
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8
RAYVOW® 50 mg Filmtabletten
RAYVOW® 100 mg Filmtabletten
RAYVOW® 50 mg Filmtabletten
Jede Filmtablette enthält 50 mg Lasmiditan (als Hemisuccinat).
RAYVOW® 100 mg Filmtabletten
Jede Filmtablette enthält 100 mg Lasmiditan (als Hemisuccinat).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette (Tablette).
RAYVOW® 50 mg Filmtabletten
Hellgraue, ovale Tablette, 8,9 x 4,9 mm, mit der Prägung “4312” auf der einen und “L‑50” auf der anderen Seite.
RAYVOW® 100 mg Filmtabletten
Hellviolette, ovale Tablette, 11,2 x 6,15 mm, mit der Prägung “4491” auf der einen und “L‑100” auf der anderen Seite.
RAYVOW ist angezeigt zur Akutbehandlung der Kopfschmerzphase von Migräne-Attacken mit oder ohne Aura bei Erwachsenen.
Dosierung
Generell beträgt die empfohlene Initialdosis bei Erwachsenen 100 mg Lasmiditan zur Akutbehandlung von Migräne-Attacken. Falls erforderlich, kann die Dosis für eine stärkere Wirksamkeit auf 200 mg erhöht oder für eine bessere Verträglichkeit auf 50 mg verringert werden.
Falls der Migräne-Kopfschmerz nach Einnahme von 50 mg oder 100 mg Lasmiditan innerhalb von 24 Stunden nach dem ersten Ansprechen erneut auftritt, kann eine zweite Dosis derselben Stärke eingenommen werden. Die Einnahme der zweiten Dosis sollte nicht innerhalb von 2 Stunden nach der ersten Einnahme erfolgen.
Es dürfen nicht mehr als 200 mg innerhalb von 24 Stunden eingenommen werden.
Wenn ein Patient auf die erste Dosis nicht anspricht, ist es unwahrscheinlich, dass eine zweite Dosis bei derselben Attacke von Nutzen ist.
Lasmiditan kann mit oder ohne Nahrung eingenommen werden.
Ältere Patienten (> 65 Jahre)
Bei älteren Patienten ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Nierenfunktionsstörung
Bei Patienten mit leichter, mittelschwerer oder schwerer Nierenfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Leberfunktionsstörung
Bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Die Anwendung von Lasmiditan wurde bei Patienten mit schwerer Leberfunktionsstörung nicht untersucht und wird daher für diese Patientengruppe nicht empfohlen (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Lasmiditan bei Kindern und Jugendlichen im Alter von 6 bis < 18 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Es gibt zur Behandlung von Migräne keinen relevanten Nutzen von Lasmiditan bei Kindern im Alter unter 6 Jahren.
Art der Anwendung
Zum Einnehmen.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Auswirkungen auf das Zentralnervensystem (ZNS) und die Verkehrstüchtigkeit
Lasmiditan ist mit Nebenwirkungen, die das ZNS betreffen, verbunden. In einer Studie im Fahrsimulator an gesunden Probanden beeinträchtigte Lasmiditan die Verkehrstüchtigkeit signifikant (siehe Abschnitt 4.7). Die Patienten sollten darauf hingewiesen werden, nach der Einnahme jeder Lasmiditan-Dosis für mindestens 8 Stunden kein Fahrzeug zu führen oder anderen Aktivitäten nachzugehen, die eine erhöhte Aufmerksamkeit erfordern, selbst wenn sie sich dazu in der Lage fühlen. Patienten, die diesen Rat nicht befolgen können, sollten Lasmiditan nicht einnehmen.
Serotonin‑Syndrom
Serotonin‑Syndrom wurde berichtet und kann unter Lasmiditan auftreten oder bei gleichzeitiger Anwendung mit anderen serotonergen Arzneimitteln [z. B. Selektive Serotonin-Wiederaufnahmehemmer (SSRIs), Serotonin‑Noradrenalin‑Wiederaufnahmehemmer (SNRIs), trizyklische Antidepressiva (TCAs) und Monoaminoxidase (MAO)-Hemmer]. Die klinische Erfahrung zur Anwendung von Lasmiditan und Triptanen in zeitlicher Nähe ist begrenzt. Die Risiken, ein Serotonin‑Syndrom zu entwickeln, können sich addieren. Zu den Symptomen des Serotonin‑Syndroms können Veränderungen des mentalen Zustands (z. B. Agitiertheit, Halluzinationen, Koma), autonome Instabilität (z. B. Tachykardie, labiler Blutdruck, Hyperthermie), neuromuskuläre Symptome (z. B. Hyperreflexie, Koordinationsstörungen) und/oder gastrointestinale Symptome (z. B. Übelkeit, Erbrechen, Durchfall) zählen. Diese Reaktionen können schwer sein. Die Symptome treten normalerweise innerhalb von Minuten bis Stunden nach Einnahme eines weiteren serotonergen Arzneimittels oder einer höheren Dosis auf. Wenn eine gleichzeitige Behandlung mit anderen serotonergen Arzneimitteln klinisch erforderlich ist, wird eine angemessene Überwachung des Patienten, insbesondere zu Beginn der Behandlung und bei Dosiserhöhungen, empfohlen. Lasmiditan ist bei Verdacht auf ein Serotonin‑Syndrom abzusetzen.
ZNS‑dämpfende Arzneimittel
Aufgrund des Potenzials von Lasmiditan, eine Sedierung sowie andere kognitive und/oder neuropsychiatrische Nebenwirkungen zu verursachen, sollte Lasmiditan in Kombination mit Alkohol oder anderen ZNS-dämpfenden Arzneimitteln mit Vorsicht angewendet werden.
Nicht bestimmungsgemäßer Arzneimittelgebrauch oder Missbrauchspotenzial
In einer Studie zum Missbrauchspotenzial zeigten Menschen mit Drogenkonsum bei Einzelgaben von 100 mg oder 200 mg Lasmiditan eine größere Vorliebe für Lasmiditan als für Placebo. In einer separaten Studie fand sich bei gesunden Probanden kein Hinweis auf einen körperlichen Entzug nach abruptem Absetzen nach 7 Tagen der Einnahme.
Die Patienten sollten auf ein Missbrauchspotenzial hin untersucht und bezüglich Anzeichen eines Lasmiditan-Missbrauchs beobachtet werden.
Kopfschmerz bei Medikamentenübergebrauch
Der übermäßige Gebrauch jeglicher Art von Arzneimitteln gegen Kopfschmerzen kann diese verschlimmern. Wenn dies festgestellt oder vermutet wird, sollte ärztlicher Rat eingeholt und die Behandlung beendet werden. Die Diagnose des Kopfschmerzes bei Medikamentenübergebrauch sollte bei Patienten angenommen werden, die trotz (oder wegen) regelmäßiger Einnahme von Arzneimitteln gegen Kopfschmerzen häufig oder täglich an Kopfschmerzen leiden.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro Tablette, d. h. es ist nahezu „natriumfrei“.
Arzneimittel, die die Herzfrequenz senken
Lasmiditan wurde mit einer Abnahme der Herzfrequenz (HF) in Verbindung gebracht. Propranolol und Lasmiditan zusammen verringerten die HF um ein mittleres Maximum von 19,3 Schlägen pro Minute (d. h. eine zusätzliche Abnahme von 5,1 Schlägen pro Minute im Vergleich zu Propranolol allein). Dies sollte bei Patienten berücksichtigt werden, bei denen diese Größenordnungen einer HF-Abnahme ein Problem darstellen könnte, einschließlich Patienten, die Arzneimittel einnehmen, die die Herzfrequenz senken.
Serotonerge Arzneimittel
Die gleichzeitige Anwendung von Lasmiditan und Arzneimitteln (z. B. SSRIs, SNRIs, TZAs), die den Serotoninspiegel erhöhen, kann das Risiko für ein Serotonin‑Syndrom erhöhen. Die klinische Erfahrung zur Anwendung von Lasmiditan und Triptanen in zeitlicher Nähe ist begrenzt. Die Risiken, ein Serotonin‑Syndrom zu entwickeln, können sich addieren. Vorsicht ist geboten. (siehe Abschnitt 4.4).
Wirkungen von Lasmiditan auf andere Arzneimittel
Die tägliche Einnahme von Lasmiditan veränderte nicht die Pharmakokinetik (PK) von Midazolam, Koffein oder Tolbutamid, die Substrate von CYP3A, CYP1A2 bzw. CYP2C9 sind. Die gleichzeitige Anwendung von Lasmiditan mit Sumatriptan (MAO-A- und OCT1-Substrat) oder Propranolol (CYP2D6-Substrat) führte zu keinen klinisch bedeutsamen Veränderungen der Exposition dieser Arzneimittel. Nach einer Einzeldosis Lasmiditan nahm die renale Kreatinin-Clearance über 24 Stunden im Vergleich zu Placebo leicht (11 %) ab, ohne Veränderung der glomerulären Filtrationsrate (GFR).
Lasmiditan ist ein in-vitro-Inhibitor von P-Glykoprotein (P-gp) und des breast cancer resistant protein (BCRP). In einer Arzneimittel-Interaktionsstudie erhöhte Lasmiditan die systemische Exposition von gleichzeitig verabreichtem Dabigatran (P-gp Substrat) um etwa 25 %. Folglich, wenn RAYVOW mit P‑gp Substraten verabreicht wird, die eine enge therapeutische Breite aufweisen (wie z. B. Digoxin), kann eine Erhöhung der systemischen Exposition des gleichzeitig verabreichten Arzneimittels klinisch bedeutsam sein (siehe Abschnitt 5.2). In derselben Studie wurde keine signifikante Veränderung der PK von Rosuvastatin (BCRP-Substrat) beobachtet, wenn es gleichzeitig mit Lasmiditan verabreicht wurde.
Wirkungen anderer Arzneimittel auf Lasmiditan
Bei gleichzeitiger Anwendung von Lasmiditan mit Dabigatranetexilat, Rosuvastatin, Sumatriptan oder Propranolol wurde keine Veränderung der PK von Lasmiditan beobachtet. Aufgrund der Clearance-Wege des Metabolismus ist es unwahrscheinlich, dass CYP-Inhibitoren oder -Induktoren die Lasmiditan-Exposition beeinflussen. Es wurde keine Veränderung der PK von Lasmiditan bei gleichzeitiger Anwendung mit Topiramat (CYP3A4-Induktor und CYP2C19-Inhibitor) beobachtet.
Schwangerschaft
Es liegen nur begrenzte Daten zur Anwendung von Lasmiditan bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Die Auswirkungen von Lasmiditan auf die Entwicklung des menschlichen Fötus sind nicht bekannt. Die Einnahme von RAYVOW während der Schwangerschaft wird nicht empfohlen.
Stillzeit
Lasmiditan und/oder seine Metaboliten wurden in die Milch von stillenden Ratten ausgeschieden (Abschnitt 5.3). Es liegen keine Daten über das Vorhandensein von Lasmiditan in der menschlichen Muttermilch, die Auswirkungen von Lasmiditan auf das gestillte Kind oder die Auswirkungen von Lasmiditan auf die Milchproduktion vor.
Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit RAYVOW verzichtet werden soll / die Behandlung mit RAYVOW zu unterbrechen ist. Dabei soll sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau berücksichtigt werden. Die Exposition des Neugeborenen kann minimiert werden, indem das Stillen für 24 Stunden nach der Behandlung vermieden wird.
Fertilität
Es ist nicht bekannt, ob Lasmiditan die menschliche Fortpflanzungsfähigkeit beeinflusst. Tierexperimentelle Studien zeigen keine Auswirkungen auf die Fertilität (siehe Abschnitt 5.3).
Lasmiditan hat großen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Die Verkehrstüchtigkeit wurde unter Verwendung einer computergestützten Fahrsimulation bewertet. Der primäre Endpunkt war der Unterschied zu Placebo in der Standardabweichung der Seitabweichung (SDLP), einem Maß für die Verkehrstüchtigkeit. Einzelgaben von 50 mg, 100 mg oder 200 mg Lasmiditan beeinträchtigten die Verkehrstüchtigkeit der Probanden 90 Minuten nach der Einnahme signifikant. In einer anderen Studie mit 100 mg oder 200 mg Lasmiditan erreichte die Verkehrstüchtigkeit 8 Stunden oder später nach Einnahme von RAYVOW in keiner der beiden Dosierungen die Schwelle für eine Beeinträchtigung der Verkehrstüchtigkeit.
Die Patienten sollten darauf hingewiesen werden, nach jeder Einnahme von Lasmiditan für mindestens 8 Stunden keinen Aktivitäten nachzugehen, die eine erhöhte Vorsicht erfordern, wie das Bedienen von Maschinen oder das Führen von Fahrzeugen, selbst wenn sie sich dazu in der Lage fühlen. Patienten, die diesen Rat nicht befolgen können, sollten Lasmiditan nicht einnehmen (siehe Abschnitt 4.4).
Zusammenfassung des Sicherheitsprofils
Die am häufigsten auftretenden Nebenwirkungen sind Benommenheit (19,9 %), Schläfrigkeit (7,8 %), Erschöpfung (7,7 %), Parästhesie (6,8 %), Übelkeit (4,9 %), Schwindel (2,6 %), Hypästhesie (2,5 %) und Muskelschwäche (2,3 %). Die meisten unerwünschten Ereignisse zeigten eine Dosisabhängigkeit.
Tabellarische Auflistung der Nebenwirkungen
In der folgenden Tabelle sind die Nebenwirkungen nach den MedDRA-Systemorganklassen und ihrer Häufigkeit aufgeführt. Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt. Die Häufigkeitseinstufungen sind: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000).
Tabelle 1. Nebenwirkungen
Systemorganklasse |
Sehr häufig |
Häufig |
Gelegentlich |
Selten |
Erkrankungen des Immunsystems |
Überempfindlichkeits- |
|||
Psychiatrische Erkrankungen |
Schlafstörungen |
Verwirrtheit |
||
Erkrankungen des Nervensystems |
Benommenheit |
Koordinationsstörungen |
Lethargie |
Serotonin- |
Augenerkrankungen |
Sehstörungen |
|||
Erkrankungen des Ohrs und des |
Schwindel |
|||
Herzerkrankungen |
Palpitationen |
|||
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Dyspnoe |
|||
Erkrankungen des Gastrointestinaltrakts |
Erbrechen |
|||
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Muskelschwäche |
Muskelkrampf |
||
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Unwohlsein |
Beschwerden in der Brust |
Beschreibung ausgewählter Nebenwirkungen
Herzfrequenz-Abnahme
In klinisch-pharmakologischen Studien wurde Lasmiditan mit einer Abnahme der Herzfrequenz um 5 bis 10 Schläge/min assoziiert, verglichen mit einer Abnahme um 2 bis 5 Schlägen/min bei Placebo. Die Inzidenz einer Bradykardie (< 50 Schläge/min und eine Abnahme gegenüber dem Ausgangswert ≥ 15 Schläge/min) betrug bei Patienten, die mit Lasmiditan behandelt wurden, 7 % bei 50 mg, 3 % bei 100 mg, 4 % bei 200 mg und 1 % bei Behandlung mit Placebo.
Blutdruck-Anstieg
Die einmalige Einnahme von Lasmiditan kann zu einem vorübergehenden Anstieg des Blutdrucks führen. Bei nicht-älteren gesunden Probanden wurde eine Stunde nach Einnahme von 200 mg Lasmiditan ein mittlerer Anstieg des systolischen und diastolischen Blutdrucks um ca. 2 bis 3 mm Hg, verglichen mit dem Anstieg von 1 mm Hg nach Einnahme von Placebo, beobachtet. Bei gesunden Freiwilligen über 65 Jahren betrug der mittlere Anstieg des systolischen Blutdrucks eine Stunde nach der Einnahme von 200 mg Lasmiditan 7 mm Hg gegenüber dem Ausgangswert. Die Einnahme von Placebo führte zu einem mittleren Anstieg von 4 mm Hg. Zwei Stunden nach der Einnahme zeigte sich kein Anstieg des mittleren Blutdrucks unter Lasmiditan im Vergleich zu Placebo. Die klinischen Daten zur Anwendung von Lasmiditan bei Patienten mit ischämischer Herzkrankheit sind begrenzt.
Überempfindlichkeitsreaktion
Überempfindlichkeitsreaktionen, einschließlich Angioödeme, Hautausschläge und Lichtempfindlichkeitsreaktionen, traten bei Patienten auf, die mit Lasmiditan behandelt wurden. In klinischen Studien wurde bei 0,1 % der mit Lasmiditan behandelten Patienten im Vergleich zu keinem Patienten in der Placebo-Gruppe von Überempfindlichkeit berichtet; alle Ereignisse waren leicht bis mittelschwer und traten innerhalb von Minuten bis zu einem Tag nach der Einnahme von Lasmiditan ein. Wenn eine schwerwiegende Überempfindlichkeitsreaktion auftritt, ist eine geeignete Therapie einzuleiten und die Anwendung von Lasmiditan zu beenden.
Benommenheit
In klinischen Studien war Benommenheit die häufigste Nebenwirkung, die von 19,9 % der Patienten berichtet wurde. Im Allgemeinen verlief dies leicht bis mittelschwer (schwere Benommenheit 1,2 %) und war selbstlimitierend mit einer medianen Zeit von 0,7 Stunden bis zum Beginn und einer medianen Dauer von 2 Stunden. Es wurden keine Unfälle oder Verletzungen bei den Patienten gemeldet, die über Benommenheit berichteten. Die Häufigkeit der Berichte über Benommenheit und andere häufige unerwünschte Ereignisse nimmt in der Regel bei wiederholter Einnahme ab.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem anzuzeigen:
Bundesinstitut für Arzneimittel und
Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: https://www.bfarm.de
Es liegen nur begrenzte Erfahrungen aus klinischen Studien mit einer Lasmiditan-Überdosierung vor. Meldungen von Überdosierungen enthielten Nebenwirkungen ähnlich denen, die bei niedrigeren Dosierungen beobachtet wurden, einschließlich Benommenheit, Schläfrigkeit, Erschöpfung, Parästhesie und Hypästhesie. Diese waren jedoch nicht mit einer Zunahme des Schweregrades oder der Häufigkeit verbunden. Da unerwünschte Wirkungen im Fall einer Überdosierung möglich sind, sollten Patienten jedoch auf Symptome von Nebenwirkungen überwacht und eine geeignete symptomatische Behandlung eingeleitet werden. Es ist kein Antidot gegen eine Lasmiditan-Überdosierung bekannt.
Pharmakotherapeutische Gruppe: Analgetika, Migränemittel, ATC- Code: N02CC08
Wirkmechanismus
Lasmiditan ist ein hochaffiner, zentral penetrierender 5-Hydroxytriptamin 1F (5-HT1F) -Rezeptoragonist. Der exakte Wirkmechanismus ist nicht bekannt, aber die therapeutischen Wirkungen von Lasmiditan bei der Behandlung von Migräne beinhalten vermutlich agonistische Wirkungen am 5-HT1F‑Rezeptor, eine Verringerung der Freisetzung von Neuropeptiden und eine Hemmung von Schmerzwegen, einschließlich des Trigeminusnervs.
Pharmakodynamische Wirkungen
In in-vitro-Bindungsstudien zeigte Lasmiditan eine > 440‑fache Selektivität für den 5‑HT1F-Rezeptor gegenüber den 5‑HT1B - und 5‑HT1D-Rezeptoren. Lasmiditan verengt ex vivo nicht die Koronararterien des Menschen, die inneren Brustarterien (Aa. mammaria interna) des Menschen oder die mittleren Meningealarterien (Aa. meningea media) des Menschen, wahrscheinlich aufgrund seiner niedrigen Affinität zum vasokonstriktiven 5‑HT1B -Rezeptor.
Kardiale Elektrophysiologie
In einer umfangreichen QT-Studie zeigte Lasmiditan im Vergleich zu Placebo eine Abnahme der Herzfrequenz um 6 Schläge pro Minute, und die Einnahme einer supra-therapeutischen Dosis von 400 mg deutete auf eine Verlängerung der QTc-Zeit bei Frauen hin. Subgruppenanalysen legten geschlechtsspezifische Unterschiede nahe, da bei der weiblichen Subgruppe eine deutlichere QTc-Zeit-Verlängerung beobachtet wurde. Da die empfohlene Höchstdosis jedoch auf 200 mg begrenzt ist, wird kein klinisch relevanter Effekt erwartet.
Klinische Wirksamkeit und Sicherheit
Die Wirksamkeit und Sicherheit von Lasmiditan wurde in drei randomisierten, placebokontrollierten, doppelblinden Studien der Phase 3 bei erwachsenen Patienten (N = 5 910) untersucht. Die Studien schlossen Patienten im Alter von 18 Jahren und älter mit 3 - 8 Migräne-Attacken pro Monat und einer mindestens mittelschwer beeinträchtigenden Migräne (Migraine Disability Assessment (MIDAS) Score ≥ 11) ein.
Studien zu singulärer Migräne-Attacke
Die Population, die in die Studien mit einer einzigen Attacke (SAMURAI und SPARTAN) aufgenommen wurde, war überwiegend weiblich (84 %) mit einem Durchschnittsalter von 42,3 Jahren. Die Patienten hatten in den 3 Monaten vor der Aufnahme in die Studie durchschnittlich 5,2 Migräneattacken pro Monat und einen mittleren MIDAS-Gesamtscore von 31,7. In der SAMURAI, aber nicht in der SPARTAN, waren Patienten mit bekannter koronarer Herzkrankheit, klinisch signifikanter Arrhythmie oder unkontrolliertem Bluthochdruck ausgeschlossen. Zusätzlich zur Migräne lagen bei 78,3 % der Patienten ≥ 1 kardiovaskulärer Risikofaktor, einschließlich Alter > 40 Jahre (54,2 %), niedriges HDL-Cholesterin (31,1 %), Bluthochdruck/Hypertonie (21,3 %), derzeitiger Raucher (14,3 %), hohes Gesamtcholesterin (10,9 %) und Diabetes (5,9 %) in der Anamnese vor. 21,7 % der Patienten erhielten eine medikamentöse Migräne-Prophylaxe und 37 % hatten innerhalb von 3 Monaten vor Studienbeginn ein Triptan eingenommen. Die am meisten belastenden Symptome (most bothersome symptoms = MBS) waren Photophobie (50,3 %), gefolgt von Übelkeit (22,2 %) und Phonophobie (20,6 %). In diesen Studien war eine zweite Einnahme des Studienarzneimittels oder eines anderen Arzneimittels 2 bis 24 Stunden nach der Erstbehandlung bei persistierender oder rezidivierender Migräne erlaubt.
Der primäre und ein wesentlicher sekundärer Endpunkt in beiden Studien waren der Anteil der Patienten ohne Schmerzen und der Anteil der Patienten ohne ihr jeweils am meisten belastendes Symptom verglichen mit Placebo 2 Stunden nach der Einnahme.
Beide Studien erreichten den primären und die wesentlichen sekundären Endpunkte. Alle Lasmiditan-Dosierungen zeigten eine statistisch signifikante und klinisch bedeutsame Verbesserung des Anteils der Patienten, die Schmerzfreiheit, MBS-Freiheit und Schmerzlinderung (definiert als eine Verringerung der Schmerzstärke von mäßig oder stark zu Studienbeginn auf leicht oder keine, oder von leicht auf keine) 2 Stunden nach der Behandlung zeigten (siehe Tabelle 2). Der Zeitpunkt des Einsetzens der Schmerzfreiheit ist in Abbildung 1 dargestellt; das Einsetzen der Schmerzlinderung folgte dem gleichen Muster wie die Schmerzfreiheit bei 50 mg und 100 mg, wobei ein zusätzliches Separieren von Placebo zu einem früheren Zeitpunkt von 30 Minuten für die 200 mg-Dosis beobachtet wurde (17,7 % für 200 mg vs. 11,6 % für Placebo, p = 0,004 bei SAMURAI, 18,6 % bei 200 mg vs. 14,7 % bei Placebo, p = 0,014 bei SPARTAN).
Tabelle 2. SAMURAI und SPARTAN: Zusammenfassung der Wirksamkeitsdaten
SAMURAI |
SPARTAN |
||||||
Lasmiditan |
Placebo |
Lasmiditan |
Placebo |
||||
100 mg |
200 mg |
50 mg |
100 mg |
200 mg |
|||
Schmerzfreiheit nach 2 Stunden | |||||||
N |
503 |
518 |
524 |
556 |
532 |
528 |
540 |
Responder (%) |
28,2 |
32,2 |
15,3 |
28,6 |
31,4 |
38,8 |
21,3 |
p-Wert |
< 0,001 |
< 0,001 |
0,006 |
< 0,001 |
< 0,001 |
||
MBS-Freiheit nach 2 Stunden | |||||||
N |
469 |
481 |
488 |
512 |
500 |
483 |
514 |
Responder (%) |
40,9 |
40,7 |
29,5 |
40,8 |
44,2 |
48,7 |
33,5 |
p-Wert |
< 0,001 |
< 0,001 |
0,018 |
< 0,001 |
< 0,001 |
||
Schmerzlinderung nach 2 Stunden | |||||||
N |
562 |
555 |
554 |
598 |
571 |
565 |
576 |
Responder (%) |
54,1 |
54,6 |
39,2 |
55,5 |
59,7 |
60,7 |
44,9 |
p-Wert |
< 0,001 |
< 0,001 |
< 0,001 |
< 0,001 |
< 0,001 |
||
Abbildung 1. Prozentsatz der Patienten, die innerhalb von 2 Stunden eine Migräne-Schmerzfreiheit erreichen in SAMURAI und SPARTAN.
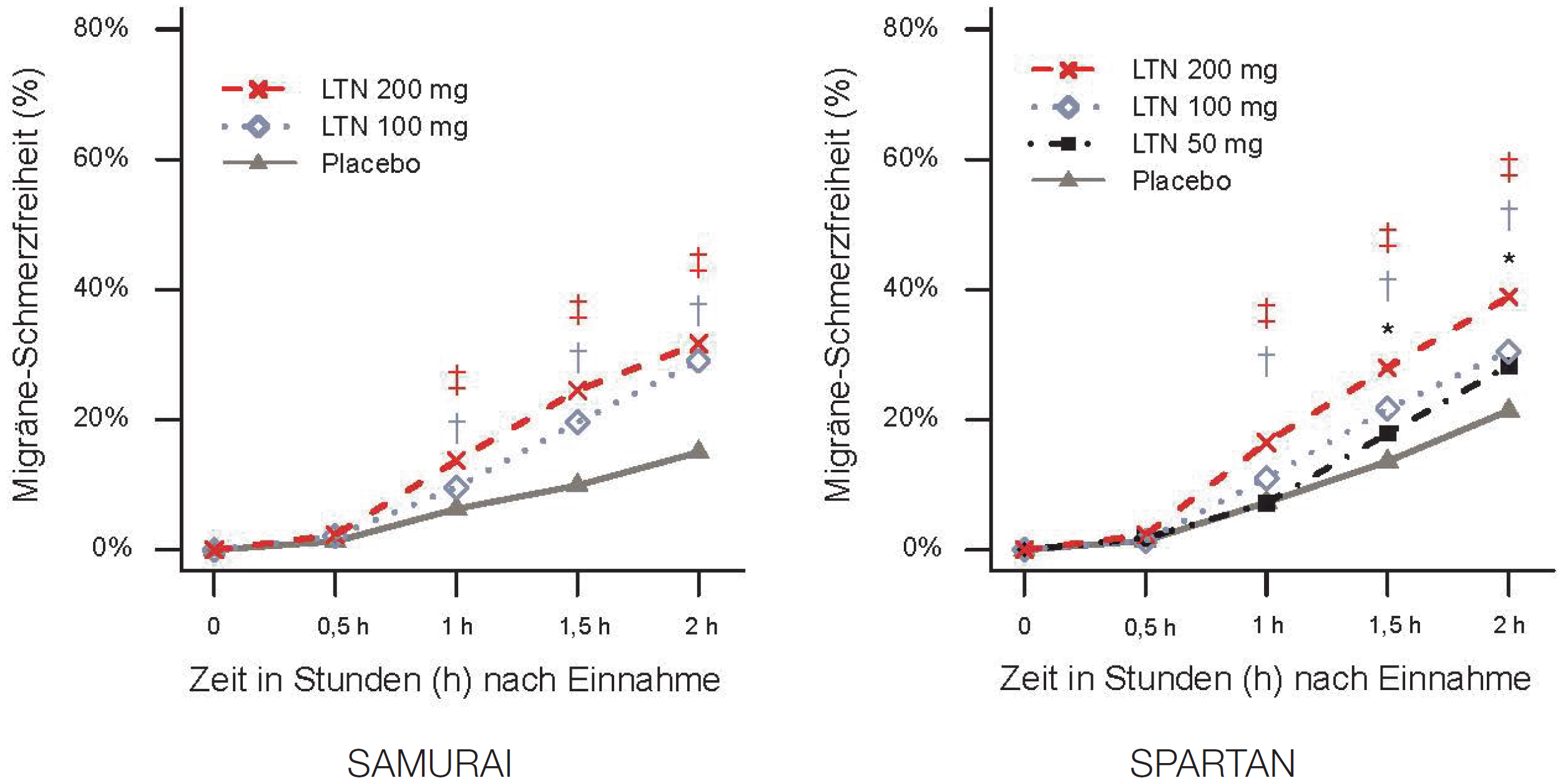
‡ Statistische Signifikanz für 200 mg LTN vs. Placebo; † Statistische Signifikanz für 100 mg LTN vs. Placebo; * Statistische Signifikanz für 50 mg LTN vs Placebo |
Abkürzungen: LTN = Lasmiditan |
Studie zur Beständigkeit der Wirkung
In einer Studie zur Beurteilung der Beständigkeit der Wirkung (CENTURION) wurden 4 Migräne-Attacken der Patienten mit 100 mg, 200 mg Lasmiditan oder einer Kontrolle behandelt. In der Kontrollgruppe erhielten die Patienten Placebo, oder eine Einzelgabe von 50 mg Lasmiditan zur Behandlung ihrer dritten oder vierten Attacke. Die eingeschlossene Population war überwiegend weiblich (84 %) mit einem Durchschnittsalter von 41,4 Jahren. Die Patienten hatten in den 3 Monaten vor Aufnahme in die Studie durchschnittlich 4,9 Migräneattacken pro Monat und einen mittleren MIDAS-Gesamtscore von 31,9. Die Studie schloss Patienten mit Herz-Kreislauf-Erkrankungen nicht aus, und 58,5 % der Patienten wiesen zusätzlich zur Migräne ≥ 1 kardiovaskulären Risikofaktor, einschließlich Alter > 40 Jahre (52,8 %), hohes Gesamtcholesterin (10,8 %), arterieller Hypertonus (16,9 %) und Diabetes in der Anamnese (3,1 %) auf. 28,8 % der Patienten erhielten aktuell vorbeugende Arzneimittel gegen Migräne, und 65,0 % hatten zuvor ein Triptan eingenommen. Das am meisten belastende Symptom war Photophobie (39,7 %), gefolgt von Übelkeit (31,9 %) und Phonophobie (19,3 %).
Die primären Endpunkte waren im Vergleich zu Placebo der Anteil an Patienten, die nach ihrer ersten Attacke 2 Stunden nach der Einnahme schmerzfrei waren, und der Anteil an Patienten, die bei mindestens 2 von 3 Attacken 2 Stunden nach Einnahme schmerzfrei waren.
Die Studie erreichte ihre primären und alle wesentlichen sekundären Endpunkte. Beide Dosierungen von 100 mg und 200 mg Lasmiditan zeigten eine statistisch signifikante und klinisch bedeutsame Verbesserung des Anteils der Patienten, die Schmerzfreiheit, Schmerzlinderung (eine Verringerung der Schmerzstärke von mäßig oder stark zu Studienbeginn auf leicht oder keine, oder von leicht auf keine), MBS-Freiheit 2 Stunden nach der Behandlung und anhaltende Schmerzfreiheit nach 24 Stunden (siehe Tabelle 3) erreichten. Der Zeitpunkt des Einsetzens der Schmerzfreiheit ist in Abbildung 2 dargestellt; die Schmerzlinderung folgte dem gleichen Muster wie die Schmerzfreiheit bei 50 mg und 100 mg, und wurde zu einem früheren Zeitpunkt von 30 Minuten bei der 200 mg-Dosis beobachtet (22,4 % für 200 mg vs. 14,0 % für Placebo, p = 0,001).
Beide Dosierungen zeigten eine beständige Wirkung mit einer statistisch signifikanten und klinisch bedeutsamen Verbesserung des Anteils der Patienten, die bei mindestens 2 von 3 Attacken Schmerzfreiheit und Schmerzlinderung erreichten (siehe Tabelle 3).
Tabelle 3. CENTURION: Zusammenfassung der Wirksamkeitsdaten
Lasmiditan |
|||
100 mg |
200 mg |
Placebo |
|
Endpunkte für einzelne Attacken (ITT) |
N = 419 |
N = 434 |
N = 443 |
Schmerzfreiheit 2 Stunden nach Einnahme während der ersten Attacke |
|||
Responder (%) |
25,8 |
29,3 |
8,4 |
p-Wert versus Placebo |
< 0,001 |
< 0,001 |
|
Schmerzlinderung 2 Stunden nach Einnahme während der ersten Attacke |
|||
Responder (%) |
65,4 |
65,2 |
41,3 |
p-Wert versus Placebo |
< 0,001 |
< 0,001 |
|
Anhaltende Schmerzfreiheit für bis zu 24 Stunden während der ersten |
|||
Responder (%) |
13,6 |
17,3 |
4,3 |
p-Wert versus Placebo |
< 0,001 |
< 0,001 |
|
MBS-Freiheit 2 Stunden nach der Einnahme während der ersten Attacke |
N = 376 |
N = 395 |
N = 396 |
Responder (%) |
40,4 |
39,0 |
28,0 |
p-Wert versus Placebo |
< 0,001 |
0,001 |
|
Endpunkte der Beständigkeit (ITT Beständigkeit) |
|||
Schmerzfreiheit 2 Stunden nach Einnahme bei mindestens 2 von 3 |
N = 340 |
N = 336 |
N = 373 |
Responder (%) |
14,4 |
24,4 |
4,3 |
p-Wert versus Placebo |
< 0,001 |
< 0,001 |
|
Schmerzlinderung 2 Stunden nach Einnahme bei mindestens 2 von 3 |
N = 332 |
N = 333 |
N = 320 |
Responder (%) |
62,3 |
66,7 |
36,9 |
p-Wert versus Placebo |
< 0,001 |
< 0,001 |
|
Abkürzungen: ITT = intent to treat (z. Dt.: Absicht zu behandeln) | |||
Abbildung 2. Prozentsatz der Patienten, die in CENTURION innerhalb von 2 Stunden eine Migräne-Schmerzfreiheit erreichen.
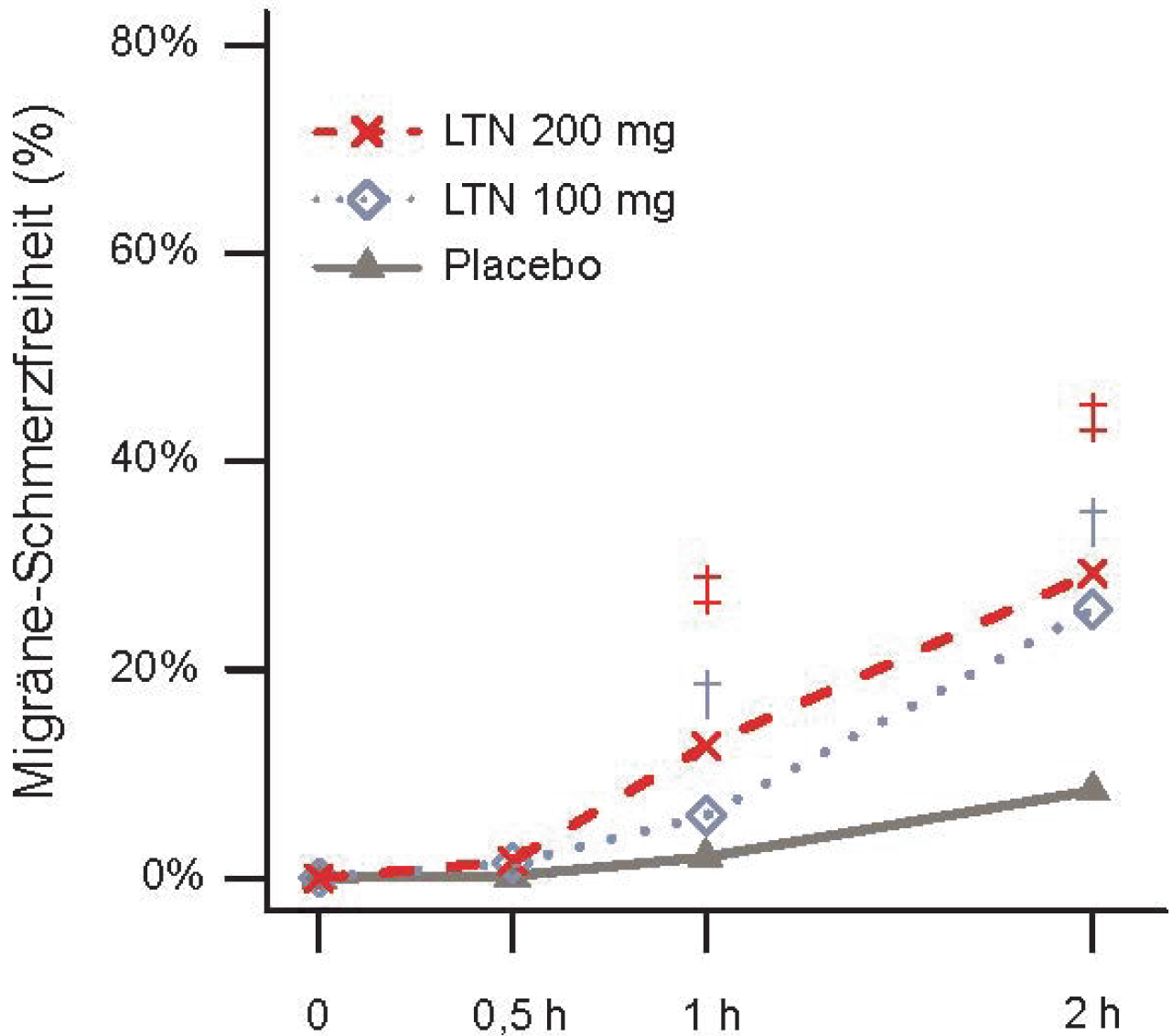
‡ Statistische Signifikanz für 200 mg LTN vs. Placebo; † Statistische Signifikanz für 100 mg LTN vs. Placebo |
Abkürzungen: LTN = Lasmiditan |
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für RAYVOW eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in der Behandlung von Migräne gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Resorption
Nach oraler Gabe wird Lasmiditan schnell mit einer medianen tmax von 1,8 Stunden resorbiert. Bei Patienten mit Migräne unterschied sich die Pharmakokinetik (PK) Lasmiditans während einer Migräne-Attacke nicht während der interiktalen Phase. Über den klinischen Dosisbereich von 50 bis 200 mg wird die absolute Bioverfügbarkeit basierend auf den Ergebnissen der Populations-PK-Analyse auf 50 % bis 58 % geschätzt. Die gleichzeitige Gabe von Lasmiditan mit einer fettreichen Mahlzeit erhöhte die mittleren Cmax- und AUC-Werte von Lasmiditan um 22 % bzw. 19 % und verzögerte die mediane tmax um 1 Stunde. Es wird nicht erwartet, dass dieser Unterschied in der Exposition klinisch bedeutsam ist. Lasmiditan wurde in klinischen Wirksamkeitsstudien unabhängig von der Nahrungsaufnahme eingenommen.
Verteilung
Die humane Plasmaproteinbindung von Lasmiditan beträgt ca. 55 % bis 60 % und liegt konzentrationsunabhängig zwischen 15 und 500 ng/ml. Das geschätzte mittlere Verteilungsvolumen betrug 304 l.
Biotransformation
Lasmiditan unterliegt einer hepatischen und extrahepatischen Metabolisierung hauptsächlich durch non-CYP-Enzyme, wobei die Ketonreduktion zu S-M8 den Hauptweg darstellt. Die folgenden Enzyme waren nicht an der Metabolisierung von Lasmiditan beteiligt: MAO-A, MAO-B, Flavin-Monooxygenase 3, CYP450-Reduktase, Xanthinoxidase, Alkoholdehydrogenase, Aldehyddehydrogenase und Aldo-Keto-Reduktasen.
Lasmiditan wird auch am Piperidinring zu M7 oxidiert. Im Vergleich zu Lasmiditan sind die Metaboliten pharmakologisch inaktiv. Lasmiditan ist in-vitro ein Substrat von P-gp.
Lasmiditan und seine Hauptmetaboliten sind in-vitro-Induktoren von CYP-Enzymen. Lasmiditan hemmt CYP2D6 in-vitro. Lasmiditan und sein Hauptmetabolit sind keine Inhibitoren von MAO-A. Lasmiditan hemmt in-vitro P-gp, BCRP- und OCT1-Effluxtransporter. Lasmiditan hemmt in-vitro die Nierentransporter OCT2, MATE1 und MATE2-K.
Eine Arzneimittel-Interaktionsstudie zeigt an, dass Lasmiditan ein schwacher P-gp-Inhibitor ist (siehe Abschnitt 4.5).
Elimination
Lasmiditan wurde mit einem geometrischen Mittelwert von t½ von ungefähr 5,7 Stunden eliminiert. Bei täglicher Dosierung wurde keine Akkumulation von Lasmiditan beobachtet. Die geschätzte mittlere Gesamtkörper-Clearance betrug 66,2 l/h. Lasmiditan zeigt im Allgemeinen eine lineare PK über den klinischen Dosisbereich von 50 bis 200 mg. Lasmiditan wird hauptsächlich verstoffwechselt ausgeschieden. Die renale Ausscheidung ist ein Nebenweg der Lasmiditan-Clearance, wobei etwa 3 % der Dosis als unverändertes Lasmiditan im Urin vorliegt. Metabolit S-M8 machte ungefähr 66 % der Dosis im Urin aus, wobei der Großteil der Ausscheidung innerhalb von 48 Stunden nach der Einnahme erfolgte.
Besondere Patientengruppen
Alter, Geschlecht, ethnische Herkunft und Körpergewicht
Alter, Geschlecht, ethnische Herkunft und Körpergewicht hatten in einer populationspharmakokinetischen Analyse von Lasmiditan keinen signifikanten Einfluss auf die Exposition. In einer Studie hatte das Geschlecht einen Einfluss auf die PK von Lasmiditan mit einer höheren Cmax (~ 20 – 30 %) und AUC (~ 30 %) bei Frauen im Vergleich zu Männern, unabhängig davon, ob Lasmiditan nach einer Mahlzeit oder nüchtern eingenommen wurde. Eine Dosisanpassung aufgrund von Alter, Geschlecht, ethnischer Herkunft oder Körpergewicht ist nicht erforderlich.
Nierenfunktionsstörung
Die Anwendung von Lasmiditan bei Patienten mit schwerer Nierenfunktionsstörung (eGFR < 30 ml/min/1,73 m2) zeigte im Vergleich zu Patienten mit normaler Nierenfunktion eine um 18 % höhere Exposition der AUC (0 - ∞) und eine 13 % höhere Cmax. Es wurden keine klinisch relevanten Unterschiede in der Exposition beobachtet. Bei Patienten mit leichter, mittelschwerer oder schwerer Nierenfunktionsstörung ist eine Dosisanpassung nicht erforderlich.
Leberfunktionsstörung
Bei Patienten mit leichter und mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse A und B) war die Lasmiditan-Exposition um 11 % und 35 % höher [AUC (0 - ∞)] als bei Patienten mit normaler Leberfunktion. Die Cmax war bei Patienten mit leichter und mittelschwerer Leberfunktionsstörung um 19 % und 33 % höher. Dieser Unterschied in der Exposition wird als nicht klinisch signifikant angesehen. Bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung ist eine Dosisanpassung nicht erforderlich. Die Anwendung von Lasmiditan wurde bei Patienten mit schwerer Leberfunktionsstörung nicht untersucht und wird daher für diese Patientengruppe nicht empfohlen.
Die Kanzerogenität wurde in einer zweijährigen Studie an Ratten und einer sechsmonatigen Studie an transgenen Mäusen untersucht. Bei männlichen Ratten wurde ein Anstieg von Hypophysentumor bedingten Todesfällen beobachtet. Die Relevanz dieser Ergebnisse im Hinblick auf das menschliche Risiko ist unbekannt. Bei Mäusen wurden keine Hinweise auf Kanzerogenität gefunden.
Lasmiditan war nicht genotoxisch, basierend auf den Ergebnissen des Ames-Tests bei Bakterien, einer Chromosomenaberrationsstudie an Ovarialzellen des chinesischen Hamsters und Mikronukleustests bei Mäusen.
Entwicklungs- und Reproduktionstoxizität
Studien mit Ratten zeigten keine Auswirkungen auf die männliche oder weibliche Fertilität.
In Studien zur embryofetalen Entwicklung traten bei Ratten und Kaninchen ein verringertes fetales Körpergewicht und Skelettvariationen auf; bei Kaninchen kam es zu einem leichten Anstieg Abort-Rate nach Implantation (embryofetale Mortalität) und in geringer Häufigkeit zu kardiovaskulären Defekten (Fehlbildungen). Die Exposition bei den Dosierungen ohne beobachtete Nebenwirkungen von 175 mg/kg/Tag (Ratten) und 75 mg/kg/Tag (Kaninchen) war etwa 37‑ bzw. 1,5‑fach höher als eine 200 mg-Dosis beim Menschen.
In einer prä- und postnatalen Studie an Ratten traten bei Expositionen mit der höchsten untersuchten Dosis von 225 mg/kg/Tag eine verlängerte Trächtigkeit und Geburt, sowie eine erhöhte Anzahl totgeborener Jungtiere und eine erhöhte Häufigkeit postnataler Todesfälle auf. Bei dieser hohen Exposition wurde während der gesamten F1-Reifungsphase eine Abnahme des mittleren Körpergewichts der F1-Jungtiere während der Vorentwöhnungsphase bei beiden Geschlechtern ohne Erholung aufrechterhalten. Die Exposition bei der Dosis von 150 mg/kg/Tag ohne beobachtete Wirkung war geschätzt > 19‑fach höher als eine 200 mg-Dosis beim Menschen.
Alle Effekte traten bei maternal toxischen Expositionen auf, welche die menschliche Exposition bei einer klinischen Dosis von 200 mg überstiegen.
Tierexperimentelle Studien haben gezeigt, dass Lasmiditan und/oder seine Metaboliten in die Milch von säugenden Ratten ausgeschieden wurden.
Tablettenkern:
Mikrokristalline Cellulose
Croscarmellose‑Natrium
Magnesiumstearat (Ph. Eur.) [pflanzlich]
Vorverkleisterte Stärke
Natriumdodecylsulfat
Filmüberzug (50 mg und 200 mg):
Poly(vinylalkohol)
Titandioxid (E 171)
Macrogol 3350
Talkum
Eisen (II, III)-oxid (E 172)
Filmüberzug (100 mg):
Poly(vinylalkohol)
Titandioxid (E 171)
Macrogol 3350
Talkum
Eisen (Ⅱ, Ⅲ)-oxid (E 172)
Eisen (Ⅲ)-oxid (E 172)
Nicht zutreffend
3 Jahre
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Perforierte Einzeldosis-Blisterpackungen aus Polychlortrifluorethylen/Polyvinylchlorid (PCTFE/PVC), versiegelt mit einer Aluminiumfolie in Packungen mit 2 x 1, 4 x 1, 6 x 1, 12 x 1 und 16 x 1 Filmtablette.
Perforierte Einzeldosis-Blisterpackungen aus Polyvinylchlorid (PVC), versiegelt mit einer Aluminiumfolie in Packungen mit 2 x 1, 4 x 1, 6 x 1, 12 x 1 und 16 x 1 Filmtablette.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Eli Lilly Nederland B.V.
Orteliuslaan 1000
3528 BD Utrecht
Niederlande
RAYVOW® 50 mg Filmtabletten
EU/1/21/1587/001
EU/1/21/1587/002
EU/1/21/1587/003
EU/1/21/1587/004
EU/1/21/1587/005
EU/1/21/1587/006
EU/1/21/1587/007
EU/1/21/1587/008
EU/1/21/1587/009
EU/1/21/1587/010
RAYVOW® 100 mg Filmtabletten
EU/1/21/1587/011
EU/1/21/1587/012
EU/1/21/1587/013
EU/1/21/1587/014
EU/1/21/1587/015
EU/1/21/1587/016
EU/1/21/1587/017
EU/1/21/1587/018
EU/1/21/1587/019
EU/1/21/1587/020
Datum der Erteilung der Zulassung: 17. August 2022
Januar 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu/ verfügbar.
Verschreibungspflichtig
RAYVOW® 50 mg Filmtabletten
Inhalt |
PZN |
2 Filmtabletten |
17363557 |
6 Filmtabletten |
17363586 |
RAYVOW® 100 mg Filmtabletten
Inhalt |
PZN |
2 Filmtabletten |
17363592 |
6 Filmtabletten |
17363646 |
Organon Healthcare GmbH
Tel. + 0800 3384 726
(+49 (0) 8920 400 2210)
dpoc.germany@organon.com