
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Winrevair® 45 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Winrevair® 60 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Winrevair 45 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Jede Durchstechflasche enthält 45 mg Sotatercept. Nach Rekonstitution enthält jeder Milliliter Lösung 50 mg Sotatercept.
Winrevair 60 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Jede Durchstechflasche enthält 60 mg Sotatercept. Nach Rekonstitution enthält jeder Milliliter Lösung 50 mg Sotatercept.
Sotatercept ist ein rekombinantes homodimeres Fusionsprotein, das aus der extrazellulären Domäne des humanen Aktivinrezeptors Typ IIA (ActRIIA) besteht, die mit der Fc-Domäne des humanen Immunglobulins G1 (IgG1) verknüpft ist. Das Fusionsprotein wird in Ovarialzellen des chinesischen Hamsters (CHO) durch rekombinante DNA-Technologie hergestellt.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung (Pulver zur Herstellung einer Injektionszubereitung).
Pulver: weißes bis cremefarbenes Pulver.
Lösungsmittel: klares, farbloses Wasser für Injektionszwecke.
Winrevair ist, in Kombination mit anderen Therapien gegen pulmonale arterielle Hypertonie (PAH), für die Behandlung von PAH bei erwachsenen Patienten mit der WHO-Funktionsklasse (FK) II, III und IV angezeigt (siehe Abschnitt 5.1).
Die Behandlung mit Winrevair sollte nur von einem in der Diagnose und Behandlung von PAH erfahrenen Arzt eingeleitet und überwacht werden.
Dosierung
Winrevair wird einmal alle 3 Wochen als subkutane Einzelinjektion in Abhängigkeit vom Körpergewicht des Patienten gegeben.
Empfohlene Initialdosis
Hämoglobinwerte (Hb-Werte) und die Thrombozytenzahl sollten vor der ersten Dosis bestimmt werden (siehe Abschnitt 4.4). Es ist kontraindiziert, die Behandlung zu beginnen, wenn die Thrombozytenzahl dauerhaft < 50 x 109/l beträgt (siehe Abschnitt 4.3).
Die Behandlung wird mit einer Einzeldosis von 0,3 mg/kg (siehe Tabelle 1) initiiert.
Tabelle 1: Injektionsvolumen für eine Dosis von 0,3 mg/kg
Gewichtsbereich des Patienten (kg) |
Injektionsvolumen (ml)* |
Set Typ |
30,0 – 40,8 |
0,2 |
Set enthält |
40,9 – 57,4 |
0,3 |
|
57,5 – 74,1 |
0,4 |
|
74,2 – 90,8 |
0,5 |
|
90,9 – 107,4 |
0,6 |
|
107,5 – 124,1 |
0,7 |
|
124,2 – 140,8 |
0,8 |
|
140,9 – 157,4 |
0,9 |
|
157,5 – 174,1 |
1,0 |
Set enthält |
174,2 – 180,0 |
1,1 |
|
* Die Konzentration der rekonstituierten Lösung beträgt 50 mg/ml (siehe Abschnitt 6.6) | ||
Empfohlene Zieldosis
Drei Wochen nach der Initial-Einzeldosis von 0,3 mg/kg sollte die Dosis nach Bestätigung eines akzeptablen Hb-Wertes und einer akzeptablen Thrombozytenzahl (siehe Abschnitt 4.2 „Dosisanpassungen aufgrund eines Hämoglobinanstiegs oder eines Abfalls der Thrombozytenzahl“) auf die empfohlene Zieldosis von 0,7 mg/kg erhöht werden. Die Behandlung ist mit 0,7 mg/kg alle 3 Wochen fortzusetzen, sofern keine Dosisanpassungen erforderlich sind.
Tabelle 2: Injektionsvolumen für eine Dosis von 0,7 mg/kg
Gewichtsbereich des Patienten (kg) |
Injektionsvolumen (ml)* |
Set Typ |
30,0 – 31,7 |
0,4 |
Set enthält |
31,8 – 38,9 |
0,5 |
|
39,0 – 46,0 |
0,6 |
|
46,1 – 53,2 |
0,7 |
|
53,3 – 60,3 |
0,8 |
|
60,4 – 67,4 |
0,9 |
|
67,5 – 74,6 |
1,0 |
Set enthält |
74,7 – 81,7 |
1,1 |
|
81,8 – 88,9 |
1,2 |
|
89,0 – 96,0 |
1,3 |
Set enthält |
96,1 – 103,2 |
1,4 |
|
103,3 – 110,3 |
1,5 |
|
110,4 – 117,4 |
1,6 |
|
117,5 – 124,6 |
1,7 |
|
124,7 – 131,7 |
1,8 |
|
131,8 – 138,9 |
1,9 |
Set enthält |
139,0 – 146,0 |
2,0 |
|
146,1 – 153,2 |
2,1 |
|
153,3 – 160,3 |
2,2 |
|
160,4 – 167,4 |
2,3 |
|
167,5 und höher |
2,4 |
|
* Die Konzentration der rekonstituierten Lösung beträgt 50 mg/ml (siehe Abschnitt 6.6) | ||
Dosisanpassungen aufgrund eines Hämoglobinanstiegs oder eines Abfalls der Thrombozytenzahl
Der Hb-Wert und die Thrombozytenzahl sind während der ersten 5 Dosen oder länger, wenn die Werte instabil sind, zu überwachen. Danach sollten Hb-Wert und Thrombozytenzahl alle 3 bis 6 Monate überprüft und die Dosis bei Bedarf angepasst werden (siehe Abschnitte 4.4 und 4.8).
Die Behandlung ist um 3 Wochen zu verschieben (d. h. Verzögerung um eine Dosis), wenn eines der folgenden Ereignisse auftritt:
Hb-Wert steigt um > 1,24 mmol/l (2 g/dl) gegenüber der vorherigen Dosis und liegt über der Obergrenze des Normalwertes (upper limit of normal, ULN).
Hb-Wert steigt um > 2,48 mmol/l (4 g/dl) gegenüber dem Ausgangswert.
Hb-Wert steigt um > 1,24 mmol/l (2 g/dl) über ULN.
Die Thrombozytenzahl fällt < 50 x 109/l.
Vor Wiederaufnahme der Behandlung müssen der Hb-Wert und die Thrombozytenzahl erneut bestimmt werden.
Bei Behandlungsverzögerungen von > 9 Wochen sollte die Behandlung erneut mit 0,3 mg/kg begonnen und die Dosis nach Bestätigung akzeptabler Hb-Werte und Thrombozytenzahlen auf 0,7 mg/kg erhöht werden.
Bei Behandlungsverzögerungen von > 9 Wochen aufgrund von Thrombozytenzahlen, die konstant < 50 x 109/l liegen, muss der Arzt vor Wiederaufnahme der Behandlung eine erneute Nutzen-Risiko-Abwägung für den Patienten vornehmen.
Versäumte Dosis
Wenn eine Dosis versäumt wurde, ist sie so bald wie möglich nachzuholen. Wird die versäumte Dosis nicht innerhalb von 3 Tagen nach dem geplanten Termin angewendet, ist der Zeitplan anzupassen, um die 3-wöchigen Dosierungsintervalle einzuhalten.
Ältere Menschen
Bei älteren Patienten ≥ 65 Jahre ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Nierenfunktionsbeeinträchtigung
Bei einer Nierenfunktionsbeeinträchtigung ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Es liegen begrenzte Daten zur Anwendung von Sotatercept bei PAH-Patienten mit schwerer Nierenfunktionsbeeinträchtigung (geschätzte Glomeruläre Filtrationsrate [eGFR] < 30 ml/min/1,73 m2) vor.
Leberfunktionsbeeinträchtigung
Bei einer Leberfunktionsbeeinträchtigung (Child-Pugh-Klassifikation A bis C) ist keine Dosisanpassung erforderlich. Sotatercept wurde bei Patienten mit beeinträchtigter Leberfunktion nicht untersucht (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Winrevair bei Kindern und Jugendlichen unter 18 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Winrevair ist nur für den einmaligen Gebrauch bestimmt.
Es sollte vor der Anwendung rekonstituiert werden. Das rekonstituierte Arzneimittel ist eine klare bis opaleszierende und farblose bis leicht bräunlich-gelbe Lösung.
Winrevair sollte mittels einer subkutanen Injektion in den Bauch (mindestens 5 cm vom Nabel entfernt), Oberarm oder Oberschenkel angewendet werden. Es sollte nicht an Stellen injiziert werden, die vernarbt, empfindlich oder verletzt sind. Bei zwei aufeinanderfolgenden Injektionen sollte nicht dieselbe Injektionsstelle verwendet werden.
Winrevair Pulver und Lösungsmittel zur Herstellung einer Injektionslösung ist für die Anwendung unter Anleitung von medizinischem Fachpersonal bestimmt. Patienten und Pflegekräfte können das Arzneimittel anwenden, wenn dies für angemessen erachtet wird und wenn sie vom medizinischen Fachpersonal hinsichtlich der Rekonstitution, der Zubereitung, des Abmessens und der Injektion von Winrevair Pulver und Lösungsmittel zur Herstellung einer Injektionslösung geschult werden.
Das medizinische Fachpersonal sollte bei einem Folgebesuch zeitnah nach einer Schulung bestätigen, dass der Patient oder die Pflegekraft die Schritte korrekt durchführen kann. Das medizinische Fachpersonal sollte außerdem eine erneute Bestätigung der Anwendungstechnik des Patienten oder der Pflegekraft in Betracht ziehen, wenn die Dosis angepasst wird oder der Patient ein anderes Set benötigt, wenn der Patient eine Erythrozytose entwickelt (siehe Abschnitt 4.4) oder zu jedem anderen Zeitpunkt nach Ermessen des medizinischen Fachpersonals.
Detaillierte Anweisungen zur ordnungsgemäßen Zubereitung und Anwendung von Winrevair finden Sie in Abschnitt 6.6.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Patienten mit einer konstanten Thrombozytenzahl < 50 x 109/l vor Beginn der Behandlung.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Erythrozytose
Erhöhungen der Hb-Werte wurden bei Patienten während der Behandlung mit Sotatercept beobachtet. Eine schwere Erythrozytose kann das Risiko für thromboembolische Ereignisse und ein Hyperviskositätssyndrom erhöhen. Vorsicht ist geboten bei Patienten mit Erythrozytose, bei denen ein erhöhtes Risiko für thromboembolische Ereignisse besteht. Der Hb-Wert ist vor jeder der ersten 5 Dosen oder länger, wenn die Werte instabil sind, und danach alle 3 bis 6 Monate zu überwachen, um festzustellen, ob Dosisanpassungen erforderlich sind (siehe Abschnitte 4.2 und 4.8). Wenn ein Patient eine Erythrozytose entwickelt, sollte der Arzt eine Neubewertung der Anwendungstechnik des Patienten oder der Pflegekraft in Betracht ziehen.
Schwere Thrombozytopenie
Bei einigen Patienten, die Sotatercept anwendeten, wurde eine verringerte Thrombozytenzahl, einschließlich schwerer Thrombozytopenie (Thrombozytenzahl < 50 x 109/l), beobachtet. Bei Patienten, die zusätzlich eine Prostazyklin-Infusion erhielten, wurde häufiger über Thrombozytopenie berichtet (21,5 % bis 24,5 %) als bei Patienten, die keine Prostazyklin-Infusion erhielten (0,0 % bis 3,1 %) (siehe Abschnitt 4.8). Eine schwere Thrombozytopenie kann das Risiko von Blutungsereignissen erhöhen. Die Thrombozytenzahl ist vor jeder der ersten 5 Dosen oder länger, wenn die Werte instabil sind, und danach alle 3 bis 6 Monate zu überwachen, um festzustellen, ob Dosisanpassungen erforderlich sind (siehe Abschnitt 4.2).
Schwerwiegende Blutungen
In klinischen Studien wurden bei 4,3 % bis 7,0 % der Patienten unter der Behandlung mit Sotatercept schwerwiegende Blutungen (einschließlich gastrointestinaler, intrakranieller Blutungen) beobachtet (siehe Abschnitt 4.8).
Bei Patienten mit schwerwiegenden Blutungsereignissen war die Wahrscheinlichkeit höher, dass sie eine Prostazyklin-Hintergrundtherapie und/oder Antithrombotika erhielten, eine niedrige Thrombozytenzahl hatten oder 65 Jahre oder älter waren. Patienten sollten über jegliche Anzeichen und Symptome eines Blutverlusts informiert werden. Ein Arzt sollte Blutungsereignisse entsprechend bewerten und behandeln. Sotatercept ist nicht anzuwenden, wenn beim Patienten eine schwerwiegende Blutung auftritt.
Einschränkung der klinischen Daten
In den klinischen Studien waren keine Teilnehmer eingeschlossen, deren PAH mit dem humanen Immundefizienzvirus (HIV), portaler Hypertonie, Bilharziose oder pulmonaler venöser Verschlusskrankheit (PVOD) assoziiert war.
Anti-Doping-Test
Die Anwendung des Arzneimittels Winrevair kann bei Dopingkontrollen zu positiven Ergebnissen führen.
Sonstige Bestandteile mit bekannter Wirkung
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d. h., es ist nahezu „natriumfrei“.
Dieses Arzneimittel enthält 0,20 mg Polysorbat 80 pro ml der rekonstituierten Lösung. Polysorbate können allergische Reaktionen hervorrufen.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter wird ein Schwangerschaftstest vor Beginn der Behandlung empfohlen. Frauen im gebärfähigen Alter sollten während der Behandlung und für mindestens 4 Monate nach der letzten Dosis, wenn die Behandlung abgesetzt wird, eine wirksame Verhütungsmethode anwenden (siehe Abschnitt 5.3).
Schwangerschaft
Bisher liegen keine Daten zur Anwendung von Sotatercept bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (Anstieg der Postimplantationsverluste, Verringerung des Körpergewichts des Fetus und eine Verzögerung bei der Ossifikation) (siehe Abschnitt 5.3).
Die Anwendung von Winrevair während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen.
Stillzeit
Es ist nicht bekannt, ob Sotatercept/Metaboliten in die Muttermilch übergehen. Ein Risiko für das Neugeborene/Kind kann nicht ausgeschlossen werden.
Das Stillen soll während der Behandlung und für 4 Monate nach der letzten Behandlungsdosis unterbrochen werden.
Fertilität
Basierend auf Tierversuchen kann Sotatercept die weibliche und männliche Fertilität beeinträchtigen (siehe Abschnitt 5.3).
Sotatercept hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die am häufigsten berichteten Nebenwirkungen entweder in STELLAR oder ZENITH waren Epistaxis (45,3 %), Kopfschmerzen (26,7 %), Teleangiektasie (25,6 %), Diarrhoe (25,6 %), erhöhtes Hämoglobin (15,1 %), Thrombozytopenie (15,1 %), Schwindelgefühl (14,7 %), Rückenschmerzen (14 %), Ausschlag (12,3 %) und Zahnfleischbluten (10,5 %).
Die am häufigsten berichteten schwerwiegenden Nebenwirkungen waren Thrombozytopenie (< 1,2 %), Epistaxis (< 1,2 %) und Schwindelgefühl (< 1,2 %).
Die häufigsten Nebenwirkungen, die zum Abbruch führten, waren Epistaxis und Teleangiektasie.
Tabellarische Auflistung der Nebenwirkungen
Die Sicherheit von Sotatercept wurde in den pivotalen placebokontrollierten Studien STELLAR und ZENITH bewertet, die 163 bzw. 86 Patienten mit PAH einschlossen, die mit Sotatercept behandelt wurden (siehe Abschnitt 5.1). Die mediane Behandlungsdauer mit Sotatercept betrug 313 Tage bei STELLAR und 434,5 Tage bei ZENITH.
Tabelle 3 zeigt die Nebenwirkungen, die in placebokontrollierten klinischen Studien und nach der Markeinführung mit Sotatercept berichtet wurden. Diese sind in der nachstehenden Tabelle nach MedDRA-Systemorganklasse und Häufigkeit aufgeführt. Die Häufigkeiten sind definiert als sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000) und sehr selten (< 1/10 000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 3: Nebenwirkungen
Systemorganklassen |
Häufigkeit |
Nebenwirkungen |
Infektionen und parasitäre Erkrankungen |
Häufig |
Harnwegsinfektionen |
Erkrankungen des Blutes und des Lymphsystems |
Sehr häufig |
Thrombozytopenie1,2 |
Erkrankungen des Nervensystems |
Sehr häufig |
Schwindelgefühl |
Herzerkrankungen |
Nicht bekannt |
Perikarderguss1 |
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Sehr häufig |
Epistaxis |
Gelegentlich |
Intrapulmonaler Shunt3 |
|
Erkrankungen des Gastroinstestinaltrakts |
Sehr häufig |
Diarrhoe |
Erkrankungen der Haut und des Unterhautgewebes |
Sehr häufig |
Teleangiektasie1 |
Häufig |
Erythem |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Sehr häufig |
Rückenschmerzen4 |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Häufig |
Jucken an der Injektionsstelle |
Untersuchungen |
Häufig |
Erhöhter Blutdruck1,5 |
1 Siehe Beschreibung ausgewählter Nebenwirkungen | ||
Beschreibung ausgewählter Nebenwirkungen
Erhöhte Hämoglobin-Werte
In STELLAR wurden erhöhte Hb-Werte („Hämoglobin erhöht“ und „Polyzythämie“) bei 8,6 % der Patienten unter Sotatercept berichtet. Basierend auf Labordaten traten moderate Anstiege der Hb-Werte (> 1,24 mmol/l [2 g/dl] über der oberen Normgrenze, ULN) bei 15,3 % der Patienten unter Sotatercept auf.
In ZENITH wurden erhöhte Hb-Werte bei 15,1 % der Patienten unter Sotatercept berichtet. Basierend auf Labordaten traten moderate Erhöhungen der Hb-Werte bei 7,1 % der Patienten unter Sotatercept auf.
Anstiege der Hb-Werte wurden durch Dosisanpassungen kontrolliert (siehe Abschnitte 4.2 und 4.4).
Thrombozytopenie
In STELLAR wurde Thrombozytopenie („Thrombozytopenie“ und „verminderte Thrombozytenzahl“) bei 10,4 % der Patienten unter Sotatercept berichtet. Bei 2,5 % der Patienten unter Sotatercept trat eine starke Verringerung der Thrombozytenzahl < 50 x 109/l auf. Bei Patienten, die zusätzlich eine Prostazyklin-Infusion erhielten, wurde häufiger über Thrombozytopenie berichtet (21,5 %) als bei Patienten, die keine Prostazyklin-Infusion erhielten (3,1 %).
In ZENITH wurde Thrombozytopenie bei 15,1 % der Patienten unter Sotatercept berichtet. Eine starke Verringerung der Thrombozytenzahl < 50 x 109/l trat bei 6,0 % der Patienten unter Sotatercept auf. Thrombozytopenie wurde nur bei Patienten berichtet, die auch eine Prostazyklin-Infusion erhielten (24,5 %).
Thrombozytopenie konnte durch Dosisanpassungen kontrolliert werden (siehe Abschnitte 4.2 und 4.4).
Teleangiektasien
In STELLAR wurde Teleangiektasie bei 16,6 % der Patienten unter Sotatercept beobachtet. Die mediane Zeit bis zum Auftreten betrug 18,6 Wochen. Therapieabbrüche aufgrund von Teleangiektasie traten bei 1 % der Patienten in der Sotatercept-Gruppe auf.
In ZENITH wurde Teleangiektasie bei 25,6 % der Patienten unter Sotatercept beobachtet. Die mediane Zeit bis zum Auftreten betrug 12,8 Wochen. Es gab keine Therapieabbrüche aufgrund von Teleangiektasie in der Sotatercept-Gruppe.
Erhöhter Blutdruck
In STELLAR wurde erhöhter Blutdruck bei 4,3 % der Patienten unter Sotatercept berichtet. Bei Patienten unter Sotatercept stieg der mittlere systolische Blutdruck gegenüber dem Ausgangswert um 2,2 mmHg und der diastolische Blutdruck um 4,9 mmHg nach 24 Wochen.
In ZENITH wurde erhöhter Blutdruck bei 2,3 % der Patienten unter Sotatercept berichtet. Bei Patienten unter Sotatercept stieg der mittlere systolische Blutdruck gegenüber dem Ausgangswert um 3,1 mmHg und der diastolische Blutdruck um 5,1 mmHg nach 24 Wochen.
Perikarderguss
Fälle von neu aufgetretenen oder sich verschlechternden Perikardergüssen (einschließlich Perikardtamponade) wurden bei mit Sotatercept behandelten Patienten berichtet, trotz verbesserter oder stabiler PAH-Hämodynamik. Die meisten Fälle wurden bei Patienten mit PAH im Zusammenhang mit Bindegewebserkrankungen, mit bereits bestehendem Perikarderguss oder beidem berichtet; die meisten erhielten zudem Prostazyklin-Analoga.
Ältere Patienten
Mit Ausnahme von Blutungsereignissen (eine kollektive Gruppe von unerwünschten Ereignissen mit klinischer Bedeutung) gab es keine Unterschiede in der Sicherheit zwischen den Subgruppen der < 65-Jährigen und der ≥ 65-Jährigen.
In STELLAR traten Blutungsereignisse häufiger in der älteren Sotatercept-Subgruppe auf (52 % versus 31,9 % bei Patienten < 65 Jahren); es gab jedoch kein nennenswertes Ungleichgewicht zwischen den Alterskategorien für ein spezifisches Blutungsereignis. Bei 3,6 % der Patienten < 65 Jahren und bei 8,0 % der Patienten ≥ 65 Jahre, die Sotatercept erhielten, kam es zu schwerwiegenden Blutungen.
In ZENITH traten Blutungsereignisse häufiger in der älteren Sotatercept-Subgruppe auf (73,3 % versus 60,7 % bei Patienten < 65 Jahre). Bei 3,6 % der Patienten < 65 Jahre und bei 13,3 % der Patienten ≥ 65 Jahre, die Sotatercept erhielten, kam es zu schwerwiegenden Blutungen.
Langzeit-Sicherheitsdaten
Langzeit-Sicherheitsdaten stehen aus gepoolten klinischen Phase-II- und Phase-III-Studien (n = 431) zur Verfügung. Die mediane Dauer der Exposition betrug 657 Tage. Das Sicherheitsprofil ähnelte im Allgemeinen dem in der pivotalen STELLAR-Studie beobachteten Sicherheitsprofil. Intrapulmonaler Rechts-Links-Shunt wurde bei Teilnehmern berichtet, die trotz verbesserter PAH-Hämodynamik eine Verschlechterung der Hypoxämie entwickelten.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: www.bfarm.de, anzuzeigen.
Bei einer Phase-I-Studie mit gesunden Freiwilligen, kam es bei einem Teilnehmer, der Sotatercept in einer Dosierung von 1 mg/kg erhielt, zu einem Anstieg des Hb-Wertes in Verbindung mit symptomatischer Hypertonie; dies besserte sich mit Phlebotomie.
Im Falle einer Überdosierung bei Patienten mit PAH müssen diese engmaschig auf Anstiege des Hb-Wertes und Blutdrucks überwacht und gegebenenfalls unterstützende Maßnahmen ergriffen werden (siehe Abschnitte 4.2 und 4.4). Sotatercept ist während der Hämodialyse nicht dialysierbar.
Pharmakotherapeutische Gruppe: Antihypertensiva, Antihypertensiva zur Behandlung der pulmonalen arteriellen Hypertonie. ATC-Code: C02KX06
Wirkmechanismus
Sotatercept ist ein Inhibitor des Aktivin-Signalwegs mit hoher Selektivität für Aktivin A, ein dimeres Glykoprotein, das zur Liganden-Superfamilie des Transforming-Wachstumsfaktors-β (transforming growth factor, TGF-β) gehört. Aktivin A bindet an den Aktivin-Rezeptor Typ IIA (ActRIIA), der Schlüsselsignale bei Inflammation, Zellproliferation, Apoptose und Gewebehomöostase reguliert.
Bei PAH-Patienten sind die Aktivin-A-Spiegel erhöht. Die Bindung von Aktivin an ActRIIA fördert die proliferative Signalübertragung, während die Signalübertragung des antiproliferativen knochenmorphogenetischen Proteinrezeptors Typ II (bone morphogenetic protein receptor type II, BMPRII) abnimmt. Das Ungleichgewicht der ActRIIA-BMPRII-Signalübertragung, die der PAH zugrunde liegt, führt zu einer Hyperproliferation von vaskulären Zellen, was zu einer pathologischen Umgestaltung der Pulmonalarterienwand, einer Verengung des Arterienlumens, einem Anstieg des pulmonalen Gefäßwiderstands und zu einem erhöhten Lungenarteriendruck und rechtsventrikulärer Dysfunktion führt.
Sotatercept besteht aus einem rekombinanten homodimeren Aktivin-Rezeptor-Typ-IIA-Fc (ActRIIA-Fc)-Fusionsprotein, das als Ligandenfalle fungiert, die überschüssiges Aktivin A und andere Liganden für ActRIIA abfängt, um die Aktivin-Signalübertragung zu hemmen. Infolgedessen gleicht Sotatercept die pro-proliferative (ActRIIA/Smad2/3-vermittelt) und anti-proliferative (BMPRII/Smad1/5/8-vermittelt) Signalgebung aus, um die vaskuläre Proliferation zu modulieren.
Pharmakodynamische Wirkungen
In einer klinischen Phase-II-Studie (PULSAR) wurde der pulmonale Gefäßwiderstand (pulmonary vascular resistance, PVR) bei Patienten mit PAH nach 24-wöchiger Behandlung mit Sotatercept untersucht. Die Abnahme des PVR gegenüber dem Ausgangswert war in den Gruppen unter Sotatercept mit 0,7 mg/kg und 0,3 mg/kg signifikant größer im Vergleich zur Placebo-Gruppe. Die Placebo-bereinigte mittlere Differenz der kleinsten Quadrate (least squares, LS) gegenüber dem Ausgangswert betrug −269,4 dyn*Sekunde/cm5 (95 % KI: −365,8; −173,0) unter Sotatercept mit 0,7 mg/kg und −151,1 dyn*Sekunde/cm5 (95 % KI: −249,6; −52,6) unter Sotatercept mit 0,3 mg/kg.
In Rattenmodellen von PAH reduzierte ein Sotatercept-Analogon die Expression entzündungsfördernder Markerproteine an der Lungenarterienwand, reduzierte die Rekrutierung von Leukozyten, hemmte die Proliferation von Endothelzellen und glatten Muskelzellen und förderte die Apoptose der Zellen in erkrankten Gefäßen. Diese zellulären Veränderungen waren mit dünneren Gefäßwänden, einem umgekehrten arteriellen und rechtsventrikulären Umbau und einer verbesserten Hämodynamik verbunden.
Immunogenität
Anti-Drug-Antikörper (ADA) wurden bei 27 % der Patienten in STELLAR und bei 43 % der Patienten in ZENITH nachgewiesen. Es wurde kein Hinweis auf eine Auswirkung von ADA auf die Pharmakokinetik, Wirksamkeit oder Sicherheit beobachtet.
Klinische Wirksamkeit und Sicherheit
STELLAR
Die Wirksamkeit von Sotatercept wurde in der pivotalen STELLAR-Studie bei erwachsenen Patienten mit PAH untersucht. STELLAR war eine doppelblinde, Placebo-kontrollierte, multizentrische klinische Studie mit parallelen Gruppen, in der 323 Patienten mit PAH (WHO-FK II oder III) zu Sotatercept (Initialdosis von 0,3 mg/kg, gesteigert auf eine Zieldosis von 0,7 mg/kg) (n = 163) oder Placebo (n = 160), einmal alle 3 Wochen subkutan angewendet, 1:1 randomisiert wurden. Die Patienten setzten ihre jeweilige Behandlung im langfristigen doppelblinden Behandlungszeitraum fort, bis alle Patienten Woche 24 abgeschlossen hatten.
Teilnehmer an dieser Studie waren Erwachsene mit einem medianen Alter von 48,0 Jahren (Altersbereich: 18 bis 82 Jahre), von denen 16,7 % der Patienten ≥ 65 Jahre alt waren. Das mediane Gewicht betrug 68,2 kg (Gewichtsbereich 38,0 bis 141,3 kg); 89,2 % der Teilnehmer hatten weiße Hautfarbe und 79,3 % waren keine Hispanoamerikaner oder Lateinamerikaner; 79,3 % waren weiblich. Die häufigsten PAH-Ätiologien waren idiopathische PAH (58,5 %), vererbbare PAH (18,3 %) und PAH im Zusammenhang mit Bindegewebserkrankungen (14,9 %), PAH im Zusammenhang mit einer einfachen angeborenen Herzerkrankung mit korrigierten systemisch-pulmonalen Shunts (5 %) oder Medikamenten- oder Toxin-induzierte PAH (3,4 %). Die mittlere Zeit zwischen der PAH-Diagnose bis zum Screening betrug 8,76 Jahre.
Die meisten Teilnehmer erhielten entweder eine dreifache (61,3 %) oder eine doppelte (34,7 %) PAH-Hintergrundtherapie, und mehr als ein Drittel (39,9 %) erhielt Prostazyklin-Infusionen. Die Anteile der Teilnehmer in der WHO-FK II betrugen 48,6 % und in der WHO-FK III 51,4 %. Die STELLAR-Studie schloss Patienten aus, bei denen eine mit HIV assoziierte PAH, eine mit portaler Hypertonie assoziierte PAH, eine mit Bilharziose assoziierte PAH und eine PVOD diagnostiziert wurden.
Der primäre Wirksamkeitsendpunkt war die Veränderung der 6-Minuten-Gehstrecke (6-Minute Walk Distance, 6MWD) gegenüber dem Ausgangswert in Woche 24. In der Sotatercept-Behandlungsgruppe betrug die Placebo-bereinigte mediane Veränderung gegenüber dem Ausgangswert in Woche 24 bei der 6MWD 40,8 Meter (95 % KI: 27,5; 54,1; p < 0,001). Der Median der Placebo-bereinigten Veränderungen bei der 6MWD in Woche 24 wurde ebenfalls in Sub-Gruppen ausgewertet. Der Behandlungseffekt war in den verschiedenen Sub-Gruppen konsistent, einschließlich Geschlecht, PAH-Diagnosegruppe, Hintergrundtherapie zu Studienbeginn, Prostazyklin-Infusionstherapie zu Studienbeginn, WHO-FK und PVR zu Studienbeginn.
Zu den sekundären Endpunkten zählten Verbesserungen bei der klinischen Verbesserung mehrerer Komponenten (multicomponent improvement, MCI), PVR, N-terminalem natriuretischem Peptid vom Pro-B-Typ (NT-proBNP), WHO-FK, Zeit bis zum Tod oder dem ersten Auftreten von klinischen Verschlechterungen.
Die MCI war ein vordefinierter Endpunkt, gemessen als Anteil der Patienten, die alle drei der folgenden Kriterien in Woche 24 im Vergleich zum Ausgangswert erreichten: Verbesserung der 6MWD (Anstieg ≥ 30 m), Verbesserung des NT-proBNP (Abnahme von NT-proBNP ≥ 30 % oder Aufrechterhaltung/Erreichen eines NT-proBNP-Spiegels < 300 ng/l) und Verbesserung der WHO-FK oder Aufrechterhaltung der WHO-FK II.
Die Krankheitsprogression wurde anhand der Zeit bis zum Tod oder dem ersten Auftreten einer klinischen Verschlechterung gemessen. Klinische Verschlechterungsereignisse umfassten eine verschlechterungsbedingte Aufnahme in eine Lungen- und/oder Herztransplantationsliste, die Notwendigkeit, eine Notfalltherapie mit einer zugelassenen PAH-Hintergrundtherapie einzuleiten, oder die Notwendigkeit, die Dosis der Prostazyklin-Infusion um ≥ 10 % zu erhöhen, die Notwendigkeit einer atrialen Septostomie, Krankenhauseinweisung wegen Verschlechterung der PAH (≥ 24 Stunden) oder Verschlechterung der PAH (Verschlechterung der WHO-FK und Abnahme der 6MWD um ≥ 15 %, wobei beide Ereignisse gleichzeitig oder zu unterschiedlichen Zeiten aufgetreten sein konnten). Klinische Verschlechterungsereignisse und Todesfälle wurden erfasst, bis der letzte Patient den Besuch in Woche 24 abgeschlossen hatte (Daten bis zum Datenschnitt; mediane Expositionsdauer 33,6 Wochen).
In Woche 24 zeigten 38,9 % der mit Sotatercept behandelten Patienten eine Verbesserung des MCI gegenüber 10,1 % in der Placebo-Gruppe (p < 0,001). Der mediane Behandlungsunterschied hinsichtlich PVR zwischen der Sotatercept- und der Placebo-Gruppe betrug −234,6 dyn*Sekunde/cm5 (95 %-KI: −288,4; −180,8; p < 0,001). Der mediane Behandlungsunterschied hinsichtlich NT-proBNP zwischen der Sotatercept- und der Placebo-Gruppe betrug −441,6 pg/ml (95 %-KI: −573,5; −309,6; p < 0,001). Eine Verbesserung der WHO-FK gegenüber dem Ausgangswert trat bei 29 % der Patienten unter Sotatercept auf, gegenüber 13,8 % unter Placebo (p < 0,001).
Die Behandlung mit Sotatercept führte im Vergleich zu Placebo zu einer Reduktion des Auftretens von Todesfällen oder klinischen Verschlechterungsereignissen um 82 % (HR 0,182; 95 %-KI: 0,075; 0,441; p < 0,001) (siehe Tabelle 4). Der Behandlungseffekt von Sotatercept gegenüber Placebo begann in Woche 10 und hielt für die Dauer der Studie an.
Tabelle 4: Todesfälle oder klinische Verschlechterungsereignisse
Sotatercept |
Placebo |
|
Gesamtzahl der Studienteilnehmer, bei denen der Tod oder mindestens ein klinisches Verschlechterungsereignis aufgetreten ist, n (%) |
7 (4,3) |
29 (18,1) |
Bewertung von Tod oder erstmaligem Auftreten von klinischen Verschlechterungsereignissen*, n (%) |
||
Tod |
2 (1,2) |
6 (3,8) |
Verschlechterungsbedingte Aufnahme in eine Lungen- und/oder Herztransplantationsliste |
1 (0,6) |
1 (0,6) |
Notwendigkeit einer atrialen Septostomie |
0 (0,0) |
0 (0,0) |
PAH-spezifische Hospitalisierung (≥ 24 Stunden) |
0 (0,0) |
8 (5,0) |
Verschlechterung der PAH† |
4 (2,5) |
15 (9,4) |
* Bei einem Studienteilnehmer kann mehr als eine Beurteilung für das erste Ereignis einer klinischen Verschlechterung erfasst werden. Es gab zwei Teilnehmer, die Placebo erhielten, und keinen Teilnehmer, der Sotatercept erhielt, bei dem mehr als eine Beurteilung für das erste Ereignis einer klinischen Verschlechterung aufgezeichnet wurde. Diese Analyse schloss die Komponente „Notwendigkeit, eine Notfalltherapie mit einer zugelassenen PAH-Therapie einzuleiten oder die Dosis der Prostazyklin-Infusion um 10 % oder mehr zu erhöhen“ aus. | ||
† Eine Verschlechterung der PAH wird dadurch definiert, dass beide der folgenden Ereignisse gleichzeitig auftreten, auch wenn sie im Vergleich zu ihren Ausgangswerten zu unterschiedlichen Zeitpunkten begannen: (a) Verschlechterung der WHO-Funktionsklasse (II zu III, III zu IV, II zu IV usw.); und (b) Abnahme der 6MWD um ≥ 15 % (bestätigt durch zwei 6MWTs im Abstand von mindestens 4 Stunden, aber nicht mehr als einer Woche). | ||
N = Anzahl der Probanden in der FAS-Population; n = Anzahl der Probanden in der Kategorie. Prozentsätze werden als (n/N)*100 berechnet. | ||
ZENITH
Die Wirksamkeit von Sotatercept wurde in der ZENITH-Studie bei erwachsenen Patienten mit PAH WHO-FK III oder IV mit hohem Mortalitätsrisiko untersucht. ZENITH war eine doppelblinde, placebokontrollierte, multizentrische klinische Parallelgruppenstudie, in der 172 Patienten im Verhältnis 1:1 randomisiert entweder Sotatercept (Anfangsdosis von 0,3 mg/kg, erhöht auf Zieldosis 0,7 mg/kg) (n = 86) oder Placebo (n = 86), einmal alle 3 Wochen angewendet, erhielten. Patienten, bei denen kein Ereignis des primären kombinierten Endpunktes auftrat, verblieben in der doppelblinden, placebokontrollierten Behandlungsphase, während Patienten, bei denen es zu einer Hospitalisierung aufgrund einer PAH-Verschlechterung von ≥ 24 Stunden kam, für die Aufnahme in die offene Langzeit-Nachbeobachtungs-Studie (long-term follow-up, LTFU) SOTERIA berechtigt waren.
Teilnehmer in der ZENITH-Studie waren Erwachsene mit einem medianen Alter von 57,5 Jahren (Spanne: 18 bis 75 Jahre), von denen 29,1 % ≥ 65 Jahre alt waren; 86,6 % der Teilnehmer waren Weiße, und 87,8 % waren nicht Hispanoamerikaner oder Latinos; und 76,7 % waren weiblich. Die untersuchten PAH-Ätiologien waren idiopathische PAH (50,0 %), PAH im Zusammenhang mit Bindegewebserkrankungen (connective tissue diseases, CTD) (27,9 %), vererbbare PAH (10,5 %), Medikamenten- oder Toxin-induzierte PAH (6,4 %) und PAH im Zusammenhang mit korrigierten angeborenen Shunts (5,2 %). Die durchschnittliche Zeit ab der PAH-Diagnose bis zum Screening betrug 7,68 Jahre. Die Teilnehmer erhielten entweder eine dreifache (72,1 %) oder doppelte (27,9 %) PAH-Hintergrundtherapie und 59,3 % erhielten eine Prostazyklin-Infusion. Die Anteile an Teilnehmern mit WHO-FK III betrugen 74,4 % und mit WHO-FK IV 25,6 %. Der REVEAL Lite 2-Risikoscore lag bei < 9 für 2,3 % der Teilnehmer, bei 9 bis 10 für 67,4 % der Teilnehmer und bei ≥ 11 für 30,2 % der Teilnehmer. Die ZENITH-Studie schloss Patienten mit HIV-assoziierter PAH, PAH im Zusammenhang mit portaler Hypertonie, Lungenvenenverschluss (pulmonary veno-occlusive disease, PVOD) oder pulmonal-kapillärer Hämangiomatose oder offensichtlichen Anzeichen einer kapillären und/oder venösen Beteiligung aus.
Der primäre Wirksamkeitsendpunkt war die Zeit bis zum ersten Auftreten von Tod jeglicher Ursache, von Lungentransplantation oder einer Hospitalisierung von ≥ 24 Stunden aufgrund einer Verschlechterung der PAH. Die Behandlung mit Sotatercept führte zu einer Reduktion um 76 % (Hazard Ratio, HR: 0,24; 95 % KI: 0,13; 0,43; p < 0,0001) des Auftretens des ersten Ereignisses des primären kombinierten Endpunkts (siehe Tabelle 5). Bis zum Datenstichtag (data cutoff) hatten weniger Teilnehmer in der Sotatercept-Behandlungsgruppe (15 [17,4 %]) ein Ereignis des primären zusammengesetzten Endpunkts als in der Placebogruppe (47 [54,7 %]).
Die mediane Zeit bis zum ersten Ereignis betrug 9,6 Monate (95 % KI: 6,2; 14,8) in der Placebogruppe. Die Kaplan-Meier-Kurven begannen sich etwa in Woche 5 zu trennen, und die Trennung nahm im weiteren Verlauf der Studie zu (siehe Abbildung 1). Der Behandlungseffekt war über die verschiedenen Subgruppen hinweg konsistent, einschließlich Alter, Geschlecht, PAH-Subtyp (CTD-assoziiert versus nicht CTD-assoziiert), WHO-FK, doppelte versus dreifache Hintergrundtherapie zu Studienbeginn, Prostazyklin-Infusionstherapie zu Studienbeginn, PVR zu Studienbeginn und eGFR zu Studienbeginn.
Tabelle 5: Komponenten des primären Endpunkts
Sotatercept |
Placebo |
Hazard Ratio |
|
(N = 86) |
(N = 86) |
(95% KI) |
|
n (%) |
n (%) |
p-Werta |
|
Anzahl (%) der Teilnehmer mit ≥ 1 primärem Ereignis |
15 (17,4) |
47 (54,7) |
0,24 (0,13; 0,43) |
während oder nach ZENITH | |||
Teilnehmer mit erstem Ereignis des kombinierten Endpunktes, nach Komponenteb |
|||
Tod jeglicher Ursachec |
6 (7,0) |
3 (3,5) |
|
Teilnehmer mit jeglichen Ereignissen der Komponenten des kombinierten Endpunktesd |
|||
Tod jeglicher Ursachec,e |
7 (8,1) |
13 (15,1) |
|
a Die primäre Analyse des kombinierten Endpunktes umfasst das erste Auftreten eines begutachteten Morbiditäts-Mortalitäts-Ereignisses bis zum Datenstichtag. Alle vor dem Stichtag eingetretenen Todesfälle sind enthalten, unabhängig von der Beurteilung und davon, ob sie während oder nach ZENITH auftraten. | |||
Abbildung 1: Kaplan-Meier-Kurve der Zeit bis zum ersten Ereignis von Tod jeglicher Ursache, Lungentransplantation oder Hospitalisierung von ≥ 24 Stunden aufgrund einer Verschlechterung der PAH
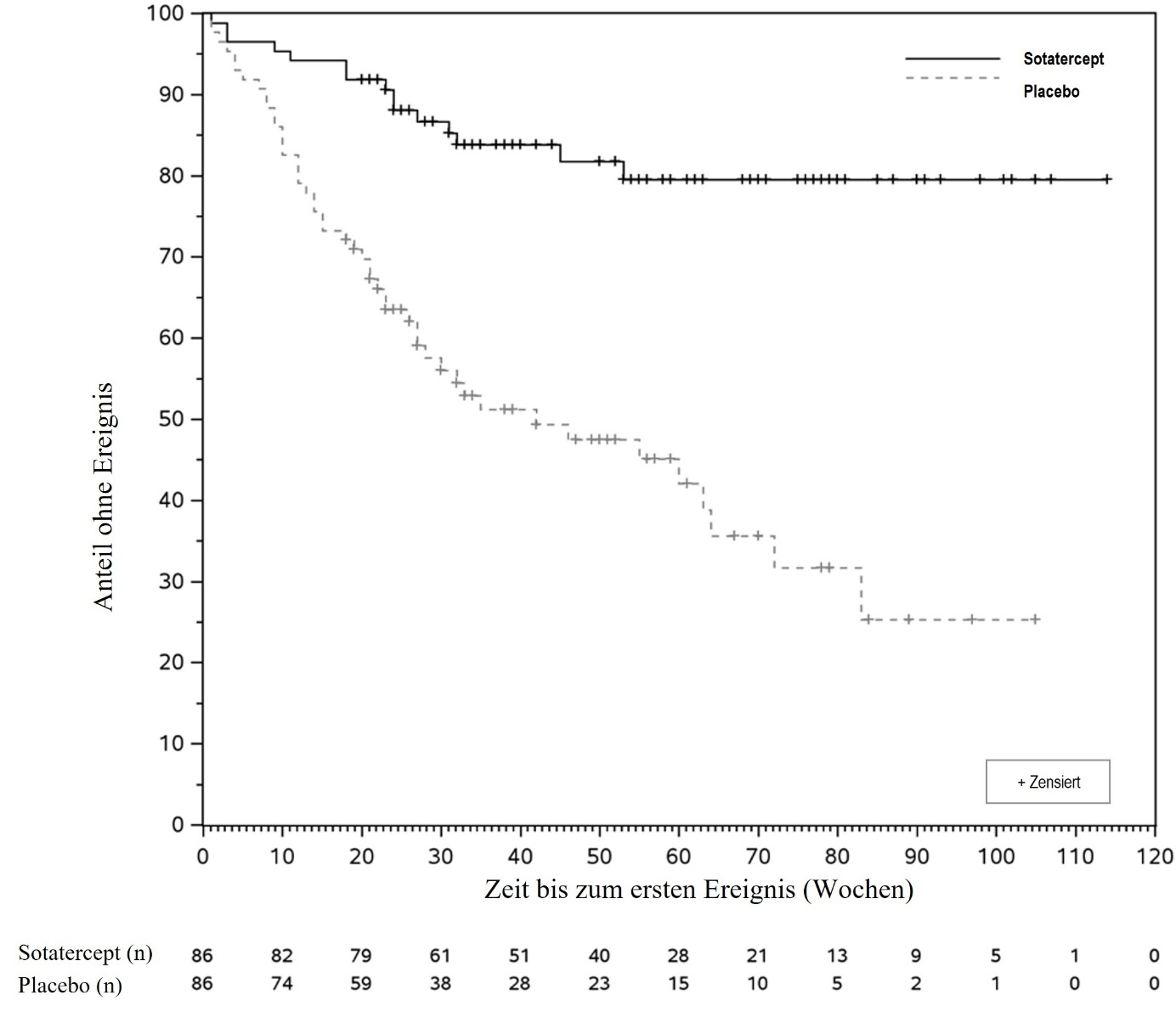
n = Anzahl der Teilnehmer unter Risiko
Basierend auf dem Ergebnis des primären Endpunkts wurde die Studie aufgrund der günstigen Wirksamkeit bei der Interimsanalyse gestoppt. Die primäre Analyse des ersten sekundären Endpunkts in der hierarchischen Teststrategie, das Gesamtüberleben (overall survival, OS), umfasste alle Todesfälle bis zum Datenstichtag, mit Ausnahme derjenigen, die nach einer Lungentransplantation oder nach der Aufnahme in eine Langzeit-Nachbeobachtungs-Studie auftraten (siehe Tabelle 5). Der Punktschätzer für die OS-Hazard-Ratio deutet darauf hin, dass die Sotatercept-Behandlungsgruppe mehr profitierte als die Placebo-Gruppe (HR: 0,42; 95 % KI: 0,17; 1,07; p = 0,0313), jedoch wurde die Grenze für die statistische Signifikanz bei der Interimsanalyse (p < 0,0021) nicht erreicht.
Andere sekundäre Endpunkte umfassten Verbesserungen hinsichtlich transplantationsfreien Überlebens, NT-proBNP, mittlerem pulmonalarteriellem Druck (PAP), PVR, 6MWD, Herzzeitvolumen und WHO-FK.
Der Punktschätzer für das transplantationsfreie Überleben deutet darauf hin, dass die Sotatercept-Behandlungsgruppe mehr profitierte als die Placebo-Gruppe (HR: 0,34; 95 % KI: 0,15; 0,78). In Woche 24 betrug der mediane Behandlungsunterschied im NT-proBNP zwischen der Sotatercept- und der Placebo-Gruppe −2339,1 pg/ml (95 % KI: (−3378,7 bis −1299,4). Der mediane Unterschied im mittleren PAP zwischen der Sotatercept- und der Placebo-Behandlungsgruppe betrug −21,2 mmHg (95 % KI: −27,8 bis −14,6). Der mediane Unterschied im PVR zwischen der Sotatercept- und der Placebo-Behandlungsgruppe betrug −339,6 dyn*sec/cm5 (95 % KI: −511,1 bis −168,1). Der mediane Unterschied in der 6MWD zwischen der Sotatercept- und der Placebo-Behandlungsgruppe betrug 63,0 m (95 % KI: 23,2 bis 102,7). Der mediane Unterschied im Herzzeitvolumen zwischen der Sotatercept- und der Placebo-Behandlungsgruppe betrug 0,5 l/min (95 % KI: −0,2 bis 1,2). Eine Verbesserung der WHO-FK gegenüber dem Ausgangswert trat bei 55,8 % der Patienten unter Sotatercept gegenüber 27,9 % unter Placebo auf.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Winrevair eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in der Behandlung der pulmonalen arteriellen Hypertonie gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Bei Patienten mit PAH in den Phase-II- und Phase-III-Studien PULSAR, SPECTRA und STELLAR betrugen das geometrische Mittel (%Variationskoeffizient, %VK) der Steady-State-AUC und die Steady-State-Spitzenkonzentration (Cmax) bei einer Dosis von 0,7 mg/kg alle 3 Wochen 171,3 µg × Tag/ml (34,2 %) bzw. 9,7 µg/ml (30 %). Die AUC und Cmax von Sotatercept steigen proportional zur Dosis. Der Steady State wird nach etwa 15 Wochen Behandlung erreicht. Das Akkumulationsverhältnis der AUC von Sotatercept betrug ungefähr 2,2. Die Sotatercept-Exposition bei Teilnehmern in der Phase-III-ZENITH-Studie war mit den oben genannten Daten konsistent.
Resorption
Die s.c.(subkutane)-Darreichung hat eine absolute Bioverfügbarkeit von ungefähr 66 % basierend auf einer populationspharmakokinetischen Analyse. Die maximale Sotatercept-Konzentration wird nach einer medianen Zeit bis zur maximalen Wirkstoffkonzentration (Tmax) von etwa 7 Tagen (Bereich von 2 bis 8 Tagen) nach mehrfacher Dosierung alle 4 Wochen erreicht.
Verteilung
Das zentrale Verteilungsvolumen (%VK) von Sotatercept beträgt ungefähr 3,6 l (24,7 %). Das periphere Verteilungsvolumen (%VK) beträgt etwa 1,7 l (73,3 %).
Biotransformation
Sotatercept wird durch allgemeine Proteinabbauprozesse katabolisiert.
Elimination
Die Clearance von Sotatercept beträgt etwa 0,18 l/Tag. Das geometrische Mittel der terminalen Halbwertszeit (%VK) beträgt etwa 21 Tage (33,8 %).
Besondere Patientengruppen
Alter, Geschlecht und ethnische Zugehörigkeit
Es wurden keine klinisch signifikanten Unterschiede in der Pharmakokinetik (PK) von Sotatercept aufgrund von Alter (18 bis 81 Jahre), Geschlecht oder ethnischer Zugehörigkeit (82,9 % Kaukasier, 3,1 % Schwarze, 7,1 % Asiaten und 6,9 % andere) beobachtet.
Körpergewicht
Die Clearance und das zentrale Verteilungsvolumen von Sotatercept nehmen mit zunehmendem Körpergewicht zu. Das empfohlene gewichtsbasierte Dosierungsschema führt zu einer konsistenten Sotatercept-Exposition.
Nierenfunktionsbeeinträchtigung
Die Sotatercept-Pharmakokinetik war bei PAH-Patienten mit leichter bis mittelschwerer Nierenfunktionsbeeinträchtigung (eGFR im Bereich von 30 bis 89 ml/min/1,73 m2) vergleichbar mit Patienten mit normaler Nierenfunktion (eGFR ≥ 90 ml/min/1,73 m2). Schwere Nierenfunktionsbeeinträchtigung (eGFR im Bereich von 15 bis 30 ml/min/1,73 m2, n = 3) hatte keinen Einfluss auf die PK von Sotatercept. Darüber hinaus ist die Sotatercept-PK bei Nicht-PAH Patienten mit terminaler Nierenerkrankung (end-stage renal disease; ESRD) (eGFR < 15 ml/min/1,73 m2) und Patienten mit normaler Nierenfunktion vergleichbar. Sotatercept ist während der Hämodialyse nicht dialysierbar. Zur Anwendung von Sotatercept bei PAH-Patienten mit schwerer Nierenfunktionsbeeinträchtigung (eGFR < 30 ml/min/1,73 m2) liegen nur begrenzte Daten vor.
Leberfunktionsbeeinträchtigung
Sotatercept wurde bei PAH-Patienten mit Leberfunktionsbeeinträchtigung (Child-Pugh-Klassifikation A bis C) nicht untersucht. Es ist nicht zu erwarten, dass eine Beeinträchtigung der Leber den Metabolismus von Sotatercept beeinflusst, da Sotatercept über einen zellulären Katabolismus metabolisiert wird.
Mit Sotatercept wurden keine Karzinogenitäts- oder Mutagenitätsstudien durchgeführt.
Toxizität bei wiederholter Gabe
Bei Ratten dauerten die längsten subkutanen Toxizitätsstudien 3 Monate und bei Affen 9 Monate. Bei Ratten umfassten die unerwünschten Befunde eine Degeneration der ableitenden Gänge/Hoden, Nebennierenkongestion/-nekrose und membranproliferative Glomerulonephritis und tubulointerstitielle Nephritis in den Nieren. Die Veränderungen der Niere waren nach einer 1-monatigen Erholungsphase nicht reversibel. Zu den unerwünschten Veränderungen bei Affen gehörten eine erhöhte interstitielle Matrix an der kortikomedulären Verbindung, eine verringerte Größe der glomerulären Büschel, Glomerulonephritis und tubulointerstitielle Nephritis in den Nieren. Nierenveränderungen bei Affen bildeten sich nach einer 3-monatigen Erholungsphase teilweise zurück. Beim No Observed Adverse Effect Level (NOAEL) bei Ratten und Affen betrug die Sotatercept-Exposition ≤ 2-Fache der klinischen Exposition bei der empfohlenen Maximaldosis beim Menschen (maximum recommended human dose, MRHD). Weitere Befunde, die an den Grenzen der klinischen Exposition bei Affen auftraten, waren entzündliche Infiltrate in der Leber, lymphoide Depletion in der Milz und entzündliche Infiltrate im Plexus choroideus.
Reproduktionstoxizität
In einer Fertilitätsstudie bei weiblichen Ratten wurde die Dauer des Östruszyklus verlängert, die Schwangerschaftsrate verringert und es kam zu einem Anstieg der Prä- und Postimplanationsverluste und zu einer Verringerung der Wurfgröße. Beim NOAEL für weibliche Fertilitätsendpunkte betrug die Sotatercept-Exposition das 2-Fache der klinischen AUC bei der MRHD.
Bei männlichen Ratten gab es nicht reversible histologische Veränderungen in den efferenten Gängen, Hoden und Nebenhoden. Histomorphologische Veränderungen in Rattenhoden korrelierten mit einem verminderten Fertilitätsindex, der sich während der 13-wöchigen behandlungsfreien Periode umkehrte. Ein NOAEL für testikuläre histologische Veränderungen wurde nicht ermittelt und der NOAEL für funktionelle Veränderungen der männlichen Fertilität liefert eine systemische Exposition, die das 2-Fache der klinischen Exposition bei der MRHD beträgt.
In Studien zur embryo-fetalen Entwicklungstoxizität führten die Auswirkungen bei Ratten und Kaninchen zu einer Verringerung der Anzahl lebender Feten und des Körpergewichts der Feten, zu Verzögerungen bei der Ossifikation sowie zu vermehrten Resorptionen und Postimplantationsverlusten. Nur bei Ratten gab es auch Skelettvariationen (erhöhte Anzahl überzähliger Rippen und Veränderungen in der Anzahl der Brust- oder Lendenwirbel). Beim NOAEL betrugen die Sotatercept-Expositionen bei Ratten und Kaninchen das 2-Fache bzw. das 0,4-Fache der klinischen Exposition bei der MRHD.
In einer prä- und postnatalen Entwicklungsstudie bei Ratten wurden keine unerwünschten Wirkungen in Zusammenhang mit Sotatercept bei Jungtieren der ersten Tochtergeneration (F1) von Muttertieren, die während der Trächtigkeit mit geschätzten Expositionen bis zum 2-Fachen der MRHD behandelt wurden, beobachtet. Bei F1-Jungtieren von Muttertieren, denen während der Laktation eine Dosis verabreicht wurde, korrelierte eine Abnahme des Jungtiergewichts mit Verzögerungen bei der Geschlechtsreife. Der NOAEL für Auswirkungen auf Wachstum und Entwicklung der Jungtiere ergibt eine systemische Exposition, die dem 0,6-Fachen der klinischen Exposition bei der MRHD entspricht.
Pulver:
Citronensäure-Monohydrat (E 330)
Natriumcitrat (E 331)
Polysorbat 80 (E 433)
Saccharose
Lösungsmittel:
Wasser für Injektionszwecke
Dieses Arzneimittel darf zur Anwendung, außer mit den in Abschnitt 6.6 aufgeführten, nicht mit anderen Arzneimitteln gemischt werden.
Ungeöffnete Durchstechflasche
4 Jahre
Nach Rekonstitution
Die biochemische und biophysikalische Stabilität nach Rekonstitution wurde für 4 Stunden bei 30 °C nachgewiesen.
Aus mikrobiologischer Sicht sollte das Arzneimittel sofort oder nicht später als 4 Stunden nach der Rekonstitution angewendet werden.
Wenn es nicht sofort angewendet wird, liegen die Aufbewahrungszeiten und -bedingungen während des Gebrauchs vor der Anwendung in der Verantwortung des Anwenders.
Im Kühlschrank lagern (2 °C – 8 °C). Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
Winrevair 45 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Durchstechflasche aus Typ-I-Glas mit 2 ml Fassungsvermögen, verschlossen mit einem Brombutyl-Gummistopfen mit Polymer-Beschichtung und einer Aluminiumversiegelung mit hellgrünem Flip-off-Verschluss aus Polypropylen, die 45 mg Sotatercept enthält.
Fertigspritze (Typ-I-Glaskartusche, verschlossen mit einem Brombutyl-Gummistopfen) mit 1 ml Lösungsmittel.
Winrevair 60 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Durchstechflasche aus Typ-I-Glas mit 2 ml Fassungsvermögen, verschlossen mit einem Brombutyl-Gummistopfen mit Polymer-Beschichtung und einer Aluminiumversiegelung mit weinrotem Flip-off-Verschluss aus Polypropylen, die 60 mg Sotatercept enthält.
Fertigspritze (Typ-I-Glaskartusche, verschlossen mit einem Brombutyl-Gummistopfen) mit 1,3 ml Lösungsmittel.
Winrevair Pulver und Lösungsmittel zur Herstellung einer Injektionslösung ist erhältlich als:
Sets mit 1 Durchstechflasche mit 45 mg Pulver, 1 Fertigspritze mit 1,0 ml Lösungsmittel, 1 Dosierspritze mit 0,1-ml-Graduierung, 1 Durchstechflaschen-Adapter (13 mm), 1 Kanüle und 4 Alkoholtupfern.
Sets mit 2 Durchstechflaschen mit 45 mg Pulver, 2 Fertigspritzen mit 1,0 ml Lösungsmittel, 1 Dosierspritze mit 0,1-ml-Graduierung, 2 Durchstechflaschen-Adapter (13 mm), 1 Kanüle und 8 Alkoholtupfern.
Sets mit 1 Durchstechflasche mit 60 mg Pulver, 1 Fertigspritze mit 1,3 ml Lösungsmittel, 1 Dosierspritze mit 0,1-ml-Graduierung, 1 Durchstechflaschen-Adapter (13 mm), 1 Kanüle und 4 Alkoholtupfern.
Sets mit 2 Durchstechflaschen mit 60 mg Pulver, 2 Fertigspritzen mit 1,3 ml Lösungsmittel, 1 Dosierspritze mit 0,1-ml-Graduierung, 2 Durchstechflaschen-Adapter (13 mm), 1 Kanüle und 8 Alkoholtupfern.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Auswahl des passenden Produktsets
Wenn das Gewicht eines Patienten die Verwendung von zwei 45-mg- oder zwei 60-mg-Durchstechflaschen erfordert, sollte ein 2-Durchstechflaschen-Set anstelle von zwei 1-Durchstechflaschen-Sets verwendet werden, um die Notwendigkeit mehrerer Injektionen zu vermeiden (siehe Abschnitt 6.5).
Anleitungen zur Rekonstitution und Anwendung
Winrevair Pulver und Lösungsmittel zur Herstellung einer Injektionslösung sollte vor der Verwendung rekonstituiert und je nach Gewicht des Patienten als einzelne Injektion angewendet werden (siehe Abschnitt 4.2).
Eine detaillierte Schritt-für-Schritt-Anleitung zur Zubereitung und Anwendung des Arzneimittels finden Sie in der separaten Gebrauchsanweisungsbroschüre, die dem Set beiliegt. Nachfolgend finden Sie einen Überblick der Rekonstitutions- und Anwendungsanleitungen.
Rekonstitution
Entnehmen Sie das Set aus dem Kühlschrank und warten Sie 15 Minuten, damit die Fertigspritze(n) und das Arzneimittel vor der Zubereitung Raumtemperatur annehmen können.
Überprüfen Sie die Durchstechflasche, um sicherzustellen, dass das Arzneimittel das Verfalldatum nicht überschritten hat. Das Pulver sollte weiß bis cremefarben sein und kann in Form eines unversehrten Pulverkuchens oder in Bruchstücken vorliegen.
Entfernen Sie den Deckel von der Durchstechflasche mit dem Pulver und wischen Sie den Gummistopfen mit einem Alkoholtupfer ab.
Setzen Sie den Durchstechflaschen-Adapter auf die Durchstechflasche.
Untersuchen Sie die Fertigspritze visuell auf Schäden oder Unversehrtheit. Überprüfen Sie visuell das sterile Wasser in der Fertigspritze, um sicherzustellen, dass keine sichtbaren Partikel vorhanden sind.
Brechen Sie die Schutzkappe der Fertigspritze ab und verbinden Sie die Spritze mit dem Durchstechflaschen-Adapter.
Injizieren Sie das gesamte sterile Wasser aus der verbundenen Fertigspritze in die Durchstechflasche mit dem Pulver.
Die mit der 45-mg-Durchstechflasche gelieferte Fertigspritze enthält 1,0 ml steriles Wasser.
Die mit der 60-mg-Durchstechflasche gelieferte Fertigspritze enthält 1,3 ml steriles Wasser.
Nach der Rekonstitution kann die 45-mg-Durchstechflasche höchstens eine Dosis von 0,9 ml des Arzneimittels liefern und die 60-mg-Durchstechflasche höchstens eine Dosis von 1,2 ml des Arzneimittels liefern. Die Endkonzentration nach der Rekonstitution ist 50 mg/ml.
Schwenken Sie die Durchstechflasche vorsichtig, um das Arzneimittel zu rekonstituieren. Bitte nicht schütteln oder kräftig bewegen.
Lassen Sie die Durchstechflasche bis zu 3 Minuten stehen, damit sich mögliche Luftblasen auflösen.
Überprüfen Sie visuell die rekonstituierte Lösung. Bei ausreichender Auflösung sollte die rekonstituierte Lösung klar bis opaleszierend und farblos bis leicht bräunlich-gelb sein und keine Klumpen oder Pulver aufweisen.
Schrauben Sie die Spritze vom Durchstechflaschen-Adapter ab und entsorgen Sie die entleerte Spritze.
Wenn ein Set mit 2 Durchstechflaschen verschrieben wurde, wiederholen Sie die Schritte aus diesem Abschnitt zur Vorbereitung der zweiten Durchstechflasche.
Verwenden Sie die rekonstituierte Lösung so schnell wie möglich, jedoch nicht später als 4 Stunden nach der Rekonstitution.
Vorbereitung der Dosierspritze
Bevor Sie die Dosierspritze vorbereiten, überprüfen Sie die rekonstituierte Lösung visuell. Die rekonstituierte Lösung sollte klar bis opaleszierend und farblos bis leicht bräunlich-gelb sein und keine Klumpen oder Pulver aufweisen.
Wischen Sie den Durchstechflaschen-Adapter mit einem Alkoholtupfer ab.
Entnehmen Sie die Dosierspritze aus ihrer Verpackung und setzen Sie die Spritze auf den Durchstechflaschen-Adapter.
Drehen Sie die Spritze und die Durchstechflasche auf den Kopf und entnehmen Sie das entsprechende Volumen für die Injektion, basierend auf dem Gewicht des Patienten.
Wenn die Dosismenge die Verwendung von zwei Durchstechflaschen erfordert, entnehmen Sie den gesamten Inhalt der ersten Durchstechflasche und überführen Sie den gesamten Inhalt langsam in die zweite Durchstechflasche, um die Dosisgenauigkeit sicherzustellen.
Drehen Sie die Spritze und die Durchstechflasche auf den Kopf und entnehmen Sie die erforderliche Menge an Arzneimittel.
Drücken Sie bei Bedarf den Kolben hinein, um überschüssiges Arzneimittel oder Luft aus der Spritze zu entfernen.
Schrauben Sie die Spritze vom Durchstechflaschen-Adapter ab und befestigen Sie die Kanüle an der Dosierspritze.
Anwendung
Winrevair ist als subkutane Einzelinjektion anzuwenden.
Wählen Sie die Injektionsstelle am Bauch (mindestens 5 cm vom Nabel entfernt), Oberschenkel oder Oberarm und wischen Sie sie mit einem Alkoholtupfer ab. Wählen Sie für jede Injektion eine neue Stelle aus, die nicht vernarbt, empfindlich oder verletzt ist.
Bei der Anwendung durch den Patienten oder die Pflegekraft sind diese dahingehend zu instruieren, nur den Bauch oder den Oberschenkel als Einstichstelle zu verwenden (siehe „Gebrauchsanweisungsbroschüre“).
Führen Sie eine subkutane Injektion durch.
Entsorgen Sie die entleerte Spritze. Verwenden Sie die Spritze nicht erneut.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Anweisungen zur Rückverfolgbarkeit biologischer Arzneimittel finden Sie in Abschnitt 4.4.
Merck Sharp & Dohme B.V.
Waarderweg 39
2031 BN Haarlem
Niederlande
EU/1/24/1850/001
EU/1/24/1850/002
EU/1/24/1850/003
EU/1/24/1850/004
Datum der Erteilung der Zulassung: 22. August 2024
März 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig
Für weitere Informationen zu diesem Präparat wenden Sie sich bitte an die deutsche Vertretung des Zulassungsinhabers:
MSD Sharp & Dohme GmbH
Levelingstr. 4a
81673 München
Tel.: +49 (0) 89 20 300 4500
E-Mail: medinfo@msd.de
RCN: 000028056-DE; 000028946-DE
FACH-9000368-0003