
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Ebglyss 250 mg Injektionslösung in einer Fertigspritze
Ebglyss 250 mg Injektionslösung im Fertigpen
Ebglyss 250 mg Injektionslösung in einer Fertigspritze
Jede Einweg-Fertigspritze enthält 250 mg Lebrikizumab in 2 ml Lösung (125 mg/ml).
Ebglyss 250 mg Injektionslösung im Fertigpen
Jeder Einweg-Fertigpen enthält 250 mg Lebrikizumab in 2 ml Lösung (125 mg/ml).
Lebrikizumab wird mittels rekombinanter DNA-Technologie in Ovarialzellen des chinesischen Hamsters (CHO-Zellen) hergestellt.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Ebglyss 250 mg Injektionslösung in einer Fertigspritze enthält 0,6 mg Polysorbat 20 (E 432).
Jede Ebglyss 250 mg Injektionslösung im Fertigpen enthält 0,6 mg Polysorbat 20 (E 432).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung (Injektion)
Klare bis schillernde, farblose bis leicht gelbliche oder leicht bräunliche Lösung, frei von sichtbaren Partikeln.
Ebglyss wird angewendet für die Behandlung von mittelschwerer bis schwerer atopischer Dermatitis bei Erwachsenen und
Jugendlichen ab 12 Jahren mit einem Körpergewicht von mindestens 40 kg, die für eine systemische Therapie in Betracht kommen.
Die Behandlung sollte von einem Arzt eingeleitet werden, der in der Diagnose und Behandlung der atopischen Dermatitis erfahren ist.
Dosierung
Die empfohlene Lebrikizumab-Dosis ist 500 mg (zwei 250-mg-Injektionen) jeweils in Woche 0 und Woche 2, gefolgt von 250 mg, die alle zwei Wochen bis Woche 16 subkutan verabreicht werden.
Bei Patienten, die nach 16 Wochen Behandlung kein klinisches Ansprechen gezeigt haben, sollte ein Abbruch der Behandlung in Betracht gezogen werden. Bei Patienten mit einem anfänglich partiellen Ansprechen kann sich der Zustand durch eine
fortgesetzte Behandlung alle zwei Wochen bis zu Woche 24 weiter verbessern.
Sobald ein klinisches Ansprechen erreicht ist, beträgt die empfohlene Erhaltungsdosis von Lebrikizumab 250 mg alle vier Wochen.
Lebrikizumab kann mit oder ohne topische Kortikosteroide (TCS) angewendet werden. Topische Calcineurin-Inhibitoren (TCI) können angewendet werden, sollten aber nur auf die Problemzonen wie Gesicht, Hals, intertriginöse Bereiche und Genitalbereich beschränkt bleiben.
Versäumte Dosis
Wenn eine Dosis vergessen wurde, sollte die Dosis so bald wie möglich nachgeholt werden. Danach sollte die Dosisgabe zum nächsten geplanten Zeitpunkt wieder aufgenommen werden.
Besondere Patientengruppen
Ältere Patienten (≥ 65 Jahre)
Bei älteren Patienten ist keine Dosisanpassung empfohlen (siehe Abschnitt 5.2).
Eingeschränkte Nieren- oder Leberfunktion
Bei Patienten mit eingeschränkter Nieren- oder Leberfunktion ist keine Dosisanpassung empfohlen (siehe Abschnitt 5.2).
Körpergewicht
Eine körpergewichtsbezogene Dosisanpassung ist nicht empfohlen (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Lebrikizumab bei Kindern im Alter von 6 Monaten bis < 12 Jahren oder Jugendlichen im Alter von 12 bis 17 Jahren und mit einem Gewicht von weniger als 40 kg ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Subkutane Anwendung.
Lebrikizumab wird als subkutane Injektion in den Oberschenkel oder Bauch verabreicht außerhalb eines 5 cm großen Bereiches um den Bauchnabel. Wenn eine andere Person die Injektion verabreicht, kann die Injektion auch in den Oberarm erfolgen.
Für die anfängliche 500-mg-Dosis werden zwei 250-mg-Injektionen nacheinander an verschiedenen Injektionsstellen verabreicht.
Es wird empfohlen, die Injektionsstelle bei jeder Injektion zu wechseln. Lebrikizumab sollte nicht in Hautbereiche injiziert werden, die berührungsempfindlich oder geschädigt sind bzw. blaue Flecken oder Narben aufweisen.
Ein Patient kann sich Lebrikizumab selbst injizieren oder die Pflegeperson des Patienten kann Lebrikizumab verabreichen, wenn der behandelnde Arzt dies für angemessen hält. Patienten und/oder Pflegepersonen sind vor der Verabreichung von Lebrikizumab entsprechend zu unterweisen. Ausführliche Anweisungen zur Verabreichung sind am Ende der Gebrauchsinformation angegeben.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Überempfindlichkeit
Wenn eine systemische Überempfindlichkeitsreaktion (unmittelbar oder verzögert) auftritt, muss Lebrikizumab abgesetzt und eine geeignete Therapie eingeleitet werden.
Konjunktivitis
Bei Patienten, die mit Lebrikizumab behandelt werden und eine Konjunktivitis entwickeln, die nach der Standardbehandlung nicht abklingt, ist eine ophthalmologische Abklärung erforderlich (siehe Abschnitt 4.8).
Helminthose
Patienten mit bekannten Helminthosen wurden von der Teilnahme an klinischen Studien ausgeschlossen. Es ist nicht bekannt, ob Lebrikizumab die Immunantwort gegen Helminthosen durch Hemmung der IL-13-Signalübertragung beeinflusst.
Patienten mit vorbestehenden Helminthosen sollten vor Beginn der Lebrikizumab-Therapie behandelt werden. Wenn Patienten während der Lebrikizumab-Therapie infiziert werden und nicht auf eine Behandlung mit Anthelminthika ansprechen, sollte die
Lebrikizumab-Therapie unterbrochen werden, bis die Infektion abgeklungen ist.
Impfungen
Vor Beginn der Therapie mit Lebrikizumab wird die Durchführung aller altersgemäßen Impfungen gemäß den aktuellen Impfrichtlinien empfohlen. Lebendimpfstoffe und attenuierte Lebendimpfstoffe sollten nicht gleichzeitig mit Lebrikizumab verabreicht werden, da die klinische Sicherheit und Wirksamkeit nicht nachgewiesen sind. Die Immunantwort auf Totimpfstoffe wurde bei einem Diphtherie-Tetanus-Pertussis(azellulär)-Kombinationsimpfstoff (Tdap) und einem Meningokokken-Polysaccharid-Impfstoff untersucht (siehe Abschnitt 4.5).
Sonstige Bestandteile
Dieses Arzneimittel enthält 0,6 mg Polysorbat 20 (E 432) pro Fertigspritze oder Fertigpen mit 250 mg, entsprechend 0,3 mg/ml. Polysorbate können allergische Reaktionen hervorrufen.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Lebendimpfstoffe
Die Sicherheit und Wirksamkeit der gleichzeitigen Anwendung von Lebrikizumab mit Lebendimpfstoffen und attenuierten Lebendimpfstoffen wurde nicht untersucht. Lebendimpfstoffe und attenuierte Lebendimpfstoffe sollten nicht gleichzeitig mit
Lebrikizumab verabreicht werden.
Totimpfstoffe
Die Immunantworten auf Totimpfstoffe wurden in einer Studie (ADopt-VA) untersucht, in der erwachsene Patienten mit atopischer Dermatitis mit 500 mg Lebrikizumab in Woche 0 und 2 gefolgt von 250 mg Lebrikizumab alle zwei Wochen behandelt wurden. Nach 12-wöchiger Verabreichung von Lebrikizumab wurden die Patienten mit einem Diphtherie-Tetanus-Pertussis(azellulär)-Kombinationsimpfstoff (Tdap) (T-Zell-abhängig) und einem Meningokokken-Polysaccharid-Impfstoff (T-Zell-unabhängig) geimpft. Die Immunantworten wurden 4 Wochen später untersucht. Die Antikörperantworten auf beide Totimpfstoffe waren durch die gleichzeitige Behandlung mit Lebrikizumab nicht negativ beeinflusst. In der Studie wurden keine unerwünschten Wechselwirkungen zwischen den Totimpfstoffen und Lebrikizumab festgestellt. Daher können Patienten, die Lebrikizumab erhalten, gleichzeitig inaktivierte oder Totimpfstoffe erhalten. Für Informationen zu Lebendimpfstoffen siehe Abschnitt 4.4.
Begleittherapien
Da Lebrikizumab ein monoklonaler Antikörper ist, sind keine pharmakokinetischen Wechselwirkungen zu erwarten.
Schwangerschaft
Bisher liegen nur sehr begrenzte Erfahrungen mit der Anwendung von Lebrikizumab bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Abschnitt 5.3). Aus Vorsichtsgründen soll eine Anwendung von Lebrikizumab während der Schwangerschaft vermieden werden.
Stillzeit
Es ist nicht bekannt, ob Lebrikizumab in die Muttermilch übergeht oder nach der Einnahme systemisch absorbiert wird. Es ist bekannt, dass mütterliches IgG in der Muttermilch vorhanden ist. Ein Risiko für das Neugeborene/Kind kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob die Behandlung mit Lebrikizumab zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Tierexperimentelle Studien haben keine Beeinträchtigung der Fertilität gezeigt (siehe Abschnitt 5.3).
Lebrikizumab hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die häufigsten Nebenwirkungen sind Konjunktivitis (6,9 %), Reaktionen an der Injektionsstelle (2,6 %), allergische Konjunktivitis (1,8 %) und trockenes Auge (1,4 %).
Tabellarische Auflistung der Nebenwirkungen
Im Rahmen des klinischen Studienprogramms zur atopischen Dermatitis erhielten insgesamt 1720 Patienten Lebrikizumab, von denen 891 Patienten über mindestens ein Jahr lang mit Lebrikizumab therapiert wurden. Soweit nicht anders angegeben, basieren die Häufigkeiten auf den gepoolten Daten von 4 randomisierten, doppelblinden Studien an Patienten mit mittelschwerer bis schwerer atopischer Dermatitis, in denen 783 Patienten während des placebokontrollierten Zeitraums (die ersten 16 Behandlungswochen) subkutan mit Lebrikizumab behandelt wurden.
In Tabelle 1 sind die in klinischen Studien beobachteten Nebenwirkungen nach Systemorganklasse und Häufigkeit aufgeführt, unter Verwendung folgender Kategorien: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1 000, <1/100); selten (≥1/10 000, <1/1 000); sehr selten (<1/10 000). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 1. Auflistung der Nebenwirkungen
MedDRA Systemorganklasse |
Häufigkeit |
Nebenwirkung |
Infektionen und parasitäre Erkrankungen |
Häufig |
Konjunktivitis |
Erkrankungen des Blutes und des Lymphsystems |
Gelegentlich |
Eosinophilie |
Augenerkrankungen |
Häufig |
Allergische Konjunktivitis |
Gelegentlich |
Keratitis |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Häufig |
Reaktion an der Injektionsstelle |
Beschreibung ausgewählter Nebenwirkungen
Konjunktivitis und assoziierte Nebenwirkungen
Konjunktivitis, allergische Konjunktivitis, Blepharitis und Keratitis wurden in den ersten 16 Behandlungswochen häufiger bei
Patienten berichtet, die mit Lebrikizumab behandelt wurden (6,9 %, 1,8 %, 0,8 % bzw. 0,6 %), als unter Placebo (1,8 %, 0,7 %, 0,2 % bzw. 0,3 %).
Während der Erhaltungsbehandlung (Woche 16–52) lag die Inzidenz von Konjunktivitis und allergischer Konjunktivitis unter Lebrikizumab bei 5,0 % bzw. 5,9 %.
In allen klinischen Studien kam es bei den mit Lebrikizumab behandelten Patienten in 0,7 % bzw. 0,3 % der Fälle zu einem Abbruch der Behandlung aufgrund von Konjunktivitis bzw. allergischer Konjunktivitis. Schwere Fälle von Konjunktivitis und allergischer Konjunktivitis traten in 0,1 % bzw. 0,2 % der Fälle auf. Bei 72 % der Patienten kam es zu einer Besserung, bei 57 % davon innerhalb von 90 Tagen.
Eosinophilie
Bei Patienten, die mit Lebrikizumab behandelt wurden, war der durchschnittliche Anstieg der Eosinophilenzahl gegenüber Baseline größer als bei Patienten, die mit Placebo behandelt wurden. Unter Lebrikizumab kam es bei 20,3 % der Patienten zu einem Anstieg der Eosinophilenzahl im Vergleich zu 11,7 % unter Placebo. Im Allgemeinen war der Anstieg gegenüber Baseline bei den mit Lebrikizumab behandelten Patienten geringfügig oder moderat ausgeprägt und nur vorübergehend. Eosinophilie > 5000 Zellen/µl wurde bei 0,4 % der mit Lebrikizumab und bei keinem der mit Placebo behandelten Patienten beobachtet. Eosinophilie als Nebenwirkung wurde bei 0,6 % der mit Lebrikizumab behandelten Patienten und mit einer vergleichbaren Häufigkeit unter den mit Placebo behandelten Patienten während des ersten Behandlungszeitraums gemeldet. Die Eosinophilie führte nicht zum
Behandlungsabbruch und es wurden keine mit Eosinophilen zusammenhängenden Erkrankungen gemeldet.
Reaktion an der Injektionsstelle
Reaktionen an der Injektionsstelle (einschließlich Schmerzen und Erythem) wurden häufiger bei Patienten berichtet, die Lebrikizumab (2,6 %) erhielten, als unter Placebo (1,5 %). Die Mehrzahl (95 %) der Reaktionen an der Injektionsstelle war leicht oder moderat und nur wenige Patienten (< 0,5 %) brachen die Lebrikizumab-Behandlung ab.
Herpes zoster
Herpes zoster wurde bei 0,6 % der mit Lebrikizumab behandelten Patienten und bei keinem der Patienten in der Placebo-Gruppe berichtet. Alle berichteten Herpes-zoster-Ereignisse waren leicht oder moderat und keines führte zu einem dauerhaften Abbruch der Behandlung.
Langzeitsicherheit
Die Sicherheit von Lebrikizumab wurde bei Erwachsenen und Jugendlichen mit bis zu 3 Jahren kontinuierlicher Behandlung untersucht. Insgesamt wurden 1 153 Patienten in die ADjoin-Verlängerungsstudie aufgenommen, entweder direkt oder aus den Studien ADvocate-1, ADvocate-2 und ADore mit bis zu 1 Jahr Exposition oder aus den Studien ADhere und ADopt-VA mit bis zu 16 Wochen Exposition. 771 Patienten schlossen 2 Jahre zusätzliche Lebrikizumab-Behandlung in ADjoin ab. Das Sicherheitsprofil blieb bei einer längeren Exposition gegenüber Lebrikizumab gleich, und es wurden keine neuen Sicherheitsbedenken beobachtet.
Kinder und Jugendliche
Jugendliche im Alter von 12 bis 17 Jahren
Die Sicherheit von Lebrikizumab wurde bei 372 Patienten im Alter von 12 bis 17 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis untersucht, darunter 270 Patienten, die mindestens ein Jahr lang Lebrikizumab erhielten. Das Sicherheitsprofil von Lebrikizumab bei diesen Patienten war vergleichbar mit dem Sicherheitsprofil bei Erwachsenen mit atopischer Dermatitis.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
In klinischen Studien wurden intravenöse Einzeldosen von bis zu 10 mg pro kg Körpergewicht und subkutane Mehrfachdosen von bis zu 500 mg ohne dosislimitierende Toxizität an Menschen verabreicht. Es gibt keine spezifische Behandlung für eine Lebrikizumab-Überdosierung. Im Falle einer Überdosierung ist der Patient auf Anzeichen oder Symptome von Nebenwirkungen zu überwachen und sofort eine geeignete symptomatische Behandlung einzuleiten.
Pharmakotherapeutische Gruppe: Andere Dermatika, Mittel zur Behandlung der Dermatitis, exkl. Corticosteroide, ATC-Code: D11AH10.
Wirkmechanismus
Lebrikizumab ist ein monoklonaler Immunglobulin (IgG4) Antikörper, der mit hoher Affinität an Interleukin(IL)‐13 bindet und die IL‐13-Signalübertragung über den heterodimeren Rezeptorkomplex IL‐4-Rezeptor-alpha(IL‐4Rα)/IL-13-Rezeptor-alpha-1(IL‐13Rα1) selektiv hemmt, wodurch die nachgelagerten Wirkungen von IL‐13 gehemmt werden. Es wird erwartet, dass die Hemmung der IL-13-Signaltransduktion bei Krankheiten von Nutzen sein wird, bei denen IL-13 einen zentralen Mediator in der Pathogenese der jeweiligen Krankheit darstellt. Lebrikizumab verhindert nicht die Bindung von IL-13 an den IL-13-Rezeptor alpha 2 (IL-13Rα2 oder Decoy-Rezeptor), was die Internalisierung von IL-13 in die Zelle ermöglicht.
Pharmakodynamische Wirkungen
In klinischen Studien zu Lebrikizumab reduzierte Lebrikizumab die Konzentrationen von Periostin im Serum, Gesamt-Immunglobulin E (IgE), CC-Chemokin-Ligand(CCL)17 [thymus and activation-regulated chemokine (TARC)], CCL18 [pulmonary and activation-regulated chemokine (PARC)] und CCL13 [monocyte chemotactic protein-4 (MCP-4)]. Die Abnahme der Typ‑2‑Entzündungsmediatoren liefert indirekte Hinweise auf eine Hemmung des IL-13-Signalwegs durch Lebrikizumab.
Immunogenität
Anti-Drug-Antikörper (ADA) wurden häufig nachgewiesen. Es wurden keine Hinweise auf Auswirkungen von ADA auf die Pharmakokinetik, Wirksamkeit oder Sicherheit festgestellt.
Klinische Wirksamkeit und Sicherheit
Erwachsene und Jugendliche mit atopischer Dermatitis
Die Wirksamkeit und Sicherheit von Lebrikizumab als Monotherapie (ADvocate-1, ADvocate-2) und mit begleitenden TCS (ADhere) wurden in drei randomisierten, doppelblinden, placebokontrollierten pivotalen Studien bei 1062 Erwachsenen und Jugendlichen (im Alter von 12 bis 17 Jahren und mit einem Gewicht von ≥ 40 kg) mit mittelschwerer bis schwerer atopischer Dermatitis, definiert als Eczema Area and Severity Index (EASI) ≥ 16, Investigator's Global Assessment (IGA) ≥ 3 und betroffener Körperoberfläche (BSA) ≥ 10 %, untersucht. Patienten, die in die drei Studien aufgenommen wurden, hatten zuvor ein unzureichendes Ansprechen auf topische Arzneimittel oder topische Behandlungen kamen aus medizinischen Gründen nicht in Frage.
In allen drei Studien erhielten die Patienten eine Anfangsdosis von 500 mg Lebrikizumab (zwei 250‑mg-Injektionen) in Woche 0 und 2, gefolgt von 250 mg alle zwei Wochen (Q2W) bis Woche 16 oder entsprechend Placebo im Verhältnis 2:1. In ADhere erhielten die Studienpatienten auch begleitend TCS mit niedriger bis mittlerer Wirkstärke oder TCI auf aktive Läsionen. Die Patienten durften nach Ermessen des Prüfarztes eine Rescue-Therapie zur Behandlung unzumutbarer Symptome der atopischen Dermatitis erhalten. Bei Patienten, die eine systemische Rescue-Therapie benötigten, wurde die Studienmedikation abgesetzt.
Patienten, die IGA 0 oder 1 oder eine mindestens 75%ige Reduktion des EASI (EASI 75) ohne Erhalt einer Rescue-Therapie erreichten, wurden erneut randomisiert, um verblindet bis zu 52 Wochen (i) Lebrikizumab 250 mg Q2W, (ii) Lebrikizumab 250 mg alle 4 Wochen (Q4W) oder (iii) entsprechend Placebo zu erhalten.
In ADvocate-1 und 2 wurden Patienten, die in Woche 16 IGA 0 oder 1 oder EASI 75 nicht erreichten oder die vor Woche 16 eine Rescue-Therapie erhielten, in einen Escape-Arm aufgenommen und erhielten unverblindet Lebrikizumab 250 mg Q2W bis Woche 52.
In ADvocate-1 und ADvocate-2 wurde den Patienten nach Abschluss der 52-wöchigen Studie bzw. nach Abschluss der 16-wöchigen Studie in ADhere die Möglichkeit angeboten, die Behandlung in einer separaten Langzeit-Verlängerungsstudie (ADjoin) fortzusetzen.
Endpunkte
In allen drei Studien waren die co-primären Endpunkte der Anteil der Patienten mit IGA 0 oder 1 („erscheinungsfrei“ oder „fast
erscheinungsfrei“) mit einer Reduktion um ≥ 2 Punkte gegenüber Baseline und der Anteil der Patienten, der eine Reduktion des EASI-Werts um mindestens 75 % (EASI 75) von Baseline bis Woche 16 erreichte. Zu den wichtigsten sekundären Endpunkten (für Multiplizität korrigiert) gehörten der Prozentsatz der Patienten, der eine Reduktion des EASI-Werts um mindestens 90 % (EASI 90), der Prozentsatz der Patienten, der eine Verbesserung um mindestens 4 Punkte gegenüber Baseline auf der numerischen Pruritus-Bewertungsskala (Pruritus-NRS), der Prozentsatz der Patienten, der eine Verbesserung um mindestens 4 Punkte gegenüber Baseline im Dermatology Life Quality Index (DLQI) und in der Beeinträchtigung des Schlafes durch Pruritus (Schlafverlust-Skala) erreichte. Letztere ist eine vom Patienten berichtete, tägliche Skala, die das Ausmaß der Beeinträchtigung des Schlafes durch Pruritus in der vorangegangenen Nacht auf einer 5-Punkte-Likert-Skala misst. Ein zusätzlicher sekundärer Endpunkt (nicht für Multiplizität korrigiert) war die Veränderung des POEM-Wertes (Patient Oriented Eczema Measure) gegenüber Baseline.
Patienten
Baseline-Merkmale
An den Monotherapiestudien ADvocate-1 und ADvocate-2 nahmen 424 bzw. 427 Patienten teil. Das Durchschnittsalter lag bei 35,8 Jahren, das Durchschnittsgewicht bei 77,1 kg, 49,9 % waren weiblich, 63,7 % waren weiß, 22,6 % asiatisch und 9,9 % schwarz, 12,0 % waren Jugendliche (12 bis 17 Jahre). Insgesamt hatten 61,5 % der Patienten zu Beginn der Studie einen IGA-Wert von 3 (mittelschwere atopische Dermatitis), 38,5 % der Patienten einen IGA-Wert von 4 (schwere atopische Dermatitis) und 54,8 % der Patienten hatten zuvor eine systemische Behandlung erhalten. Der mittlere EASI-Wert bei Baseline lag bei 29,6, der mittlere NRS-Wert für Pruritus bei 7,2 und der mittlere DLQI bei 15,5.
An der Studie mit begleitender TCS-Therapie ADhere nahmen 211 Patienten teil, das Durchschnittsalter lag bei 37,2 Jahren, das Durchschnittsgewicht bei 76,2 kg, 48,8 % waren weiblich, 61,6 % waren weiß, 14,7 % asiatisch und 13,3 % schwarz, 21,8 % waren Jugendliche. In dieser Studie hatten 69,2 % der Patienten zu Beginn der Studie einen IGA-Wert von 3 (mittelschwere atopische Dermatitis), 30,8 % der Patienten einen IGA-Wert von 4 (schwere atopische Dermatitis) und 47,4 % der Patienten hatten zuvor eine systemische Behandlung erhalten. Der mittlere EASI-Wert bei Baseline lag bei 27,3, der mittlere NRS-Wert für Pruritus bei 7,1 und der mittlere DLQI bei 14,4.
Klinisches Ansprechen
Monotherapiestudien (ADvocate-1 und ADvocate-2) – Induktionsphase, Woche 0–16
In ADvocate-1 und ADvocate-2 erreichten in Woche 16 unter Lebrikizumab 250 mg Q2W signifikant mehr Patienten IGA 0 oder 1 mit einer Verbesserung um ≥ 2 Punkte gegenüber Baseline, EASI 75, EASI 90, eine Verbesserung um ≥ 4 Punkte auf der Pruritus-NRS und im DLQI im Vergleich zu Placebo (siehe Tabelle 2).
In beiden Monotherapiestudien reduzierte Lebrikizumab den schwersten Pruritus während der letzten 24 Stunden im Vergleich zu Placebo, gemessen anhand der prozentualen Veränderung auf der Pruritus-NRS gegenüber Baseline bereits in der ersten Behandlungswoche im Vergleich zu Placebo. Die Verbesserung des Pruritus trat zusammen mit einer Besserung der Hautentzündung im Zusammenhang mit atopischer Dermatitis und der Lebensqualität auf.
Tabelle 2. Ergebnisse zur Wirksamkeit der Lebrikizumab-Monotherapie in ADvocate-1 und ADvocate-2 in Woche 16
ADvocate-1 |
ADvocate-2 |
|||
Woche 16 |
||||
|
Placebo N = 141 |
LEB N = 283 |
Placebo N = 146 |
LEB N = 281 |
|
IGA 0 oder 1, %a |
12,7 |
43,1*** |
10,8 |
33,2*** |
EASI 75, %b |
16,2 |
58,8*** |
18,1 |
52,1*** |
EASI 90, %b |
9,0 |
38,3*** |
9,5 |
30,7*** |
Pruritus-NRS (Verbesserung um ≥ 4 Punkte), %c |
13,0 |
45,9*** |
11,5 |
39,8*** |
DLQI (Erwachsene) (Verbesserung um ≥ 4 Punkte), %d |
33,8 |
75,6*** |
33,6 |
66,3*** |
LEB = Lebrikizumab; N = Anzahl an Patienten.
a Patienten mit einem IGA von 0 oder 1 („erscheinungsfrei“ oder „fast erscheinungsfrei“) mit einer Reduktion um ≥ 2 Punkte gegenüber Baseline auf einer IGA-Skala von 0–4.
b Patienten mit einer Reduktion des EASI um75 % beziehungsweise 90 % von Baseline bis Woche 16.
c Der Prozentsatz wird relativ zur Anzahl der Patienten mit einer Baseline-Pruritus-NRS-Wert ≥ 4 berechnet.
d Der Prozentsatz wird relativ zur Anzahl der Patienten mit einem Baseline-DLQI-Wert ≥ 4 berechnet.
*** p < 0,001 vs. Placebo
In den beiden Studien benötigten weniger Patienten, die auf Lebrikizumab randomisiert wurden, eine Rescue-Therapie (topische Kortikosteroide, systemische Kortikosteroide, Immunsuppressiva) als Patienten, die nach Randomisierung Placebo erhielten (14,7 % vs. 36,6 % über beide Studien hinweg).
Monotherapiestudien (ADvocate-1 und ADvocate-2) – Erhaltungszeitraum, Wochen 16–52
Zur Beurteilung der Aufrechterhaltung des Ansprechens wurden 157 Patienten aus ADvocate-1 und 134 Patienten aus ADvocate-2, die mit Lebrikizumab 250 mg Q2W behandelt wurden und in Woche 16 IGA 0 oder 1 bzw. EASI 75 ohne topische oder systemische Rescue-Therapie erreichten, erneut verblindet im Verhältnis 2:2:1 randomisiert. Die Patienten erhielten über weitere 36 Wochen verblindet entweder (i) Lebrikizumab 250 mg Q2W, (ii) Lebrikizumab 250 mg Q4W oder (iii) Placebo. Die Gesamtdauer der Studie betrug 52 Wochen (siehe Tabelle 3).
Tabelle 3. Ergebnisse zur Wirksamkeit der Lebrikizumab-Monotherapie in ADvocate-1 und ADvocate-2 in Woche 52 bei Patienten, die in ADvocate-1 und ADvovate-2 in Woche 16 auf die Behandlung ansprachen (gepoolte Analyse)
ADvocate-1 und ADvocate-2 (gepoolt) |
||
Woche 52 |
||
Placebod |
LEB 250 mg |
|
IGA 0 oder 1, %a |
47,9 |
76,9** |
EASI 75, %b |
66,4 |
81,7* |
EASI 90, %b |
41,9 |
66,4** |
Pruritus-NRS (Verbesserung um ≥ 4 Punkte), %c |
66,3 |
84,7 |
a Patienten mit IGA 0/1 mit einer Verbesserung um ≥ 2 Punkte gegenüber Baseline in Woche 16, die in Woche 52 weiterhin IGA 0/1 mit einer Verbesserung um ≥ 2 Punkte aufwiesen.
b Patienten, die in Woche 16 EASI 75 erreichten und in Woche 52 weiterhin EASI 75 aufwiesen, oder Patienten, die in Woche 16 EASI 75 erreichten und in Woche 52 EASI 90 aufwiesen.
c Der Prozentsatz wird relativ zur Anzahl der Patienten mit einer Baseline-Pruritus-NRS-Wert ≥ 4 berechnet.
d Patienten, die in Woche 16 auf Lebrikizumab 250 mg Q2W ansprachen (IGA 0 oder 1 oder EASI 75) und dann auf Placebo randomisiert wurden.
*p < 0,05; ** p < 0,01 versus Placebo.
Von den Patienten, die Lebrikizumab während der Induktionsphase erhielten und die unverblindete Behandlung mit Lebrikizumab 250 mg Q2W bis Woche 52 im Escape-Arm fortsetzten, erreichten 58 % EASI 75 und 28 % IGA 0 oder 1 mit einer Verbesserung um ≥ 2 Punkte gegenüber Baseline in Woche 52 von ADvocate-1 und ADvocate-2 (gepoolt).
Monotherapiestudien (ADvocate-1 und ADvocate-2) – Langzeit-Verlängerung (ADjoin), Woche 52–152
Die Aufrechterhaltung des klinischen Ansprechens wurde bei Patienten beurteilt, die in Woche 16 IGA 0 oder 1 oder EASI 75 ohne topische oder systemische Rescue-Therapie erreichten, die die 52 Wochen Lebrikizumab-Monotherapie in ADvocate-1 oder ADvocate-2 abschlossen und die die Behandlung in der 100-wöchigen Verlängerungsstudie ADjoin fortsetzten (Abbildung 1).
Abbildung 1. IGA 0 oder 1, EASI 75 und Pruritus-NRS in Woche 16 bei Behandlungsrespondern, die Woche 52 in ADvocate-1 und ADvocate-2 abschlossen und die in ADjoin* mit Lebrikizumab 250 mg Q4W behandelt wurden
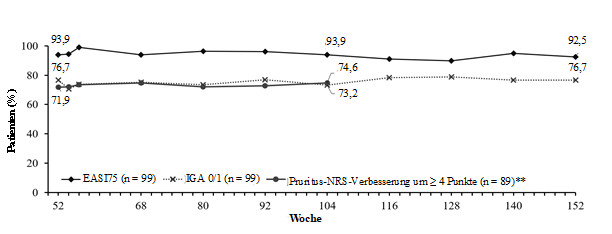
* Fehlende Daten wurden als multiple Imputation nach der MCMC-Methode (Markov Chain-Monte Carlo) ergänzt.
** Nur bei Patienten mit einem Pruritus-NRS-Wert ≥ 4 bei Baseline. Pruritus wurde nur im ersten Jahr der ADjoin-Studie beurteilt.
Studie mit begleitender TCS-Therapie (ADhere), Woche 0 bis 16
In ADhere erreichte von Baseline bis Woche 16 ein signifikant größerer Anteil der Patienten, die nach Randomisierung Lebrikizumab 250 mg Q2W + TCS erhielten, IGA 0 oder 1, EASI 75 und Verbesserungen um ≥ 4 Punkte auf der Pruritus-NRS und im DLQI als unter Placebo + TCS (siehe Tabelle 4).
Tabelle 4. Ergebnisse zur Wirksamkeit der Lebrikizumab-Kombinationstherapie mit TCS in Woche 16 in ADhere
ADhere |
||
Woche 16 |
||
|
Placebo + TCS N = 66 |
LEB |
|
IGA 0 oder 1, %a |
22,1 |
41,2* |
EASI 75, %b |
42,2 |
69,5*** |
EASI 90, %b |
21,7 |
41,2** |
Pruritus-NRS (Verbesserung um ≥ 4 Punkte), %c |
31,9 |
50,6* |
DLQI (Erwachsene) (Verbesserung um ≥ 4 Punkte), %d |
58,7 |
77,4* |
a Patienten mit einem IGA von 0 oder 1 („erscheinungsfrei“ oder „fast erscheinungsfrei“) mit einer Reduktion um ≥ 2 Punkte gegenüber Baseline auf einer IGA-Skala von 0–4.
b Patienten mit einer Reduktion des EASI um 75 % beziehungsweise 90 % von Baseline bis Woche 16.
c Der Prozentsatz wird relativ zur Anzahl der Patienten mit einem Baseline-Pruritus NRS-Wert ≥ 4 berechnet.
d Der Prozentsatz wird relativ zur Anzahl der Patienten mit einem Baseline-DLQI-Wert ≥ 4 berechnet.
* p < 0,05; **p < 0,01; ***p < 0,001 vs. Placebo.
In ADhere wendeten Patienten, die von Woche 0 bis 16 Lebrikizumab 250 mg Q2W+TCS erhielten, seltener TCS mit hoher Wirkstärke als Rescue-Therapie an, als Patienten, die Placebo + TCS erhielten (1,4 % bzw. 4,5 %).
Studie mit begleitender TCS-Therapie (ADhere) – Langzeit-Verlängerung (ADjoin), Woche 16–116
Das klinische Ansprechen wurde bei Patienten aufrechterhalten, die in der Studie ADhere in Woche 16 IGA 0 oder 1 oder EASI 75 erreichten und die die Behandlung mit Lebrikizumab 250 mg Q4W in der 100-wöchigen Verlängerungsstudie ADjoin fortsetzten.
Klinisches Ansprechen bei Patienten, die mit Ciclosporin nicht ausreichend kontrolliert sind, die es nicht vertragen oder für die es medizinisch nicht angeraten ist (ADvantage)
Die Studie ADvantage beurteilte die Wirksamkeit von Lebrikizumab im Vergleich zu Placebo bei Erwachsenen und Jugendlichen (im Alter von ≥ 12 bis < 18 Jahren und mit einem Körpergewicht von ≥ 40 kg) mit mittelschwerer bis schwerer atopischer Dermatitis, die gleichzeitig mit TCS behandelt wurden und die unter Ciclosporin nicht ausreichend kontrolliert waren oder für die Ciclosporin medizinisch nicht angeraten war.
Insgesamt wurden 331 Patienten aufgenommen und das Durchschnittsalter betrug 33,8 Jahre. 52,9 % waren männlich, 93,7 % Weiße und Jugendliche machten 11,8 % der Patientenpopulation aus. Zur Baseline betrugen der mittlere EASI 28,1 und der mittlere Pruritus-NRS-Wert 6,9. 38,7 % der Patienten hatten einen IGA-Wert von 4. Insgesamt hatten 53,2 % der Teilnehmer zuvor Ciclosporin erhalten (siehe Tabelle 5).
Tabelle 5. Wirksamkeitsergebnisse der Kombinationstherapie von Lebrikizumab und TCS in Woche 16 der Studie ADvantage
ADvantage |
||
Woche 16 |
||
Placebo + TCS |
LEB |
|
EASI 75, %a |
40,8 |
68,4*** |
IGA 0 oder 1, %b |
24,5 |
42,0** |
EASI 90, %c |
20,8 |
42,9*** |
Pruritus-NRS (Verbesserung um ≥ 4 Punkte), %d |
29,7 |
49,9* |
DLQI (Erwachsene) (Verbesserung um ≥ 4 Punkte), %e |
69,6 |
78,0 |
a Patienten mit einer Reduktion des EASI um 75 % von Baseline bis Woche 16.
b Patienten mit einem IGA von 0 oder 1 („erscheinungsfrei“ oder „fast erscheinungsfrei“) mit einer Reduktion um ≥ 2 Punkte gegenüber Baseline auf einer IGA-Skala von 0–4.
c Patienten mit einer Reduktion des EASI um 90 % von Baseline bis Woche 16.
d Der Prozentsatz wird relativ zur Anzahl der Patienten mit einem Baseline-Pruritus NRS-Wert ≥ 4 berechnet.
e Der Prozentsatz wird relativ zur Anzahl der Patienten mit einem Baseline-DLQI-Wert ≥ 4 berechnet.
* p < 0,05; ** p < 0,01; *** p < 0,001 versus Placebo. p-Werte für IGA 0 oder 1, EASI 90, Pruritus-NRS und DLQI sind nominal.
Weitere Patient-reported outcomes
In beiden Monotherapiestudien (ADvocate-1 und ADvocate-2) und in den TCS-Studien (ADhere und ADvantage) verbesserte
Lebrikizumab 250 mg Q2W signifikant den POEM-Wert und die Beeinträchtigung des Schlafs durch Pruritus (Schlafverlust-Skala) in Woche 16 im Vergleich zu Placebo.
Jugendliche (von 12 bis 17 Jahren)
In den Monotherapiestudien ADvocate-1 und ADvocate-2 lag das Durchschnittsalter der jugendlichen Patienten bei 14,6 Jahren, das Durchschnittsgewicht bei 68,2 kg und 56,9 % waren weiblich. In diesen Studien hatten 63,7 % einen IGA-Ausgangswert von 3 (mittelschwere atopische Dermatitis), 36,3 % hatten einen IGA-Ausgangswert (Baseline) von 4 (schwere atopische Dermatitis) und 47,1 % hatten zuvor eine systemische Behandlung erhalten. In der TCS-Studie (ADhere) lag das Durchschnittsalter der jugendlichen Patienten bei 14,6 Jahren, das Durchschnittsgewicht bei 62,2 kg und 50,0 % waren weiblich. In dieser Studie hatten 76,1 %
einen IGA-Ausgangswert von 3 (mittelschwere atopische Dermatitis), 23,9 % einen IGA-Ausgangswert von 4 (schwere atopische Dermatitis) und 23,9 % hatten zuvor eine systemische Behandlung erhalten.
Die Ergebnisse zur Wirksamkeit in Woche 16 bei jugendlichen Patienten sind in Tabelle 6 dargestellt.
Tabelle 6. Ergebnisse zur Wirksamkeit der Lebrikizumab-Monotherapie in ADvocate-1, ADvocate-2 und der Lebrikizumab-Kombinationstherapie mit TCS in ADhere der jugendlichen Patienten in Woche 16
ADvocate-1 |
ADvocate-2 |
ADhere |
||||
Woche 16 |
||||||
|
Placebo N = 18 |
LEB |
Placebo N = 17 |
LEB |
Placebo + TCS N = 14 |
LEB |
|
IGA 0 oder 1, %a |
22,2 |
48,6 |
5,9 |
44,1** |
28,6 |
57,3 |
EASI 75, %a |
22,2 |
62,2** |
12,0 |
61,7** |
57,1 |
88,0* |
EASI 90, %a |
16,7 |
45,9* |
6,1 |
34,3* |
28,6 |
55,1 |
Pruritus NRS (Verbesserung um ≥ 4-Punkte), %b |
22,8 |
54,3* |
0,3 |
42,1 |
13,8 |
45,8 |
a In Woche 16, Patienten mit einem IGA von 0 oder 1 („erscheinungsfrei“ oder „fast erscheinungsfrei“) mit einer Reduktion um ≥ 2 Punkte gegenüber Baseline auf einer IGA-Skala von 0–4 oder Reduktion des EASI um 75 % oder 90 % von Baseline bis Woche 16. | ||||||
Bei jugendlichen Patienten, die mit Lebrikizumab bzw. Lebrikizumab + TCS behandelt wurden, wurden klinisch bedeutsame Verbesserungen des Schweregrads der Erkrankung erzielt und das Ansprechen blieb bis Woche 52 erhalten. Zusätzliche Daten aus der einarmigen ADore-Studie mit Lebrikizumab bei 206 Jugendlichen untermauern die Wirksamkeit von Lebrikizumab bei jugendlichen Patienten bis zu 52 Behandlungswochen.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Lebrikizumab eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in atopischer Dermatitis gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Resorption
Nach einer subkutanen Dosis von 250 mg Lebrikizumab wurden die maximalen Serumkonzentrationen etwa 7 bis 8 Tage nach der Dosis erreicht.
Nach den Aufsättigungsdosen von 500 mg in Woche 0 und Woche 2 wurden mit der ersten Dosis von 250 mg Q2W in Woche 4 Steady-State-Serumkonzentrationen erreicht.
Basierend auf einer populationspharmakokinetischen (PK) Analyse betrugen die voraussichtlichen Steady-State-Talkonzentrationen (CTal,ss) nach der subkutanen Gabe von Lebrikizumab 250 mg Q2W und Q4W bei Patienten mit atopischer Dermatitis (Median und 5.-95. Perzentile) 87 (46–159) µg/ml bzw. 36 (18–68) µg/ml.
Basierend auf einer Populations-PK-Analyse wurde die absolute Bioverfügbarkeit auf 86 % geschätzt. Die Injektionsstelle hatte
keinen signifikanten Einfluss auf die Resorption von Lebrikizumab.
Verteilung
Basierend auf einer Populations-PK-Analyse betrug das Gesamtverteilungsvolumen im Steady-State 5,14 l.
Biotransformation
Da Lebrikizumab ein Protein ist, wurden keine Metabolisierungsstudien durchgeführt. Es wird erwartet, dass Lebrikizumab über
katabole Prozesse auf die gleiche Weise wie endogenes IgG zu kleinen Peptiden und einzelnen Aminosäuren abgebaut wird.
Elimination
In der Populations-PK-Analyse betrug die Clearance 0,154 l/Tag und war unabhängig von der Dosis. Die mittlere Eliminationshalbwertszeit betrug etwa 24,5 Tage.
Linearität/Nicht-Linearität
Lebrikizumab zeigte eine lineare Pharmakokinetik mit dosisproportionalem Anstieg der Exposition über einen Dosisbereich von 37,5 bis 500 mg bei Verabreichung als subkutane Injektion an AD-Patienten oder gesunde Freiwillige.
Besondere Patientengruppen
Geschlecht, Alter und ethnische Zugehörigkeit
Geschlecht, Alter (12 bis 93 Jahre) und ethnische Zugehörigkeit hatten keinen signifikanten Einfluss auf die Pharmakokinetik von Lebrikizumab.
Eingeschränkte Nieren- und Leberfunktion
Es wurden keine spezifischen klinisch-pharmakologischen Studien zur Beurteilung der Auswirkungen einer eingeschränkten
Nieren- oder Leberfunktion auf die Pharmakokinetik von Lebrikizumab durchgeführt. Es wird nicht erwartet, dass Lebrikizumab als monoklonaler Antikörper in nennenswertem Umfang über die Nieren oder die Leber eliminiert wird. Populations‑PK‑Analysen zeigen, dass Marker der Nieren- oder Leberfunktion die Pharmakokinetik von Lebrikizumab nicht beeinflussten.
Körpergewicht
Die Lebrikizumab-Exposition war bei Patienten mit höherem Körpergewicht niedriger, dies hatte jedoch keine bedeutsamen Auswirkungen auf die klinische Wirksamkeit.
Kinder und Jugendliche
Basierend auf der Populations-PK-Analyse wiesen Jugendliche im Alter von 12 bis 17 Jahren mit atopischer Dermatitis geringfügig höhere Lebrikizumab-Serum-Talkonzentrationen auf als Erwachsene, was mit ihrem geringeren Körpergewicht zusammenhing.
Basierend auf den konventionellen Studien zur Toxizität bei wiederholter Gabe (einschließlich sicherheitspharmakologischer Endpunkte) und Reproduktions- und Entwicklungstoxizität, lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Das mutagene Potenzial von Lebrikizumab wurde nicht untersucht; es ist jedoch nicht zu erwarten, dass monoklonale Antikörper die DNA oder Chromosomen verändern.
Mit Lebrikizumab wurden keine Studien zur Karzinogenität durchgeführt. Die Auswertung der verfügbaren Erkenntnisse in Bezug auf IL-13-Hemmung und der tierexperimentellen Toxikologiedaten zu Lebrikizumab deutet nicht auf ein karzinogenes Potenzial für Lebrikizumab hin.
Bei geschlechtsreifen Affen wurden nach langfristigen intravenösen (Weibchen) oder subkutanen (Männchen) Behandlungen mit Lebrikizumab keine Auswirkungen auf Fruchtbarkeitsparameter beobachtet. Lebrikizumab hatte keine Auswirkungen auf die
embryo-fötale oder postnatale Entwicklung.
Histidin
Essigsäure 99 % (E 260)
Saccharose
Polysorbat 20 (E 432)
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
3 Jahre
Nach der Entnahme aus dem Kühlschrank muss Ebglyss innerhalb von 7 Tagen (bis zu 30 °C) verwendet oder verworfen werden. Nach der Lagerung außerhalb des Kühlschranks nicht wieder in den Kühlschrank geben.
Im Kühlschrank lagern (2 °C – 8 °C).
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ebglyss 250 mg Injektionslösung in einer Fertigspritze
2 ml Lösung in einer 2,25 ml-Klarglas-Fertigspritze (Glastyp 1) mit kleinem Rundflansch mit integrierter („staked“) 27-Gauge-Dünnwand-Kanüle (12,7 mm) aus rostfreiem Stahl, verschlossen mit einem laminierten Kolben aus Bromobutylelastomer, versehen mit einem starren Nadelschutz und montiert in einer passiven Sicherheitsvorrichtung.
Packungsgrößen:
1 Fertigspritze
2 Fertigspritzen
3 Fertigspritzen
Mehrfachpackung mit 4 (2 Packungen mit je 2) Einzeldosis-Fertigspritzen
Mehrfachpackung mit 5 (5 Packungen mit je 1) Einzeldosis-Fertigspritzen
Mehrfachpackung mit 6 (3 Packungen mit je 2) Einzeldosis-Fertigspritzen
Ebglyss 250 mg Injektionslösung im Fertigpen
2 ml Lösung in einer 2,25 ml-Klarglas-Spritze (Glastyp 1) in einem Fertigpen mit extrakleinem Rundflansch mit integrierter („staked“) 27-Gauge-Dünnwand-Kanüle (8 mm) aus rostfreiem Stahl, verschlossen mit einem laminierten Kolben aus Bromobutylelastomer und versehen mit einem starren Nadelschutz.
Packungsgrößen:
1 Fertigpen
2 Fertigpens
3 Fertigpens
Mehrfachpackung mit 4 (2 Packungen mit je 2) Einzeldosis-Fertigpens
Mehrfachpackung mit 5 (5 Packungen mit je 1) Einzeldosis-Fertigpens
Mehrfachpackung mit 6 (3 Packungen mit je 2) Einzeldosis-Fertigpens
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Ausführliche Anweisungen für die Verabreichung von Ebglyss in einer Fertigspritze oder in einem Fertigpen sind am Ende der
Packungsbeilage angeführt.
Die Lösung sollte eine klare bis schillernde, farblose bis leicht gelbliche oder leicht bräunliche Lösung und frei von sichtbaren Partikeln sein. Wenn die Lösung trüb oder verfärbt ist oder sichtbare Partikel enthält, darf sie nicht verwendet werden.
Die Fertigspritze oder den Fertigpen vor hohen Temperaturen und direktem Sonnenlicht schützen. Nicht schütteln.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Almirall, S.A.
Ronda General Mitre, 151
08022 Barcelona
Spanien
EU/1/23/1765/001
EU/1/23/1765/002
EU/1/23/1765/003
EU/1/23/1765/004
EU/1/23/1765/005
EU/1/23/1765/006
EU/1/23/1765/007
EU/1/23/1765/008
EU/1/23/1765/009
EU/1/23/1765/010
EU/1/23/1765/011
EU/1/23/1765/012
Datum der Erteilung der Zulassung: 16. November 2023
12/2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.