
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Vabysmo® 120 mg/ml Injektionslösung
Vabysmo® 120 mg/ml Injektionslösung in einer Fertigspritze
Ein ml Lösung enthält 120 mg Faricimab.
Fertigspritze
Jede Fertigspritze enthält 21 mg Faricimab in 0,175 ml Lösung. Dies ergibt eine verwendbare Menge zur Abgabe einer Einzeldosis von 0,05 ml Lösung, die 6 mg Faricimab enthält.
Durchstechflasche
Jede Durchstechflasche enthält 28,8 mg Faricimab in 0,24 ml Lösung. Dies ergibt eine verwendbare Menge zur Abgabe einer Einzeldosis von 0,05 ml Lösung, die 6 mg Faricimab enthält.
Faricimab ist ein humanisierter Antikörper, der in einer Zellkultur aus Säugetierzellen (Ovarialzellen des chinesischen Hamsters) durch rekombinante DNA-Technologie hergestellt wird.
Sonstige Bestandteile mit bekannter Wirkung
Jede 0,05‑ml‑Lösung enthält 0,02 mg Polysorbat und 0,07 mg Natrium.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung (Injektion)
Klare bis opaleszente, farblose bis bräunlich-gelbe Lösung mit einem pH‑Wert von 5,5 und einer Osmolalität von 270 ‑ 370 mOsm/kg.
Vabysmo wird angewendet zur Behandlung von erwachsenen Patienten mit:
neovaskulärer (feuchter) altersabhängiger Makuladegeneration (nAMD),
einer Visusbeeinträchtigung infolge eines diabetischen Makulaödems (DMÖ),
einer Visusbeeinträchtigung aufgrund eines Makulaödems infolge eines retinalen Venenverschlusses (RVV) (Venenastverschluss [VAV] oder Zentralvenenverschluss [ZVV]).
Dieses Arzneimittel muss von einem qualifizierten Arzt mit Erfahrung in der Durchführung intravitrealer Injektionen appliziert werden.
Dosierung
Neovaskuläre (feuchte) altersabhängige Makuladegeneration (nAMD)
Die empfohlene Dosis beträgt 6 mg (0,05 ml Lösung), angewendet als intravitreale Injektion alle 4 Wochen (monatlich) für die ersten 3 Dosen.
Anschließend wird 16 und/oder 20 Wochen nach Einleitung der Behandlung eine Beurteilung der Krankheitsaktivität basierend auf den anatomischen und/oder visuellen Befunden empfohlen, um die Behandlung an den individuellen Patienten anpassen zu können. Bei Patienten ohne Krankheitsaktivität ist eine Verabreichung von Faricimab alle 16 Wochen (4 Monate) zu erwägen. Bei Patienten mit Krankheitsaktivität ist eine Behandlung alle 8 Wochen (2 Monate) oder 12 Wochen (3 Monate) zu erwägen. Wenn sich die anatomischen und/oder visuellen Befunde ändern, ist das Behandlungsintervall entsprechend anzupassen, und eine Intervallverkürzung ist vorzunehmen, wenn sich anatomische und/oder visuelle Befunde verschlechtern (siehe Abschnitt 5.1). Für Behandlungsintervalle von 8 Wochen oder weniger zwischen den Injektionen gibt es nur begrenzte Sicherheitsdaten (siehe Abschnitt 4.4). Die Kontrolle zwischen den Injektionsterminen ist in Abhängigkeit vom Zustand des Patienten und nach Ermessen des Arztes zu planen, es besteht jedoch keine Notwendigkeit einer monatlichen Kontrolle zwischen den Injektionen.
Visusbeeinträchtigung infolge eines diabetischen Makulaödems (DMÖ) und
Makulaödem infolge eines retinalen Venenverschlusses (RVV)
Die empfohlene Dosis beträgt 6 mg (0,05 ml Lösung), angewendet als intravitreale Injektion alle 4 Wochen (monatlich); 3 oder mehr aufeinanderfolgende monatliche Injektionen können erforderlich sein.
Anschließend wird die Behandlung in Abhängigkeit von der Krankheitsaktivität („Treat and Extend“) individuell angepasst. Basierend auf der ärztlichen Beurteilung der anatomischen und/oder visuellen Befunde des Patienten kann das Dosierungsintervall in Schritten von bis zu 4 Wochen verlängert werden. Wenn sich die anatomischen und/oder visuellen Befunde ändern, ist das Behandlungsintervall entsprechend anzupassen, und eine Intervallverkürzung ist vorzunehmen, wenn sich anatomische und/oder visuelle Befunde verschlechtern (siehe Abschnitt 5.1). Behandlungsintervalle mit weniger als 4 Wochen und mehr als 4 Monaten zwischen den Injektionen wurden nicht untersucht. Die Kontrolle zwischen den Injektionsterminen ist in Abhängigkeit vom Zustand des Patienten und nach Ermessen des Arztes zu planen, es besteht jedoch keine Notwendigkeit einer monatlichen Kontrolle zwischen den Injektionen.
Umstellung von Anti-VEGF-Therapien auf Vabysmo
Bei Patienten, die zuvor mit Anti-VEGF-Arzneimitteln behandelt wurden und zu Vabysmo wechseln, kann sich das Behandlungsschema von dem für nicht vorbehandelte Patienten unterscheiden. Die Behandlungsintervalle sind auf Grundlage der Beurteilung des Arztes hinsichtlich der visuellen und/oder anatomischen Ergebnisse des Patienten festzulegen (siehe Abschnitt 5.1).
Bei Patienten mit stabilen visuellen und anatomischen Ergebnissen können die vorherigen Behandlungsintervalle nach der ersten Injektion von Vabysmo beibehalten oder verlängert werden, z. B. mit einem „Treat and Extend“-Dosierungsschema. In der anfänglichen Umstellungsphase liegen nur begrenzte Daten für die Umstellung auf „Treat and Extend“-Intervalle von mehr als 12 Wochen vor.
Bei Patienten mit suboptimalen visuellen und/oder anatomischen Ergebnissen kann die Behandlung mit Vabysmo mit 1 Injektion alle 4 Wochen (monatlich) für bis zu 3 aufeinanderfolgende Dosen beginnen, gefolgt von einer Anpassung der Injektionsintervalle, wie z. B. mit einem „Treat and Extend“-Dosierungsschema.
Wenn sich die visuellen und/oder anatomischen Ergebnisse verschlechtern, sollte das Behandlungsintervall nach Ermessen des Arztes entsprechend verkürzt werden. Das Intervall zwischen zwei Injektionen sollte nicht weniger als 1 Monat betragen.
Dauer der Behandlung
Dieses Arzneimittel ist für die Langzeitbehandlung bestimmt. Wenn die visuellen und/oder anatomischen Befunde darauf hinweisen, dass der Patient von einer fortgesetzten Behandlung nicht profitiert, ist die Behandlung abzubrechen.
Verspätete oder versäumte Dosis
Wenn eine Dosis verspätet verabreicht oder ausgelassen wurde, ist der Patient zum nächstmöglichen Termin vom Arzt zu untersuchen und die Dosierung nach Ermessen des Arztes weiterzuführen.
Besondere Patientengruppen
Ältere Patienten
Bei Patienten ≥ 65 Jahre ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Zu nAMD- und RVV-Patienten ≥ 85 Jahre liegen nur begrenzte Sicherheitsdaten vor (siehe Abschnitt 4.4).
Nierenfunktionsstörung
Bei Patienten mit Nierenfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Leberfunktionsstörung
Bei Patienten mit Leberfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Kinder und Jugendliche
In den Indikationen nAMD, DMÖ und RVV gibt es in der pädiatrischen Bevölkerungsgruppe keine relevante Verwendung für dieses Arzneimittel.
Art der Anwendung
Nur zur intravitrealen Anwendung. Jede Fertigspritze oder Durchstechflasche ist nur für die Behandlung eines einzigen Auges zu verwenden.
Vabysmo ist vor der Anwendung visuell auf Schwebstoffe und Verfärbungen zu prüfen. Bei Vorhandensein von Schwebstoffen oder Verfärbungen darf die Fertigspritze oder Durchstechflasche nicht verwendet werden.
Das intravitreale Injektionsverfahren muss unter aseptischen Bedingungen durchgeführt werden. Dies beinhaltet eine chirurgische Händedesinfektion, ein steriles Abdecktuch sowie ein steriles Augenlid-Spekulum (oder Äquivalent). Vor dem intravitrealen Verfahren ist eine sorgfältige Anamnese des Patienten hinsichtlich Überempfindlichkeitsreaktionen durchzuführen (siehe Abschnitt 4.8). Vor der Injektion ist eine adäquate Anästhesie vorzunehmen und ein topisches Breitspektrum-Mikrobizid zur Desinfektion der periokularen Haut, des Augenlids und der Augenoberfläche anzuwenden.
Fertigspritze
Die Fertigspritze enthält einen Überschuss an Volumen. Das überschüssige Volumen muss vor der Injektion der empfohlenen Dosis verworfen werden. Die Injektion des gesamten Volumens der Fertigspritze kann zu einer Überdosierung führen.
Um Luftblasen zusammen mit dem überschüssigen Arzneimittel zu entfernen, drücken Sie langsam den Spritzenkolben, bis sich der untere Rand der Kuppel des Gummistopfens auf einer Linie mit der 0,05-ml-Dosismarkierung der Spritze befindet (siehe Abschnitte 4.9 und 6.6).
Die Injektionsfilternadel (in der Packung enthalten) ist 3,5 bis 4,0 mm posterior zum Limbus in den Glaskörper einzuführen. Dabei ist der horizontale Meridian zu vermeiden und in Richtung Bulbusmitte zu zielen. Danach wird das Injektionsvolumen von 0,05 ml langsam injiziert; für nachfolgende Injektionen sind andere Stellen der Sklera zu verwenden.
Durchstechflasche
Die Injektionsnadel (30 G x ½ʺ, nicht in der Packung enthalten) ist 3,5 bis 4,0 mm posterior zum Limbus in den Glaskörper einzuführen. Dabei ist der horizontale Meridian zu vermeiden und in Richtung Bulbusmitte zu zielen. Danach wird das Injektionsvolumen von 0,05 ml langsam injiziert; für nachfolgende Injektionen sind andere Stellen der Sklera zu verwenden.
Überwachung nach der Injektion
Nach der Injektion ist nicht verwendetes Arzneimittel oder Abfallmaterial entsprechend den nationalen Anforderungen zu beseitigen.
Unmittelbar nach der intravitrealen Injektion sind die Patienten auf eine Erhöhung des intraokularen Drucks zu überwachen. Eine geeignete Überwachung kann aus einer Kontrolle der Perfusion des Sehnervenkopfes oder einer Tonometrie bestehen. Falls erforderlich soll sterile Ausrüstung zur Durchführung einer Parazentese zur Verfügung stehen.
Nach der intravitrealen Injektion sind die Patienten anzuweisen, alle Symptome, die auf eine Endophthalmitis hinweisen (z. B. Sehverlust, Augenschmerzen, Rötungen des Auges, Photophobie, verschwommenes Sehen), unverzüglich zu melden.
Hinweise zur Handhabung des Arzneimittels vor der Anwendung siehe Abschnitt 6.6.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Aktive oder vermutete okulare oder periokulare Infektionen.
Aktive intraokulare Entzündung.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Durch die intravitreale Injektion bedingte Reaktionen
Intravitreale Injektionen einschließlich jener von Faricimab wurden mit Endophthalmitis, intraokularer Entzündung, rhegmatogener Netzhautablösung, Netzhauteinriss und iatrogenem traumatischem Katarakt in Verbindung gebracht (siehe Abschnitt 4.8). Vabysmo muss immer unter ordnungsgemäßen aseptischen Bedingungen injiziert werden. Die Patienten sollen angewiesen werden, alle Symptome, wie beispielsweise Schmerzen, Sehverlust, Photophobie, verschwommenes Sehen, Mouches volantes oder Rötung, die auf eine Endophthalmitis oder eine der oben aufgeführten Nebenwirkungen hinweisen, unverzüglich zu melden, um eine sofortige und angemessene Behandlung zu ermöglichen. Patienten mit häufigeren Injektionen können ein erhöhtes Risiko für eingriffsbedingte Komplikationen haben.
Erhöhung des intraokularen Drucks
Eine vorübergehende Erhöhung des intraokularen Drucks (IOD) wurde innerhalb von 60 Minuten nach intravitrealen Injektionen einschließlich jener von Faricimab beobachtet (siehe Abschnitt 4.8). Besondere Vorsicht ist bei Patienten mit schlecht eingestelltem Glaukom geboten (Vabysmo darf nicht injiziert werden, solange der Augeninnendruck ≥ 30 mmHg beträgt). In allen Fällen müssen sowohl der IOD als auch die Perfusion des Sehnervenkopfes kontrolliert und bei Bedarf angemessen behandelt werden.
Systemische Wirkungen
Nach der intravitrealen Injektion von Inhibitoren des vaskulären endothelialen Wachstumsfaktors (vascular endothelial growth factor - VEGF) wurden systemische unerwünschte Ereignisse einschließlich arterieller thromboembolischer Ereignisse beschrieben und es besteht ein theoretisches Risiko, dass diese mit der VEGF‑Hemmung in Zusammenhang stehen. In den klinischen Studien mit Faricimab bei Patienten mit nAMD, DMÖ und RVV wurde eine niedrige Inzidenzrate arterieller thromboembolischer Ereignisse beobachtet. Diese ist ähnlich wie die Inzidenzrate, die in den anderen klinischen Studien mit Anti-VEGF-Inhibitoren bei Patienten berichtet wurde. Es gibt nur begrenzte Daten zur Sicherheit einer Behandlung mit Faricimab bei DMÖ-Patienten mit hohem Blutdruck (≥ 140/90 mmHg) und Gefäßerkrankungen sowie bei nAMD- und RVV-Patienten im Alter ≥ 85 Jahren.
Immunogenität
Da es sich um ein therapeutisches Protein handelt, besteht bei Faricimab die Möglichkeit einer Immunogenität (siehe Abschnitt 4.8). Die Patienten sollen angewiesen werden, ihren Arzt über Anzeichen oder Symptome einer intraokularen Entzündung zu informieren, wie z. B. Sehverlust, Augenschmerzen, erhöhte Lichtempfindlichkeit, Mouches volantes oder Verschlechterung einer Augenrötung, die ein klinisches Anzeichen für eine Überempfindlichkeit gegenüber Faricimab sein könnten (siehe Abschnitt 4.8).
Beidseitige Behandlung
Die Sicherheit und Wirksamkeit von Faricimab bei gleichzeitiger Anwendung in beiden Augen wurde nicht untersucht. Eine bilaterale Behandlung könnte zu bilateralen Nebenwirkungen am Auge und/oder möglicherweise zu einer erhöhten systemischen Exposition führen, was das Risiko systemischer Nebenwirkungen erhöhen könnte. Bis Daten zur bilateralen Anwendung vorliegen, stellt dies ein theoretisches Risiko für Faricimab dar.
Gleichzeitige Anwendung anderer Anti-VEGF-Arzneimittel
Es liegen keine Daten zur gleichzeitigen Anwendung von Faricimab mit anderen Anti-VEGF-Arzneimitteln am selben Auge vor. Faricimab ist nicht gleichzeitig mit anderen Anti-VEGF-Arzneimitteln zu verabreichen (systemisch oder okular).
Verwendung anderer Injektionsnadeln mit der Fertigspritze
Verwenden Sie die Fertigspritze nur mit der beiliegenden Injektionsfilternadel. Es liegen keine klinischen Daten über die Verwendung anderer Injektionsnadeln mit der Fertigspritze vor.
Aussetzen der Behandlung
In folgenden Fällen soll die Behandlung ausgesetzt werden:
Rhegmatogene Netzhautablösung, Makulaforamen Stadium 3 oder 4, Netzhautabriss; die Behandlung ist erst wieder aufzunehmen, wenn eine adäquate Korrektur durchgeführt wurde.
Behandlungsbedingte Abnahme der bestkorrigierten Sehschärfe (best corrected visual acuity - BCVA) um ≥ 30 Buchstaben im Vergleich zur letzten Untersuchung der Sehschärfe; die Behandlung soll nicht früher als zum nächsten vorgesehenen Termin fortgesetzt werden.
Ein intraokularer Druck ≥ 30 mmHg.
Eine subretinale Blutung mit Beteiligung der Fovea centralis oder, wenn die Größe der Blutung ≥ 50 % der Gesamtfläche der Läsion umfasst.
Durchgeführte oder geplante intraokulare Operation innerhalb der vergangenen oder kommenden 28 Tage; die Behandlung soll nicht früher als zum nächsten vorgesehenen Termin fortgesetzt werden.
Einriss des retinalen Pigmentepithels
Ein Einriss des retinalen Pigmentepithels (RPE) ist eine Komplikation der Abhebung des retinalen Pigmentepithels (retinal pigment epithelial detachment - PED) bei Patienten mit nAMD. Risikofaktoren für die Entwicklung eines RPE-Einrisses nach einer Anti-VEGF-Therapie wegen einer nAMD sind großflächige und/oder starke Abhebungen des retinalen Pigmentepithels. Bei Patienten mit diesen Risikofaktoren für Einrisse des RPE ist bei der Einleitung einer Therapie mit Faricimab Vorsicht geboten. RPE-Einrisse kommen häufig vor bei nAMD-Patienten mit PED, die mit Anti-VEGF-Mitteln einschließlich Faricimab intravitreal behandelt werden. In der Faricimab-Gruppe war die Rate der RPE-Einrisse höher (2,9 %) als in der Aflibercept-Gruppe (1,5 %). Die meisten Ereignisse traten während der Aufsättigungsphase auf und waren leicht bis mäßig, ohne Auswirkungen auf das Sehvermögen.
Populationen mit begrenzten Daten
Es liegen nur begrenzte Daten zur Behandlung von nAMD‑ und RVV-Patienten ≥ 85 Jahren und DMÖ‑Patienten mit Typ‑I‑Diabetes, Patienten mit einem HbA1c-Wert über 10 %, Patienten mit hohem Risiko für eine proliferative diabetische Retinopathie (DR), hohem Blutdruck (≥ 140/90 mmHg) und Gefäßerkrankungen sowie zu anhaltenden Dosierungsintervallen, die kürzer als alle 8 Wochen (Q8W) sind, und von nAMD-, DMÖ‑ und RVV-Patienten mit aktiven systemischen Infektionen, vor. Zu anhaltenden Dosierungsintervallen von 8 Wochen und weniger liegen nur begrenzte Sicherheitsinformationen vor und diese können mit einem höheren Risiko für okulare und systemische Nebenwirkungen, einschließlich schwerwiegender Nebenwirkungen, verbunden sein. Es gibt auch keine Erfahrungen mit der Behandlung mit Faricimab bei diabetischen oder RVV-Patienten mit unkontrollierter Hypertonie und Patienten mit RVV, bei denen eine vorherige Behandlung fehlgeschlagen ist. Der Arzt soll diesen Mangel an Informationen bei der Behandlung solcher Patienten berücksichtigen.
Natriumgehalt
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d. h., es ist nahezu „natriumfrei“.
Polysorbatgehalt
Dieses Arzneimittel enthält 0,02 mg Polysorbat pro 0,05‑ml‑Dosis. Patienten mit einer Überempfindlichkeit gegenüber Polysorbat sollen dieses Arzneimittel nicht anwenden.
Schulungsmaterialien
Der verschreibende Arzt muss mit dem Leitfaden für Patienten vertraut sein, der erstellt wurde, um das Bewusstsein für Anzeichen und Symptome von intraokularer Entzündung und Endophthalmitis zu gewährleisten, und muss diesen Leitfaden dem Patienten/der Betreuungsperson aushändigen und diese Ereignisse erklären.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt. Basierend auf der Biotransformation und Elimination von Faricimab (siehe Abschnitt 5.2) sind keine Wechselwirkungen zu erwarten. Faricimab ist jedoch nicht gleichzeitig mit anderen systemischen oder okularen Anti-VEGF-Arzneimitteln zu verabreichen (siehe Abschnitt 4.4).
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während der Behandlung sowie nach der letzten intravitrealen Injektion von Faricimab für mindestens 3 Monate eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Zur Anwendung von Faricimab bei Schwangeren liegen keine oder nur begrenzte Daten vor. Die systemische Exposition durch Faricimab nach okularer Anwendung ist gering, jedoch muss Faricimab aufgrund seines Wirkmechanismus (VEGF-Hemmung) als potenziell teratogen und embryo-/fetotoxisch eingeschätzt werden (siehe Abschnitt 5.3).
Faricimab darf während der Schwangerschaft nicht angewendet werden, es sei denn, der potenzielle Nutzen überwiegt das potenzielle Risiko für den Fetus.
Stillzeit
Es ist nicht bekannt, ob Faricimab in die Muttermilch übergeht. Ein Risiko für das gestillte Neugeborene/den Säugling kann nicht ausgeschlossen werden. Vabysmo darf während der Stillzeit nicht angewendet werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen abzubrechen ist oder ob die Behandlung mit Faricimab abzubrechen ist bzw. auf die Behandlung mit Faricimab verzichtet werden soll. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
In einer 6‑monatigen Studie mit Faricimab an Cynomolgus-Affen wurden keine Auswirkungen auf die Reproduktionsorgane oder die Fertilität beobachtet (siehe Abschnitt 5.3).
Vabysmo hat einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Vorübergehende Sehstörungen können nach der intravitrealen Injektion und der damit einhergehenden Augenuntersuchung auftreten. Patienten sollen kein Fahrzeug führen und keine Maschinen bedienen, bis sich ihr Sehvermögen wieder ausreichend erholt hat.
Zusammenfassung des Sicherheitsprofils
Die am häufigsten berichteten Nebenwirkungen waren Katarakt (10 %), Bindehautblutung (7 %), Glaskörperablösung (4 %), erhöhter IOD (4 %), Mouches volantes (4 %), Augenschmerzen (3 %) und Einriss des retinalen Pigmentepithels (nur nAMD) (3 %).
Die schwerwiegendsten Nebenwirkungen waren Uveitis (0,5 %), Endophthalmitis (0,4 %), Vitritis (0,4 %), Netzhauteinriss (0,2 %), rhegmatogene Netzhautablösung (0,1 %) und traumatischer Katarakt (< 0,1 %) (siehe Abschnitt 4.4).
Tabellarische Auflistung der Nebenwirkungen
Die in klinischen Studien oder aus Beobachtungen nach der Markteinführung berichteten Nebenwirkungen sind nach MedDRA-Systemorganklassen aufgelistet und entsprechend ihrer Häufigkeit nach folgender Konvention geordnet: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000) oder nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 1: Häufigkeit von Nebenwirkungen
MedDRA-Systemorganklasse |
Häufigkeitskategorie |
Augenerkrankungen | |
Katarakt |
Häufig |
Bindehautblutung |
Häufig |
Glaskörperablösung |
Häufig |
Erhöhter intraokularer Druck |
Häufig |
Mouches volantes |
Häufig |
Einriss des retinalen Pigmentepithels (nur nAMD) |
Häufig |
Augenschmerzen |
Häufig |
Hornhautabschürfung |
Gelegentlich |
Augenreizung |
Gelegentlich |
Verstärkte Tränensekretion |
Gelegentlich |
Verschwommenes Sehen |
Gelegentlich |
Augenjucken |
Gelegentlich |
Augenbeschwerden |
Gelegentlich |
Okuläre Hyperämie |
Gelegentlich |
Iritis |
Gelegentlich |
Verminderte Sehschärfe |
Gelegentlich |
Uveitis |
Gelegentlich |
Endophthalmitis |
Gelegentlich |
Fremdkörpergefühl |
Gelegentlich |
Glaskörperblutung |
Gelegentlich |
Vitritis |
Gelegentlich |
Iridozyklitis |
Gelegentlich |
Bindehauthyperämie |
Gelegentlich |
Schmerzen während eines Eingriffs |
Gelegentlich |
Netzhauteinriss |
Gelegentlich |
Rhegmatogene Netzhautablösung |
Gelegentlich |
Vorübergehend verminderte Sehschärfe |
Selten |
Traumatischer Katarakt |
Selten |
Retinale Vaskulitis* |
Nicht bekannt |
Retinale okklusive Vaskulitis* |
Nicht bekannt |
Mit Sternchen (*) gekennzeichnete Begriffe sind Nebenwirkungen, die auf der Grundlage von Spontanberichten nach der Markteinführung identifiziert wurden. Da diese Nebenwirkungen auf freiwilliger Basis von einer Population unbekannter Größe gemeldet wurden, ist es nicht immer möglich, ihre Häufigkeit verlässlich abzuschätzen.
Beschreibung ausgewählter Nebenwirkungen
Retinale Vaskulitis und retinale okklusive Vaskulitis
Seltene Fälle von retinaler Vaskulitis und/oder retinaler okklusiver Vaskulitis wurden nach der Markteinführung spontan berichtet (siehe Abschnitt 4.4). Retinale Vaskulitis und retinale okklusive Vaskulitis wurden auch bei Patienten berichtet, die mit intravitrealen Therapien behandelt wurden.
Produktklassenbezogene Nebenwirkungen
Nach intravitrealer Anwendung von VEGF-Inhibitoren besteht ein theoretisches Risiko für arterielle thromboembolische Ereignisse, einschließlich Schlaganfall und Myokardinfarkt. In den klinischen Studien mit Faricimab bei Patienten mit nAMD, DMÖ und RVV wurde eine geringe Inzidenzrate arterieller thromboembolischer Ereignisse beobachtet (siehe Abschnitt 4.4). Über die Indikationen hinweg wurden keine nennenswerten Unterschiede zwischen den mit Faricimab und den mit dem Vergleichspräparat behandelten Gruppen festgestellt.
Immunogenität
Bei Patienten, die mit Faricimab behandelt werden, besteht die Möglichkeit einer Immunantwort (siehe Abschnitt 4.4). Nach Gabe von randomisiert zugeteiltem Faricimab bis zu 112 (nAMD), 100 (DMÖ) bzw. 72 (RVV) Wochen waren bei ca. 13,8 % (nAMD), 9,6 % (DMÖ) bzw. 14,4 % (RVV) der Patienten behandlungsbedingte Anti-Faricimab-Antikörper nachweisbar. Die klinische Bedeutung von Anti-Faricimab-Antikörpern für die Sicherheit ist derzeit unklar. Die Inzidenz intraokularer Entzündungen lag bei anti-Faricimab-Antikörper-positiven Patienten bei 12/98 (12,2 %; nAMD), 15/128 (11,7 %; DMÖ) und 9/95 (9,5 %; RVV) und bei anti-Faricimab-Antikörper-negativen Patienten bei 8/562 (1,4 %; nAMD), 5/1 124 (0,4 %; DMÖ) und 10/543 (1,8 %; RVV). Die Inzidenz schwerwiegender okularer Nebenwirkungen lag bei anti-Faricimab-Antikörper-positiven Patienten bei 6/98 (6,1 %; nAMD), 14/128 (10,9 %; DMÖ) und 7/95 (7,4 %; RVV) und bei anti-Faricimab-Antikörper-negativen Patienten bei 23/562 (4,1 %; nAMD), 45/1 124 (4,0 %; DMÖ) und 34/543 (6,3 %; RVV). Anti-Faricimab-Antikörper standen nicht in Verbindung mit Auswirkungen auf die klinische Wirksamkeit oder die systemische Pharmakokinetik.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51‑59
63225 Langen
Tel: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
anzuzeigen.
Eine Überdosierung mit einem größeren als dem empfohlenen Injektionsvolumen kann zu einem Anstieg des Augeninnendrucks führen. Im Falle einer Überdosierung muss der IOD überwacht und, sofern dies vom behandelnden Arzt für erforderlich gehalten wird, eine geeignete Behandlung eingeleitet werden.
Pharmakotherapeutische Gruppe: Ophthalmika, antineovaskuläre Mittel, ATC‑Code: S01LA09
Wirkmechanismus
Faricimab ist ein humanisierter bispezifischer Immunglobulin‑G1(IgG1)-Antikörper, der durch Hemmung von zwei unterschiedlichen Signalwegen wirkt, indem er sowohl Angiopoietin‑2 (Ang‑2) als auch den vaskulären endothelialen Wachstumsfaktor A (VEGF‑A) neutralisiert.
Ang‑2 verursacht eine vaskuläre Instabilität, indem es die Destabilisierung des Endothels, den Verlust von Perizyten und die pathologische Angiogenese fördert und so die vaskuläre Leckage und Entzündung verstärkt. Außerdem sensibilisiert es die Blutgefäße für die Aktivität von VEGF‑A, was zu einer weiteren Destabilisierung der Gefäße führt. Ang‑2 und VEGF‑A erhöhen synergistisch die Gefäßpermeabilität und stimulieren die Neovaskularisation.
Durch die duale Hemmung von Ang‑2 und VEGF‑A reduziert Faricimab die Gefäßpermeabilität und Entzündung, hemmt die pathologische Angiogenese und stellt die Gefäßstabilität wieder her.
Pharmakodynamische Wirkungen
In den sechs nachstehend beschriebenen Phase‑III-Studien wurde ab Tag 7 eine Verminderung der medianen okularen Konzentrationen von freiem Ang‑2 und freiem VEGF‑A gegenüber dem Ausgangswert beobachtet.
nAMD
In den Studien TENAYA und LUCERNE wurden objektive, prä-spezifizierte visuelle und anatomische Kriterien sowie die klinische Einschätzung des behandelnden Arztes als Entscheidungsgrundlage für die Behandlung zu den Beurteilungszeitpunkten der Krankheitsaktivität (Woche 20 und Woche 24) verwendet.
Die mittlere Reduktion der zentralen Subfelddicke (Central Subfield Thickness - CST) ab Behandlungsbeginn bis zu den Kontrolluntersuchungen zum primären Endpunkt (gemittelt in den Wochen 40 ‑ 48) war vergleichbar zu der unter Aflibercept beobachteten und betrug in den Studien TENAYA und LUCERNE bei mit Faricimab behandelten Patienten bei einer Gabe bis zu alle 16 Wochen (Q16W) ‑137 µm bzw. ‑137 µm im Vergleich zu ‑129 µm bzw. ‑131 µm unter Aflibercept. Diese mittleren CST-Reduktionen blieben bis zum Ende von Jahr 2 erhalten.
In Woche 48 zeigte sich in beiden Studien eine vergleichbare Wirkung von Faricimab und Aflibercept auf die Reduktion von intraretinaler Flüssigkeit (IRF), subretinaler Flüssigkeit (SRF) und PED. Diese Effekte bei IRF, SRF und PED blieben bis Jahr 2 erhalten. Auch die Veränderung der Gesamtfläche der CNV-Läsionen und die Reduktion des CNV-Bereichs mit Leckage waren bei den Patienten in den Behandlungsarmen mit Faricimab und Aflibercept vergleichbar.
DMÖ
In den Studien YOSEMITE und RHINE waren anatomische Parameter im Zusammenhang mit dem Makulaödem Teil der Beurteilung der Krankheitsaktivität, die die Behandlungsentscheidungen leitete.
In der Studie YOSEMITE war die mittlere CST-Reduktion gegenüber dem Ausgangswert bei den Kontrolluntersuchungen zum primären Endpunkt (gemittelt in den Wochen 48 ‑ 56) numerisch größer als unter Aflibercept. Sie lag bei den Patienten, denen Faricimab alle 8 Wochen bzw. Faricimab mit einem anpassbaren Dosierungsintervall von bis zu alle 16 Wochen verabreicht wurde, bei ‑207 µm bzw. ‑197 µm, im Vergleich zu ‑170 µm bei Patienten, denen Aflibercept alle 8 Wochen verabreicht wurde; in der Studie RHINE lagen die Ergebnisse bei 196 µm, 188 µm bzw. 170 µm. Im Verlauf des zweiten Jahres wurde eine konsistente CST-Reduktion beobachtet. In beiden Studien erreichte ein größerer Anteil der Patienten in beiden Behandlungsarmen mit Faricimab im Vergleich zum Aflibercept-Behandlungsarm im Verlauf des zweiten Jahres das Nichtvorhandensein von IRF und das Nichtvorhandensein eines DMÖ (definiert als Erreichen einer CST unter 325 μm).
RVV
In den Phase‑III-Studien bei Patienten mit retinalem Venenastverschluss (VAV; BALATON) und retinalem Zentralvenenverschluss/Hemi-Zentralvenenverschluss (ZVV/HZVV; COMINO) wurde mit Faricimab alle 4 Wochen eine mittlere CST-Reduktion vom Ausgangswert bis Woche 24 beobachtet, die mit der von Aflibercept alle 4 Wochen vergleichbar war. In der Studie BALATON betrug die mittlere CST-Reduktion vom Ausgangswert bis Woche 24 311,4 μm für Faricimab alle 4 Wochen gegenüber 304,4 μm für Aflibercept alle 4 Wochen und in der Studie COMINO 461,6 μm für Faricimab gegenüber 448,8 μm für Aflibercept. Die CST-Reduktionen blieben bis Woche 72 erhalten, als die Patienten auf ein anpassbares Dosierungsschema mit Faricimab bis zu alle 16 Wochen umgestellt wurden.
In beiden Studien erreichten vergleichbare Anteile der Patienten in den beiden Behandlungsarmen mit Faricimab alle 4 Wochen und Aflibercept alle 4 Wochen im Verlauf bis Woche 24 das Nichtvorhandensein von IRF, SRF und Makulaödem (definiert als Erreichen einer CST unter 325 μm). Diese Ergebnisse blieben bis Woche 72 erhalten, als die Patienten auf ein anpassbares Dosierungsschema mit Faricimab bis zu alle 16 Wochen umgestellt wurden.
Klinische Wirksamkeit und Sicherheit
nAMD
Die Sicherheit und Wirksamkeit von Faricimab wurde in den beiden randomisierten, multizentrischen, doppelblinden, aktiv kontrollierten 2‑jährigen Nicht-Unterlegenheits-Studien TENAYA und LUCERNE bei Patienten mit nAMD untersucht. Insgesamt wurden 1 329 Patienten eingeschlossen, von denen 1 135 (85 %) Patienten die Studien bis Woche 112 abschlossen. Insgesamt erhielten 1 326 Patienten mindestens eine Dosis (664 Faricimab). Das Alter der Patienten reichte von 50 bis 99 Jahren mit einem Durchschnittsalter [Standardabweichung; SD] von 75,9 [8,6] Jahren.
In beiden Studien wurden die Patienten im Verhältnis 1:1 einem von zwei Behandlungsarmen randomisiert zugeteilt:
Faricimab 6 mg bis zu alle 16 Wochen nach vier initialen monatlichen Dosen
Aflibercept 2 mg alle 8 Wochen nach drei initialen monatlichen Dosen
Nach den ersten vier monatlichen Dosen (Wochen 0, 4, 8 und 12) erhielten die Patienten, die dem Behandlungsarm mit Faricimab randomisiert zugeteilt waren, ihre Dosis alle 16 Wochen, alle 12 Wochen oder alle 8 Wochen, basierend auf einer Beurteilung der Krankheitsaktivität in den Wochen 20 und 24. Die Krankheitsaktivität wurde anhand objektiver, prä-spezifizierter visueller (BCVA) und anatomischer (CST) Kriterien beurteilt, sowie anhand der klinischen Beurteilung des behandelnden Arztes hinsichtlich des Vorliegens einer Makulablutung oder behandlungsbedürftiger nAMD-Krankheitsaktivität (nur Woche 24). Die festgelegten Dosierungsintervalle wurden bei den Patienten bis Woche 60 ohne zusätzliche Therapie beibehalten. Ab Woche 60 wurden die Patienten im Faricimab-Behandlungsarm auf ein anpassbares Dosierungsschema umgestellt, bei dem ihr Behandlungsintervall in 4‑Wochen-Schritten verlängert (bis zu alle 16 Wochen) oder in 8‑Wochen-Schritten (bis zu alle 8 Wochen) verkürzt werden konnte, basierend auf einer automatisierten objektiven Beurteilung präspezifizierter visueller Befunde (BCVA) und anatomischer Kriterien der Krankheitsaktivität (CST und Makulablutung). Die Patienten im Aflibercept-Arm verblieben während der gesamten Studienperiode bei der Dosierung alle 8 Wochen. Beide Studien dauerten 112 Wochen.
Ergebnisse
Beide Studien zeigten die Wirksamkeit bezüglich des primären Endpunkts, der definiert war als mittlere Veränderung der BCVA gegenüber dem Ausgangswert, gemittelt über die Untersuchungstermine in Woche 40, 44 und 48 und gemessen anhand des Buchstaben-Scores der Early Treatment Diabetic Retinopathy Study (ETDRS) (Tabelle 2 und Tabelle 3). In beiden Studien hatten die mit Faricimab bis zu alle 16 Wochen behandelten Patienten nach Jahr 1 eine mittlere Veränderung der BCVA gegenüber dem Ausgangswert, welche der unter Aflibercept alle 8 Wochen nicht unterlegen war und diese Sehverbesserungen wurden bis Woche 112 aufrechterhalten. Die Verbesserungen der BCVA ab Behandlungsbeginn bis Woche 112 sind in Abbildung 1 dargestellt.
Der Anteil der Patienten unter den verschiedenen Behandlungsintervallen in Woche 112 betrug in den Studien TENAYA bzw. LUCERNE:
Alle 16 Wochen: 59 % bzw. 67 %
Alle 12 Wochen: 15 % bzw. 14 %
Alle 8 Wochen: 26 % bzw. 19 %
Tabelle 2: Wirksamkeitsergebnisse bei den Kontrolluntersuchungen zum primären Endpunkta und zum Zeitpunkt nach 2 Jahrenb in der Studie TENAYA
Wirksamkeitsergebnisse |
TENAYA |
|||
Jahr 1 |
Jahr 2 |
|||
Faricimab bis zu alle 16 Wochen |
Aflibercept alle 8 Wochen |
Faricimab bis zu alle 16 Wochen |
Aflibercept alle 8 Wochen |
|
Mittlere Veränderung der BCVA gegenüber dem Ausgangswert, gemessen anhand des ETDRS‑Buchstaben-Scores (95‑%‑KI) |
5,8 |
5,1 |
3,7 |
3,3 |
Unterschied im LS-Mean (95‑%‑KI) |
0,7 |
0,4 |
||
Anteil der Patienten mit Zuwachs von ≥ 15 Buchstaben gegenüber dem Ausgangswert (CMH‑gewichteter Anteil, 95‑%‑KI) |
20,0 % |
15,7 % |
22,5 % |
16,9 % |
Unterschied im CMH‑gewichteten %‑Wert (95‑%‑KI) |
4,3 % |
5,6 % |
||
Anteil der Patienten mit Vermeidung des Verlusts von ≥ 15 Buchstaben gegenüber dem Ausgangswert (CMH‑gewichteter Anteil, 95‑%‑KI) |
95,4 % |
94,1 % |
92,1 % |
88,6 % |
Unterschied im CMH‑gewichteten %‑Wert (95‑%‑KI) |
1,3 % |
3,4 % |
||
aDurchschnitt der Wochen 40, 44 und 48; bDurchschnitt der Wochen 104, 108 und 112
BCVA: Bestkorrigierte Sehschärfe
ETDRS: Early Treatment Diabetic Retinopathy Study
KI: Konfidenzintervall
LS: Least Squares (kleinste Fehlerquadrate)
CMH: Cochran-Mantel-Haenszel-Methode; ein statistischer Test, der eine Schätzung einer Assoziation mit einem binären Ergebnis erstellt und zur Beurteilung kategorischer Variablen verwendet wird.
Tabelle 3: Wirksamkeitsergebnisse bei den Kontrolluntersuchungen zum primären Endpunkta und zum Zeitpunkt nach 2 Jahrenb in der Studie LUCERNE
Wirksamkeitsergebnisse |
LUCERNE |
|||
Jahr 1 |
Jahr 2 |
|||
Faricimab bis zu alle 16 Wochen |
Aflibercept alle 8 Wochen |
Faricimab bis zu alle 16 Wochen |
Aflibercept alle 8 Wochen |
|
Mittlere Veränderung der BCVA gegenüber dem Ausgangswert, gemessen anhand des ETDRS‑Buchstaben-Scores (95‑%‑KI) |
6,6 |
6,6 |
5,0 |
5,2 |
Unterschied im LS-Mean (95‑%‑KI) |
0,0 |
-0,2 |
||
Anteil der Patienten mit Zuwachs von ≥ 15 Buchstaben gegenüber dem Ausgangswert (CMH‑gewichteter Anteil, 95‑%‑KI) |
20,2 % |
22,2 % |
22,4 % |
21,3 % |
Unterschied im CMH‑gewichteten %‑Wert (95‑%‑KI) |
-2,0 % |
1,1 % |
||
Anteil der Patienten mit Vermeidung des Verlusts von ≥ 15 Buchstaben gegenüber dem Ausgangswert (CMH‑gewichteter Anteil, 95‑%‑KI) |
95,8 % |
97,3 % |
92,9 % |
93,2 % |
Unterschied im CMH‑gewichteten %‑Wert (95‑%‑KI) |
-1,5 % |
-0,2 % |
||
aDurchschnitt der Wochen 40, 44 und 48; bDurchschnitt der Wochen 104, 108 und 112
BCVA: Bestkorrigierte Sehschärfe
ETDRS: Early Treatment Diabetic Retinopathy Study
KI: Konfidenzintervall
LS: Least Squares (kleinste Fehlerquadrate)
CMH: Cochran-Mantel-Haenszel-Methode; ein statistischer Test, der eine Schätzung einer Assoziation mit einem binären Ergebnis erstellt und zur Beurteilung kategorischer Variablen verwendet wird.
Abbildung 1: Mittlere Veränderung der Sehschärfe von Behandlungsbeginn bis zum Jahr 2 (Woche 112); kombinierte Daten der Studien TENAYA und LUCERNE
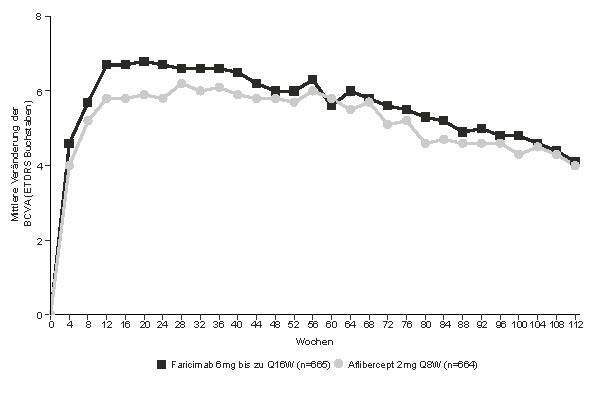
Sowohl in der Studie TENAYA als auch in der Studie LUCERNE waren die Verbesserungen der BCVA und der CST in Woche 60 gegenüber dem Ausgangswert in beiden Behandlungsarmen vergleichbar und stimmten mit jenen überein, die in Woche 48 beobachtet wurden.
In Woche 60 hatten 46 % der Patienten sowohl in der Studie TENAYA als auch in der Studie LUCERNE ein Intervall von alle 16 Wochen. Davon behielten 69 % der Patienten in beiden Studien ein Intervall von alle 16 Wochen bis Woche 112 ohne Intervallverkürzung bei.
In Woche 60 hatten 80 % der Patienten in der Studie TENAYA und 78 % der Patienten in der Studie LUCERNE ein Intervall von ≥ alle 12 Wochen (alle 16 Wochen oder alle 12 Wochen). Davon behielten 67 % bzw. 75 % der Patienten ein Intervall von ≥ alle 12 Wochen bis Woche 112 bei, ohne eine Intervallverkürzung auf unter alle 12 Wochen.
In Woche 60 hatten 33 % der Patienten sowohl in der Studie TENAYA als auch in der Studie LUCERNE ein Intervall von alle 12 Wochen. Davon behielten 3,2 % bzw. 0 % der Patienten in der Studie TENAYA bzw. in der Studie LUCERNE das Intervall von alle 12 Wochen bis Woche 112 bei.
In Woche 60 hatten 20 % der Patienten in der Studie TENAYA und 22 % der Patienten in der Studie LUCERNE ein Intervall von alle 8 Wochen. Davon behielten 34 % bzw. 30 % der Patienten in der Studie TENAYA bzw. der Studie LUCERNE die Therapie alle 8 Wochen bis Woche 112 bei.
Die Ergebnisse zur Wirksamkeit stimmten bei allen auswertbaren Subgruppen (z. B. nach Alter, Geschlecht, ethnischer Zugehörigkeit, Ausgangswert der Sehschärfe, Läsionstyp, Läsionsgröße), in jeder Studie und in der gepoolten Analyse mit den Ergebnissen in der Gesamtpopulation überein.
In allen Studien zeigte Faricimab bis zu alle 16 Wochen eine Verbesserung beim prä-spezifizierten Wirksamkeitsendpunkt der mittleren Veränderung des Gesamtscores des National Eye Institute Visual Function Questionnaire (NEI-VFQ-25) zwischen Behandlungsbeginn und Woche 48, die mit der unter Aflibercept alle 8 Wochen vergleichbar war und den Schwellenwert von 4 Punkten übertraf. Die Größenordnung dieser Veränderungen entspricht einem Zuwachs von 15 Buchstaben bei der BCVA.
Die Inzidenz okularer unerwünschter Ereignisse am Studienauge betrug 53,9 % bzw. 52,1 % und die Inzidenz nicht-okularer unerwünschter Ereignisse 73,3 % bzw. 74,3 % bis Woche 112 in den Faricimab- bzw. Aflibercept-Behandlungsarmen (siehe Abschnitte 4.4 und 4.8).
DMÖ
Die Sicherheit und Wirksamkeit von Faricimab wurde in zwei randomisierten, multizentrischen, doppelblinden, aktiv kontrollierten 2‑jährigen Nicht-Unterlegenheits-Studien (YOSEMITE und RHINE) bei Patienten mit DMÖ untersucht. Insgesamt wurden 1 891 Patienten in die zwei Studien aufgenommen, wobei 1 622 (86 %) der Patienten bis Woche 100 an den Studien teilnahmen. Insgesamt 1 887 Patienten wurden bis Woche 56 mit mindestens einer Dosis behandelt (1 262 mit Faricimab). Das Alter der Patienten reichte von 24 bis 91 Jahren mit einem Durchschnittsalter [SD] von 62,2 [9,9] Jahren. Die Gesamtpopulation umfasste sowohl anti-VEGF-naive Patienten (78 %) als auch Patienten, die vor der Studienteilnahme bereits mit einem VEGF‑Inhibitor behandelt worden waren (22 %). In beiden Studien wurden die Patienten im Verhältnis 1:1:1 einem der drei Behandlungsschemata randomisiert zugeteilt:
Faricimab 6 mg alle 8 Wochen nach den ersten 6 monatlichen Dosen.
Faricimab 6 mg bis zu alle 16 Wochen mit anpassbarem Dosierungsintervall verabreicht in 4‑, 8‑, 12‑ oder 16‑Wochen-Intervallen, nach den ersten 4 monatlichen Dosen.
Aflibercept 2 mg alle 8 Wochen nach den ersten 5 monatlichen Dosen.
In dem 16‑Wochen-Behandlungsarm mit anpassbarem Dosierungsintervall folgte die Dosierung einem standardisierten Treat-and-Extend-Ansatz. Das Dosierungsintervall konnte in 4‑Wochen-Schritten verlängert oder in 4‑ oder 8‑Wochen-Schritten verkürzt werden, basierend auf anatomischen und/oder visuellen Befunden auf Grundlage von Daten, die ausschließlich bei Injektionsterminen der Studie erhoben wurden.
Ergebnisse
Beide Studien zeigten Wirksamkeit bezüglich des primären Endpunkts, der definiert war als mittlere Veränderung der BCVA gegenüber dem Ausgangswert nach 1 Jahr (Durchschnittswert der Untersuchungstermine in Woche 48, 52 und 56), gemessen anhand des ETDRS-Buchstaben-Score. In beiden Studien hatten die mit Faricimab bis zu alle 16 Wochen behandelten Patienten eine mittlere Veränderung der BCVA gegenüber dem Ausgangswert, die der der Patienten, die 1 Jahr mit Aflibercept alle 8 Wochen behandelt wurden, nicht unterlegen war. Diese Sehverbesserungen wurden im zweiten Jahr aufrechterhalten.
Nach den ersten 4‑Monats-Dosen konnten die Patienten im Behandlungsarm mit Faricimab mit anpassbarem Dosierungsintervall von bis zu alle 16 Wochen bis zum Ende der Woche 96 insgesamt mindestens 6 und höchstens 21 Injektionen erhalten. In der Studie YOSEMITE bzw. der Studie RHINE erreichten in Woche 52 74 % bzw. 71 % der Patienten in dem 16‑Wochen-Behandlungsarm mit anpassbarem Dosierungsintervall mit Faricimab ein Dosierungsintervall von alle 16 Wochen bzw. alle 12 Wochen (53 % bzw. 51 % alle 16 Wochen, 21 % bzw. 20 % alle 12 Wochen). Von diesen Patienten erhielten 75 % in der Studie YOSEMITE und 84 % in der Studie RHINE das Dosierungsintervall von mindestens alle 12 Wochen ohne Intervallverkürzung auf weniger als alle 12 Wochen bis zum Ende der Woche 96 aufrecht; von den Patienten mit einem Dosierungsintervall von alle 16 Wochen in Woche 52 erhielten 70 % (YOSEMITE) bzw. 82 % (RHINE) das Dosierungsintervall von alle 16 Wochen ohne Intervallverkürzung bis zum Ender der Woche 96 aufrecht. In Woche 96 hatten 78 % der Patienten im Behandlungsarm mit Faricimab mit anpassbarem Dosierungsintervall von bis zu alle 16 Wochen in beiden Studien ein Dosierungsintervall von alle 16 Wochen oder alle 12 Wochen erreicht (60 % bzw. 64 % alle 16 Wochen, 18 % bzw. 14 % alle 12 Wochen). Bei 4 % bzw. 6 % der Patienten wurde das Dosierungsintervall auf alle 8 Wochen ausgeweitet und mit einem Dosierungsintervall von höchstens alle 8 Wochen bis zum Ende der Woche 96 beibehalten; lediglich 3 % bzw. 5 % der Patienten in der Studie YOSEMITE oder RHINE wurden mit einem Dosierungsintervall von alle 4 Wochen bis Ende der Woche 96 behandelt.
Detaillierte Ergebnisse aus der Analyse der Studien YOSEMITE und RHINE sind in den Tabellen 4 und 5 sowie in Abbildung 2 dargestellt.
Tabelle 4: Wirksamkeitsergebnisse bei den Kontrolluntersuchungen zum primären Endpunkt in Jahr 1a sowie in Jahr 2b in der Studie YOSEMITE
Wirksamkeitsergebnisse |
YOSEMITE |
|||||
Jahr 1 |
Jahr 2 |
|||||
Faricimab alle 8 Wochen |
Faricimab mit anpassbarem Dosierungs-intervall von bis zu alle 16 Wochen |
Aflibercept alle 8 Wochen |
Faricimab alle 8 Wochen |
Faricimab mit anpassbarem Dosierungs-intervall von bis zu alle 16 Wochen |
Aflibercept alle 8 Wochen |
|
Mittlere Veränderung der BCVA gegenüber dem Ausgangswert, gemessen anhand des ETDRS-Buchstaben-Scores (97,5‑%‑KI für Jahr 1 und 95‑%‑KI für Jahr 2) |
10,7 |
11,6 |
10,9 |
10,7 |
10,7 |
11,4 |
Unterschied im LS-Mean (97,5‑%‑KI für Jahr 1, 95‑%‑KI für Jahr 2) |
-0,2 |
0,7 |
-0,7 |
-0,7 |
||
Anteil der Patienten mit Zunahme der BCVA um mindestens 15 Buchstaben gegenüber dem Ausgangswert (CMH-gewichteter Anteil, 95‑%‑KI für Jahr 1 und Jahr 2) |
29,2 % |
35,5 % |
31,8 % |
37,2 % |
38,2 % |
37,4 % |
Unterschied im CMH-gewichteten %‑Wert (95‑%‑KI für Jahr 1 und Jahr 2) |
-2,6 % |
3,5 % |
-0,2 % |
0,2 % |
||
Anteil der Patienten mit Vermeidung eines BCVA-Verlustes von mindestens 15 Buchstaben gegenüber dem Ausgangswert (CMH-gewichteter Anteil, 95‑%‑KI für Jahr 1 und Jahr 2) |
98,1 % |
98,6 % |
98,9 % |
97,6 % |
97,8 % |
98,0 % |
Unterschied im CMH-gewichteten %‑Wert (95‑%‑KI für Jahr 1 und Jahr 2) |
-0,8 % |
-0,3 % |
-0,4 % |
-0,2 % |
||
aDurchschnitt der Wochen 48, 52 und 56; bDurchschnitt der Wochen 92, 96 und 100
BCVA: Bestkorrigierte Sehschärfe
ETDRS: Early Treatment Diabetic Retinopathy Study
LS: Least Squares (kleinste Fehlerquadrate)
KI: Konfidenzintervall
CMH: Cochran-Mantel-Haenszel-Methode; ein statistischer Test, der eine Schätzung einer Assoziation mit einem binären Ergebnis erstellt und zur Beurteilung kategorischer Variablen verwendet wird.
Hinweis: Der CMH‑gewichtete %‑Wert für den Aflibercept-Behandlungsarm wurde für den Vergleich von Faricimab alle 8 Wochen mit Aflibercept dargestellt; der entsprechende CMH‑gewichtete %‑Wert für den Vergleich von Faricimab mit anpassbarem Dosierungsintervall mit Aflibercept ist jedoch ähnlich wie der oben dargestellte Wert.
Tabelle 5: Wirksamkeitsergebnisse bei den Kontrolluntersuchungen zum primären Endpunkt in Jahr 1a sowie in Jahr 2b in der Studie RHINE
Wirksamkeitsergeb-nisse |
RHINE |
|||||
Jahr 1 |
Jahr 2 |
|||||
Faricimab alle 8 Wochen |
Faricimab mit anpass-barem Dosie-rungsinter-vall |
Afliber-cept alle 8 Wochen |
Faricimab alle 8 Wochen |
Faricimab mit anpass-barem Dosierungs-intervall von bis zu alle 16 Wochen |
Afliber-cept alle 8 Wochen |
|
Mittlere Veränderung der BCVA gegenüber dem Ausgangswert, gemessen anhand des ETDRS‑Buchstaben-Scores (97,5‑%‑KI für Jahr 1 und 95‑%‑KI für Jahr 2) |
11,8 |
10,8 |
10,3 |
10,9 |
10,1 |
9,4 |
Unterschied im LS‑Mean (97,5‑%‑KI für Jahr 1, 95‑%‑KI für Jahr 2) |
1,5 |
0,5 |
1,5 |
0,7 |
||
Anteil der Patienten mit Zunahme der BCVA um mindestens 15 Buchstaben gegenüber dem Ausgangswert (CMH‑gewichteter Anteil, 95‑%‑KI für Jahr 1 und Jahr 2) |
33,8 % |
28,5 % |
30,3 % |
39,8 % |
31,1 % |
39,0 % |
Unterschied im CMH‑gewichteten |
3,5 % |
-2,0 % |
0,8 % |
-8 % |
||
Anteil der Patienten mit Vermeidung eines BCVA-Verlustes von mindestens 15 Buchstaben gegenüber dem Ausgangswert (CMH‑gewichteter Anteil, 95‑%‑KI für Jahr 1 und Jahr 2) |
98,9 % |
98,7 % |
98,6 % |
96,6 % |
96,8 % |
97,6 % |
Unterschied im CMH‑gewichteten |
0,3 % |
0,0 % |
-1,0 % |
-0,7 % |
||
aDurchschnitt der Wochen 48, 52 und 56; bDurchschnitt der Wochen 92, 96 und 100
BCVA: Bestkorrigierte Sehschärfe
ETDRS: Early Treatment Diabetic Retinopathy Study
LS: Least Squares (kleinste Fehlerquadrate)
KI: Konfidenzintervall
CMH: Cochran-Mantel-Haenszel-Methode; ein statistischer Test, der eine Schätzung einer Assoziation mit einem binären Ergebnis erstellt und zur Beurteilung kategorischer Variablen verwendet wird.
Hinweis: Der CMH‑gewichtete %‑Wert für den Aflibercept-Behandlungsarm wurde für den Vergleich von Faricimab alle 8 Wochen mit Aflibercept dargestellt; der entsprechende CMH‑gewichtete %‑Wert für den Vergleich von Faricimab mit anpassbarem Dosierungsintervall mit Aflibercept ist jedoch ähnlich wie der oben dargestellte Wert.
Abbildung 2: Mittlere Veränderung der Sehschärfe von Behandlungsbeginn bis Jahr 2 (Woche 100) in den Studien YOSEMITE und RHINE
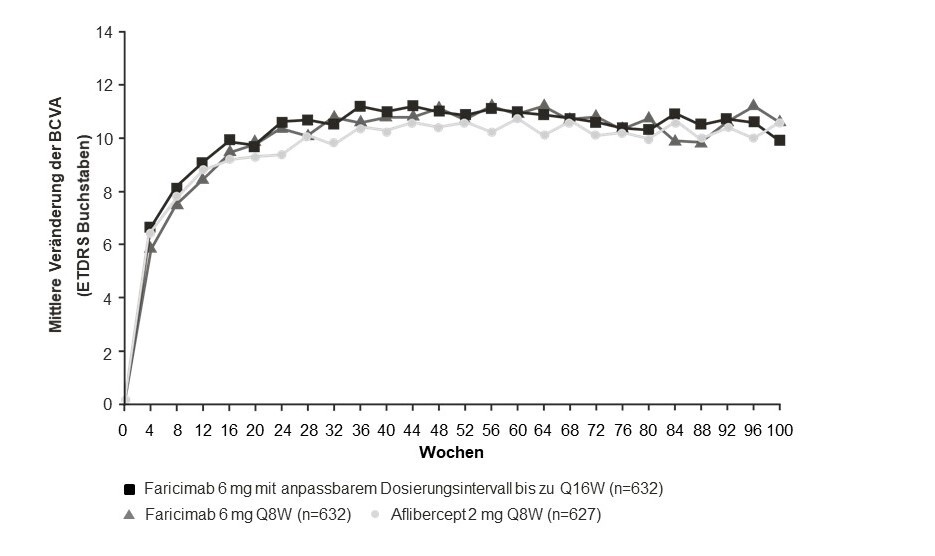
Die Ergebnisse zur Wirksamkeit bei den Patienten, die vor der Studienteilnahme anti-VEGF-naiv waren, sowie bei allen anderen auswertbaren Subgruppen (z. B. nach Alter, Geschlecht, ethnischer Zugehörigkeit, Ausgangs‑HbA1c, Ausgangswert der Sehschärfe) in jeder Studie stimmten mit den Ergebnissen in der Gesamtpopulation überein.
In allen Studien zeigten Faricimab alle 8 Wochen und Faricimab mit anpassbarem Dosierungsintervall von bis zu alle 16 Wochen Verbesserungen beim prä-spezifizierten Wirksamkeitsendpunkt der mittleren Veränderung des zusammengesetzten NEI-VFQ-25-Scores zwischen Behandlungsbeginn und Woche 52, die mit denen unter Aflibercept alle 8 Wochen vergleichbar waren und den Schwellenwert von 4 Punkten übertrafen. Faricimab alle 8 Wochen und Faricimab mit anpassbarem Dosierungsintervall von bis zu alle 16 Wochen zeigten außerdem klinisch bedeutsame Verbesserungen beim prä-spezifizierten Wirksamkeitsendpunkt der Veränderung des NEI-VFQ‑25-Scores für Aktivitäten im Nahsichtbereich, Aktivitäten im Fernsichtbereich und Autofahren zwischen Behandlungsbeginn und Woche 52, die mit denen unter Aflibercept alle 8 Wochen vergleichbar waren. Die Größenordnung dieser Veränderungen entspricht einem Zuwachs von 15 Buchstaben bei der BCVA. Bei vergleichbaren Anteilen von Patienten kam es unter Faricimab alle 8 Wochen, unter Faricimab mit anpassbarem Dosierungsintervall von bis zu alle 16 Wochen und unter Aflibercept alle 8 Wochen zu einer klinisch bedeutsamen Verbesserung beim prä-spezifizierten Wirksamkeitsendpunkt der Veränderung des zusammengesetzten Scores des NEI-VFQ-25 zwischen Behandlungsbeginn und Woche 52 um ≥ 4 Punkte. Diese Ergebnisse wurden in Woche 100 aufrechterhalten.
Ein weiterer wesentlicher Wirksamkeitsendpunkt in den DMÖ‑Studien war die Veränderung der Early Treatment Diabetic Retinopathy Study-Diabetic Retinopathy Severity Scale (ETDRS-DRSS) ab Behandlungsbeginn bis Woche 52. Von den 1 891 in die Studien YOSEMITE und RHINE aufgenommenen Patienten waren 708 bzw. 720 Patienten in Bezug auf DR‑Endpunkte auswertbar.
Die ETDRS-DRSS-Scores reichten zu Behandlungsbeginn von 10 bis 71.
Die Mehrzahl der Patienten, etwa 60 %, hatte zu Behandlungsbeginn eine mäßige bis schwere nichtproliferative DR (DRSS 43/47/53).
Der Anteil der Patienten, die eine ETDRS-DRSS-Verbesserung in Woche 52 und Woche 96 um ≥ 2 Stufen und ≥ 3 Stufen gegenüber dem Ausgangswert erreichten, sind in den Tabellen 6 und 7 zusammengefasst.
Tabelle 6: Anteil der Patienten, die in der Studie YOSEMITE in Woche 52 und in Woche 96 eine ≥ 2‑stufige und ≥ 3‑stufige Verbesserung des ETDRS-DRSS-Scores gegenüber dem Ausgangswert erreichten (auswertbare DR‑Population)
YOSEMITE |
||||||
52 Wochen |
96 Wochen |
|||||
Faricimab alle 8 Wochen |
Faricimab |
Aflibercept alle 8 Wochen |
Faricimab alle 8 Wochen |
Faricimab |
Aflibercept alle 8 Wochen |
|
Anteil der Patienten mit ≥ 2‑stufiger ETDRS-DRSS-Verbesserung gegenüber dem Ausgangswert |
46,0 % |
42,5 % |
35,8 % |
51,4 % |
42,8 % |
42,2 % |
Gewichteter Unterschied (97,5‑%‑KI für Jahr 1, 95‑%‑KI für Jahr 2) |
10,2 % |
6,1 % |
9,1 % |
0,0 % |
||
Anteil der Patienten mit ≥ 3‑stufiger ETDRS-DRSS-Verbesserung gegenüber dem Ausgangswert (CMH‑gewichteter Anteil) |
16,8 % |
15,5 % |
14,7 % |
22,4 % |
14,6 % |
20,9 % |
Gewichteter Unterschied (95‑%‑KI für Jahr 1 und Jahr 2) |
2,1 % |
0,6 % |
1,5 % |
-6,7 % |
||
ETDRS-DRSS: Early Treatment Diabetic Retinopathy Study-Diabetic Retinopathy Severity Scale
KI: Konfidenzintervall
CMH: Cochran-Mantel-Haenszel-Methode; ein statistischer Test, der eine Schätzung einer Assoziation mit einem binären Ergebnis erstellt und zur Beurteilung kategorischer Variablen verwendet wird.
Hinweis: Der CMH‑gewichtete %‑Wert für den Aflibercept-Behandlungsarm wurde für den Vergleich von Faricimab alle 8 Wochen mit Aflibercept dargestellt; der entsprechende CMH‑gewichtete %‑Wert für den Vergleich von Faricimab mit anpassbarem Dosierungsintervall mit Aflibercept ist jedoch ähnlich wie der oben dargestellte Wert.
Tabelle 7: Anteil der Patienten, die in der Studie RHINE in Woche 52 und in Woche 96 eine ≥ 2‑stufige und ≥ 3‑stufige Verbesserung des ETDRS-DRSS-Scores gegenüber dem Ausgangswert erreichten (auswertbare DR‑Population)
RHINE |
||||||
52 Wochen |
96 Wochen |
|||||
Faricimab alle 8 Wochen |
Faricimab |
Aflibercept alle 8 Wochen |
Faricimab alle 8 Wochen |
Faricimab |
Aflibercept alle 8 Wochen |
|
Anteil der Patienten mit ≥ 2‑stufiger ETDRS-DRSS-Verbesserung gegenüber dem Ausgangswert |
44,2 % |
43,7 % |
46,8 % |
53,5 % |
44,3 % |
43,8 % |
Gewichteter Unterschied (97,5‑%‑KI für Jahr 1, 95‑%‑KI für Jahr 2) |
-2,6 % |
-3,5 % |
9,7 % |
0,3 % |
||
Anteil der Patienten mit ≥ 3‑stufiger ETDRS-DRSS-Verbesserung gegenüber dem Ausgangswert (CMH‑gewichteter Anteil) |
16,7 % |
18,9 % |
19,4 % |
25,1 % |
19,3 % |
21,8 % |
Gewichteter Unterschied (95‑%‑KI für Jahr 1 und Jahr 2) |
-0,2 % |
-1,1 % |
3,3 % |
-2,7 % |
||
ETDRS-DRSS: Early Treatment Diabetic Retinopathy Study-Diabetic Retinopathy Severity Scale
KI: Konfidenzintervall
CMH: Cochran-Mantel-Haenszel-Methode; ein statistischer Test, der eine Schätzung einer Assoziation mit einem binären Ergebnis erstellt und zur Beurteilung kategorischer Variablen verwendet wird.
Hinweis: Der CMH‑gewichtete %‑Wert für den Aflibercept-Behandlungsarm wurde für den Vergleich von Faricimab alle 8 Wochen mit Aflibercept dargestellt; der entsprechende CMH‑gewichtete %‑Wert für den Vergleich von Faricimab mit anpassbarem Dosierungsintervall mit Aflibercept ist jedoch ähnlich wie der oben dargestellte Wert.
Die Behandlungseffekte in den auswertbaren Subgruppen (z. B. nach vorheriger Anti-VEGF-Therapie, Alter, Geschlecht, ethnischer Zugehörigkeit, Ausgangs‑HbA1c, Ausgangswert der Sehschärfe) in jeder Studie stimmten allgemein mit den Ergebnissen in der Gesamtpopulation überein.
Die Behandlungseffekte in den Subgruppen nach DR‑Schweregrad bei Behandlungsbeginn waren unterschiedlich und zeigten die größten ≥ 2‑stufigen DRSS-Verbesserungen bei Patienten mit mäßig schwerer und schwerer nichtproliferativer DR, wobei etwa 90 % der Patienten in allen Behandlungsarmen in beiden Studien übereinstimmend Verbesserungen erreichten.
Die Inzidenz okularer unerwünschter Ereignisse am Studienauge betrug 49,7 %, 49,2 % bzw. 45,4 %, und die Inzidenz nicht-okularer unerwünschter Ereignisse betrug 73,0 %, 74,2 % bzw. 75,7 % bis Woche 100 in den Behandlungsarmen Faricimab alle 8 Wochen, Faricimab bis zu alle 16 Wochen bzw. Aflibercept alle 8 Wochen (siehe Abschnitte 4.4 und 4.8).
1 474 Patienten, die zuvor die Studie YOSEMITE oder RHINE abgeschlossen hatten, wurden in die Studie RHONE-X aufgenommen, eine 2-jährige, multizentrische, Langzeit-Verlängerungsstudie zur Beurteilung der Langzeit-Sicherheit und -Verträglichkeit von intravitrealem Faricimab 6 mg, das mit einem personalisierten Behandlungsintervall verabreicht wurde.
Das in der Studie RHONE-X beobachtete Langzeit-Sicherheitsprofil von Faricimab stimmte mit dem der Studien YOSEMITE und RHINE überein.
RVV
Die Sicherheit und Wirksamkeit von Faricimab wurde in zwei randomisierten, multizentrischen, doppelblinden, 72-wöchigen Studien bei Patienten mit Makulaödem infolge von VAV (BALATON) oder ZVV/HZVV (COMINO) untersucht. Bis Monat 6 liegen Daten aus der aktiven Vergleichskontrolle vor.
Insgesamt wurden 1 282 Patienten (553 in BALATON und 729 in COMINO) in die beiden Studien eingeschlossen, wobei 1 276 Patienten bis Woche 24 mit mindestens einer Dosis behandelt wurden (641 mit Faricimab). Das Alter der Patienten reichte von 28 bis 93 Jahren mit einem Durchschnittsalter [SD] von 64 [10,7] Jahren bzw. von 22 bis 100 Jahren mit einem Durchschnittsalter [SD] von 65 [13,2] Jahren in BALATON bzw. COMINO.
Insgesamt schlossen 489 von 533 Patienten, die randomisiert in die Studie BALATON zugeteilt worden waren, die Studie in Woche 72 ab. 263 Patienten, die ursprünglich Faricimab randomisiert zugeteilt worden waren („vorherige Faricimab-Patienten“) und 267, die ursprünglich Aflibercept randomisiert zugeteilt worden waren („vorherige Aflibercept-Patienten“) erhielten während der Phase mit angepasstem Dosierungsschema von Faricimab mindestens eine Dosis Faricimab.
Insgesamt schlossen 656 von 729 Patienten, die randomisiert in die Studie COMINO zugeteilt worden waren, die Studie in Woche 72 ab. 353 „vorherige Faricimab-Patienten“ und 342 „vorherige Aflibercept-Patienten“ erhielten während der Phase mit angepasstem Dosierungsschema von Faricimab mindestens eine Dosis Faricimab.
In beiden Studien wurden die Patienten bis Woche 24 im Verhältnis 1:1 einem von zwei Behandlungsarmen randomisiert zugeteilt:
Faricimab 6 mg alle 4 Wochen für 6 aufeinanderfolgende monatliche Dosen
Aflibercept 2 mg alle 4 Wochen für 6 aufeinanderfolgende monatliche Dosen
Nach 6 initialen monatlichen Dosen wechselten die Patienten, die ursprünglich dem Behandlungsarm mit 2 mg Aflibercept randomisiert zugeteilt worden waren, in den Behandlungsarm mit 6 mg Faricimab, und konnten 6 mg Faricimab gemäß anpassbarem Dosierungsschema bis zu alle 16 Wochen erhalten, wobei das Dosierungsintervall in 4‑Wochen‑Schritten verlängert oder um 4-, 8- oder 12‑Wochen‑Schritte verkürzt werden konnte, basierend auf einer automatisierten objektiven Beurteilung der präspezifizierten visuellen und anatomischen Krankheitsaktivitätskriterien.
Ergebnisse
Beide Studien zeigten Wirksamkeit bezüglich des primären Endpunkts, der definiert war als Veränderung der BCVA gegenüber dem Ausgangswert in Woche 24, gemessen anhand des ETDRS-Buchstaben-Scores. In beiden Studien hatten die mit Faricimab alle 4 Wochen behandelten Patienten eine mittlere Veränderung der BCVA gegenüber dem Ausgangswert, welche der der Patienten, die mit Aflibercept alle 4 Wochen behandelt wurden, nicht unterlegen war. Diese Sehverbesserungen wurden bis Woche 72 aufrechterhalten, als die Patienten auf ein anpassbares Dosierungsschema mit Faricimab bis zu alle 16 Wochen umgestellt wurden.
Zwischen Woche 24 und Woche 68 erreichten 81,5 % bzw. 74,0 % der Patienten, die Faricimab als anpassbares Dosierungsschema bis zu alle 16 Wochen erhielten, ein Dosierungsintervall von ≥ alle 12 Wochen (alle 16 Wochen bzw. alle 12 Wochen) in BALATON bzw. COMINO. Von diesen Patienten beendeten 72,1 % bzw. 61,6 % mindestens einen Zyklus von alle 12 Wochen und behielten die Dosierung von ≥ alle 12 Wochen ohne eine Intervallverringerung unter alle 12 Wochen bis Woche 68 in BALATON bzw. COMINO bei; 1,2 % bzw. 2,5 % der Patienten erhielten nur die Dosierung alle 4 Wochen bis Woche 68 in BALATON bzw. COMINO.
In allen Studien zeigten die Patienten im Behandlungsarm mit Faricimab alle 4 Wochen in Woche 24 eine Verbesserung beim präspezifizierten Wirksamkeitsendpunkt der Veränderung des zusammengesetzten Scores NEI-VFQ-25 von Behandlungsbeginn bis Woche 24, die mit der unter Aflibercept alle 4 Wochen vergleichbar war. Faricimab alle 4 Wochen zeigte auch eine Verbesserung beim präspezifizierten Wirksamkeitsendpunkt der Veränderung des NEI VFQ-25-Scores für Aktivitäten im Nah- und Fernsichtbereich von Behandlungsbeginn bis Woche 24, die mit der unter Aflibercept alle 4 Wochen vergleichbar war. Diese Ergebnisse wurden bis Woche 72 aufrechterhalten, als alle Patienten Faricimab bis zu alle 16 Wochen als anpassbares Dosierungsschema erhielten.
Tabelle 8: Wirksamkeitsergebnisse bei der Kontrolluntersuchung zum primären Endpunkt in Woche 24 und zu Studienendea in BALATON
Wirksamkeitsergebnisse |
BALATON |
|||
24 Wochen |
72 Wochena |
|||
Faricimab alle 4 Wochen |
Aflibercept alle 4 Wochen |
Faricimab alle 4 Wochen bis Faricimab anpassbar |
Aflibercept alle 4 Wochen bis Faricimab anpassbar |
|
Mittlere Veränderung der BCVA gegenüber dem Ausgangswert, gemessen anhand des ETDRS‑Buchstaben-Scores (95‑%‑KI) |
16,9 |
17,5 |
18,1 |
18,8 |
Unterschied im LS‑Mean (95‑%‑KI) |
-0,6 |
|||
Anteil der Patienten mit Zunahme um mindestens 15 Buchstaben gegenüber dem Ausgangswert (CMH‑gewichteter Anteil, 95‑%‑KI) |
56,1 % |
60,4 % |
61,5 % |
65,8 % |
Unterschied im CMH‑gewichteten %‑Wert (95‑%‑KI) |
-4,3 % |
|||
aDurchschnitt der Wochen 64, 68 und 72
BCVA: Bestkorrigierte Sehschärfe
ETDRS: Early Treatment Diabetic Retinopathy Study
KI: Konfidenzintervall
LS: Least Squares (kleinste Fehlerquadrate)
CMH: Cochran-Mantel-Haenszel-Methode; ein statistischer Test, der eine Schätzung einer Assoziation mit einem binären Ergebnis erstellt und zur Beurteilung kategorischer Variablen verwendet wird.
Tabelle 9: Wirksamkeitsergebnisse bei der Kontrolluntersuchung zum primären Endpunkt in Woche 24 und zu Studienendea in COMINO
Wirksamkeitsergebnisse |
COMINO |
|||
24 Wochen |
72 Wochena |
|||
Faricimab alle 4 Wochen |
Aflibercept alle 4 Wochen |
Faricimab alle 4 Wochen bis Faricimab anpassbar |
Aflibercept alle 4 Wochen bis Faricimab anpassbar |
|
Mittlere Veränderung der BCVA gegenüber dem Ausgangswert, gemessen anhand des ETDRS‑Buchstaben-Scores (95‑%‑KI) |
16,9 |
17,3 |
16,9 |
17,1 |
Unterschied im LS‑Mean (95‑%‑KI) |
-0,4 |
|||
Anteil der Patienten mit Zunahme um mindestens 15 Buchstaben gegenüber dem Ausgangswert (CMH‑gewichteter Anteil, 95‑%‑KI) |
56,6 % |
58,1 % |
57,6 % |
59,5 % |
Unterschied im CMH‑gewichteten %‑Wert (95‑%‑KI) |
-1,5 % |
|||
aDurchschnitt der Wochen 64, 68 und 72
BCVA: Bestkorrigierte Sehschärfe
ETDRS: Early Treatment Diabetic Retinopathy Study
KI: Konfidenzintervall
LS: Least Squares (kleinste Fehlerquadrate)
CMH: Cochran-Mantel-Haenszel-Methode; ein statistischer Test, der eine Schätzung einer Assoziation mit einem binären Ergebnis erstellt und zur Beurteilung kategorischer Variablen verwendet wird.
Abbildung 3: Mittlere Veränderung der Sehschärfe von Behandlungsbeginn bis Woche 72 in der Studie BALATON
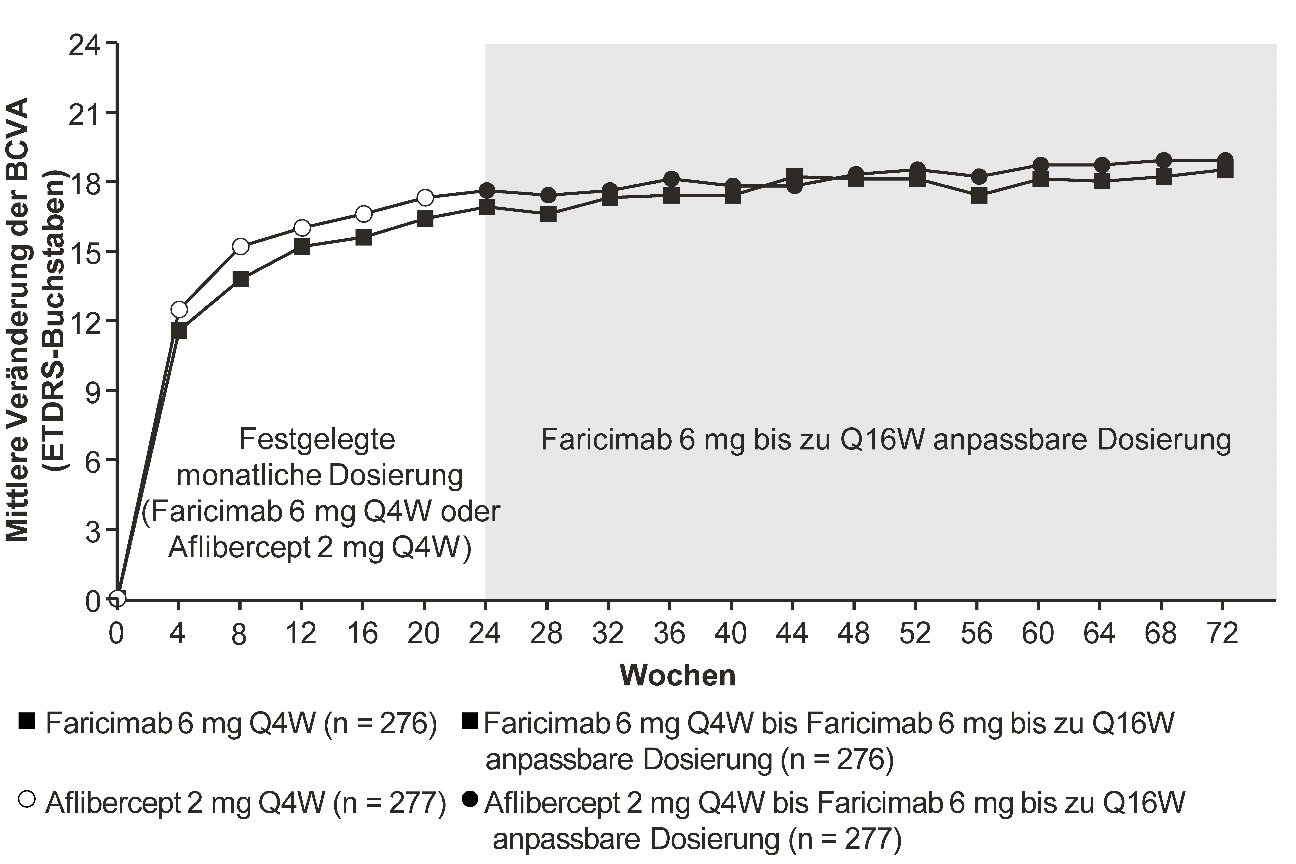
Die anpassbare Dosierung bis zu alle 16 Wochen von Faricimab 6 mg startete in Woche 24, jedoch erhielten nicht alle Patienten in Woche 24 Faricimab.
Abbildung 4: Mittlere Veränderung der Sehschärfe von Behandlungsbeginn bis Woche 72 in der Studie COMINO
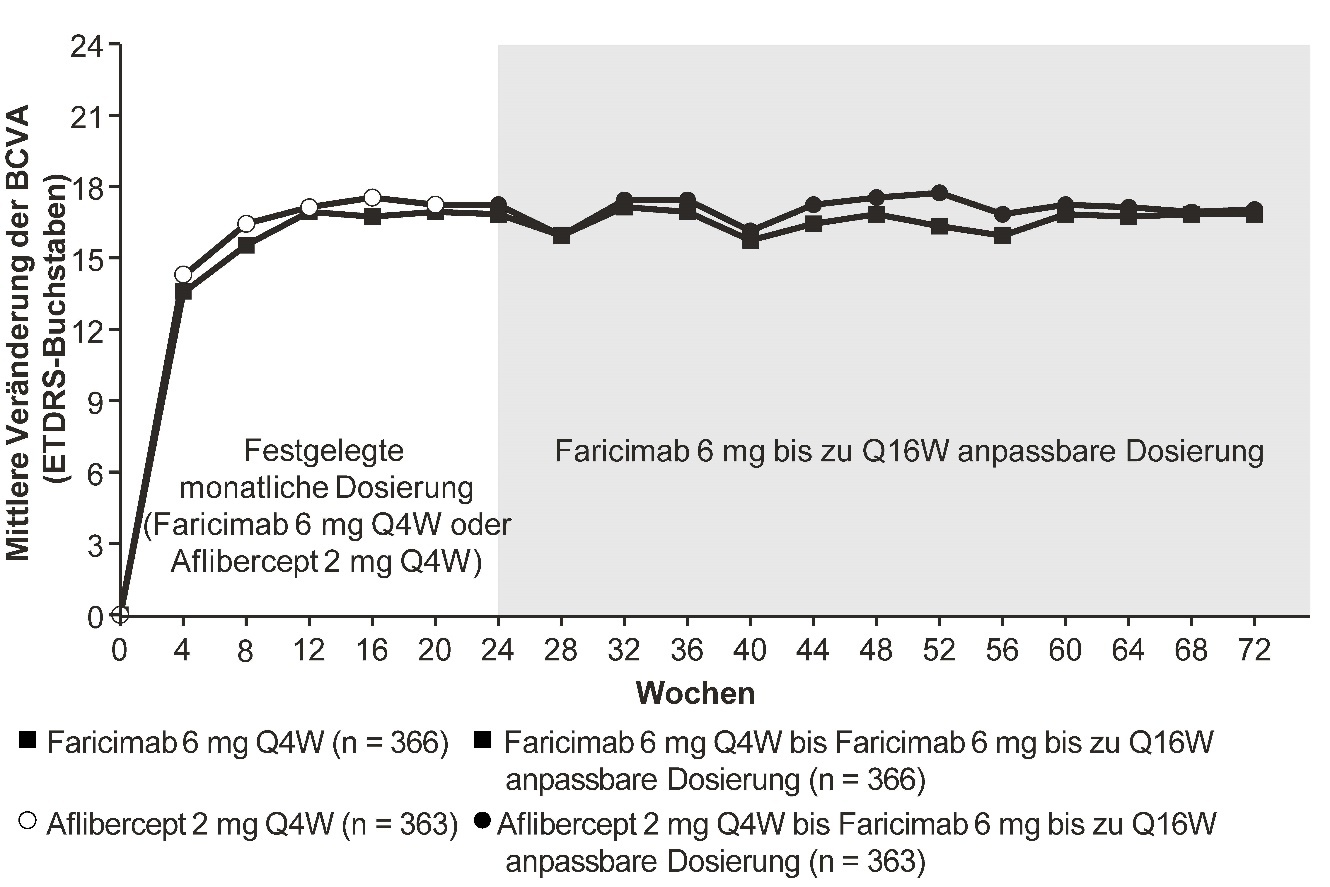
Die anpassbare Dosierung bis zu alle 16 Wochen von Faricimab 6 mg startete in Woche 24, jedoch erhielten nicht alle Patienten in Woche 24 Faricimab.
Die Inzidenz okularer unerwünschter Ereignisse am Studienauge betrug 20,1 % bzw. 24,6 % und die Inzidenz nicht-okularer unerwünschter Ereignisse 32,9 % bzw. 36,4 % bis Woche 24 in den Behandlungsarmen Faricimab alle 4 Wochen bzw. Aflibercept alle 4 Wochen (siehe Abschnitt 4.8).
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Faricimab eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen bei nAMD, DMÖ und RVV gewährt (siehe Abschnitt 4.2 bezüglich Informationen zur Anwendung bei Kindern und Jugendlichen).
Faricimab wird intravitreal angewendet, um lokale Wirkungen im Auge zu entfalten.
Resorption und Verteilung
Basierend auf einer populationspharmakokinetischen Analyse (unter Einschluss von nAMD und DMÖ, n = 2 246) wird geschätzt, dass die maximalen Konzentrationen von freiem (nicht an VEGF‑A und Ang‑2 gebundenem) Faricimab im Plasma (Cmax) etwa 2 Tage nach der Verabreichung auftreten. Die mittleren (± Standardabweichung) Cmax im Plasma werden bei nAMD- bzw. DMÖ‑Patienten auf 0,23 (0,07) µg/ml bzw. 0,22 (0,07) µg/ml geschätzt. Nach wiederholter Anwendung werden mittlere Talkonzentrationen von freiem Faricimab im Plasma für die Dosierung alle 8 Wochen von 0,002 ‑ 0,003 µg/ml vorhergesagt.
Faricimab zeigte über den Dosisbereich von 0,5 mg bis 6 mg eine dosisproportionale Pharmakokinetik (basierend auf Cmax und AUC). Nach monatlicher Gabe kam es im Glaskörper oder im Plasma zu keiner Kumulation.
Die maximalen Konzentrationen von freiem Faricimab im Plasma sind voraussichtlich etwa 600- bzw. 6 000‑mal niedriger als im Kammerwasser bzw. Glaskörper. Daher sind systemische pharmakodynamische Wirkungen unwahrscheinlich, was auch durch das Nichtvorhandensein signifikanter Veränderungen der Konzentration von freiem VEGF und Ang‑2 im Plasma nach der Behandlung mit Faricimab in klinischen Studien gestützt wird.
Eine populationspharmakokinetische Analyse ergab einen Einfluss von Alter und Körpergewicht auf die okulare bzw. systemische Pharmakokinetik von Faricimab. Der Einfluss beider Faktoren wurde als klinisch nicht bedeutsam angesehen; eine Dosisanpassung ist nicht erforderlich.
Biotransformation und Elimination
Faricimab ist ein proteinbasiertes Therapeutikum, daher sind sein Metabolismus und seine Elimination nicht vollständig charakterisiert. Es ist davon auszugehen, dass Faricimab in Lysosomen zu kleinen Peptiden und Aminosäuren katabolisiert wird, die möglicherweise auf ähnliche Weise wie endogenes IgG renal ausgeschieden werden.
Das Plasmakonzentrations-Zeit-Profil von Faricimab nahm parallel zum Konzentrations-Zeit-Profil im Glaskörper bzw. Kammerwasser ab. Die geschätzte mittlere okulare Halbwertszeit und apparente systemische Halbwertszeit von Faricimab beträgt ca. 7,5 Tage.
Eine pharmakokinetische Analyse von Patienten mit nAMD, DMÖ und RVV (n = 2 977) hat gezeigt, dass die Pharmakokinetik von Faricimab bei Patienten mit nAMD, DMÖ und RVV vergleichbar ist.
Besondere Patientengruppen
Ältere Patienten
In den sechs klinischen Phase‑III-Studien waren etwa 58 % (1 496/2 571) der Patienten, die der Behandlung mit Faricimab randomisiert zugeteilt wurden, ≥ 65 Jahre alt. Eine populationspharmakokinetische Analyse ergab einen Effekt des Alters auf die okulare Pharmakokinetik von Faricimab. Der Effekt wurde als klinisch nicht bedeutsam eingestuft. Bei Patienten ≥ 65 Jahre ist keine Dosisanpassung erforderlich (siehe Abschnitt 4.2).
Nierenfunktionsstörung
Bei Patienten mit Nierenfunktionsstörung wurden keine speziellen Studien mit Faricimab durchgeführt. Eine pharmakokinetische Analyse der Patienten in allen klinischen Studien, von denen 63 % eine Nierenfunktionsstörung hatten (leicht 38 %, mäßig 23 % und schwer 2 %), ergab keine Unterschiede in Bezug auf die systemische Pharmakokinetik von Faricimab nach intravitrealer Anwendung von Faricimab. Bei Patienten mit Nierenfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Abschnitt 4.2).
Leberfunktionsstörung
Bei Patienten mit Leberfunktionsstörung wurden keine speziellen Studien mit Faricimab durchgeführt. Bei dieser Population sind jedoch keine besonderen Abwägungen erforderlich, da der Metabolismus über Proteolyse erfolgt und nicht von der Leberfunktion abhängig ist. Bei Patienten mit Leberfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Abschnitt 4.2).
Weitere besondere Patientengruppen
Die systemische Pharmakokinetik von Faricimab wird durch die ethnische Zugehörigkeit der Patienten nicht beeinflusst. Das Geschlecht zeigte keinen klinisch relevanten Einfluss auf die systemische Pharmakokinetik von Faricimab. Es ist keine Dosisanpassung erforderlich.
Es wurden keine Studien zum kanzerogenen oder mutagenen Potenzial von Faricimab durchgeführt.
Bei trächtigen Cynomolgus-Affen lösten intravenöse Injektionen von Faricimab, die zu einer Serumexposition (Cmax) von mehr als dem 500‑Fachen der maximalen Exposition beim Menschen führen, keine Entwicklungstoxizität oder Teratogenität aus und sie hatten keine Auswirkung auf Gewicht oder Struktur der Plazenta; dennoch soll Faricimab aufgrund seiner pharmakologischen Wirkung als potenziell teratogen und embryo-/fetotoxisch angesehen werden.
Die systemische Exposition nach okularer Verabreichung von Faricimab ist sehr gering.
Histidin
Essigsäure 30 % (zur pH‑Wert-Einstellung) (E 260)
Methionin
Polysorbat 20 (E 432)
Natriumchlorid
Saccharose
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Fertigspritze: 2 Jahre
Durchstechflasche: 30 Monate
Im Kühlschrank lagern (2 °C ‑ 8 °C).
Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Die Fertigspritze in der versiegelten Blisterpackung im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Die ungeöffnete Fertigspritze oder Durchstechflasche kann vor der Anwendung bis zu 24 Stunden bei Raumtemperatur, 20 °C bis 25 °C, im Umkarton aufbewahrt werden.
Sicherstellen, dass die Injektion unmittelbar nach Vorbereitung der Dosis verabreicht wird.
Fertigspritze
Injektionslösung in einer Fertigspritze, bestehend aus einem Glaszylinder (Typ I) mit einer Dosismarkierung, einem Butylgummistopfen und einer manipulationssicheren Verschlusskappe (mit einer starren Spitzenkappe, einer Butylgummispitzenkappe und einem Luer-Lock-Adapter). Die Fertigspritze ist mit einem Spritzenkolben und einem verlängerten Fingergriff ausgestattet. Jede Fertigspritze enthält 21 mg Faricimab in 0,175 ml Lösung.
Packungsgröße: eine sterile, extradünnwandige Injektionsfilternadel (30 G × ½", 0,30 mm x 12,7 mm, 5 µm), zusammen mit einer Fertigspritze verpackt.
Die Gummispitzenkappe, der Gummistopfen, der Glaszylinder und die Injektionsfilternadel stehen in Kontakt mit dem Arzneimittel.
Durchstechflasche
0,24 ml sterile Lösung in einer Glas-Durchstechflasche mit beschichtetem Gummistopfen, versiegelt mit einer Aluminiumkappe mit gelber Flip-off-Dichtungsscheibe aus Kunststoff.
Packungsgröße: eine Durchstechflasche und eine stumpfe Transfer-Filternadel (18 G x 1½ʺ, 1,2 mm x 40 mm, 5 µm).
Nicht schütteln.
Nach der Entnahme aus dem Kühlschrank und vor der Anwendung muss Vabysmo visuell überprüft werden. Sind Partikel oder Trübungen sichtbar, darf Vabysmo nicht verwendet werden.
Fertigspritze
Die Fertigspritze ist nur für den einmaligen Gebrauch in einem Auge bestimmt. Die sterile Fertigspritze darf nur unter aseptischen Bedingungen geöffnet werden. Die Lösung muss vor der Anwendung visuell überprüft werden. Sind Partikel oder Trübungen sichtbar, darf die Fertigspritze nicht verwendet werden.
Die Fertigspritze enthält mehr als die empfohlene Dosis von 6 mg Faricimab (entspricht 0,05 ml). Jede Fertigspritze enthält 21 mg Faricimab in 0,175 ml Lösung. Das überschüssige Volumen muss vor der Injektion verworfen werden.
Nicht anwenden, wenn die Verpackung, die Fertigspritze und/oder die Injektionsfilternadel beschädigt sind oder wenn das Verfalldatum überschritten ist. Eine ausführliche Gebrauchsanweisung ist in der Packungsbeilage enthalten.
Durchstechflasche
Die Durchstechflasche enthält mehr als die empfohlene Dosis von 6 mg. Das Füllvolumen der Durchstechflasche (0,24 ml) darf nicht vollständig verwendet werden. Das überschüssige Volumen muss vor der Injektion verworfen werden. Die Injektion des gesamten Volumens der Durchstechflasche hat eine Überdosierung zur Folge. Die zu injizierende Dosis muss auf die 0,05‑ml-Dosismarkierung eingestellt werden, d. h. auf 6 mg Faricimab.
Der Inhalt der Durchstechflasche und die Transfer-Filternadel sind steril und nur zur einmaligen Anwendung bestimmt. Nicht anwenden, wenn die Verpackung, die Durchstechflasche und/oder die Filternadel beschädigt sind oder wenn das Verfalldatum überschritten ist. Eine ausführliche Gebrauchsanweisung ist in der Packungsbeilage enthalten.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Anleitung für den Gebrauch der Fertigspritze:
Das Entnehmen der Spritze aus der Blisterpackung (Schritt 1) und alle nachfolgenden Schritte sind unter aseptischen Bedingungen durchzuführen. | |
Hinweis: Die Dosis muss auf die 0,05‑ml-Dosismarkierung eingestellt werden. | |
Blisterpackung öffnen und Spritzenkappe entfernen | |
1 |
Deckel von der Blisterpackung abziehen und unter aseptischen Bedingungen die Fertigspritze entnehmen. |
2 |
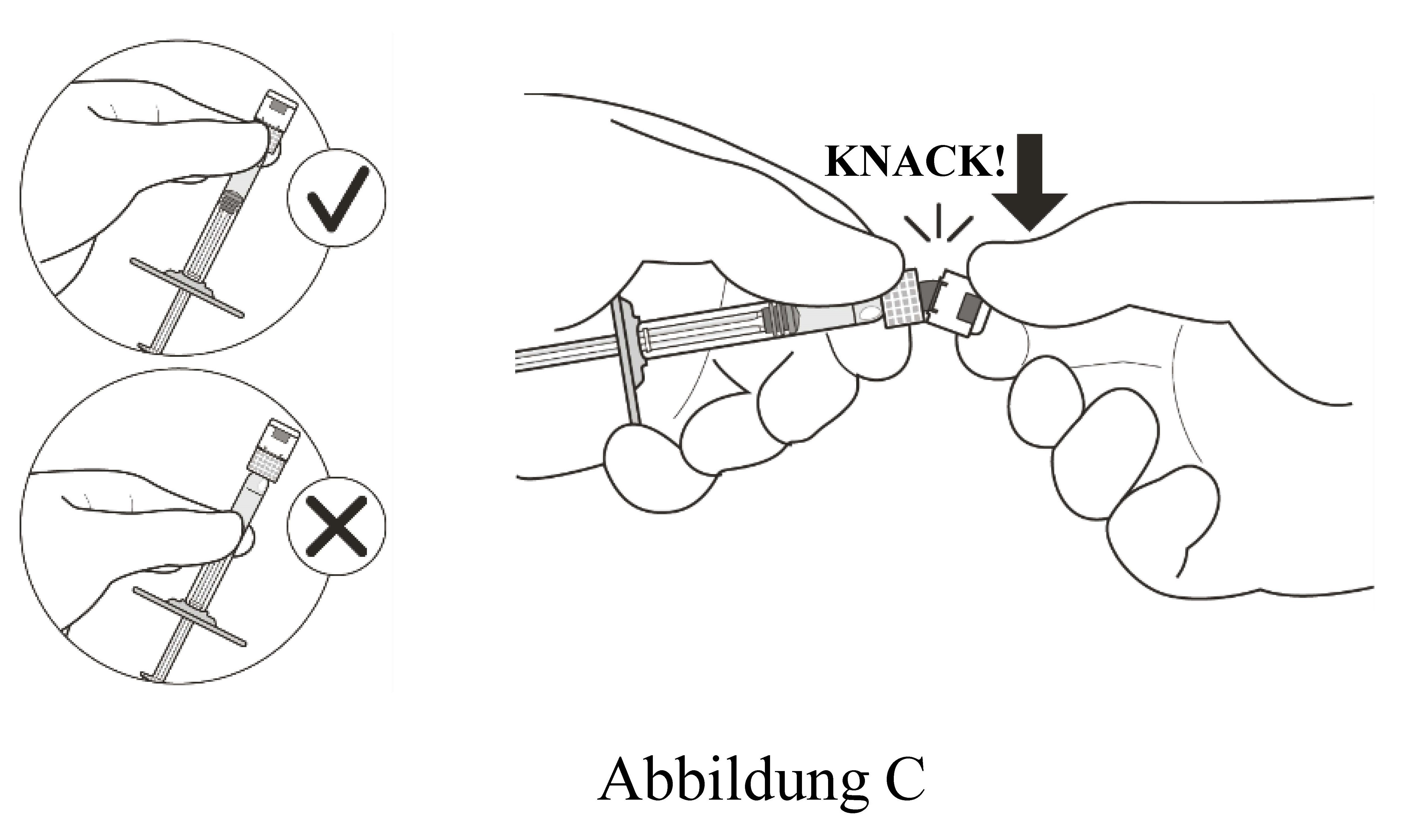
Spritze an der weißen Manschette festhalten; Spritzenkappe abbrechen (siehe Abbildung C). |
Kappe nicht abdrehen. |
|
 |
Injektionsfilternadel befestigen | |
3 |
Injektionsfilternadel unter aseptischen Bedingungen aus der Verpackung entnehmen. |
4 |
Die Injektionsfilternadel unter aseptischen Bedingungen fest am Luer-Lock der Spritze befestigen (siehe Abbildung D). |
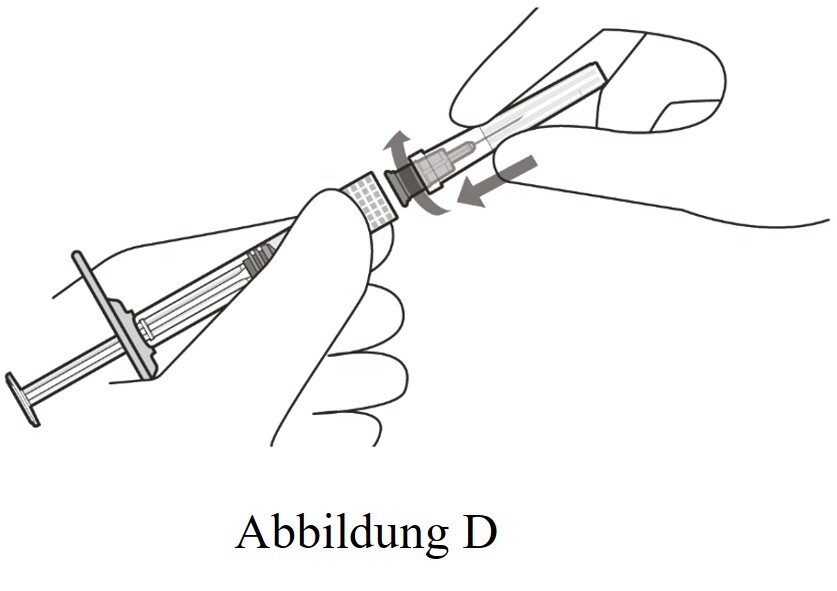 |
Zur Verabreichung nur die beigefügte Injektionsfilternadel verwenden |
5 |
Die Nadelkappe durch gerades Abziehen vorsichtig entfernen. |
Luftblasen lösen | |
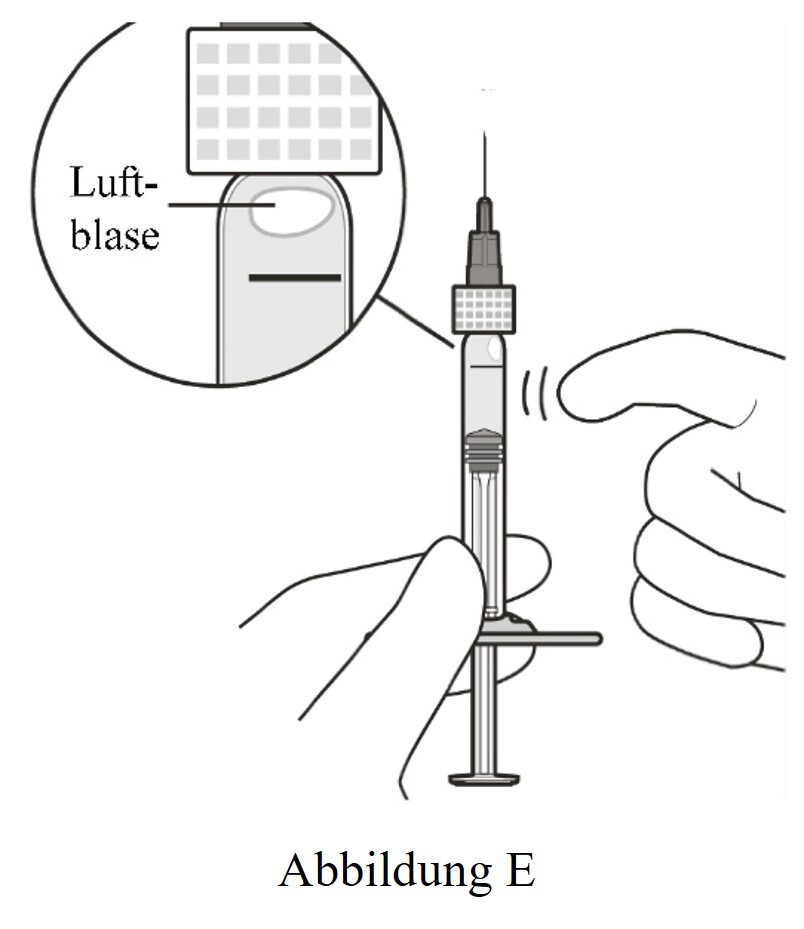
6 |
Die Spritze mit der Injektionsfilternadel nach oben halten. Die Spritze auf Luftblasen überprüfen. |
7 |
Bei vorhandenen Luftbläschen vorsichtig mit dem Finger gegen die Spritze klopfen, bis die Bläschen nach oben steigen (siehe Abbildung E). |
 | |
Dosis des Arzneimittels einstellen und Luft entfernen | ||
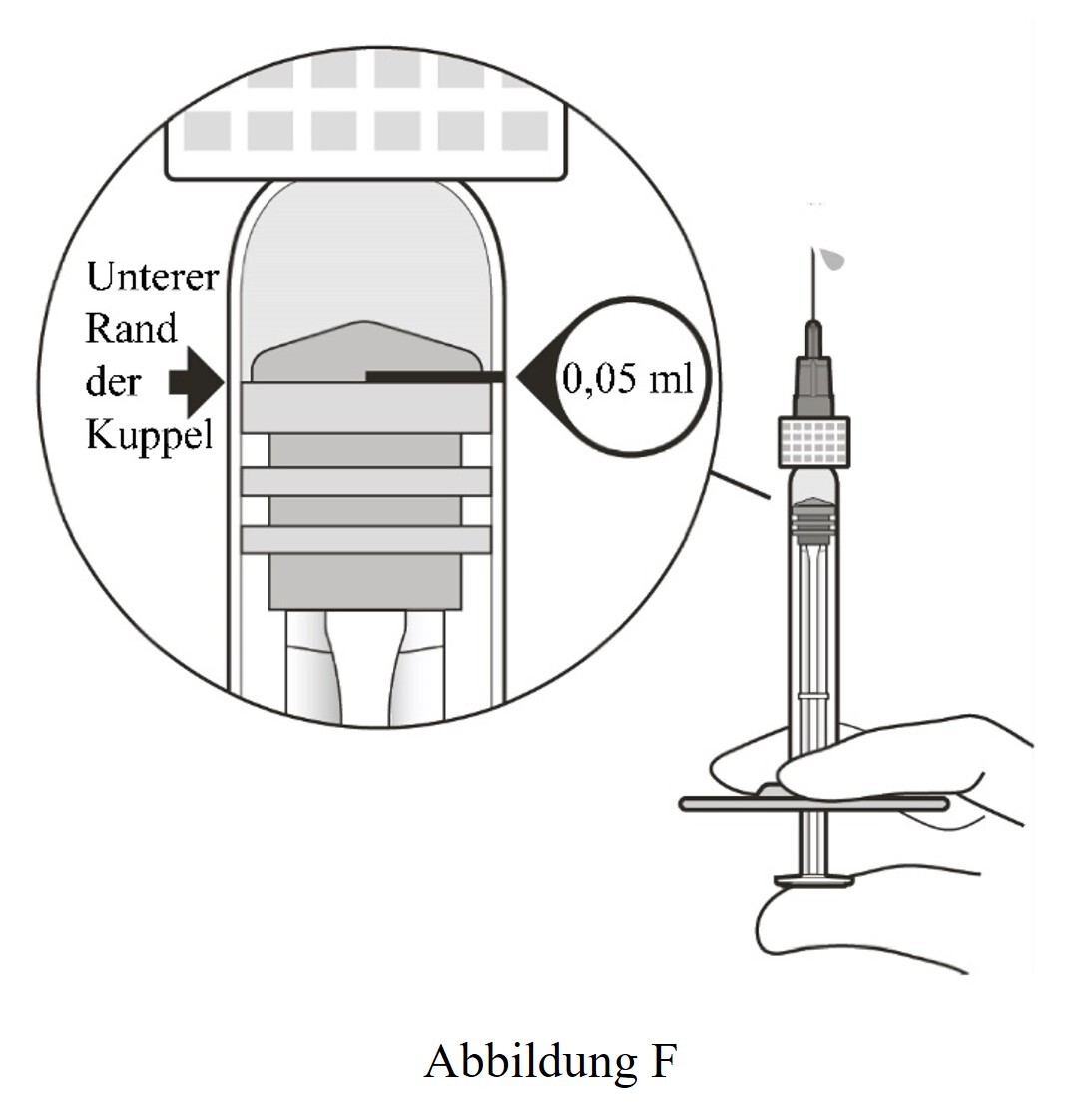
8 |
Die Spritze auf Augenhöhe halten und langsam auf den Spritzenkolben drücken, bis der untere Rand der Kuppel des Gummistopfens auf derselben Höhe liegt wie die 0,05-ml-Dosismarkierung (siehe Abbildung F). Dadurch wird Luft und überschüssige Lösung entfernt und die Dosis auf 0,05 ml eingestellt. |
|
Sicherstellen, dass die Injektion unmittelbar nach Vorbereitung der Dosis verabreicht wird. | ||
 | ||
Injektionsverfahren | |
9 |
Das Injektionsverfahren muss unter aseptischen Bedingungen durchgeführt werden. |
Roche Registration GmbH
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Deutschland
EU/1/22/1683/001
EU/1/22/1683/002
Datum der Erteilung der Zulassung: 15. September 2022
Juni 2026
Verschreibungspflichtig
Roche Pharma AG
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Telefon (07624) 14-0
Telefax (07624) 1019
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.