
Tecentriq® 1 875 mg Injektionslösung
Eine Durchstechflasche mit 15 ml Injektionslösung enthält 1 875 mg Atezolizumab.
Jeder ml Lösung enthält 125 mg Atezolizumab.
Atezolizumab ist ein im Fc‑Teil modifizierter, humanisierter monoklonaler Immunglobulin(Ig)G1-anti‑PD‑L1(programmed death ligand 1)‑Antikörper, der in Ovarialzellen des chinesischen Hamsters mittels rekombinanter Desoxyribonukleinsäure(DNA)‑Technologie hergestellt wird.
Sonstiger Bestandteil mit bekannter Wirkung
Jede 1 875-mg-Durchstechflasche Tecentriq enthält 9 mg Polysorbat 20.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektionslösung.
Klare, farblose bis leicht gelbliche Flüssigkeit. Die Lösung hat einen pH-Wert von 5,5 – 6,1 und eine Osmolalität von 359 – 459 mOsm/kg.
Urothelkarzinom (urothelial carcinoma, UC)
Tecentriq als Monotherapie wird angewendet bei erwachsenen Patienten zur Behandlung des lokal fortgeschrittenen oder metastasierten UC
nach vorheriger platinhaltiger Chemotherapie oder
die für eine Behandlung mit Cisplatin als ungeeignet angesehen werden, und deren Tumoren eine PD‑L1‑Expression ≥ 5 % aufweisen (siehe Abschnitt 5.1).
Nicht-kleinzelliges Lungenkarzinom (non‑small cell lung cancer, NSCLC) im Frühstadium
Tecentriq als Monotherapie wird angewendet zur adjuvanten Behandlung des NSCLC nach vollständiger Resektion und platinbasierter Chemotherapie bei erwachsenen Patienten mit hohem Risiko für ein Rezidiv und deren Tumoren eine PD‑L1‑Expression auf ≥ 50 % der Tumorzellen (tumour cells, TC) aufweisen und kein EGFR(epidermal growth factor receptor, epidermaler Wachstumsfaktorrezeptor)‑mutiertes oder ALK(anaplastische-Lymphomkinase)‑positives NSCLC haben (siehe Abschnitt 5.1 zu den Auswahlkriterien).
Fortgeschrittenes NSCLC
Tecentriq wird angewendet in Kombination mit Bevacizumab, Paclitaxel und Carboplatin bei erwachsenen Patienten zur Erstlinienbehandlung des metastasierten NSCLC mit nicht-plattenepithelialer Histologie. Bei Patienten mit EGFR‑mutiertem oder ALK‑positivem NSCLC ist Tecentriq in Kombination mit Bevacizumab, Paclitaxel und Carboplatin nur nach Versagen der entsprechenden zielgerichteten Therapien anzuwenden (siehe Abschnitt 5.1).
Tecentriq wird angewendet in Kombination mit nab-Paclitaxel und Carboplatin zur Erstlinienbehandlung des metastasierten NSCLC mit nicht-plattenepithelialer Histologie bei erwachsenen Patienten, die kein EGFR‑mutiertes oder ALK-positives NSCLC haben (siehe Abschnitt 5.1).
Tecentriq als Monotherapie wird angewendet bei erwachsenen Patienten zur Erstlinienbehandlung des metastasierten NSCLC, deren Tumoren eine PD-L1-Expression ≥ 50 % der TC oder ≥ 10 % bei tumorinfiltrierenden Immunzellen (immune cells, IC) aufweisen und die kein EGFR-mutiertes oder ALK‑positives NSCLC haben (siehe Abschnitt 5.1).
Tecentriq als Monotherapie wird angewendet bei erwachsenen Patienten zur Erstlinienbehandlung des fortgeschrittenen NSCLC, die für eine platinbasierte Therapie ungeeignet sind (siehe Abschnitt 5.1 zu den Auswahlkriterien).
Tecentriq als Monotherapie wird angewendet bei erwachsenen Patienten zur Behandlung des lokal fortgeschrittenen oder metastasierten NSCLC nach vorheriger Chemotherapie. Patienten mit EGFR‑mutiertem oder ALK‑positivem NSCLC sollen vor der Therapie mit Tecentriq zudem auch bereits entsprechende zielgerichtete Therapien erhalten haben (siehe Abschnitt 5.1).
Kleinzelliges Lungenkarzinom (small cell lung cancer, SCLC)
Tecentriq wird angewendet in Kombination mit Carboplatin und Etoposid bei erwachsenen Patienten zur Erstlinienbehandlung des SCLC im fortgeschrittenen Stadium (extensive-stage small cell lung cancer, ES-SCLC) (siehe Abschnitt 5.1).
Triple-negatives Mammakarzinom (triple-negative breast cancer, TNBC)
Tecentriq wird angewendet in Kombination mit nab-Paclitaxel bei erwachsenen Patienten zur Behandlung des nicht resezierbaren lokal fortgeschrittenen oder metastasierten TNBC, deren Tumoren eine PD-L1-Expression ≥ 1 % aufweisen und die keine vorherige Chemotherapie zur Behandlung der metastasierten Erkrankung erhalten haben.
Hepatozelluläres Karzinom (hepatocellular carcinoma, HCC)
Tecentriq wird angewendet in Kombination mit Bevacizumab bei erwachsenen Patienten zur Behandlung des fortgeschrittenen oder nicht resezierbaren HCC, die keine vorherige systemische Behandlung erhalten haben (siehe Abschnitt 5.1).
Tecentriq darf nur unter Aufsicht von Ärzten mit Erfahrung in der Krebstherapie eingeleitet und angewendet werden.
Patienten, die aktuell Atezolizumab intravenös erhalten, können auf die Injektionslösung von Tecentriq umgestellt werden und umgekehrt.
PD-L1-Testung bei Patienten mit UC oder TNBC oder NSCLC
Tecentriq als Monotherapie
Falls im Anwendungsgebiet angegeben, sind die Patienten für eine Behandlung mit Tecentriq basierend auf der PD-L1-Expression auf dem Tumor auszuwählen, die mittels eines validierten Tests bestätigt wurde (siehe Abschnitte 4.1 und 5.1).
Tecentriq als Kombinationstherapie
Patienten mit nicht vorbehandeltem TNBC sind für eine Behandlung anhand der Tumor-PD-L1-Expression auszuwählen, die mittels eines validierten Tests bestätigt wurde (siehe Abschnitt 5.1).
Dosierung
Die empfohlene Dosis von Tecentriq Injektionslösung beträgt 1 875 mg alle drei Wochen, wie in Tabelle 1 dargestellt.
Wird Tecentriq als Kombinationstherapie verabreicht, beachten Sie für die Arzneimittel der Kombinationstherapie bitte die jeweiligen Fachinformationen (siehe auch Abschnitt 5.1).
Tabelle 1: Empfohlene Dosierung für Tecentriq, subkutan verabreicht
Indikation |
Empfohlene Dosierung und Schema |
Dauer der Behandlung |
Tecentriq Monotherapie |
||
Erstlinien-UC |
1 875 mg alle 3 Wochen |
Bis eine Krankheitsprogression oder eine nicht kontrollierbare Toxizität auftritt. |
Metastasiertes Erstlinien-NSCLC | ||
Erstlinien-NSCLC, Patienten ungeeignet für platinbasierte Therapie | ||
NSCLC im Frühstadium |
1 875 mg alle 3 Wochen |
Für die Dauer von 1 Jahr, außer es kommt zu einem Rezidiv oder zum Auftreten inakzeptabler Toxizität. Eine Behandlungsdauer von über einem Jahr wurde nicht untersucht. |
Zweitlinien-UC |
1 875 mg alle 3 Wochen |
Bis zum Verlust des klinischen Nutzens oder bis eine nicht kontrollierbare Toxizität auftritt. |
Zweitlinien-NSCLC | ||
Tecentriq Kombinationstherapie | ||
Erstlinien-nicht-Plattenepithel-NSCLC |
Induktions- und Erhaltungsphasen: Induktionsphase für Kombinationspartner (vier oder sechs Zyklen): Erhaltungsphase (ohne Chemotherapie): Bevacizumab alle drei Wochen. |
Bis eine Krankheitsprogression oder eine nicht kontrollierbare Toxizität auftritt. Atypisches Ansprechen (d. h. eine initiale Krankheitsprogression gefolgt von einer Tumorregression) wurde unter fortgeführter Behandlung mit Tecentriq nach Krankheitsprogression beobachtet. Eine Weiterbehandlung über eine Krankheitsprogression hinaus kann nach Ermessen des Arztes in Erwägung gezogen werden. |
Erstlinien-nicht-Plattenepithel-NSCLC |
Induktions- und Erhaltungsphasen: Induktionsphase für Kombinationspartner (vier oder sechs Zyklen): |
Bis eine Krankheitsprogression oder eine nicht kontrollierbare Toxizität auftritt. Atypisches Ansprechen (d. h. eine initiale Krankheitsprogression gefolgt von einer Tumorregression) wurde unter fortgeführter Behandlung mit Tecentriq nach Krankheitsprogression beobachtet. Eine Weiterbehandlung über eine Krankheitsprogression hinaus kann nach Ermessen des Arztes in Erwägung gezogen werden. |
Erstlinien-ES-SCLC mit Carboplatin und Etoposid |
Induktions- und Erhaltungsphasen: Induktionsphase für Kombinationspartner (vier Zyklen): |
Bis eine Krankheitsprogression oder eine nicht kontrollierbare Toxizität auftritt. Atypisches Ansprechen (d. h. eine initiale Krankheitsprogression gefolgt von einer Tumorregression) wurde unter fortgeführter Behandlung mit Tecentriq nach Krankheitsprogression beobachtet. Eine Weiterbehandlung über eine Krankheitsprogression hinaus kann nach Ermessen des Arztes in Erwägung gezogen werden. |
Erstlinien-nicht resezierbares lokal fortgeschrittenes oder metastasiertes TNBC mit nab-Paclitaxel |
1 875 mg alle 3 Wochen |
Bis eine Krankheitsprogression oder eine nicht kontrollierbare Toxizität auftritt. |
Fortgeschrittenes oder nicht resezierbares HCC mit Bevacizumab |
1 875 mg alle 3 Wochen |
Bis zum Verlust des klinischen Nutzens oder bis eine nicht kontrollierbare Toxizität auftritt. |
Verspätete oder versäumte Dosen
Wenn eine geplante Dosis von Tecentriq versäumt wurde, ist diese so schnell wie möglich nachzuholen. Das Anwendungsschema muss so angepasst werden, dass das entsprechende Intervall zwischen den Dosen eingehalten wird.
Dosisanpassung während der Behandlung
Eine Dosisreduktion von Tecentriq wird nicht empfohlen.
Dosisverzögerung oder Absetzen (siehe auch Abschnitte 4.4 und 4.8)
Tabelle 2: Hinweise zur Dosisanpassung von Tecentriq
Immunvermittelte Nebenwirkung |
Schweregrad |
Anpassung der Behandlung |
Pneumonitis |
Grad 2 |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, wenn sich der Grad der Nebenwirkung innerhalb von 12 Wochen auf Grad 0 oder Grad 1 verbessert und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde |
Grad 3 oder 4 |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Hepatitis bei Patienten ohne HCC |
Grad 2: oder Bilirubinwert im Blut > 1,5 bis 3 x ULN) |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, wenn sich der Grad der Nebenwirkung innerhalb von 12 Wochen auf Grad 0 oder Grad 1 verbessert und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde |
|
Grad 3 oder 4: oder Bilirubinwert im Blut > 3 x ULN) |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Hepatitis bei Patienten mit HCC |
Wenn AST/ALT bei Behandlungsbeginn im Normbereich und Anstieg auf > 3 x bis ≤ 10 x ULN oder Wenn AST/ALT bei Behandlungsbeginn > 1 x bis ≤ 3 x ULN und Anstieg auf > 5 x bis ≤ 10 x ULN oder Wenn AST/ALT bei Behandlungsbeginn > 3 x bis ≤ 5 x ULN und Anstieg auf > 8 x bis ≤ 10 x ULN |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, wenn sich der Grad der Nebenwirkung innerhalb von 12 Wochen auf Grad 0 oder Grad 1 verbessert und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde |
|
Wenn AST-/ALT-Anstieg auf > 10 x ULN oder Gesamtbilirubin Anstieg auf > 3 x ULN |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Kolitis |
Grad 2 oder 3 Diarrhö (Anstieg um ≥ 4 Stuhlgänge/Tag gegenüber Behandlungsbeginn) oder Symptomatische Kolitis |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, wenn sich der Grad der Nebenwirkung innerhalb von 12 Wochen auf Grad 0 oder Grad 1 verbessert und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde |
Grad 4 Diarrhö oder Kolitis (lebensbedrohlich; unverzügliche Intervention indiziert) |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Hypothyreose oder Hyperthyreose |
Symptomatisch |
Behandlung mit Tecentriq unterbrechen Hypothyreose: Hyperthyreose: |
Nebenniereninsuffizienz |
Symptomatisch |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, sobald sich die Symptome innerhalb von 12 Wochen auf Grad 0 oder Grad 1 verbessert haben und die Dosis von Corticosteroiden auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde und der Patient unter der Ersatztherapie stabil ist |
Hypophysitis |
Grad 2 oder 3 |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, sobald sich die Symptome innerhalb von 12 Wochen auf Grad 0 oder Grad 1 verbessert haben und die Dosis von Corticosteroiden auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde und der Patient unter der Ersatztherapie stabil ist |
Grad 4 |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Diabetes mellitus Typ 1 |
Grad 3 oder 4 Hyperglykämie (Nüchternglucose von > 250 mg/dl oder 13,9 mmol/l) |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, sobald durch eine Insulin‑Ersatztherapie eine Kontrolle des Stoffwechsels erreicht wurde |
Ausschlag/ schwerwiegende unerwünschte Reaktionen der Haut |
Grad 3 oder Verdacht auf Stevens-Johnson-Syndrom (SJS) oder toxische epidermale Nekrolyse (TEN)1 |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, sobald sich die Symptome innerhalb von 12 Wochen auf Grad 0 oder Grad 1 verbessert haben und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde |
|
Grad 4 oder bestätigtes Stevens-Johnson-Syndrom (SJS) oder bestätigte toxische epidermale Nekrolyse (TEN)1 |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Myastheniesyndrom/ |
Grad 1 oder 2 Gesichtsparese |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, wenn das Ereignis vollständig abgeklungen ist. Wenn das Ereignis während der Unterbrechung der Behandlung mit Tecentriq nicht vollständig abgeklungen ist, Behandlung mit Tecentriq dauerhaft absetzen |
|
Alle Grade Myastheniesyndrom/Myasthenia gravis, Guillain Barré Syndrom und Meningoenzephalitis oder Grad 3 oder 4 Gesichtsparese |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Myelitis |
Grad 2, 3 oder 4 |
Behandlung mit Tecentriq dauerhaft absetzen |
Pankreatitis |
Grad 3 oder 4 Erhöhung der Serumamylase‑ oder Lipase‑Spiegel (> 2 x ULN) |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, sobald sich die Serumamylase‑ und Lipase‑Spiegel innerhalb von 12 Wochen wieder auf Grad 0 oder Grad 1 verbessert haben oder die Symptome der Pankreatitis abgeklungen sind, und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde |
Grad 4 oder jeglicher Grad rezidivierender Pankreatitis |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Myokarditis |
Grad 2 oder höher |
Behandlung mit Tecentriq dauerhaft absetzen |
Nephritis |
Grad 2 |
Behandlung mit Tecentriq unterbrechen Die Behandlung kann wieder aufgenommen werden, wenn sich der Grad der Nebenwirkung innerhalb von 12 Wochen auf Grad 0 oder Grad 1 verbessert und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde |
Grad 3 oder 4 |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Myositis |
Grad 2 oder 3 |
Behandlung mit Tecentriq unterbrechen |
Grad 4 oder Grad 3 rezidivierender Myositis |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Erkrankungen des Perikards |
Grad 1 Perikarditis |
Behandlung mit Tecentriq unterbrechen2 |
Grad 2 oder höher |
Behandlung mit Tecentriq dauerhaft absetzen |
|
Hämophagozytische Lymphohistiozytose |
Verdacht auf hämophagozytische Lymphohistiozytose1 |
Behandlung mit Tecentriq dauerhaft absetzen |
Andere immunvermittelte Nebenwirkungen |
Grad 2 oder 3 |
Behandlung unterbrechen bis sich die Nebenwirkungen innerhalb von 12 Wochen auf Grad 0 - 1 verbessert haben und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde |
Grad 4 oder wiederkehrende Grad 3 |
Behandlung mit Tecentriq dauerhaft absetzen (außer bei Endokrinopathien, die mit Hormonsubstitutionstherapie kontrolliert werden können) |
|
Sonstige Nebenwirkungen |
Schweregrad |
Anpassung der Behandlung |
Infusionsbedingte Reaktionen |
Grad 1 oder 2 |
Injektionsgeschwindigkeit verringern oder die Injektion unterbrechen. Die Behandlung kann wieder aufgenommen werden, sobald das Ereignis abgeklungen ist |
Grad 3 oder 4 |
Behandlung mit Tecentriq dauerhaft absetzen |
ALT = Alaninaminotransferase, AST = Aspartataminotransferase, ULN = upper limit of normal (obere Normalgrenze)
Anmerkung: Die Toxizität ist gemäß der aktuellen Version der Common Terminology Criteria for Adverse Events (NCI‑CTCAE) des National Cancer Instituts einzustufen.
1 Unabhängig vom Schweregrad
2 Eine gründliche kardiologische Untersuchung ist durchzuführen, um die Ätiologie zu bestimmen und um entsprechend zu behandeln.
Spezielle Patientengruppen
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Tecentriq bei Kindern und Jugendlichen unter 18 Jahren ist nicht erwiesen. Zurzeit vorliegende Daten zur intravenösen Anwendung von Tecentriq sind in den Abschnitten 4.8, 5.1 und 5.2 beschrieben, eine Dosierungsempfehlung kann jedoch nicht gegeben werden.
Ältere Patienten
Gemäß den Ergebnissen einer populationspharmakokinetischen Analyse ist bei Patienten im Alter von ≥ 65 Jahren keine Dosisanpassung von Tecentriq erforderlich (siehe Abschnitte 4.8 und 5.1).
Patienten asiatischer Herkunft
Aufgrund von erhöhten hämatologischen Toxizitäten, die in der Studie IMpower150 bei asiatischen Patienten beobachtet wurden, wird empfohlen, mit einer Anfangsdosis von 175 mg/m2 Paclitaxel alle drei Wochen zu beginnen.
Nierenfunktionsstörung
Gemäß den Ergebnissen einer populationspharmakokinetischen Analyse ist bei Patienten mit leichter bis mäßiger Nierenfunktionsstörung keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Die Daten zu Patienten mit schwerer Nierenfunktionsstörung sind zu begrenzt, um Rückschlüsse auf diese Population zuzulassen.
Leberfunktionsstörung
Gemäß den Ergebnissen einer populationspharmakokinetischen Analyse ist bei Patienten mit leichter oder mäßiger Leberfunktionsstörung keine Dosisanpassung erforderlich. Tecentriq wurde bisher nicht bei Patienten mit schwerer Leberfunktionsstörung untersucht (siehe Abschnitt 5.2).
ECOG(Eastern Cooperative Oncology Group)‑Performance‑Status ≥ 2
Patienten mit einem ECOG‑Performance‑Status ≥ 2 waren aus den klinischen Prüfungen zu TNBC, ES-SCLC, Zweitlinien‑UC und HCC ausgeschlossen (siehe Abschnitte 4.4 und 5.1).
Art der Anwendung
Es ist wichtig, die Angaben auf dem Arzneimittel zu überprüfen, um sicherzustellen, dass der Patient die richtige, ihm verschriebene Darreichungsform (intravenös oder subkutan) erhält.
Tecentriq Injektionslösung ist nicht zur intravenösen Anwendung vorgesehen und darf nur als subkutane Injektion verabreicht werden.
Vor der Anwendung ist Tecentriq Injektionslösung aus der gekühlten Lagerung zu entnehmen, damit die Lösung Raumtemperatur annehmen kann. Hinweise zur Anwendung und Handhabung der Tecentriq Injektionslösung vor der Anwendung, siehe Abschnitt 6.6.
Verabreichen Sie 15 ml Tecentriq Injektionslösung innerhalb von etwa 7 Minuten in den Oberschenkel. Die Verwendung eines subkutanen Infusionssets (z. B. Flügelkanüle/Butterfly) wird empfohlen. Verabreichen Sie dem Patienten NICHT das im Schlauch verbliebene Restvolumen.
Die Injektionsstellen sind abwechselnd nur zwischen dem linken und rechten Oberschenkel auszuwählen. Neue Injektionen sind mindestens 2,5 cm von einer vorherigen Injektionsstelle entfernt und niemals in Bereiche zu injizieren, an denen die Haut gerötet, geprellt, empfindlich oder verhärtet ist. Während der Behandlung mit Tecentriq Injektionslösung sind andere Arzneimittel zur subkutanen Anwendung vorzugsweise an anderen Stellen zu injizieren.
Überempfindlichkeit gegen Atezolizumab oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Immunvermittelte Nebenwirkungen
Die meisten immunvermittelten Nebenwirkungen während der Behandlung mit Atezolizumab waren mit Unterbrechung der Behandlung mit Atezolizumab und Einleitung einer Corticosteroid-Behandlung und/oder unterstützender Therapie reversibel. Es wurden immunvermittelte Nebenwirkungen beobachtet, die mehr als ein Körpersystem betrafen. Immunvermittelte Nebenwirkungen durch Atezolizumab können auch nach der letzten Dosis Atezolizumab auftreten.
Bei Verdacht auf immunvermittelte Nebenwirkungen müssen gründliche Untersuchungen zur Bestätigung der Ätiologie oder zum Ausschluss anderer Ursachen vorgenommen werden. Abhängig vom Schweregrad der Nebenwirkung ist die Behandlung mit Atezolizumab zu unterbrechen und Corticosteroide sind zu verabreichen. Nach Besserung auf ≤ Grad 1 sind die Corticosteroide über einen Zeitraum von ≥ 1 Monat auszuschleichen. Basierend auf limitierten Daten aus klinischen Prüfungen von Patienten, deren immunvermittelte Nebenwirkungen nicht mit einer systemischen Corticosteroid-Anwendung unter Kontrolle gebracht werden konnten, kann die Gabe anderer systemischer Immunsuppressiva in Betracht gezogen werden.
Atezolizumab muss dauerhaft abgesetzt werden, wenn eine immunvermittelte Nebenwirkung Grad 3 erneut auftritt sowie bei jeder immunvermittelten Nebenwirkung Grad 4, außer bei Endokrinopathien, die mit Hormonsubstitutionstherapie kontrolliert werden können (siehe Abschnitte 4.2 und 4.8).
Bei Patienten mit vorbestehender Autoimmunerkrankung (AIE) deuten Daten aus Beobachtungsstudien darauf hin, dass das Risiko immunvermittelter Nebenwirkungen nach einer Therapie mit Immun-Checkpoint-Inhibitoren im Vergleich zu dem Risiko bei Patienten ohne vorbestehende AIE erhöht sein kann. Darüber hinaus traten häufig Schübe der zugrunde liegenden AIE auf, die jedoch in der Mehrzahl leicht und beherrschbar waren.
Immunvermittelte Pneumonitis
Fälle von Pneumonitis, einschließlich tödlicher Fälle, wurden in klinischen Prüfungen mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind auf Anzeichen und Symptome einer Pneumonitis zu überwachen, und andere Ursachen als eine immunvermittelte Pneumonitis sind auszuschließen.
Die Behandlung mit Atezolizumab ist bei Pneumonitis von Grad 2 zu unterbrechen und eine Behandlung mit 1 – 2 mg/kg Körpergewicht (KG)/Tag Prednison oder eines Äquivalents ist einzuleiten. Wenn sich die Symptome auf ≤ Grad 1 verbessern, sind die Corticosteroide über einen Zeitraum von ≥ 1 Monat auszuschleichen. Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, wenn sich das Ereignis innerhalb von 12 Wochen auf ≤ Grad 1 verbessert hat und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde. Bei Auftreten einer Pneumonitis von Grad 3 oder 4 muss die Behandlung mit Atezolizumab dauerhaft abgesetzt werden.
Immunvermittelte Hepatitis
Fälle von Hepatitis, einige mit tödlichem Ausgang, wurden in klinischen Prüfungen mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind auf Anzeichen und Symptome einer Hepatitis zu überwachen.
Aspartataminotransferase (AST), Alaninaminotransferase (ALT) und Bilirubin sind vor Behandlungsbeginn, während der Behandlung mit Atezolizumab in regelmäßigen Abständen und wenn klinisch indiziert zu überwachen.
Bei Patienten ohne HCC ist die Behandlung mit Atezolizumab zu unterbrechen, wenn Ereignisse von Grad 2 (ALT oder AST > 3 bis 5 x ULN oder Bilirubin im Blut > 1,5 bis 3 x ULN) länger als 5 bis 7 Tage anhalten, und eine Behandlung mit 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents ist einzuleiten. Wenn sich das unerwünschte Ereignis auf ≤ Grad 1 verbessert, sind die Corticosteroide über einen Zeitraum von ≥ 1 Monat auszuschleichen.
Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, wenn sich das Ereignis innerhalb von 12 Wochen auf ≤ Grad 1 verbessert hat und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde. Die Behandlung mit Atezolizumab ist bei Ereignissen von Grad 3 oder Grad 4 dauerhaft abzusetzen (ALT oder AST > 5,0 x ULN oder Bilirubin im Blut > 3 x ULN).
Bei Patienten mit HCC ist die Behandlung mit Atezolizumab zu unterbrechen, wenn ALT oder AST von Normalwerten bei Behandlungsbeginn auf > 3 x bis ≤ 10 x ULN oder von > 1 x bis ≤ 3 x ULN bei Behandlungsbeginn auf > 5 x bis ≤ 10 x ULN oder von > 3 x bis ≤ 5 x ULN bei Behandlungsbeginn auf > 8 x bis ≤ 10 x ULN ansteigen, und länger als 5 bis 7 Tage anhalten. Eine Behandlung mit 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents ist einzuleiten. Wenn sich das Ereignis auf ≤ Grad 1 verbessert, sind die Corticosteroide über einen Zeitraum von ≥ 1 Monat auszuschleichen.
Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, wenn sich das Ereignis innerhalb von 12 Wochen auf ≤ Grad 1 verbessert hat und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde. Die Behandlung mit Atezolizumab ist dauerhaft abzusetzen, wenn ALT oder AST auf > 10 x ULN oder das Gesamtbilirubin auf > 3 x ULN ansteigt.
Immunvermittelte Kolitis
Fälle von Diarrhö oder Kolitis wurden in klinischen Prüfungen mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind auf Anzeichen und Symptome von Kolitis zu überwachen.
Die Behandlung mit Atezolizumab ist bei Grad 2 oder 3 Diarrhö (Anstieg um ≥ 4 Stuhlgänge/Tag gegenüber Behandlungsbeginn) oder Kolitis (symptomatisch) zu unterbrechen. Bei Grad 2 Diarrhö oder Kolitis ist bei anhaltenden Symptomen über 5 Tage oder bei Wiederauftreten der Symptome eine Behandlung mit 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents einzuleiten. Bei Grad 3 Diarrhö oder Kolitis ist eine Behandlung mit intravenösen Corticosteroiden (1 bis 2 mg/kg KG/Tag Methylprednisolon oder eines Äquivalents) einzuleiten. Nach einer Besserung der Symptome soll eine Behandlung mit 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents eingeleitet werden. Wenn sich die Symptome auf ≤ Grad 1 verbessern, sind die Corticosteroide über einen Zeitraum von ≥ 1 Monat auszuschleichen. Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, wenn sich das Ereignis innerhalb von 12 Wochen auf ≤ Grad 1 verbessert hat und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde. Die Behandlung mit Atezolizumab ist bei Diarrhö oder Kolitis von Grad 4 (lebensbedrohlich; unverzügliche Intervention indiziert) dauerhaft abzusetzen. Die potenzielle Komplikation einer gastrointestinalen Perforation in Verbindung mit Kolitis soll in Betracht gezogen werden.
Immunvermittelte Endokrinopathien
Hypothyreose, Hyperthyreose, Nebenniereninsuffizienz, Hypophysitis und Diabetes mellitus Typ 1, einschließlich diabetischer Ketoazidose, wurden in klinischen Prüfungen mit Atezolizumab beobachtet (siehe Abschnitt 4.8).
Die Patienten sind auf klinische Anzeichen und Symptome von Endokrinopathien zu überwachen. Die Schilddrüsenfunktion ist vor und regelmäßig während der Behandlung mit Atezolizumab zu überwachen. Für Patienten mit auffälligen Ergebnissen aus Schilddrüsenfunktionstests zu Behandlungsbeginn ist eine geeignete Behandlung in Betracht zu ziehen.
Asymptomatische Patienten mit auffälligen Ergebnissen aus Schilddrüsenfunktionstests können mit Atezolizumab behandelt werden. Bei symptomatischer Hypothyreose ist die Behandlung mit Atezolizumab zu unterbrechen und eine Schilddrüsenhormon‑Ersatztherapie nach Bedarf einzuleiten. Eine isolierte Hypothyreose kann durch eine Ersatztherapie und ohne Corticosteroide behandelt werden. Bei symptomatischem Hyperthyreose ist die Behandlung mit Atezolizumab zu unterbrechen und ein Thyreostatikum nach Bedarf anzuwenden. Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, sobald die Symptome unter Kontrolle sind und sich die Schilddrüsenfunktion verbessert.
Bei symptomatischer Nebenniereninsuffizienz ist die Behandlung mit Atezolizumab zu unterbrechen und eine Behandlung mit intravenösen Corticosteroiden (1 bis 2 mg/kg KG/Tag Methylprednisolon oder eines Äquivalents) einzuleiten. Sobald sich die Symptome verbessern, ist die Behandlung mit 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents fortzuführen. Wenn sich die Symptome auf ≤ Grad 1 verbessern, sind die Corticosteroide über einen Zeitraum von ≥ 1 Monat auszuschleichen. Die Behandlung kann wieder aufgenommen werden, wenn sich das Ereignis innerhalb von 12 Wochen auf ≤ Grad 1 verbessert hat und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde und wenn der Patient unter der Ersatztherapie (falls notwendig) stabil ist.
Bei Hypophysitis Grad 2 oder 3 ist die Behandlung mit Atezolizumab zu unterbrechen und eine Behandlung mit intravenösen Corticosteroiden (1 bis 2 mg/kg KG/Tag Methylprednisolon oder eines Äquivalents) einzuleiten. Bei Bedarf soll eine Hormonsubstitutionstherapie eingeleitet werden. Sobald sich die Symptome verbessern, ist die Behandlung mit 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents fortzuführen. Wenn sich die Symptome auf ≤ Grad 1 verbessern, sind die Corticosteroide über einen Zeitraum von ≥ 1 Monat auszuschleichen. Die Behandlung kann wieder aufgenommen werden, wenn sich das Ereignis innerhalb von 12 Wochen auf ≤ Grad 1 verbessert hat und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde und wenn der Patient unter der Ersatztherapie (falls notwendig) stabil ist. Die Behandlung mit Atezolizumab ist bei Hypophysitis Grad 4 dauerhaft abzusetzen.
Bei Diabetes mellitus Typ 1 ist eine Behandlung mit Insulin einzuleiten. Bei Hyperglykämie ≥ Grad 3 (Nüchternglucose > 250 mg/dl oder 13,9 mmol/l) ist die Behandlung mit Atezolizumab zu unterbrechen. Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, wenn durch eine Insulin‑Ersatztherapie der Stoffwechsel wieder unter Kontrolle ist.
Immunvermittelte Meningoenzephalitis
Das Auftreten von Meningoenzephalitis wurde in klinischen Prüfungen mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind auf klinische Anzeichen und Symptome einer Meningitis oder Enzephalitis zu überwachen.
Die Behandlung mit Atezolizumab ist bei Meningitis oder Enzephalitis jeglichen Grades dauerhaft abzusetzen. Eine Behandlung mit intravenösen Corticosteroiden (1 bis 2 mg/kg KG/Tag Methylprednisolon oder eines Äquivalents) ist einzuleiten. Nach einer Verbesserung der Symptome soll eine Behandlung mit 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents eingeleitet werden.
Immunvermittelte Neuropathien
Das Auftreten des Myastheniesyndroms/der Myasthenia gravis oder des Guillain‑Barré‑Syndroms, die lebensbedrohlich sein können, sowie von Gesichtsparese wurden bei Patienten beobachtet, die mit Atezolizumab behandelt wurden. Die Patienten sind auf Symptome einer motorischen oder sensorischen Neuropathie zu überwachen (siehe auch Abschnitt „Immunvermitteltes Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom“).
Myelitis wurde in klinischen Prüfungen mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind engmaschig auf Anzeichen und Symptome, die auf eine Myelitis hindeuten, zu überwachen.
Die Behandlung mit Atezolizumab ist beim Myastheniesyndrom/Myasthenia gravis oder Guillain‑Barré‑Syndrom jeglichen Grades dauerhaft abzusetzen. Die Einleitung systemischer Corticosteroide mit einer Dosis von 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents ist in Betracht zu ziehen.
Bei einer Gesichtsparese von Grad 1 oder 2 ist die Behandlung mit Atezolizumab zu unterbrechen und eine Behandlung mit systemischen Corticosteroiden (1 bis 2 mg/kg KG/Tag Prednison oder Äquivalent) in Betracht zu ziehen. Die Behandlung kann erst dann wieder aufgenommen werden, wenn das Ereignis vollständig abgeklungen ist. Die Behandlung mit Atezolizumab ist bei einer Gesichtsparese von Grad 3 oder 4 oder bei jeder anderen Neuropathie, die während der Unterbrechung der Behandlung mit Atezolizumab nicht vollständig abklingt, dauerhaft abzusetzen.
Bei Myelitis von Grad 2, 3 oder 4 muss die Behandlung mit Atezolizumab dauerhaft abgesetzt werden.
Immunvermittelte Pankreatitis
Das Auftreten von Pankreatitis, einschließlich eines Anstiegs der Amylase‑ und Lipase‑Spiegel im Serum, wurde in klinischen Prüfungen mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind engmaschig auf Anzeichen und Symptome einer akuten Pankreatitis zu überwachen.
Die Behandlung mit Atezolizumab ist bei ≥ Grad 3 erhöhten Amylase‑ oder Lipase‑Spiegeln im Serum (> 2 x ULN) oder Grad 2 oder 3 Pankreatitis zu unterbrechen und eine Behandlung mit intravenösen Corticosteroiden (1 bis 2 mg/kg KG/Tag Methylprednisolon oder eines Äquivalents) ist einzuleiten. Sobald sich die Symptome verbessern, ist die Behandlung mit 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents fortzuführen. Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, wenn sich die Amylase‑ und Lipase‑Spiegel im Serum innerhalb von 12 Wochen wieder auf ≤ Grad 1 verbessern oder die Symptome der Pankreatitis abgeklungen sind und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde. Die Behandlung mit Atezolizumab ist bei Grad-4-Pankreatitis oder bei rezidivierender Pankreatitis jeglicher Grade dauerhaft abzusetzen.
Immunvermittelte Myokarditis
Fälle von Myokarditis, einschließlich tödlicher Fälle, wurden mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind auf Anzeichen und Symptome einer Myokarditis zu überwachen (siehe auch Abschnitt „Immunvermitteltes Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom“).
Patienten mit kardialen oder kardiopulmonalen Symptomen sind auf eine mögliche Myokarditis zu untersuchen, damit bereits im frühen Stadium geeignete Maßnahmen eingeleitet werden können. Bei Verdacht auf Myokarditis ist die Behandlung mit Atezolizumab zu unterbrechen, eine sofortige Behandlung mit systemischen Corticosteroiden in einer Dosis von 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents ist einzuleiten sowie eine umgehende kardiologische Untersuchung mit diagnostischer Abklärung gemäß den aktuellen klinischen Leitlinien. Sobald die Diagnose einer Myokarditis gestellt wurde, ist die Behandlung mit Atezolizumab bei Myokarditis von Grad ≥ 2 dauerhaft abzusetzen (siehe Abschnitt 4.2).
Immunvermittelte Nephritis
Das Auftreten von Nephritis wurde in klinischen Prüfungen mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind auf Änderungen der Nierenfunktion zu überwachen.
Bei Nephritis Grad 2 ist die Behandlung mit Atezolizumab zu unterbrechen und eine Behandlung mit systemischen Corticosteroiden in einer Dosis von 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents einzuleiten. Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, wenn sich das Ereignis innerhalb von 12 Wochen auf ≤ Grad 1 verbessert hat und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde. Bei Nephritis Grad 3 oder 4 ist die Behandlung mit Atezolizumab dauerhaft abzusetzen.
Immunvermittelte Myositis
Fälle von Myositis, einschließlich tödlicher Fälle, wurden mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind auf Anzeichen und Symptome einer Myositis zu überwachen (siehe auch Abschnitt „Immunvermitteltes Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom“).
Wenn ein Patient Anzeichen und Symptome einer Myositis entwickelt, ist eine engmaschige Überwachung durchzuführen und der Patient unverzüglich zur Beurteilung und Behandlung an einen Spezialisten zu überweisen. Bei Myositis Grad 2 oder 3 ist die Behandlung mit Atezolizumab zu unterbrechen und eine Behandlung mit Corticosteroiden (1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents) einzuleiten. Wenn sich die Symptome auf ≤ Grad 1 verbessern, sind die Corticosteroide, wie klinisch indiziert, auszuschleichen. Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, wenn sich das Ereignis innerhalb von 12 Wochen auf ≤ Grad 1 verbessert hat und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag orales Prednison oder eines Äquivalents verringert wurde. Die Behandlung mit Atezolizumab ist bei Myositis Grad 4 oder rezidivierender Myositis Grad 3 dauerhaft abzusetzen oder, wenn die Corticosteroid-Dosis nicht innerhalb von 12 Wochen nach Auftreten auf ein Äquivalent von ≤ 10 mg/Tag Prednison verringert werden konnte.
Immunvermittelte schwerwiegende unerwünschte Reaktionen der Haut
Bei mit Atezolizumab behandelten Patienten wurden immunvermittelte schwerwiegende unerwünschte Reaktionen der Haut (severe cutaneous adverse reactions, SCARs) einschließlich Fälle von Stevens-Johnson-Syndrom (SJS) und toxischer epidermaler Nekrolyse (TEN) berichtet. Die Patienten sind auf schwere Hautreaktionen zu überwachen und andere Ursachen sind auszuschließen. Bei Verdacht auf eine SCAR sind die Patienten zur weiteren Diagnose und Behandlung an einen Spezialisten zu überweisen.
Basierend auf dem Schweregrad der Nebenwirkung ist die Behandlung mit Atezolizumab bei Reaktionen der Haut von Grad 3 zu unterbrechen und eine Behandlung mit systemischen Corticosteroiden in einer Dosis von 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents einzuleiten. Die Behandlung mit Atezolizumab kann wieder aufgenommen werden, sobald sich das Ereignis innerhalb von 12 Wochen auf ≤ Grad 1 verbessert hat und die Corticosteroid‑Dosis auf ≤ 10 mg/Tag Prednison oder eines Äquivalents verringert wurde. Bei Reaktionen der Haut von Grad 4 ist die Behandlung mit Atezolizumab dauerhaft abzusetzen und Corticosteroide sind anzuwenden.
Die Behandlung mit Atezolizumab ist bei Patienten mit Verdacht auf SJS oder TEN zu unterbrechen. Bei bestätigtem SJS oder TEN ist Atezolizumab dauerhaft abzusetzen.
Vorsicht ist geboten, wenn die Anwendung von Atezolizumab bei einem Patienten in Erwägung gezogen wird, bei dem bereits während einer vorherigen Behandlung mit anderen immunstimulierenden Krebsmedikamenten eine schwere oder lebensbedrohliche kutane Nebenwirkung aufgetreten ist.
Immunvermittelte Erkrankungen des Perikards
Erkrankungen des Perikards, einschließlich Perikarditis, Perikarderguss und Herzbeuteltamponade, von denen einige zum Tod führten, wurden unter Atezolizumab beobachtet (siehe Abschnitt 4.8). Die Patienten sind auf klinische Anzeichen und Symptome von Erkrankungen des Perikards zu überwachen.
Bei Verdacht auf Perikarditis Grad 1 ist die Behandlung mit Atezolizumab zu unterbrechen und umgehend eine kardiologische Untersuchung mit diagnostischer Abklärung gemäß den aktuellen klinischen Leitlinien einzuleiten. Bei Verdacht auf eine Erkrankung des Perikards ≥ Grad 2 ist die Behandlung mit Atezolizumab zu unterbrechen, eine sofortige Behandlung mit systemischen Corticosteroiden in einer Dosierung von 1 bis 2 mg/kg KG/Tag Prednison oder eines Äquivalents einzuleiten sowie eine umgehende kardiologische Untersuchung mit diagnostischer Abklärung gemäß den aktuellen klinischen Leitlinien zu veranlassen. Sobald die Diagnose einer Erkrankung des Perikards gestellt ist, muss die Behandlung mit Atezolizumab bei einer Erkrankung des Perikards ≥ Grad 2 dauerhaft abgebrochen werden (siehe Abschnitt 4.2).
Hämophagozytische Lymphohistiozytose
Bei Patienten, die Atezolizumab erhielten, wurde über hämophagozytische Lymphohistiozytose (HLH), einschließlich Fälle mit tödlichem Ausgang, berichtet (siehe Abschnitt 4.8). Eine HLH ist in Betracht zu ziehen, wenn das Erscheinungsbild des Zytokin-Freisetzungssyndroms atypisch ist oder länger andauert. Die Patienten sind auf klinische Anzeichen und Symptome einer HLH zu überwachen. Bei Verdacht auf HLH muss Atezolizumab dauerhaft abgesetzt werden und die Patienten sind zur weiteren Diagnose und Behandlung an einen Spezialisten zu überweisen.
Immunvermitteltes Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom
Fälle von Myokarditis-Myositis-Myasthenia-gravis-Overlap-Syndrom (das sich als Überlappung von zwei oder allen drei Erkrankungen manifestiert), von denen einige tödlich verliefen, wurden nach der Markteinführung und in klinischen Studien mit Atezolizumab beobachtet. Eine frühzeitige Erkennung und aggressive Behandlung sind entscheidend, um die damit verbundene Morbidität und das Mortalitätsrisiko zu senken.
Es ist wichtig, überlappende Symptome zu erkennen, die die Diagnose und Behandlung erschweren und ein höheres Mortalitätsrisiko mit sich bringen können. Patienten, die Anzeichen oder Symptome einer Erkrankung aufweisen, sollten auch auf die beiden anderen Erkrankungen hin überwacht und entsprechend der klinischen Indikation behandelt werden. Bezüglich der Empfehlungen zur Dosisanpassung für die einzelnen Erkrankungen (Myokarditis, Myositis, Myasthenia gravis) wird auf Abschnitt 4.2, Tabelle 2 verwiesen. Dabei sind die jeweils strengsten Empfehlungen zu befolgen.
Andere immunvermittelte Nebenwirkungen
Angesichts des Wirkmechanismus von Atezolizumab können andere immunvermittelte Nebenwirkungen auftreten, einschließlich nicht-infektiöser Zystitis, Uveitis und autoimmunhämolytischer Anämie.
Alle vermuteten immunvermittelten Nebenwirkungen sollen bewertet werden, um andere Ursachen auszuschließen. Die Patienten sollen auf Anzeichen und Symptome immunvermittelter Nebenwirkungen überwacht und je nach Schweregrad der Nebenwirkung mit Behandlungsmodifikationen und Corticosteroiden, wie klinisch indiziert, behandelt werden (siehe Abschnitt 4.2 und Abschnitt 4.8).
Infusionsbedingte Reaktionen
Infusionsbedingte Reaktionen, einschließlich Anaphylaxie, wurden mit Atezolizumab beobachtet (siehe Abschnitt 4.8). Bei Patienten mit infusionsbedingten Reaktionen von Grad 1 oder 2 ist die Injektionsgeschwindigkeit zu reduzieren oder die Behandlung zu unterbrechen. Bei Patienten mit infusionsbedingten Reaktionen von Grad 3 oder 4 ist Atezolizumab dauerhaft abzusetzen. Patienten mit infusionsbedingten Reaktionen von Grad 1 oder 2 können Atezolizumab unter engmaschiger Überwachung weiterhin erhalten; eine Prämedikation mit einem Antipyretikum und Antihistaminikum kann in Erwägung gezogen werden.
Krankheitsspezifische Vorsichtsmaßnahmen
Anwendung von Atezolizumab in Kombination mit Bevacizumab, Paclitaxel und Carboplatin bei metastasiertem NSCLC mit nicht-plattenepithelialer Histologie
Vor Einleitung der Behandlung mit dem Vierfach-Regime, bestehend aus Atezolizumab, Bevacizumab, Paclitaxel und Carboplatin, müssen Ärzte die kombinierten Risiken sorgfältig abwägen (siehe Abschnitt 4.8).
Anwendung von Atezolizumab in Kombination mit nab-Paclitaxel beim metastasierten TNBC
Treten während der Behandlung mit Atezolizumab kombiniert mit nab-Paclitaxel Neutropenien und periphere Neuropathien auf, so können diese durch Unterbrechung der Behandlung mit nab-Paclitaxel reversibel sein. Ärzte müssen die Fachinformation von nab-Paclitaxel bezüglich spezifischer Vorsichtsmaßnahmen und Kontraindikationen beachten.
Anwendung von Atezolizumab bei bisher unbehandelten Patienten mit UC, die als ungeeignet für eine Cisplatin‑Therapie eingestuft wurden
Die Krankheitsmerkmale zu Behandlungsbeginn und die Prognosen in der Studienpopulation der IMvigor210-Kohorte-1 waren generell vergleichbar mit denen von Patienten in der Klinik, die als ungeeignet für eine Cisplatin‑Therapie eingestuft werden, für die aber eine Carboplatin‑basierte Kombinations‑Chemotherapie infrage kommt. Für die Subgruppe von Patienten, die für keinerlei Chemotherapie geeignet sind, ist die Datenlage ungenügend. Daher soll Atezolizumab bei diesen Patienten nach sorgfältiger Abwägung des individuellen Nutzen‑Risiko‑Verhältnisses mit Vorsicht eingesetzt werden.
Anwendung von Atezolizumab in Kombination mit Bevacizumab, Paclitaxel und Carboplatin
Patienten mit NSCLC und eindeutiger Tumorinfiltration in die großen Blutgefäße des Brustkorbs oder eindeutiger Kavitation pulmonaler Läsionen, wie mittels bildgebender Verfahren festgestellt, wurden aus der zulassungsrelevanten klinischen Prüfung IMpower150 ausgeschlossen, nachdem einige Fälle tödlicher pulmonaler Hämorrhagie beobachtet wurden. Pulmonale Hämorrhagie ist ein bekannter Risikofaktor einer Behandlung mit Bevacizumab.
Aufgrund fehlender Daten soll Atezolizumab bei diesen Patientenpopulationen nach sorgfältiger Abwägung des Nutzen‑Risiko‑Verhältnisses für den Patienten mit Vorsicht eingesetzt werden.
Anwendung von Atezolizumab in Kombination mit Bevacizumab, Paclitaxel und Carboplatin bei EGFR-mutierten Patienten mit NSCLC, die unter Erlotinib + Bevacizumab progredient wurden
In der Studie IMpower150 wurden keine Daten zur Wirksamkeit von Atezolizumab in Kombination mit Bevacizumab, Paclitaxel und Carboplatin bei EGFR-mutierten Patienten, die zuvor unter Erlotinib + Bevacizumab progredient wurden, erhoben.
Anwendung von Atezolizumab in Kombination mit Bevacizumab bei HCC
Die Daten von HCC-Patienten mit Child-Pugh-Score-B-Lebererkrankung, die mit Atezolizumab in Kombination mit Bevacizumab behandelt wurden, sind sehr begrenzt und es liegen derzeit keine Daten für HCC-Patienten mit Child-Pugh-Score-C-Lebererkrankung vor.
Patienten, die mit Bevacizumab behandelt werden, haben ein erhöhtes Blutungsrisiko und bei Patienten mit HCC, die mit Atezolizumab in Kombination mit Bevacizumab behandelt wurden, wurden Fälle von schweren gastrointestinalen Blutungen, einschließlich tödlicher Ereignisse, berichtet. Bei Patienten mit HCC soll vor Beginn der Behandlung mit Atezolizumab in Kombination mit Bevacizumab ein Screening auf Ösophagusvarizen und deren anschließende Behandlung entsprechend der klinischen Praxis durchgeführt werden. Bevacizumab soll bei Patienten, bei denen unter der Kombinationsbehandlung Blutungen von Grad 3 oder 4 auftreten, dauerhaft abgesetzt werden. Bitte beachten Sie die Fachinformation von Bevacizumab.
Diabetes mellitus kann während der Behandlung mit Atezolizumab in Kombination mit Bevacizumab auftreten. Ärzte sollen den Blutzuckerspiegel je nach klinischer Indikation vor und regelmäßig während der Behandlung mit Atezolizumab in Kombination mit Bevacizumab überwachen.
Anwendung von Atezolizumab als Monotherapie zur Erstlinienbehandlung des metastasierten NSCLC
Ärzte sollen den verzögerten Beginn der Atezolizumab-Wirkung vor Einleitung der Erstlinienbehandlung als Monotherapie bei Patienten mit NSCLC berücksichtigen. Bei Atezolizumab wurde im Vergleich zu Chemotherapie eine erhöhte Anzahl an Todesfällen innerhalb von 2,5 Monaten nach der Randomisierung, gefolgt von einem Langzeitüberlebensvorteil, beobachtet. Es konnten keine spezifischen Faktoren, die mit frühen Todesfällen in Verbindung stehen, identifiziert werden (siehe Abschnitt 5.1).
Von klinischen Prüfungen ausgeschlossene Patienten
Patienten mit folgenden Erkrankungen waren von der Teilnahme an den klinischen Prüfungen ausgeschlossen: Autoimmunerkrankung in der Vorgeschichte, Pneumonitis in der Vorgeschichte, aktive Gehirnmetastasen, Patienten mit einem ECOG‑Performance‑Status ≥ 2 (außer Patienten mit fortgeschrittenem NSCLC, die für eine platinbasierte Therapie ungeeignet sind), HIV, Infektion mit Hepatitis B oder Hepatitis C (bei Patienten ohne HCC), Patienten mit erheblichen kardiovaskulären Erkrankungen und Patienten mit unzureichender hämatologischer Funktion bzw. unzureichender Funktion von Endorganen. Patienten, denen innerhalb von 28 Tagen vor Aufnahme in die Studie ein attenuierter Lebendimpfstoff verabreicht wurde, sowie Patienten, die innerhalb von 4 Wochen vor Aufnahme in die Studie systemische immunstimulierende Arzneimittel oder innerhalb von 2 Wochen vor Aufnahme in die Studie systemische Immunsuppressiva oder innerhalb von 2 Wochen vor Einleitung der Studienbehandlung therapeutische orale oder intravenöse Antibiotika erhalten hatten, waren von der Teilnahme an den klinischen Prüfungen ausgeschlossen.
Sonstiger Bestandteil mit bekannter Wirkung
Dieses Arzneimittel enthält Polysorbat 20. Jede 1 875-mg-Durchstechflasche Tecentriq Injektionslösung enthält 9 mg Polysorbat 20, entsprechend 0,6 mg/ml. Polysorbat 20 kann allergische Reaktionen hervorrufen.
Patientenpass
Der verordnende Arzt muss die Risiken einer Therapie mit Tecentriq mit dem Patienten besprechen. Dem Patienten wird ein Patientenpass ausgehändigt, und er wird aufgefordert, diesen immer bei sich zu tragen.
Es wurden keine formalen pharmakokinetischen Interaktionsstudien mit Atezolizumab durchgeführt. Da Atezolizumab durch katabolischen Abbau aus dem Blutkreislauf eliminiert wird, sind keine metabolischen Arzneimittelwechselwirkungen zu erwarten.
Eine Verwendung systemischer Corticosteroide oder Immunsuppressiva vor Behandlungsbeginn mit Atezolizumab soll aufgrund möglicher Beeinträchtigungen der pharmakodynamischen Aktivität und der Wirksamkeit von Atezolizumab vermieden werden. Systemische Corticosteroide oder andere Immunsuppressiva können jedoch nach Beginn der Therapie mit Atezolizumab zur Behandlung von immunvermittelten Nebenwirkungen angewendet werden (siehe Abschnitt 4.4).
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während und für 5 Monate nach der Behandlung mit Atezolizumab eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Es liegen keine Daten zur Anwendung von Atezolizumab bei Schwangeren vor. Bisher wurden keine Entwicklungs‑ und Reproduktionsstudien mit Atezolizumab durchgeführt. In tierexperimentellen Studien wurde nachgewiesen, dass die Hemmung des PD‑L1/PD‑1‑Signalweges in Schwangerschaft‑Mausmodellen zu einer immunvermittelten Abstoßungsreaktion gegen den sich entwickelnden Fetus und zum fetalen Tod führen kann (siehe Abschnitt 5.3). Diese Ergebnisse deuten auf ein potenzielles Risiko hin, dass die Verabreichung von Atezolizumab während der Schwangerschaft, basierend auf dem Wirkmechanismus, eine fruchtschädigende Wirkung, einschließlich erhöhter Abort‑ oder Totgeburtsraten, haben kann.
Von humanen Immunglobulinen G1 (IgG1) ist bekannt, dass sie die Plazentaschranke passieren; daher besteht die Möglichkeit, dass Atezolizumab, ein IgG1, von der Mutter auf den sich entwickelnden Fetus übergehen kann.
Atezolizumab darf während der Schwangerschaft nicht angewendet werden, es sei denn, eine Behandlung mit Atezolizumab ist aufgrund des klinischen Zustandes der Frau erforderlich.
Stillzeit
Es ist nicht bekannt, ob Atezolizumab in die Muttermilch übergeht. Atezolizumab ist ein monoklonaler Antikörper, der erwartungsgemäß in der menschlichen Vormilch und in geringen Konzentrationen auch in der Muttermilch enthalten ist. Ein Risiko für das Neugeborene/den Säugling kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen oder die Behandlung mit Tecentriq zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Es sind keine klinischen Daten über mögliche Auswirkungen von Atezolizumab auf die Fertilität verfügbar. Es wurden keine Toxizitätsstudien zur Untersuchung des Einflusses von Atezolizumab auf die Reproduktion und auf die Entwicklung durchgeführt. Jedoch zeigten Daten einer 26‑wöchigen Toxizitätsstudie mit Mehrfachdosen von Atezolizumab eine Auswirkung auf den Menstruationszyklus, bei einer geschätzten mittleren Exposition (AUC), die dem 6‑Fachen der AUC von Patienten in der empfohlenen Dosierung entsprach. Dieser Effekt war reversibel (siehe Abschnitt 5.3). Eine Auswirkung auf die männlichen Fortpflanzungsorgane wurde nicht beobachtet.
Tecentriq hat geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Patienten, bei denen eine Ermüdung (Fatigue) auftritt, sind anzuweisen, bis zum Abklingen der Symptome kein Fahrzeug zu führen und keine Maschinen zu bedienen (siehe Abschnitt 4.8).
Zusammenfassung des Sicherheitsprofils
Die Sicherheit von Atezolizumab als Monotherapie basiert auf gepoolten Daten von 5 039 Patienten mit unterschiedlichen Tumorarten, denen Atezolizumab intravenös verabreicht wurde, und 247 Patienten mit NSCLC, denen Atezolizumab subkutan verabreicht wurde. Die häufigsten Nebenwirkungen (> 10 %) waren Ermüdung (Fatigue) (29,3 %), verminderter Appetit (20,1 %), Ausschlag (19,7 %), Übelkeit (18,8 %), Husten (18,2 %), Diarrhö (18,1 %), Fieber (17,9 %), Dyspnoe (16,6 %), Arthralgie (16,2 %), Pruritus (13,3 %), Asthenie (13 %), Rückenschmerzen (12,2 %), Erbrechen (11,7 %), Harnwegsinfektion (11 %) und Kopfschmerzen (10,2 %).
Die Sicherheit von intravenös verabreichtem Atezolizumab als Kombinationstherapie mit anderen Arzneimitteln wurde bei 4 535 Patienten mit unterschiedlichen Tumorarten untersucht. Die häufigsten Nebenwirkungen (≥ 20 %) waren Anämie (36,8 %), Neutropenie (36,6 %), Übelkeit (35,5 %), Ermüdung (Fatigue) (33,1 %), Alopezie (28,1 %), Ausschlag (27,8 %), Diarrhö (27,6 %), Thrombozytopenie (27,1 %), Obstipation (25,8 %), verminderter Appetit (24,7 %) und periphere Neuropathie (24,4 %).
Das Sicherheitsprofil von Tecentriq Injektionslösung war im Allgemeinen vergleichbar mit dem bekannten Sicherheitsprofil der intravenösen Darreichungsform, mit einer zusätzlichen Nebenwirkung: Reaktionen an der Injektionsstelle (4,5 % im Behandlungsarm mit Tecentriq subkutan gegenüber 0 % im Behandlungsarm mit Atezolizumab intravenös).
Anwendung von Atezolizumab im adjuvanten NSCLC-Setting
Das Sicherheitsprofil von Atezolizumab im adjuvanten Setting in der Patientenpopulation mit nicht-kleinzelligem Lungenkarzinom (NSCLC) (IMpower010) stimmte im Allgemeinen mit dem Sicherheitsprofil der gepoolten Monotherapie bei fortgeschrittener Erkrankung überein. Allerdings lag die Häufigkeit immunvermittelter Nebenwirkungen von Atezolizumab in der IMpower010 bei 51,7 % im Vergleich zu 38,4 % in der gepoolten Monotherapiepopulation mit fortgeschrittener Erkrankung. Im adjuvanten Setting wurden keine neuen immunvermittelten Nebenwirkungen festgestellt.
Anwendung von Atezolizumab in Kombination mit Bevacizumab, Paclitaxel und Carboplatin
In der Erstlinienstudie beim NSCLC (IMpower150) wurde eine insgesamt höhere Frequenz von unerwünschten Ereignissen beim Vierfach-Regime bestehend aus Atezolizumab, Bevacizumab, Paclitaxel und Carboplatin im Vergleich zu Atezolizumab, Paclitaxel und Carboplatin beobachtet, einschließlich Ereignissen von Grad 3 und 4 (63,6 % im Vergleich zu 57,5 %), Ereignissen von Grad 5 (6,1 % im Vergleich zu 2,5 %), unerwünschten Ereignissen von besonderem Interesse bezogen auf Atezolizumab (52,4 % im Vergleich zu 48,0 %) sowie unerwünschten Ereignissen, die zum Abbruch von einer in der Studie erhaltenen Therapie führten (33,8 % im Vergleich zu 13,3 %). Übelkeit, Diarrhö, Stomatitis, Ermüdung (Fatigue), Fieber, mukosale Entzündung, verminderter Appetit, vermindertes Gewicht, Hypertonie und Proteinurie wurden bei Patienten, die Atezolizumab in Kombination mit Bevacizumab, Paclitaxel und Carboplatin erhielten, häufiger berichtet (≥ 5 % Unterschied). Andere klinisch signifikante unerwünschte Ereignisse, die öfter unter Atezolizumab, Bevacizumab, Paclitaxel und Carboplatin beobachtet wurden, waren Epistaxis, Hämoptyse und zerebrovaskulärer Insult, einschließlich tödlicher Ereignisse.
Weitere Angaben zu schwerwiegenden Nebenwirkungen sind in Abschnitt 4.4 aufgeführt.
Tabellarische Auflistung der Nebenwirkungen
Die Nebenwirkungen (adverse reactions = ARs) sind gemäß MedDRA nach Systemorganklasse (system organ class = SOC) und Häufigkeitskategorien in Tabelle 3 für Atezolizumab (intravenös und subkutan) als Monotherapie oder als Kombinationstherapie aufgelistet. Nebenwirkungen, die bekanntermaßen unter der Therapie mit Atezolizumab als Monotherapie bzw. unter Chemotherapie allein auftreten können, können ebenso unter der Kombinationstherapie auftreten, auch wenn diese nicht in den klinischen Prüfungen zur Untersuchung der Kombinationstherapie beobachtet wurden. Die folgenden Häufigkeitskategorien wurden verwendet: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Die Nebenwirkungen werden für jede Häufigkeitsgruppe nach abnehmendem Schweregrad aufgeführt.
Tabelle 3: Zusammenfassung der Nebenwirkungen bei Patienten, die mit Atezolizumab behandelt wurden
Atezolizumab Monotherapie |
Atezolizumab als Kombinationstherapie |
|||
Infektionen und parasitäre Erkrankungen | ||||
Sehr häufig |
Harnwegsinfektiona |
Lungeninfektionb |
||
Häufig |
Sepsisaj |
|||
Selten |
Zytomegalievirus-Infektion |
Zytomegalievirus-Infektion |
||
Erkrankungen des Blutes und des Lymphsystems | ||||
Sehr häufig |
Anämieax |
Anämieax, Thrombozytopenied, Neutropeniee, Leukopenief |
||
Häufig |
Thrombozytopenied, Neutropeniee |
Lymphopenieg |
||
Selten |
Hämophagozytische Lymphohistiozytose, autoimmunhämolytische Anämieaw |
Hämophagozytische Lymphohistiozytose, autoimmunhämolytische Anämieaw |
||
Erkrankungen des Immunsystems | ||||
Häufig |
Infusionsbedingte Reaktionh |
Infusionsbedingte Reaktionh |
||
Selten |
Sarkoidoseas |
|||
Endokrine Erkrankungen | ||||
Sehr häufig |
Hypothyreosei |
|||
Häufig |
Hypothyreosei, Hyperthyreosej |
Hyperthyreosej |
||
Gelegentlich |
Diabetes mellitusk, Nebenniereninsuffizienzl, Hypophysitism |
Hypophysitism |
||
Stoffwechsel- und Ernährungsstörungen | ||||
Sehr häufig |
Verminderter Appetit |
Verminderter Appetit |
||
Häufig |
Hypokaliämieae, Hyponatriämieaf, Hyperglykämie, Hypoalbuminämie, Hypophosphatämie, Hypokalzämie |
Hypokaliämieae, Hyponatriämieaf, Hypomagnesiämien, Hypoalbuminämie, Hypophosphatämie, Hypokalzämie |
||
Erkrankungen des Nervensystems | ||||
Sehr häufig |
Kopfschmerzen |
Periphere Neuropathieo, Kopfschmerzen |
||
Häufig |
Periphere Neuropathieo |
Synkope, Schwindelgefühl |
||
Gelegentlich |
Guillain‑Barré‑Syndromp, Meningoenzephalitisq |
|||
Selten |
Myastheniesyndromr, Gesichtsparese, Myelitis |
Gesichtsparese |
||
Augenerkrankungen | ||||
Gelegentlich |
Uveitisat |
|||
Selten |
Uveitisat |
|||
Herzerkrankungen | ||||
Häufig |
Erkrankungen des Perikardsao |
|||
Gelegentlich |
Erkrankungen des Perikardsao |
|||
Selten |
Myokarditiss |
|||
Gefäßerkrankungen | ||||
Sehr häufig |
Hypertonieai |
|||
Häufig |
Hypotonie |
|||
Erkrankungen der Atemwege, des Brustraums und Mediastinums | ||||
Sehr häufig |
Dyspnoe, Husten |
Dyspnoe, Husten, Nasopharyngitisam |
||
Häufig |
Pneumonitist, Hypoxieag, Nasopharyngitisam |
Dysphonie |
||
Erkrankungen des Gastrointestinaltrakts | ||||
Sehr häufig |
Übelkeit, Erbrechen, Diarrhöu |
Übelkeit, Erbrechen, Diarrhöu, Obstipation |
||
Häufig |
Kolitisv, Abdominalschmerzen, Dysphagie, Schmerzen im Oropharynxw, Mundtrockenheit |
Stomatitis, Dysgeusie, Kolitisv |
||
Gelegentlich |
Pankreatitisx |
|||
Selten |
Zöliakie |
Zöliakie |
||
Leber- und Gallenerkrankungen | ||||
Häufig |
Erhöhte AST, erhöhte ALT, Hepatitisy |
Erhöhte AST, erhöhte ALT |
||
Erkrankungen der Haut und des Unterhautgewebes | ||||
Sehr häufig |
Ausschlagz, Pruritus |
Ausschlagz, Pruritus, Alopezieah |
||
Häufig |
Trockene Hautaq |
|||
Gelegentlich |
Schwerwiegende unerwünschte Reaktionen der Hautak, Psoriasisan, Lichen-Erkrankungenar |
Schwerwiegende unerwünschte Reaktionen der Hautak, Psoriasisan |
||
Selten |
Pemphigoid |
Pemphigoid, Lichen-Erkrankungenar |
||
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | ||||
Sehr häufig |
Arthralgie, Rückenschmerzen |
Arthralgie, Schmerzen des Muskel- und Skelettsystemsaa, Rückenschmerzen |
||
Häufig |
Schmerzen des Muskel- und Skelettsystemsaa, Arthritisau |
Arthritisau |
||
Gelegentlich |
Myositisab, Tenosynovitisav |
Tenosynovitisav |
||
Erkrankungen der Nieren und Harnwege | ||||
Häufig |
Erhöhtes Kreatinin im Blutc |
Proteinurieac, erhöhtes Kreatinin im Blutc |
||
Gelegentlich |
Nephritisad |
|||
Nicht bekannt |
Nicht-infektiöse Zystitisal |
|||
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | ||||
Sehr häufig |
Fieber, Ermüdung (Fatigue), Asthenie |
Fieber, Ermüdung (Fatigue), Asthenie, peripheres Ödem |
||
Häufig |
Grippeähnliche Erkrankung, Schüttelfrost, Reaktion an der Injektionsstelleap |
|||
Untersuchungen | ||||
Häufig |
Gamma‑Glutamyltransferase erhöht |
Erhöhte alkalische Phosphatase im Blut, Gamma‑Glutamyltransferase erhöht |
||
Gelegentlich |
Kreatinphosphokinase im Blut erhöht |
|||
a Schließt Berichte von Harnwegsinfektion, Zystitis, Pyelonephritis, Harnwegsinfektion durch Escherichia, bakterieller Harnwegsinfektion, Niereninfektion, akuter Pyelonephritis, chronische Pyelonephritis, Pyelitis, Nierenabszess, Infektion des Harntrakts durch Streptokokken, Urethritis, Harnwegsinfektion durch Pilze, Harnwegsinfektion durch Pseudomonas ein.
b Schließt Berichte von Pneumonie, Bronchitis, Infektionen der unteren Atemwege, infektiösem Pleuraerguss, Tracheobronchitis, atypischer Pneumonie, Lungenabszess, infektiöser Exazerbation einer chronisch obstruktiven Lungenerkrankung, parakanzeröser Pneumonie, Pyopneumothorax, Pleurainfektion, Pneumonie nach einem Eingriff ein.
c Schließt Berichte von erhöhtem Kreatinin im Blut, Hyperkreatininämie ein.
d Schließt Berichte von Immunthrombozytopenie (gemeldet in Studien außerhalb des gepoolten Datensatzes), Thrombozytopenie, verminderter Thrombozytenzahl ein.
e Schließt Berichte von Neutropenie, erniedrigter Neutrophilenzahl, febriler Neutropenie, neutropenischer Sepsis, Granulozytopenie ein.
f Schließt Berichte von erniedrigter Leukozytenzahl, Leukopenie ein.
g Schließt Berichte von Lymphopenie, erniedrigter Lymphozytenzahl ein.
h Schließt Berichte über infusionsbedingte Reaktionen, Zytokin-Freisetzungssyndrom, Überempfindlichkeit, Anaphylaxie ein.
i Schließt Berichte von Schilddrüsenantikörper-positiver autoimmuner Hypothyreose, Immunthyreoiditis, erniedrigtem Thyreotropin im Blut, erhöhtem Thyreotropin im Blut, Euthyroid-Sick-Syndrom, Struma, Hypothyreose, immunvermittelter Unterfunktion der Schilddrüse, immunvermittelter Thyreoiditis, Myxödem, primärer Hypothyreose, Schilddrüsenerkrankung, erniedrigtem Schilddrüsenhormon, anomalen Schilddrüsenfunktionstests, Thyreoiditis, akuter Thyreoiditis, erniedrigtem Thyroxin, erniedrigtem freien Thyroxin, erhöhtem freien Thyroxin, erhöhtem Thyroxin, erniedrigtem Trijodthyronin, erhöhtem Trijodthyronin, anomalem freien Trijodthyronin, erniedrigtem freien Trijodthyronin, erhöhtem freien Trijodthyronin, stummer Thyreoiditis ein.
j Schließt Berichte von Hyperthyreose, Basedow-Krankheit, endokriner Ophthalmopathie, Exophthalmus ein.
k Schließt Berichte von Diabetes mellitus, Diabetes mellitus Typ 1, diabetischer Ketoazidose, Ketoazidose ein.
l Schließt Berichte von Nebenniereninsuffizienz, erniedrigtem Corticotropin im Blut, Glucocorticoid-Mangel, primärer Nebenniereninsuffizienz, sekundärer Nebennierenrindeninsuffizienz ein.
m Schließt Berichte von Hypophysitis, Hypopituitarismus, sekundärer Nebennierenrindeninsuffizienz, Störung der Temperaturregulation ein.
n Schließt Berichte von Hypomagnesiämie, erniedrigtem Magnesium im Blut ein.
o Schließt Berichte von peripherer Neuropathie, Autoimmun-Neuropathie, peripherer sensorischer Neuropathie, Polyneuropathie, Herpes zoster, peripherer motorischer Neuropathie, neuralgischer Schulteramyotrophie, peripherer sensomotorischer Neuropathie, toxischer Neuropathie, axonaler Neuropathie, lumbosakraler Plexopathie, Arthropathia neuropathica, peripherer Nerveninfektion, Neuritis, immunvermittelter Neuropathie ein.
p Schließt Berichte von Guillain‑Barré‑Syndrom, aufsteigender schlaffer Lähmung, demyelinisierender Polyneuropathie ein.
q Schließt Berichte von Enzephalitis, autoimmuner Enzephalitis, Meningitis, aseptischer Meningitis, Photophobie ein.
r Schließt Berichte von Myasthenia gravis ein.
s Schließt Berichte von Myokarditis, Autoimmunmyokarditis, immunvermittelter Myokarditis ein.
t Schließt Berichte von Pneumonitis, Lungeninfiltration, Bronchiolitis, immunvermittelter Lungenerkrankung, immunvermittelter Pneumonitis, interstitieller Lungenerkrankung, Alveolitis, Opazität in der Lunge, Lungenfibrose, Lungentoxizität, strahlenbedingter Pneumonitis ein.
u Schließt Berichte von Diarrhö, Stuhldrang, häufigen Darmentleerungen, gastrointestinaler Hypermotilität ein.
v Schließt Berichte von Kolitis, autoimmuner Kolitis, Kolitis ischämisch, mikroskopischer Kolitis, Kolitis ulcerosa, Diversionskolitis, eosinophiler Kolitis, immunvermittelter Enterokolitis ein.
w Schließt Berichte von Schmerzen im Oropharynx, Beschwerden im Oropharynx, Rachenreizung ein.
x Schließt Berichte von Autoimmunpankreatitis, Pankreatitis, akuter Pankreatitis, erhöhter Lipase, erhöhter Amylase ein.
y Schließt Berichte von Aszites, autoimmuner Hepatitis, hepatischer Zytolyse, Hepatitis, akuter Hepatitis, toxischer Hepatitis, Lebertoxizität, immunvermittelter Hepatitis, Lebererkrankung, arzneimittelbedingtem Leberschaden, Leberversagen, Steatosis hepatis, Leberläsion, Leberschädigung, Ösophagusvarizen mit Blutung, ösophageale Varizen, spontaner bakterieller Peritonitis ein.
z Schließt Berichte von Akne, Blasen, Dermatitis, akneiformer Dermatitis, allergischer Dermatitis, Medikamentenausschlag, Ekzem, infiziertem Ekzem, Erythem, Erythem des Augenlids, Ausschlag am Augenlid, fixem Exanthem, Follikulitis, Furunkel, Handekzem, immunvermittelter Dermatitis, Lippenbläschen, Blutblasen am Mund, palmar-plantarem Erythrodysästhesiesyndrom, Pemphigoid, Ausschlag, erythematösem Hautausschlag, makulösem Ausschlag, makulo-papulösem Ausschlag, morbiliformer Ausschlag, papulösem Ausschlag, papulosquamösem Ausschlag, Ausschlag mit Juckreiz, pustulösem Ausschlag, blasigem Hautausschlag, Dermatitis am Skrotum, seborrhoischer Dermatitis, Exfoliation der Haut, Hauttoxizität, Hautulkus, Ausschlag am Gefäßzugang ein.
aa Schließt Berichte von Schmerzen des Muskel- und Skelettsystems, Myalgie, Knochenschmerzen ein.
ab Schließt Berichte von Myositis, Rhabdomyolyse, Polymyalgia rheumatica, Dermatomyositis, Muskelabszess und nachweisbarem Myoglobin im Urin, Myopathie, Polymyositis ein.
ac Schließt Berichte von Proteinurie, nachweisbarem Protein im Urin, Hämoglobinurie, Anomalie des Urins, nephrotischem Syndrom, Albuminurie ein.
ad Schließt Berichte von Nephritis, autoimmuner Nephritis, Purpura-Schönlein-Henoch-Nephritis, paraneoplastischer Glomerulonephritis, tubulo-interstitieller Nephritis ein.
ae Schließt Berichte von Hypokaliämie, erniedrigtem Kalium im Blut ein.
af Schließt Berichte von Hyponatriämie, erniedrigtem Natrium im Blut ein.
ag Schließt Berichte von Hypoxie, erniedrigter Sauerstoffsättigung, pO2 erniedrigt ein.
ah Schließt Berichte von Alopezie, Madarosis, Alopecia areata, Alopecia totalis, Hypotrichose ein.
ai Schließt Berichte von Hypertonie, erhöhtem Blutdruck, hypertensiver Krise, erhöhtem systolischen Blutdruck, diastolischer Hypertonie, inadäquat eingestelltem Blutdruck, hypertonischer Retinopathie, hypertensiver Nephropathie, essentieller Hypertonie, orthostatischer Hypertonie ein.
aj Schließt Berichte von Sepsis, septischem Schock, Urosepsis, neutropenischer Sepsis, pulmonaler Sepsis, bakterieller Sepsis, Klebsiella-Sepsis, Abdominalsepsis, Candidasepsis, Escherichia-Sepsis, Sepsis durch Pseudomonas, Sepsis durch Staphylokokken ein.
ak Schließt Berichte von bullöser Dermatitis, exfoliativem Hautausschlag, Erythema multiforme, Dermatitis exfoliativa, generalisierte Dermatitis exfoliativa, toxischem Hautausschlag, Stevens-Johnson-Syndrom, Arzneimittelwirkung mit Eosinophilie und systemischen Symptomen, Epidermolysis acuta toxica, kutaner Vaskulitis ein.
al Schließt Berichte von nicht-infektiöser und immunvermittelter Zystitis ein.
am Schließt Berichte von Nasopharyngitis, verstopfter Nase und Rhinorrhoe ein.
an Schließt Berichte von Psoriasis, psoriasiformer Dermatitis ein.
ao Schließt Berichte von Perikarditis, Perikarderguss, Herzbeuteltamponade und konstriktive Perikarditis ein.
ap Wurde in einer Studie außerhalb des gepoolten Datensatzes berichtet (in Zusammenhang mit subkutaner Anwendung). Die Häufigkeit basiert auf der Exposition gegenüber Tecentriq Injektionslösung in IMscin001 und IMscin002 und schließt Berichte von Reaktionen an der Injektionsstelle, Schmerzen an der Injektionsstelle, Erythem an der Injektionsstelle und Ausschlag an der Injektionsstelle ein.
aq Schließt Berichte von trockener Haut, Xerose ein.
ar Schließt Berichte von lichenoider Keratose, Lichen sclerosus und Lichen planus ein.
as Schließt Berichte von Sarkoidose, Lungensarkoidose und Sarkoidose der Lymphknoten ein.
atSchließt Berichte von Uveitis, Iridozyklitis und Iritis ein.
au Schließt Berichte von Arthritis, Gelenkschwellung, Osteoarthritis, rheumatoider Arthritis, Polyarthritis, Osteoarthritis der Wirbelsäule, autoimmuner Arthritis, immunvermittelter Arthritis, Spondylitis, Gelenkerguss, Arthropathie, Oligoarthritis, rheumatischer Erkrankung ein.
av Schließt Berichte von Tendinitis, Sehnenschmerzen, Tenosynovitis und Synovitis ein.
aw Schließt Berichte von autoimmunhämolytischer Anämie und hämolytischer Anämie ein.
ax Schließt Berichte von Anämie, erniedrigtem Hämoglobin und erniedrigter Erythrozytenzahl ein.
Beschreibung ausgewählter Nebenwirkungen
Die im Folgenden aufgeführten Daten geben Informationen zu klinisch signifikanten Nebenwirkungen bei Atezolizumab als Monotherapie in klinischen Prüfungen (siehe Abschnitt 5.1). Angaben zu den signifikanten Nebenwirkungen von Atezolizumab als Kombinationstherapie sind dargestellt, sofern klinisch relevante Unterschiede im Vergleich zu Atezolizumab als Monotherapie beobachtet wurden. Die Behandlungsempfehlungen bei diesen Nebenwirkungen sind in den Abschnitten 4.2 und 4.4 beschrieben.
Immunvermittelte Pneumonitis
Eine Pneumonitis trat bei 3,0 % (151/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Drei dieser Patienten erlitten ein tödliches Ereignis. Die mediane Zeit bis zum Auftreten betrug 3,7 Monate (Bereich: 3 Tage bis 29,8 Monate). Die mediane Dauer betrug 1,7 Monate (Bereich: 0 Tage bis 27,8+ Monate; + kennzeichnet einen zensierten Wert). Die Pneumonitis führte bei 41 (0,8 %) Patienten zum Absetzen von Atezolizumab. Eine Pneumonitis, die eine Anwendung von Corticosteroiden erforderlich machte, trat bei 1,8 % (92/5 039) der mit Atezolizumab als Monotherapie behandelten Patienten auf.
Immunvermittelte Hepatitis
Eine Hepatitis trat bei 1,7 % (88/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Unter den 88 Patienten erlitten drei ein tödliches Ereignis. Die mediane Zeit bis zum Auftreten betrug 1,4 Monate (Bereich: 0 Tage bis 26,3 Monate). Die mediane Dauer betrug 1 Monat (Bereich: 0 Tage bis 52,1+ Monate; + kennzeichnet einen zensierten Wert). Die Hepatitis führte bei 46 (0,9 %) Patienten zum Absetzen von Atezolizumab. Eine Hepatitis, die eine Anwendung von Corticosteroiden erforderlich machte, trat bei 2,6 % (130/5 039) der mit Atezolizumab als Monotherapie behandelten Patienten auf.
Immunvermittelte Kolitis
Eine Kolitis trat bei 1,2 % (62/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 4,5 Monate (Bereich: 15 Tage bis 36,4 Monate). Die mediane Dauer betrug 1,4 Monate (Bereich: 3 Tage bis 50,2+ Monate; + kennzeichnet einen zensierten Wert). Die Kolitis führte bei 24 (0,5 %) Patienten zum Absetzen von Atezolizumab. Eine Kolitis, die eine Anwendung von Corticosteroiden erforderlich machte, trat bei 0,6 % (30/5 039) der mit Atezolizumab als Monotherapie behandelten Patienten auf.
Immunvermittelte Endokrinopathien
Thyreoidale Erkrankungen
Eine Hypothyreose trat bei 8,5 % (427/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 4,2 Monate (Bereich: 0 Tage bis 38,5 Monate). Eine Hypothyreose trat bei 17,4 % (86/495) der Patienten auf, die Atezolizumab als Monotherapie im adjuvanten NSCLC-Setting erhielten. Die mediane Zeit bis zum Auftreten betrug 4,0 Monate (Bereich: 22 Tage bis 11,8 Monate).
Ein Hyperthyreose trat bei 2,4 % (121/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 2,7 Monate (Bereich: 0 Tage bis 24,3 Monate). Ein Hyperthyreose trat bei 6,5 % (32/495) der Patienten auf, die Atezolizumab als Monotherapie im adjuvanten NSCLC-Setting erhielten. Die mediane Zeit bis zum Auftreten betrug 2,8 Monate (Bereich: 1 Tag bis 9,9 Monate).
Nebenniereninsuffizienz
Eine Nebenniereninsuffizienz trat bei 0,5 % (25/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 6,2 Monate (Bereich: 3 Tage bis 21,4 Monate). Eine Nebenniereninsuffizienz führte bei 5 (0,1 %) Patienten zum Absetzen von Atezolizumab. Eine Nebenniereninsuffizienz, die eine Anwendung von Corticosteroiden erforderlich machte, trat bei 0,4 % (20/5 039) der mit Atezolizumab als Monotherapie behandelten Patienten auf.
Hypophysitis
Hypophysitis trat bei 0,2 % (9/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 5,3 Monate (Bereich: 21 Tage bis 13,7 Monate). Bei 6 (0,1 %) Patienten war die Anwendung von Corticosteroiden erforderlich und die Behandlung mit Atezolizumab wurde bei 1 (< 0,1 %) Patienten abgesetzt.
Hypophysitis trat bei 1,4 % (15/1 093) der Patienten auf, die Atezolizumab in Kombination mit Paclitaxel, gefolgt von Atezolizumab, dosisdichtem Doxorubicin oder Epirubicin und Cyclophosphamid, erhielten. Die mediane Zeit bis zum Auftreten betrug 3,8 Monate (Bereich: 2,4 bis 10,7 Monate). Bei 11 Patienten (1,0 %) war die Anwendung von Corticosteroiden erforderlich. Die Behandlung mit Atezolizumab wurde bei 7 (0,6 %) Patienten abgesetzt.
Hypophysitis trat bei 0,8 % (3/393) der Patienten auf, die Atezolizumab zusammen mit Bevacizumab, Paclitaxel und Carboplatin erhielten. Die mediane Zeit bis zum Auftreten betrug 7,7 Monate (Bereich: 5,0 bis 8,8 Monate). Bei 2 Patienten war die Anwendung von Corticosteroiden erforderlich.
Hypophysitis trat bei 0,4 % (2/473) der Patienten auf, die Atezolizumab zusammen mit nab-Paclitaxel und Carboplatin erhielten. Die mediane Zeit bis zum Auftreten betrug 5,2 Monate (Bereich: 5,1 bis 5,3 Monate). Bei beiden Patienten war die Anwendung von Corticosteroiden erforderlich.
Diabetes mellitus
Diabetes mellitus trat bei 0,6 % (30/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 5,5 Monate (Bereich: 3 Tage bis 29,0 Monate). Diabetes mellitus führte bei < 0,1 % (3/5 039) Patienten zum Absetzen von Atezolizumab. Bei 4 Patienten (< 0,1 %) war die Anwendung von Corticosteroiden erforderlich.
Diabetes mellitus trat bei 2,0 % (10/493) der Patienten mit HCC auf, die Atezolizumab in Kombination mit Bevacizumab erhielten. Die mediane Zeit bis zum Auftreten betrug 4,4 Monate (Bereich: 1,2 Monate bis 8,3 Monate). Diabetes mellitus führte bei keinem Patienten zum Absetzen von Atezolizumab.
Immunvermittelte Meningoenzephalitis
Eine Meningoenzephalitis trat bei 0,4 % (22/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 15 Tage (Bereich: 0 Tage bis 12,5 Monate). Die mediane Dauer betrug 24 Tage (Bereich: 6 Tage bis 14,5+ Monate; + kennzeichnet einen zensierten Wert).
Eine Meningoenzephalitis, die eine Anwendung von Corticosteroiden erforderlich machte, trat bei 0,2 % (12/5 039) der mit Atezolizumab behandelten Patienten auf. Acht Patienten (0,2 %) mussten Atezolizumab absetzen.
Immunvermittelte Neuropathien
Guillain‑Barré‑Syndrom und demyelinisierende Polyneuropathie
Guillain‑Barré‑Syndrom und demyelinisierende Polyneuropathie traten bei 0,1 % (6/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 4,1 Monate (Bereich: 18 Tage bis 8,1 Monate). Die mediane Dauer lag bei 8,0 Monaten (Bereich: 18 Tage bis 24,5+ Monate; + kennzeichnet einen zensierten Wert). Das Guillain‑Barré‑Syndrom führte bei einem Patienten (< 0,1 %) zum Absetzen der Behandlung mit Atezolizumab. Ein Guillain‑Barré‑Syndrom, das eine Anwendung von Corticosteroiden erforderlich machte, trat bei < 0,1 % (3/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten.
Immunvermittelte Gesichtsparese
Eine Gesichtsparese trat bei < 0,1 % (1/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die Zeit bis zum Auftreten betrug 29 Tage. Die Dauer lag bei 1,1 Monaten. Das Ereignis erforderte keine Anwendung von Corticosteroiden und führte nicht zum Absetzen von Atezolizumab.
Immunvermittelte Myelitis
Eine Myelitis trat bei < 0,1 % (1/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die Zeit bis zum Auftreten betrug 3 Tage. Das Ereignis erforderte die Anwendung von Corticosteroiden, führte jedoch nicht zum Absetzen von Atezolizumab.
Myastheniesyndrom
Myasthenia gravis trat bei < 0,1 % (2/5 039) Patienten auf (einschließlich eines tödlichen Ereignisses), die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 2,6 Monate (Bereich: 1,2 Monate bis 4 Monate).
Immunvermittelte Pankreatitis
Eine Pankreatitis, einschließlich erhöhter Amylase‑ und Lipasespiegel, trat bei 0,8 % (40/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 5 Monate (Bereich: 0 Tage bis 24,8 Monate). Die mediane Dauer betrug 24 Tage (Bereich: 3 Tage bis 40,4+ Monate; + kennzeichnet einen zensierten Wert). Eine Pankreatitis führte bei 3 (< 0,1 %) Patienten zum Absetzen der Behandlung mit Atezolizumab. Eine Pankreatitis, die eine Anwendung von Corticosteroiden erforderlich machte, trat bei 0,2 % (8/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten.
Immunvermittelte Myokarditis
Eine Myokarditis trat bei < 0,1 % (5/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Von diesen 5 Patienten kam es bei einem im adjuvanten NSCLC-Setting zu einem tödlichen Ereignis. Die mediane Zeit bis zum Auftreten betrug 3,7 Monate (Bereich: 1,5 bis 4,9 Monate). Die mediane Dauer betrug 14 Tage (Bereich: 12 Tage bis 2,8 Monate). Eine Myokarditis führte bei 3 (< 0,1 %) Patienten zum Absetzen von Atezolizumab. Bei 3 (< 0,1 %) Patienten war die Anwendung von Corticosteroiden erforderlich.
Immunvermittelte Nephritis
Eine Nephritis trat bei 0,2 % (11/5 039) der Patienten auf, die Atezolizumab erhielten. Die mediane Zeit bis zum Auftreten betrug 5,1 Monate (Bereich: 3 Tage bis 17,5 Monate). Nephritis führte bei 5 (≤ 0,1 %) Patienten zum Absetzen von Atezolizumab. Bei 5 (0,1 %) Patienten war die Anwendung von Corticosteroiden erforderlich.
Immunvermittelte Myositis
Eine Myositis trat bei 0,6 % (32/5 039) der Patienten auf, die Atezolizumab als Monotherapie erhielten. Die mediane Zeit bis zum Auftreten betrug 3,5 Monate (Bereich: 12 Tage bis 11,5 Monate). Die mediane Dauer betrug 3,2 Monate (Bereich: 9 Tage bis 51,1+ Monate; + kennzeichnet einen zensierten Wert). Eine Myositis führte bei 6 Patienten (0,1 %) zum Absetzen von Atezolizumab. Bei 10 Patienten (0,2 %) war die Anwendung von Corticosteroiden erforderlich.
Immunvermittelte schwerwiegende unerwünschte Reaktionen der Haut
Schwerwiegende unerwünschte Reaktionen der Haut (SCARs) traten bei 0,6 % (30/5 039) der Patienten auf, die eine Monotherapie mit Atezolizumab erhielten. Von den 30 Patienten erlitt einer ein tödliches Ereignis. Die mediane Zeit bis zum Auftreten betrug 4,8 Monate (Bereich: 3 Tage bis 15,5 Monate). Die mediane Dauer betrug 2,4 Monate (Bereich: 1 Tag bis 37,5+ Monate; + kennzeichnet einen zensierten Wert). Bei 3 (< 0,1 %) Patienten führten SCARs zum Absetzen von Atezolizumab. Bei 0,2 % (9/5 039) der Patienten, die eine Monotherapie mit Atezolizumab erhielten, traten SCARs auf, die den Einsatz von systemischen Corticosteroiden erforderten.
Immunvermittelte Erkrankungen des Perikards
Erkrankungen des Perikards traten bei 1 % (49/5 039) der Patienten auf, die eine Monotherapie mit Atezolizumab erhielten. Die mediane Zeit bis zum Auftreten betrug 1,4 Monate (Bereich: 6 Tage bis 17,5 Monate). Die mediane Dauer betrug 2,5 Monate (Bereich: 0 bis 51,5+ Monate; + kennzeichnet einen zensierten Wert). Erkrankungen des Perikards führten bei 3 (< 0,1 %) Patienten zum Absetzen von Tecentriq. Erkrankungen des Perikards, die die Anwendung von Corticosteroiden erforderten, traten bei 0,2 % (7/5 039) der Patienten auf.
Wirkungen auf die Klasse der Immuncheckpoint-Inhibitoren
Es wurden Fälle der folgenden Nebenwirkungen während der Behandlung mit anderen Immuncheckpoint-Inhibitoren berichtet, die auch während der Behandlung mit Atezolizumab auftreten können: exokrine Pankreasinsuffizienz.
Immunogenität
Subkutane Darreichungsform
In IMscin001 war die Inzidenz therapiebedingter Anti-Atezolizumab-Antikörper bei Patienten, die mit Tecentriq subkutan und intravenös behandelt wurden, nach einer medianen Behandlungsdauer von 2,8 Monaten vergleichbar (19,5 % [43/221] bzw. 13,9 % [15/108]). Die Inzidenz therapiebedingter Anti-rHuPH20-Antikörper bei Patienten, die mit Tecentriq subkutan behandelt wurden, betrug 5,4 % (12/224). Die klinische Relevanz der Entwicklung von Anti-rHuPH20-Antikörpern nach einer Behandlung mit Tecentriq Injektionslösung ist nicht bekannt.
Intravenöse Darreichungsform
In mehreren Phase-II- und -III-Studien entwickelten 13,1 % bis 54,1 % der Patienten therapiebedingte Antikörper gegen den Wirkstoff (antidrug antibodies, ADA). Patienten, die therapiebedingte ADA entwickelten, wiesen bei Behandlungsbeginn tendenziell schlechtere Gesundheits- und Krankheitsmerkmale auf. Diese Unausgewogenheiten in den Gesundheits- und Krankheitsmerkmalen zu Behandlungsbeginn können die Interpretation von pharmakokinetischen (PK) Analysen sowie Wirksamkeits- und Sicherheitsanalysen verzerren. Um die Auswirkung von ADA auf die Wirksamkeit zu beurteilen, wurden explorative Analysen durchgeführt, die um die Unausgewogenheiten bei den Gesundheits- und Krankheitsmerkmalen zu Behandlungsbeginn bereinigt waren. Diese Analysen schlossen eine mögliche Abschwächung des Wirksamkeitsvorteils bei Patienten, die ADA entwickelten, im Vergleich zu Patienten, die keine ADA entwickelten, nicht aus. Die mediane Zeit bis zum Auftreten von ADA reichte von 3 Wochen bis 5 Wochen.
In gepoolten Datensätzen von Patienten, die Atezolizumab als Monotherapie (n = 3 460) beziehungsweise als Kombinationstherapie (n = 2 285) erhalten haben, wurden für die ADA-positive Population im Vergleich zur ADA-negativen Population die folgenden Anteile unerwünschter Ereignisse (adverse events, AEs) beobachtet: (bei Monotherapie) Grad 3 - 4 AEs 46,2 % vs. 39,4 %, schwerwiegende unerwünschte Ereignisse (serious adverse events, SAEs) 39,6 % vs. 33,3 %, AEs, die zum Abbruch der Behandlung führten, 8,5 % vs. 7,8 %; (bei Kombinationstherapie) Grad 3 - 4 AEs 63,9 % vs. 60,9 %, SAEs 43,9 % vs. 35,6 %, AEs, die zum Abbruch der Behandlung führten, 22,8 % vs. 18,4 %. Jedoch kann man anhand der verfügbaren Daten keine sicheren Schlussfolgerungen über mögliche Muster von Nebenwirkungen ziehen.
Kinder und Jugendliche
Die Sicherheit von Atezolizumab bei Kindern und Jugendlichen ist nicht ausreichend belegt. In einer klinischen Prüfung mit 69 pädiatrischen Patienten (< 18 Jahre) wurden keine neuen Sicherheitssignale beobachtet und das Sicherheitsprofil war vergleichbar mit dem von Erwachsenen.
Ältere Patienten
Bezüglich Sicherheit wurden insgesamt keine Unterschiede zwischen Patienten im Alter von < 65, 65 – 74 und 75 – 84 Jahren festgestellt, die Atezolizumab (intravenös oder subkutan) als Monotherapie erhielten. Die Daten für Patienten im Alter von ≥ 85 Jahren sind zu begrenzt, um aussagekräftige Schlussfolgerungen über diese Population zu ziehen.
In der Studie IMpower150 war ein Alter von ≥ 65 Jahren bei Patienten, die Atezolizumab in Kombination mit Bevacizumab, Carboplatin und Paclitaxel erhielten, mit einem erhöhten Risiko für die Entwicklung von unerwünschten Ereignissen verbunden. Die Daten für Patienten im Alter von ≥ 75 Jahren aus den Studien IMpower150, IMpower133, IMpower110 und IMscin001 sind zu begrenzt, um Rückschlüsse zuzulassen. In der IPSOS-Studie zur Erstlinienbehandlung des NSCLC, bei Patienten, die für eine platinbasierte Therapie ungeeignet sind, gab es bezüglich der Sicherheit insgesamt keine Unterschiede im Sicherheitsprofil der Erstlinien-Atezolizumab-Monotherapie zwischen den Alterssubgruppen der Patienten.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen‑Risiko‑Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51‑59
63225 Langen
Tel: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
anzuzeigen.
Es liegen keine Informationen zu Überdosierungen von Atezolizumab vor.
Im Fall einer Überdosierung sind die Patienten engmaschig auf Anzeichen oder Symptome von Nebenwirkungen zu überwachen und eine geeignete symptomatische Behandlung ist einzuleiten.
Pharmakotherapeutische Gruppe: Antineoplastische Mittel, monoklonale Antikörper und Antikörper-Wirkstoff-Konjugate, PD-1/PDL-1 (Programmed Cell Death-1-Rezeptor/Programmed Cell Death-Ligand-1)-Inhibitoren. ATC‑Code: L01FF05.
Tecentriq Injektionslösung enthält den Wirkstoff Atezolizumab, der für die therapeutische Wirkung dieses Arzneimittels verantwortlich ist und rekombinante humane Hyaluronidase (rHuPH20), ein Enzym, das bei subkutaner Verabreichung zur Steigerung der Dispersion und Resorption von co-formulierten Wirkstoffen verwendet wird.
Wirkmechanismus
PD‑L1 (programmed death‑ligand 1) kann auf Tumorzellen und/oder tumorinfiltrierenden Immunzellen exprimiert werden und zur Inhibierung der antitumoralen Immunantwort im Mikromilieu des Tumors beitragen. Bindet PD‑L1 an die Rezeptoren PD‑1 und B7.1 auf T‑Zellen und Antigen‑präsentierenden Zellen, wird die zytotoxische T‑Zell‑Aktivität, die T‑Zell‑Proliferation und die Zytokin‑Produktion unterdrückt.
Atezolizumab ist ein im Fc‑Teil modifizierter, humanisierter monoklonaler Immunglobulin G1(IgG1)‑Antikörper, der direkt an PD‑L1 bindet und zu einer dualen Blockade der PD‑1‑ und B7.1‑Rezeptoren führt. Dadurch wird die PD‑L1/PD‑1‑vermittelte Hemmung der Immunantwort aufgehoben, wobei die antitumorale Immunantwort reaktiviert wird, ohne dass eine Antikörper‑induzierte zelluläre Zytotoxizität ausgelöst wird. Atezolizumab hat keinen Einfluss auf die PD‑L2/PD‑1‑Interaktion, sodass die PD‑L2/PD‑1‑vermittelten inhibitorischen Signale fortbestehen.
Klinische Wirksamkeit und Sicherheit
Urothelkarzinom
Intravenöse Darreichungsform
IMvigor211 (GO29294): Randomisierte Studie an Patienten mit lokal fortgeschrittenem oder metastasiertem UC, die zuvor mit Chemotherapie behandelt wurden
Eine unverblindete, multizentrische, internationale, randomisierte Studie der Phase III (IMvigor211) wurde zur Bewertung der Wirksamkeit und Sicherheit von Atezolizumab im Vergleich zu Chemotherapie (Vinflunin, Docetaxel oder Paclitaxel gemäß Ermessen des Prüfarztes) bei Patienten mit lokal fortgeschrittenem oder metastasiertem UC durchgeführt, die während oder im Anschluss an ein platinhaltiges Chemotherapieregime eine Krankheitsprogression erlitten. Von der Studie ausgeschlossen waren Patienten mit einer Autoimmunerkrankung in der Vorgeschichte, aktiven oder Corticosteroid‑abhängigen Gehirnmetastasen, einer Verabreichung von attenuierten Lebendimpfstoffen innerhalb von 28 Tagen vor Studieneinschluss, einer Verabreichung von systemischen Immunstimulanzien innerhalb von 4 Wochen oder von systemischen Immunsuppressiva innerhalb von 2 Wochen vor Einschluss. Tumorbewertungen wurden in den ersten 54 Wochen alle 9 Wochen und anschließend alle 12 Wochen durchgeführt. Tumorproben wurden prospektiv auf PD‑L1‑Expression auf tumorinfiltrierenden Immunzellen (IC) getestet und die Ergebnisse für die Einteilung in PD‑L1‑Expression‑Subgruppen für die nachfolgend beschriebenen Analysen verwendet.
Insgesamt wurden 931 Patienten in die Studie aufgenommen. Die Patienten wurden im Verhältnis 1:1 randomisiert und erhielten entweder Atezolizumab oder Chemotherapie. Die Randomisierung war stratifiziert nach Chemotherapie (Vinflunin vs. Taxan), PD‑L1‑Expression-Status auf den IC (< 5 % vs. ≥ 5 %), Anzahl der prognostischen Risikofaktoren (0 vs. 1 – 3) und Lebermetastasen (ja vs. nein). Zu den prognostischen Risikofaktoren gehörten ein zeitlicher Abstand zur vorherigen Chemotherapie von < 3 Monaten, ECOG‑Performance‑Status > 0 und Hämoglobin < 10 g/dl.
Atezolizumab wurde in einer Fixdosis von 1 200 mg alle 3 Wochen als intravenöse Infusion verabreicht. Eine Dosisreduktion war nicht zulässig. Die Patienten wurden bis zum Verlust des klinischen Nutzens gemäß Bewertung des Prüfarztes oder bis zum Auftreten inakzeptabler Toxizität behandelt. Vinflunin wurde in einer Dosierung von 320 mg/m2 an Tag 1 jedes 3‑Wochen‑Zyklus als intravenöse Infusion verabreicht, bis eine Krankheitsprogression oder inakzeptable Toxizität auftrat. Paclitaxel wurde in einer Dosierung von 175 mg/m2 an Tag 1 jedes 3‑Wochen‑Zyklus als 3‑stündige intravenöse Infusion verabreicht, bis eine Krankheitsprogression oder inakzeptable Toxizität auftrat. Docetaxel wurde in einer Dosierung von 75 mg/m2 an Tag 1 jedes 3‑Wochen‑Zyklus als intravenöse Infusion verabreicht, bis eine Krankheitsprogression oder inakzeptable Toxizität auftrat. Bei allen behandelten Patienten betrug die mediane Dauer der Behandlung 2,8 Monate im Atezolizumab‑Arm, 2,1 Monate in den Vinflunin‑ und Paclitaxel‑Armen und 1,6 Monate im Docetaxel‑Arm.
Die demographische Verteilung sowie die Krankheitsmerkmale bei Behandlungsbeginn der Primäranalyse‑Population waren zwischen den Behandlungsarmen gut ausgewogen. Das mediane Alter lag bei 67 Jahren (Bereich: 31 bis 88); 77,1 % der Patienten waren männlich. Die Mehrheit der Patienten war kaukasischer Herkunft (72,1 %); 53,9 % der Patienten im Chemotherapie‑Arm erhielten Vinflunin. 71,4 % der Patienten hatten mindestens einen schlechten prognostischen Risikofaktor und 28,8 % hatten zu Behandlungsbeginn Lebermetastasen. Der ECOG‑Performance‑Status betrug zu Behandlungsbeginn 0 (45,6 %) oder 1 (54,4 %). Bei 71,1 % der Patienten lag der Primärtumor in der Blase, und 25,4 % der Patienten hatten ein UC der oberen Harnwege. 24,2 % der Patienten hatten nur eine vorherige platinhaltige adjuvante oder neoadjuvante Therapie erhalten, und es war innerhalb von 12 Monaten zu einer Krankheitsprogression gekommen.
Der primäre Wirksamkeitsendpunkt in der Studie IMvigor211 ist das Gesamtüberleben (overall survival, OS). Die sekundären Wirksamkeitsendpunkte sind die anhand der RECIST‑Kriterien (Response Evaluation Criteria in Solid Tumors) v1.1 vom Prüfarzt bewertete objektive Ansprechrate (objective response rate, ORR), das progressionsfreie Überleben (progression free survival, PFS) sowie die Dauer des Ansprechens (duration of response, DOR). Vergleiche bezüglich OS zwischen Behandlungs‑ und Kontrollarm innerhalb der IC2/3‑, IC1/2/3‑ und ITT‑Populationen (Intention‑to‑treat, d. h. all comers, alle Patienten, die die Einschlusskriterien erfüllen) wurden nach folgendem hierarchischem festgelegtem Verfahren getestet, das auf einem stratifizierten zweiseitigen Log‑Rank‑Test (5 %) basierte: Schritt 1) IC2/3‑Population; Schritt 2) IC1/2/3‑Population; Schritt 3) All‑comers‑Population. Die OS‑Ergebnisse für Schritt 2 und Schritt 3 konnten nur formal auf statistische Signifikanz getestet werden, wenn das Ergebnis im vorangegangenen Schritt statistisch signifikant war.
Die mediane Nachbeobachtungszeit des Überlebens beträgt 17 Monate. Die Primäranalyse der Studie IMvigor211 erreichte den primären Endpunkt für OS nicht. Atezolizumab zeigte keinen statistisch signifikanten Überlebensvorteil im Vergleich zu Chemotherapie bei Patienten mit vorbehandeltem, lokal fortgeschrittenem oder metastasiertem UC. Gemäß der vorgegebenen hierarchischen Testreihenfolge wurde die IC2/3‑Population zuerst geprüft und zeigte ein Hazard Ratio (HR) für OS von 0,87 (95‑%‑KI: 0,63; 1,21; medianes OS 11,1 vs. 10,6 Monate für Atezolizumab bzw. Chemotherapie). Der stratifizierte Log‑Rank‑p‑Wert betrug 0,41 und somit wurden die Ergebnisse in dieser Population als nicht statistisch signifikant gewertet. Die Konsequenz war, dass keine formalen Tests auf statistische Signifikanz des OS in den IC1/2/3‑ oder All‑comers‑Populationen vorgenommen werden konnten. Die Ergebnisse dieser Analysen gelten als explorativ. Die wesentlichen Ergebnisse der All‑comer‑Population sind in Tabelle 4 zusammengefasst. Die Kaplan‑Meier‑Kurve für OS in der All‑comer‑Population ist in Abbildung 1 dargestellt.
Eine explorative, aktualisierte Analyse zum Überleben wurde in der ITT-Population mit einer medianen Nachbeobachtungszeit des Überlebens von 34 Monaten durchgeführt. Das mediane OS betrug im Atezolizumab-Arm 8,6 Monate (95‑%‑KI: 7,8; 9,6) und im Chemotherapie-Arm 8,0 Monate (95‑%‑KI: 7,2; 8,6), mit einer Hazard Ratio von 0,82 (95‑%‑KI: 0,71; 0,94). In Übereinstimmung mit dem bei der Primäranalyse beobachteten Trend bezüglich 12-Monats-OS-Raten wurden in der ITT-Population bei Patienten im Atezolizumab-Arm im Vergleich zu Patienten im Chemotherapie-Arm numerisch höhere 24-Monats- und 30-Monats-OS-Raten beobachtet. Der Prozentsatz der noch lebenden Patienten betrug in Monat 24 (KM-Schätzer) im Chemotherapie-Arm 12,7 % und im Atezolizumab-Arm 22,5 %. In Monat 30 (KM-Schätzer) betrug der Prozentsatz der noch lebenden Patienten im Chemotherapie-Arm 9,8 % und im Atezolizumab-Arm 18,1 %.
Tabelle 4: Zusammenfassung der Wirksamkeit in der All‑comer‑Population (IMvigor211)
Wirksamkeitsendpunkte |
Atezolizumab |
Chemotherapie |
Primäre Wirksamkeitsendpunkte | ||
Gesamtüberleben (OS)* | ||
Anz. der Todesfälle (%) |
324 (69,4 %) |
350 (75,4 %) |
Mediane Zeit bis zum Ereignis (Monate) |
8,6 |
8,0 |
95‑%‑KI |
7,8; 9,6 |
7,2; 8,6 |
Stratifizierteǂ Hazard Ratio (95‑%‑KI) |
0,85 (0,73; 0,99) |
|
12‑Monats‑Gesamtüberleben (%)** |
39,2 % |
32,4 % |
Sekundäre und explorative Endpunkte | ||
Vom Prüfarzt bewertetes PFS (RECIST v1.1) | ||
Anz. der Ereignisse (%) |
407 (87,2 %) |
410 (88,4 %) |
Mediane Dauer des PFS (Monate) |
2,1 |
4,0 |
95‑%‑KI |
2,1; 2,2 |
3,4; 4,2 |
Stratifizierte Hazard Ratio (95‑%‑KI) |
1,10 (0,95; 1,26) |
|
Vom Prüfarzt bewertete ORR (RECIST v1.1) |
n = 462 |
n = 461 |
Anz. der Patienten mit bestätigtem Ansprechen (%) |
62 (13,4 %) |
62 (13,4 %) |
95‑%‑KI |
10,45; 16,87 |
10,47; 16,91 |
Anz. mit vollständigem Ansprechen (%) |
16 (3,5 %) |
16 (3,5 %) |
Anz. mit teilweisem Ansprechen (%) |
46 (10,0 %) |
46 (10,0 %) |
Anz. mit stabiler Erkrankung |
92 (19,9 %) |
162 (35,1 %) |
Vom Prüfarzt bewertete DOR (RECIST v1.1) |
n = 62 |
n = 62 |
Median in Monaten *** |
21,7 |
7,4 |
95‑%‑KI |
13,0; 21,7 |
6,1; 10,3 |
KI = Konfidenzintervall; DOR = duration of response (Dauer des Ansprechens); ORR = objective response rate (objektive Ansprechrate); OS = overall survival (Gesamtüberleben); PFS = progression‑free survival (progressionsfreies Überleben); RECIST = Response Evaluation Criteria in Solid Tumours v1.1.
* Eine Analyse des Gesamtüberlebens in der Gesamtpopulation wurde basierend auf dem stratifizierten Log‑Rank‑Test durchgeführt und das Ergebnis nur zu deskriptiven Zwecken angegeben (p = 0,0378); gemäß prä-spezifizierter Testhierarchie kann der p‑Wert für die OS‑Analyse in der All‑comer‑Population nicht als statistisch signifikant betrachtet werden.
ǂ Stratifiziert nach Chemotherapie (Vinflunin vs. Taxan), Status auf IC (< 5 % vs. ≥ 5 %), Anzahl prognostischer Risikofaktoren (0 vs. 1 – 3) und Lebermetastasen (ja vs. nein).
** Basierend auf Kaplan‑Meier-Schätzung
*** Ansprechen hielt bei 63 % der Responder im Atezolizumab‑Arm an und bei 21 % der Responder im Chemotherapie‑Arm.
Abbildung 1: Kaplan‑Meier‑Kurve für das Gesamtüberleben (IMvigor211)
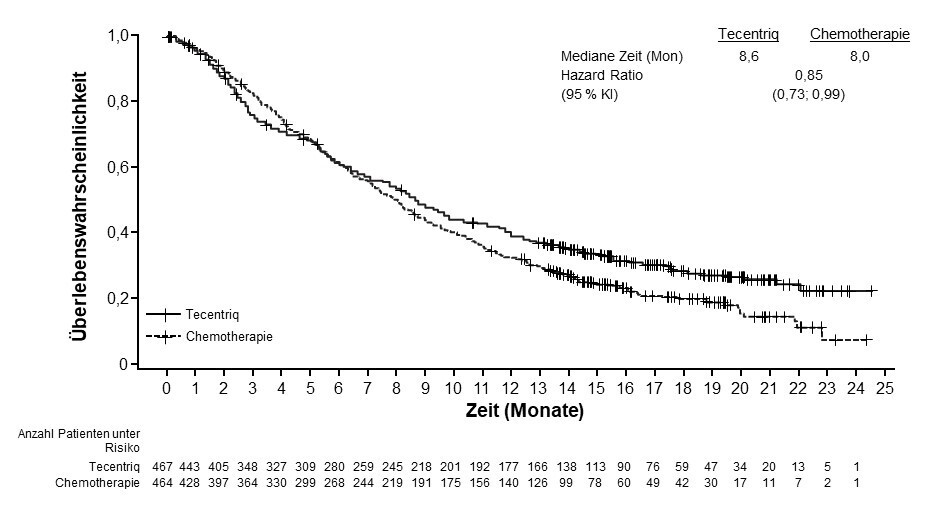
IMvigor210 (GO29293): Einarmige Studie an nicht vorbehandelten UC‑Patienten, die nicht für eine Cisplatin‑Therapie geeignet sind, und an UC‑Patienten, die bereits mit Chemotherapie behandelt wurden
Eine multizentrische, internationale, einarmige klinische Prüfung der Phase II mit zwei Kohorten, IMvigor210, wurde bei Patienten mit lokal fortgeschrittenem oder metastasiertem UC (auch als urotheliales Blasenkarzinom bezeichnet) durchgeführt.
In die Studie wurden insgesamt 438 Patienten eingeschlossen und in zwei Kohorten aufgeteilt. In Kohorte 1 wurden vorher nicht behandelte Patienten mit lokal fortgeschrittenem oder metastasiertem UC eingeschlossen, die für eine cisplatinbasierte Chemotherapie ungeeignet waren oder bei denen mindestens 12 Monate nach Behandlung mit einem platinhaltigen neoadjuvanten oder adjuvanten Chemotherapie‑Regime eine Krankheitsprogression auftrat. In Kohorte 2 wurden Patienten eingeschlossen, die mindestens ein platinbasiertes Chemotherapie‑Regime zur Behandlung des lokal fortgeschrittenen oder metastasierten UC erhalten hatten oder deren Erkrankung innerhalb von 12 Monaten nach einer Behandlung mit einem platinhaltigen neoadjuvanten oder adjuvanten Chemotherapie‑Regime progredient war.
In Kohorte 1 wurden 119 Patienten mit 1 200 mg Atezolizumab als intravenöse Infusion alle 3 Wochen bis zur Krankheitsprogression behandelt. Das mediane Alter betrug 73 Jahre. Die meisten Patienten waren männlich (81 %) und der Großteil der Patienten war kaukasischer Herkunft (91 %).
Kohorte 1 umfasste 45 Patienten (38 %) mit ECOG‑Performance‑Status 0, 50 Patienten (42 %) mit ECOG‑Performance‑Status 1 und 24 Patienten (20 %) mit ECOG‑Performance‑Status 2, 35 Patienten (29 %) ohne Risikofaktoren nach Bajorin (ECOG‑Performance‑Status ≥ 2 und viszerale Metastasen), 66 Patienten (56 %) mit einem Risikofaktor nach Bajorin und 18 Patienten (15 %) mit zwei Risikofaktoren nach Bajorin, 84 Patienten (71 %) mit Nierenfunktionsstörung (glomeruläre Filtrationsrate [GFR] < 60 ml/min) und 25 Patienten (21 %) mit Lebermetastasen.
Der primäre Wirksamkeitsendpunkt in Kohorte 1 war die bestätigte objektive Ansprechrate (ORR), bewertet durch eine unabhängige Prüfeinrichtung (Independent Review Facility = IRF) unter Verwendung der RECIST‑Kriterien (Response Evaluation Criteria in Solid Tumors) v1.1.
Die Primäranalyse wurde durchgeführt, als alle Patienten für mindestens 24 Wochen nachbeobachtet worden waren. Die mediane Dauer der Behandlung betrug 15,0 Wochen, und die mediane Nachbeobachtungszeit des Überlebens betrug 8,5 Monate bei allen Patienten (all comers). Zwar wurden klinisch relevante IRF bewertete ORR gemäß RECIST v1.1 nachgewiesen, jedoch wurde die statistische Signifikanz für den primären Endpunkt – im Vergleich zu einer prä‑spezifizierten historischen Kontrollansprechrate von 10 % – nicht erreicht. Die bestätigten ORR gemäß IRF‑RECIST v1.1 betrugen 21,9 % (95‑%‑KI: 9,3; 40,0) für Patienten mit einer PD‑L1‑Expression ≥ 5 %, 18,8 % (95‑%‑KI: 10,9; 29,0) für Patienten mit PD‑L1‑Expression ≥ 1 % und 19,3 % (95‑%‑KI: 12,7; 27,6) bei allen Patienten (all comers). Die mediane Dauer des Ansprechens (DOR) wurde weder in einer PD‑L1‑Expression‑Subgruppe noch bei allen Patienten (all comers) erreicht. Das OS war mit einer Ereignis‑pro‑Patient‑Rate von circa 40 % noch nicht aussagekräftig. Das mediane OS für alle Patienten‑Subgruppen (PD‑L1‑Expression ≥ 5 % und ≥ 1 %) und alle Patienten (all comers) lag bei 10,6 Monaten.
Eine aktualisierte Analyse wurde für Kohorte 1 nach einer medianen Nachbeobachtungszeit des Überlebens von 17,2 Monaten durchgeführt und ist in Tabelle 5 zusammengefasst. Die DOR wurde weder in einer PD‑L1‑Expression‑Subgruppe noch bei allen Patienten (all comers) erreicht.
Tabelle 5: Zusammenfassung der aktualisierten Wirksamkeit (IMvigor210, Kohorte 1)
Wirksamkeitsendpunkt |
PD‑L1-Expression von |
PD‑L1-Expression von ≥ 1 % bei IC |
Alle Patienten (all comers) |
ORR (bewertet durch IRF; RECIST v1.1) |
n = 32 |
n = 80 |
n = 119 |
Anzahl der Patienten mit Ansprechen (%) |
9 (28,1 %) |
19 (23,8 %) |
27 (22,7 %) |
95‑%‑KI |
13,8; 46,8 |
15,0; 34,6 |
15,5; 31,3 |
Anzahl mit vollständigem Ansprechen (%) |
4 (12,5 %) |
8 (10,0 %) |
11 (9,2 %) |
Anzahl mit teilweisem Ansprechen (%) |
5 (15,6 %) |
11 (13,8 %) |
16 (13,4 %) |
DOR (bewertet durch IRF; RECIST v1.1) |
n = 9 |
n = 19 |
n = 27 |
Patienten mit Ereignis (%) |
3 (33,3 %) |
5 (26,3 %) |
8 (29,6 %) |
Median (Monate) (95‑%‑KI) |
NE (11,1; NE) |
NE (NE) |
NE (14,1; NE) |
PFS (bewertet durch IRF; RECIST v1.1) |
n = 32 |
n = 80 |
n = 119 |
Patienten mit Ereignis (%) |
24 (75,0 %) |
59 (73,8 %) |
88 (73,9 %) |
Median (Monate) (95‑%‑KI) |
4,1 (2,3; 11,8) |
2,9 (2,1; 5,4) |
2,7 (2,1; 4,2) |
OS |
n = 32 |
n = 80 |
n = 119 |
Patienten mit Ereignis (%) |
18 (56,3 %) |
42 (52,5 %) |
59 (49,6 %) |
Median (Monate) (95‑%‑KI) |
12,3 (6,0; NE) |
14,1 (9,2; NE) |
15,9 (10,4; NE) |
1‑Jahresrate des Gesamtüberlebens (%) |
52,4 % |
54,8 % |
57,2 % |
KI = Konfidenzintervall; DOR = duration of response (Dauer des Ansprechens); IC = tumour‑infiltrating immune cells (tumorinfiltrierende Immunzellen); IRF = independent review facility (unabhängige Prüfeinrichtung); NE = not estimable (nicht schätzbar); ORR = objective response rate (objektive Ansprechrate); OS = overall survival (Gesamtüberleben); PFS = progression‑free survival (progressionsfreies Überleben); RECIST = Response Evaluation Criteria in Solid Tumours v1.1.
Zum Zeitpunkt der finalen Analyse für Kohorte 1 hatten die Patienten eine mediane Nachbeobachtungszeit des Überlebens von 96,4 Monaten. Das mediane OS betrug 12,3 Monate (95‑%‑KI: 6,0; 49,8) bei Patienten mit einer PD-L1-Expression ≥ 5 % (Patienten gemäß dem Anwendungsgebiet).
Ko‑primäre Wirksamkeitsendpunkte in Kohorte 2 waren die bestätigte, IRF bewertete ORR unter Verwendung von RECIST v1.1 und die Prüfarzt bewertete ORR unter Verwendung der modifizierten RECIST‑Kriterien (mRECIST). Es wurden 310 Patienten mit Atezolizumab 1 200 mg als intravenöse Infusion alle 3 Wochen bis zum Verlust des klinischen Nutzens behandelt. Die Primäranalyse von Kohorte 2 wurde durchgeführt, als alle Patienten für mindestens 24 Wochen nachbeobachtet worden waren. In der Studie wurden die ko‑primären Endpunkte aus Kohorte 2 erreicht. Die Studie zeigte eine statistisch signifikante, IRF bewertete ORR unter Verwendung von RECIST v1.1 und eine statistisch signifikante Prüfarzt bewertete ORR unter Verwendung von mRECIST, verglichen mit einer prä‑spezifizierten historischen Kontrollansprechrate von 10 %.
Eine Analyse für Kohorte 2 wurde auch nach einer medianen Nachbeobachtungszeit des Überlebens von 21,1 Monaten durchgeführt. Die bestätigten ORR gemäß IRF‑RECIST v1.1 betrugen 28,0 % (95‑%‑KI: 19,5; 37,9) bei Patienten mit PD‑L1‑Expression ≥ 5 %, 19,3 % (95‑%‑KI: 14,2; 25,4) bei Patienten mit PD‑L1‑Expression ≥ 1 % und 15,8 % (95‑%‑KI: 11,9; 20,4) bei allen Patienten (all comers). Die bestätigten ORR gemäß Prüfarzt bewerteter mRECIST betrugen 29,0 % (95‑%‑KI: 20,4; 38,9) bei Patienten mit PD‑L1‑Expression ≥ 5 %, 23,7 % (95‑%‑KI: 18,1; 30,1) bei Patienten mit PD‑L1‑Expression ≥ 1 % und 19,7 % (95‑%‑KI: 15,4; 24,6) bei allen Patienten (all comers). Die Rate des vollständigen Ansprechens gemäß IRF‑RECIST v1.1 in der Gesamtpopulation (all comer) betrug 6,1 % (95‑%‑KI: 3,7; 9,4). In Kohorte 2 wurde die mediane DOR in keiner PD‑L1‑Expression‑Subgruppe oder bei allen Patienten erreicht; diese wurde jedoch bei Patienten mit PD‑L1‑Expression < 1 % erreicht (13,3 Monate; 95‑%‑KI: 4.2; NE). Die OS Rate nach 12 Monaten betrug bei allen Patienten (all comers) 37 %.
Zum Zeitpunkt der finalen Analyse für Kohorte 2 hatten die Patienten eine mediane Nachbeobachtungszeit des Überlebens von 46,2 Monaten. Das mediane OS betrug 11,9 Monate (95‑%‑KI: 9,0; 22,8) bei Patienten mit einer PD-L1-Expression ≥ 5 %, 9,0 Monate (95‑%‑KI: 7,1; 11,1) bei Patienten mit einer PD-L1-Expression ≥ 1 % und 7,9 Monate (95‑%‑KI: 6,7; 9,3) bei allen Patienten.
IMvigor130 (WO30070): Studie der Phase III mit Atezolizumab Monotherapie und in Kombination mit einer platinbasierten Chemotherapie an Patienten mit unbehandeltem lokal fortgeschrittenem oder metastasiertem Urothelkarzinom
IMvigor130, eine multizentrische, randomisierte, placebokontrollierte, teilweise verblindete (nur Arme A und C) Studie der Phase III wurde durchgeführt, um die Wirksamkeit und Sicherheit einer Atezolizumab + platinbasierten Kombinations-Chemotherapie (d. h. entweder Cisplatin oder Carboplatin mit Gemcitabin), Arm A, oder Atezolizumab-Monotherapie (Arm B, unverblindeter Arm) im Vergleich zu Placebo + platinbasierter Kombinations-Chemotherapie (Arm C) bei Patienten mit lokal fortgeschrittenem oder metastasiertem UC zu untersuchen, die keine vorherige systemische Behandlung im metastasierten Setting erhalten haben. Die ko-primären Wirksamkeitsendpunkte wurden in einem hierarchisch festgelegten Verfahren analysiert und waren Prüfarzt bewertetes progressionsfreies Überleben (PFS) in Arm A gegenüber Arm C und Gesamtüberleben (OS) in Arm A gegenüber C und anschließend Arm B gegenüber C. Das Gesamtüberleben war für den Vergleich von Arm A gegenüber Arm C statistisch nicht signifikant, so dass keine weiteren formalen Analysen gemäß der vordefinierten hierarchischen Analysenreihenfolge durchgeführt werden konnten.
Basierend auf der Empfehlung eines unabhängigen Datenkontrollkomitees (iDMC) wurde nach einem frühen Review der Überlebensdaten der Einschluss von Patienten, deren Tumoren eine niedrige PD-L1-Expression aufwiesen (weniger als 5 % der Immunzellen wurden mittels Immunhistochemie mit dem VENTANA PD-L1-[SP142]-Test positiv auf PD-L1 gefärbt), in den Atezolizumab-Monotherapie-Behandlungsarm gestoppt, nachdem in dieser Subgruppe bei einer ungeplanten frühen Analyse ein verringertes Gesamtüberleben beobachtet wurde. Dies geschah jedoch, nachdem die überwiegende Mehrheit der Patienten bereits aufgenommen war.
Von den 719 Patienten, die in den Behandlungsarm mit Atezolizumab-Monotherapie (n = 360) bzw. Chemotherapie allein (n = 359) aufgenommen wurden, waren 50 bzw. 43 Patienten nach den Galsky-Kriterien nicht für Cisplatin geeignet und hatten Tumoren mit hoher PD-L1-Expression (≥ 5 % der Immunzellen hatten nach Immunhistochemie mit dem VENTANA PD-L1-[SP142]-Test eine positive Färbung auf PD-L1). In einer explorativen Analyse in dieser Patientenuntergruppe betrug die nicht stratifizierte HR für OS 0,56 (95‑%‑KI: 0,34; 0,91). Das mediane OS betrug 18,6 Monate (95‑%‑KI: 14,0; 49,4) im Atezolizumab-Monotherapiearm vs. 10,0 Monate (95‑%‑KI: 7,4; 18,1) im Chemotherapiearm (siehe Abbildung 2).
Abbildung 2: Kaplan-Meier-Kurve für das Gesamtüberleben bei Patienten, die für eine Behandlung mit Cisplatin ungeeignet sind und deren Tumoren eine hohe PD-L1-Expression aufweisen (Arm B vs. Arm C)
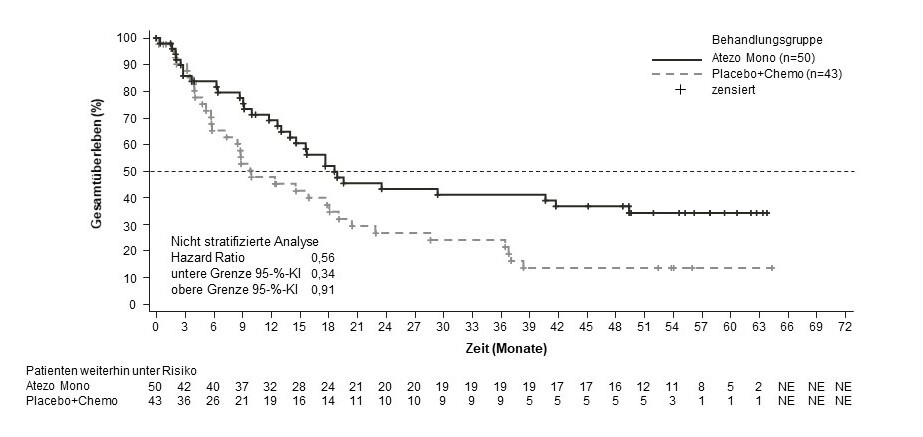
Nicht‑kleinzelliges Lungenkarzinom
Adjuvante Behandlung des NSCLC im Frühstadium
Intravenöse Darreichungsform
IMpower010 (GO29527): Randomisierte Studie der Phase III an Patienten mit reseziertem NSCLC nach cisplatinbasierter Chemotherapie
Eine unverblindete, multizentrische, randomisierte Studie der Phase III, GO29527 (IMpower010), wurde durchgeführt, um die Wirksamkeit und Sicherheit von Atezolizumab bei der adjuvanten Behandlung von Patienten mit NSCLC im Stadium IB (Tumore ≥ 4 cm) ‑ IIIA (gemäß Staging-System der Union for International Cancer Control/American Joint Committee on Cancer, 7. Version) zu untersuchen.
Die folgenden Auswahlkriterien definieren Patienten mit hohem Risiko für ein Rezidiv gemäß dem Anwendungsgebiet und entsprechen der Patientenpopulation mit Stadium II - IIIA gemäß der 7. Version des Staging-Systems:
Tumorgröße ≥ 5 cm; oder Tumoren jeglicher Größe, die entweder mit einem N1- oder N2-Status einhergehen; oder Tumoren, die in thorakale Strukturen eindringen (direktes Eindringen in die parietale Pleura, die Brustwand, das Zwerchfell, den Nervus phrenicus, die mediastinale Pleura, den parietalen Herzbeutel, das Mediastinum, das Herz, die großen Gefäße, die Luftröhre, den Nervus laryngeus recurrens, die Speiseröhre, den Wirbelkörper oder die Carina); oder Tumoren, die den Hauptbronchus < 2 cm distal der Carina, aber ohne Beteiligung der Carina betreffen; oder Tumoren, die mit einer Atelektase oder obstruktiven Pneumonitis der gesamten Lunge einhergehen; oder Tumoren mit separatem/n Knoten im gleichen oder einem anderen ipsilateralen Lappen als dem primären.
Die Studie schloss keine Patienten mit N2‑Status ein, bei denen der Tumor in das Mediastinum, das Herz, die großen Gefäße, die Luftröhre, den Nervus laryngeus recurrens, die Speiseröhre, den Wirbelkörper oder die Carina eingedrungen war, oder bei denen ein separater/separate Tumorknoten in einem anderen ipsilateralen Lappen vorhanden war.
Insgesamt wurden 1 280 Patienten in die Studie aufgenommen, bei denen der Tumor vollständig entfernt wurde und die für bis zu 4 Zyklen einer cisplatinbasierten Chemotherapie in Frage kamen. Die cisplatinbasierten Chemotherapieschemata sind in Tabelle 6 dargestellt.
Tabelle 6: Adjuvante Chemotherapieschemata (IMpower010)
Adjuvante cisplatinbasierte Chemotherapie: |
Vinorelbin 30 mg/m2 intravenös, Tag 1 und 8 |
Docetaxel 75 mg/m2 intravenös, Tag 1 | |
Gemcitabin 1 250 mg/m2 intravenös, Tag 1 und 8 | |
Pemetrexed 500 mg/m2 intravenös, Tag 1 (Nicht-Plattenepithel) |
Nach Abschluss der cisplatinbasierten Chemotherapie (bis zu vier Zyklen) wurden insgesamt 1 005 Patienten im Verhältnis 1:1 randomisiert und erhielten entweder Atezolizumab (Arm A) oder die bestmögliche unterstützende Behandlung (best supportive care, BSC) (Arm B). Atezolizumab wurde in einer Fixdosis von 1 200 mg alle 3 Wochen als intravenöse Infusion über 16 Zyklen verabreicht, es sei denn, es kam zu einem Rezidiv oder zu inakzeptabler Toxizität. Die Randomisierung erfolgte stratifiziert nach Geschlecht, Krankheitsstadium, Histologie und PD-L1-Expression.
Patienten mit einer Autoimmunerkrankung in der Anamnese oder Patienten, die innerhalb von 28 Tagen vor der Randomisierung einen abgeschwächten Lebendimpfstoff oder innerhalb von 4 Wochen vor der Randomisierung systemische Immunstimulanzien oder innerhalb von 2 Wochen vor der Randomisierung systemische Immunsuppressiva erhalten hatten, wurden ausgeschlossen. Tumorbewertungen wurden zu Randomisierungsbeginn und während des ersten Jahres nach Zyklus 1, Tag 1 alle 4 Monate und dann alle 6 Monate bis Jahr 5 und danach jährlich durchgeführt.
Die demographischen Charakteristika der ITT-Population und deren Krankheitscharakteristika zu Behandlungsbeginn waren zwischen den Behandlungsarmen ausgeglichen. Das mediane Alter betrug 62 Jahre (Bereich: 26 - 84), und 67 % der Patienten waren männlich. Die Mehrheit der Patienten waren kaukasischer (73 %) und 24 % asiatischer Herkunft. Die meisten Patienten waren Raucher oder ehemalige Raucher (78 %). Der ECOG-Performance-Status betrug bei den Patienten zu Behandlungsbeginn 0 (55 %) oder 1 (44 %). Insgesamt hatten 12 % der Patienten ein NSCLC im Stadium IB, 47 % im Stadium II und 41 % im Stadium IIIA. Der Prozentsatz der Patienten, deren Tumoren eine PD-L1-Expression von ≥ 1 % bzw. ≥ 50 % der TC aufwiesen, bewertet mit dem VENTANA PD-L1-(SP263)-Test, betrug 55 % bzw. 26 %.
Der primäre Wirksamkeitsnachweis war das vom Prüfarzt bewertete krankheitsfreie Überleben (disease-free survival, DFS). Das DFS war definiert als die Zeit von der Randomisierung bis zum Auftreten eines der folgenden Ereignisse: erstes dokumentiertes Rezidiv, neues primäres NSCLC oder Tod jeglicher Ursache, je nachdem was zuerst eintrat. Das primäre Wirksamkeitsziel war die Bewertung des DFS in der Patientenpopulation mit PD-L1 ≥ 1 % der TC im Stadium II - IIIA. Wichtige sekundäre Wirksamkeitsziele waren die Bewertung des DFS in der Patientenpopulation mit PD-L1 ≥ 50 % der TC im Stadium II - IIIA und das Gesamtüberleben (OS) in der ITT-Population.
Zum Zeitpunkt der DFS-Zwischenanalyse hatte die Studie ihren primären Endpunkt erreicht. In der Analyse von Patienten mit PD-L1 ≥ 50 % der TC im Stadium II - IIIA und ohne EGFR-Mutationen oder ALK-Translokationen (n = 209) wurde eine Verbesserung des DFS im Atezolizumab-Arm im Vergleich zum BSC-Arm beobachtet. Die Ergebnisse waren zum Zeitpunkt der finalen DFS-Analyse mit einer medianen Nachbeobachtungszeit von 65 Monaten konsistent.
Die wichtigsten Wirksamkeitsergebnisse zu DFS und OS für die Patientenpopulation mit PD-L1 ≥ 50 % der TC im Stadium II - IIIA und ohne EGFR-Mutationen oder ALK-Translokationen sind in Tabelle 7 zusammengefasst. Die Kaplan-Meier-Kurve für das DFS ist in Abbildung 3 dargestellt.
Tabelle 7: Zusammenfassung der Wirksamkeit in der Patientenpopulation mit PD-L1-Expression ≥ 50 % der TC im Stadium II - IIIA und ohne EGFR-Mutationen oder ALK-Translokationen (IMpower010)
Wirksamkeitsendpunkt |
Arm A |
Arm B |
Prüfarzt bewertetes DFS* |
n = 106 |
n = 103 |
Anzahl der Ereignisse (%) |
34 (32,1%) |
55 (53,4%) |
Mediane Dauer des DFS (Monate) |
NE |
42,9 |
95-%-KI |
(NE) |
(32,0; NE) |
Stratifizierteǂ Hazard Ratio (95-%-KI) |
0,52 (0,33; 0,80) |
|
OS* |
n = 106 |
n = 103 |
Anzahl der Ereignisse (%) |
22 (20,8%) |
41 (39,8%) |
Mediane OS (Monate) |
NE |
87,1 |
95-%-KI |
(NE) |
(72,0; NE) |
Stratifizierteǂ Hazard Ratio (95-%-KI) |
0,47 (0,28; 0,80) |
|
DFS = disease-free survival (krankheitsfreies Überleben); KI = Konfidenzintervall; NE = not estimable (nicht schätzbar)
* Aktualisierte DFS und OS Analyse zum Zeitpunkt des klinischen Stichtages 26. Januar 2024
ǂ Stratifiziert nach Stadium, Geschlecht und Histologie
Abbildung 3: Kaplan-Meier-Kurve des krankheitsfreien Überlebens in der Population mit PD-L1-Expression ≥ 50 % der TC im Stadium II - IIIA und ohne EGFR-Mutationen oder ALK-Translokationen (IMpower010)
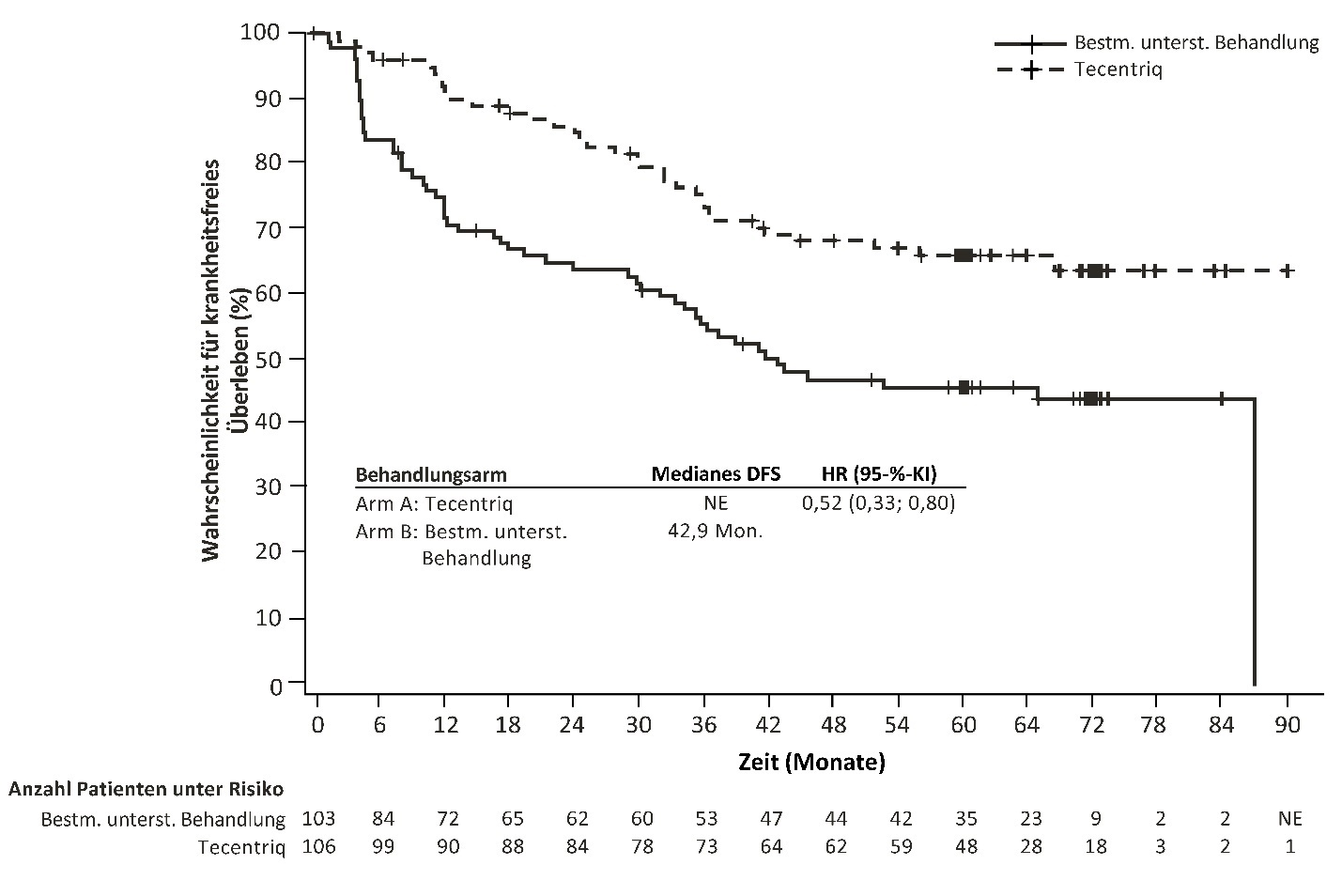
Die beobachtete Verbesserung des krankheitsfreien Überlebens im Atezolizumab-Arm im Vergleich zum BSC-Arm zeigte sich konsistent in der Mehrzahl der vordefinierten Subgruppen in der Patientenpopulation mit PD-L1 ≥ 50 % der TC im Stadium II - IIIA und ohne EGFR-Mutationen oder ALK-Translokationen, darunter sowohl Patienten mit Nicht-Plattenepithel-NSCLC (nicht stratifizierte HR von 0,40; 95-%-KI: 0,23; 0,70; medianes DFS NE vs. 36,8 Monate) als auch Patienten mit Plattenepithel-NSCLC (nicht stratifizierte HR von 0,67; 95-%-KI: 0,34; 1,32; medianes DFS nicht abschätzbar).
Erstlinienbehandlung des fortgeschrittenen NSCLC
Intravenöse Darreichungsform
IMpower150 (GO29436): Randomisierte Studie der Phase III bei chemotherapienaiven Patienten mit metastasiertem NSCLC mit nicht-plattenepithelialer Histologie, in Kombination mit Paclitaxel und Carboplatin mit oder ohne Bevacizumab
Eine unverblindete, multizentrische, internationale, randomisierte Studie der Phase III, IMpower150, wurde zur Untersuchung der Wirksamkeit und Sicherheit von Atezolizumab in Kombination mit Paclitaxel und Carboplatin, mit oder ohne Bevacizumab, bei chemotherapienaiven Patienten mit metastasiertem NSCLC mit nicht-plattenepithelialer Histologie durchgeführt.
Patienten mit einer Autoimmunerkrankung in der Anamnese, Patienten, die mit einem attenuierten Lebendimpfstoff innerhalb von 28 Tagen vor der Randomisierung geimpft wurden, systemische Immunstimulanzien innerhalb von 4 Wochen oder systemische Immunsuppressiva innerhalb von 2 Wochen vor der Randomisierung erhalten haben, mit aktiven oder unbehandelten ZNS-Metastasen, eindeutiger Tumorinfiltration in die großen Blutgefäße des Brustkorbs oder eindeutiger Kavitation pulmonaler Läsionen, wie mittels bildgebender Verfahren festgestellt, wurden von der Studie ausgeschlossen. Eine Beurteilung des Tumors wurde während der ersten 48 Wochen alle 6 Wochen nach Zyklus 1, Tag 1 durchgeführt und danach alle 9 Wochen. Tumorproben wurden auf die PD-L1-Expression auf Tumorzellen (TC) und tumorinfiltrierenden Immunzellen (IC) untersucht und die Ergebnisse verwendet, um die PD-L1-Expressions-Subgruppen für die unten beschriebenen Analysen zu definieren.
Insgesamt wurden 1 202 Patienten eingeschlossen, im Verhältnis 1:1:1 randomisiert und einem der in Tabelle 8 beschriebenen Behandlungsschemata zugeteilt. Die Randomisierung wurde nach Geschlecht, Vorhandensein von Lebermetastasen und PD-L1-Tumorexpression auf TC und IC stratifiziert.
Tabelle 8: Intravenöse Behandlungsschemata (IMpower150)
Behandlungs-schema |
Induktion |
Erhaltung |
A |
Atezolizumaba (1 200 mg) + Paclitaxel (200 mg/m2)b,c + Carboplatinc (AUC 6) |
Atezolizumaba (1 200 mg) |
B |
Atezolizumaba (1 200 mg) + Bevacizumabd (15 mg/kg KG) + Paclitaxel (200 mg/m2)b,c + Carboplatinc (AUC 6) |
Atezolizumaba (1 200 mg) + Bevacizumabd (15 mg/kg KG) |
C |
Bevacizumabd (15 mg/kg KG) + Paclitaxel (200 mg/m2)b,c + Carboplatinc (AUC 6) |
Bevacizumabd (15 mg/kg KG) |
a Atezolizumab wird bis zum Verlust des klinischen Nutzens gemäß Bewertung des Prüfarztes angewendet
b Die Anfangsdosis von Paclitaxel für Patienten asiatischer Herkunft betrug aufgrund der höheren Anzahl hämatologischer Toxizitäten bei Patienten aus asiatischen Ländern, im Vergleich zu Patienten aus nicht-asiatischen Ländern, 175 mg/m2
c Paclitaxel und Carboplatin werden bis zum Abschluss von 4 oder 6 Zyklen oder fortschreitender Erkrankung oder bis zum Auftreten inakzeptabler Toxizität, je nachdem was zuerst eintritt, angewendet
d Bevacizumab wird bis zum Fortschreiten der Erkrankung oder dem Auftreten inakzeptabler Toxizität angewendet
Die demographischen Merkmale und die Merkmale der Grunderkrankung der Studienpopulation zum Behandlungsbeginn waren zwischen den Behandlungsarmen ausgeglichen. Das mediane Alter betrug 63 Jahre (Bereich: 31 bis 90) und 60 % der Patienten waren männlich. Die Mehrheit der Patienten war kaukasischer Herkunft (82 %). Ungefähr 10 % der Patienten hatten eine bekannte EGFR-Mutation, 4 % hatten bekannte ALK-Translokationen, 14 % hatten Lebermetastasen zum Behandlungsbeginn und die meisten Patienten waren Raucher oder ehemalige Raucher (80 %). Der ECOG-Performance-Status betrug zu Behandlungsbeginn 0 (43 %) oder 1 (57 %). 51 % der Tumoren der Patienten hatten eine PD-L1-Expression von ≥ 1 % TC oder ≥ 1 % IC und 49 % der Tumoren der Patienten hatten eine PD-L1-Expression von < 1 % TC und < 1 % IC.
Zum Zeitpunkt der finalen Analyse in Bezug auf das PFS hatten die Patienten eine mediane Nachbeobachtungszeit von 15,3 Monaten. Die ITT-Population, einschließlich Patienten mit EGFR-Mutationen oder ALK-Translokationen, die bereits eine Behandlung mit Tyrosinkinase-Inhibitoren erhalten hatten, zeigte eine klinisch bedeutsame Verbesserung des PFS in Arm B verglichen mit Arm C (HR von 0,61; 95‑%‑KI: 0,52; 0,72, medianes PFS 8,3 vs. 6,8 Monate).
Zum Zeitpunkt der OS-Interimsanalyse hatten die Patienten eine mediane Nachbeobachtungszeit von 19,7 Monaten. Die wesentlichen Ergebnisse dieser Analyse, wie auch jene der aktualisierten PFS-Analyse in der ITT-Population sind in Tabellen 9 und 10 zusammengefasst. Die Kaplan‑Meier‑Kurve für das Gesamtüberleben in der ITT-Population ist in Abbildung 4 dargestellt. Die Abbildung 5 fasst die Ergebnisse für das OS in den ITT- und PD-L1-Subgruppen zusammen. Aktualisierte Ergebnisse in Bezug auf das PFS sind in den Abbildungen 6 und 7 dargestellt.
Tabelle 9: Zusammenfassung der aktualisierten Wirksamkeitsergebnisse in der ITT-Population (IMpower150)
Wirksamkeitsendpunkt |
Arm A |
Arm B |
Arm C |
Sekundäre Endpunkte# |
|||
Vom Prüfarzt bewertetes PFS (RECIST v1.1)* |
n = 402 |
n = 400 |
n = 400 |
Anzahl der Ereignisse (%) |
330 (82,1 %) |
291 (72,8 %) |
355 (88,8 %) |
Mediane Dauer des PFS (Monate) |
6,7 |
8,4 |
6,8 |
95‑%‑KI |
(5,7; 6,9) |
(8,0; 9,9) |
(6,0; 7,0) |
Stratifizierte Hazard Ratio‡^ (95‑%‑KI) |
0,91 (0,78; 1,06) |
0,59 (0,50; 0,69) |
--- |
12‑Monats‑PFS (%) |
24 |
38 |
20 |
OS-Interimsanalyse* |
n = 402 |
n = 400 |
n = 400 |
Anzahl der Todesfälle (%) |
206 (51,2 %) |
192 (48,0 %) |
230 (57,5 %) |
Stratifizierte Hazard Ratio‡^ (95‑%‑KI) |
0,85 (0,71; 1,03) |
0,76 (0,63; 0,93) |
--- |
6-Monats-OS (%) |
84 |
85 |
81 |
12-Monats-OS (%) |
66 |
68 |
61 |
Vom Prüfarzt bewertetes bestes Gesamtansprechen3* (RECIST v1.1) |
n = 401 |
n = 397 |
n = 393 |
Anzahl der Patienten mit Ansprechen (%) |
163 (40,6 %) |
224 (56,4 %) |
158 (40,2 %) |
95‑%‑KI |
(35,8; 45,6) |
(51,4; 61,4) |
(35,3; 45,2) |
Anzahl mit vollständigem Ansprechen (%) |
8 (2,0 %) |
11 (2,8 %) |
3 (0,8 %) |
Anzahl mit teilweisem Ansprechen (%) |
155 (38,7 %) |
213 (53,7 %) |
155 (39,4 %) |
Vom Prüfarzt bewertete DOR* (RECIST v1.1) |
n = 163 |
n = 224 |
n = 158 |
Median in Monaten |
8.3 |
11,5 |
6,0 |
95‑%‑KI |
(7,1; 11,8) |
(8,9; 15,7) |
(5,5; 6,9) |
# Die primären Wirksamkeitsendpunkte waren PFS und OS und wurden in der ITT-Wildtyp-Population analysiert; das heißt ohne Patienten mit EGFR-Mutationen oder ALK-Translokationen
1 Basierend auf dem stratifizierten Log‑Rank‑Test
2 Für Informationszwecke; in der ITT-Population, Vergleiche zwischen Arm B und Arm C sowie zwischen Arm A und Arm C wurden noch nicht formal gemäß prä-spezifizierter Testhierarchie geprüft
3 Bestes Gesamtansprechen für vollständiges und teilweises Ansprechen
‡ Stratifiziert nach Geschlecht, Vorhandensein von Lebermetastasen und PD-L1-Tumorexpression auf TC und IC
^ Arm C ist die Vergleichsgruppe für alle Hazard Ratios
* Aktualisierte PFS-Analyse und OS-Interimsanalyse zum Zeitpunkt des klinischen Stichtages 22. Januar 2018
PFS = progression‑free survival (progressionsfreies Überleben); RECIST = Response Evaluation Criteria in Solid Tumours v1.1.
KI = Konfidenzintervall; DOR = duration of response (Dauer des Ansprechens); OS = overall survival (Gesamtüberleben).
Tabelle 10: Zusammenfassung der aktualisierten Wirksamkeitsergebnisse für Arm A vs. Arm B in der ITT-Population (IMpower150)
Wirksamkeitsendpunkt |
Arm A |
Arm B |
|
Vom Prüfarzt bewertetes PFS (RECIST v1.1)* |
n = 402 |
n = 400 |
|
Anzahl der Ereignisse (%) |
330 (82,1 %) |
291 (72,8 %) |
|
Mediane Dauer des PFS (Monate) |
6,7 |
8,4 |
|
95‑%‑KI |
(5,7; 6,9) |
(8,0; 9,9) |
|
Stratifizierte Hazard Ratio‡^ (95‑%‑KI) |
0,67 (0,57; 0,79) |
||
OS-Interimsanalyse* |
n = 402 |
n = 400 |
|
Anzahl der Todesfälle (%) |
206 (51,2 %) |
192 (48,0 %) |
|
Stratifizierte Hazard Ratio‡^ (95‑%‑KI) |
0,90 (0,74; 1,10) |
||
1 Basierend auf dem stratifizierten Log‑Rank‑Test
2 Für Informationszwecke; in der ITT-Population wurden keine Vergleiche zwischen Arm A und Arm B in der prä-spezifizierten Testhierarchie eingeschlossen
‡ Stratifiziert nach Geschlecht, Vorhandensein von Lebermetastasen und PD-L1-Expression auf TC und IC
* Aktualisierte PFS-Analyse und OS-Interimsanalyse zum Zeitpunkt des klinischen Stichtages 22. Januar 2018
^ Arm A ist die Vergleichsgruppe für alle Hazard Ratios
Abbildung 4: Kaplan-Meier-Kurve für das Gesamtüberleben in der ITT-Population (IMpower150)
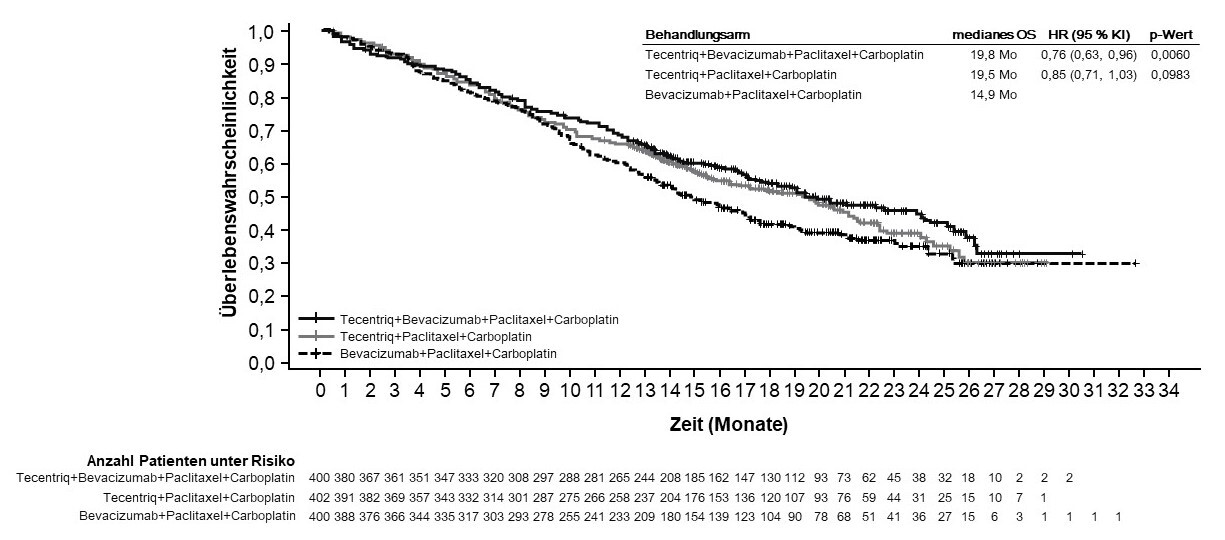
Abbildung 5: Forest‑Diagramm zum Gesamtüberleben nach PD‑L1‑Expression in der ITT‑Population, Arm B vs. C (IMpower150)
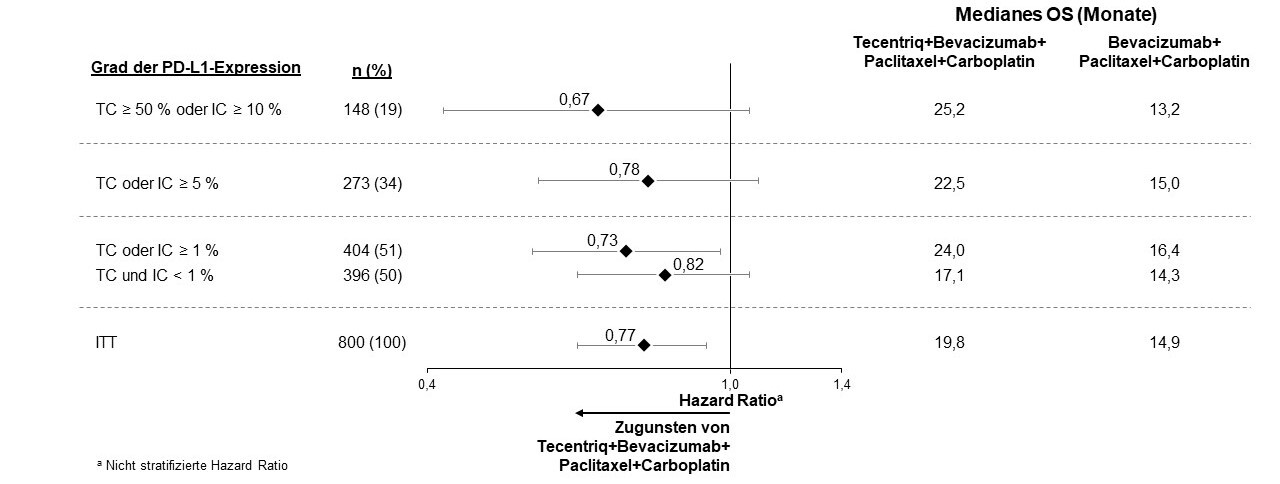
Abbildung 6: Kaplan-Meier-Kurve für das PFS in der ITT-Population (IMpower150)
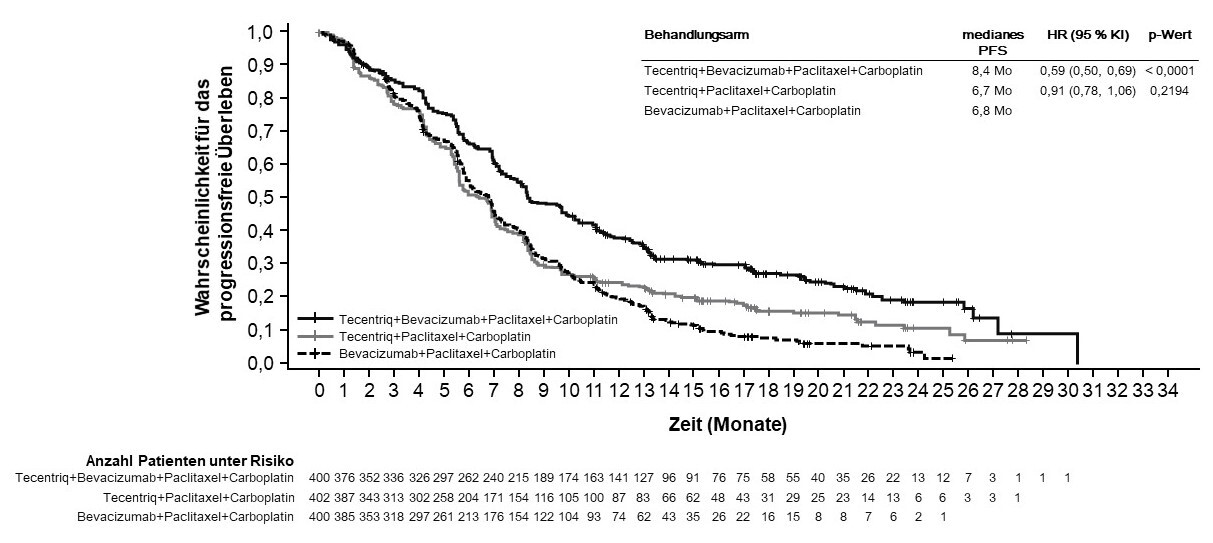
Abbildung 7: Forest-Diagramm zum progressionsfreien Überleben nach PD-L1-Expression in der ITT-Population, Arm B vs. C (IMpower150)
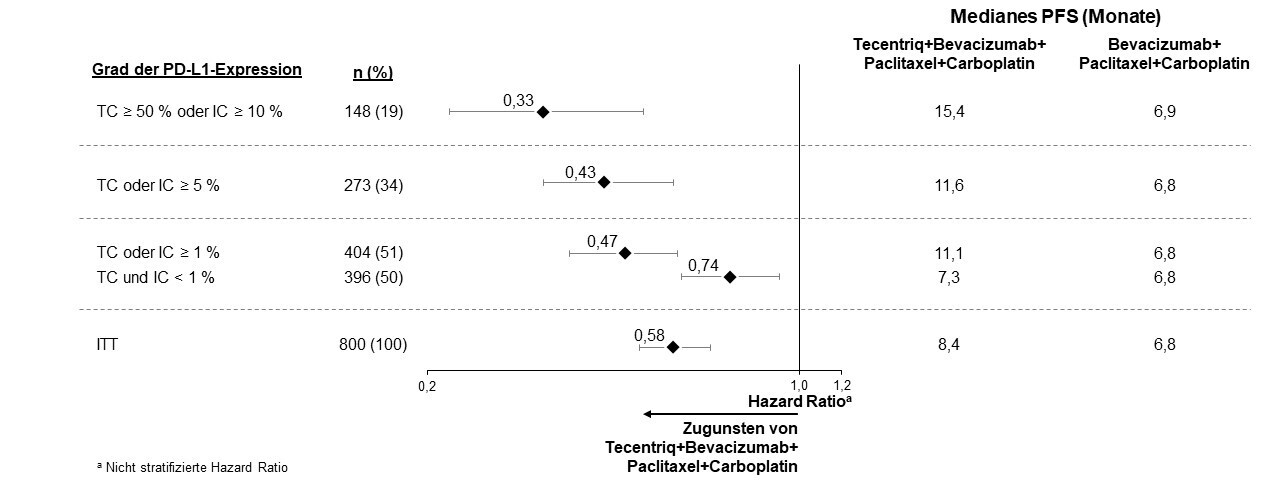
In Arm B, verglichen mit Arm C, zeigten prä-spezifizierte Subgruppenanalysen in der OS-Interimsanalyse eine Verbesserung des OS bei Patienten mit EGFR-Mutationen oder ALK-Translokationen (Hazard Ratio [HR] von 0,54; 95‑%‑KI: 0,29; 1,03, medianes OS nicht erreicht vs. 17,5 Monate) sowie bei Patienten mit Lebermetastasen (HR von 0,52; 95‑%‑KI: 0,33; 0,82, medianes OS 13,3 Monate vs. 9,4 Monate). Eine Verbesserung des PFS wurde ebenfalls bei Patienten mit EGFR-Mutationen oder ALK-Translokationen (HR von 0,55; 95‑%‑KI: 0,35; 0,87, medianes PFS 10 vs. 6,1 Monate) sowie bei Patienten mit Lebermetastasen (HR von 0,41; 95‑%‑KI: 0,26; 0,62, medianes PFS 8,2 vs. 5,4 Monate) gezeigt. Die OS-Ergebnisse waren bei Patienten im Alter von < 65 und ≥ 65 ähnlich. Die Daten für Patienten im Alter von ≥ 75 Jahren sind zu begrenzt, um Rückschlüsse auf diese Population zuzulassen. Für alle Subgruppenanalysen gilt, dass keine formalen statistischen Untersuchungen geplant waren.
IMpower130 (GO29537): Randomisierte Studie der Phase III bei chemotherapienaiven Patienten mit metastasiertem NSCLC mit nicht-plattenepithelialer Histologie, in Kombination mit nab-Paclitaxel und Carboplatin
Eine unverblindete, randomisierte Studie der Phase III, GO29537 (IMpower130), wurde zur Untersuchung der Wirksamkeit und Sicherheit von Atezolizumab in Kombination mit nab-Paclitaxel und Carboplatin bei chemotherapienaiven Patienten mit metastasiertem NSCLC mit nicht-plattenepithelialer Histologie durchgeführt. Patienten mit EGFR-Mutationen oder ALK-Translokationen sollen im Vorfeld bereits eine Behandlung mit Tyrosinkinase-Inhibitoren erhalten haben.
Das Tumorstadium der Patienten wurde gemäß des American Joint Committee on Cancer (AJCC), 7. Edition bewertet. Von der Studie ausgeschlossen waren Patienten mit einer Autoimmunerkrankung in der Anamnese, sowie Patienten, die mit einem attenuierten Lebendimpfstoff innerhalb von 28 Tagen vor der Randomisierung geimpft worden waren oder Immunstimulanzien innerhalb von 4 Wochen oder systemische Immunsuppressiva innerhalb von 2 Wochen vor der Randomisierung erhalten hatten oder aktive oder unbehandelte ZNS-Metastasen hatten. Ebenfalls nicht geeignet waren Patienten, die eine vorherige Behandlung mit CD137-Agonisten oder Immun-Checkpoint-Inhibitoren (anti-PD-1 und anti-PD-L1 therapeutische Antikörper) erhalten hatten. Patienten, die eine vorherige anti-CTLA-4-Behandlung erhalten hatten, konnten eingeschlossen werden, sofern sie ihre letzte Dosis mindestens 6 Wochen vor der Randomisierung erhalten hatten und keine schweren immunvermittelten unerwünschten Ereignisse durch die CTLA-4-Behandlung auftraten (NCI CTCAE Grade 3 und 4). Eine Beurteilung des Tumors wurde während der ersten 48 Wochen alle 6 Wochen nach Zyklus 1 durchgeführt und danach alle 9 Wochen. Tumorproben wurden auf die PD-L1-Expression auf Tumorzellen (TC) und tumorinfiltrierende Immunzellen (IC) untersucht und die Ergebnisse verwendet, um die PD-L1-Expressions-Subgruppen für die unten beschriebenen Analysen zu definieren.
Die eingeschlossenen Patienten, inklusive der Patienten mit EGFR-Mutationen oder ALK-Translokationen, wurden im Verhältnis 2:1 randomisiert und einem der in Tabelle 11 beschriebenen Behandlungsschemata zugeteilt. Die Randomisierung wurde nach Geschlecht, Vorhandensein von Lebermetastasen und PD-L1-Expression auf TC und IC stratifiziert. Die Patienten, die Behandlungsschema B erhielten, konnten bei Krankheitsprogression das Behandlungsschema wechseln (Crossover) und Atezolizumab als Monotherapie erhalten.
Tabelle 11: Intravenöse Behandlungsschemata (IMpower130)
Behandlungs-schema |
Induktion |
Erhaltung |
|
A |
Atezolizumab (1 200 mg)a + nab‑Paclitaxel (100 mg/m2)b,c + Carboplatin (AUC 6)c |
Atezolizumab (1 200 mg)a |
|
B |
Nab‑Paclitaxel (100 mg/m2)b,c + Carboplatin (AUC 6)c |
Bestmögliche unterstützende Behandlung oder Pemetrexed |
|
a Atezolizumab wird bis zum Verlust des klinischen Nutzens gemäß Bewertung des Prüfarztes angewendet
b Nab-Paclitaxel wird an den Tagen 1, 8 und 15 jedes Zyklus angewendet
c Nab-Paclitaxel und Carboplatin werden bis zum Abschluss von 4 - 6 Zyklen oder fortschreitender Erkrankung oder bis zum Auftreten inakzeptabler Toxizität, je nachdem was zuerst eintritt, angewendet
Die demographischen Merkmale und die Merkmale der Grunderkrankung der Studienpopulation (n = 679) zum Behandlungsbeginn waren zwischen den Behandlungsarmen ausgeglichen. Das mediane Alter betrug 64 Jahre (Bereich: 18 bis 86 Jahre). Die Mehrheit der Patienten war männlich (59 %) und kaukasischer Herkunft (90 %). Zum Behandlungsbeginn hatten 14,7 % der Patienten Lebermetastasen und die meisten Patienten waren Raucher oder ehemalige Raucher (90 %). Die Mehrheit der Patienten hatte einen ECOG-Performance-Status von 1 zum Behandlungsbeginn (59 %) und eine PD-L1-Expression < 1 % (ungefähr 52 %). Von 107 Arm B Patienten, die nach der Induktionstherapie entweder eine stabile Erkrankung, ein teilweises oder ein vollständiges Ansprechen als Therapieantwort aufwiesen, erhielten 40 Patienten eine Pemetrexed Wechselerhaltungstherapie.
Die Primäranalyse wurde bei allen Patienten, außer bei Patienten mit EGFR-Mutationen oder ALK-Translokationen, durchgeführt. Diese Population wurde als ITT-WT definiert (n = 679). Die Patienten hatten eine mediane Überlebens-Nachbeobachtungszeit von 18,6 Monaten und zeigten im Vergleich zur Kontrollgruppe ein verbessertes OS und PFS unter Atezolizumab, nab-Paclitaxel und Carboplatin. Die wesentlichen Ergebnisse dieser Analyse sind in Tabelle 12 zusammengefasst und die Kaplan‑Meier‑Kurven für das OS und das PFS sind in Abbildung 8 bzw. 10 dargestellt. Die explorativen Ergebnisse zu OS und PFS in Abhängigkeit von der PD-L1-Expression sind in den Abbildungen 9 bzw. 11 zusammengefasst. Patienten mit Lebermetastasen zeigten kein verbessertes PFS oder OS unter Anwendung von Atezolizumab, nab-Paclitaxel und Carboplatin, im Vergleich zu nab-Paclitaxel und Carboplatin (HR von 0,93; 95‑%‑KI: 0,59; 1,47 für PFS und HR von 1,04; 95‑%‑KI: 0,63; 1,72 für OS).
Im nab-Paclitaxel und Carboplatin Arm erhielten 59 % der Patienten nach Krankheitsprogression eine geeignete Krebs-Immuntherapie, wovon 41 % der Patienten Atezolizumab im Rahmen der Crossover-Behandlung erhielten, verglichen mit 7,3 % der Patienten im Atezolizumab, nab-Paclitaxel und Carboplatin Arm.
In einer explorativen Auswertung mit längerer Nachbeobachtungszeit (Median 24,1 Monate) blieb das mediane OS in beiden Armen unverändert im Vergleich zur Primäranalyse (HR von 0,82; 95‑%‑KI: 0,67; 1,01).
Tabelle 12: Zusammenfassung der Wirksamkeit von IMpower130 in der Primäranalyse‑Population (ITT-WT Population)
Wirksamkeitsendpunkte |
Arm A |
Arm B |
Ko-primäre Endpunkte |
||
OS |
n = 451 |
n = 228 |
Anzahl der Todesfälle (%) |
226 (50,1 %) |
131 (57,5 %) |
Mediane Zeit bis zum Ereignis (Monate) |
18,6 |
13,9 |
95‑%‑KI |
(16,0; 21,2) |
(12,0; 18,7) |
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,79 (0,64; 0,98) |
|
p-Wert |
0,033 |
|
12‑Monats‑OS (%) |
63 |
56 |
Vom Prüfarzt bewertetes PFS (RECIST v1.1) |
n = 451 |
n = 228 |
Anzahl der Ereignisse (%) |
347 (76,9 %) |
198 (86,8 %) |
Mediane Dauer des PFS (Monate) |
7,0 |
5,5 |
95‑%‑KI |
(6,2; 7,3) |
(4,4; 5,9) |
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,64 (0,54; 0,77) |
|
p-Wert |
< 0,0001 |
|
12‑Monats‑PFS (%) |
29 % |
14 % |
Andere Endpunkte |
||
Vom Prüfarzt bewertete ORR (RECIST v1.1)^ |
n = 447 |
n = 226 |
Anzahl der Patienten mit Ansprechen (%) |
220 (49,2 %) |
72 (31,9 %) |
95‑%‑KI |
(44,5; 54,0) |
(25,8; 38,4) |
Anzahl mit vollständigem Ansprechen (%) |
11 (2,5 %) |
3 (1,3 %) |
Anzahl mit teilweisem Ansprechen (%) |
209 (46,8 %) |
69 (30,5 %) |
Vom Prüfarzt bewertete, bestätigte DOR (RECIST v1.1)^ |
n = 220 |
n = 72 |
Median in Monaten |
8,4 |
6,1 |
95‑%‑KI |
(6,9; 11,8) |
(5,5; 7,9) |
‡ Stratifiziert nach Geschlecht und PD‑L1-Expression auf TC und IC
^ Die bestätigten ORR und DOR sind explorative Endpunkte
PFS = progression‑free survival (progressionsfreies Überleben); RECIST = Response Evaluation Criteria in Solid Tumors v1.1.; KI = Konfidenzintervall; ORR = objective response rate (objektive Ansprechrate); DOR = duration of response (Dauer des Ansprechens); OS = overall survival (Gesamtüberleben)
Abbildung 8: Kaplan-Meier-Kurven zum Gesamtüberleben (IMpower130)
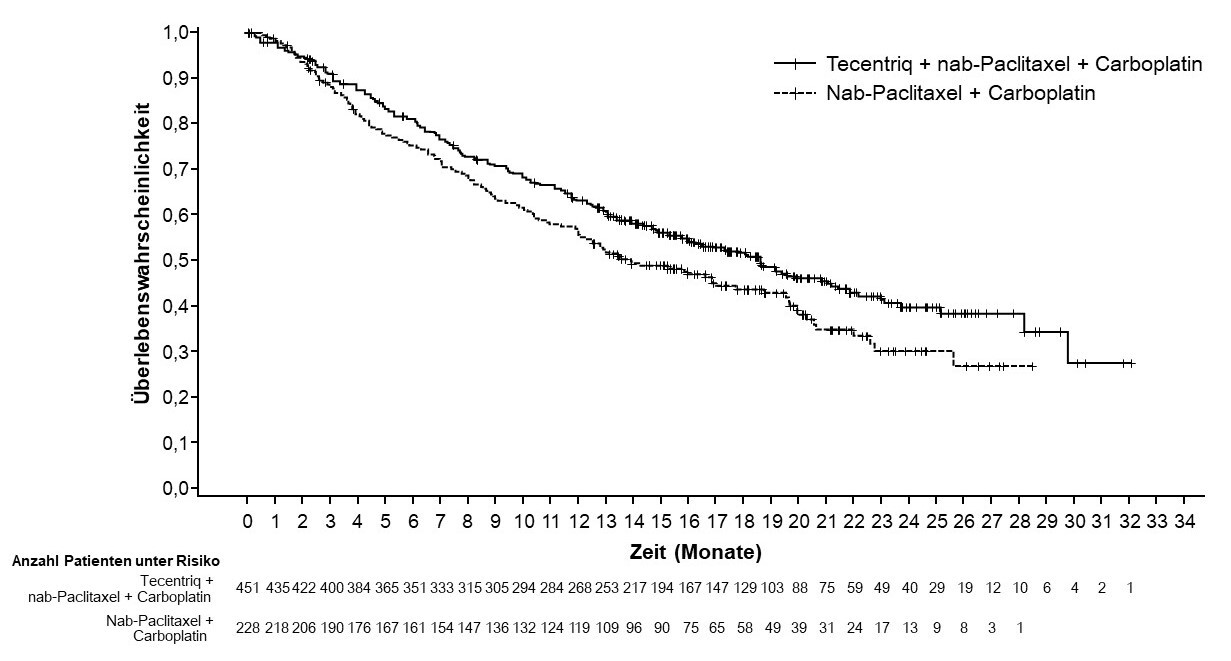
Abbildung 9: Forest‑Diagramm zum Gesamtüberleben nach PD‑L1‑Expression (IMpower130)
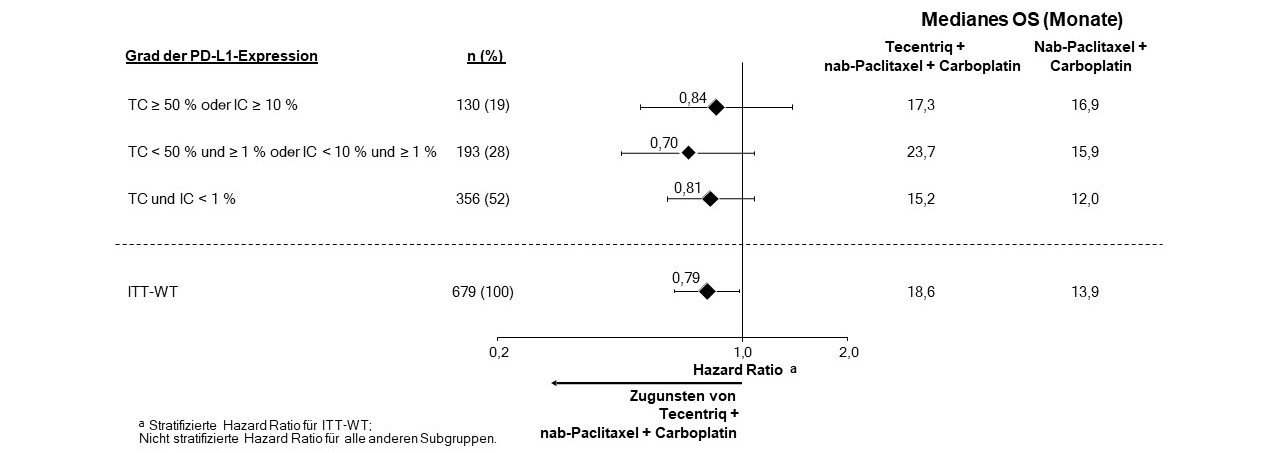
Abbildung 10: Kaplan-Meier-Kurven zum progressionsfreien Überleben (IMpower130)
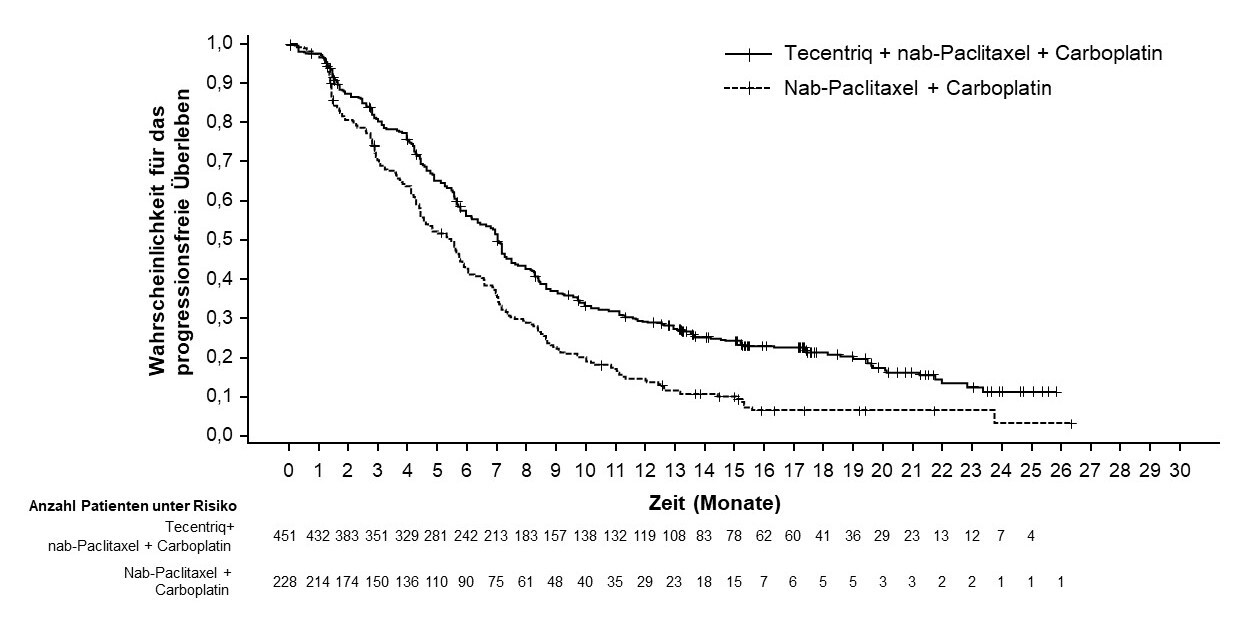
Abbildung 11: Forest-Diagramm zum progressionsfreien Überleben nach PD‑L1-Expression (IMpower130)
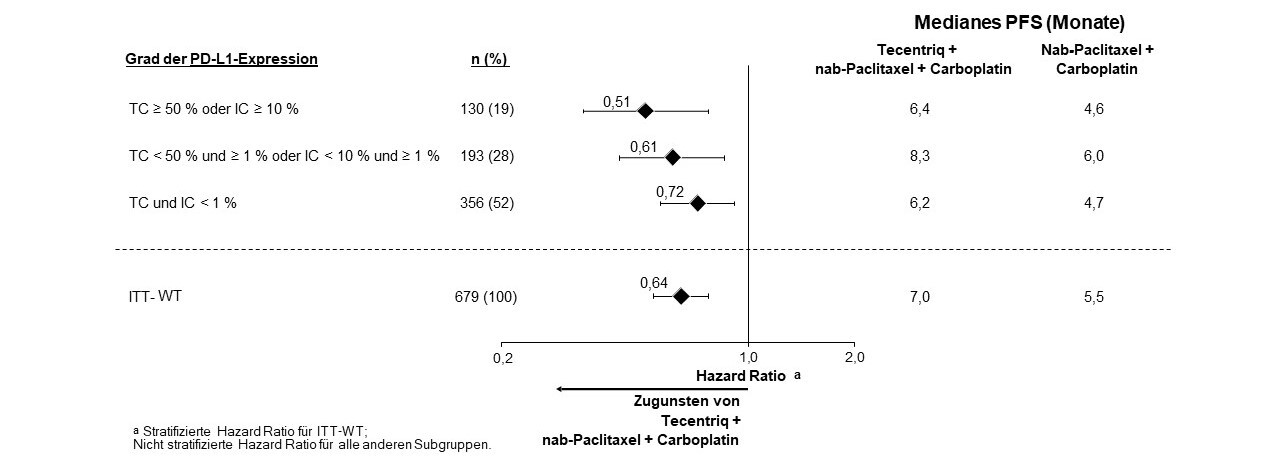
IMpower110 (GO29431): Randomisierte Studie der Phase III bei chemotherapienaiven Patienten mit metastasiertem NSCLC
Eine unverblindete, multizentrische, randomisierte Studie der Phase III, IMpower110, wurde durchgeführt, um die Wirksamkeit und Sicherheit von Atezolizumab bei chemotherapienaiven Patienten mit metastasiertem NSCLC zu untersuchen. Die Patienten hatten eine PD-L1-Expression ≥ 1 % TC (≥ 1 % PD‑L1 gefärbte Tumorzellen) oder ≥ 1 % IC (PD-L1 gefärbte tumorinfiltrierende Immunzellen, die ≥ 1 % der Tumorfläche abdecken) basierend auf dem VENTANA PD-L1-(SP142)-Test.
Insgesamt wurden 572 Patienten im Verhältnis 1:1 randomisiert, um Atezolizumab (Arm A) oder Chemotherapie (Arm B) zu erhalten. Atezolizumab wurde in einer Fixdosis von 1 200 mg alle 3 Wochen als intravenöse Infusion bis zum durch den Prüfarzt festgestellten Verlust des klinischen Nutzens oder dem Auftreten einer inakzeptablen Toxizität verabreicht. Die Chemotherapie-Schemata sind in Tabelle 13 beschrieben. Die Randomisierung erfolgte stratifiziert nach Geschlecht, ECOG-Performance-Status, Histologie und PD-L1-Tumorexpression auf TC und IC.
Tabelle 13: Chemotherapie-Behandlungsschemata für die intravenöse Behandlung (IMpower110)
Behandlungs-schema |
Induktion |
Erhaltung |
B (Nicht‑Platten-epithel) |
Cisplatina (75 mg/m²) + Pemetrexeda (500 mg/m²) ODER Carboplatina(AUC 6) + Pemetrexeda (500 mg/m²) |
Pemetrexedb,d (500 mg/m²) |
B (Plattenepithel) |
Cisplatina (75 mg/m²) + Gemcitabina,c (1 250 mg/m2) ODER Carboplatina (AUC 5) + Gemcitabina,c (1 000 mg/m2) |
Bestmögliche unterstützende Behandlungd |
a Cisplatin, Carboplatin, Pemetrexed und Gemcitabin werden bis zum Abschluss von 4 oder 6 Zyklen oder bis zum Fortschreiten der Erkrankung oder bis zum Auftreten inakzeptabler Toxizität angewendet
b Pemetrexed wird als Erhaltungstherapie alle 21 Tage bis zum Fortschreiten der Erkrankung oder dem Auftreten inakzeptabler Toxizität angewendet
c Gemcitabin wird an den Tagen 1 und 8 jedes Zyklus angewendet
d Es war kein Crossover vom Kontrollarm (platinbasierte Chemotherapie) in den Atezolizumab-Arm (Arm A) erlaubt
Patienten mit einer Autoimmunerkrankung in der Anamnese, Patienten, die mit einem attenuierten Lebendimpfstoff innerhalb von 28 Tagen vor der Randomisierung geimpft wurden, systemische Immunstimulanzien innerhalb von 4 Wochen oder systemische Immunsuppressiva innerhalb von 2 Wochen vor der Randomisierung erhalten haben, mit aktiven oder unbehandelten ZNS-Metastasen, wurden von der Studie ausgeschlossen. Eine Beurteilung des Tumors wurde während der ersten 48 Wochen alle 6 Wochen nach Zyklus 1, Tag 1 durchgeführt und danach alle 9 Wochen.
Die demographischen Merkmale und die Merkmale der Grunderkrankung der Patienten mit einer PD‑L1-Expression ≥ 1 % TC oder ≥ 1 % IC, die keine EGFR-Mutationen oder ALK-Translokationen aufweisen (n = 554), waren zwischen den Behandlungsarmen ausgeglichen. Das mediane Alter betrug 64,5 Jahre (Bereich: 30 bis 87) und 70 % der Patienten waren männlich. Die Mehrheit der Patienten war kaukasischer Herkunft (84 %) und asiatisch (14 %). Die meisten Patienten waren Raucher oder ehemalige Raucher (87 %) und der ECOG-Performance-Status betrug zu Beginn 0 (36 %) oder 1 (64 %). Insgesamt hatten 69 % der Patienten eine Nicht-Plattenepithel-Erkrankung und 31 % der Patienten eine Plattenepithel-Erkrankung. Die demographischen Merkmale und die Merkmale der Grunderkrankung der Patienten mit einer hohen PD-L1-Expression (PD-L1 ≥ 50 % TC oder ≥ 10 % IC), die keine EGFR-Mutationen oder ALK-Translokationen aufweisen (n = 205), waren insgesamt repräsentativ für die breitere Studienpopulation und zwischen den Behandlungsarmen ausgeglichen.
Der primäre Endpunkt war das OS. Zum Zeitpunkt der OS-Interimsanalyse zeigte sich bei Patienten mit hoher PD-L1-Expression, die randomisiert einer Behandlung mit Atezolizumab (Arm A) zugeteilt worden waren, nicht eingeschlossen die Patienten mit EGFR-Mutationen oder ALK-Translokationen (n = 205), eine statistisch signifikante Verbesserung des OS im Vergleich zu Chemotherapie (Arm B) (HR von 0,59; 95‑%‑KI: 0,40; 0,89; medianes OS von 20,2 Monaten vs. 13,1 Monaten), mit einem zweiseitigen p-Wert von 0,0106. Die mediane Nachbeobachtungszeit zum Überleben betrug 15,7 Monate bei Patienten mit hoher PD-L1-Expression.
Für diese Patienten blieb das mediane OS in einer explorativen OS-Analyse mit längerer Nachbeobachtungszeit (Median: 31,3 Monate) für den Atezolizumab-Arm unverändert (20,2 Monate) gegenüber der primären OS-Interimsanalyse und betrug für den Chemotherapie-Arm 14,7 Monate (HR von 0,76; 95‑%‑KI: 0,54; 1,09). Die wichtigsten Ergebnisse der explorativen Analyse sind in Tabelle 14 zusammengefasst. Die Kaplan-Meier-Kurven für OS und PFS bei Patienten mit hoher PD-L1-Expression sind in den Abbildungen 12 und 13 dargestellt. Im Atezolizumab-Arm starben innerhalb der ersten 2,5 Monate mehr Patienten (16/107; 15,0 %) als im Chemotherapie-Arm (10/98; 10,2 %). Es konnten keine spezifischen Faktoren, die mit frühen Todesfällen in Verbindung stehen, identifiziert werden.
Tabelle 14: Zusammenfassung der Wirksamkeit bei Patienten mit hoher PD-L1-Expression ≥ 50 % TC oder ≥ 10 % IC (IMpower110)
Wirksamkeitsendpunkte |
Arm A |
Arm B |
|
Primärer Wirksamkeitsendpunkt |
|||
Gesamtüberleben (OS) |
n = 107 |
n = 98 |
|
Anzahl der Todesfälle (%) |
64 (59,8 %) |
64 (65,3 %) |
|
Mediane Zeit bis zum Ereignis (Monate) |
20,2 |
14,7 |
|
95‑%‑KI |
(17,2; 27,9) |
(7,4; 17,7) |
|
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,76 (0,54; 1,09) |
||
12‑Monats-Gesamtüberleben (%) |
66,1 |
52,3 |
|
Sekundäre Wirksamkeitsendpunkte |
|||
Vom Prüfarzt bewertetes PFS (RECIST v1.1) |
n = 107 |
n = 98 |
|
Anzahl der Ereignisse (%) |
82 (76,6 %) |
87 (88,8 %) |
|
Mediane Dauer des PFS (Monate) |
8,2 |
5,0 |
|
95‑%‑KI |
(6,8; 11,4) |
(4,2; 5,7) |
|
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,59 (0,43; 0,81) |
||
12‑Monats-PFS (%) |
39,2 |
19,2 |
|
Vom Prüfarzt bewertete ORR (RECIST v1.1) |
n = 107 |
n = 98 |
|
Anzahl der Patienten mit Ansprechen (%) |
43 (40,2 %) |
28 (28,6 %) |
|
95‑%‑KI |
(30,8; 50,1) |
(19,9; 38,6) |
|
Anzahl mit vollständigem Ansprechen (%) |
1 (0,9 %) |
2 (2,0 %) |
|
Anzahl mit teilweisem Ansprechen (%) |
42 (39,3 %) |
26 (26,5 %) |
|
Vom Prüfarzt bewertete DOR (RECIST v1.1) |
n = 43 |
n = 28 |
|
Median in Monaten |
38,9 |
8,3 |
|
95‑%-KI |
(16,1; NE) |
(5,6; 11,0) |
|
‡ Stratifiziert nach Geschlecht und ECOG-Performance-Status (0 vs. 1)
PFS = progression‑free survival (progressionsfreies Überleben); RECIST = Response Evaluation Criteria in Solid Tumours v1.1; KI = Konfidenzintervall; ORR = objective response rate (objektive Ansprechrate); DOR = duration of response (Dauer des Ansprechens); OS = overall survival (Gesamtüberleben); NE = not estimable (nicht schätzbar).
Abbildung 12: Kaplan-Meier-Kurve des Gesamtüberlebens bei Patienten mit hoher PD-L1-Expression ≥ 50 % TC oder ≥ 10 % IC (IMpower110)
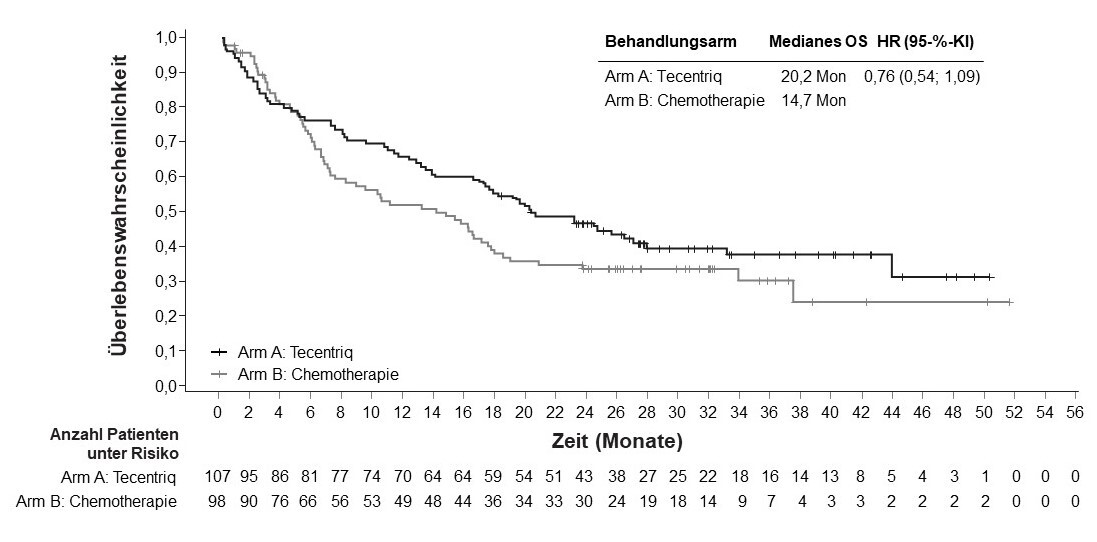
Abbildung 13: Kaplan-Meier-Kurve des progressionsfreien Überlebens bei Patienten mit hoher PD-L1-Expression ≥ 50 % TC oder ≥ 10 % IC (IMpower110)
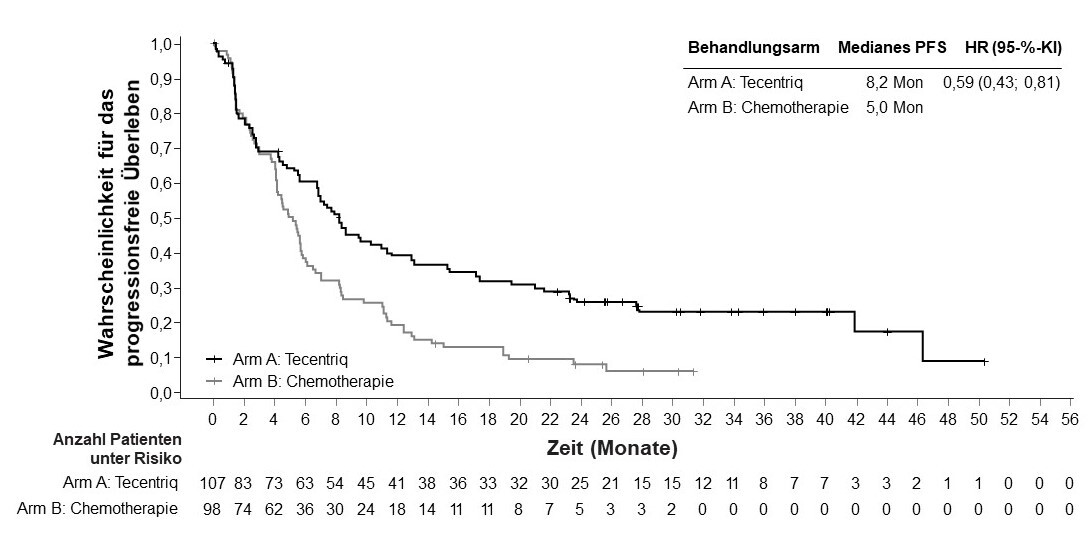
Die beobachtete Verbesserung im OS im Atezolizumab-Arm im Vergleich zum Chemotherapie-Arm wurde bei Patienten mit hoher PD-L1-Expression durchgängig in den Subgruppen nachgewiesen, einschließlich bei Patienten mit Nicht-Plattenepithel-NSCLC (Hazard Ratio [HR] von 0,62; 95‑%‑KI: 0,40; 0,96; medianes OS 20,2 vs. 10,5 Monate) und bei Patienten mit Plattenepithel-NSCLC (HR von 0,56; 95‑%‑KI: 0,23; 1,37; medianes OS nicht erreicht vs. 15,3 Monate). Die Daten für Patienten im Alter von ≥ 75 Jahren und für Patienten, die lebenslange Nichtraucher waren, sind zu begrenzt, um Rückschlüsse auf diese Subgruppen zuzulassen.
IPSOS-Studie (MO29872): Randomisierte Phase-III-Studie bei Patienten mit nicht vorbehandeltem, lokal fortgeschrittenem inoperablem oder metastasiertem NSCLC, die für eine platinbasierte Chemotherapie ungeeignet sind
Eine unverblindete, randomisierte, kontrollierte Phase-III-Studie, MO29872 (IPSOS), wurde durchgeführt, um die Wirksamkeit und Sicherheit von Atezolizumab im Vergleich zu einer Monochemotherapie (Vinorelbin oder Gemcitabin nach Wahl des Prüfarztes) bei nicht vorbehandelten Patienten mit fortgeschrittenem oder rezidiviertem (Stadium IIIB [gemäß der 7. Version des AJCC], nicht für multimodale Therapien geeignet) oder metastasiertem (Stadium IV) NSCLC zu untersuchen, die für eine platinbasierte Chemotherapie als ungeeignet eingestuft wurden.
Die folgenden Auswahlkriterien definieren Patienten, die ungeeignet für eine platinbasierte Chemotherapie und in dem Anwendungsgebiet eingeschlossen sind: Patienten im Alter von > 80 Jahren oder mit einem ECOG-Performance-Status (PS) von 3 oder Patienten mit einem ECOG-PS 2 in Kombination mit relevanten Komorbiditäten oder ältere Patienten (≥70 Jahre) in Kombination mit relevanten Komorbiditäten. Relevante Komorbiditäten sind Herzerkrankungen, Erkrankungen des Nervensystems, psychiatrische Erkrankungen, Gefäßerkrankungen, Erkrankungen der Nieren, Stoffwechsel- und Ernährungsstörungen oder Erkrankungen der Lunge, die nach Einschätzung des behandelnden Arztes eine Kontraindikation für eine Behandlung mit einer platinbasierten Therapie sind.
Von der Studie ausgeschlossen waren Patienten unter 70 Jahren mit einem ECOG-PS von 0 oder 1; Patienten mit aktiven oder unbehandelten ZNS-Metastasen; Patienten, die innerhalb von 4 Wochen vor der Randomisierung einen abgeschwächten Lebendimpfstoff oder systemische Immunstimulanzien oder systemische Immunsuppressiva erhalten haben. Patienten mit EGFR-Mutationen oder ALK-Translokationen waren ebenfalls von der Studie ausgeschlossen. Die Patienten wurden unabhängig von ihrem PD-L1-Status des Tumors in die Studie eingeschlossen.
Die Patienten wurden im Verhältnis 2:1 randomisiert und erhielten entweder Atezolizumab (Arm A) oder Chemotherapie (Arm B). Atezolizumab wurde in einer Fixdosis von 1 200 mg alle 3 Wochen als intravenöse Infusion verabreicht. Die Chemotherapieschemata sind in Tabelle 15 beschrieben. Die Behandlung wurde solange fortgeführt, bis eine Krankheitsprogression gemäß RECIST v1.1 oder eine nicht kontrollierbare Toxizität auftrat. Die Randomisierung erfolgte stratifiziert nach Histologie (Plattenepithel/Nicht-Plattenepithel), PD-L1-Expression (PD-L1-IHC-Status, bewertet mit dem VENTANA PD-L1-(SP142)-Test: TC3 oder IC3 vs. TC0/1/2 und IC0/1/2 vs. nicht bekannt) sowie Hirnmetastasen (ja/nein).
Tabelle 15: Behandlungsschemata (IPSOS)
Behandlungsschema |
||
A |
Atezolizumab 1 200 mg als intravenöse Infusion an Tag 1 eines jeden 21-Tage-Zyklus. |
|
B |
Vinorelbin: intravenöse Infusion von 25 – 30 mg/m2 oder oral 60 – 80 mg/m2 an den Tagen 1 und 8 eines jeden 21-Tage-Zyklus oder an den Tagen 1, 8 und 15 eines jeden 28-Tage-Zyklus oder wöchentliche Gabe oder |
|
Insgesamt wurden 453 Patienten in die Studie aufgenommen (ITT-Population). Die Mehrheit der Patienten war kaukasischer Herkunft (65,8 %) und männlich (72,4 %). Das mediane Alter der Patienten betrug 75 Jahre und 72,8 % der Patienten waren 70 Jahre oder älter. Der Anteil der Patienten mit einem ECOG-PS von 0, 1, 2 bzw. 3 betrug 1,5 %, 15,0 %, 75,9 % bzw. 7,5 %. Insgesamt hatten 13,7 % der Patienten eine Erkrankung im Stadium IIIB, die nicht für eine multimodale Behandlung geeignet war, und 86,3 % hatten eine Erkrankung im Stadium IV. Der prozentuale Anteil der Patienten mit Tumoren mit einer PD-L1-Expression TC < 1 %, 1 – 49 % und ≥ 50 %, bewertet mit dem VENTANA PD-L1-(SP263)-Test, betrug 46,8 %, 28,7 % bzw. 16,6 %, während bei 7,9 % der Patienten der PD-L1-Expressionsstatus nicht bekannt war.
Der primäre Endpunkt der Studie war das Gesamtüberleben (OS). Zum Zeitpunkt der finalen OS-Analyse betrug die mediane Nachbeobachtung 41,0 Monate. Die Ergebnisse zur Wirksamkeit sind in Tabelle 16 und Abbildung 14 dargestellt.
Tabelle 16: Zusammenfassung der Wirksamkeit bei NSCLC Patienten, die für eine platinbasierte Chemotherapie ungeeignet sind (IPSOS)
Wirksamkeitsendpunkt |
Atezolizumab |
Chemotherapie |
Primärer Wirksamkeitsendpunkt | ||
Gesamtüberleben (OS) | ||
Anz. der Ereignisse (%) |
249 (82,5 %) |
130 (86,1 %) |
Mediane Zeit bis zum Ereignis (Monate) (95‑%‑KI*) |
10,3 (9,4; 11,9) |
9,2 (5,9; 11,2) |
Stratifizierte Hazard Ratio (95‑%‑KI) ǂ |
0,78 (0,63; 0,97) |
|
Sekundäre Endpunkte | ||
Vom Prüfarzt bewertetes PFS (RECIST 1.1) | ||
Anz. der Ereignisse (%) |
276 (91,4 %) |
138 (91,4 %) |
Mediane Dauer des PFS (Monate) (95‑%‑KI) |
4,2 (3,7; 5,5) |
4,0 (2,9; 5,4) |
Stratifizierte Hazard Ratio (95‑%‑KI) ǂ |
0,87 (0,70; 1,07) |
|
ORR (RECIST 1.1) |
||
Anz. der Patienten mit bestätigtem |
51 (16,9 %) |
12 (7,9 %) |
DOR (RECIST 1.1) |
||
Median in Monaten (95-%-KI) |
14,0 (8,1; 20,3) |
7,8 (4,8; 9,7) |
KI = Konfidenzintervall; DOR = duration of response (Dauer des Ansprechens); ORR = objective response rate (objektive Ansprechrate); OS = overall survival (Gesamtüberleben); PFS = progression‑free survival (progressionsfreies Überleben); RECIST = Response Evaluation Criteria in Solid Tumours v1.1.
ǂ Geschätzte Hazard Ratio und 95-%-KI mittels Cox-Modell mit Behandlungsgruppe als Kovariate. Für die stratifizierte Analyse wurden der histologische Subtyp, der PD-L1-IHC-Status und Hirnmetastasen (ja/nein) als Stratifizierungsfaktoren hinzugefügt.
Abbildung 14: Kaplan-Meier-Kurven zum Gesamtüberleben für NSCLC Patienten, die für eine platinbasierte Chemotherapie ungeeignet sind (IPSOS)
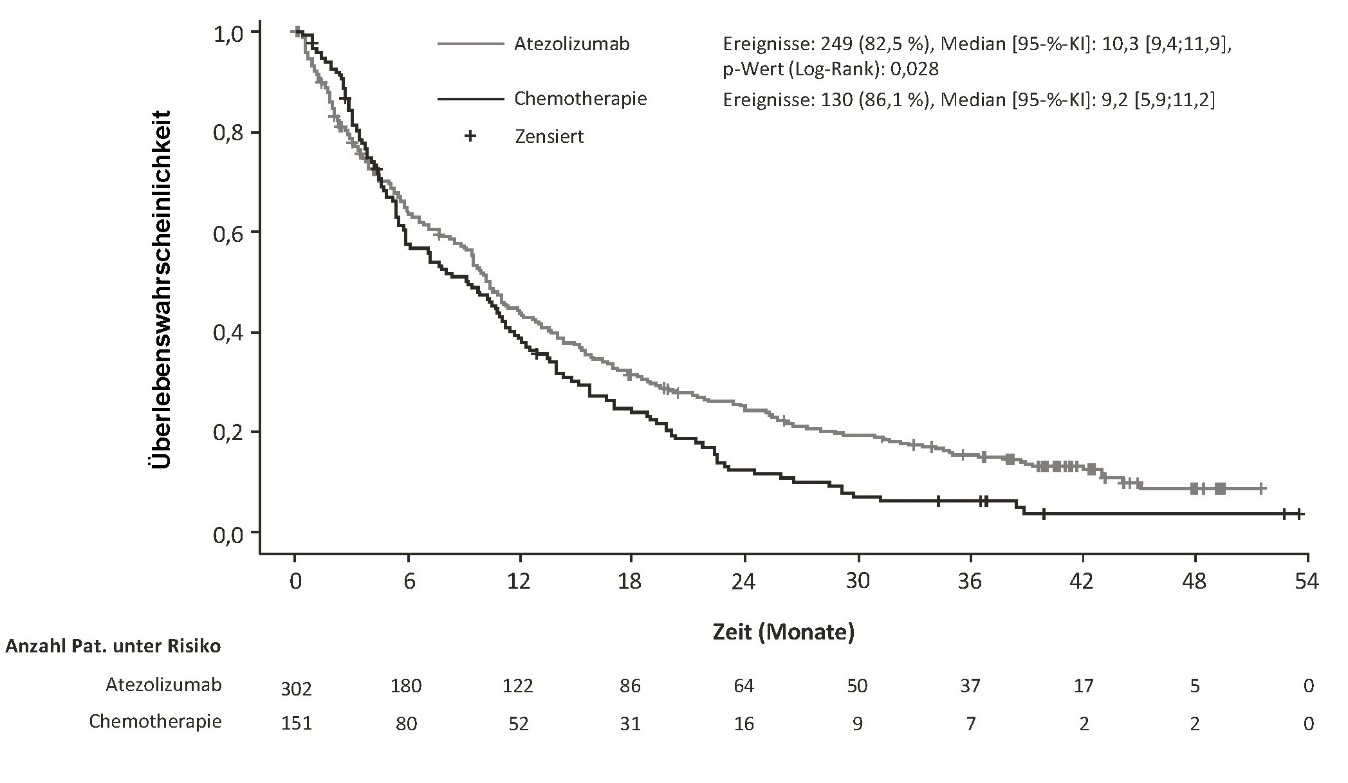
Zweitlinienbehandlung des NSCLC
Subkutane Darreichungsform
IMscin001 (BP40657): Randomisierte Phase-Ib/III-Studie bei Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC, die zuvor mit einer platinhaltigen Chemotherapie behandelt wurden
Eine unverblindete, multizentrische, internationale, randomisierte Phase-Ib/III-Studie, BP40657 (IMscin001), wurde durchgeführt, um die Pharmakokinetik, Wirksamkeit und Sicherheit von Tecentriq subkutan im Vergleich zu Atezolizumab intravenös bei Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC zu untersuchen, die noch keine Krebsimmuntherapie (KIT) erhalten hatten und bei denen eine vorherige platinbasierte Therapie fehlgeschlagen ist. IMscin001 wurde konzipiert, um die Nicht-Unterlegenheit der Serum-Ctrough von Atezolizumab in Zyklus 1 (vor Verabreichung von Zyklus 2) und die modellbasierte vorhergesagte AUC von 0 bis 21 Tagen in Zyklus 1 von Atezolizumab subkutan im Vergleich zu Atezolizumab intravenös (ko-primärer Endpunkt) nachzuweisen. Sekundäre Endpunkte schlossen Wirksamkeit (PFS, ORR, OS, DOR) und Sicherheit ein.
In Teil 2 (Phase III) wurden insgesamt 371 Patienten eingeschlossen und 2:1 randomisiert, um entweder 1 875 mg Tecentriq subkutan Q3W oder 1 200 mg Atezolizumab intravenös Q3W zu erhalten. Eine Dosisreduktion war nicht erlaubt.
Patienten wurden ausgeschlossen, wenn sie eine Autoimmunerkrankung in der Anamnese, aktive oder Corticosteroid-pflichtige Hirnmetastasen hatten, eine Impfung mit einem attenuierten Lebendimpfstoff oder systemische Immunstimulanzien innerhalb von 4 Wochen bzw. systemische Immunsuppressiva innerhalb von 2 Wochen vor der Randomisierung erhalten hatten.
Das mediane Alter lag bei 64 Jahren (Bereich: 27 bis 85), und 69 % der Patienten waren männlich. Die Mehrheit der Patienten war kaukasischer Herkunft (67 %). Etwa zwei Drittel der Patienten (65 %) hatten eine nicht-plattenepitheliale Erkrankung, 5 % hatten eine bekannte EGFR-Mutation, 2 % hatten bekannte ALK-Translokationen, 40 % waren PD-L1-positiv (TC ≥ 1 % und/oder IC ≥ 1 %), 16 % hatten zu Behandlungsbeginn nicht-aktive ZNS-Metastasen, 26 % hatten einen ECOG-PS von 0,74 % hatten einen ECOG-PS von 1 und die meisten Patienten waren Raucher oder ehemalige Raucher (70 %). 80 % hatten eine vorherige therapeutische Behandlung erhalten.
Zum Zeitpunkt der Primäranalyse betrug die mediane Nachbeobachtungszeit des Überlebens 4,7 Monate und die OS-Ergebnisse waren unreif. Im Behandlungsarm mit Tecentriq subkutan traten 86 (35 %) Todesfälle und im Behandlungsarm mit Atezolizumab intravenös 37 (30 %) Todesfälle auf. Eine aktualisierte Post-hoc-Analyse wurde 9 Monate nach der primären Analyse mit einer medianen Nachbeobachtungszeit des Überlebens von 9,5 Monaten durchgeführt. Die Wirksamkeitsergebnisse der aktualisierten Analysen sind in Tabelle 17 zusammengefasst.
Tabelle 17: Zusammenfassung der aktualisierten Wirksamkeitsanalysen (IMscin001)
Wirksamkeitsendpunkt |
Tecentriq subkutan |
Tecentriq intravenös |
Vom Prüfarzt bewertete ORR (RECIST v1.1)* |
n = 245 |
n = 124 |
Anzahl Patienten mit bestätigtem Ansprechen (%) |
27 (11,0 %) |
13 (10,5 %) |
95‑%‑KI |
(7,39; 15,63) |
(5,70; 17,26) |
Vom Prüfarzt bewertetes PFS (RECIST v1.1)* |
n = 247 |
n = 124 |
Anzahl der Ereignisse (%) |
219 (88,7 %) |
107 (86,3 %) |
Median (Monate) (95-%-KI) |
2.8 (2,7; 4,1) |
2,9 (1,8; 4,2) |
OS* |
n = 247 |
n = 124 |
Anzahl der Ereignisse (%) |
144 (58,3 %) |
79 (63,7 %) |
Median (Monate) (95-%-KI) |
10,7 (8,5; 13,8) |
10,1 (7,5; 12,1) |
KI = Konfidenzintervall; ORR = objective response rate (objektive Ansprechrate); OS = overall survival (Gesamtüberleben); PFS = progression‑free survival (progressionsfreies Überleben); RECIST = Response Evaluation Criteria in Solid Tumours v1.1.
* Deskriptive Analysen
Intravenöse Darreichungsform
OAK (GO28915): Randomisierte Studie der Phase III an Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC, die bereits mit Chemotherapie behandelt wurden
Eine unverblindete, multizentrische, internationale, randomisierte Studie der Phase III, OAK, wurde zur Bewertung der Wirksamkeit und Sicherheit von Atezolizumab im Vergleich zu Docetaxel bei Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC durchgeführt, die während oder im Anschluss an ein platinhaltiges Regime eine Krankheitsprogression erlitten. Von der Studie ausgeschlossen waren Patienten mit einer Autoimmunerkrankung in der Vorgeschichte, aktiven oder Corticosteroid‑abhängigen Gehirnmetastasen, einer Verabreichung von attenuierten Lebendimpfstoffen innerhalb von 28 Tagen vor Studieneinschluss, einer Verabreichung von systemischen Immunstimulanzien innerhalb von 4 Wochen oder von systemischen Immunsuppressiva innerhalb von 2 Wochen vor Einschluss. Tumorbewertungen wurden in den ersten 36 Wochen alle 6 Wochen und anschließend alle 9 Wochen durchgeführt. Tumorproben wurden prospektiv auf PD‑L1‑Expression auf den Tumorzellen (TC) und tumorinfiltrierenden Immunzellen (IC) getestet.
Insgesamt wurden 1 225 Patienten in die Studie aufgenommen, von denen die ersten 850 randomisierten Patienten gemäß Analysenplan in die Primäranalyse zur Wirksamkeit eingeschlossen wurden. Die Randomisierung wurde nach PD‑L1‑Expression‑Status auf den IC, der Anzahl der bisherigen Chemotherapie‑Regime und nach Histologie stratifiziert. Die Patienten wurden im Verhältnis 1:1 randomisiert und erhielten entweder Atezolizumab oder Docetaxel.
Atezolizumab wurde in einer Fixdosis von 1 200 mg alle 3 Wochen als intravenöse Infusion verabreicht. Eine Dosisreduktion war nicht gestattet. Die Patienten wurden bis zum Verlust des klinischen Nutzens gemäß Bewertung des Prüfarztes behandelt. Docetaxel wurde in einer Dosierung von 75 mg/m2 an Tag 1 jedes 3‑Wochen‑Zyklus als intravenöse Infusion verabreicht, bis eine Krankheitsprogression auftrat. Bei allen behandelten Patienten betrug die mediane Dauer der Behandlung 2,1 Monate im Docetaxel‑Arm und 3,4 Monate im Atezolizumab‑Arm.
Die demographische Verteilung sowie die Krankheitsmerkmale bei Behandlungsbeginn der Primäranalyse‑Population waren zwischen den Behandlungsarmen gut ausgewogen. Das mediane Alter lag bei 64 Jahren (Bereich: 33 bis 85); 61 % der behandelten Patienten waren männlich. Die Mehrheit der Patienten war kaukasischer Herkunft (70 %). Ungefähr drei Viertel der Patienten hatten ein Karzinom mit nicht‑plattenepithelialer Histologie (74 %), bei 10 % war das Vorliegen einer EGFR‑Mutation bekannt, bei 0,2 % war eine ALK‑Translokation bekannt, 10 % hatten zu Behandlungsbeginn ZNS‑Metastasen und die meisten Patienten waren aktuelle oder ehemalige Raucher (82 %). Die Patienten hatten zu Behandlungsbeginn einen ECOG-Performance‑Status von 0 (37 %) oder 1 (63 %). 75 % der Patienten hatten nur ein vorangehendes platinbasiertes Chemotherapie‑Regime erhalten.
Der primäre Wirksamkeitsendpunkt war das Gesamtüberleben. Die wesentlichen Ergebnisse dieser Studie mit einer medianen Nachbeobachtungszeit des Überlebens von 21 Monaten sind in Tabelle 18 zusammengefasst. Kaplan‑Meier‑Kurven für das Gesamtüberleben in der Intention‑to‑Treat(ITT)‑Population sind in Abbildung 15 dargestellt. Abbildung 16 enthält eine Zusammenfassung der Ergebnisse zum Gesamtüberleben in der ITT‑Population und in den PD‑L1‑Subgruppen, die einen Nutzen von Atezolizumab in Bezug auf das Gesamtüberleben in allen Subgruppen, einschließlich derjenigen mit PD‑L1‑Expression < 1 % auf TC und IC, zeigt.
Tabelle 18: Zusammenfassung der Wirksamkeit in der Primäranalyse‑Population (all comers)* (OAK)
Wirksamkeitsendpunkt |
Atezolizumab |
Docetaxel |
Primärer Wirksamkeitsendpunkt |
||
Gesamtüberleben (OS) |
||
Anzahl der Todesfälle (%) |
271 (64 %) |
298 (70 %) |
Mediane Zeit bis zum Ereignis (Monate) |
13,8 |
9,6 |
95‑%‑KI |
(11,8; 15,7) |
(8,6; 11,2) |
Stratifizierteǂ Hazard Ratio (95‑%‑KI) |
0,73 (0,62; 0,87) |
|
p‑Wert** |
0,0003 |
|
12‑Monats‑Gesamtüberleben (%)*** |
218 (55 %) |
151 (41 %) |
18‑Monats‑Gesamtüberleben (%)*** |
157 (40 %) |
98 (27 %) |
Sekundäre Endpunkte | ||
Vom Prüfarzt bewertetes PFS (RECIST v1.1) | ||
Anzahl der Ereignisse (%) |
380 (89 %) |
375 (88 %) |
Mediane Dauer des PFS (Monate) |
2,8 |
4,0 |
95‑%‑KI |
(2,6; 3,0) |
(3,3; 4,2) |
Stratifizierte Hazard Ratio (95‑%‑KI) |
0,95 (0,82; 1,10) |
|
Vom Prüfarzt bewertete ORR (RECIST v1.1) | ||
Mit einem Ansprechen (%) |
58 (14 %) |
57 (13 %) |
95‑%‑KI |
(10,5; 17,3) |
(10,3; 17,0) |
Vom Prüfarzt bewertete DOR (RECIST v1.1) |
n = 58 |
n = 57 |
Median in Monaten |
16,3 |
6,2 |
95‑%‑KI |
(10,0; NE) |
(4,9; 7,6) |
KI = Konfidenzintervall; DOR = duration of response (Dauer des Ansprechens); NE = not estimable (nicht schätzbar); ORR = objective response rate (objektive Ansprechrate); OS = overall survival (Gesamtüberleben); PFS = progression‑free survival (progressionsfreies Überleben); RECIST = Response Evaluation Criteria in Solid Tumours v1.1.
* Die Primäranalyse‑Population besteht aus den ersten 850 randomisierten Patienten
ǂ Stratifiziert nach PD‑L1‑Expression auf tumorinfiltrierenden Immunzellen, der Anzahl bisheriger Chemotherapie‑Regime und der Histologie
** Basierend auf dem stratifizierten Log‑Rank‑Test
*** Basierend auf Kaplan‑Meier‑Schätzung
Abbildung 15: Kaplan‑Meier‑Kurve zum Gesamtüberleben in der Primäranalyse‑Population (all comers) (OAK)
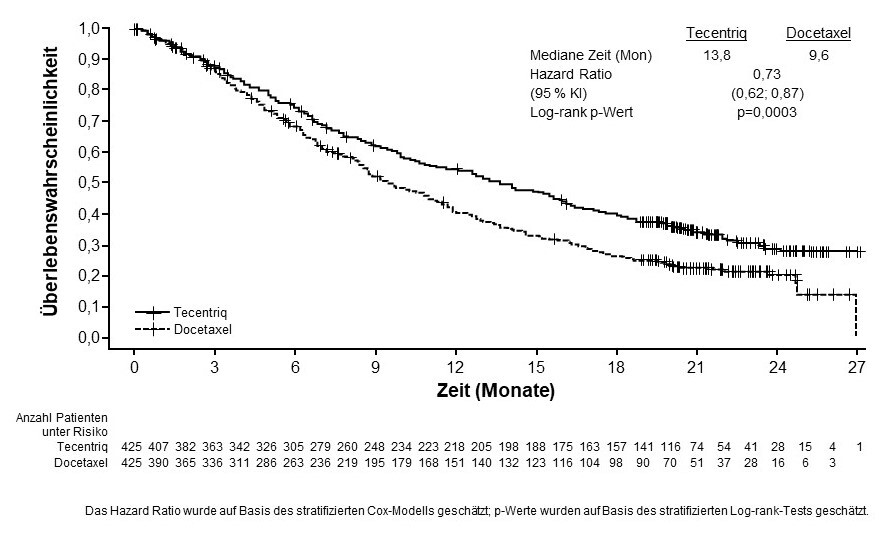
Abbildung 16: Forest‑Diagramm zum Gesamtüberleben bei PD‑L1‑Expression in der Primäranalyse‑Population (OAK)
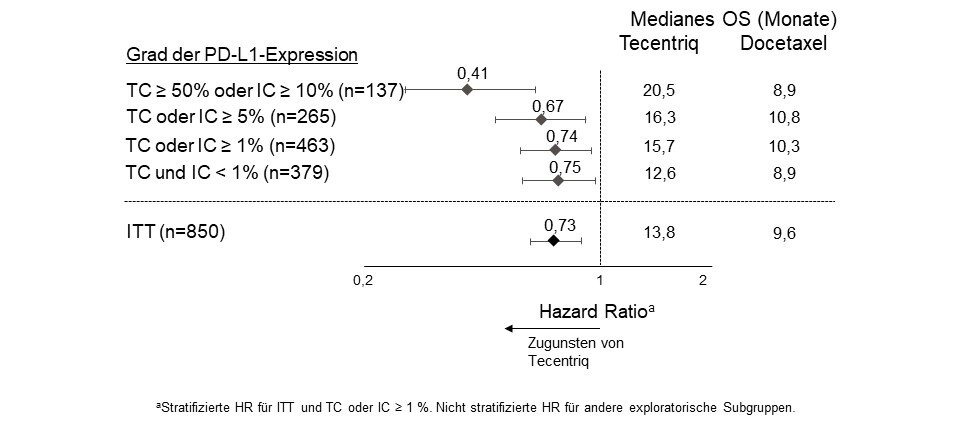
Eine Verbesserung im Gesamtüberleben unter Atezolizumab im Vergleich zu Docetaxel wurde sowohl bei Patienten mit Nicht‑Plattenepithel‑NSCLC (Hazard Ratio [HR] von 0,73; 95‑%‑KI: 0,60; 0,89; medianes OS 15,6 vs. 11,2 Monate für Atezolizumab bzw. Docetaxel) als auch bei Patienten mit Plattenepithel‑NSCLC (HR von 0,73; 95‑%‑KI: 0,54; 0,98; medianes OS 8,9 vs. 7,7 Monate für Atezolizumab bzw. Docetaxel) beobachtet. Die beobachtete Verbesserung im OS wurde durchgängig bei allen Patienten‑Subgruppen nachgewiesen, einschließlich derjenigen mit Gehirnmetastasen zu Behandlungsbeginn (HR von 0,54; 95‑%‑KI: 0,31; 0,94; medianes OS 20,1 vs. 11,9 Monate für Atezolizumab bzw. Docetaxel) und bei Patienten, die nie geraucht haben (HR von 0,71; 95‑%‑KI: 0,47; 1,08; medianes OS 16,3 vs. 12,6 Monate für Atezolizumab bzw. Docetaxel). Demgegenüber zeigten Patienten mit einer EGFR‑Mutation kein verbessertes Gesamtüberleben unter Atezolizumab gegenüber Docetaxel (HR von 1,24; 95‑%‑KI: 0,71; 2,18; medianes OS 10,5 vs. 16,2 Monate für Atezolizumab bzw. Docetaxel).
Eine verlängerte Zeit bis zur Verschlechterung der vom Patienten berichteten Schmerzen im Brustraum wurde unter Atezolizumab im Vergleich zu Docetaxel (HR von 0,71; 95‑%‑KI: 0,49; 1,05; Median in keinem Arm erreicht) beobachtet, gemessen anhand des EORTC QLQ‑LC13. Die Zeit bis zur Verschlechterung anderer Lungenkrebssymptome (z. B. Husten, Dyspnoe, Schmerzen in Armen/Schultern), gemessen anhand des EORTC QLQ‑LC13, war zwischen Atezolizumab und Docetaxel ähnlich. Aufgrund des unverblindeten Studiendesigns sind die Ergebnisse mit Vorsicht zu interpretieren.
POPLAR (GO28753): Randomisierte Studie der Phase II bei Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC, die bereits mit Chemotherapie behandelt wurden
Eine multizentrische, internationale, randomisierte, unverblindete, kontrollierte Studie der Phase II, POPLAR, wurde bei Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC durchgeführt, die während oder nach einem platinhaltigen Chemotherapie‑Regime, unabhängig von einer PD‑L1‑Expression, eine Krankheitsprogression erlitten. Der primäre Wirksamkeitsendpunkt war das Gesamtüberleben. Insgesamt wurden 287 Patienten im Verhältnis 1:1 randomisiert und erhielten entweder Atezolizumab (1 200 mg als intravenöse Infusion alle 3 Wochen bis zum Verlust des klinischen Nutzens) oder Docetaxel (75 mg/m2 als intravenöse Infusion an Tag 1 jedes 3‑Wochen‑Zyklus bis zur Krankheitsprogression). Die Randomisierung erfolgte stratifiziert nach PD‑L1‑Expression‑Status auf Immunzellen, der Anzahl der bisherigen Chemotherapie‑Regime und der Histologie. Eine aktualisierte Analyse mit insgesamt 200 beobachteten Todesfällen und einer medianen Nachbeobachtungszeit des Überlebens von 22 Monaten ergab ein medianes OS von 12,6 Monaten bei Patienten, die mit Atezolizumab behandelt wurden vs. 9,7 Monaten bei Patienten, die mit Docetaxel behandelt wurden (HR von 0,69; 95‑%‑KI: 0,52; 0,92). Die ORR betrug 15,3 % vs. 14,7 % und die mediane DOR lag bei 18,6 Monaten vs. 7,2 Monaten für Atezolizumab bzw. Docetaxel.
Kleinzelliges Lungenkarzinom
Intravenöse Darreichungsform
IMpower133 (GO30081): Randomisierte Studie der Phase I/III bei Patienten mit chemotherapienaivem SCLC im fortgeschrittenen Stadium in Kombination mit Carboplatin und Etoposid
Eine randomisierte, multizentrische, doppelblinde, placebokontrollierte Studie der Phase I/III, IMpower133, wurde zur Untersuchung der Wirksamkeit und Sicherheit von Atezolizumab in Kombination mit Carboplatin und Etoposid bei Patienten mit chemotherapienaivem ES-SCLC durchgeführt.
Patienten mit aktiven oder unbehandelten ZNS-Metastasen oder einer Autoimmunerkrankung in der Anamnese oder die mit einem attenuierten Lebendimpfstoff innerhalb von 4 Wochen vor der Randomisierung geimpft wurden oder die systemische Immunsuppressiva innerhalb von 1 Woche vor der Randomisierung erhalten hatten, wurden von der Studie ausgeschlossen. Eine Beurteilung des Tumors wurde während der ersten 48 Wochen alle 6 Wochen nach Zyklus 1, Tag 1 durchgeführt und danach alle 9 Wochen. Bei Patienten, die die festgelegten Kriterien erfüllten und einwilligten, nach Krankheitsprogression weiterbehandelt zu werden, wurden bis zum Behandlungsabbruch alle 6 Wochen Beurteilungen des Tumors vorgenommen.
Insgesamt wurden 403 Patienten eingeschlossen, im Verhältnis 1:1 randomisiert und einem der in Tabelle 19 beschriebenen Behandlungsschemata zugeteilt. Die Randomisierung wurde nach Geschlecht, ECOG-Performance-Satus und Vorhandensein von Hirnmetastasen stratifiziert.
Tabelle 19: Intravenöses Behandlungsschema (IMpower133)
Behandlungs-schema |
Induktion |
Erhaltung |
A |
Atezolizumab (1 200 mg)a + Carboplatin (AUC 5)b + Etoposid (100 mg/m2)b,c |
Atezolizumab (1 200 mg)a |
B |
Placebo + Carboplatin (AUC 5)b + Etoposid (100 mg/m2)b,c |
Placebo |
a Atezolizumab wurde bis zum Verlust des klinischen Nutzens gemäß Bewertung des Prüfarztes angewendet
b Carboplatin und Etoposid wurden bis zum Abschluss von 4 Zyklen oder fortschreitender Erkrankung oder bis zum Auftreten inakzeptabler Toxizität, je nachdem was zuerst eintrat, angewendet
c Etoposid wurde an Tag 1, 2 und 3 jedes Zyklus angewendet
Die demographischen Merkmale und die Merkmale der Grunderkrankung der Studienpopulation zum Behandlungsbeginn waren zwischen den Behandlungsarmen ausgeglichen. Das mediane Alter betrug 64 Jahre (Bereich: 26 bis 90 Jahre) und 10 % der Patienten waren ≥ 75 Jahre. Die Mehrheit der Patienten war männlich (65 %), kaukasischer Herkunft (80 %) und 9 % der Patienten hatten Hirnmetastasen. Die meisten Patienten waren Raucher oder ehemalige Raucher (97 %). Der ECOG-Performance-Status betrug zu Behandlungsbeginn 0 (35 %) oder 1 (65 %).
Zum Zeitpunkt der Primäranalyse hatten die Patienten eine mediane Nachbeobachtungszeit von 13,9 Monaten. Eine statistisch signifikante Verbesserung des OS wurde bei Atezolizumab in Kombination mit Carboplatin und Etoposid im Vergleich zum Kontrollarm beobachtet (HR von 0,70; 95‑%‑KI: 0,54; 0,91; medianes OS von 12,3 Monaten vs. 10,3 Monate). In der finalen explorativen OS-Analyse mit längerer Nachbeobachtung (median: 22,9 Monate) war das mediane OS in beiden Armen unverändert im Vergleich zur primären OS-Interimanalyse. Die Ergebnisse bezüglich PFS, ORR und DOR aus der Primäranalyse wie auch die explorativen Ergebnisse der finalen OS-Analyse sind in Tabelle 20 zusammengefasst. Die Kaplan‑Meier‑Kurven für das OS und das PFS sind in den Abbildungen 17 und 18 dargestellt. Die Daten zu Patienten mit Hirnmetastasen sind zu begrenzt, um Rückschlüsse auf diese Population zuzulassen.
Tabelle 20: Zusammenfassung der Wirksamkeit (IMpower133)
Wesentliche Wirksamkeitsendpunkte |
Arm A |
Arm B |
|
Ko-primäre Endpunkte |
|||
OS-Analyse* |
n = 201 |
n = 202 |
|
Anzahl der Todesfälle (%) |
142 (70,6 %) |
160 (79,2 %) |
|
Mediane Zeit bis zum Ereignis (Monate) |
12,3 |
10,3 |
|
95‑%‑KI |
(10,8; 15,8) |
(9,3; 11,3) |
|
Stratifizierte Hazard Ratio ‡ (95‑%‑KI) |
0,76 (0,60; 0,95) |
||
p-Wert |
0,0154*** |
||
12-Monats-OS (%) |
51,9 |
39,0 |
|
Vom Prüfarzt bewertetes PFS (RECIST v1.1)** |
n = 201 |
n = 202 |
|
Anzahl der Ereignisse (%) |
171 (85,1 %) |
189 (93,6 %) |
|
Mediane Dauer des PFS (Monate) |
5,2 |
4,3 |
|
95‑%‑KI |
(4,4; 5,6) |
(4,2; 4,5) |
|
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,77 (0,62; 0,96) |
||
p-Wert |
0,0170 |
||
6-Monats-PFS (%) |
30,9 |
22,4 |
|
Andere Endpunkte |
|||
Vom Prüfarzt bewertete ORR (RECIST 1.1)** ^ |
n = 201 |
n = 202 |
|
Anzahl der Patienten mit Ansprechen (%) |
121 (60,2 %) |
130 (64,4 %) |
|
95‑%‑KI |
(53,1; 67,0) |
(57,3; 71,0) |
|
Anzahl mit vollständigem Ansprechen (%) |
5 (2,5 %) |
2 (1,0 %) |
|
Anzahl mit teilweisem Ansprechen (%) |
116 (57,7 %) |
128 (63,4 %) |
|
Vom Prüfarzt bewertete DOR (RECIST 1.1)** ^ |
n = 121 |
n = 130 |
|
Median in Monaten |
4,2 |
3,9 |
|
95‑%‑KI |
(4,1; 4,5) |
(3,1; 4,2) |
|
PFS = progression‑free survival (progressionsfreies Überleben); RECIST = Response Evaluation Criteria in Solid Tumours v1.1., KI = Konfidenzintervall; ORR = objective response rate (objektive Ansprechrate); OS = overall survival (Gesamtüberleben).
‡ Stratifiziert nach Geschlecht und ECOG-Performance-Status
* Explorative OS-Analyse zum klinischen Stichtag 24. Januar 2019
** PFS-, ORR- und DOR-Analysen zum klinischen Stichtag 24. April 2018
*** nur zu deskriptiven Zwecken
^ Bestätigte ORR und DOR sind explorative Endpunkte
Abbildung 17: Kaplan-Meier-Kurve zum Gesamtüberleben (IMpower133)
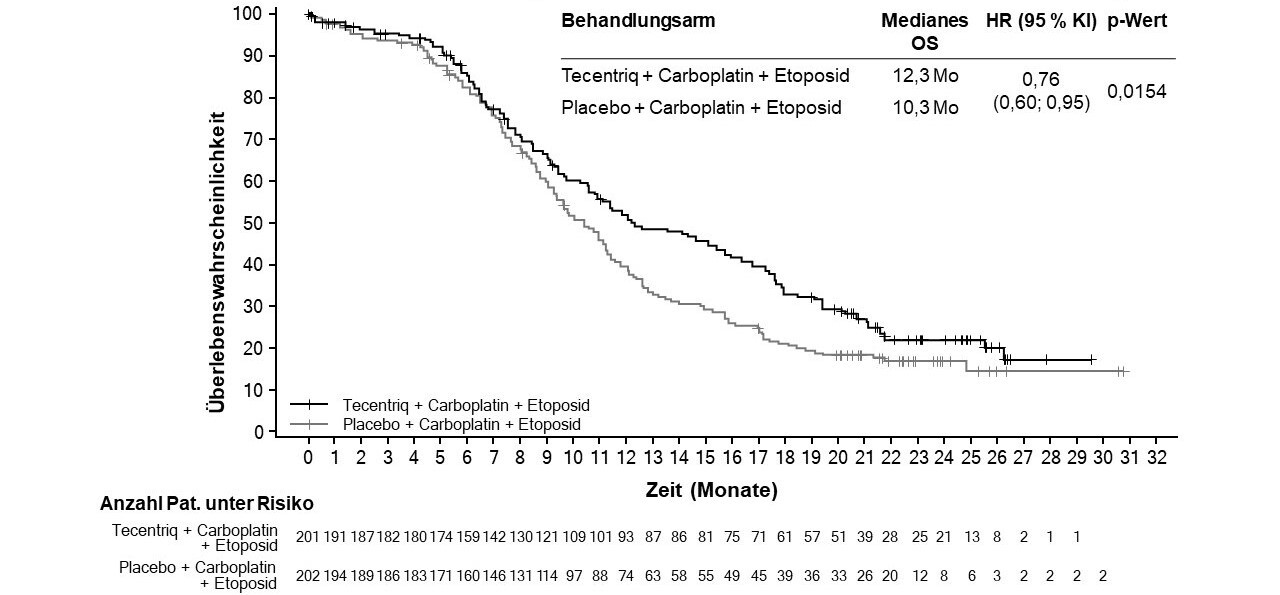
Abbildung 18: Kaplan-Meier-Kurve zum progressionsfreien Überleben (IMpower133)
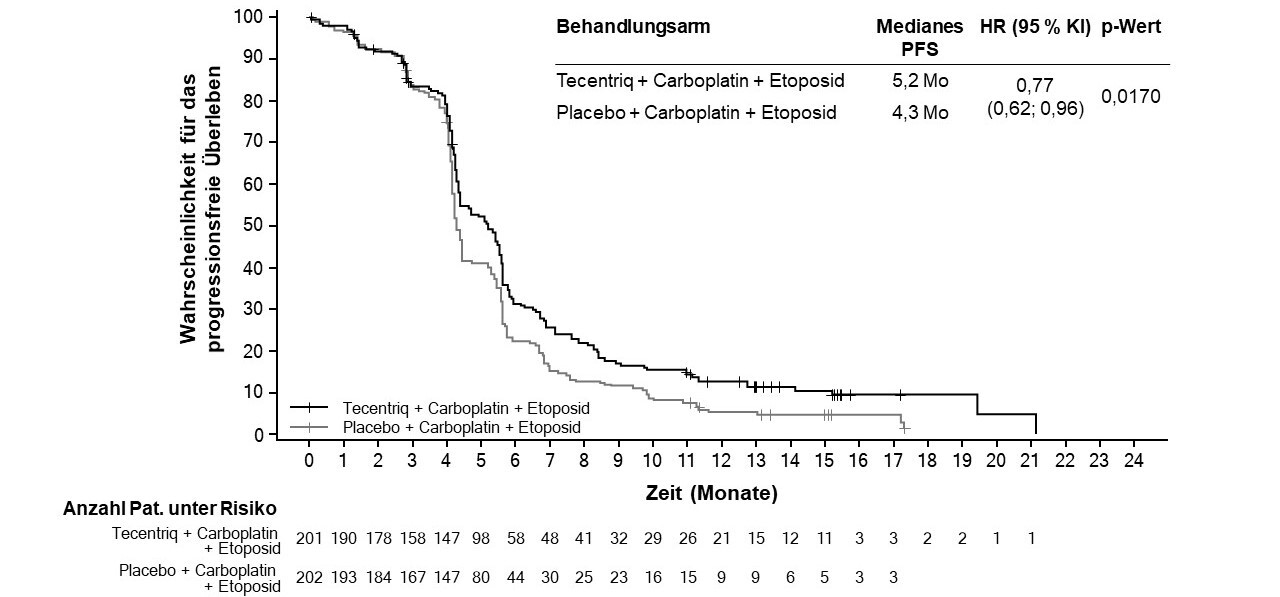
Triple-negatives Mammakarzinom
Intravenöse Darreichungsform
IMpassion130 (WO29522): Randomisierte Studie der Phase III bei Patienten mit lokal fortgeschrittenem oder metastasiertem TNBC ohne vorherige Behandlung der metastasierten Erkrankung
Eine doppelblinde, zweiarmige, multizentrische, internationale, randomisierte, placebokontrollierte Studie der Phase III, IMpassion130, wurde durchgeführt, um die Wirksamkeit und Sicherheit von Atezolizumab in Kombination mit nab-Paclitaxel bei Patienten mit nicht resezierbarem lokal fortgeschrittenem oder metastasiertem TNBC, die keine vorherige Chemotherapie zur Behandlung ihrer metastasierten Erkrankung erhalten hatten, zu beurteilen. Die Patienten mussten für eine Taxan-Monotherapie geeignet sein (d. h. keine schnelle klinische Progression, keine lebensbedrohlichen viszeralen Metastasen, keine Notwendigkeit für eine schnelle Symptom- und/oder Krankheitskontrolle). Sie waren ausgeschlossen, wenn sie innerhalb der letzten 12 Monate mit einer vorherigen Chemotherapie im neoadjuvanten oder adjuvanten Setting behandelt worden waren, eine Autoimmunerkrankung in der Vorgeschichte hatten, innerhalb der letzten 4 Wochen vor der Randomisierung mit einem attenuierten Lebendimpfstoff geimpft worden waren, innerhalb der letzten 4 Wochen vor der Randomisierung systemische Immunstimulanzien oder innerhalb der letzten 2 Wochen vor der Randomisierung systemische Immunsuppressiva erhalten hatten, oder unbehandelte, symptomatische oder Corticosteroid-abhängige Hirnmetastasen hatten. Tumor-Bewertungen wurden während der ersten 12 Monate nach Zyklus 1, an Tag 1 alle 8 Wochen durchgeführt (± 1 Woche) und danach alle 12 Wochen (± 1 Woche).
Insgesamt wurden 902 Patienten in die Studie eingeschlossen und stratifiziert nach Vorhandensein von Lebermetastasen, vorheriger Taxanbehandlung und nach PD‑L1‑Expressionsstatus auf tumorinfiltrierenden Immunzellen (IC) (PD‑L1 gefärbte tumorinfiltrierende Immunzellen [IC] < 1 % der Tumorfläche vs. ≥ 1 % der Tumorfläche), bewertet mit dem VENTANA PD-L1-(SP142)-Test.
Die Patienten wurden entweder einer Behandlung mit Atezolizumab 840 mg oder Placebo, als intravenöse Infusionen an den Tagen 1 und 15 eines jeden 28‑tägigen Behandlungszyklus, kombiniert mit nab-Paclitaxel (100 mg/m²) als intravenöse Infusion an den Tagen 1, 8 und 15 jedes 28‑tägigen Behandlungszyklus, randomisiert zugeteilt. Die Patienten wurden bis zur radiologischen Krankheitsprogression nach RECIST v1.1 oder dem Auftreten inakzeptabler Toxizitäten behandelt. Die Behandlung mit Atezolizumab konnte weitergeführt werden, wenn nab-Paclitaxel wegen Auftreten inakzeptabler Toxizitäten abgebrochen wurde. Die mediane Anzahl an Behandlungszyklen in jedem Behandlungsarm war für Atezolizumab 7 und für nab-Paclitaxel 6.
Die demographischen Charakteristika der Studienpopulation und deren Krankheitscharakteristika zu Behandlungsbeginn waren zwischen den Behandlungsarmen ausgeglichen. Die meisten Patienten waren Frauen (99,6 %), 67,5 % waren kaukasischen und 17,8 % asiatischen Ursprungs. Das mediane Alter betrug 55 Jahre (Bereich: 20 – 86). Der ECOG-Performance-Status zu Behandlungsbeginn betrug 0 (58,4 %) oder 1 (41,3 %). Zu Behandlungsbeginn hatten insgesamt 41 % der eingeschlossenen Patienten eine PD-L1-Expression ≥ 1 %, 27 % hatten Lebermetastasen und 7 % asymptomatische Hirnmetastasen. Etwa die Hälfte der Patienten erhielten Taxane (51 %) oder Anthrazykline (54 %) als (neo‑)adjuvante Behandlung. Patientendemographien und die Tumorerkrankung von Patienten mit PD-L1-Expression ≥ 1 % bei Behandlungsbeginn waren insgesamt für die breitere Studienpopulation repräsentativ.
Die ko-primären Wirksamkeitsendpunkte schlossen Prüfarzt bewertetes progressionsfreies Überleben (PFS) nach RECIST v1.1 und Gesamtüberleben (OS), jeweils sowohl in der ITT-Population als auch bei Patienten mit PD-L1-Expression ≥ 1 %, ein. Sekundäre Wirksamkeitsendpunkte schlossen die objektive Ansprechrate (ORR) und die Ansprechdauer (DOR) nach RECIST v1.1 ein.
Die Ergebnisse zum PFS, zur ORR und zur DOR aus der Studie IMpassion130 für Patienten mit PD-L1-Expression ≥ 1 % zum Zeitpunkt der finalen Analyse zum PFS und nach einer medianen Nachbeobachtung von 13 Monaten sind in Tabelle 21 und entsprechender Kaplan-Meier-Kurve des PFS in Abbildung 19 zusammengefasst. Patienten mit PD-L1-Expression < 1 % zeigten keine Verbesserung im PFS, wenn Atezolizumab in Kombination mit nab-Paclitaxel angewendet wurde (HR von 0,94; 95‑%‑KI: 0,78; 1,13).
Die finale OS-Analyse wurde bei Patienten mit einer PD-L1-Expression ≥ 1 % nach einer medianen Nachbeobachtung von 19,12 Monaten durchgeführt. Ergebnisse zum OS sind in Tabelle 21 und entsprechender Kaplan-Meier-Kurve in Abbildung 20 dargestellt. Patienten mit PD-L1-Expression < 1 % zeigten keine Verbesserung im OS, wenn Atezolizumab in Kombination mit nab-Paclitaxel angewendet wurde (HR von 1,02; 95‑%‑KI: 0,84; 1,24).
Explorative Subgruppenanalysen wurden bei Patienten mit PD-L1-Expression ≥ 1 % durchgeführt, wobei eine vorherige (neo‑)adjuvante Behandlung, BRCA-(breast cancer)-1/2-Mutationen und asymptomatische Hirnmetastasen zu Behandlungsbeginn untersucht wurden.
Bei Patienten, die eine vorherige (neo‑)adjuvante Behandlung erhalten hatten (n = 242), betrug die Hazard Ratio für primäres (finales) PFS 0,79 und für finales OS 0,77, wohingegen bei Patienten, die keine vorherige (neo‑)adjuvante Behandlung erhalten hatten (n = 127), die Hazard Ratio für primäres (finales) PFS 0,44 und für finales OS 0,54 betrug.
In der Studie IMpassion130 trugen von 614 getesteten Patienten 89 (15 %) eine pathogene BRCA1/2-Mutation. In der PD-L1-positiven und BRCA1/2-mutierten Subgruppe erhielten 19 Patienten Atezolizumab kombiniert mit nab-Paclitaxel und 26 Patienten Placebo kombiniert mit nab-Paclitaxel. Basierend auf einer explorativen Analyse und unter Anerkennung der geringen Anzahl an Patienten scheint das Vorhandensein von BRCA1/2-Mutationen keinen Einfluss auf den klinischen Nutzen von Atezolizumab kombiniert mit nab-Paclitaxel in Bezug auf das PFS zu haben.
Bei Patienten mit asymptomatischen Hirnmetastasen zu Behandlungsbeginn konnte die Wirksamkeit nicht nachgewiesen werden. Die Anzahl an Patienten war jedoch begrenzt. Das mediane PFS betrug 2,2 Monate im Behandlungsarm Atezolizumab in Kombination mit nab-Paclitaxel (n = 15) verglichen mit 5,6 Monate im Behandlungsarm Placebo in Kombination mit nab-Paclitaxel (n = 11) (HR von 1,40; 95‑%‑KI: 0,57; 3,44).
Tabelle 21: Zusammenfassung der Wirksamkeit bei Patienten mit PD-L1-Expression ≥ 1 % (IMpassion130)
Wirksamkeitsendpunkte |
Atezolizumab + nab-Paclitaxel |
Placebo + nab-Paclitaxel |
Primäre Wirksamkeitsendpunkte |
n = 185 |
n = 184 |
Prüfarzt bewertetes PFS (RECIST v1.1) – Primäranalyse3 | ||
Anzahl der Ereignisse (%) |
138 (74,6 %) |
157 (85,3 %) |
Mediane Dauer des PFS (Monate) |
7,5 |
5,0 |
95‑%‑KI |
(6,7; 9,2) |
(3,8; 5,6) |
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,62 (0,49; 0,78) |
|
p-Wert1 |
< 0,0001 |
|
12-Monats-PFS (%) |
29,1 |
16,4 |
Prüfarzt bewertetes PFS (RECIST v1.1) –aktualisierte explorative Analyse4 | ||
Anzahl der Ereignisse (%) |
149 (80,5 %) |
163 (88,6 %) |
Mediane Dauer des PFS (Monate) |
7,5 |
5,3 |
95‑%‑KI |
(6,7; 9,2) |
(3,8; 5,6) |
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,63 (0,50 – 0,80) |
|
p-Wert1 |
< 0,0001 |
|
12-Monats-PFS (%) |
30,3 |
17,3 |
OS1, 2, 5 |
||
Anzahl der Todesfälle (%) |
120 (64,9 %) |
139 (75,5 %) |
Mediane Zeit bis zum Ereignis (Monate) |
25,4 |
17,9 |
95‑%‑KI |
(19,6; 30,7) |
(13,6; 20,3) |
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,67 (0,53; 0,86) |
|
Sekundäre und explorative Endpunkte | ||
Prüfarzt bewertete ORR (RECIST v1.1)3 |
n = 185 |
n = 183 |
Anzahl mit Ansprechen (%) |
109 (58,9 %) |
78 (42,6 %) |
95‑%‑KI |
(51,5; 66,1) |
(35,4; 50,1) |
Anz. mit vollständigem Ansprechen (%) |
19 (10,3 %) |
2 (1,1 %) |
Anz. mit teilweisem Ansprechen (%) |
90 (48,6 %) |
76 (41,5 %) |
Anz. mit stabiler Erkrankung |
38 (20,5 %) |
49 (26,8 %) |
Prüfarzt bewertete DOR3 |
n = 109 |
n = 78 |
Median in Monaten |
8,5 |
5,5 |
95‑%‑KI |
(7,3; 9,7) |
(3,7; 7,1) |
‡ Stratifiziert nach Vorhandensein von Lebermetastasen und vorheriger Behandlung mit Taxanen. | ||
Abbildung 19: Kaplan-Meier-Kurve des progressionsfreien Überlebens bei Patienten mit PD-L1-Expression ≥ 1 % (IMpassion130)
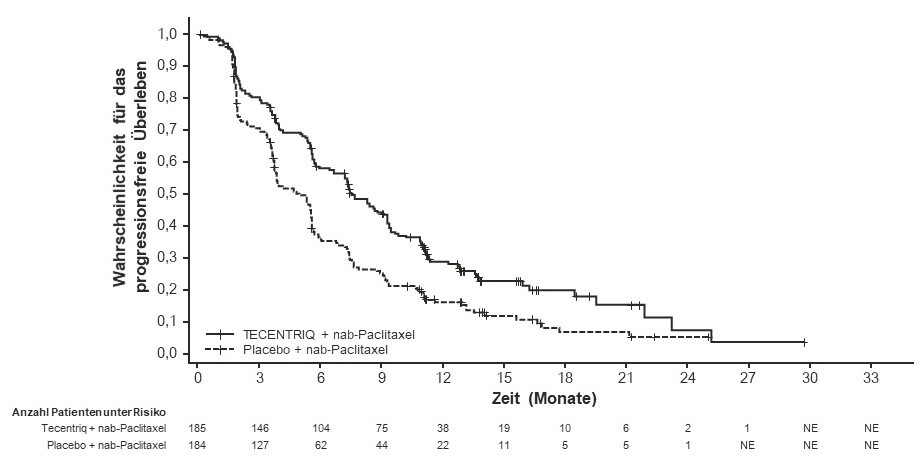
Abbildung 20: Kaplan-Meier-Kurve des Gesamtüberlebens bei Patienten mit PD-L1-Expression ≥ 1 % (IMpassion130)
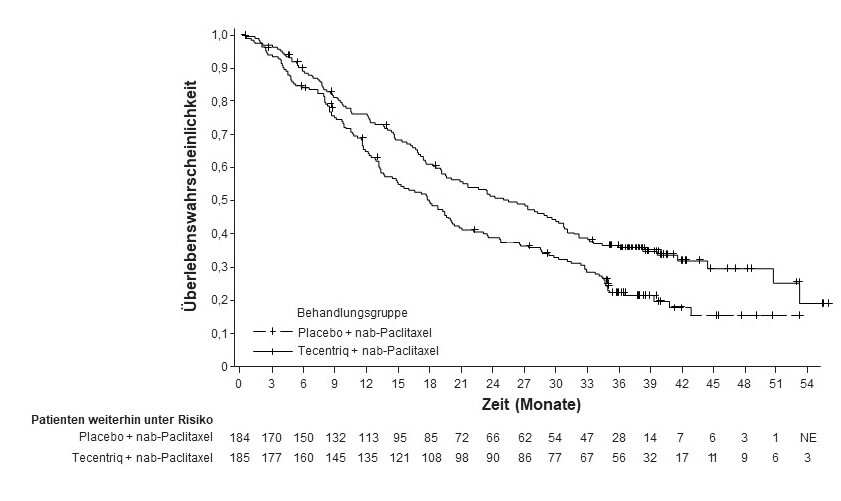
Die Dauer bis zu einer Verschlechterung des patientenberichteten allgemeinen Gesundheitszustandes/der gesundheitsbezogenen Lebensqualität (health-related quality of life, HRQoL), was einem Abfall ≥ 10 Punkte gegenüber dem Ausgangswert entspricht, gemessen anhand des EORTC QLQ-C30, war in beiden Behandlungsarmen ähnlich. Das deutet darauf hin, dass die HRQoL über eine vergleichbare Dauer während der Behandlung bei allen Patienten aufrecht erhalten wurde.
Hepatozelluläres Karzinom
Intravenöse Darreichungsform
IMbrave150 (YO40245): Randomisierte Studie der Phase III in Kombination mit Bevacizumab bei Patienten mit nicht resezierbarem HCC, die keine vorherige systemische Behandlung erhalten haben
Eine randomisierte, multizentrische, internationale, unverblindete Studie der Phase III, IMbrave150, wurde durchgeführt, um die Wirksamkeit und Sicherheit von Atezolizumab in Kombination mit Bevacizumab bei Patienten mit lokal fortgeschrittenem oder metastasiertem und/oder nicht resezierbarem HCC, die keine vorherige systemische Behandlung erhalten hatten, zu bewerten. Insgesamt wurden 501 Patienten randomisiert (2:1), die entweder Atezolizumab (1 200 mg) und 15 mg/kg KG Bevacizumab alle 3 Wochen als intravenöse Infusion, oder 400 mg Sorafenib oral zweimal pro Tag erhielten. Die Randomisierung wurde nach geographischen Regionen, makrovaskulärer Invasion und/oder extrahepatischer Ausbreitung, Alpha-Fetoprotein (AFP) zu Behandlungsbeginn und ECOG-Performance-Status stratifiziert. In beiden Armen wurden die Patienten bis zum Verlust des klinischen Nutzens oder dem Auftreten einer inakzeptablen Toxizität behandelt. Die Patienten konnten die Behandlung mit entweder Atezolizumab oder Bevacizumab abbrechen (z. B. aufgrund von Nebenwirkungen) und die Behandlung als Monotherapie solange fortführen bis unter der Monotherapie ein Verlust des klinischen Nutzens oder eine inakzeptable Toxizität auftrat.
In die Studie wurden erwachsene Patienten eingeschlossen, deren Erkrankung nicht durch einen operativen Eingriff und/oder lokoregionale Therapien behandelt werden konnte oder danach fortgeschritten ist, die einen Child-Pugh-Score A, ECOG 0/1 aufwiesen und die keine vorherige systemische Behandlung erhalten hatten. Blutungen (einschließlich tödlicher Ereignisse) sind bekannte Nebenwirkungen von Bevacizumab und Blutungen im oberen Gastrointestinaltrakt sind eine häufige und lebensbedrohliche Komplikation bei Patienten mit HCC. Daher mussten die Patienten innerhalb von 6 Monaten vor Behandlungsbeginn auf das Vorhandensein von Varizen untersucht werden und wurden ausgeschlossen, wenn sie innerhalb von 6 Monaten vor Behandlungsbeginn Varizenblutungen, unbehandelte oder unvollständig behandelte Varizen mit Blutungen oder mit einem hohen Blutungsrisiko hatten. Bei Patienten mit aktiver Hepatitis B war ein HBV-DNA-Spiegel < 500 IE/ml innerhalb von 28 Tagen vor Einleitung der Studienbehandlung und eine Standard-Anti-HBV-Behandlung über mindestens 14 Tage vor Studieneintritt und über die gesamte Studiendauer erforderlich.
Patienten wurden auch ausgeschlossen, wenn sie mäßige oder schwere Aszites, eine hepatische Enzephalopathie in der Anamnese, ein bekanntes fibrolamelläres HCC, ein sarkomatoides HCC, ein kombiniertes Cholangiokarzinom und HCC, eine aktive HBV/HCV-Koinfektion oder Autoimmunerkrankungen in der Anamnese hatten, eine Impfung mit einem attenuierten Lebendimpfstoff oder systemische Immunstimulanzien innerhalb von 4 Wochen bzw. systemische Immunsuppressiva innerhalb von 2 Wochen vor der Randomisierung erhalten hatten oder unbehandelte oder Corticosteroid-abhängige Hirnmetastasen hatten. In den ersten 54 Wochen wurden alle 6 Wochen Tumoruntersuchungen nach Zyklus 1, Tag 1 durchgeführt, danach alle 9 Wochen.
Die demographischen Merkmale und die Merkmale der Grunderkrankung der Studienpopulation zu Behandlungsbeginn waren zwischen den Behandlungsarmen ausgeglichen. Das mediane Alter betrug 65 Jahre (Bereich: 26 bis 88 Jahre) und 83 % waren männlich. Die Mehrheit der Patienten waren asiatischer (57 %) und kaukasischer Herkunft (35 %). 40 % kamen aus Asien (ohne Japan), während 60 % aus dem Rest der Welt stammten. Ungefähr 75 % der Patienten hatten eine makrovaskuläre Invasion und/oder eine extrahepatische Ausbreitung und 37 % hatten einen Baseline-AFP-Wert ≥ 400 ng/ml. Der ECOG-Performance-Status betrug zu Behandlungsbeginn 0 (62 %) oder 1 (38 %). Die primären Risikofaktoren für die Entwicklung eines HCC waren bei 48 % der Patienten eine Infektion mit dem Hepatitis-B-Virus, bei 22 % der Patienten eine Infektion mit dem Hepatitis-C-Virus und bei 31 % der Patienten eine nicht-virale Erkrankung. Das HCC wurde bei 82 % der Patienten als Barcelona-Clinic-Liver-Cancer-(BCLC)-Stadium C, bei 16 % der Patienten als -Stadium B und bei 3 % der Patienten als -Stadium A eingestuft.
Die ko-primären Wirksamkeitsendpunkte waren OS und IRF bewertetes PFS gemäß RECIST v1.1. Zum Zeitpunkt der primären Analyse hatten die Patienten eine mediane Nachbeobachtungszeit von 8,6 Monaten. Die Daten zeigten eine statistisch signifikante Verbesserung des OS und des PFS, gemäß IRF-RECIST v1.1 bei Atezolizumab + Bevacizumab im Vergleich zu Sorafenib. Eine statistisch signifikante Verbesserung wurde auch bei der bestätigten objektiven Ansprechrate (ORR) gemäß IRF-RECIST v1.1 und HCC-modifiziertem RECIST (mRECIST) beobachtet. Die wesentlichen Wirksamkeitsergebnisse aus der Primäranalyse sind in Tabelle 22 zusammengefasst.
Es wurde eine aktualisierte, deskriptive Wirksamkeitsanalyse mit einer medianen Nachbeobachtungszeit des Überlebens von 15,6 Monaten durchgeführt. Das mediane OS betrug 19,2 Monate (95‑%‑KI: 17,0; 23,7) im Atezolizumab + Bevacizumab-Arm gegenüber 13,4 Monaten (95‑%‑KI: 11,4; 16,9) im Sorafenib-Arm mit einer HR von 0,66 (95‑%‑KI: 0,52; 0,85). Das mediane IRF bewertete PFS gemäß RECIST v1.1 betrug 6,9 Monate (95‑%‑KI: 5,8; 8,6) im Atezolizumab + Bevacizumab-Arm gegenüber 4,3 Monaten (95‑%‑KI: 4,0; 5,6) im Sorafenib-Arm mit einer HR von 0,65 (95‑%‑KI: 0,53; 0,81).
Die IRF bewertete ORR gemäß RECIST v1.1 betrug 29,8 % (95‑%‑KI: 24,8; 35,0) im Atezolizumab + Bevacizumab-Arm und 11,3 % (95‑%‑KI: 6,9; 17,3) im Sorafenib-Arm. Die mediane IRF bewertete Dauer des Ansprechens (DOR) gemäß RECIST v1.1 bei den bestätigten Respondern betrug 18,1 Monate (95‑%‑KI: 14,6: nicht schätzbar) im Atezolizumab + Bevacizumab-Arm im Vergleich zu 14,9 Monaten (95‑%‑KI: 4,9; 17,0) im Sorafenib-Arm.
Kaplan-Meier-Kurven für OS (aktualisierte Analyse) und PFS (Primäranalyse) sind in den Abbildungen 21 bzw. 22 dargestellt.
Tabelle 22: Zusammenfassung der Wirksamkeit (IMbrave150 Primäranalyse)
Wesentliche Wirksamkeitsendpunkte |
Atezolizumab + Bevacizumab |
Sorafenib |
OS |
n = 336 |
n = 165 |
Anzahl der Todesfälle (%) |
96 (28,6 %) |
65 (39,4 %) |
Mediane Zeit bis zum Ereignis (Monate) |
NE |
13,2 |
95‑%‑KI |
(NE, NE) |
(10,4; NE) |
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,58 (0,42; 0,79) |
|
p-Wert1 |
0,0006 |
|
6-Monats-OS (%) |
84,8 % |
72,3 % |
IRF bewertetes PFS gemäß RECIST 1.1 |
n = 336 |
n = 165 |
Anzahl der Ereignisse (%) |
197 (58,6 %) |
109 (66,1 %) |
Mediane Dauer des PFS (Monate) |
6,8 |
4,3 |
95‑%‑KI |
(5,8; 8,3) |
(4,0; 5,6) |
Stratifizierte Hazard Ratio‡ (95‑%‑KI) |
0,59 (0,47; 0,76) |
|
p-Wert1 |
< 0,0001 |
|
6-Monats-PFS |
54,5 % |
37,2 % |
IRF bewertete ORR gemäß RECIST 1.1 |
n = 326 |
n = 159 |
Anzahl Patienten mit bestätigtem Ansprechen (%) |
89 (27,3 %) |
19 (11,9 %) |
95‑%‑KI |
(22,5; 32,5) |
(7,4; 18,0) |
p-Wert2 |
< 0,0001 |
|
Anz. mit vollständigem Ansprechen (%) |
18 (5,5 %) |
0 |
Anz. mit teilweisem Ansprechen (%) |
71 (21,8 %) |
19 (11,9 %) |
Anz. mit stabiler Erkrankung (%) |
151 (46,3 %) |
69 (43,4 %) |
IRF bewertete DOR gemäß RECIST 1.1 |
n = 89 |
n = 19 |
Median in Monaten |
NE |
6,3 |
95‑%‑KI |
(NE; NE) |
(4,7; NE) |
Bereich (Monate) |
(1,3+; 13,4+) |
(1,4+; 9,1+) |
IRF bewertete ORR, HCC mRECIST |
n = 325 |
n = 158 |
Anzahl Patienten mit bestätigtem Ansprechen (%) |
108 (33,2 %) |
21 (13,3 %) |
95‑%‑KI |
(28,1; 38,6) |
(8,4; 19,6) |
p-Wert2 |
< 0,0001 |
|
Anz. mit vollständigem Ansprechen (%) |
33 (10,2 %) |
3 (1,9 %) |
Anz. mit teilweisem Ansprechen (%) |
75 (23,1 %) |
18 (11,4 %) |
Anz. mit stabiler Erkrankung (%) |
127 (39,1 %) |
66 (41,8 %) |
IRF bewertete DOR, HCC mRECIST |
n = 108 |
n = 21 |
Median in Monaten |
NE |
6,3 |
95‑%‑KI |
(NE; NE) |
(4,9; NE) |
Bereich (Monate) |
(1,3+; 13,4+) |
(1,4+; 9,1+) |
‡ Stratifiziert nach geographischer Region (Asien ohne Japan vs. Rest der Welt), makrovaskulärer Invasion und/oder extrahepatischer Ausbreitung (Vorhandensein vs. Abwesenheit) und Baseline-AFP (< 400 vs. ≥ 400 ng/ml) | ||
Abbildung 21: Kaplan-Meier-Kurve des OS in der ITT-Population (IMbrave150 aktualisierte Analyse)
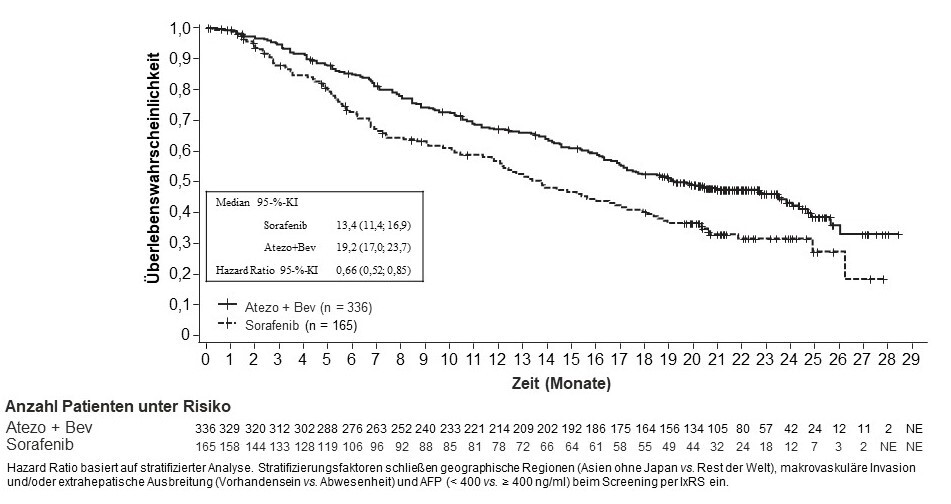
Abbildung 22: Kaplan‑Meier-Kurve des IRF-PFS gemäß RECIST v1.1 in der ITT-Population (IMbrave150 Primäranalyse)
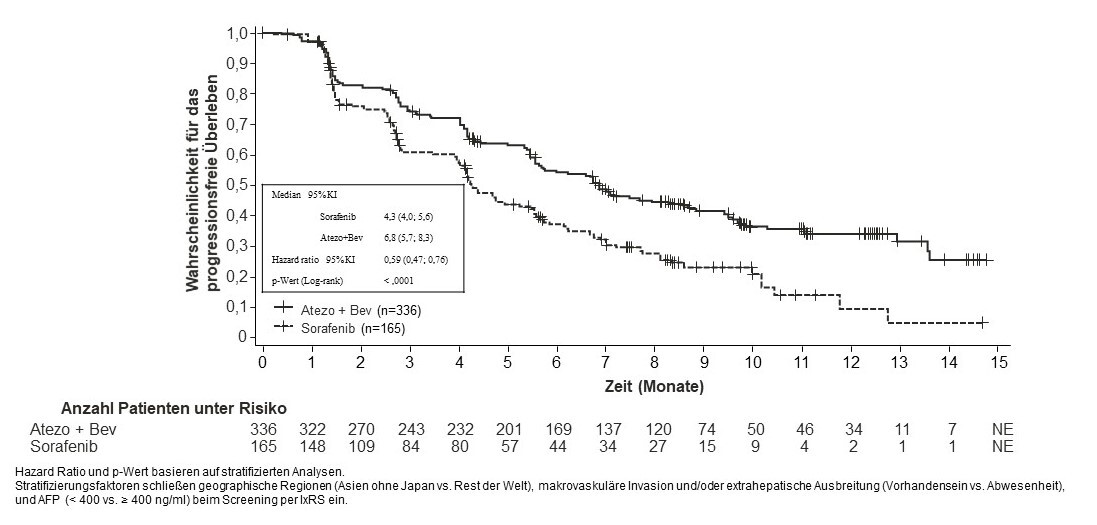
Wirksamkeit bei älteren Patienten
Bezüglich Sicherheit wurden insgesamt keine Unterschiede zwischen Patienten im Alter von ≥ 65 Jahren und jüngeren Patienten festgestellt, die Atezolizumab als Monotherapie erhielten. In der Studie IMpower150 stand das Alter von ≥ 65 mit einer verringerten Wirkung von Atezolizumab bei Patienten, die Atezolizumab in Kombination mit Carboplatin und Paclitaxel erhielten, in Verbindung.
Die Daten für Patienten im Alter von ≥ 75 Jahren aus den Studien IMpower150, IMpower133, IMpower110 und IMscin001 sind zu begrenzt, um Rückschlüsse auf diese Population zuzulassen.
Kinder und Jugendliche
Intravenöse Darreichungsform
Eine frühe, multizentrische, unverblindete Studie wurde mit pädiatrischen (< 18 Jahre, n = 69) und jungen erwachsenen Patienten (18 – 30 Jahre, n = 18) mit rezidivierenden oder progressiven soliden Tumoren sowie mit Hodgkin- und Non-Hodgkin-Lymphomen durchgeführt, um die Sicherheit und Pharmakokinetik von Atezolizumab zu untersuchen. Die Patienten wurden mit 15 mg/kg KG Atezolizumab intravenös alle drei Wochen behandelt (siehe Abschnitt 5.2).
Subkutane Darreichungsform
Es wurden keine speziellen Studien mit Tecentriq Injektionslösung bei Kindern und Jugendlichen durchgeführt.
Modellbasierte vorhergesagte Expositionskennzahlen von Atezolizumab nach Anwendung von 1 875 mg Q3W Tecentriq subkutan und Atezolizumab intravenös (1 200 mg Q3W) in der IMscin001-Studie sind in Tabelle 23 dargestellt.
Atezolizumab Zyklus 1 Ctrough (d. h. vor der Dosisgabe in Zyklus 2) zeigte die Nicht-Unterlegenheit von Atezolizumab in Tecentriq Injektionslösung gegenüber Atezolizumab intravenös, mit einem geometrischen Mittelwert (geometric mean ratio, GMR) von 1,05 (90‑%‑KI: 0,88 - 1,24).
Der GMR für Zyklus 1 für die modellbasierte vorhergesagte AUC von 0 bis 21 Tagen (AUC0-21d) betrug 0,87 (90‑%‑KI: 0,83 - 0,92).
Das maximale systemische Akkumulationsverhältnis nach Gabe von 1 875 mg Q3W von Tecentriq Injektionslösung beträgt 2,2.
Die modellbasierten vorhergesagten Ctrough- und AUC-Werte im Steady-State waren bei Tecentriq subkutan und Atezolizumab intravenös vergleichbar (siehe Tabelle 23). Eine pharmakokinetische Analyse legt nahe, dass der Steady State nach mehrfacher Verabreichung nach 6 bis 9 Wochen erreicht wird.
Tabelle 23: Modellbasierte vorhergesagte Exposition von Atezolizumab (geometrisches Mittel mit 5ter bis 95ter Perzentile) nach subkutaner oder intravenöser Anwendung von Atezolizumab
Parameter |
Tecentriq subkutan |
Atezolizumab intravenös |
Ctrough im Steady-Statea |
205 |
179 |
AUC im Steady-Statea |
6 163 |
6 107 |
a modellbasierte vorhergesagte Exposition basierend auf einer populationspharmakokinetischen Analyse
Resorption
Tecentriq Injektionslösung wird als subkutane Injektion verabreicht.
Basierend auf einer populationspharmakokinetischen Analyse der Daten des Zyklus 1 aus dem randomisierten Teil der IMscin001-Studie betrug die absolute Bioverfügbarkeit 61 % und die Resorptionsrate erster Ordnung (ka) 0,37 (1/Tag).
Das geometrische Mittel der maximalen Serumkonzentration (Cmax) von Atezolizumab betrug 189 µg/ml und die mediane Zeit bis zur maximalen Serumkonzentration (Tmax) 4,5 Tage.
Verteilung
Eine populationspharmakokinetische Analyse zeigte, dass bei einem typischen Patienten das Verteilungsvolumen im zentralen Kompartiment 3,28 l und das Volumen im Steady State 6,91 l beträgt.
Biotransformation
Der Metabolismus von Atezolizumab wurde nicht direkt untersucht. Antikörper werden hauptsächlich durch Katabolismus eliminiert.
Elimination
Eine populationspharmakokinetische Analyse zeigte, dass die Clearance von Atezolizumab 0,200 l/Tag und die typische terminale Eliminationshalbwertszeit 27 Tage beträgt.
Besondere Patientengruppen
Basierend auf der Populationspharmakokinetik und auf Expositionsanalysen zum Ansprechen haben Alter (21 – 89 Jahre), Region, ethnische Zugehörigkeit, Nierenfunktionsstörung, leichte Leberfunktionsstörung, Grad der PD‑L1‑Expression oder ECOG‑Performance‑Status keine Auswirkung auf die Pharmakokinetik von Atezolizumab. Körpergewicht, Geschlecht, positiver ADA‑Status, Albuminspiegel und Tumorlast haben eine statistisch signifikante, aber keine klinisch relevante Auswirkung auf die Pharmakokinetik von Atezolizumab. Es werden keine Dosisanpassungen empfohlen.
Ältere Patienten
Es wurden keine spezifischen Studien mit Atezolizumab bei älteren Patienten durchgeführt. Die Auswirkung des Alters auf die Pharmakokinetik von Atezolizumab wurde in einer populationsbezogenen PK-Analyse bewertet. Basierend auf den Daten von Patienten im Alter von 21 bis 89 Jahren (n = 472) und einem medianen Alter von 62 Jahren wurde das Alter nicht als signifikante, die Pharmakokinetik von intravenös verabreichtem Atezolizumab beeinflussende Kovariable identifiziert. Es wurde kein klinisch bedeutsamer Unterschied in der Pharmakokinetik von Atezolizumab bei Patienten im Alter von < 65 Jahren (n = 274), zwischen 65 bis 75 Jahren (n = 152) und im Alter von > 75 Jahren (n = 46) beobachtet (siehe Abschnitt 4.2).
Bei Patienten < 65 Jahre (n = 138), Patienten zwischen 65 – 75 Jahren (n = 89) und Patienten > 75 Jahren (n = 19) wurde kein klinisch relevanter Unterschied in der PK von Atezolizumab subkutan beobachtet.
Kinder und Jugendliche
Die pharmakokinetischen Ergebnisse einer frühen, multizentrischen, unverblindeten Studie, die mit pädiatrischen (< 18 Jahre, n = 69) und jungen erwachsenen Patienten (18 – 30 Jahre, n = 18) durchgeführt wurde, zeigen, dass Clearance und Verteilungsvolumen von intravenös verabreichtem Atezolizumab zwischen pädiatrischen Patienten, welche 15 mg/kg KG Atezolizumab alle drei Wochen erhielten und jungen Erwachsenen, welche 1 200 mg Atezolizumab alle drei Wochen intravenös erhielten, nach Normierung auf das Körpergewicht, vergleichbar waren. Dabei war die Exposition bei pädiatrischen Patienten mit abnehmendem Körpergewicht tendenziell niedriger. Diese Unterschiede standen nicht in Zusammenhang mit einer Abnahme der Atezolizumab-Konzentrationen unter die angestrebte therapeutische Exposition. Da die Datenlage für Kinder < 2 Jahre begrenzt ist, kann daraus keine definitive Schlussfolgerung gezogen werden.
Es wurden keine speziellen Studien mit Tecentriq Injektionslösung bei Kindern und Jugendlichen durchgeführt.
Nierenfunktionsstörung
Es wurden keine spezifischen Studien zur Anwendung von Atezolizumab intravenös bei Patienten mit Nierenfunktionsstörung durchgeführt. In der populationspharmakokinetischen Analyse wurden keine klinisch bedeutsamen Unterschiede in der Clearance von Atezolizumab bei Patienten mit leichter (geschätzter glomerulärer Filtrationsrate [eGFR] 60 bis 89 ml/min/1,73 m2; n = 208) oder moderater (eGFR 30 bis 59 ml/min/1,73 m2; n = 116) Nierenfunktionsstörung im Vergleich zu Patienten mit normaler Nierenfunktion (eGFR höher als oder gleich 90 ml/min/1,73 m2; n = 140) festgestellt. Nur wenige Patienten hatten eine schwere Nierenfunktionsstörung (eGFR 15 bis 29 ml/min/1,73 m2; n = 8) (siehe Abschnitt 4.2). Die Auswirkung einer schweren Nierenfunktionsstörung auf die Pharmakokinetik von Atezolizumab ist nicht bekannt.
Bei Patienten mit leichter (eGFR 60 bis 89 ml/min/1,73 m2; n = 111) oder mittelschwerer (eGFR 30 bis 59 ml/min/1,73 m2; n = 32) Nierenfunktionsstörung wurden keine klinisch bedeutsamen Unterschiede in der Clearance von subkutan verabreichtem Atezolizumab im Vergleich zu Patienten mit normaler (eGFR größer oder gleich 90 ml/min/1,73 m2; n = 103) Nierenfunktion festgestellt.
Leberfunktionsstörung
Es wurden keine spezifischen Studien zur Anwendung von Atezolizumab bei Patienten mit Leberfunktionsstörung durchgeführt. In der populationspharmakokinetischen Analyse wurden keine klinisch bedeutsamen Unterschiede in der Clearance von Atezolizumab intravenös oder subkutan verabreicht bei Patienten mit leichter Leberfunktionsstörung (Bilirubin ≤ ULN und AST > ULN oder Bilirubin > 1,0 bis 1,5 × ULN und beliebige AST) oder mäßiger Leberfunktionsstörung (Bilirubin > 1,5 bis 3 x ULN und beliebige AST) im Vergleich zu Patienten mit normaler Leberfunktion (Bilirubin ≤ ULN und AST ≤ ULN) beobachtet. Zu Patienten mit schwerer Leberfunktionsstörung (Bilirubin > 3 x ULN und beliebige AST) liegen keine Daten vor. Leberfunktionsstörungen wurden nach den Kriterien für Leberfunktionsstörungen des National Cancer Institute Organ Dysfunction Working Group (NCI-ODWG) definiert (siehe Abschnitt 4.2). Die Auswirkung einer schweren Leberfunktionsstörung (Bilirubin > 3 × ULN und beliebige AST) auf die Pharmakokinetik von Atezolizumab ist nicht bekannt.
Kanzerogenität
Es wurden keine Kanzerogenitätsstudien zum Nachweis eines kanzerogenen Potenzials von Atezolizumab durchgeführt.
Mutagenität
Es wurden keine Mutagenitätsstudien zum Nachweis eines mutagenen Potenzials von Atezolizumab durchgeführt. Jedoch ist nicht davon auszugehen, dass monoklonale Antikörper zu Veränderungen der DNA oder Chromosomen führen.
Fertilität
Es wurden keine Studien zur Beeinflussung der Fertilität durch Atezolizumab durchgeführt. Die Studie zur langfristigen Toxizität beinhaltete jedoch die Beurteilung der männlichen und weiblichen Fortpflanzungsorgane von Cynomolgus‑Affen. Die wöchentliche Verabreichung von Atezolizumab intravenös an weibliche Affen in Dosen, die etwa dem 6‑Fachen der AUC von Patienten in der empfohlenen Dosierung entsprachen, verursachte einen unregelmäßigen Menstruationszyklus und einen Mangel an neu gebildeten Gelbkörpern in den Eierstöcken, was reversibel war. Eine Auswirkung auf die männlichen Fortpflanzungsorgane wurde nicht festgestellt.
Teratogenität
Es wurden keine tierexperimentellen Studien zur Reproduktionstoxizität oder Teratogenität mit Atezolizumab durchgeführt. Tierexperimentelle Studien haben gezeigt, dass die Hemmung des PD‑L1/PD‑1‑Signalweges eine immunvermittelte Abstoßung des sich entwickelnden Fetus, die zu dessen Tod führt, hervorrufen kann. Die Anwendung von Atezolizumab könnte demnach den menschlichen Fetus schädigen, dies kann eine embryonale Letalität mit einschließen.
Subkutane Darreichungsform
Hyaluronidase kommt in den meisten Geweben des menschlichen Körpers vor. Präklinische Daten zur rekombinanten humanen Hyaluronidase lassen auf Grundlage konventioneller Studien zur Toxizität bei wiederholter Gabe einschließlich der Endpunkte zur Sicherheitspharmakologie keine besonderen Gefahren für den Menschen erkennen. Studien zur Reproduktionstoxizität mit rHuPH20 zeigten embryofetale Toxizität bei Mäusen bei hoher systemischer Exposition, aber kein teratogenes Potenzial.
Rekombinante humane Hyaluronidase (rHuPH20)
Histidin
Essigsäure 30 %
Methionin
Polysorbat 20 (E 432)
Saccharose
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Ungeöffnete Durchstechflasche
3 Jahre
Vorbereitete Spritze
Nach der Überführung aus der Durchstechflasche in die Spritze ist Tecentriq Injektionslösung ab dem Zeitpunkt der Zubereitung bis zu 30 Tage bei 2 °C bis 8 °C und bis zu 8 Stunden bei ≤ 30 °C bei diffusem Tageslicht physikalisch und chemisch stabil.
Aus mikrobiologischer Sicht ist die Lösung sofort nach der Überführung aus der Durchstechflasche in die Spritze zu verwenden, da das Arzneimittel keine antimikrobiellen Konservierungsmittel oder bakteriostatischen Substanzen enthält. Falls die Lösung nicht unmittelbar verwendet wird, liegen die Aufbewahrungszeiten und ‑bedingungen vor der Anwendung in der Verantwortung des Anwenders und sollen normalerweise 24 Stunden bei 2 °C bis 8 °C nicht überschreiten, es sei denn, die Zubereitung wurde unter kontrollierten und validierten aseptischen Bedingungen durchgeführt.
Im Kühlschrank lagern (2 °C ‑ 8 °C).
Nicht einfrieren.
Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Vorbereitung der Spritze, siehe Abschnitt 6.3.
Durchstechflasche der Glasart I mit Butylgummi‑Stopfen und einem Aluminiumsiegel mit einem violetten Flip-off-Deckel aus Plastik mit 15 ml Injektionslösung.
Packung mit 1 Durchstechflasche.
Vorbereitung der Spritze
Tecentriq Injektionslösung ist vor der Anwendung visuell zu überprüfen, um sicherzustellen, dass keine Partikel oder Verfärbungen vorhanden sind.
Tecentriq Injektionslösung ist eine gebrauchsfertige Lösung, die NICHT verdünnt oder mit anderen Arzneimitteln gemischt werden darf. Nicht schütteln.
Tecentriq Injektionslösung ist nur zur einmaligen Anwendung bestimmt und muss von medizinischem Fachpersonal vorbereitet werden.
Es wurden keine Inkompatibilitäten zwischen Tecentriq Injektionslösung und Polypropylen (PP), Polycarbonat (PC), Edelstahl (SS), Polyvinylchlorid (PVC) und Polyurethanen (PU) beobachtet.
Tecentriq Injektionslösung enthält keine antimikrobiellen Konservierungsmittel oder bakteriostatisch wirkenden Substanzen.
Nehmen Sie die Durchstechflasche aus der gekühlten Lagerung und warten Sie, bis die Lösung Raumtemperatur angenommen hat.
Entnehmen Sie den gesamten Inhalt der Tecentriq Injektionslösung mit einer sterilen Spritze und Transfernadel aus der Durchstechflasche (18 G empfohlen).
Entfernen Sie die Transfernadel und bringen Sie ein subkutanes Infusionsset (z. B. Flügelkanüle/Butterfly) an, das eine 23 ‑ 25 G Edelstahl-Injektionsnadel enthält. Verwenden Sie zur Verabreichung ein subkutanes Infusionsset mit einem Restvolumen von NICHT mehr als 0,5 ml.
Befüllen Sie die subkutane Infusionsleitung mit der Arzneimittellösung, um die Luft aus der Infusionsleitung zu entfernen, und beenden Sie das Befüllen, bevor die Flüssigkeit die Nadel erreicht.
Stellen Sie sicher, dass die Spritze nach dem Entlüften und Entfernen des überschüssigen Volumens genau 15 ml der Lösung enthält.
Verabreichen Sie die Lösung sofort, um ein Verstopfen der Nadel zu vermeiden. Bewahren Sie die vorbereitete Spritze, die an das bereits vorbereitete subkutane Infusionsset angeschlossen wurde, NICHT auf.
Wenn die Dosis nicht sofort verabreicht wird, siehe unten „Aufbewahrung der Spritze“.
Aufbewahrung der Spritze
Wenn die Dosis nicht unmittelbar verabreicht werden soll, ziehen Sie den gesamten Inhalt der Tecentriq Injektionslösung unter aseptischen Bedingungen aus der Durchstechflasche in die Spritze auf, unter Berücksichtigung des Dosisvolumens (15 ml) und des Entlüftungsvolumens für das subkutane Infusionsset. Ersetzen Sie die Transfernadel durch eine Spritzenverschlusskappe. Schließen Sie KEIN subkutanes Infusionsset zur Aufbewahrung an.
Wenn die Spritze im Kühlschrank aufbewahrt wird, soll sie vor der Anwendung Raumtemperatur angenommen haben.
Beseitigung
Die Freisetzung von Tecentriq in die Umwelt ist so gering wie möglich zu halten. Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Roche Registration GmbH
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Deutschland
EU/1/17/1220/003
Datum der Erteilung der Zulassung: 21. September 2017
Datum der letzten Verlängerung der Zulassung: 25. April 2022
Juni 2026
Verschreibungspflichtig
Roche Pharma AG
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Telefon (07624) 14-0
Telefax (07624) 1019
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel‑Agentur https://www.ema.europa.eu verfügbar.