 mg (n = 24) untersucht. 97 % der Patienten (n = 108) erhielten gleichzeitig Methotrexat. Primärer Endpunkt war der Anteil der Patienten mit zufriedenstellendem humoralen Ansprechen, definiert als ≥ 2‑facher Anstieg der Antikörperkonzentration gegenüber Baseline bis Woche 4 bei mindestens 6 der 12 Pneumokokken-Antigene (1, 3, 4, 5, 6B, 7F, 9V, 14, 18C, 19A, 19F und 23F). Die Ergebnisse in Woche 4 zeigten bei 67,5 % (95 %-CI: 57,4; 77,5) bzw. 56,5 % (95 %-CI: 36,3; 76,8) der mit 15 mg bzw. 30 mg Upadacitinib behandelten Patienten ein zufriedenstellendes humorales Ansprechen.
Klinische Wirksamkeit und Sicherheit
Rheumatoide Arthritis
Die Wirksamkeit und Sicherheit von 15 mg Upadacitinib einmal täglich wurden in fünf randomisierten, doppelblinden, multizentrischen Phase-III-Studien bei Patienten mit mittelschwerer bis schwerer aktiver rheumatoider Arthritis, die die ACR/EULAR-Klassifikationskriterien von 2010 erfüllten, untersucht (siehe Tabelle 4). Patienten, die 18 Jahre und älter waren, konnten an den Studien teilnehmen. Das Vorliegen von mindestens 6 druckschmerzhaften und 6 geschwollenen Gelenken sowie der Nachweis einer systemischen Entzündung auf Basis der hsCRP-Erhöhung waren bei Baseline erforderlich. Vier Studien beinhalteten Langzeit-Fortsetzungsphasen von bis zu 5 Jahren und eine Studie (SELECT-COMPARE) beinhaltete eine Langzeit-Fortsetzungsphase von bis zu 10 Jahren.
Die primäre Analyse all dieser Studien umfasste alle randomisierten Studienteilnehmer, die mindestens eine Dosis von Upadacitinib oder Placebo erhielten. Für kategoriale Endpunkte wurde die Non-Responder-Imputation angewendet.
In allen Phase-III-Studien war die Wirksamkeit, die mit 15 mg Upadacitinib einmal täglich beobachtet wurde, im Allgemeinen ähnlich der, die mit 30 mg Upadacitinib einmal täglich beobachtet wurde.
Tabelle 4 Zusammenfassung der klinischen Studien
Studientitel |
Population (n) |
Behandlungsarme |
Wichtige Endpunkte |
SELECT-EARLY |
MTX-naiva |
Monotherapie |
|
SELECT-MONOTHERAPY |
MTX-IRb |
Monotherapie |
|
SELECT-NEXT |
csDMARD-IRc |
In Kombination mit csDMARDs |
|
SELECT-COMPARE |
MTX-IRd |
In Kombination mit MTX |
|
SELECT-BEYOND |
bDMARD‑IRe |
In Kombination mit csDMARDs |
|
Abkürzungen: ACR20 (oder 50) = Verbesserung um ≥ 20 % (oder ≥ 50 %) gemäß American College of Rheumatology; bDMARD = biologisches DMARD; CRP = C-reaktives Protein; DAS28 = Disease Activity Score 28 Gelenke; mTSS = modifizierter Total-Sharp-Score; csDMARD = konventionelles synthetisches DMARD; HAQ-DI = Health Assessment Questionnaire-Disability Index; SF-36 PCS = körperliche Domäne des short‑form‑36-Gesundheitsfragebogens; CDAI = Clinical Disease Activity Index; FACIT-F = Functional Assessment of Chronic Illness Therapy-Fatigue Score; IR = inadequate responder (Patient mit unzureichendem Ansprechen); MTX = Methotrexat; n = Anzahl randomisierter Patienten | |||
Klinisches Ansprechen
Remission und niedrige Krankheitsaktivität
In den Studien erreichte ein signifikant größerer Anteil an Patienten unter 15 mg Upadacitinib im Vergleich zu Placebo, MTX oder Adalimumab eine niedrige Krankheitsaktivität (DAS28-CRP ≤ 3,2) und eine klinische Remission (DAS28-CRP < 2,6) (Tabelle 5). Im Vergleich zu Adalimumab wurde in der Studie SELECT-COMPARE mit Upadacitinib nach 12 Wochen signifikant häufiger niedrige Krankheitsaktivität erreicht. Insgesamt wurden niedrige Krankheitsaktivität und klinische Remission über alle Patientenpopulationen hinweg mit und ohne MTX vergleichbar oft erreicht. Nach drei Jahren blieben 297 von 651 ursprünglich mit 15 mg Upadacitinib randomisierten Patienten (45,6 %) bzw. 111 von 327 ursprünglich mit Adalimumab randomisierten Patienten (33,9 %) in der Studie SELECT-COMPARE und 216 von 317 ursprünglich mit 15 mg Upadacitinib randomisierten Patienten (68,1 %) bzw. 149 von 315 ursprünglich mit MTX-Monotherapie randomisierten Patienten (47,3 %) in der Studie SELECT-EARLY. Bei den Patienten, die ihre ursprünglich zugewiesene Behandlung beibehielten, blieben die niedrige Krankheitsaktivität und die klinische Remission über 3 Jahre erhalten.
ACR-Ansprechen
In allen Studien erzielten mehr Patienten, die mit 15 mg Upadacitinib behandelt wurden, nach 12 Wochen ein ACR20-, ACR50- und ACR70-Ansprechen als Patienten, die mit Placebo, MTX oder Adalimumab behandelt wurden (Tabelle 5). Bei allen Parametern war die Dauer bis zum Eintreten der Wirkung kurz, wobei höhere ACR20-Ansprechraten bereits nach 1 Woche verzeichnet wurden. Es wurde ein anhaltendes Ansprechen beobachtet (mit und ohne MTX) und die ACR20/50/70-Ansprechraten konnten bei den Patienten, die ihre ursprünglich zugewiesene Behandlung beibehielten, bis zu 3 Jahre aufrechterhalten werden.
Die Behandlung mit 15 mg Upadacitinib als Monotherapie oder in Kombination mit csDMARDs, führte zu einer Verbesserung bei allen ACR-Komponenten, einschließlich der Anzahl druckschmerzhafter und geschwollener Gelenke, der allgemeinen Beurteilung der Krankheitsaktivität durch den Arzt (PhGA) und den Patienten (PGA), des HAQ-DI, der Schmerzen und des hsCRP.
Tabelle 5 Ansprechen und Remission
Studie |
SELECT-EARLY |
SELECT-MONO |
SELECT-NEXT |
SELECT-COMPARE |
SELECT-BEYOND |
||||||||
MTX |
UPA |
MTX |
UPA |
PBO |
UPA |
PBO |
UPA |
ADA |
PBO |
UPA |
|||
N |
314 |
317 |
216 |
217 |
221 |
221 |
651 |
651 |
327 |
169 |
164 |
||
Woche |
|||||||||||||
LDA DAS28-CRP ≤ 3,2 (% der Patienten) | |||||||||||||
12a/14b |
28 |
53g |
19 |
45e |
17 |
48e |
14 |
45e,h |
29 |
14 |
43e |
||
24c/26d |
32 |
60f |
18 |
55g,h |
39 |
||||||||
48 |
39 |
59g |
50h |
35 |
|||||||||
CR DAS28-CRP < 2,6 (% der Patienten) | |||||||||||||
12a/14b |
14 |
36g |
8 |
28e |
10 |
31e |
6 |
29e,h |
18 |
9 |
29g |
||
24c/26d |
18 |
48e |
9 |
41g,h |
27 |
||||||||
48 |
29 |
49g |
38i |
28 |
|||||||||
ACR20 (% der Patienten) | |||||||||||||
12a/14b |
54 |
76g |
41 |
68e |
36 |
64e |
36 |
71e,j |
63 |
28 |
65e |
||
24c/26d |
59 |
79g |
36 |
67g,i |
57 |
||||||||
48 |
57 |
74g |
65i |
54 |
|||||||||
ACR50 (% der Patienten) | |||||||||||||
12a/14b |
28 |
52g |
15 |
42g |
15 |
38g |
15 |
45g,h |
29 |
12 |
34g |
||
24c/26d |
33 |
60e |
21 |
54g,h |
42 |
||||||||
48 |
43 |
63g |
49i |
40 |
|||||||||
ACR70 (% der Patienten) | |||||||||||||
12a/14b |
14 |
32g |
3 |
23g |
6 |
21g |
5 |
25g,h |
13 |
7 |
12 |
||
24c/26d |
18 |
44g |
10 |
35g,h |
23 |
||||||||
48 |
29 |
51g |
36h |
23 |
|||||||||
CDAI ≤ 10 (% der Patienten) | |||||||||||||
12a/14b |
30 |
46g |
25 |
35l |
19 |
40e |
16 |
40e,h |
30 |
14 |
32g |
||
24c/26d |
38 |
56g |
22 |
53g,h |
38 |
||||||||
48 |
43 |
60g |
47h |
34 |
|||||||||
Abkürzungen: ACR20 (bzw. 50/70) = Verbesserung um ≥ 20 % (bzw. ≥ 50/70 %) gemäß American College of Rheumatology; ADA = Adalimumab; CDAI = Clinical Disease Activity Index; CR = clinical remission (klinische Remission); CRP = C-reaktives Protein; DAS28 = Disease Activity Score 28 Gelenke; IR = inadequate responder (Patient mit unzureichendem Ansprechen); LDA = low disease activity (niedrige Krankheitsaktivität); MTX = Methotrexat; PBO = Placebo; UPA = Upadacitinib | |||||||||||||
Radiologisches Ansprechen
Die Hemmung der radiologischen Progression wurde anhand des modifizierten Total-Sharp-Scores (mTSS) und seiner Komponenten, dem Erosionsscore und dem Score für Gelenkspaltverengung in Woche 24/26 sowie in Woche 48 in den Studien SELECT-EARLY und SELECT-COMPARE untersucht.
Die Behandlung mit 15 mg Upadacitinib in Kombination mit MTX führte im Vergleich zu Placebo in der Studie SELECT-COMPARE und als Monotherapie im Vergleich zu MTX in der Studie SELECT-EARLY zu einer signifikant größeren Hemmung der radiologischen Progression (Tabelle 6). Die Daten vom Erosionsscore und vom Score für Gelenkspaltverengung waren konsistent mit den Gesamtscores. Der Anteil der Patienten ohne radiologische Progression (mTSS-Veränderung ≤ 0) war in beiden Studien unter 15 mg Upadacitinib signifikant höher. Die Hemmung der radiologischen Progression blieb in beiden Studien bei den Patienten, die ihre ursprünglich zugewiesene Behandlung mit 15 mg Upadacitinib beibehielten, bis zur Woche 96 erhalten (basierend auf den verfügbaren Ergebnissen von 327 Patienten in der Studie SELECT-COMPARE und 238 Patienten in der Studie SELECT-EARLY).
Tabelle 6 Radiologische Veränderungen
Studie |
SELECT- |
SELECT- |
||||
Behandlungsarm |
MTX |
UPA |
PBOa |
UPA |
ADA 40 mg |
|
Modifizierter Total-Sharp-Score, mittlere Veränderung gegenüber Baseline | ||||||
Woche 24b/26c |
0,7 |
0,1f |
0,9 |
0,2g |
0,1 |
|
Woche 48 |
1,0 |
0,03e |
1,7 |
0,3e |
0,4 |
|
Anteil der Patienten ohne radiologische Progressiond | ||||||
Woche 24b/26c |
77,7 |
87,5f |
76,0 |
83,5f |
86,8 |
|
Woche 48 |
74,3 |
89,9e |
74,1 |
86,4e |
87,9 |
|
Abkürzungen: ADA = Adalimumab; IR = inadequate responder (Patient mit unzureichendem Ansprechen); MTX = Methotrexat; PBO = Placebo; UPA = Upadacitinib | ||||||
Ansprechen der körperlichen Funktionsfähigkeit und gesundheitsbezogene Ergebnisse
Die Behandlung mit 15 mg Upadacitinib, in Monotherapie oder in Kombination mit csDMARDs, führte gemessen anhand des HAQ-DI zu einer signifikant größeren Verbesserung der körperlichen Funktionsfähigkeit im Vergleich zu allen Vergleichspräparaten (siehe Tabelle 7). Die Verbesserung des HAQ-DI blieb bei den Patienten, die ihre ursprünglich zugewiesene Behandlung mit 15 mg Upadacitinib beibehielten, über drei Jahre erhalten, basierend auf den verfügbaren Ergebnissen der Studien SELECT-COMPARE und SELECT-EARLY.
Tabelle 7 Mittlere Veränderung des HAQ-DI gegenüber Baselinea,b
Studie |
SELECT-EARLY |
SELECT-MONO |
SELECT-NEXT |
SELECT- |
SELECT-BEYOND |
||||||
Behandlungsarm |
MTX |
UPA |
MTX |
UPA |
PBO |
UPA |
PBO |
UPA |
ADA |
PBO |
UPA |
N |
313 |
317 |
216 |
216 |
220 |
216 |
648 |
644 |
324 |
165 |
163 |
Score bei Baseline, Mittelwert |
1,6 |
1,6 |
1,5 |
1,5 |
1,4 |
1,5 |
1,6 |
1,6 |
1,6 |
1,6 |
1,7 |
Woche |
-0,5 |
-0,8h |
-0,3 |
-0,7g |
-0,3 |
-0,6g |
-0,3 |
-0,6g,i |
-0,5 |
-0,2 |
-0,4g |
Woche |
-0,6 |
-0,9g |
-0,3 |
-0,7h,i |
-0,6 |
||||||
Abkürzungen: ADA = Adalimumab; HAQ-DI = Health Assessment Questionnaire-Disability Index; IR = inadequate responder (Patient mit unzureichendem Ansprechen); MTX = Methotrexat; PBO = Placebo; UPA = Upadacitinib | |||||||||||
In den Studien SELECT-MONOTHERAPY, SELECT-NEXT und SELECT-COMPARE führte die Behandlung mit 15 mg Upadacitinib zu einer signifikant größeren Verbesserung der mittleren Dauer der Morgensteifigkeit im Vergleich zu Placebo oder MTX.
In den klinischen Studien erzielten die mit Upadacitinib behandelten Patienten im Vergleich zu Placebo und MTX signifikante Verbesserungen der vom Patienten berichteten Lebensqualität, gemessen anhand der körperlichen Domäne des short-form-36-Gesundheitsfragebogens (SF-36). Außerdem berichteten Patienten, die mit Upadacitinib behandelt wurden, im Vergleich zu Patienten, die mit Placebo behandelt wurden, von einer signifikanten Verbesserung bzgl. der Fatigue, gemessen anhand des Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F).
Psoriasis-Arthritis
Die Wirksamkeit und Sicherheit von 15 mg Upadacitinib einmal täglich wurden in zwei randomisierten, doppelblinden, multizentrischen, placebokontrollierten Phase‑III-Studien bei Patienten ab 18 Jahren mit mittelschwerer bis schwerer aktiver Psoriasis-Arthritis untersucht. Alle Patienten wiesen seit mindestens 6 Monaten eine aktive Psoriasis-Arthritis gemäß CASPAR (Classification Criteria for Psoriatic Arthritis), mindestens 3 druckschmerzhafte Gelenke und mindestens 3 geschwollene Gelenke sowie eine aktive Plaque-Psoriasis oder eine Plaque-Psoriasis in der Anamnese auf. Primärer Endpunkt war in beiden Studien der Anteil der Patienten, die in Woche 12 ein ACR20-Ansprechen erreichten.
SELECT‑PsA 1 war eine 24‑wöchige Studie mit 1 705 Patienten mit unzureichendem Ansprechen auf oder Unverträglichkeit gegen mindestens ein nicht biologisches DMARD. Bei Baseline erhielten 1 393 Patienten (82 %) begleitend mindestens ein nicht biologisches DMARD, 1 084 (64 %) nur eine begleitende MTX-Behandlung und 311 (18 %) eine Monotherapie. Die Patienten erhielten einmal täglich 15 mg oder 30 mg Upadacitinib, Adalimumab oder Placebo. In Woche 24 wurden alle zu Placebo randomisierten Patienten verblindet auf 15 mg oder 30 mg Upadacitinib einmal täglich umgestellt. Die Studie SELECT‑PsA 1 beinhaltete eine Langzeit-Fortsetzungsphase von bis zu 5 Jahren.
SELECT‑PsA 2 war eine 24‑wöchige Studie mit 642 Patienten mit unzureichendem Ansprechen auf oder Unverträglichkeit gegen mindestens ein biologisches DMARD. Bei Baseline erhielten 296 Patienten (46 %) begleitend mindestens ein nicht biologisches DMARD, 222 (35 %) nur eine begleitende MTX-Behandlung und 345 (54 %) eine Monotherapie. Die Patienten erhielten einmal täglich 15 mg oder 30 mg Upadacitinib oder Placebo. In Woche 24 wurden alle zu Placebo randomisierten Patienten verblindet auf 15 mg oder 30 mg Upadacitinib einmal täglich umgestellt. Die Studie SELECT-PsA 2 beinhaltete eine Langzeit-Fortsetzungsphase von bis zu 3 Jahren.
Klinisches Ansprechen
In beiden Studien erreichte zu Woche 12 ein statistisch signifikant größerer Anteil an Patienten unter 15 mg Upadacitinib ein ACR20-Ansprechen als unter Placebo (Tabelle 8). Bei allen Parametern war die Dauer bis zum Eintreten der Wirkung kurz, wobei höhere ACR20-Ansprechraten bereits nach 2 Wochen verzeichnet wurden.
Die Behandlung mit 15 mg Upadacitinib führte im Vergleich zu Placebo zu einer Verbesserung bei allen ACR-Komponenten, einschließlich der Anzahl druckschmerzhafter/schmerzhafter und geschwollener Gelenke, der allgemeinen Beurteilung der Krankheitsaktivität durch den Arzt (PhGA) und den Patienten (PGA), des HAQ‑DI, des Schmerzes und des hsCRP.
In der Studie SELECT‑PsA 1 zeigte 15 mg Upadacitinib im Vergleich zu Adalimumab eine Nichtunterlegenheit in Bezug auf den Anteil der Patienten, die in Woche 12 ein ACR20-Ansprechen erreichten; eine Überlegenheit gegenüber Adalimumab konnte jedoch nicht nachgewiesen werden.
In beiden Studien wurde hinsichtlich der primären und wichtigsten sekundären Endpunkte bei Monotherapie oder in Kombination mit Methotrexat ein vergleichbares Ansprechen beobachtet.
Die Wirksamkeit von 15 mg Upadacitinib wurde unabhängig von der untersuchten Subgruppe nachgewiesen, u. a. BMI bei Baseline, hsCRP bei Baseline und Anzahl vorheriger nicht biologischer DMARDs (≤ 1 oder > 1).
Tabelle 8 Klinisches Ansprechen in den Studien SELECT‑PsA 1 und SELECT‑PsA 2
Studie |
SELECT‑PsA 1 |
SELECT-PsA 2 |
|||
Behandlungsarm |
PBO |
UPA |
ADA |
PBO |
UPA |
N |
423 |
429 |
429 |
212 |
211 |
ACR20, % der Patienten (95 %‑CI) | |||||
Woche 12 |
36 |
71 (66; 75)f |
65 (61; 70) |
24 (18; 30) |
57 (50; 64) |
Unterschied gegenüber Placebo |
35 (28; 41)d,e |
- |
33 (24; 42)d,e |
||
Woche 24 |
45 |
73 (69; 78) |
67 (63; 72) |
20 (15; 26) |
59 (53; 66) |
Woche 56 |
74 (70; 79) |
69 (64; 73) |
60 (53; 66) |
||
ACR50, % der Patienten (95 %‑CI) | |||||
Woche 12 |
13 |
38 (33; 42) |
38 (33; 42) |
5 (2; 8) |
32 (26; 38) |
Woche 24 |
19 |
52 (48; 57) |
44 (40; 49) |
9 (6; 13) |
38 (32; 45) |
Woche 56 |
60 (55; 64) |
51 (47; 56) |
41 (34; 47) |
||
ACR70, % der Patienten (95 %‑CI) | |||||
Woche 12 |
2 (1; 4) |
16 (12; 19) |
14 (11; 17) |
1 (0; 1) |
9 (5; 12) |
Woche 24 |
5 (3; 7) |
29 (24; 33) |
23 (19; 27) |
1 (0; 2) |
19 (14; 25) |
Woche 56 |
41 (36; 45) |
31 (27; 36) |
24 (18; 30) |
||
MDA, % der Patienten (95 %‑CI) | |||||
Woche 12 |
6 (4; 9) |
25 (21; 29) |
25 (21; 29) |
4 (2; 7) |
17 (12; 22) |
Woche 24 |
12 |
37 (32; 41)e |
33 (29; 38) |
3 (1; 5) |
25 (19; 31)e |
Woche 56 |
45 (40; 50) |
40 (35; 44) |
29 (23; 36) |
||
Abklingen der Enthesitis (LEI = 0), % der Patienten (95 %‑CI)a | |||||
Woche 12 |
33 |
47 (42; 53) |
47 (41; 53) |
20 (14; 27) |
39 (31; 47) |
Woche 24 |
32 |
54 (48; 60)e |
47 (42; 53) |
15 (9; 21) |
43 (34; 51) |
Woche 56 |
59 (53; 65) |
54 (48; 60) |
43 (34; 51) |
||
Abklingen der Daktylitis (LDI = 0), % der Patienten (95 %‑CI)b | |||||
Woche 12 |
42 |
74 (66; 81) |
72 (64; 80) |
36 (24; 48) |
64 (51; 76) |
Woche 24 |
40 |
77 (69; 84) |
74 (66; 82) |
28 (17; 39) |
58 (45; 71) |
Woche 56 |
75 (68; 82) |
74 (66; 82) |
51 (38; 64) |
||
PASI75, % der Patienten (95 %‑CI)c | |||||
Woche 16 |
21 |
63 (56; 69)e |
53 (46; 60) |
16 (10; 22) |
52 (44; 61)e |
Woche 24 |
27 |
64 (58; 70) |
59 (52; 65) |
19 (12; 26) |
54 (45; 62) |
Woche 56 |
65 (59; 72) |
61 (55; 68) |
52 (44; 61) |
||
PASI90, % der Patienten (95 %‑CI)c | |||||
Woche 16 |
12 |
38 (32; 45) |
39 (32; 45) |
8 (4; 13) |
35 (26; 43) |
Woche 24 |
17 |
42 (35; 48) |
45 (38; 52) |
7 (3; 11) |
36 (28; 44) |
Woche 56 |
49 (42; 56) |
47 (40; 54) |
41 (32; 49) |
||
|
Abkürzungen: ACR20 (bzw. 50/70) = Verbesserung um ≥ 20 % (bzw. ≥ 50/70 %) gemäß American College of Rheumatology; ADA = Adalimumab; bDMARD = biologisches krankheitsmodifizierendes Antirheumatikum (biologic disease-modifying anti-rheumatic drug); IR = inadequate responder (Patient mit unzureichendem Ansprechen); MDA = minimal disease activity (minimale Krankheitsaktivität); PASI75 (bzw. 90) = Verbesserung des Psoriasis Area and Severity Index um ≥ 75 % (bzw. ≥ 90 %); PBO = Placebo; UPA = Upadacitinib a Bei Patienten mit Enthesitis bei Baseline (n = 241, 270 bzw. 265 in der Studie SELECT‑PsA 1 und n = 144 bzw. 133 in der Studie SELECT‑PsA 2) | |||||
Radiologisches Ansprechen
In der Studie SELECT‑PsA 1 wurde die Hemmung der Progression struktureller Schäden radiologisch gemessen und als Veränderung des modifizierten Total-Sharp-Scores (mTSS) und seiner Komponenten, des Erosionsscores und des Scores für Gelenkspaltverengung in Woche 24 gegenüber Baseline ausgedrückt.
Die Behandlung mit 15 mg Upadacitinib führte im Vergleich zu Placebo in Woche 24 zu einer statistisch signifikant größeren Hemmung der radiologischen Progression (Tabelle 9). Die Erosionsscores und die Scores für Gelenkspaltverengung waren konsistent mit den Gesamtscores. Der Anteil der Patienten ohne radiologische Progression (mTSS-Veränderung ≤ 0,5) war in Woche 24 unter 15 mg Upadacitinib im Vergleich zu Placebo höher.
Tabelle 9 Radiologische Veränderungen in der Studie SELECT‑PsA 1
Behandlungsarm |
PBO |
UPA |
ADA |
Modifizierter Total-Sharp-Score, mittlere Veränderung gegenüber Baseline (95 %-CI) | |||
Woche 24 |
0,25 (0,13; 0,36) |
-0,04 (-0,16; 0,07)c |
0,01 (-0,11; 0,13) |
Woche 56a |
0,44 (0,29; 0,59) |
-0,05 (-0,20; 0,09) |
-0,06 (-0,20; 0,09) |
Anteil der Patienten ohne radiologische Progressionb, % (95 %‑CI) | |||
Woche 24 |
92 (89; 95) |
96 (94; 98) |
95 (93; 97) |
Woche 56a |
89 (86; 92) |
97 (96; 99) |
94 (92; 97) |
Abkürzungen: ADA = Adalimumab; PBO = Placebo; UPA = Upadacitinib | |||
Ansprechen der körperlichen Funktionsfähigkeit und gesundheitsbezogene Ergebnisse
In der Studie SELECT‑PsA 1 zeigten mit 15 mg Upadacitinib behandelte Patienten in Woche 12 gegenüber Baseline mittels HAQ‑DI bewertet eine statistisch signifikante Verbesserung der körperlichen Funktionsfähigkeit (‑0,42 [95 %‑CI: ‑0,47; ‑0,37]) im Vergleich zu Patienten, die Placebo erhielten (‑0,14 [95 %‑CI: ‑0,18; ‑0,09]). Die Verbesserung bei Patienten unter Adalimumab betrug ‑0,34 (95 %‑CI: ‑0,38; ‑0,29). In der Studie SELECT‑PsA 2 zeigten mit 15 mg Upadacitinib behandelte Patienten in Woche 12 gegenüber Baseline eine statistisch signifikante Verbesserung des HAQ‑DI (‑0,30 [95 %‑CI: ‑0,37; ‑0,24]) im Vergleich zu Patienten, die Placebo erhielten (‑0,10 [95 %‑CI: ‑0,16; ‑0,03]). In beiden Studien wurde die Verbesserung der körperlichen Funktionsfähigkeit bis Woche 56 aufrechterhalten.
Die gesundheitsbezogene Lebensqualität wurde mittels SF‑36 Version 2 beurteilt. In beiden Studien verzeichneten Patienten, die 15 mg Upadacitinib erhielten, in Woche 12 eine statistisch signifikant größere Verbesserung bei der körperlichen Domäne des SF‑36 gegenüber Baseline als Patienten, die Placebo erhielten. Die Verbesserungen gegenüber Baseline wurden in beiden Studien bis Woche 56 aufrechterhalten.
Patienten, die 15 mg Upadacitinib erhielten, verzeichneten in Woche 12 im Vergleich zu Placebo in beiden Studien eine statistisch signifikante Verbesserung der Fatigue gegenüber Baseline, gemessen anhand des FACIT‑F-Scores. Die Verbesserungen gegenüber Baseline wurden in beiden Studien bis Woche 56 aufrechterhalten.
Bei Baseline der Studie SELECT‑PsA 1 hatten 31 % der Patienten eine Psoriasis-Spondylitis, in der Studie SELECT‑PsA 2 waren es 34 % der Patienten mit Psoriasis-Spondylitis. Patienten mit Psoriasis-Spondylitis, die mit 15 mg Upadacitinib behandelt wurden, zeigten in Woche 24 im Vergleich zu Placebo Verbesserungen beim Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) gegenüber Baseline. Die Verbesserungen gegenüber Baseline wurden in beiden Studien bis Woche 56 aufrechterhalten.
Axiale Spondyloarthritis
Nicht röntgenologische axiale Spondyloarthritis
Die Wirksamkeit und Sicherheit von 15 mg Upadacitinib einmal täglich wurden in einer randomisierten, doppelblinden, multizentrischen, placebokontrollierten Studie bei Patienten ab 18 Jahren mit aktiver nicht röntgenologischer axialer Spondyloarthritis untersucht. Bei der Studie SELECT‑AXIS 2 (nr‑axSpA) handelte es sich um eine 52‑wöchige placebokontrollierte Studie mit 314 Patienten mit aktiver nicht röntgenologischer axialer Spondyloarthritis und unzureichendem Ansprechen auf mindestens zwei NSAR oder Unverträglichkeit oder Kontraindikation gegen NSAR. Patienten müssen objektive Anzeichen von Entzündungen aufgewiesen haben, die durch erhöhtes C‑reaktives Protein (CRP) (definiert als > ULN) und/oder in der Magnetresonanztomografie (MRT) als Sakroiliitis angezeigt wurden, und durften keine eindeutigen radiologischen Hinweise auf strukturelle Schäden an den Iliosakralgelenken haben. Patienten wiesen eine aktive Erkrankung auf, definiert als ein Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) von ≥ 4 und ein Patient’s Assessment of Total Back Pain Score von ≥ 4, basierend auf einer numerischen Ratingskala (NRS) von 0 bis 10, bei dem Screening- und Baselinebesuch. Bei Baseline hatten die Patienten durchschnittlich 9,1 Jahre lang Symptome einer nicht röntgenologischen axialen Spondyloarthritis und 29,1 % der Patienten erhielten begleitend ein csDMARD. 32,9 % der Patienten wiesen ein unzureichendes Ansprechen oder eine Unverträglichkeit gegen eine bDMARD-Therapie auf. Die Patienten erhielten einmal täglich 15 mg Upadacitinib oder Placebo. In Woche 52 wurden alle zu Placebo randomisierten Patienten auf 15 mg Upadacitinib einmal täglich umgestellt. Der primäre Endpunkt war der Anteil der Patienten, die in Woche 14 ein ASAS40-Ansprechen gemäß der Assessment of SpondyloArthritis International Society erreichten. Die Studie beinhaltete eine langfristige Fortsetzung um bis zu 2 Jahre. Von den Patienten, die ursprünglich zu Upadacitinib randomisiert wurden, setzten 75 % (117/156) in SELECT-AXIS 2 (nr-axSpA) die Therapie über 2 Jahre fort.
Klinisches Ansprechen
In der Studie SELECT‑AXIS 2 (nr‑axSpA) erreichte in Woche 14 ein signifikant größerer Anteil an Patienten unter 15 mg Upadacitinib ein ASAS40-Ansprechen als unter Placebo (Tabelle 10).
Ein numerischer Unterschied zwischen den Behandlungsarmen wurde zu allen Zeitpunkten von Woche 2 bis Woche 14 beobachtet.
Die Behandlung mit 15 mg Upadacitinib führte in Woche 14 im Vergleich zu Placebo zu einer Verbesserung bei den einzelnen ASAS-Komponenten (allgemeine Beurteilung der Krankheitsaktivität durch den Patienten, Beurteilung der Rückenschmerzen insgesamt, Entzündung und Funktionsfähigkeit) und anderen Messungen der Krankheitsaktivität, einschließlich des hsCRP.
Die Wirksamkeit von 15 mg Upadacitinib zeigte sich unabhängig von den Untergruppen, einschließlich Geschlecht, BMI bei Baseline, Dauer der Symptome der nicht röntgenologischen axialen Spondyloarthritis, hsCRP bei Baseline, Sakroiliitis auf dem MRT und vorherige Anwendung von bDMARDs.
Tabelle 10 Klinisches Ansprechen in der Studie SELECT‑AXIS 2 (nr‑axSpA)
Behandlungsarm |
PBO |
UPA 15 mg |
N |
157 |
156 |
ASAS40, % der Patienten (95 %‑CI)a | ||
Woche 14 |
22,5 (16,0; 29,1) |
44,9 (37,1; 52,7) |
Unterschied gegenüber Placebo (95 %‑CI) |
22,2 (12,1; 32,3)b |
|
Woche 52 |
42,7 (34,9; 50,4) |
62,8 (55,2; 70,4)d |
ASAS20, % der Patienten (95 %‑CI)a | ||
Woche 14 |
43,8 (36,0; 51,5) |
66,7 (59,3; 74,1)b |
Partielle Remission gemäß ASAS, % der Patienten (95 %‑CI) | ||
Woche 14 |
7,6 (3,5; 11,8) |
18,6 (12,5; 24,7)c |
BASDAI50, % der Patienten (95 %‑CI) | ||
Woche 14 |
22,1 (15,5; 28,6) |
42,3 (34,6; 50,1)b |
Veränderung des ASDAS-CRP gegenüber Baseline (95 %‑CI) | ||
Woche 14 |
-0,71 (-0,85; -0,56) |
-1,36 (-1,50; -1,21)b |
Inaktive Erkrankung gemäß ASDAS, % der Patienten (95 %‑CI) | ||
Woche 14 |
5,2 (1,7; 8,7) |
14,1 (8,6; 19,6)c |
Niedrige Krankheitsaktivität gemäß ASDAS, % der Patienten (95 %‑CI) | ||
Woche 14 |
18,3 (12,2; 24,4) |
42,3 (34,6; 50,1)b |
Abkürzungen: ASAS20 (bzw. ASAS40) = Verbesserung gemäß den Kriterien der Assessment of SpondyloArthritis International Society um ≥ 20 % (bzw. ≥ 40 %); ASDAS‑CRP = C-reaktives Protein im Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; PBO = Placebo; UPA = Upadacitinib | ||
Die Wirksamkeit blieb über 2 Jahre erhalten, wie anhand der in Tabelle 10 dargestellten Endpunkte beurteilt wurde.
Ansprechen der körperlichen Funktionsfähigkeit und gesundheitsbezogene Ergebnisse
Patienten, die mit 15 mg Upadacitinib behandelt wurden, zeigten im Vergleich zu Placebo in Woche 14 gegenüber Baseline eine signifikante Verbesserung der körperlichen Funktionsfähigkeit gemäß Beurteilung anhand BASFI.
Patienten, die mit 15 mg Upadacitinib behandelt wurden, zeigten im Vergleich zu Placebo in Woche 14 signifikante Verbesserungen der Rückenschmerzen insgesamt und der nächtlichen Rückenschmerzen.
Patienten, die mit 15 mg Upadacitinib behandelt wurden, zeigten im Vergleich zu Placebo in Woche 14 signifikante Verbesserungen der gesundheitsbezogenen Lebensqualität und der allgemeinen Gesundheit, gemessen anhand des ASQoL-Fragebogens und des ASAS-Gesundheitsindex.
Die Verbesserungen des BASFI, der Rückenschmerzen insgesamt und der nächtlichen Rückenschmerzen, des ASQoL-Fragebogens und des ASAS-Gesundheitsindex wurden über 2 Jahre aufrechterhalten.
Objektive Entzündungsparameter
Die Anzeichen einer Entzündung wurden mittels MRT beurteilt und als Veränderung des Spondyloarthritis Research Consortium of Canada (SPARCC) Score für die Iliosakralgelenke gegenüber Baseline ausgedrückt. In Woche 14 wurde bei Patienten, die mit 15 mg Upadacitinib behandelt wurden, eine signifikante Verbesserung der Entzündungsanzeichen in den Iliosakralgelenken im Vergleich zu Placebo beobachtet. Die mittels MRT beurteilte Verbesserung der Entzündung blieb über 2 Jahre erhalten.
Ankylosierende Spondylitis (AS, röntgenologische axiale Spondyloarthritis)
Die Wirksamkeit und Sicherheit von 15 mg Upadacitinib einmal täglich wurden in zwei randomisierten, doppelblinden, multizentrischen, placebokontrollierten Studien bei Patienten ab 18 Jahren mit aktiver ankylosierender Spondylitis, definiert als ein Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) Score von ≥ 4 und ein Patient’s Assessment of Total Back Pain Score von ≥ 4, untersucht. Beide Studien beinhalteten eine Langzeit-Fortsetzungsphase von bis zu 2 Jahren.
SELECT‑AXIS 1 war eine 14‑wöchige, placebokontrollierte Studie mit 187 Patienten mit ankylosierender Spondylitis und unzureichendem Ansprechen auf mindestens zwei NSAR oder Unverträglichkeit oder Kontraindikation gegenüber NSAR, die bisher nicht mit biologischen DMARDs behandelt worden waren. Bei Baseline hatten die Patienten durchschnittlich 14,4 Jahre lang Symptome einer ankylosierenden Spondylitis und etwa 16 % der Patienten erhielten begleitend ein csDMARD. Die Patienten erhielten einmal täglich 15 mg Upadacitinib oder Placebo. In Woche 14 wurden alle zu Placebo randomisierten Patienten auf 15 mg Upadacitinib einmal täglich umgestellt. Der primäre Endpunkt war der Anteil der Patienten, die in Woche 14 ein ASAS40-Ansprechen gemäß der Assessment of SpondyloArthritis International Society erreichten.
SELECT‑AXIS 2 (AS) war eine 14-wöchige, placebokontrollierte Studie mit 420 Patienten mit ankylosierender Spondylitis, die zuvor mit bDMARDs behandelt worden waren (bei 77,4 % war entweder ein TNF-Blocker oder ein Interleukin-17-Inhibitor (IL-17i) unwirksam; bei 30,2 % lag eine Unverträglichkeit vor; bei 12,9 % waren zuvor zwei bDMARDs verabreicht worden, zeigten jedoch keine mangelnde Wirksamkeit gegenüber diesen). Bei Baseline hatten die Patienten durchschnittlich 12,8 Jahre lang Symptome einer ankylosierenden Spondylitis und etwa 31 % der Patienten erhielten begleitend ein csDMARD. Die Patienten erhielten einmal täglich 15 mg Upadacitinib oder Placebo. In Woche 14 wurden alle zu Placebo randomisierten Patienten auf 15 mg Upadacitinib einmal täglich umgestellt. Der primäre Endpunkt war der Anteil der Patienten, die in Woche 14 ein ASAS40-Ansprechen gemäß der Assessment of SpondyloArthritis International Society erreichten.
Von den Patienten, die ursprünglich zu Upadacitinib randomisiert wurden, setzten 72 % (67/93) in SELECT-AXIS 1 und 77 % (163/211) in SELECT-AXIS 2 (AS) die Therapie über 2 Jahre fort.
Klinisches Ansprechen
In beiden Studien erreichte in Woche 14 ein signifikant größerer Anteil an Patienten unter 15 mg Upadacitinib ein ASAS40-Ansprechen als unter Placebo (Tabelle 11). Ein numerischer Unterschied zwischen den Behandlungsarmen hinsichtlich ASAS40 wurde ab Woche 2 in SELECT‑AXIS 1 und ab Woche 4 in SELECT‑AXIS 2 (AS) beobachtet.
Die Behandlung mit 15 mg Upadacitinib führte in Woche 14 im Vergleich zu Placebo zu Verbesserungen in den einzelnen ASAS-Domänen (allgemeine Beurteilung der Krankheitsaktivität durch den Patienten, Beurteilung der Rückenschmerzen insgesamt, Entzündung und Funktionsfähigkeit) und anderer Parameter der Krankheitsaktivität, darunter auch hsCRP.
Die Wirksamkeit von 15 mg Upadacitinib wurde unabhängig von der untersuchten Subgruppe nachgewiesen, u. a. Geschlecht, BMI bei Baseline, Dauer der AS‑Symptome, hsCRP bei Baseline sowie vorherige Anwendung von bDMARDs.
Tabelle 11 Klinisches Ansprechen
Studie |
SELECT‑AXIS 1 |
SELECT‑AXIS 2 (AS) |
||
Behandlungsarm |
PBO |
UPA 15 mg |
PBO |
UPA 15 mg |
N |
94 |
93 |
209 |
211 |
ASAS40, % der Patienten (95 %‑CI)a,b | ||||
Woche 14 |
25,5 (16,7; 34,3) |
51,6 (41,5; 61,8) |
18,2 (13,0; 23,4) |
44,5 (37,8; 51,3) |
Unterschied gegenüber Placebo (95 %-CI) |
26,1 (12,6; 39,5)c |
26,4 (17,9; 34,9)c |
||
ASAS20, % der Patienten (95 %‑CI)a | ||||
Woche 14 |
40,4 (30,5; 50,3) |
64,5 (54,8; 74,2)e |
38,3 (31,7; 44,9) |
65,4 (59,0; 71,8)c |
Partielle Remission gemäß ASAS, % der Patienten (95 %‑CI) | ||||
Woche 14 |
1,1 (0,0; 3,1) |
19,4 (11,3; 27,4)c |
4,3 (1,6; 7,1) |
17,5 (12,4; 22,7)c |
BASDAI50, % der Patienten (95 %‑CI) | ||||
Woche 14 |
23,4 (14,8; 32,0) |
45,2 (35,0; 55,3)d |
16,7 (11,7; 21,8) |
43,1 (36,4; 49,8)c |
Veränderung des ASDAS-CRP gegenüber Baseline (95 %‑CI) | ||||
Woche 14 |
-0,54 (-0,71; -0,37) |
-1,45 (-1,62; -1,28)c |
-0,49 (-0,62; -0,37) |
-1,52 (-1,64; -1,39)c |
Inaktive Erkrankung gemäß ASDAS, % der Patienten (95 %‑CI) | ||||
Woche 14 |
0 |
16,1 (8,7; 23,6)e |
1,9 (0,1; 3,8) |
12,8 (8,3; 17,3)c |
Niedrige Krankheitsaktivität gemäß ASDAS, % der Patienten (95 %‑CI) | ||||
Woche 14 |
10,6 (4,4; 16,9) |
49,5 (39,3; 59,6)f |
10,1 (6,0; 14,2) |
44,1 (37,4; 50,8)c |
Bedeutende Verbesserung gemäß ASDAS, % der Patienten (95 %‑CI) | ||||
Woche 14 |
5,3 (0,8; 9,9) |
32,3 (22,8; 41,8)e |
4,8 (1,9; 7,7) |
30,3 (24,1; 36,5)e |
a Ein ASAS20 (ASAS40)-Ansprechen ist definiert als eine Verbesserung um ≥ 20 % (≥ 40 %) und eine absolute Verbesserung gegenüber Baseline um ≥ 1 (≥ 2) Einheit(en) (Spannweite 0 bis 10) in ≥ 3 von 4 Bereichen (allgemeine Beurteilung durch den Patienten, Rückenschmerzen insgesamt, Funktionsfähigkeit und Entzündung) und keine Verschlechterung in der potenziell verbleibenden Domäne (definiert als Verschlechterung um ≥ 20 % und ≥ 1 Einheit für ASAS20 bzw. um > 0 Einheiten für ASAS40). | ||||
In beiden Studien blieb die Wirksamkeit über 2 Jahre erhalten, wie anhand der in Tabelle 11 dargestellten Endpunkte beurteilt wurde.
Ansprechen der körperlichen Funktionsfähigkeit und gesundheitsbezogene Ergebnisse
In beiden Studien zeigten Patienten, die mit 15 mg Upadacitinib behandelt wurden, im Vergleich zu Placebo in Woche 14 gegenüber Baseline eine signifikante Verbesserung der körperlichen Funktionsfähigkeit gemäß Beurteilung anhand des Bath Ankylosing Spondylitis Functional Index (BASFI). Die Verbesserung des BASFI wurde über 2 Jahre aufrechterhalten.
In der Studie SELECT‑AXIS 2 (AS) zeigten Patienten, die mit 15 mg Upadacitinib behandelt wurden, im Vergleich zu Placebo in Woche 14 signifikante Verbesserungen der Rückenschmerzen insgesamt und der nächtlichen Rückenschmerzen. Die Verbesserungen der Rückenschmerzen insgesamt und der nächtlichen Rückenschmerzen wurden über 2 Jahre aufrechterhalten.
In der Studie SELECT‑AXIS 2 (AS) zeigten Patienten, die mit 15 mg Upadacitinib behandelt wurden, im Vergleich zu Placebo in Woche 14 signifikante Verbesserungen der gesundheitsbezogenen Lebensqualität und des allgemeinen Gesundheitszustands, gemessen anhand des ASQoL-Fragebogens bzw. des ASAS-Gesundheitsindex. Die Verbesserungen des ASQoL-Fragebogens und des ASAS-Gesundheitsindex wurden über 2 Jahre aufrechterhalten.
Enthesitis
In der Studie SELECT‑AXIS 2 (AS) zeigten Patienten mit vorbestehender Enthesitis (n = 310), die mit 15 mg Upadacitinib behandelt wurden, eine signifikante Verbesserung der Enthesitis im Vergleich zu Placebo, gemessen anhand der Veränderung des Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) in Woche 14 gegenüber Baseline. Die Verbesserung der Enthesitis wurde über 2 Jahre aufrechterhalten.
Beweglichkeit der Wirbelsäule
In der Studie SELECT‑AXIS 2 (AS) zeigten Patienten, die mit 15 mg Upadacitinib behandelt wurden, im Vergleich zu Placebo eine signifikante Verbesserung der Beweglichkeit der Wirbelsäule, gemessen anhand der Veränderung des Bath Ankylosing Spondylitis Metrology Index (BASMI) in Woche 14 gegenüber Baseline. Die Verbesserung des BASMI wurde über 2 Jahre aufrechterhalten.
Objektive Entzündungsparameter
Die Anzeichen einer Entzündung wurden mittels MRT beurteilt und als Veränderung des SPARCC-Scores für die Wirbelsäule gegenüber Baseline ausgedrückt. In beiden Studien wurde in Woche 14 bei Patienten, die mit 15 mg Upadacitinib behandelt wurden, eine signifikante Verbesserung der Entzündungszeichen der Wirbelsäule im Vergleich zu Placebo beobachtet. Die mittels MRT beurteilte Verbesserung der Entzündung blieb über 2 Jahre erhalten.
Riesenzellarteriitis
Die Wirksamkeit und Sicherheit von 15 mg Upadacitinib einmal täglich wurden in SELECT-GCA, einer randomisierten, doppelblinden, multizentrischen, placebokontrollierten Phase‑III-Studie, bei Patienten im Alter von 50 Jahren oder älter mit neu auftretender oder rezidivierender Riesenzellarteriitis untersucht. SELECT-GCA (Periode 1) war eine 52‑wöchige Studie, in der 428 Patienten in dem Verhältnis 2:1:1 randomisiert und einmal täglich 15 mg Upadacitinib, 7,5 mg Upadacitinib oder Placebo verabreicht wurde. Alle Patienten bekamen eine Kortikosteroid-Basismedikation (Prednison oder Prednisolon). Der Behandlungsarm mit Upadacitinib erhielt eine vorgegebene Ausschleichtherapie mit Kortikosteroiden, mit dem Ziel 0 mg bis Woche 26 zu erreichen; der Behandlungsarm mit Placebo erhielt eine vorgegebene Ausschleichtherapie mit Kortikosteroiden, mit dem Ziel 0 mg bis Woche 52 zu erreichen. Der primäre Endpunkt war der Anteil der Patienten, die zu Woche 52 eine anhaltende Remission erreicht hatten, definiert als Fehlen von Anzeichen und Symptomen einer Riesenzellarteriitis von Woche 12 bis Woche 52 und die Einhaltung der protokolldefinierten Ausschleichtherapie mit Kortikosteroiden. Patienten, die die Studienbehandlung vorzeitig abbrachen (Upadacitinib oder Placebo) oder die eine Untersuchung versäumt hatten, wurden als Non-Responder gezählt. Die Studie umfasste eine 52‑wöchige verblindete Verlängerung (Periode 2) für eine Gesamtstudiendauer von bis zu 2 Jahren.
Klinisches Ansprechen
15 mg Upadacitinib und eine 26‑wöchige Kortikosteroid-Ausschleichtherapie zeigten eine Überlegenheit bei der Erreichung einer kortikosteroidfreien anhaltenden Remission zu Woche 52 im Vergleich zu Placebo und einer 52‑wöchigen Kortikosteroid-Ausschleichtherapie (Tabelle 12). Die Ergebnisse für jede Komponente der anhaltenden Remission und der anhaltenden Remission zu Woche 52 sind konsistent mit denen der kombinierten Endpunkte. Bei anhaltender Remission zu Woche 52 (primärer Endpunkt) wurde ein ähnlicher Prozentsatz der Patienten in beiden Behandlungsarmen aufgrund eines vorzeitigen Abbruchs der Studienbehandlung (Placebo: 19,6 %; Upadacitinib 15 mg: 20,1 %) oder einer versäumten Untersuchung (Placebo: 0,9 %; Upadacitinib 15 mg: 0,5 %) als Non-Responder eingestuft.
Die Behandlungseffekte in den Untergruppen (Geschlecht, Alter, ethnische Zugehörigkeit, vorherige Anwendung eines Interleukin-6-Inhibitors, neu auftretende oder rezidivierende Riesenzellarteriitis, Kortikosteroid-Ausgangsdosis und Riesenzellarteriitis mit oder ohne Polymyalgia rheumatica) entsprachen den Ergebnissen in der gesamten Studienpopulation.
Bei einem signifikant geringeren Anteil der mit 15 mg Upadacitinib und 26‑wöchiger Kortikosteroid-Ausschleichtherapie behandelten Patienten kam es bis Woche 52 zu mindestens einem Riesenzellarteriitis-Schub im Vergleich zu den mit Placebo und 52‑wöchiger Kortikosteroid-Ausschleichtherapie behandelten Patienten. Darüber hinaus war das Risiko eines Schubs im Behandlungsarm mit Upadacitinib signifikant niedriger als mit Placebo, gemessen an der Zeit bis zum ersten Schub bis Woche 52 (Tabelle 12).
Tabelle 12 Klinisches Ansprechen in der Studie SELECT-GCA (Periode 1)
Behandlungsarm |
PBO + 52‑wöchige Kortikosteroid-Ausschleichtherapie |
UPA 15 mg + 26‑wöchige Kortikosteroid-Ausschleichtherapie |
Behandlungsunterschied (95 %-CI) |
Anhaltende Remission zu Woche 52a |
29,0 % |
46,4 % |
17,1 %e |
Anhaltende komplette Remission zu Woche 52b |
16,1 % |
37,1 % |
20,7 %f |
Komplette Remission zu Woche 52c |
19,6 % |
50,2 % |
30,3 %f |
Komplette Remission zu Woche 24c |
36,1 % |
57,2 % |
20,8 %f |
Zeit bis zum ersten RZA-Schub bis Woche 52d |
0,57e,g |
||
Patienten mit einem oder mehreren RZA-Schüben bis Woche 52d |
55,6 % |
34,3 % |
0,47e,h |
Abkürzungen: ESR = Erythrozytensedimentationsrate; RZA = Riesenzellarteriitis; hsCRP = hochempfindliches C-reaktives Protein; PBO = Placebo; UPA = Upadacitinib | |||
Kumulative Kortikosteroiddosis
Bei den Patienten, die eine 52‑wöchige Nachbeobachtung abgeschlossen haben, war die kumulative Kortikosteroid-Exposition in Woche 52 signifikant niedriger bei Patienten, die 15 mg Upadacitinib und eine 26‑wöchige Kortikosteroid-Ausschleichtherapie erhielten, im Vergleich zu Placebo und einer 52‑wöchigen Kortikosteroid-Ausschleichtherapie (im Median 1 615 mg gegenüber 2 882 mg). Der Vergleich der kumulativen Kortikosteroiddosis zwischen dem Behandlungsarm mit Upadacitinib und Placebo wird von der unterschiedlichen vorgegebenen Kortikosteroid-Ausschleichtherapie im Behandlungsarm mit Upadacitinib gegenüber Placebo beeinflusst.
Gesundheitsbezogene Ergebnisse
Die Fatigue wurde anhand des FACIT-Fatigue‑Scores beurteilt. Bei Patienten, die 15 mg Upadacitinib und eine 26‑wöchige Kortikosteroid-Ausschleichtherapie erhielten, kam es zu einer signifikant stärkeren Verbesserung des FACIT-Fatigue‑Score (4,0; 95 %‑CI: 1,33; 6,76) in Woche 52 gegenüber dem Ausgangswert und im Vergleich zu Placebo und einer 52‑wöchigen Kortikosteroid-Ausschleichtherapie.
Die gesundheitsbezogene Lebensqualität wurde anhand des SF-36 beurteilt. Bei Patienten, die 15 mg Upadacitinib und eine 26‑wöchige Kortikosteroid-Ausschleichtherapie erhielten, kam es zu einer signifikant stärkeren Verbesserung im Physical Component Summary‑Score des SF-36 (3,75; 95 %‑CI: 1,39; 6,11) in Woche 52 gegenüber dem Ausgangswert und im Vergleich zu Placebo und einer 52‑wöchigen Kortikosteroid-Ausschleichtherapie.
Erhaltung des Ansprechens
Insgesamt 223 Patienten nahmen an der verblindeten Verlängerungsphase (Periode 2) der SELECT-GCA-Studie teil. Patienten, die in Periode 1 ursprünglich zu Upadacitinib 15 mg randomisiert worden waren und bei denen eine anhaltende Remission für mindestens 24 Wochen vor Woche 52 erreicht wurde (definiert durch das Fehlen von Anzeichen und Symptomen der Riesenzellarteriitis sowie Kortikosteroidfreiheit), wurden erneut zu Upadacitinib 15 mg (N = 68) oder Placebo (N = 35) randomisiert.
Unter den Patienten, die zu Woche 52 eine anhaltende Remission erreicht hatten, hielten 71,7 % der Patienten, die auf eine Behandlung mit Upadacitinib 15 mg re-randomisiert wurden, bis Woche 104 eine kortikosteroidfreie Remission aufrecht, verglichen mit 31,4 % der Patienten, die zu Placebo re-randomisiert wurden.
Unter den Patienten, die zu Placebo re-randomisiert wurden, erlebten 59,5 % einen Schub der Riesenzellarteriitis, verglichen mit 7,4 % der Patienten, die zu Upadacitinib 15 mg re-randomisiert wurden. Die mediane Zeit bis zum Schub betrug 113 Tage bei den Patienten, die auf Placebo re-randomisiert wurden und war größer als 365 Tage (Median nicht erreicht) bei Patienten, die auf Upadacitinib 15 mg re-randomisiert wurden.
Atopische Dermatitis
Die Wirksamkeit und Sicherheit von 15 mg und 30 mg Upadacitinib einmal täglich wurden in drei randomisierten, doppelblinden, multizentrischen Phase‑III-Studien (MEASURE UP 1, MEASURE UP 2 und AD UP) mit insgesamt 2 782 Patienten (ab 12 Jahren) untersucht. Upadacitinib wurde bei 542 (344 in der Primäranalyse) Jugendlichen und 2 240 Erwachsenen mit mittelschwerer bis schwerer atopischer Dermatitis (AD) untersucht, die mit topischen Arzneimitteln nicht ausreichend kontrolliert werden konnten. Bei Baseline mussten die Patienten alle der folgenden Kriterien erfüllen: einen Score von ≥ 3 auf einer Skala von 0 bis 4 mit ansteigendem Schweregrad des Investigator’s Global Assessment (vIGA‑AD, allgemeine Beurteilung der AD durch den Prüfer) (anhand von Erythem, Induration/Papeln sowie Nässen/Verkrusten), einen EASI-Score (Eczema Area and Severity Index, Index zu Schwere und Ausbreitung eines Ekzems) von ≥ 16 (zusammengefasster Score zur Beurteilung von Ausmaß und Schweregrad von Erythem, Ödem/Papeln, Kratzen und Lichenifikation an 4 verschiedenen Körperstellen), eine Beteiligung der Körperoberfläche (BSA) von ≥ 10 % und einen wöchentlichen Durchschnittswert des schlimmsten Pruritus auf einer numerischen Ratingskala (NRS) von ≥ 4.
In allen drei Studien erhielten die Patienten 16 Wochen lang einmal täglich 15 mg bzw. 30 mg Upadacitinib oder ein entsprechendes Placebo. In der Studie AD UP erhielten die Patienten zusätzlich topische Kortikosteroide (TCS). Nach Beendigung der doppelblinden Phase erhielten die Patienten, die ursprünglich zu Upadacitinib randomisiert worden waren, weiterhin bis Woche 260 die gleiche Dosis. Die Patienten im Behandlungsarm mit Placebo wurden im Verhältnis 1:1 neu randomisiert und erhielten bis Woche 260 15 mg oder 30 mg Upadacitinib.
Merkmale bei Baseline
In den Monotherapiestudien (MEASURE UP 1 und 2) hatten 50,0 % der Patienten bei Baseline einen vIGA‑AD-Score von 3 (mittelschwer) und 50,0 % von 4 (schwer). Der mittlere EASI-Score bei Baseline betrug 29,3, der mittlere wöchentliche Durchschnittswert auf der NRS zum schlimmsten Pruritus war 7,3. In der Studie mit begleitenden TCS (AD UP) hatten 47,1 % der Patienten bei Baseline einen vIGA‑AD-Score von 3 (mittelschwer) und 52,9 % von 4 (schwer). Der mittlere EASI-Score bei Baseline betrug 29,7, der mittlere wöchentliche Durchschnittswert auf der NRS zum schlimmsten Pruritus 7,2.
Klinisches Ansprechen
Studien mit Monotherapie (MEASURE UP 1 und MEASURE UP 2) und mit gleichzeitiger Anwendung von TCS (AD UP)
Ein signifikant größerer Anteil der mit 15 mg oder 30 mg Upadacitinib behandelten Patienten erreichte in Woche 16 einen vIGA‑AD-Score von 0 oder 1, EASI75 oder eine Verbesserung des NRS-Scores zum schlimmsten Pruritus um ≥ 4 Punkte. Auch wurden schnelle Verbesserungen des Erscheinungsbilds der Haut und des Juckreizes erreicht (siehe Tabelle 13).
Abbildung 1 zeigt den Anteil der Patienten in MEASURE UP 1 und 2, die bis Woche 16 ein EASI75-Ansprechen bzw. eine mittlere prozentuale Veränderung des NRS-Scores zum schlimmsten Pruritus gegenüber Baseline erreichten.
Tabelle 13 Ergebnisse zur Wirksamkeit von Upadacitinib
Studie |
MEASURE UP 1 |
MEASURE UP 2 |
AD UP |
||||||
Behandlungsarm |
PBO |
UPA |
UPA |
PBO |
UPA |
UPA |
PBO + TCS |
UPA 15 mg + TCS |
UPA 30 mg + TCS |
Anzahl randomisierter Studienteilnehmer |
281 |
281 |
285 |
278 |
276 |
282 |
304 |
300 |
297 |
Endpunkte Woche 16, % Responder (95 %‑CI) | |||||||||
vIGA-AD-Score von 0/1a,b |
8 |
48d |
62d |
5 |
39d |
52d |
11 |
40d |
59d |
EASI75a |
16 |
70d |
80d |
13 |
60d |
73d |
26 |
65d |
77d |
EASI90a |
8 |
53d |
66d |
5 |
42d |
58d |
13 |
43d |
63d |
EASI100a |
2 |
17d |
27d |
1 |
14d |
19d |
1 |
12e |
23d |
NRS‑Score zum schlimmsten Pruritusc |
12 |
52d |
60d |
9 |
42d |
60d |
15 |
52d |
64d |
Frühe Endpunkte, % Responder (95 %-CI) | |||||||||
EASI75a |
4 |
38d |
47d |
4 |
33d |
44d |
7 |
31d |
44d |
NRS-Score zum schlimmsten Pruritus |
0 |
15d |
20d |
1 |
7d |
16d |
3 |
12d |
19d |
Abkürzungen: UPA = Upadacitinib (RINVOQ); PBO = Placebo | |||||||||
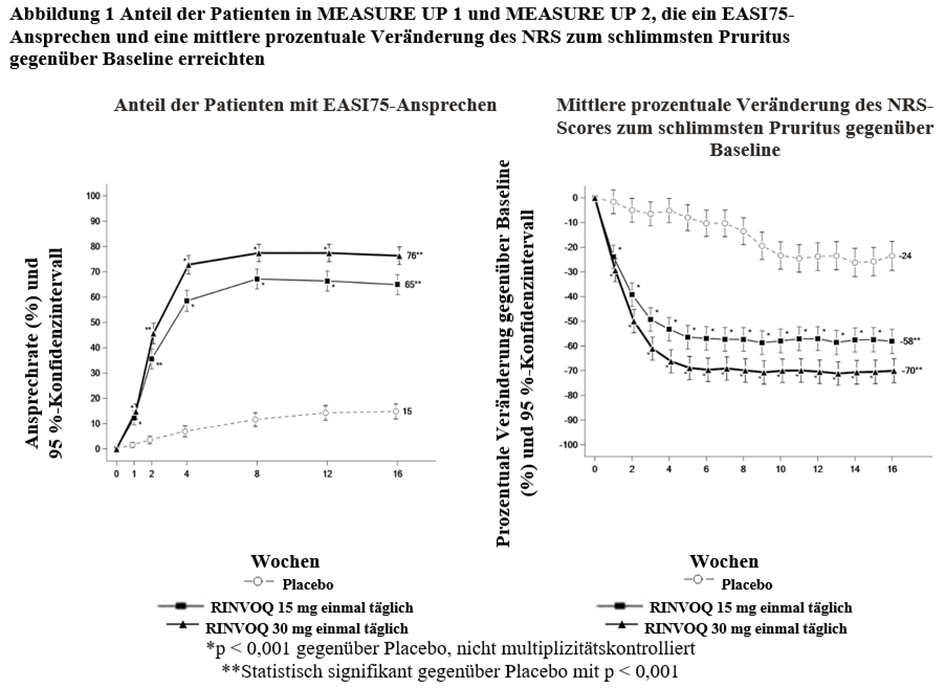
Die Behandlungseffekte in Untergruppen (Gewicht, Alter, Geschlecht, ethnische Zugehörigkeit und vorherige systemische Behandlung mit Immunsuppressiva) entsprachen den Ergebnissen in der gesamten Studienpopulation.
Die Ergebnisse in Woche 16 wurden bei Patienten, die mit 15 mg oder 30 mg Upadacitinib behandelt wurden, bis Woche 52 beibehalten.
Lebensqualität/Patientenfragebögen (patient‑reported outcomes, PROs)
Tabelle 14 Ergebnisse der Patientenfragebögen zu Upadacitinib in Woche 16
Studie |
MEASURE UP 1 |
MEASURE UP 2 |
||||
Behandlungsarm |
PBO |
UPA |
UPA |
PBO |
UPA |
UPA |
Anzahl randomisierter Studienteilnehmer |
281 |
281 |
285 |
278 |
276 |
282 |
% Responder (95 %-CI) | ||||||
ADerm‑SS Skin Pain |
15 |
54e |
63e |
13 |
49e |
65e |
ADerm‑IS Sleep |
13 |
55e |
66e |
12 |
50e |
62e |
DLQI 0/1c |
4 |
30e |
41e |
5 |
24e |
38e |
HADS Anxiety < 8 und HADS Depression < 8d |
14 |
46e |
49e |
11 |
46e |
56e |
Abkürzungen: UPA = Upadacitinib (RINVOQ); PBO = Placebo; DLQI = Dermatology Life Quality Index (Index zur Lebensqualität bei Hauterkrankungen); HADS = Hospital Anxiety and Depression Scale (Skala für Angstzustände und Depression) | ||||||
Colitis ulcerosa
Die Wirksamkeit und Sicherheit von Upadacitinib wurden in drei multizentrischen, doppelblinden, placebokontrollierten klinischen Phase‑III-Studien untersucht: zwei Replikateinleitungsstudien – UC‑1 (U‑ACHIEVE Induction) und UC‑2 (U‑ACCOMPLISH) – und eine Erhaltungsstudie UC‑3 (U‑ACHIEVE Maintenance). Darüber hinaus wurden Sicherheit und Wirksamkeit von Upadacitinib in einer Langzeit-Verlängerungsstudie, UC-4 (U-ACTIVATE), untersucht.
Die Aktivität der Erkrankung basierte auf dem adaptierten Mayo-Score (aMS, Mayo-Scoring-System ohne die allgemeine Beurteilung durch den Arzt), der zwischen 0 und 9 lag und drei Subscores mit jeweils einem Wert von 0 (normal) bis 3 (äußerst schwer) umfasst: Subscore zur Stuhlfrequenz (SFS), Subscore zu rektalen Blutungen (RBS) und einen zentral überprüften endoskopischen Subscore (ES).
Einleitungsstudien (UC‑1 und UC‑2)
In den Studien UC‑1 und UC‑2 wurden 988 Patienten (473 bzw. 515 Patienten) im Verhältnis 2:1 randomisiert und 8 Wochen lang mit 45 mg Upadacitinib einmal täglich oder Placebo behandelt und in die Auswertung zur Wirksamkeit eingeschlossen. Alle in die Studie aufgenommenen Patienten hatten eine mittelschwere bis schwere aktive Colitis ulcerosa, definiert als aMS von 5 bis 9 mit einem ES von 2 oder 3, und ein vorheriges Therapieversagen, einschließlich unzureichendem Ansprechen, Verlust des Ansprechens oder Unverträglichkeit gegenüber einer vorherigen konventionellen und/oder Biologikatherapie. Bei 52 % (246/473) bzw. 51 % (262/515) der Patienten wurde ein vorheriges Versagen mindestens einer Biologikatherapie (vorheriges Versagen der Biologikatherapie) beobachtet. Ein vorheriges Versagen einer konventionellen Therapie, aber nicht einer Biologikatherapie (kein vorheriges Versagen der Biologikatherapie), wurde bei 48 % (227/473) bzw. 49 % (253/515) der Patienten beobachtet.
Bei Baseline erhielten in UC‑1 und UC‑2 39 % bzw. 37 % der Patienten Kortikosteroide, 1,1 % bzw. 0,6 % Methotrexat und 68 % bzw. 69 % Aminosalicylate. Eine gleichzeitige Behandlung mit Thiopurin war während der Studien nicht erlaubt. Die Aktivität der Erkrankung war bei 61 % bzw. 60 % der Patienten mittelschwer (aMS ≥ 5, ≤ 7) und bei 39 % bzw. 40 % schwer (aMS > 7).
Der primäre Endpunkt war die klinische Remission gemäß aMS in Woche 8. Tabelle 15 zeigt den primären Endpunkt sowie die wichtigsten sekundären Endpunkte, darunter klinisches Ansprechen, Mukosaheilung, histologisch-endoskopische Mukosaheilung und tiefe Mukosaheilung.
Tabelle 15 Anteil der Patienten, die in Woche 8 in den Einleitungsstudien UC‑1 und UC‑2 die primären und die wichtigsten sekundären Endpunkte zur Wirksamkeit erreichten
UC‑1 |
UC‑2 |
|||||
Endpunkt |
PBO |
UPA |
Behandlungsunterschied |
PBO |
UPA |
Behandlungsunterschied |
Klinische Remissiona |
4,8 % |
26,1 % |
21,6 %* |
4,1 % |
33,5 % |
29,0 %* |
Vorheriges Versagen der Biologikatherapie+ |
0,4 % |
17,9 % |
17,5 % |
2,4 % |
29,6 % |
27,1 % |
Kein vorheriges Versagen der Biologikatherapie+ |
9,2 % |
35,2 % |
26,0 % |
5,9 % |
37,5 % |
31,6 % |
Klinisches Ansprechenb |
27,3 % |
72,6 % |
46,3 %* |
25,4 % |
74,5 % |
49,4 %* |
Vorheriges Versagen der Biologikatherapie+ |
12,8 % |
64,4 % |
51,6 % |
19,3 % |
69,4 % |
50,1 % |
Kein vorheriges Versagen der Biologikatherapie+ |
42,1 % |
81,8 % |
39,7 % |
31,8 % |
79,8 % |
48,0 % |
Mukosaheilungc |
7,4 % |
36,3 % |
29,3 %* |
8,3 % |
44,0 % |
35,1 %* |
Vorheriges Versagen der Biologikatherapie+ |
1,7 % |
27,0 % |
25,3 % |
4,8 % |
37,1 % |
32,3 % |
Kein vorheriges Versagen der Biologikatherapie+ |
13,2 % |
46,8 % |
33,6 % |
12,0 % |
51,2 % |
39,2 % |
Histologisch-endoskopische Mukosaheilungd |
6,6 % |
30,1 % |
23,7 %* |
5,9 % |
36,7 % |
30,1 %* |
Vorheriges Versagen der Biologikatherapie+ |
1,4 % |
22,7 % |
21,3 % |
4,6 % |
30,7 % |
26,1 % |
Kein vorheriges Versagen der Biologikatherapie+ |
11,8 % |
38,2 % |
26,4 % |
7,2 % |
42,9 % |
35,7 % |
Tiefe Mukosaheilunge |
1,3 % |
10,7 % |
9,7 %* |
1,7 % |
13,5 % |
11,3 %* |
Vorheriges Versagen der Biologikatherapie+ |
0 |
6,5 % |
6,5 % |
1,1 % |
9,2 % |
8,1 % |
Kein vorheriges Versagen der Biologikatherapie+ |
2,6 % |
15,4 % |
12,8 % |
2,4 % |
17,9 % |
15,5 % |
Abkürzungen: PBO = Placebo; UPA = Upadacitinib; aMS = adaptierter Mayo-Score, basierend auf dem Mayo Scoring System (ohne Physician's Global Assessment), mit Werten von 0 bis 9 und drei Subscores, die jeweils mit 0 (normal) bis 3 (schwerste Ausprägung) bewertet wurden: Subscore zur Stuhlhäufigkeit (SFS), Subscore zu rektalen Blutungen (RBS) und ein zentral bewerteter endoskopischer Subscore (ES). | ||||||
Krankheitsaktivität und Symptome
Der partielle adaptierte Mayo-Score (paMS) setzt sich aus SFS und RBS zusammen. Symptomatisches Ansprechen gemäß paMS wird definiert als Verringerung um ≥ 1 Punkt und ≥ 30 % gegenüber Baseline und eine Verringerung des RBS um ≥ 1 oder ein absoluter RBS-Wert von ≤ 1. Eine statistisch signifikante Verbesserung gemäß paMS gegenüber Placebo zeigte sich bereits in Woche 2 (UC-1: 60,1 % gegenüber 27,3 % und UC-2: 63,3 % gegenüber 25,9 %).
Verlängerte Einleitungsphase
Insgesamt 125 Patienten in UC‑1 und UC‑2, die nach 8‑wöchiger Behandlung mit 45 mg Upadacitinib einmal täglich kein klinisches Ansprechen erreichten, traten in eine 8‑wöchige, offene, verlängerte Einleitungsphase ein. Nach weiteren 8 Wochen der Behandlung (insgesamt 16 Wochen) mit 45 mg Upadacitinib einmal täglich erreichten 48,3 % der Patienten ein klinisches Ansprechen gemäß aMS. Von den Patienten, die auf die Behandlung mit 45 mg Upadacitinib einmal täglich über 16 Wochen ansprachen, behielten 35,7 % bzw. 66,7 % der Patienten das klinische Ansprechen gemäß aMS bis Woche 52 unter einer Erhaltungstherapie mit 15 mg bzw. 30 mg Upadacitinib einmal täglich bei und 19,0 % bzw. 33,3 % erreichten eine klinische Remission gemäß aMS.
Erhaltungsstudie (UC‑3)
Die Auswertung zur Wirksamkeit von UC‑3 wurde bei 451 Patienten durchgeführt, die mit einer 8‑wöchigen Einleitungstherapie mit 45 mg Upadacitinib einmal täglich ein klinisches Ansprechen gemäß aMS erreichten. Die Patienten wurden randomisiert und erhielten bis zu 52 Wochen lang einmal täglich 15 mg Upadacitinib, 30 mg Upadacitinib oder Placebo.
Der primäre Endpunkt war die klinische Remission gemäß aMS in Woche 52. Tabelle 16 zeigt die wichtigsten sekundären Endpunkte, darunter die Aufrechterhaltung der klinischen Remission, kortikosteroidfreie klinische Remission, Mukosaheilung, histologisch-endoskopische Mukosaheilung und tiefe Mukosaheilung.
Tabelle 16 Anteil der Patienten, die in Woche 52 die primären und die wichtigsten sekundären Endpunkte in der Erhaltungsstudie UC‑3 erreichten
PBO |
UPA |
UPA |
Behandlungsunterschied |
Behandlungsunterschied |
|
Klinische Remissiona |
12,1 % |
42,3 % |
51,7 % |
30,7 %* |
39,0 %* |
Vorheriges Versagen der Biologikatherapie+ |
7,5 % |
40,5 % |
49,1 % |
33,0 % |
41,6 % |
Kein vorheriges Versagen der Biologikatherapie+ |
17,6 % |
43,9 % |
54,0 % |
26,3 % |
36,3 % |
Aufrechterhaltung der klinischen Remissionb |
N = 54 |
N = 47 |
N = 58 |
37,4 %* |
47,0 %* |
Vorheriges Versagen der Biologikatherapie |
N = 22 |
N = 17 |
N = 20 |
62,8 % |
59,4 % |
Kein vorheriges Versagen der Biologikatherapie |
N = 32 |
N = 30 |
N = 38 |
21,3 % |
39,9 % |
Kortikosteroidfreie klinische Remissionc |
N = 54 |
N = 47 |
N = 58 |
35,4 %* |
45,1 %* |
Vorheriges Versagen der Biologikatherapie |
N = 22 |
N = 17 |
N = 20 |
57,0 % |
59,4 % |
Kein vorheriges Versagen der Biologikatherapie |
N = 32 |
N = 30 |
N = 38 |
21,3 % |
37,2 % |
Mukosaheilungd |
14,5 % |
48,7 % |
61,6 % |
34,4 %* |
46,3 %* |
Vorheriges Versagen der Biologikatherapie+ |
7,8 % |
43,3 % |
56,1 % |
35,5 % |
48,3 % |
Kein vorheriges Versagen der Biologikatherapie+ |
22,5 % |
53,6 % |
66,6 % |
31,1 % |
44,1 % |
Histologisch-endoskopische Mukosaheilunge |
11,9 % |
35,0 % |
49,8 % |
23,8 %* |
37,3 %* |
Vorheriges Versagen der Biologikatherapie+ |
5,2 % |
32,9 % |
47,6 % |
27,7 % |
42,4 % |
Kein vorheriges Versagen der Biologikatherapie+ |
20,0 % |
36,9 % |
51,8 % |
16,9 % |
31,8 % |
Tiefe Mukosaheilungf |
4,7 % |
17,6 % |
19,0 % |
13,0 %* |
13,6 %* |
Vorheriges Versagen der Biologikatherapie+ |
2,5 % |
17,2 % |
16,1 % |
14,7 % |
13,6 % |
Kein vorheriges Versagen der Biologikatherapie+ |
7,5 % |
18,0 % |
21,6 % |
10,6 % |
14,2 % |
Abkürzungen: PBO = Placebo; UPA = Upadacitinib; aMS = adaptierter Mayo-Score, basierend auf dem Mayo Scoring System (ohne Physician's Global Assessment), mit Werten von 0 bis 9 und drei Subscores, die jeweils mit 0 (normal) bis 3 (schwerste Ausprägung) bewertet wurden: Subscore zur Stuhlhäufigkeit (SFS), Subscore zu rektalen Blutungen (RBS) und ein zentral bewerteter endoskopischer Subscore (ES). | |||||
Krankheitssymptome
Eine symptomatische Remission gemäß paMS, definiert als SFS ≤ 1 und RBS = 0, erreichten im Zeitverlauf bis Woche 52 mehr Patienten, die mit 15 mg und 30 mg Upadacitinib einmal täglich behandelt wurden, als unter Placebo (Abbildung 2).
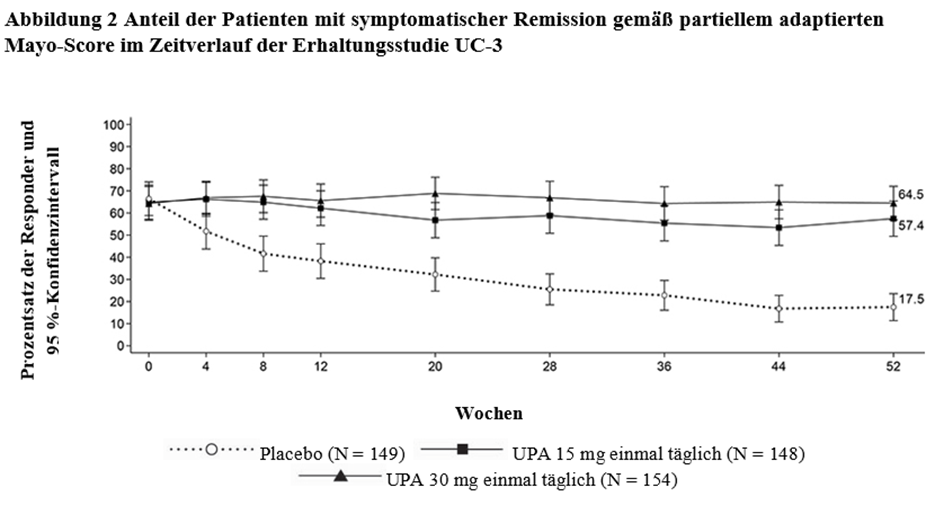
Endoskopische Beurteilung
Die endoskopische Remission (Normalisierung des endoskopischen Erscheinungsbildes der Schleimhaut) wurde als ES von 0 definiert. In Woche 8 erreichte ein signifikant größerer Anteil der Patienten, die mit 45 mg Upadacitinib einmal täglich behandelt wurden, eine endoskopische Remission im Vergleich zu Placebo (UC-1: 13,7 % gegenüber 1,3 %, UC-2: 18,2 % gegenüber 1,7 %). In UC‑3 erreichte ein signifikant größerer Anteil der Patienten, die mit 15 mg und 30 mg Upadacitinib einmal täglich behandelt wurden, im Vergleich zu Placebo in Woche 52 eine endoskopische Remission (24,2 % und 25,9 % vs. 5,6 %). Die Aufrechterhaltung der Muskosaheilung in Woche 52 (ES ≤ 1 ohne Friabilität) wurde unter denjenigen Patienten, die am Ende der Einleitungstherapie eine Muskosaheilung erreichten, im Vergleich zu Placebo bei einem signifikant größeren Anteil der Patienten beobachtet, die mit 15 mg und 30 mg Upadacitinib einmal täglich behandelt wurden (61,6 % und 69,5 % vs. 19,2 %).
Lebensqualität
Patienten, die mit Upadacitinib behandelt wurden, zeigten im Vergleich zu Placebo eine signifikant größere und klinisch bedeutsame Verbesserung der gesundheitsbezogenen Lebensqualität, gemessen anhand des Inflammatory-Bowel-Disease-Questionnaire(IBDQ)-Gesamtscores. Verbesserungen zeigten sich in allen 4 Domänen-Scores: systemische Symptome (einschließlich Fatigue), soziale Funktion, emotionale Funktion und Darmsymptome (einschließlich Bauchschmerzen und Stuhldrang). Die Veränderungen des IBDQ-Gesamtscores in Woche 8 gegenüber dem Ausgangswert betrugen mit 45 mg Upadacitinib einmal täglich im Vergleich zu Placebo 55,3 bzw. 21,7 in der Studie UC-1 und 52,2 bzw. 21,1 in der Studie UC-2. Veränderungen des IBDQ-Gesamtscores gegenüber Baseline betrugen in Woche 52 bei Patienten unter 15 mg Upadacitinib und 30 mg Upadacitinib einmal täglich 49,2 und 58,9 und unter Placebo 17,9.
Langzeit-Verlängerungsstudie (UC-4)
Patienten, die nach einem Jahr eine klinische Remission gemäß aMS in UC-3 erreichten, konnten in der Verlängerungsstudie (UC-4) mit derselben Dosis fortfahren. Zu Beginn der Studie UC-4 befanden sich mit 15 mg bzw. 30 mg Upadacitinib 96 bzw. 146 Patienten in klinischer Remission und 49 bzw. 82 Patienten in endoskopischer Remission. Diese Population überschneidet sich teilweise, aber nicht vollständig, mit der in der obigen Tabelle aufgeführten Population, die den Anteil der Patienten darstellt, die in der Erhaltungsstudie UC-3 die Endpunkte in Woche 52 erreichten. Von den Patienten, die nach einem Jahr eine Remission gemäß aMS in UC-3 erreichten und für die Daten aus 96 Wochen vorlagen, konnten 55/70 (78,6 %) bzw. 75/89 (84,3 %) die klinische Remission und 22/34 (64,7 %) bzw. 40/54 (74,1 %) die endoskopische Remission nach 96 Wochen zusätzlicher Behandlung mit 15 mg bzw. 30 mg Upadacitinib aufrecht halten.
Bei Patienten, die nach Abschluss von UC-3 (ein Jahr) in die Verlängerungsstudie aufgenommen wurden und über Daten aus 96 Wochen verfügten, blieb die Verbesserung des IBDQ-Gesamtscores und IBDQ-Domainscores bis Woche 96 in UC-4 erhalten.
Das Sicherheitsprofil von Upadacitinib bei langfristiger Behandlung entsprach dem in der placebokontrollierten Phase.
Morbus Crohn
Die Wirksamkeit und Sicherheit von Upadacitinib wurden in drei multizentrischen, doppelblinden, placebokontrollierten Phase‑III-Studien untersucht: zwei Einleitungsstudien, CD‑1 (U‑EXCEED) und CD‑2 (U‑EXCEL), gefolgt von einer 52‑wöchigen Langzeit-Fortsetzungsstudie mit Erhaltungstherapie, CD‑3 (U‑ENDURE). Die koprimären Endpunkte waren die klinische Remission und das endoskopische Ansprechen in Woche 12 bei CD‑1 und CD‑2 und in Woche 52 bei CD‑3.
Die eingeschlossenen Patienten waren zwischen 18 und 75 Jahre alt und hatten mittelschweren bis schweren aktiven Morbus Crohn (MC), definiert als eine durchschnittliche tägliche Stuhlfrequenz (SF) mit sehr weichem oder flüssigem Stuhl von ≥ 4 und/oder ein durchschnittlicher täglicher abdominaler Schmerz-Score bzw. Score für Bauchschmerzen (BS) von ≥ 2 sowie ein von einem zentralen Prüfer bestätigter Simple Endoscopic Score for CD (SES‑CD) von ≥ 6 (bzw. ≥ 4 bei isolierter Erkrankung des Ileums) unter Ausschluss der verengenden Komponente. Patienten mit symptomatischen Darmstrikturen wurden von den CD-Studien ausgeschlossen.
Einleitungsstudien (CD‑1 und CD‑2)
In CD‑1 und CD‑2 wurden 1 021 Patienten (495 bzw. 526 Patienten) im Verhältnis 2:1 randomisiert und 12 Wochen lang mit 45 mg Upadacitinib einmal täglich oder Placebo behandelt.
In CD‑1 hatten alle Patienten auf eine Behandlung mit einem oder mehreren Biologika unzureichend angesprochen oder diese nicht vertragen (vorheriges Versagen der Biologikatherapie). Von diesen Patienten hatten 61 % (301/495) auf eine Behandlung mit zwei oder mehr Biologika unzureichend angesprochen oder diese nicht vertragen.
In CD‑2 hatten 45 % (239/526) der Patienten auf eine Behandlung mit einem oder mehreren Biologika unzureichend angesprochen oder diese nicht vertragen (vorheriges Versagen der Biologikatherapie) und 55 % (287/526) hatten auf konventionelle Therapien unzureichend angesprochen oder diese nicht vertragen, aber nicht auf eine Behandlung mit einem Biologikum (kein vorheriges Versagen der Biologikatherapie).
Bei Baseline erhielten in CD‑1 und CD‑2 34 % bzw. 36 % der Patienten Kortikosteroide, 7 % bzw. 3 % Immunmodulatoren und 15 % bzw. 25 % Aminosalicylate.
In beiden Studien begannen Patienten, die bei Baseline Kortikosteroide erhielten, ab Woche 4 mit einem ausschleichenden Kortikosteroid-Behandlungsschema.
Beide Studien beinhalteten eine 12‑wöchige Fortsetzungsphase mit 30 mg Upadacitinib einmal täglich für Patienten, die mit 45 mg Upadacitinib einmal täglich behandelt wurden und in Woche 12 kein klinisches Ansprechen gemäß SF/Score für BS erreichten (≥ 30 % Abnahme der durchschnittlichen täglichen SF mit sehr weichem oder flüssigem Stuhl und/oder ≥ 30 % Abnahme des durchschnittlichen täglichen Scores für BS und beide nicht höher als bei Baseline).
Klinische Krankheitsaktivität und Symptome
In CD‑1 und CD‑2 erreichte ein signifikant größerer Anteil der mit 45 mg Upadacitinib behandelten Patienten den koprimären Endpunkt der klinischen Remission in Woche 12 im Vergleich zu Placebo (Tabelle 17). Der Wirkeintritt war schnell und wurde bereits in Woche 2 erreicht (Tabelle 17).
In beiden Studien zeigte sich bei Patienten, die 45 mg Upadacitinib erhielten, eine signifikant größere Verbesserung der Fatigue gegenüber Baseline, gemessen anhand des FACIT‑F-Scores in Woche 12 im Vergleich zu Placebo.
Endoskopische Beurteilung
In CD‑1 und CD‑2 erreichte ein signifikant größerer Anteil der mit 45 mg Upadacitinib behandelten Patienten den koprimären Endpunkt des endoskopischen Ansprechens in Woche 12 im Vergleich zu Placebo (Tabelle 17). In CD‑1 und CD‑2 erreichte ein größerer Anteil der mit 45 mg Upadacitinib behandelten Patienten (14 % bzw. 19 %) einen SES‑CD von 0–2 im Vergleich zu Placebo (0 % bzw. 5 %).
Tabelle 17 Anteil der Patienten, die in den Einleitungsstudien CD‑1 und CD‑2 die primären und weiteren Endpunkte zur Wirksamkeit erreichten
Studie |
CD‑1 |
CD‑2 |
||||
Behandlungsarm |
PBO |
UPA |
Behandlungs- |
PBO |
UPA |
Behandlungs- |
Koprimäre Endpunkte in Woche 12 | ||||||
Klinische Remissiona |
14 % |
40 % |
26 % |
22 % |
51 % |
29 % |
Vorheriges Versagen der Biologikatherapie |
N = 78 |
N = 161 |
33 % |
|||
Kein vorheriges Versagen der Biologikatherapie |
N = 98 |
N = 189 |
26 % |
|||
Endoskopisches Ansprechenb |
4 % |
35 % |
31 % |
13 % |
46 % |
33 % |
Vorheriges Versagen der Biologikatherapie |
N = 78 |
N = 161 |
29 % |
|||
Kein vorheriges Versagen der Biologikatherapie |
N = 98 |
N = 189 |
36 % |
|||
Weitere Endpunkte in Woche 12 | ||||||
Klinische Remission gemäß CDAIc |
21 % |
39 % |
18 % |
29 % |
49 % |
21 % |
Klinisches Ansprechen (CR‐100)d |
27 % |
51 % |
23 % |
37 % |
57 % |
20 % |
Kortikosteroidfreie klinische Remissiona,e |
N = 60 |
N = 108 |
30 % |
N = 64 |
N = 126 |
33 % |
Endoskopische Remissionf |
2 % |
19 % |
17 % |
7 % |
29 % |
22 % |
Mukosaheilungg |
N = 171 |
N = 322 |
17 % |
N = 174 |
N = 349 |
20 % |
Endpunkte zum frühen Wirkeintritt | ||||||
Klinische Remission in Woche 4a |
9 % |
32 % |
23 % |
15 % |
36 % |
21 % |
CR‐100 in Woche 2d |
12 % |
33 % |
21 % |
20 % |
32 % |
12 % |
Abkürzungen: PBO = Placebo; UPA = Upadacitinib | ||||||
Erhaltungsstudie (CD‑3)
Die Auswertung zur Wirksamkeit von CD‑3 wurde bei 502 Patienten durchgeführt, die mit der 12‑wöchigen Einleitungstherapie mit 45 mg Upadacitinib einmal täglich ein klinisches Ansprechen gemäß SF/Score für BS erreichten. Die Patienten wurden erneut randomisiert und erhielten 52 Wochen lang entweder ein Erhaltungstherapieschema mit 15 mg oder 30 mg Upadacitinib einmal täglich oder Placebo.
Klinische Krankheitsaktivität und Symptome
In Woche 52 erreichte ein signifikant größerer Anteil der Patienten, die mit 15 mg und 30 mg Upadacitinib behandelt wurden, den koprimären Endpunkt der klinischen Remission im Vergleich zu Placebo (Abbildung 3, Tabelle 18).
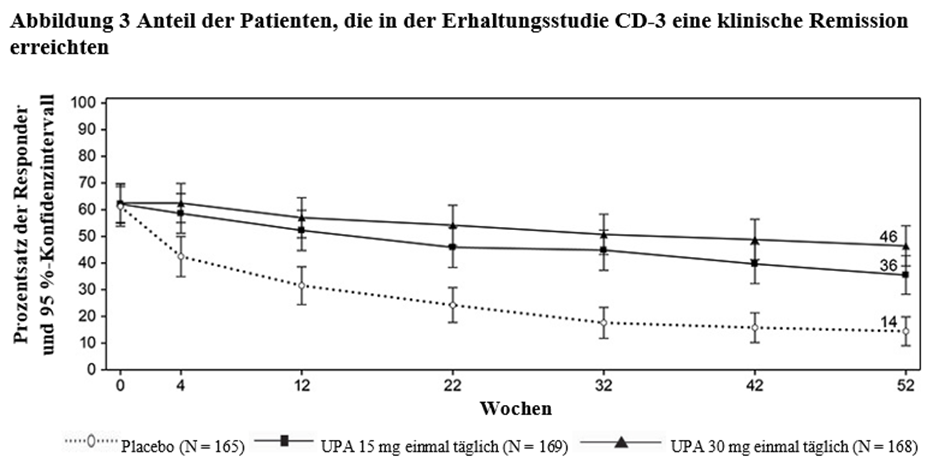
In Woche 52 verzeichneten Patienten, die mit 30 mg Upadacitinib behandelt wurden, im Vergleich zu Placebo eine signifikant größere Verbesserung der Fatigue gegenüber Baseline, gemessen anhand des FACIT‑F-Scores.
Tabelle 18 Anteil der Patienten, die in Woche 52 die primären und weiteren Endpunkte zur Wirksamkeit der Erhaltungsstudie CD‑3 erreichten
Behandlungsarm |
PBO+ |
UPA |
UPA |
Behandlungsunterschied |
Behandlungsunterschied |
Koprimäre Endpunkte | |||||
Klinische Remissiona |
14 % |
36 % |
46 % |
22 % |
32 % |
Vorheriges Versagen der Biologikatherapie |
N = 126 |
N = 124 |
N = 127 |
24 % |
34 % |
Kein vorheriges Versagen der Biologikatherapie |
N = 39 |
N = 45 |
N = 41 |
12 % |
26 % |
Endoskopisches Ansprechenb |
7 % |
28 % |
40 % |
21 % |
34 % |
Vorheriges Versagen der Biologikatherapie |
N = 126 |
N = 124 |
N = 127 |
19 % |
35 % |
Kein vorheriges Versagen der Biologikatherapie |
N = 39 |
N = 45 |
N = 41 |
22 % |
26 % |
Weitere Endpunkte | |||||
Klinische Remission gemäß CDAIc |
15 % |
37 % |
48 % |
24 % |
33 % |
Klinisches Ansprechen (CR‐100)d |
15 % |
41 % |
51 % |
27 % |
36 % |
Kortikosteroidfreie klinische Remissiona,e |
14 % |
35 % |
45 % |
21 % |
30 % |
Aufrechterhaltung der klinischen Remissiona,f |
N = 101 |
N = 105 |
N = 105 |
32 % |
40 % |
Endoskopische Remissiong |
5 % |
19 % |
29 % |
14 % |
24 % |
Mukosaheilungh |
N = 164 |
N = 167 |
N = 168 |
10 % |
21 % |
Tiefe Remissiona,i |
4 % |
14 % |
23 % |
10 % |
18 % |
Abkürzungen: PBO = Placebo; UPA = Upadacitinib | |||||
Patienten, die in Woche 12 in CD‑1 und CD‑2 kein klinisches SF/BS-Ansprechen auf die Einleitungstherapie mit Upadacitinib zeigten (122 Patienten), erhielten 30 mg Upadacitinib einmal täglich für weitere 12 Wochen. Von diesen Patienten erreichten 53 % in Woche 24 ein klinisches Ansprechen. Von den Patienten, die auf den verlängerten Behandlungszeitraum ansprachen und weiterhin eine Erhaltungstherapie mit 30 mg Upadacitinib erhielten, erreichten 25 % eine klinische Remission und 22 % ein endoskopisches Ansprechen in Woche 52.
Endoskopische Beurteilung
In CD‑3 erreichte in Woche 52 ein signifikant größerer Anteil der mit 15 mg und 30 mg Upadacitinib behandelten Patienten den koprimären Endpunkt des endoskopischen Ansprechens im Vergleich zu Placebo (Tabelle 18). Zusätzlich zu den in Tabelle 18 beschriebenen endoskopischen Endpunkten erreichte in Woche 52 ein größerer Anteil der mit 15 mg und 30 mg Upadacitinib behandelten Patienten (11 % bzw. 21 %) einen SES‑CD von 0–2 im Vergleich zu Placebo (3 %). Eine kortikosteroidfreie endoskopische Remission bei Patienten, die bei Baseline Steroide anwendeten, wurde in Woche 52 bei einem größeren Anteil der mit 15 mg und 30 mg Upadacitinib behandelten Patienten (17 % bzw. 25 %) im Vergleich zu Placebo (3 %) erreicht.
Abklingen der extraintestinalen Manifestationen
In Woche 52 wurde bei einem größeren Anteil der mit 15 mg Upadacitinib behandelten Patienten (25 %) und bei einem signifikant größeren Anteil der mit 30 mg Upadacitinib behandelten Patienten (36 %) im Vergleich zu Placebo (15 %) ein Abklingen der extraintestinalen Manifestationen beobachtet.
Notfallbehandlung
In CD‑3 konnten Patienten, die auf die Erhaltungstherapie unzureichend oder nicht mehr ansprachen, eine Notfallbehandlung mit 30 mg Upadacitinib erhalten. Von den Patienten, die in die Gruppe mit 15 mg Upadacitinib randomisiert wurden und mindestens 12 Wochen lang eine Notfallbehandlung mit 30 mg Upadacitinib erhielten, erreichten 84 % (76/90) ein klinisches SF/BS-Ansprechen und 48 % (43/90) eine klinische Remission 12 Wochen nach Beginn der Notfallbehandlung.
Ergebnisse zur gesundheitsbezogenen Lebensqualität
Patienten, die mit Upadacitinib behandelt wurden, erreichten im Vergleich zu Patienten unter Placebo eine größere Verbesserung der gesundheitsbezogenen Lebensqualität (HRQOL), gemessen anhand des Inflammatory-Bowel-Disease-Questionnaire(IBDQ)-Gesamtscores. Verbesserungen zeigten sich in allen 4 Domänen-Scores: systemische Symptome (einschließlich Fatigue) und Darmsymptome (einschließlich Bauchschmerzen und Stuhldrang) sowie soziale und emotionale Funktion. Veränderungen des IBDQ-Gesamtscores gegenüber Baseline betrugen in Woche 12 unter 45 mg Upadacitinib einmal täglich 46,0 im Vergleich zu 21,6 unter Placebo in CD‑1 bzw. 46,3 im Vergleich zu 24,4 in CD‑2. Veränderungen des IBDQ‑Gesamtscores gegenüber Baseline betrugen in Woche 52 bei mit 15 mg bzw. 30 mg Upadacitinib einmal täglich behandelten Patienten 59,3 bzw. 64,5 und 46,4 unter Placebo.
Kinder und Jugendliche
Insgesamt 542 Jugendliche im Alter von 12 bis 17 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis wurden in den drei globalen Phase‑III-Studien randomisiert, von denen 344 in der Primäranalyse untersucht wurden. Jugendliche in der Primäranalyse wurden randomisiert und erhielten entweder 15 mg (N = 114) bzw. 30 mg (N = 114) Upadacitinib oder ein entsprechendes Placebo (N = 116) als Monotherapie oder in Kombination mit topischen Kortikosteroiden. Die Wirksamkeit war bei Jugendlichen und Erwachsenen vergleichbar. Das Sicherheitsprofil bei Jugendlichen war in der Regel mit dem bei Erwachsenen vergleichbar, wobei die Raten einiger unerwünschter Ereignisse, u. a. Neutropenie und Herpes zoster, dosisabhängig höher waren. Unter beiden Dosierungen war die Neutropenierate bei Jugendlichen etwas höher als bei Erwachsenen. Unter beiden Dosierungen war die Herpes-zoster-Rate bei Erwachsenen höher als bei Jugendlichen.
Tabelle 19 Ergebnisse zur Wirksamkeit von Upadacitinib bei Jugendlichen in Woche 16
Studie |
MEASURE UP 1 |
MEASURE UP 2 |
AD UP |
|||||||||
Behandlungsgruppe |
PBO |
UPA |
UPA |
PBO |
UPA |
UPA |
PBO + |
UPA |
UPA |
|||
Anzahl randomisierter jugendlicher Studienteilnehmer |
40 |
42 |
42 |
36 |
33 |
35 |
40 |
39 |
37 |
|||
% Responder (95 %-CI) | ||||||||||||
vIGA-AD 0/1a,b |
8 |
38 |
69 |
3 |
42 |
62 |
8 |
31 |
65 |
|||
EASI75a |
8 |
71 |
83 |
14 |
67 |
74 |
30 |
56 |
76 |
|||
NRS‑Score zum schlimmsten Pruritusc |
15 |
45 |
55 |
3 |
33 |
50 |
13 |
42 |
55 |
|||
Abkürzungen: UPA = Upadacitinib (RINVOQ); PBO = Placebo | ||||||||||||
Die Europäische Arzneimittel-Agentur hat für RINVOQ eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in der Behandlung der chronischen idiopathischen Arthritis (einschließlich rheumatoider Arthritis, Psoriasis-Arthritis, Spondyloarthritis und juveniler idiopathischer Arthritis), der atopischen Dermatitis, Colitis ulcerosa und Morbus Crohn gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Plasmaexposition von Upadacitinib ist im gesamten therapeutischen Dosisbereich proportional zur Dosis. Innerhalb von 4 Tagen werden Steady‑State-Konzentrationen im Plasma erreicht, wobei es nach mehrfacher Anwendung einmal täglich zu einer minimalen Akkumulation kommt.
Resorption
Nach oraler Anwendung der Upadacitinib-Retardformulierung wird Upadacitinib mit einer mittleren Tmax von 2 bis 4 Stunden resorbiert. Die Einnahme von Upadacitinib zusammen mit einer fettreichen Mahlzeit hatte keine klinisch relevante Auswirkung auf die Upadacitinib-Exposition (AUC um 29 % und Cmax um 39 % auf 60 % erhöht). In klinischen Studien wurde Upadacitinib unabhängig von den Mahlzeiten angewendet (siehe Abschnitt 4.2). In vitro ist Upadacitinib ein Substrat für die Effluxtransporter P-gp und BCRP.
Verteilung
Upadacitinib wird zu 52 % an Plasmaproteine gebunden. Upadacitinib verteilt sich gleichermaßen auf Zellbestandteile von Plasma und Blut, wie das Blut-Plasma-Verhältnis von 1,0 zeigt.
Metabolismus
Der Metabolismus von Upadacitinib wird durch CYP3A4 und möglicherweise in geringem Maße auch durch CYP2D6 vermittelt. Die pharmakologische Wirkung von Upadacitinib wird dem Grundmolekül zugeschrieben. In einer Humanstudie mit radioaktiv markiertem Wirkstoff entfielen 79 % der gesamten Radioaktivität im Plasma auf unverändertes Upadacitinib, während der Hauptmetabolit (Produkt aus Monooxidation und nachfolgender Glucuronidierung) 13 % der gesamten Radioaktivität im Plasma ausmachte. Für Upadacitinib wurden keine aktiven Metaboliten identifiziert.
Elimination
Nach Anwendung einer Einzeldosis von [14C]-Upadacitinib-Lösung mit sofortiger Freisetzung wurde Upadacitinib vorwiegend als unverändertes Grundmolekül im Urin (24 %) und im Stuhl (38 %) ausgeschieden. Etwa 34 % der Upadacitinib-Dosis wurden als Metaboliten ausgeschieden. Die mittlere terminale Eliminationshalbwertszeit von Upadacitinib lag im Bereich von 9 bis 14 Stunden.
Besondere Patientengruppen
Niereninsuffizienz
Die AUC von Upadacitinib war im Vergleich zu Studienteilnehmern mit normaler Nierenfunktion bei Studienteilnehmern mit leichter Niereninsuffizienz (geschätzte glomeruläre Filtrationsrate 60–89 ml/min/1,73 m2) um 18 %, mit mittelschwerer Niereninsuffizienz (geschätzte glomeruläre Filtrationsrate 30–59 ml/min/1,73 m2) um 33 % und mit schwerer Niereninsuffizienz (geschätzte glomeruläre Filtrationsrate 15–29 ml/min/1,73 m2) um 44 % höher. Die Cmax von Upadacitinib war bei Studienteilnehmern mit normaler und beeinträchtigter Nierenfunktion vergleichbar. Eine leichte oder mittelschwere Niereninsuffizienz hat keine klinisch relevante Auswirkung auf die Upadacitinib-Exposition (siehe Abschnitt 4.2).
Leberinsuffizienz
Eine leichte (Child-Pugh A) und mittelschwere (Child-Pugh B) Leberinsuffizienz hat keinen klinisch relevanten Einfluss auf die Exposition von Upadacitinib. Die AUC von Upadacitinib war bei Studienteilnehmern mit leichter und mittelschwerer Leberinsuffizienz im Vergleich zu Studienteilnehmern mit normaler Leberfunktion um 28 % bzw. 24 % höher. Die Cmax von Upadacitinib war im Vergleich zu Studienteilnehmern mit normaler Leberfunktion bei Studienteilnehmern mit leichter Leberinsuffizienz unverändert und bei Studienteilnehmern mit mittelschwerer Leberinsuffizienz um 43 % höher. Upadacitinib wurde bei Patienten mit schwerer (Child-Pugh C) Leberinsuffizienz nicht untersucht.
Kinder und Jugendliche
Die Pharmakokinetik von Upadacitinib wurde bei Kindern und Jugendlichen mit rheumatoider Arthritis, Psoriasis-Arthritis, axialer Spondyloarthritis, Colitis ulcerosa und Morbus Crohn noch nicht untersucht (siehe Abschnitt 4.2).
Die Pharmakokinetik und Steady-State-Konzentrationen von Upadacitinib sind bei Erwachsenen und Jugendlichen im Alter von 12 bis 17 Jahren mit atopischer Dermatitis vergleichbar. Die Dosierungsempfehlung bei Jugendlichen von 30 kg bis < 40 kg wurde mittels pharmakokinetischer Modellierung und Simulation von Populationen bestimmt. Es liegen keine klinischen Daten zur Exposition bei Jugendlichen mit einem Körpergewicht < 40 kg vor.
Die Pharmakokinetik von Upadacitinib bei Kindern (< 12 Jahre alt) mit atopischer Dermatitis ist nicht erwiesen.
Intrinsische Faktoren
Alter, Geschlecht, Körpergewicht und ethnische Zugehörigkeit hatten keine klinisch relevante Auswirkung auf die Exposition von Upadacitinib. Die Pharmakokinetik von Upadacitinib ist bei Patienten mit rheumatoider Arthritis, Psoriasis-Arthritis, axialer Spondyloarthritis, Riesenzellarteriitis, atopischer Dermatitis, Colitis ulcerosa und Morbus Crohn vergleichbar.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Bei Expositionen (basierend auf AUC), die bei männlichen und weiblichen Sprague-Dawley-Ratten etwa 4- bzw. 10-mal höher waren als die klinische Dosis von 15 mg, 2‑ bzw. 5‑mal höher als die klinische Dosis von 30 mg und 1,7‑ bzw. 4‑mal höher als die klinische Dosis von 45 mg, war Upadacitinib in einer 2-jährigen Karzinogenitätsstudie an Sprague-Dawley-Ratten nicht karzinogen. Upadacitinib war in einer 26-wöchigen Karzinogenitätsstudie an transgenen CByB6F1-Tg(HRAS)2Jic-Mäusen nicht karzinogen.
Upadacitinib war auf Grundlage der Ergebnisse von In-vitro- und In-vivo-Tests für Genmutationen und Chromosomenaberrationen weder mutagen noch genotoxisch.
In einer Studie zur Fertilität und frühen embryonalen Entwicklung hatte Upadacitinib bei Expositionen (basierend auf AUC) von bis zu etwa dem 17‑ bzw. 34‑Fachen der empfohlenen Höchstdosis für Menschen (maximum recommended human dose, MRHD) von 45 mg bei männlichen bzw. weiblichen Ratten keine Auswirkungen auf die Fertilität. In dieser Fertilitätsstudie mit Ratten wurden dosisabhängige Zunahmen der fetalen Resorptionen verbunden mit Postimplantationsverlusten auf die entwicklungstoxischen/teratogenen Wirkungen von Upadacitinib zurückgeführt. Bei Expositionen unter der klinischen Exposition (basierend auf der AUC) wurden keine Nebenwirkungen beobachtet. Postimplantationsverluste wurden bei einer Exposition, die das 9-Fache der klinischen Exposition bei der MRHD von 45 mg (basierend auf der AUC) betrug, beobachtet.
In Tierstudien zur embryonalen und fetalen Entwicklung war Upadacitinib sowohl bei Ratten als auch bei Kaninchen teratogen. Upadacitinib führte bei Ratten zu Anstiegen von Skelettmissbildungen beim 1,6‑, 0,8‑ bzw. 0,6‑Fachen der klinischen Exposition (basierend auf AUC) bei den 15‑, 30‑ bzw. 45‑mg-Dosen (MRHD). Bei Kaninchen wurde eine erhöhte Inzidenz von kardiovaskulären Missbildungen beim 15‑, 7,6‑ bzw. 6‑Fachen der klinischen Exposition bei den 15‑, 30‑ bzw. 45‑mg-Dosen (basierend auf AUC) beobachtet.
Nach Verabreichung von Upadacitinib an laktierende Ratten war der zeitliche Verlauf der Konzentrationen von Upadacitinib in der Milch in der Regel parallel zu denjenigen im Plasma, wobei die Exposition in der Milch im Verhältnis zum mütterlichen Plasma etwa 30‑fach höher war. Bei etwa 97 % des Upadacitinib-bezogenen Substanzmaterials in der Milch handelte es sich um das Grundmolekül Upadacitinib.
Tablettenkern
Mikrokristalline Cellulose
Hypromellose
Mannitol (Ph. Eur.)
Weinsäure (Ph. Eur.)
Hochdisperses Siliciumdioxid
Magnesiumstearat (Ph. Eur.)
Filmüberzug
Poly(vinylalkohol)
Macrogol
Talkum
Titandioxid (E171)
Eisen(II,III)-oxid (E172) (nur RINVOQ 15 mg Retardtabletten)
Eisen(III)-oxid (E172)
Eisen(III)-hydroxid-oxid x H2O (E172) (nur RINVOQ 45 mg Retardtabletten)
Nicht zutreffend.
RINVOQ 15 mg Retardtabletten
Retardtabletten in Blisterpackungen: 2 Jahre
Retardtabletten in Flaschen: 3 Jahre
RINVOQ 30 mg Retardtabletten
Retardtabletten in Blisterpackungen: 2 Jahre
Retardtabletten in Flaschen: 3 Jahre
RINVOQ 45 mg Retardtabletten
Retardtabletten in Blisterpackungen: 2 Jahre
Retardtabletten in Flaschen: 3 Jahre
Für dieses Arzneimittel sind bezüglich der Temperatur keine besonderen Lagerungsbedingungen erforderlich.
In der Original-Blisterpackung oder -Flasche aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Flasche fest verschlossen halten.
RINVOQ 15 mg Retardtabletten
Polyvinylchlorid/Polyethylen/Poly(chlortrifluorethylen)/Aluminium-Blisterpackungen (Kalenderpackungen) in Packungen, die 28 oder 98 Retardtabletten enthalten, oder in Mehrfachpackungen, die 84 (3 Packungen mit 28) Retardtabletten enthalten.
HDPE-Flaschen mit Trockenmittel und Polypropylenverschluss im Umkarton, die 30 Retardtabletten enthalten.
Packungsgrößen: 1 Flasche (30 Retardtabletten) oder 3 Flaschen (90 Retardtabletten)
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
RINVOQ 30 mg Retardtabletten
Polyvinylchlorid/Polyethylen/Poly(chlortrifluorethylen)/Aluminium-Blisterpackungen (Kalenderpackungen) in Packungen, die 28 oder 98 Retardtabletten enthalten.
HDPE-Flaschen mit Trockenmittel und Polypropylenverschluss im Umkarton, die 30 Retardtabletten enthalten.
Packungsgrößen: 1 Flasche (30 Retardtabletten) oder 3 Flaschen (90 Retardtabletten)
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
RINVOQ 45 mg Retardtabletten
Polyvinylchlorid/Polyethylen/Poly(chlortrifluorethylen)/Aluminium-Blisterpackungen (Kalenderpackungen) in Packungen, die 28 Retardtabletten enthalten.
HDPE-Flaschen mit Trockenmittel und Polypropylenverschluss im Umkarton, die 28 Retardtabletten enthalten.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
AbbVie Deutschland GmbH & Co. KG
Knollstraße
67061 Ludwigshafen
Deutschland
EU/1/19/1404/001
EU/1/19/1404/002
EU/1/19/1404/003
EU/1/19/1404/004
EU/1/19/1404/005
EU/1/19/1404/006
EU/1/19/1404/007
EU/1/19/1404/008
EU/1/19/1404/009
EU/1/19/1404/010
EU/1/19/1404/011
Datum der Erteilung der Zulassung: 16. Dezember 2019
Datum der letzten Verlängerung der Zulassung: 19. September 2024
Juni 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
Behördlich genehmigtes Schulungsmaterial zu diesem Arzneimittel ist durch Scannen des QR-Codes auf der Fachinformation mit einem Smartphone verfügbar. Die gleichen Informationen finden Sie auch unter der folgenden Internetadresse:
arzneimittelsuche.abbvie.de/results.html?q=rinvoq&nodeType=damSearchTag

|