
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
SOTYKTU 6 mg Filmtabletten
Jede Filmtablette enthält 6 mg Deucravacitinib.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 44 mg Lactose (siehe Abschnitt 4.4).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette (Tablette)
Rosafarbene, runde, bikonvexe Filmtablette mit einem Durchmesser von 8 mm und dem Aufdruck „BMS 895“ und „6 mg“ auf einer Seite in zwei Zeilen und ohne Aufdruck auf der anderen Seite.
Plaque-Psoriasis
SOTYKTU wird angewendet zur Behandlung erwachsener Patienten mit mittelschwerer bis schwerer Plaque‑Psoriasis, die für eine systemische Therapie infrage kommen.
Psoriasis-Arthritis
SOTYKTU wird allein oder in Kombination mit Methotrexat (MTX) angewendet zur Behandlung erwachsener Patienten mit aktiver Psoriasis-Arthritis (PsA), die auf eine vorangegangene krankheitsmodifizierende antirheumatische (disease-modifying antirheumatic drugs, DMARD)-Therapie unzureichend angesprochen oder diese nicht vertragen haben (siehe Abschnitt 5.1).
Die Behandlung sollte unter der Anleitung und Überwachung eines Arztes eingeleitet werden, der Erfahrung in der Diagnose und Behandlung von Erkrankungen hat, für die SOTYKTU indiziert ist.
Dosierung
Die empfohlene Dosis ist 6 mg zum Einnehmen einmal täglich.
Wenn ein Patient nach 24 Wochen keine Anzeichen für einen therapeutischen Nutzen aufweist, sollte ein Abbruch der Behandlung in Erwägung gezogen werden. Das Ansprechen des Patienten auf die Behandlung sollte regelmäßig beurteilt werden.
Besondere Patientengruppen
Ältere Patienten
Bei älteren Patienten ab einem Alter von 65 Jahren ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Die klinische Erfahrung bei Patienten ≥ 75 Jahre ist sehr begrenzt und Deucravacitinib sollte in dieser Patientengruppe mit Vorsicht angewendet werden.
Nierenfunktionsstörung
Bei Patienten mit Nierenfunktionsstörung, einschließlich dialysepflichtiger Patienten mit terminaler Niereninsuffizienz (end‑stage renal disease, ESRD), ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Leberfunktionsstörung
Bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Die Anwendung von Deucravacitinib wird bei Patienten mit schwerer Leberfunktionsstörung nicht empfohlen (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Deucravacitinib bei Kindern und Jugendlichen unter einem Alter von 18 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Zum Einnehmen.
Die Tabletten können unabhängig von den Mahlzeiten eingenommen werden. Die Tabletten sollten im Ganzen geschluckt werden und dürfen nicht zerstoßen, zerschnitten oder gekaut werden.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Klinisch bedeutsame aktive Infektionen (z. B. aktive Tuberkulose, siehe Abschnitt 4.4).
Infektionen
Deucravacitinib kann das Infektionsrisiko erhöhen (siehe Abschnitt 4.8).
Die Behandlung mit Deucravacitinib darf bei Patienten mit jeglichen klinisch bedeutsamen aktiven Infektionen erst nach Abklingen oder angemessener Behandlung der Infektion eingeleitet werden (siehe Abschnitt 4.3). Vorsicht ist geboten, wenn die Anwendung von Deucravacitinib bei Patienten mit chronischen Infektionen oder rezidivierenden Infektionen in der Vorgeschichte in Erwägung gezogen wird.
Mit Deucravacitinib behandelte Patienten sollten angewiesen werden, ärztlichen Rat einzuholen, wenn Anzeichen oder Symptome auftreten, die auf eine Infektion hindeuten. Wenn bei einem Patienten eine klinisch bedeutsame Infektion auftritt oder der Patient auf die Standardtherapie nicht anspricht, sollte der Patient sorgfältig überwacht werden, und Deucravacitinib sollte erst nach Abklingen der Infektion gegeben werden.
Untersuchung auf Tuberkulose vor der Behandlung
Vor Einleitung der Behandlung mit Deucravacitinib sollten die Patienten auf Tuberkulose (TB)‑Infektionen untersucht werden. Deucravacitinib darf Patienten mit aktiver TB nicht gegeben werden (siehe Abschnitt 4.3). Die Behandlung einer latenten TB sollte vor der Gabe von Deucravacitinib begonnen werden. Bei Patienten mit latenter oder aktiver TB in der Anamnese, bei denen kein angemessener Behandlungszyklus bestätigt werden kann, sollte vor der Einleitung der Deucravacitinib‑Therapie eine Anti‑TB‑Therapie in Erwägung gezogen werden. Patienten, die Deucravacitinib erhalten, sollten auf Anzeichen und Symptome einer aktiven TB überwacht werden.
Bösartige Erkrankungen
In klinischen Studien zu Deucravacitinib wurden bösartige Erkrankungen, einschließlich Lymphome und nicht‑melanozytärer Hautkrebs (NMSC), beobachtet.
Es ist nicht bekannt, ob die Hemmung der Tyrosinkinase 2 (TYK2) mit den Nebenwirkungen, die bei einer Hemmung der Januskinasen (JAK) beobachtet wurden, in Zusammenhang steht. In einer großangelegten, randomisierten, aktiv kontrollierten Studie zu einem JAK‑Hemmer bei Patienten mit rheumatoider Arthritis (RA) im Alter von 50 Jahren und älter mit mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurde bei der Behandlung mit einem JAK‑Hemmer im Vergleich zu Tumornekrosefaktor (TNF)‑Hemmern eine höhere Rate bösartiger Erkrankungen, insbesondere Lungenkrebs, Lymphome und NMSC, beobachtet.
Es liegen nur begrenzte klinische Daten für die Beurteilung eines potenziellen Zusammenhangs zwischen der Exposition gegenüber Deucravacitinib und der Entstehung von bösartigen Erkrankungen vor. Derzeit werden Untersuchungen zur Langzeitsicherheit durchgeführt. Die Risiken und der Nutzen einer Deucravacitinib‑Behandlung sollten vor Einleitung der Therapie bei Patienten abgewogen werden.
Schwere kardiovaskuläre Ereignisse (Major adverse cardiovascular events, MACE), tiefe Venenthrombose (TVT) und Lungenembolie (LE)
Es ist nicht bekannt, ob die TYK2-Hemmung mit den Nebenwirkungen, die bei einer JAK-Hemmung beobachtet wurden, in Zusammenhang steht. In einer großangelegten, randomisierten, aktiv kontrollierten Studie zu einem JAK‑Hemmer bei Patienten mit RA im Alter von 50 Jahren und älter mit mindestens einem zusätzlichen kardiovaskulären Risikofaktor wurden eine höhere MACE-Rate, definiert als kardiovaskulärer Tod, nicht-tödlicher Myokardinfarkt und nicht-tödlicher Schlaganfall, sowie eine dosisabhängige höhere Rate einer tiefen Venenthrombose, einschließlich TVT und LE, bei der Behandlung mit einem JAK‑Hemmer im Vergleich zu TNF‑Hemmern beobachtet.
In klinischen Studien mit Deucravacitinib wurde kein erhöhtes Risiko für MACE, TVT und LE beobachtet. Untersuchungen zur Langzeitsicherheit von Deucravacitinib werden derzeit noch durchgeführt. Die Risiken und der Nutzen einer Deucravacitinib‑Behandlung sollten vor Einleitung der Therapie bei Patienten abgewogen werden.
Impfungen
Vor Einleitung der Therapie mit Deucravacitinib ist eine Vervollständigung aller altersangemessenen Impfungen gemäß den aktuellen Impfempfehlungen zu erwägen. Die Anwendung von Lebendimpfstoffen bei mit Deucravacitinib behandelten Patienten sollte vermieden werden. Das Ansprechen auf Lebend‑ oder Totimpfstoffe wurde nicht untersucht.
Sonstige Bestandteile
Lactose
Dieses Arzneimittel enthält Lactose. Patienten mit der seltenen hereditären Galactose‑Intoleranz, völligem Lactasemangel oder Glucose‑Galactose‑Malabsorption dürfen dieses Arzneimittel nicht anwenden.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Tablette, d. h. es ist nahezu „natriumfrei“.
Klinische Studien deuten darauf hin, dass Deucravacitinib bei gleichzeitiger Verabreichung mit den folgenden anderen Arzneimitteln keine klinisch relevanten Arzneimittelwechselwirkungen aufweist und daher keine Dosisanpassungen notwendig sind.
Wirkung von Deucravacitinib auf andere Arzneimittel
Deucravacitinib hat keine bedeutsamen Auswirkungen auf die Plasmaexposition von Rosuvastatin (Substrat von BCRP und OATP), Methotrexat (Substrat von BCRP und renalen Transportern), Mycophenolat‑Mofetil (MMF) (Substrat von CES1 und CES2), Metformin (Substrat des renalen MATE2K‑abhängigen und des hepatischen OCT1‑abhängigen Arzneimitteltransports) oder oralen Kontrazeptiva (Norethisteronacetat und Ethinylestradiol).
Wirkung anderer Arzneimittel auf Deucravacitinib
Arzneimittel, die Inhibitoren oder Induktoren von CYP‑Enzymen oder ‑Transportern sind, wie Ciclosporin (dualer Inhibitor von P‑gp/Breast Cancer Resistance Protein [BCRP]), Fluvoxamin (starker CYP1A2‑Inhibitor), Ritonavir (mittelstarker CYP1A2‑Induktor), Diflunisal (UGT1A9‑Inhibitor), Pyrimethamin (OCT1‑Inhibitor), Famotidin (H2‑Rezeptor‑Antagonist) oder Rabeprazol (Protonenpumpeninhibitor) haben keine bedeutsamen Auswirkungen auf die Deucravacitinib‑Plasmaexposition (siehe Abschnitt 5.2).
Schwangerschaft
Bisher liegen nur begrenzte Erfahrungen mit der Anwendung von Deucravacitinib bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Abschnitt 5.3). Aus Vorsichtsgründen soll eine Anwendung von Deucravacitinib während der Schwangerschaft vermieden werden.
Stillzeit
Deucravacitinib und sein Metabolit (BMT‑153261) werden in die Muttermilch ausgeschieden (siehe Abschnitt 5.2). Die geschätzte relative tägliche Dosis für das Kind entspricht 12 % der Dosis der Mutter.
Es ist nicht bekannt, ob Deucravacitinib Auswirkungen auf Neugeborene/Kinder hat.
Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Deucravacitinib verzichtet werden soll/die Behandlung zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Die Auswirkungen von Deucravacitinib auf die Fertilität des Menschen wurden nicht untersucht. Tierexperimentelle Studien deuten nicht auf direkte oder indirekte schädliche Wirkungen in Bezug auf die Fertilität hin (siehe Abschnitt 5.3).
Deucravacitinib hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die am häufigsten berichtete Nebenwirkung waren Infektionen der oberen Atemwege (18,9 % in Studien zu Plaque‑Psoriasis und 15,1 % in Studien zu Psoriasis-Arthritis). Insgesamt entsprach das Sicherheitsprofil, das bei mit Deucravacitinib behandelten Patienten mit Psoriasis-Arthritis beobachtet wurde, dem bei Patienten mit Plaque-Psoriasis beobachteten Sicherheitsprofil. Das über einen längeren Zeitraum erfasste Sicherheitsprofil von Deucravacitinib war mit den vorangegangenen Erfahrungen vergleichbar und stimmte mit diesen überein.
Tabellarische Auflistung der Nebenwirkungen
Die Nebenwirkungen von Deucravacitinib aus klinischen Studien sind aufgeführt (siehe Tabelle 1) nach MedDRA‑Systemorganklasse und gemäß den folgenden Konventionen: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1 000, < 1/100); selten (≥ 1/10 000, < 1/1 000); sehr selten (< 1/10 000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 1: Liste der Nebenwirkungen
Systemorganklasse |
Häufigkeit |
Nebenwirkung |
Infektionen und parasitäre Erkrankungen |
Sehr häufig |
Infektionen der oberen Atemwegea |
Häufig |
Herpes simplex Infektionenb |
|
Gelegentlich |
Pneumonie |
|
Erkrankungen des Gastrointestinaltrakts |
Häufig |
Orale Ulzerationenc |
Erkrankungen der Haut und des Unterhautgewebes |
Häufig |
Akneiformer Ausschlagd |
Untersuchungen |
Häufig |
Kreatinphosphokinase im Blut erhöht |
a Infektionen der oberen Atemwege umfassen Nasopharyngitis, Infektion der oberen Atemwege, virale Infektion der oberen Atemwege, Pharyngitis, Sinusitis, akute Sinusitis, Rhinitis, Tonsillitis, Peritonsillarabszess, Laryngitis, Tracheitis, Pharyngotonsillitis und Rhinotracheitis. | ||
Beschreibung ausgewählter Nebenwirkungen
Infektionen
In POETYK PSO‑1 und POETYK PSO‑2 (siehe Abschnitt 5.1) traten in den ersten 16 Wochen bei 29,1 % der Patienten in der Deucravacitinib‑Gruppe Infektionen auf (116,0 Ereignisse pro 100 Patientenjahre), verglichen mit 21,5 % der Patienten in der Placebogruppe (83,7 Ereignisse pro 100 Patientenjahre). Der Großteil der Infektionen war nicht schwerwiegend und leicht bis mittelschwer ausgeprägt und führte nicht zum Absetzen von Deucravacitinib. Die Inzidenz schwerwiegender Infektionen betrug in der Deucravacitinib‑Gruppe 0,6 % (2,0 Ereignisse pro 100 Patientenjahre) und in der Placebogruppe 0,5 % (1,6 Ereignisse pro 100 Patientenjahre).
Die Infektionsrate in der Deucravacitinib‑Gruppe stieg bis einschließlich Woche 52 nicht an (95,4 Ereignisse pro 100 Patientenjahre). Die Rate schwerwiegender Infektionen in der Deucravacitinib‑Gruppe stieg bis einschließlich Woche 52 nicht an (1,7 Ereignisse pro 100 Patientenjahre).
Insgesamt entsprach die Infektionsrate, einschließlich schwerwiegender Infektionen, die bei mit Deucravacitinib behandelten Patienten mit Psoriasis-Arthritis beobachtet wurde, der bei Patienten mit Plaque-Psoriasis beobachteten Infektionsrate.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen‑Risiko‑Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung anzuzeigen am:
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: http://www.bfarm.de
Deucravacitinib wurde gesunden Probanden in Einzeldosen von bis 40 mg (> das 6‑Fache der für den Menschen empfohlenen Dosis von 6 mg/Tag) und in mehreren Dosen von bis zu 24 mg/Tag (12 mg zweimal täglich) über einen Zeitraum von 14 Tagen gegeben, ohne dass eine dosislimitierende Toxizität aufgetreten ist.
Im Falle einer Überdosis wird empfohlen, den Patienten auf Anzeichen und Symptome von Nebenwirkungen zu überwachen und unverzüglich eine geeignete symptomatische Behandlung einzuleiten. Durch eine Dialyse wird Deucravacitinib nicht in wesentlichem Umfang aus dem systemischen Kreislauf entfernt (siehe Abschnitt 5.2).
Pharmakotherapeutische Gruppe: Immunsuppressiva, ATC‑Code: L04AF07
Wirkmechanismus
Deucravacitinib hemmt selektiv das Enzym TYK2 (TYK2 gehört zur JAK‑Familie). Deucravacitinib bindet an die regulatorische Domäne von TYK2 und bewirkt die Stabilisierung einer hemmenden Wechselwirkung zwischen der regulatorischen und der katalytischen Domäne des Enzyms. Dies führt zu einer allosterischen Hemmung der rezeptorvermittelten Aktivierung von TYK2 und der Zellfunktionen, die dieser nachgelagert sind. TYK2 vermittelt die Signaltransduktion von Interleukin‑23 (IL‑23), Interleukin‑12 (IL‑12) und Typ‑I‑Interferonen (IFN). Bei diesen handelt es sich um natürlich vorkommende Zytokine, die an Entzündungs‑ und Immunreaktionen beteiligt sind. Deucravacitinib hemmt die Freisetzung von proinflammatorischen Zytokinen und Chemokinen.
Pharmakodynamische Wirkungen
Bei Patienten mit Psoriasis reduzierte Deucravacitinib die mit Psoriasis assoziierte Genexpression in psoriatischer Haut; dies schloss die Genexpression von durch den IL‑23‑Signalweg und den Typ‑1‑IFN‑Signalweg regulierten Genen ein. Bei Patienten mit Psoriasis und Psoriasis-Arthritis reduzierte Deucravacitinib nach 16 Wochen einmal täglicher Behandlung zirkulierendes IL‑17A, IL‑19 und β‑Defensin. Bei Patienten mit Psoriasis-Arthritis wurde eine Reduktion von zirkulierenden Spiegeln weiterer Biomarker beobachtet, einschließlich C‑reaktives Protein (CRP), Matrix-Metalloproteinase-3 (MMP‑3), Matrix-Metalloproteinase-1 (MMP-1), Abbauprodukt von Typ-I-Kollagen (C1M) und TNF‑alpha.
Klinische Wirksamkeit und Sicherheit
Plaque-Psoriasis
Die Wirksamkeit und Sicherheit von Deucravacitinib wurden in zwei multizentrischen, randomisierten, doppelblinden, Placebo‑ und Apremilast‑kontrollierten klinischen Studien (POETYK PSO‑1 und POETYK PSO‑2) bei Patienten untersucht, die mindestens 18 Jahre alt waren, an mittelschwerer bis schwerer Plaque‑Psoriasis litten und für eine systemische Therapie oder Phototherapie infrage kamen. Bei den Patienten waren ≥ 10 % der Körperoberfläche (KOF) betroffen, sie hatten einen Psoriasis Area and Severity Index (PASI)‑Score von ≥ 12 und einen Wert für die static Physician’s Global Assessment (sPGA) ≥ 3 (mittelschwer oder schwer) auf einer 5‑Punkte‑Skala für den allgemeinen Schweregrad der Erkrankung.
In POETYK PSO‑1 und POETYK PSO‑2 wurden insgesamt 1 686 Patienten untersucht, von denen 843 nach der Randomisierung Deucravacitinib 6 mg einmal täglich, 422 Apremilast 30 mg zweimal täglich und 421 Placebo erhielten.
In beiden Studien wurden Patienten, die Placebo erhielten, in Woche 16 auf Deucravacitinib umgestellt; diese Behandlung wurde dann bis Woche 52 fortgesetzt. Patienten, die nach der Randomisierung Apremilast erhielten und in Woche 24 kein PASI 50‑ (POETYK PSO‑1) oder PASI 75‑Ansprechen (POETYK PSO‑2) erreichten, wurden auf Deucravacitinib umgestellt und setzten die Behandlung bis Woche 52 fort. In POETYK PSO‑1 wurden Patienten, die nach der Randomisierung Deucravacitinib erhielten, bis Woche 52 behandelt. In POETYK PSO‑2 wurden mit Deucravacitinib behandelte Patienten, die in Woche 24 PASI 75 erreichten, im Verhältnis 1:1 erneut randomisiert, um Deucravacitinib zu erhalten (Erhaltungstherapie), oder wurden auf Placebo umgestellt (Absetzen).
Die Krankheitsmerkmale bei Studienbeginn waren bei der Studienpopulation in beiden Studien gleich: Die meisten Patienten waren männlich (67 %), das mittlere Alter betrug ca. 47 Jahre, wobei die meisten Patienten 40 bis 64 Jahre alt waren. 10 % der Patienten waren ≥ 65 Jahre alt. Insgesamt war der mediane PASI‑Score 18,7 und die mediane betroffene KOF betrug 20 %. Der sPGA‑Score zu Studienbeginn war bei 79,8 % der Patienten 3 (mittelschwer) und bei 20,2 % 4 (schwer). Der mediane Dermatology Life Quality Index (DLQI)‑Score war 11. Insgesamt 18,4 % der Studienteilnehmer hatten Psoriasis‑Arthritis in der Anamnese.
In beiden Studien hatten 40 % der Patienten zuvor eine Phototherapie erhalten, 42,4 % waren mit keinerlei systemischer Therapie vorbehandelt worden (einschließlich Behandlungen mit Biologika und/oder Nicht-Biologika), 41 % hatten zuvor eine systemische Behandlung mit Nicht-Biologika erhalten, und 34,8 % hatten zuvor eine Biologika-Therapie erhalten (16,1 % TNF‑, 4,9 % IL‑12/23‑, 16,6 % IL‑17‑ und 4,4 % IL‑23‑Inhibitoren).
Die co‑primären Endpunkte in den zwei Studien waren die Anteile von Patienten, die Folgendes erreichten: 1) mindestens eine 75%ige Verbesserung der PASI‑Scores (PASI 75) gegenüber dem Studienbeginn und 2) einen sPGA‑Score von klar oder fast klar (0 oder 1) in Woche 16 im Vergleich zu Placebo.
In der Studie POETYK PSO‑1 wurde in Woche 16 ein PASI 75 in der Deucravacitinib-Gruppe bei 58,4 %, in der Apremilast-Gruppe bei 35,1 % und in der Placebo-Gruppe bei 12,7 % der Patienten erreicht. Ein sPGA von klar oder fast klar in Woche 16 wurde bei 53,6 %, 32,1 % bzw. 7,2 % der Patienten in der Deucravacitinib‑, Apremilast‑ bzw. Placebo‑Gruppe erreicht. Für diese co-primären Endpunkte wurde die Überlegenheit von Deucravacitinib gegenüber Placebo nachgewiesen. In der Studie POETYK PSO‑2 wurden damit übereinstimmende Ergebnisse beobachtet.
In Tabelle 2 sind die Hauptwirksamkeitsergebnisse für die co-primären Endpunkte und andere Endpunkte aufgeführt.
Tabelle 2: Hauptwirksamkeitsergebnisse bei Erwachsenen mit Plaque‑Psoriasis
POETYK PSO‑1 |
POETYK PSO‑2 |
|||||
Endpunkt |
Deucravacitinib |
Apremilast |
Placebo |
Deucravacitinib |
Apremilast |
Placebo |
sPGA 0/1 | ||||||
Woche 16 |
178 (53,6) |
54 (32,1)d |
12 (7,2)a,d |
253 (49,5) |
86 (33,9)d |
22 (8,6)a,d |
Woche 24 |
195 (58,7) |
52 (31,0)d |
- |
251 (49,8)b |
75 (29,5)d |
- |
sPGA 0 | ||||||
Woche 16 |
58 (17,5) |
8 (4,8)d |
1 (0,6)d |
80 (15,7) |
16 (6,3)e |
3 (1,2)d |
PASI 75 | ||||||
Woche 16 |
194 (58,4) |
59 (35,1)d |
21 (12,7)a,d |
271 (53,0) |
101 (39,8)e |
24 (9,4)a,d |
Woche 24 |
230 (69,3) |
64 (38,1)d |
- |
296 (58,7)b |
96 (37,8)d |
- |
PASI 90 | ||||||
Woche 16 |
118 (35,5) |
33 (19,6)e |
7 (4,2)d |
138 (27,0) |
46 (18,1)f |
7 (2,7)d |
Woche 24 |
140 (42,2) |
37 (22,0)d |
- |
164 (32,5)b |
50 (19,7)d |
- |
PASI 100 | ||||||
Woche 16 |
47 (14,2) |
5 (3,0)d |
1 (0,6)d |
52 (10,2) |
11 (4,3)f |
3 (1,2)d |
Kopfhautspezifischer PGA 0/1c |
(N = 209) |
(N = 110) |
(N = 121) |
(N = 305) |
(N = 166) |
(N = 173) |
Woche 16 |
147 (70,3) |
43 (39,1)d |
21 (17,4)d |
182 (59,7) |
61 (36,7)d |
30 (17,3)d |
Es wurde eine Imputation für Non-Responder (Non‑Responder Imputation, NRI) angewendet; Patienten, die die Behandlung oder die Studie vor dem Endpunkt abbrachen oder deren Daten fehlten, wurden als Non‑Responder gewertet. | ||||||
Bei der Untersuchung der Faktoren Alter, Geschlecht, Abstammung, Körpergewicht, Dauer der Erkrankung, Schweregrad der Erkrankung bei Studienbeginn und vorherige Behandlung mit biologisch oder nicht biologisch hergestellten Wirkstoffen wurden keine Unterschiede hinsichtlich des Ansprechens auf Deucravacitinib zwischen diesen Untergruppen festgestellt.
Ansprechen im Zeitverlauf
Deucravacitinib zeigte einen schnellen Wirkeintritt, wobei das maximale PASI 75‑Ansprechen bis Woche 24 erreicht (POETYK PSO‑1 und POETYK PSO‑2) und bis Woche 52 aufrechterhalten (POETYK PSO‑1) wurde (siehe Abbildung 1).
Abbildung 1: PASI 75‑Ansprechen (NRI) bis Woche 52 nach Besuchstermin in POETYK PSO‑1
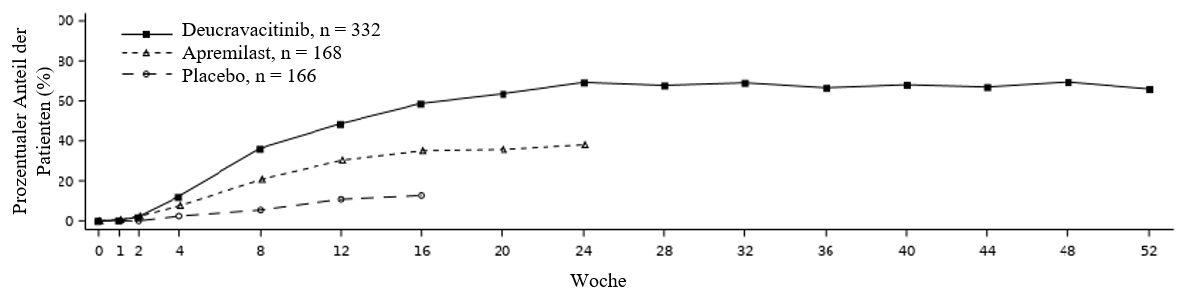
Aufrechterhaltung und Beständigkeit des Ansprechens
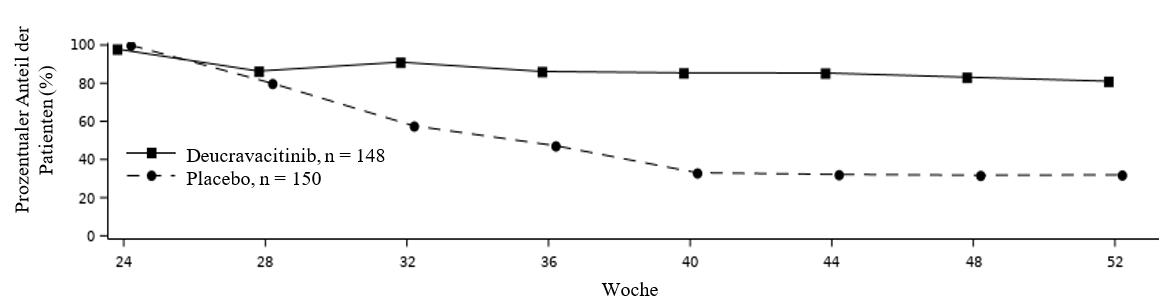
Um die Aufrechterhaltung und Beständigkeit des Ansprechens in POETYK PSO‑2 zu bewerten, wurden Patienten, die nach der ursprünglichen Randomisierung Deucravacitinib erhielten und in Woche 24 ein PASI 75‑Ansprechen erreicht hatten, erneut randomisiert, um entweder die Behandlung mit Deucravacitinib fortzusetzen oder Placebo zu erhalten. Bei Patienten, die in Woche 24 Responder waren und nach der erneuten Randomisierung Placebo erhielten, betrug die mediane Zeit bis zum Verlust des PASI 75‑Ansprechens ca. 12 Wochen. Abbildung 2 zeigt das PASI 75‑Ansprechen in den zwei Studienarmen von Woche 24 bis 52.
Abbildung 2: PASI 75‑Ansprechen (NRI) nach erneuter Randomisierung in Woche 24 in POETYK PSO‑2
Von den Patienten berichtete Ergebnisse
Bei den mit Deucravacitinib behandelten Patienten wurden im Vergleich zu Placebo in Woche 16 und im Vergleich zu Apremilast in Woche 16 und Woche 24 signifikant größere Verbesserungen der gesundheitsbezogenen Lebensqualität, gemessen anhand des Dermatology Life Quality Index (DLQI), sowie der von den Patienten berichteten Psoriasis‑Symptome (Juckreiz, Schmerzen, brennendes Gefühl, stechendes Gefühl und Spannen der Haut) und ‑Anzeichen (Trockenheit, Rissbildung, Schuppung, Ablösen oder Abblättern der Haut, Rötung und Blutung), gemessen anhand des Psoriasis Symptoms and Signs Diary (PSSD), beobachtet. Die Verbesserung dieser Arten von Ansprechen bei Patienten, die eine kontinuierliche Behandlung mit Deucravacitinib erhielten, wurde in POETYK PSO‑1 bis Woche 52 aufrechterhalten.
Tabelle 3: Von den Patienten berichtete Ergebnisse in POETYK PSO‑1 und POETYK PSO‑2
POETYK PSO‑1 |
POETYK PSO‑2 |
|||||
Deucravacitinib |
Apremilast |
Placebo |
Deucravacitinib |
Apremilast |
Placebo |
|
DLQI |
N = 322 |
N = 161 |
N = 160 |
N = 495 |
N = 247 |
N = 246 |
Woche 16, n (%) |
132 (41,0) |
46 (28,6)a |
17 (10,6)b |
186 (37,6) |
57 (23.1)b |
24 (9,8)b |
Woche 24, n (%) |
155 (48,1) |
39 (24,2)b |
- |
205 (41,4) |
53 (21,5)b |
- |
PSSD‑Symptom-Score |
N = 306 |
N = 158 |
N = 151 |
N = 466 |
N = 233 |
N = 239 |
Woche 16, Mittelwert (SF) |
-26,7 (1,8) |
-17,8 (2,2)b |
-3,6 (2,1)b |
-28,3 (1,1) |
-21,1 (1,4)b |
-4,7 (1,4)b |
Woche 24, Mittelwert (SF) |
-31,9 (2,0) |
-20,7 (2,4)b |
- |
-29,1 (1,1) |
-21,4 (1,5)b |
- |
PSSD‑Anzeichen‑Score |
N = 306 |
N = 158 |
N = 151 |
N = 466 |
N = 233 |
N = 239 |
Woche 16, Mittelwert (SF) |
-28,9 (1,8) |
-20,0 (2,2)b |
-5,3 (2,1)a |
-31,9 (1) |
-23,8 (1,4)b |
-7,1 (1,4)b |
Woche 24, Mittelwert (SF) |
-33,8 (2,0) |
-22,5 (2,4)b |
- |
-32,4 (1,1) |
-24,2 (1,5)b |
- |
*Patienten mit einem Score von ≥ 2 bei Studienbeginn | ||||||
Psoriasis-Arthritis
Die Wirksamkeit und Sicherheit von Deucravacitinib wurden in zwei multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studien (POETYK PsA-1 und POETYK PsA-2) bei Patienten untersucht, die mindestens 18 Jahre alt waren und an aktiver Psoriasis-Arthritis (PsA) (≥ 3 geschwollene Gelenke, ≥ 3 druckschmerzempfindliche Gelenke, C‑reaktives Protein (CRP)-Wert von ≥ 3 mg/l) und an aktiver Plaque-Psoriasis litten oder Plaque-Psoriasis in der Anamnese hatten. In beiden Studien hatten die Patienten bei Studienbeginn eine seit mindestens 3 Monaten diagnostizierte Psoriasis-Arthritis und erfüllten beim Screening die Klassifikationskriterien für Psoriasis-Arthritis (CASPAR). In POETYK PsA-1 lag bei den Patienten außerdem mindestens eine in der Röntgenuntersuchung nachgewiesene Knochenerosion an Händen und/oder Füßen vor.
Die Wirksamkeit wurde in den Studien PsA‑1 (n = 670) und PsA‑2 (n = 624) bei insgesamt 1 294 Patienten bis Woche 16 untersucht. In PsA‑1 wurden die Patienten randomisiert und erhielten 16 Wochen lang entweder Deucravacitinib 6 mg einmal täglich oder Placebo. In Woche 16 wurden die ursprünglich der Placebo-Gruppe zugewiesenen Patienten auf Deucravacitinib 6 mg einmal täglich umgestellt. Die Patienten wurden bis Woche 52 nachbeobachtet. In PsA‑2 wurden die Patienten randomisiert und erhielten entweder Deucravacitinib 6 mg einmal täglich, Placebo oder Apremilast 30 mg zweimal täglich (als Sicherheitsreferenzarm). Der Zeitraum für die placebokontrollierte Wirksamkeitsbewertung betrug 16 Wochen. In Woche 16 wurden die ursprünglich der Placebo-Gruppe zugewiesenen Patienten in PsA‑2 auf Deucravacitinib 6 mg einmal täglich umgestellt, und die zu Deucravacitinib oder Apremilast zugewiesenen Patienten setzten ihre zugewiesene Behandlung bis Woche 52 fort.
Die Krankheitsmerkmale bei Studienbeginn aus gepoolten Daten zeigten eine mediane Krankheitsdauer von 4,0 Jahren, und die meisten Patienten hatten eine Polyarthritis (75,7 %), gefolgt von Oligoarthritis (13,5 %). Bei 8,4 % der Patienten war überwiegend das distale Interphalangealgelenk betroffen, während 15,5 % eine periphere und psoriatische Spondyloarthritis aufwiesen. Dactylitis und Enthesitis waren bei 30,8 % bzw. 61,8 % der Patienten vorhanden, und 49,1 % hatten eine psoriatische Hautbeteiligung von ≥ 3 % der Körperoberfläche (Body Surface Area, BSA) und einen Static Physician’s Global Assessment (sPGA)-Wert von mindestens 2.
In beiden Studien waren die meisten Patienten zuvor nicht mit Biologika behandelt worden, und alle wiesen ein unzureichendes Ansprechen oder eine Unverträglichkeit gegenüber oder einen Verlust des Ansprechens auf nichtsteroidale Antirheumatika [NSAR], konventionelle synthetische DMARDs [csDMARDs] oder Apremilast auf. Zusätzlich wurden in PsA‑2 Patienten aufgenommen, die bereits mit Anti‑TNF alpha behandelt worden waren. In den Analysen der gepoolten Studien erhielten 56,3 % der Patienten gleichzeitig Methotrexat (MTX), 10,0 % erhielten gleichzeitig andere csDMARDs und 33,7 % erhielten keine csDMARDs gleichzeitig.
In beiden Studien war der primäre Endpunkt der prozentuale Anteil der Patienten, die in Woche 16 ein American College of Rheumatology (ACR) 20-Ansprechen erreichten.
Klinisches Ansprechen
In beiden Studien führte die Behandlung mit Deucravacitinib in Woche 16 im Vergleich zu Placebo zu einer signifikanten Verbesserung der Krankheitsaktivität, gemessen anhand von ACR20/50/70 und der minimalen Krankheitsaktivität (minimal disease activity, MDA) (siehe Tabelle 4). Das klinische Ansprechen bei den mit Deucravacitinib behandelten Patienten verbesserte sich nach Woche 16 weiter, und die Ergebnisse wurden bis Woche 52 aufrechterhalten.
Tabelle 4: Wirksamkeitsergebnisse bei Erwachsenen mit Psoriasis-Arthritis
POETYK PsA-1 |
POETYK PsA-2 |
|||||
Deucravacitinib |
Placebo |
Unterschied zu Placebo |
Deucravacitinib |
Placebo |
Unterschied zu Placebo |
|
ACR20-Ansprechen | ||||||
Woche 16 (%) |
54,2 |
34,1 |
20,0 |
54,2 |
39,4 |
14,8 |
Woche 52 (%)* |
76,3 |
71,9 |
||||
ACR50-Ansprechen | ||||||
Woche 16 (%) |
24,7 |
13,5 |
11,2 |
28,8 |
16,3 |
12,5 |
Woche 52 (%)* |
48,9 |
47,0 |
||||
ACR70-Ansprechen | ||||||
Woche 16 (%) |
11,6 |
5,4 |
6,2 |
10,6 |
5,4 |
5,2 |
Woche 52 (%)* |
30,2 |
30,0 |
||||
Ansprechen hinsichtlich der minimalen Krankheitsaktivität (MDA)** | ||||||
Woche 16 (%) |
19,0 |
10,2 |
8,9 |
25,6 |
14,7 |
10,9 |
Woche 52 (%)* |
40,7 |
48,3 |
||||
Bis Woche 16 wurde eine Imputation von Non-Respondern (Non-Responder Imputation, NRI) angewendet. Nach 16 Wochen werden die beobachteten Daten ohne Imputation dargestellt. | ||||||
In PsA‑1 und PsA‑2 wurde in Woche 16 bei den mit Deucravacitinib behandelten Patienten eine Verbesserung in allen ACR-Komponenten festgestellt, einschließlich der Beurteilung der Schmerzen durch den Patienten (Schmerz-VAS), sowie eine weitere Verbesserung nach Woche 16 mit Aufrechterhaltung bis Woche 52.
Die Anzahl an Patienten mit axialer Beteiligung/prädominanter Spondylitis war zu gering für eine aussagekräftige Beurteilung.
In POETYK PsA-1 erreichten Patienten, die Deucravacitinib erhielten, im Vergleich zu Placebo in Woche 16 eine nominal signifikante Verbesserung des Disease Activity Scores (28 Gelenke) anhand von CRP (DAS28-CRP) (-1,33 bzw. -0,83). In POETYK PsA‑2 wurde in Woche 16 ebenfalls eine ähnliche Verbesserung beobachtet (-1,28 bzw. -0,80).
Bei mit SOTYKTU behandelten Patienten mit gleichzeitig bestehender Plaque‑Psoriasis wurde eine signifikante Verbesserung im Vergleich zu Placebo beobachtet, gemessen anhand des Psoriasis Area Severity Index (PASI 75) in Woche 16, und die Ergebnisse wurden in beiden Studien bis Woche 52 aufrechterhalten.
Ansprechen im Hinblick auf die körperliche Funktionsfähigkeit
In beiden Studien zeigten mit Deucravacitinib behandelte Patienten im Vergleich zu Placebo eine statistisch signifikante Verbesserung der körperlichen Funktionsfähigkeit gegenüber Studienbeginn, gemessen anhand des Health Assessment Questionnaire Disability Index (HAQ‑DI). Der adjustierte mittlere Unterschied (95 %‑KI) der Veränderung des HAQ‑DI gegenüber dem Ausgangswert im Vergleich zu Placebo betrug in Woche 16 -0,17 (-0,24; -0,09) in der Studie PsA-1 und -0,11 (‑0,18; ‑0,04) in der Studie PsA-2. Der Anteil der HAQ‑DI-Responder (Verbesserung gegenüber Studienbeginn um ≥ 0,35) in Woche 16 in PsA‑1 und PsA‑2 betrug 51,3 % bzw. 48,9 % bei Patienten, die Deucravacitinib erhielten, und 38,8 % bzw. 43,2 % bei Patienten, die Placebo erhielten. Das Ansprechen wurde in beiden Studien bis Woche 52 aufrechterhalten.
Weitere gesundheitsbezogene Ergebnisse
Die mit Deucravacitinib behandelten Patienten zeigten in Woche 16 im Vergleich zu Placebo Verbesserungen des SF‑36 Physical Component Summary (PCS)‑Scores. In Woche 16 wurde unter Deucravacitinib im Vergleich zu Placebo eine signifikante Verbesserung des PCS‑Scores beobachtet (adjustierter mittlerer Unterschied (95 %‑KI) gegenüber dem Ausgangswert: 2,3 (1,3; 3,4), p < 0,0001 in POETYK PsA-1 und 2,0 (1,0; 3,1), p = 0,0002 in POETYK PsA-2). Die Verbesserungen wurden in beiden Studien bis Woche 52 aufrechterhalten.
Ältere Menschen
Von den 1 519 Patienten mit Plaque‑Psoriasis, die in klinischen Studien mit Deucravacitinib behandelt wurden, waren 152 Patienten mindestens 65 Jahre alt, darunter 21 Patienten, die mindestens 75 Jahre alt waren (siehe Abschnitt 4.2). Unter den 1 312 Patienten mit Psoriasis-Arthritis, die in klinischen Studien mit Deucravacitinib behandelt wurden, waren 171 Patienten mindestens 65 Jahre alt und 22 Patienten mindestens 75 Jahre alt.
Hinsichtlich Exposition, Sicherheit oder Wirksamkeit wurden zwischen älteren und jüngeren Patienten, die Deucravacitinib erhielten, insgesamt keine Unterschiede beobachtet.
Kinder und Jugendliche
Die Europäische Arzneimittel‑Agentur hat für SOTYKTU eine Zurückstellung von der Verpflichtung zur Vorlage der Ergebnisse von Studien in einer oder mehreren pädiatrischen Altersklassen zur Behandlung von Plaque-Psoriasis und Psoriasis-Arthritis gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Deucravacitinib zeigte eine fast vollständige orale Resorption, einen dosisabhängigen Anstieg der Exposition und keine offensichtliche zeitabhängige Pharmakokinetik.
Resorption
Nach oraler Einnahme der Tabletten wurde Deucravacitinib schnell und fast vollständig resorbiert. Der mediane Tmax‑Wert lag im Bereich von 2 bis 3 Stunden, und die absolute orale Bioverfügbarkeit betrug bei gesunden Probanden 99 %. Nach einmal täglicher Gabe wurde eine mäßige Akkumulation (< 1,4‑fach im Steady‑State) beobachtet.
Mahlzeiten
Deucravacitinib kann ohne Rücksicht auf Mahlzeiten oder Arzneimittel, die den pH‑Wert im Magen regulieren (H2‑Rezeptor‑Blocker und Protonenpumpeninhibitoren), angewendet werden. Die gleichzeitige Einnahme von Nahrung oder Arzneimitteln, die den pH‑Wert im Magen regulieren, hatte keine Auswirkungen auf die Gesamtexposition (AUC[INF]) von Deucravacitinib.
Verteilung
Das Verteilungsvolumen im Steady-State (Vss) beträgt 140 l; das ist mehr als das Gesamtkörperwasser [42 l] und deutet auf eine extravaskuläre Verteilung hin. Deucravacitinib ist zu 81,6 % an menschliche Plasmaproteine gebunden, und zwar hauptsächlich an humanes Serumalbumin.
Deucravacitinib wird zwischen Plasma und Bestandteilen roter Blutkörperchen verteilt, mit einem Konzentrationsverhältnis von Blut zu Plasma von 1,26.
Übergang in die Muttermilch
Nach der oralen Verabreichung einer Einzeldosis Deucravacitinib von 9 mg an acht gesunde, stillende Frauen waren Deucravacitinib und sein Hauptmetabolit BMT-153261 in der Muttermilch messbar. Die Spitzenkonzentrationen in der Milch wurden schnell erreicht, mit medianen Tmax-Werten von etwa 1 Stunde bei Deucravacitinib und 6 Stunden bei BMT‑153261. Die geschätzten Verhältnisse von Milch zu Plasma für Cmax und AUC betrugen 1,5 bzw. 3,2 für Deucravacitinib und waren höher für BMT‑153261 (Cmax-Verhältnis 13; AUC-Verhältnis 16). Innerhalb von 24 Stunden nach der Verabreichung wurden 93 % von Deucravacitinib und 80 % von BMT‑153261 in die Muttermilch ausgeschieden. Das Stillen hatte wenig bis keinen Einfluss auf die Plasmaexposition von Deucravacitinib.
Biotransformation
Beim Menschen wird Deucravacitinib über vier primäre Stoffwechselwege metabolisiert: N‑Demethylierung an der Triazol-Gruppe durch Cytochrom P‑450 (CYP) 1A2 unter Bildung des Hauptmetaboliten BMT‑153261, Cyclopropylcarboxamid‑Hydrolyse durch Carboxylesterase 2 (CSE2) unter Bildung des Hauptmetaboliten BMT‑158170, N‑Glucuronidierung durch Uridin‑Glucuronyl‑Transferase (UGT) unter Bildung von BMT‑334616 und Monooxidation durch CYP2B6/2D6 an der deuterierten Methylgruppe unter Bildung von M11.
Im Steady‑State macht Deucravacitinib den größten zirkulierenden Anteil aus, und zwar 49 % der gemessenen, verbindungsbezogenen Komponenten. Es wurden zwei zirkulierende Hauptmetaboliten, BMT‑153261 und BMT‑158170, identifiziert, die mit der Muttersubstanz Deucravacitinib vergleichbare Halbwertszeiten aufweisen. BMT‑153261 hat eine mit der Mutterverbindung vergleichbare Wirkstärke, und BMT‑158170 ist nicht pharmakologisch wirksam. Die Exposition gegenüber zirkulierendem BMT‑153261 ist deutlich geringer als die Exposition gegenüber der Mutterverbindung. Daher wird die überwiegende pharmakologische Aktivität der Mutterverbindung Deucravacitinib zugeschrieben.
Außerdem wurden keine Metaboliten, die nur beim Menschen auftreten, und keine langlebigen zirkulierenden Metaboliten identifiziert.
Elimination
Deucravacitinib wird über mehrere Wege eliminiert, darunter Phase‑I‑ und Phase‑II‑Metabolismus, sowie durch direkte renale und fäkale Ausscheidung. Darüber hinaus trug kein einzelnes Enzym zu mehr als 26 % der Gesamt‑Clearance bei. Deucravacitinib wird extensiv metabolisiert: 59 % der oral angewendeten [14C]‑Deucravacitinib‑Dosis werden als Metaboliten im Urin (37 % der Dosis) und im Stuhl (22 % der Dosis) ausgeschieden. Unverändertes Deucravacitinib in Urin und Stuhl machte 13 % bzw. 26 % der Dosis aus.
Die effektive Halbwertszeit von Deucravacitinib 6 mg bei gesunden erwachsenen Menschen beträgt ca. 10 Stunden, mit einer Gesamt‑Clearance von 15,3 l/h (VK 27 %). Basierend auf populationspharmakokinetischen Analysen ist die effektive Halbwertszeit von Deucravacitinib bei Patienten mit Psoriasis und Psoriasis-Arthritis ähnlich wie bei gesunden Erwachsenen und liegt zwischen 10 und 11 Stunden. Die Deucravacitinib‑Clearance bei Psoriasis-Patienten ist ähnlich wie bei gesunden Erwachsenen. Die Deucravacitinib‑Clearance bei Patienten mit Psoriasis-Arthritis ist im Vergleich zu gesunden Erwachsenen moderat niedriger (um 29 %). Deucravacitinib ist ein Substrat von Efflux‑Transportern, P‑Glykoprotein (P‑gp) und Breast Cancer Resistance Protein (BCRP) und vom Aufnahmetransporter OCT1. Aufgrund der hohen passiven Permeabilität, der hohen oralen Bioverfügbarkeit und der niedrigen Affinität zu diesen Transportern ist der Beitrag dieser Transporter zur Pharmakokinetik von Deucravacitinib minimal.
Deucravacitinib ist kein Substrat der Transporter OATP, NTCP, OAT1, OAT3, OCT2, MATE1 oder MATE2K.
Linearität/Nicht‑Linearität
Die Pharmakokinetik von als Tabletten angewendeten Deucravacitinib‑Einzeldosen war über den Dosisbereich von 3 mg bis 36 mg hinweg linear.
Wechselwirkungen
Wirkung von Deucravacitinib auf andere Arzneimittel
In‑vitro‑Studien haben keine Hinweise dafür geliefert, dass Deucravacitinib und seine zirkulierenden Hauptmetaboliten bei klinisch relevanten Expositionen die wichtigsten CYP‑Enzyme (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4), UGT (1A1, 1A4, 1A6, 1A9, 2B7), CES2 und Arzneimitteltransporter (P‑gp, BCRP, OATP1B1, OATP1B3, BSEP, MRP2, OAT1, OAT3, OCT1, OCT2, MATE1 und MATE2K) hemmen. Darüber hinaus ist Deucravacitinib kein Induktor von CYP1A2, CYP2B6 und CYP3A4 (siehe Abschnitt 4.5).
Besondere Patientengruppen
Ältere Patienten
Die mittlere Exposition gegenüber Deucravacitinib im Steady‑State (Cavg,ss), welche auf einer populationspharmakokinetischen Analyse basiert, war bei Patienten im Alter von 65‑74 Jahren [n = 87 von 1 387 (6,3 %) bei Psoriasis und n = 155 von 1 325 (11,7 %) bei Psoriasis-Arthritis] um bis zu 31 % und bei Patienten im Alter von 75‑84 Jahren [n = 13 von 1 387 (0,94 %) bei Psoriasis und n = 23 von 1 325 (1,7 %) bei Psoriasis-Arthritis] um bis zu 53 % erhöht. Es liegen begrenzte Daten zur Exposition bei Patienten im Alter von ≥ 85 Jahren vor.
Patienten mit Nierenfunktionsstörung
Eine Nierenfunktionsstörung hat keine klinisch bedeutsamen Auswirkungen auf die Exposition gegenüber Deucravacitinib (siehe Abschnitt 4.2). Dies basiert auf einer dedizierten Studie, bei der die geschätzte glomeruläre Filtrationsrate (eGFR) mithilfe einer Gleichung für die Änderung der Ernährung bei Nierenerkrankungen (Modification of Diet in Renal Disease, MDRD) bestimmt wurde. Im Vergleich zur Gruppe mit normaler Nierenfunktion war über die Gruppen mit Nierenfunktionsstörung (leicht [eGFR: ≥ 60 bis < 90 ml/min], mittelschwer [eGFR: ≥ 30 bis < 60 ml/min], schwer [eGFR: < 30 ml/min] und ESRD [eGFR: < 15 ml/min]) hinweg Cmax von Deucravacitinib um bis zu 15 % verändert und AUC[INF] um bis zu 48 % erhöht. Im Vergleich zur Gruppe mit normaler Nierenfunktion war Cmax von BMT‑153261 über die Gruppen mit Nierenfunktionsstörung hinweg um bis zu 34 % erhöht und AUC[INF] um bis zu 84 % erhöht.
Durch eine Dialyse wird Deucravacitinib nicht in wesentlichem Umfang aus dem systemischen Kreislauf entfernt (5,4 % der Dosis pro Dialyse entfernt).
Patienten mit Leberfunktionsstörung
Eine leichte (Child‑Pugh‑Klasse A) und mittelschwere (Child‑Pugh‑Klasse B) Leberfunktionsstörung hat keine klinisch bedeutsamen Auswirkungen auf die Exposition gegenüber Deucravacitinib (siehe Abschnitt 4.2). Im Vergleich zur Gruppe mit normaler Leberfunktion waren Gesamt‑Cmax und ‑AUC[INF] von Deucravacitinib in den Gruppen mit leichter und mittelschwerer Leberfunktionsstörung um bis zu 10 % bzw. 40 % erhöht, während Cmax und AUC[INF] von ungebundenem Deucravacitinib um bis zu 26 % bzw. 60 % erhöht waren. Bei Erwachsenen mit schwerer (Child‑Pugh‑Klasse C) Leberfunktionsstörung war Gesamt‑Cmax von Deucravacitinib mit entsprechenden gesunden Erwachsenen vergleichbar, während Gesamt‑AUC um 43 % höher war. Bei diesen Erwachsenen waren Cmax und AUC[INF] von ungebundenem Deucravacitinib um 62 % bzw. 131 % erhöht. Die Anwendung von Deucravacitinib wird bei Patienten mit schwerer Leberfunktionsstörung nicht empfohlen (siehe Abschnitt 4.2).
Der AUC[0‑T]‑Wert von BMT‑153261 war bei Patienten mit leichter, mittelschwerer und schwerer Leberfunktionsstörung im Vergleich zu Patienten mit normaler Leberfunktion um 19 %, 53 % bzw. 76 % verringert, während der Cmax‑Wert von BMT‑153261 bei Patienten mit leichter, mittelschwerer und schwerer Leberfunktionsstörung um 25 %, 59 % bzw. 79 % verringert war.
Geschlecht
Basierend auf pharmakokinetischen Populationsmodellen und Simulationen wird erwartet, dass bei Frauen im Vergleich zu Männern die mittlere Steady‑State‑Exposition gegenüber Deucravacitinib (Cmax,ss und Cavg,ss) bis zu 30 % höher ist.
Körpergewicht
Basierend auf pharmakokinetischen Populationsmodellen und Simulationen wird erwartet, dass bei Patienten mit geringerem Körpergewicht (< 60 kg) das geometrische Mittel der Steady‑State‑Exposition gegenüber Deucravacitinib (Cmaxss) bis zu 37,4 % und (Cavgss) bis zu 26,6 % höher ist. Bei Patienten mit höherem Körpergewicht (> 90 kg) wird ein geringeres geometrisches Mittel der Steady‑State‑Exposition gegenüber Deucravacitinib von bis zu 24,8 % (Cmaxss) und bis zu 19,6 % (Cavgss) (im Vergleich zu Patienten mit einem Körpergewicht von 60‑90 kg) erwartet.
Intrinsische Faktoren
Abstammung und ethnische Zugehörigkeit hatten keine klinisch bedeutsamen Auswirkungen auf die Exposition gegenüber Deucravacitinib.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Reproduktions- und Entwicklungstoxizität, Genotoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Toxizität bei wiederholter Gabe
In der Studie zur chronischen Toxizität bei Ratten wurden bei Expositionen (AUC) auf dem Lowest Observed Effect Level (LOEL), die etwa dem 7‑Fachen der für den Menschen empfohlenen Dosis (Recommended Human Dose, RHD) entsprachen, geringere Lymphozytenzahlen, eine Abnahme der Zellularität des Knochenmarks sowie der Zellularität des Lymphgewebes in Geweben des Immunsystems festgestellt. Diese Wirkungen waren nicht mit klinischen Anzeichen einer Immunsuppression (z. B. Infektionen) assoziiert. Bei Expositionen (AUC) auf dem Lowest Observed Effect Level (LOEL) von etwa dem 33‑Fachen der RHD wurden reduzierte Thrombozytenzahlen und verringerte Parameter der Erythrozytenmasse beobachtet. In der Studie zur chronischen Toxizität bei Affen wurden bei Expositionen (AUC) auf dem Lowest Observed Effect Level (LOEL) von etwa dem 6‑Fachen der RHD klinische und mikroskopische Hautveränderungen und verringerte Parameter der Erythrozytenmasse festgestellt.
Entwicklungs‑ und Reproduktionstoxizität
Bei männlichen und weiblichen Ratten hatte Deucravacitinib bei Expositionen (AUC) von bis zu ca. dem 178‑ und 136‑Fachen der RHD keine Auswirkungen auf die Fertilität bzw. die frühe Embryonalentwicklung.
Deucravacitinib war bei Expositionen des Muttertieres (AUC) von bis zu ca. dem 211‑Fachen der RHD bei Ratten oder dem 72‑/16‑Fachen (gesamt/ungebunden) der RHD bei Kaninchen weder für den Embryo tödlich noch hatte es teratogene Wirkungen.
In einer Studie zur prä‑ und postnatalen Entwicklung bei Ratten wurden während des Zeitraums vor dem Absetzen bei Expositionen des Muttertieres (AUC) von etwa dem 87‑Fachen der RHD vorübergehend niedrigere Körpergewichte der Jungtiere festgestellt. Dieser Effektwurde im Zeitraum nach dem Absetzen vollständig wieder aufgehoben.
Nach Verabreichung von radioaktiv markiertem Deucravacitinib an laktierende Ratten waren Deucravacitinib und/oder seine Metaboliten in der Milch vorhanden, mit Konzentrationsverhältnissen von Milch zu Plasma von 2,7 bis 30,9.
Tablettenkern
Hypromelloseacetatsuccinat
Lactose
Mikrokristalline Cellulose
Croscarmellose‑Natrium
Siliciumdioxid-Hydrat
Magnesiumstearat
Filmüberzug
Poly(vinylalkohol)
Titandioxid (E171)
Macrogol
Talkum
Eisen(III)‑oxid (E172)
Eisen(III)‑hydroxid-oxid × H2O (E172)
Nicht zutreffend.
3 Jahre.
Für dieses Arzneimittel sind keine besonderen Aufbewahrungsbedingungen erforderlich.
Durchsichtige Blisterpackung aus Polyvinylchlorid/Polychlortrifluorethylen (PVC/PCTFE) mit durchdrückbarer Aluminiumfolie mit 7 oder 14 Filmtabletten pro Blisterpackung (Kalender‑ oder Nicht-Kalender-Blisterpackung).
Packungsgrößen: 7, 14, 28 und 84 Filmtabletten.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Bristol-Myers Squibb Pharma EEIG
Plaza 254
Blanchardstown Corporate Park 2
Dublin 15, D15 T867
Irland
EU/1/23/1718/001
EU/1/23/1718/002
EU/1/23/1718/003
EU/1/23/1718/004
EU/1/23/1718/005
EU/1/23/1718/006
EU/1/23/1718/007
EU/1/23/1718/008
Datum der Erteilung der Zulassung: 24. März 2023
April 2026 (2)
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig
Bristol-Myers Squibb GmbH & Co. KGaA
Arnulfstraße 29
80636 München
Medizinische Information
Telefon: 0800 0752002
e-Mail: medwiss.info@bms.com
www.bmsmedinfo.de