
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Piasky® 340 mg Injektions-/Infusionslösung
Jede 2-ml-Durchstechflasche enthält 340 mg Crovalimab.
Jeder ml Injektions-/Infusionslösung enthält 170 mg Crovalimab.
Crovalimab ist ein humanisierter monoklonaler Antikörper, der mittels rekombinanter DNA-Technologie in Ovarialzellen des chinesischen Hamsters (CHO-Zellen) hergestellt wird.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Injektions-/Infusionslösung (Injektion/Infusion).
Klare bis stark opaleszente und nahezu farblose bis bräunlich-gelbe Lösung. Die Lösung hat einen pH-Wert von ca. 5,8 und eine Osmolalität von ca. 297 mOsm/kg.
Piasky als Monotherapie wird angewendet zur Behandlung von erwachsenen und pädiatrischen Patienten ab 12 Jahren mit einem Gewicht von mindestens 40 kg mit paroxysmaler nächtlicher Hämoglobinurie (PNH):
Bei Patienten mit Hämolyse mit klinischen Symptomen, die auf eine hohe Krankheitsaktivität hinweisen.
Bei Patienten, die nach mindestens 6 Monaten Behandlung mit einem Inhibitor der Komplementkomponente 5 (C5) klinisch stabil sind.
Die Behandlung ist unter der Aufsicht eines Arztes einzuleiten, der in der Behandlung von hämatologischen Erkrankungen erfahren ist.
Dosierung
Das empfohlene Dosierungsschema besteht aus einer Initialdosis, die als intravenöse Infusion (an Tag 1) verabreicht wird, gefolgt von vier weiteren wöchentlichen Initialdosen, die (an den Tagen 2, 8, 15 und 22) als subkutane Injektion verabreicht werden. Die Erhaltungsdosis wird erstmals an Tag 29 und dann alle 4 Wochen als subkutane Injektion verabreicht. Die zu verabreichenden Dosen basieren auf dem Körpergewicht des Patienten, wie in Tabelle 1 dargestellt.
Bei Patienten, die von einer Behandlung mit einem anderen Komplementinhibitor umgestellt werden, ist die erste intravenöse Initialdosis von Piasky zum Zeitpunkt der nächsten geplanten Gabe des Komplementinhibitors zu verabreichen (siehe Abschnitt 4.4 bzgl. weiterer Informationen zu einer Umstellung bei der Behandlung mit Inhibitoren der Komplementkomponente C5). Die Verabreichung der nachfolgenden subkutanen Initial- und Erhaltungsdosen von Piasky erfolgt entsprechend dem in Tabelle 1 angegebenen Schema.
Tabelle 1: Dosierungsschema von Piasky basierend auf dem Körpergewicht
Körpergewicht |
≥ 40 kg bis < 100 kg |
≥ 100 kg |
|
Initialdosis Tag 2, 8, 15, 22 |
1 000 mg (intravenös) 340 mg (subkutan) |
1 500 mg (intravenös) 340 mg (subkutan) |
|
Erhaltungsdosis Tag 29 und danach Q4Wa |
680 mg (subkutan) |
1 020 mg (subkutan) |
a Q4W = alle 4 Wochen
Das Dosierungsschema darf in Ausnahmefällen innerhalb eines 2-tägigen Zeitraums vom geplanten Anwendungstag abweichen (außer an Tag 1 und Tag 2). In einem solchen Fall ist die weitere Dosis gemäß dem normalen Behandlungsplan zu verabreichen.
Dauer der Behandlung
Piasky ist für die Langzeitbehandlung vorgesehen, es sei denn, das Absetzen des Arzneimittels ist klinisch indiziert (siehe Abschnitt 4.4).
Verspätete oder versäumte Dosen
Falls eine geplante Dosis vollständig oder ein Teil einer geplanten Dosis von Piasky versäumt wurde, sollte die fehlende Dosis oder der Rest der geplanten Dosis so bald wie möglich vor dem Tag der nächsten geplanten Dosis verabreicht werden. Die nächste Dosis ist dann am regulär geplanten Verabreichungstag anzuwenden. Nehmen Sie nicht zwei Dosen oder verabreichen Sie nicht mehr als die verschriebene Dosis am selben Tag, wenn Sie die vorherige Anwendung vergessen haben.
Dosisanpassungen
Eine Anpassung der Erhaltungsdosis ist erforderlich, wenn sich das Körpergewicht des Patienten im Verlauf der Behandlung um 10 % oder mehr verändert und dauerhaft über oder unter 100 kg liegt (für die empfohlene Dosis siehe Tabelle 1). Dementsprechend ist das Körpergewicht des Patienten in regelmäßigen Abständen zu überwachen.
Besondere Patientengruppen
Ältere Patienten
Bei Patienten im Alter von ≥ 65 Jahren ist keine Dosisanpassung erforderlich. Die Erfahrungen mit Crovalimab in dieser Patientenpopulation sind jedoch begrenzt. (siehe Abschnitt 5.2).
Nierenfunktionsstörung
Bei Patienten mit leichter, mäßiger oder schwerer Nierenfunktionsstörung wird keine Dosisanpassung empfohlen (siehe Abschnitt 5.2).
Leberfunktionsstörung
Bei Patienten mit leichter Leberfunktionsstörung wird keine Dosisanpassung empfohlen. Crovalimab wurde bei Patienten mit mäßiger bis schwerer Leberfunktionsstörung nicht untersucht und es können keine Dosierungsempfehlungen gegeben werden (siehe Abschnitt 5.2).
Kinder und Jugendliche
Bei Jugendlichen ab 12 Jahren mit einem Körpergewicht von ≥ 40 kg ist keine Dosisanpassung von Crovalimab erforderlich. Die Sicherheit und Wirksamkeit von Crovalimab bei Kindern unter 12 Jahren und Kindern mit einem Körpergewicht < 40 kg ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Piasky wird als intravenöse Infusion (erste Dosis) und als subkutane Injektion (weitere Dosen) verabreicht.
Intravenöse Verabreichung
Piasky ist für die intravenöse Verabreichung unter angemessener aseptischer Technik zuzubereiten. Piasky muss von medizinischem Fachpersonal verdünnt und als intravenöse Infusion über 60 Minuten ± 10 Minuten (1 000 mg) oder 90 Minuten ± 10 Minuten (1 500 mg) verabreicht werden. Piasky darf nicht als intravenöse Druck- oder Bolusinjektion angewendet werden.
Hinweise zur Verdünnung des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
Die Infusion von Crovalimab kann verlangsamt oder unterbrochen werden, wenn der Patient eine Reaktion im Zusammenhang mit einer Infusion entwickelt. Die Infusion ist sofort abzubrechen, wenn bei dem Patienten eine schwerwiegende Überempfindlichkeitsreaktion auftritt (siehe Abschnitt 4.4).
Subkutane Verabreichung
Piasky muss unverdünnt angewendet und mit angemessener aseptischer Technik zubereitet werden. Es wird empfohlen, Piasky in den Bauch zu injizieren. Bei jeder Injektion in den Bauch ist die Injektionsstelle zu wechseln. Die Injektionen dürfen niemals in Leberflecken, Narben, blaue Flecken oder Bereiche injiziert werden, in denen die Haut empfindlich, geschwollen, gerötet, verhärtet oder verletzt ist.
Anwendung durch den Patienten und/oder die Betreuungsperson
Nach einer entsprechenden Einweisung in die subkutane Injektionstechnik kann der Patient Piasky selbst verabreichen oder die Betreuungsperson kann Piasky ohne ärztliche Aufsicht verabreichen, wenn der behandelnde Arzt dies für angemessen hält.
Eine umfassende Anleitung zur Anwendung von Piasky befindet sich am Ende der Packungsbeilage.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Patienten mit nicht abgeklungener Neisseria meningitidis-Infektion.
Patienten, die derzeit nicht gegen Neisseria meningitidis geimpft sind, es sei denn, sie erhalten bis 2 Wochen nach der Impfung eine geeignete Antibiotikaprophylaxe (siehe Abschnitt 4.4).
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Schwerwiegende Meningokokken-Infektion
Aufgrund seines Wirkmechanismus kann die Anwendung von Crovalimab die Anfälligkeit des Patienten für Meningokokken-Infektionen (Septikämie und/oder Meningitis) erhöhen. Bei Patienten, die mit terminalen Komplementinhibitoren behandelt wurden, wurden Fälle von schwerwiegenden oder tödlichen Meningokokken-Infektionen/-Sepsis berichtet, was eine bekannte Klassenwirkung ist.
Meningokokken-Infektionen können schnell lebensbedrohlich oder tödlich werden, wenn sie nicht rechtzeitig erkannt und behandelt werden. Um das Infektionsrisiko zu reduzieren, müssen alle Patienten mindestens 2 Wochen vor der ersten Crovalimab-Dosis mit einem tetravalenten Meningokokken-Impfstoff geimpft werden. Wenn bei einem ungeimpften Patienten eine sofortige Behandlung mit Crovalimab angezeigt ist, sollte die erforderliche Impfung so schnell wie möglich verabreicht werden, und die Patienten sollten vom Zeitpunkt des Beginns der Crovalimab-Behandlung bis 2 Wochen nach der Impfung eine Antibiotikaprophylaxe erhalten. Zur Vorbeugung gegen die häufig pathogenen Meningokokken-Serogruppen werden, sofern verfügbar, Impfstoffe gegen die Serogruppen A, C, Y, W und B empfohlen. Die Patienten müssen ihren Impfstatus gemäß den geltenden lokalen Impfleitlinien auf dem aktuellen Stand halten. Wenn der Patient von einer anderen Behandlung mit einem terminalen Komplementinhibitor umgestellt wird, hat der Arzt zu überprüfen, ob die Meningokokken-Impfung gemäß den lokalen Richtlinien für die Impfung aktuell ist. Die Impfung kann das Komplementsystem weiter aktivieren. Infolgedessen kann es bei Patienten mit komplementvermittelten Erkrankungen, einschließlich PNH, zu einer vorübergehenden Verschlechterung der Anzeichen und Symptome der Grunderkrankung, wie z. B. Hämolyse, kommen. Daher sind die Patienten nach der empfohlenen Impfung engmaschig auf Krankheitssymptome zu überwachen.
Möglicherweise reicht die Impfung nicht aus, um eine Meningokokken-Infektion zu verhindern. Die prophylaktische Anwendung antibakterieller Wirkstoffe ist auf der Grundlage lokaler Empfehlungen abzuwägen. Alle Patienten sind auf frühe Anzeichen einer Meningokokken-Infektion zu überwachen, bei Verdacht auf eine Infektion sofort zu untersuchen und bei Bedarf mit geeigneten Antibiotika zu behandeln. Die Patienten sind über diese Anzeichen und Symptome aufzuklären sowie über die zu unternehmenden Schritte zu informieren, damit sie sofort ärztliche Hilfe in Anspruch nehmen können. Die Ärzte müssen den Nutzen und die Risiken einer Behandlung mit Piasky mit den Patienten besprechen und ihnen einen Leitfaden für Patienten/Betreuungspersonen und einen Patientenpass aushändigen (siehe unten „Schulungsmaterialien“). Gemäß den jährlichen Erinnerungen hat das medizinische Fachpersonal sicherzustellen, dass die Impfungen der Patienten auf dem neuesten Stand sind.
Andere systemische Infektionen
Aufgrund seines Wirkmechanismus muss Crovalimab bei Patienten mit aktiven systemischen Infektionen mit Vorsicht angewendet werden. Patienten können eine erhöhte Infektionsanfälligkeit aufweisen, insbesondere gegenüber Neisseria spp. und anderen verkapselten Bakterien. Impfungen zur Prophylaxe von Infektionen mit Streptococcus pneumoniae und Haemophilus influenzae Typ b (Hib) müssen entsprechend den lokalen Richtlinien verabreicht werden.
Wenn lokale Richtlinien Impfungen zur Prävention von Infektionen mit Streptococcus pneumoniae und Haemophilus influenzae Typ b (Hib) vorschreiben, müssen diese mindestens 2 Wochen vor der ersten Gabe von Crovalimab erfolgen. Wenn bei einem ungeimpften Patienten eine sofortige Behandlung mit Crovalimab indiziert ist, ist die erforderliche Impfung so schnell wie möglich zu verabreichen, und der Patient sollte prophylaktisch Antibiotika erhalten, und zwar ab dem Zeitpunkt, zu dem die Behandlung mit Crovalimab beginnt, bis 2 Wochen nach der Impfung oder gemäß dem lokalen Behandlungsstandard, je nachdem, welcher Zeitraum länger ist.
Wenn Piasky bei Patienten mit aktiven systemischen Infektionen verabreicht wird, müssen die Patienten engmaschig auf Anzeichen und Symptome einer Verschlechterung der Infektion überwacht werden. Patienten wurden von klinischen Studien mit Crovalimab ausgeschlossen, wenn sie innerhalb von 14 Tagen vor Beginn der Behandlung eine aktive systemische bakterielle, virale oder Pilzinfektion hatten.
Patienten sollten Informationen aus der Packungsbeilage erhalten, um ihr Bewusstsein für die Anzeichen und Symptome potenzieller schwerwiegender Infektionen zu schärfen.
Typ-III-Immunkomplex-Reaktionen
Eine Bildung von Immunkomplexen tritt bei Patienten auf, die zwischen C5-Inhibitoren wechseln, welche an verschiedene Epitope binden (siehe Abschnitt 4.5). Bei manchen Patienten kann die Bildung dieser Komplexe zu Immunkomplex-vermittelten Reaktionen vom Typ III führen, die auch als Typ-III-Immunkomplex-Reaktionen bezeichnet werden. Bei Patienten, die zuvor noch nie mit einem C5-Inhibitor behandelt wurden, oder bei Patienten, bei denen der Wirkstoff einer vorherigen C5-Inhibitor-Behandlung vom Körper ausgeschieden wurde (d. h. seit der letzten Dosis sind mindestens 5,5 Halbwertszeiten der vorherigen Behandlung vergangen), besteht kein Risiko für Typ-III-Immunkomplex-Reaktionen. In klinischen Studien mit Crovalimab wurde über Nebenwirkungen von Immunkomplex-vermittelten Reaktionen des Typs III berichtet (siehe Abschnitt 4.8).
Anzeichen und Symptome von Typ-III-Immunkomplex-Reaktionen, die in klinischen Studien beobachtet wurden, waren Arthralgie und andere Erkrankungen des Bewegungsapparats und des Bindegewebes, Ausschlag und andere Erkrankungen der Haut und der Unterhaut, Fieber, Asthenie/Ermüdung (Fatigue), gastrointestinale Beschwerden, Kopfschmerzen und axonale Neuropathie. Typ-III-Immunkomplex-Reaktionen können sich auch als Nierenanomalien manifestieren, was jedoch in klinischen Studien mit Crovalimab nicht beobachtet wurde.
Aufgrund der in klinischen Studien beobachteten Zeit bis zum Auftreten von Typ-III-Immunkomplex-Reaktionen wird empfohlen, die Patienten nach der Umstellung von Eculizumab oder Ravulizumab auf Crovalimab (oder umgekehrt) während der ersten 30 Tage auf das Auftreten von Symptomen von Typ-III-Immunkomplex-Reaktionen zu überwachen. Bei leichten oder mäßigen Typ-III-Immunkomplex-Reaktionen kann die Anwendung einer symptomatischen Behandlung (z. B. topische Corticosteroide, Antihistaminika, Antipyretika und/oder Analgetika) in Betracht gezogen werden. Bei schweren Reaktionen kann eine orale oder parenterale Corticosteroidtherapie eingeleitet und wie klinisch indiziert ausgeschlichen werden.
Reaktionen im Zusammenhang mit einer Infusion und injektionsbedingte Reaktionen
Die Anwendung von Crovalimab kann je nach Art der Anwendung zu Reaktionen im Zusammenhang mit einer Infusion oder systemischen injektionsbedingten Reaktionen führen. Hierzu können allergische Reaktionen oder Überempfindlichkeitsreaktionen (einschließlich Anaphylaxie), aber auch eine Reihe anderer Symptome wie Kopfschmerzen oder Muskelschmerzen zählen.
Im Falle einer schweren Reaktion im Zusammenhang mit einer Infusion nach intravenöser Verabreichung von Piasky ist die Behandlung zu unterbrechen und eine geeignete medizinische Behandlung ist einzuleiten. Im Falle einer schwerwiegenden injektionsbedingten Reaktion nach subkutaner Verabreichung oder jeglicher Inzidenz einer schweren allergischen Reaktion nach intravenöser oder subkutaner Verabreichung müssen Patienten/Betreuungspersonen sofort ärztlichen Rat einholen, eine geeignete medizinische Behandlung ist einzuleiten. Die Patienten sollten mit ihrem Arzt besprechen, ob die Behandlung mit Piasky fortgesetzt werden kann.
Schwerwiegende Hämolyse nach Absetzen der Behandlung bei PNH-Patienten
Im Falle eines Absetzens von Piasky müssen Patienten, die nicht auf eine andere Behandlung der PNH umgestellt werden, engmaschig auf Anzeichen und Symptome einer schwerwiegenden intravaskulären Hämolyse überwacht werden, die anhand eines erhöhten Laktatdehydrogenase (LDH)-Spiegels zusammen mit einer plötzlichen Abnahme der PNH-Klongröße oder des Hämoglobins, oder dem Wiederauftreten von Symptomen wie Ermüdung (Fatigue), Hämoglobinurie, Abdominalschmerzen, Atemnot (Dyspnoe), schweren unerwünschten vaskulären Ereignissen (einschließlich Thrombose), Dysphagie oder erektiler Dysfunktion identifiziert werden können. Treten nach dem Absetzen Anzeichen und Symptome einer Hämolyse auf, einschließlich eines erhöhten LDH-Wertes, ist die Wiederaufnahme einer geeigneten Behandlung zu erwägen.
Immunogenität mit daraus resultierendem Verlust der Exposition und Wirksamkeit
Patienten können Anti-Wirkstoff-Antikörper (antidrug antibodies, ADA) entwickeln, die die Crovalimab-Exposition beeinträchtigen können. Die Entwicklung von ADA kann einen Verlust der Crovalimab-Exposition bewirken, was in der Folge zu einem Verlust der Wirksamkeit von Crovalimab führen kann. In klinischen Studien wurde bei Patienten, die mit Crovalimab behandelt wurden, ein Wirksamkeitsverlust und ein Expositionsverlust infolge der Entwicklung von ADA beobachtet. Die Patienten sind routinemäßig auf klinische Anzeichen eines Expositions- und eines Wirksamkeitsverlusts, einschließlich schwerwiegender intravaskulärer Hämolyse, zu überwachen. Im Falle einer persistierenden schwerwiegenden intravaskulären Hämolyse trotz konformer Behandlung mit Crovalimab sind die Patienten umgehend zu untersuchen, um die Ätiologie zu beurteilen und die Möglichkeit in Betracht zu ziehen, dass die Entwicklung von ADA zu einem Verlust der Exposition und Wirksamkeit führt. Eine Nutzen-Risiko-Bewertung der Fortführung der Crovalimab-Therapie ist durchzuführen und ein Wechsel zu einer anderen Therapie zu erwägen. Patienten/Betreuungspersonen sind anzuweisen, sofort einen Arzt aufzusuchen, wenn der Patient Anzeichen einer Verschlechterung der PNH entwickelt. Siehe Abschnitte 4.8 und 5.1.
Schulungsmaterialien
Alle Angehörigen der Gesundheitsberufe, von denen zu erwarten ist, dass sie Piasky verschreiben, anwenden oder die Verabreichung überwachen, müssen sicherstellen, dass sie den Leitfaden für medizinisches Fachpersonal erhalten haben und mit ihm vertraut sind. Das medizinische Fachpersonal erhält jährlich eine Erinnerung, um sicherzustellen, dass die Impfungen der Patienten auf dem neuesten Stand sind. Der verschreibende Arzt muss den Patienten und/oder ihren Betreuungspersonen den Nutzen und die Risiken der Therapie mit Piasky erklären und mit ihnen besprechen und sicherstellen, dass der Leitfaden für Patienten/Betreuungspersonen und der Patientenpass ausgehändigt werden.
Angehörige der Gesundheitsberufe müssen die Patienten und/oder ihre Betreuungspersonen anweisen, den Patientenpass mit Informationen über die wichtigsten Anzeichen und Symptome von Meningokokken-Infektionen und schweren allergischen Reaktionen stets bei sich zu tragen und bei Auftreten von Symptomen einer Meningokokken-Infektion und/oder schweren allergischen Reaktionen einen Notarzt zu rufen.
Crovalimab und andere C5-Inhibitoren binden verschiedene Epitope an C5, sodass sich Immunkomplexe aus den von C5 überbrückten Antikörpern bilden können, wenn beide im Blutkreislauf vorhanden sind. Diese Immunkomplexe, die auch als Drug-Target-Drug-Complexes (DTDCs) bezeichnet werden, können eine oder mehrere Einheiten von C5 enthalten, die sowohl an Crovalimab als auch an einen anderen C5-Inhibitor gebunden sind, und es wird erwartet, dass sie innerhalb von etwa 8 Wochen (im Fall von Eculizumab) ausgeschieden werden. Bei einem Wechsel von C5-Inhibitoren mit verlängerter Halbwertszeit wie Ravulizumab können die Immunkomplexe nach einem längeren Zeitraum ausgeschieden werden. Bei einigen Patienten führt die Bildung dieser Komplexe zu Typ-III-Immunkomplex-Reaktionen (siehe Abschnitte 4.4 und 4.8). Bei Patienten, die von einer Therapie mit einem anderen C5-Inhibitor umgestellt werden, wird aufgrund der Bildung von Immunkomplexen ein vorübergehender Anstieg der Clearance beobachtet, was eine schnellere Elimination von Crovalimab bewirkt. Dieser transiente Anstieg der Clearance ist jedoch klinisch nicht relevant und erfordert keine Dosisanpassung bei Patienten, die von einem anderen C5-Inhibitor umgestellt werden.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Es wird nicht erwartet, dass Crovalimab pharmakokinetische Wechselwirkungen mit anderen Arzneimitteln zeigt, die die Metabolisierung von Cytochrom-P450 (CYP)-Enzymen beeinflussen, da sich die Clearance-Signalwege von Immunglobulinen G (IgGs) von denen kleiner Moleküle unterscheiden.
Schwangerschaft
Es liegen keine Daten über die Anwendung von Crovalimab bei Schwangeren vor.
Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Abschnitt 5.3). Es ist bekannt, dass humanes IgG die Plazentaschranke nach dem ersten Trimester der Schwangerschaft passiert. Aufgrund seines Wirkmechanismus kann Crovalimab potenziell eine terminale Komplementhemmung im fetalen Kreislauf verursachen.
Daher kann die Anwendung von Piasky bei schwangeren Frauen in Betracht gezogen werden, wenn der klinische Zustand der Frau eine Behandlung mit Crovalimab erfordert.
Stillzeit
Es ist nicht bekannt, ob Crovalimab in die Muttermilch übergeht. Es ist bekannt, dass humanes IgG1 in die Muttermilch übergeht. Ein Risiko für den gestillten Säugling kann nicht ausgeschlossen werden.
Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob die Behandlung mit Piasky zu unterbrechen ist, wobei der Nutzen des Stillens für den Säugling und der Nutzen der Behandlung für die Mutter zu berücksichtigen sind.
Fertilität
Es liegen keine klinischen Daten zur Wirkung von Crovalimab auf die Fertilität beim Menschen vor. Tierexperimentelle Daten aus Studien zur Toxizität bei wiederholter Gabe zeigten keine Auswirkungen auf die männlichen oder weiblichen Fortpflanzungsorgane (siehe Abschnitt 5.3).
Piasky hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die häufigsten beobachteten Nebenwirkungen waren Immunkomplex-vermittelte Reaktionen des Typs III (18,9 % bei Patienten, die von einer Behandlung mit einem anderen C5-Inhibitor auf Crovalimab umgestellt wurden), Infektionen der oberen Atemwege (18,6 %), Fieber (13,5 %), Kopfschmerzen (10,9 %) und Reaktionen im Zusammenhang mit einer Infusion (10,2 %). Die am häufigsten beobachteten schwerwiegenden Nebenwirkungen waren Immunkomplex-vermittelte Reaktionen des Typs III (4,0 % bei Patienten, die von einer Behandlung mit einem anderen C5-Inhibitor auf Crovalimab umgestellt wurden) und Pneumonie (1,5 %).
Die Sicherheitsergebnisse der 44 Patienten in der COMPOSER-Studie, bei denen die mediane Behandlungsdauer 4,69 Jahre betrug (Bereich: 0,4 – 6,3 Jahre), zeigten keine zusätzlichen Sicherheitsbedenken im Zusammenhang mit der Langzeitanwendung von Crovalimab.
Tabellarische Auflistung der Nebenwirkungen
Die Sicherheit von Crovalimab bei Patienten mit PNH wurde in drei Phase-III-Studien, COMMODORE 2 (BO42162), COMMODORE 3 (YO42311) und COMMODORE 1 (BO42161), sowie einer Phase-I/II-Studie (COMPOSER, BP39144) untersucht.
Sofern nicht anders angegeben, sind in Tabelle 2 die Nebenwirkungen aufgeführt, die im Zusammenhang mit der Anwendung von Crovalimab in einer gepoolten Auswertung von 393 Patienten, die an den Phase-III-Studien teilnahmen, berichtet wurden. Die mediane Behandlungsdauer mit Crovalimab betrug basierend auf der gepoolten Analyse von 393 Patienten 64 Wochen (Bereich: 0,1 – 136,4 Wochen).
Die Nebenwirkungen sind nach MedDRA-Systemorganklasse aufgelistet. Die entsprechende Häufigkeitskategorie für jede Nebenwirkung basiert auf der folgenden Konvention: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000). Innerhalb jeder Häufigkeitskategorie sind die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 2: Zusammenfassung der Nebenwirkungen, die bei mit Piasky behandelten Patienten auftraten
Systemorganklassen gemäß MedDRA-Datenbank |
Nebenwirkungen (MedDRA) |
Häufigkeitskategorie |
Infektionen und parasitäre Erkrankungen |
Infektion der oberen Atemwege |
Sehr häufig |
Pneumonie |
Häufig |
|
Atemwegsinfektion | ||
Harnwegsinfektion | ||
Nasopharyngitis | ||
Sepsis |
Gelegentlich |
|
Septischer Schock | ||
Bakteriämie | ||
Pyelonephritis | ||
Erkrankungen des Immunsystems |
Immunkomplex-vermittelte Reaktion (Typ III)* |
Sehr häufig |
Überempfindlichkeit |
Häufig |
|
Erkrankungen des Nervensystems |
Kopfschmerzen |
Sehr häufig |
Erkrankungen des Gastrointestinaltrakts |
Abdominalschmerz |
Häufig |
Diarrhö | ||
Erkrankungen der Haut und des Unterhautgewebes |
Ausschlag |
Häufig |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Arthralgie |
Häufig |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber |
Sehr häufig |
Asthenie |
Häufig |
|
Ermüdung (Fatigue) | ||
Reaktion an der Injektionsstelle |
Gelegentlich |
|
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen |
Reaktion im Zusammenhang mit einer Infusion |
Sehr häufig |
Injektionsbedingte Reaktion |
Häufig |
* Die Typ-III-Immunkomplex-vermittelte Reaktion (auch als Typ-III-Immunkomplex-Reaktion bezeichnet) ist auf Patienten beschränkt, die von einem anderen C5-Inhibitor zu Crovalimab oder von Crovalimab zu einem anderen C5-Inhibitor wechseln. Die Häufigkeit von Typ-III-Immunkomplex-Reaktionen wird für eine Untergruppe von n = 201 Patienten berichtet, die von der Behandlung mit einem anderen C5-Inhibitor auf Crovalimab umgestellt wurden, wobei die Inzidenzraten unter Verwendung dieser n = 201 Patienten als Nenner berechnet werden. Siehe unten.
Beschreibung ausgewählter Nebenwirkungen
Typ-III-Immunkomplex-Reaktionen (siehe Abschnitte 4.4 und 4.5)
In allen Phase-III-Studien kam es bei 19,4 % (39 von 201) der Patienten, die von einer Behandlung mit Eculizumab oder Ravulizumab auf Crovalimab umgestellt wurden, zu einer Typ-III-Immunkomplex-Reaktion (die als Typ-III-Immunkomplex-vermittelte Reaktion berichtet wurde). Von diesen 39 Patienten erlitten 2 Patienten nach Absetzen von Crovalimab und Umstellung auf Ravulizumab eine zweite Typ-III-Immunkomplex-Reaktion. Die häufigsten gemeldeten Anzeichen und Symptome waren Arthralgie und Ausschlag, und andere berichtete Symptome schlossen Fieber, Kopfschmerzen, Myalgie, Abdominalschmerzen, Asthenie/Ermüdung (Fatigue) und axonale Neuropathie ein. Die mediane Zeit bis zum Einsetzen einer Typ-III-Immunkomplex-Reaktion bei Patienten, die von der Behandlung mit Eculizumab oder Ravulizumab auf Crovalimab umgestellt wurden, betrug 1,6 Wochen (Bereich: 0,7 – 4,4 Wochen), wobei 5,1 % der Patienten (2 von 39) eine Typ-III-Immunkomplex-Reaktion mit einer Zeit bis zum Einsetzen erlitten, die 4 Wochen überschritt. Die meisten Fälle von Typ-III-Immunkomplex-Reaktionen waren vorübergehend und die mediane Dauer betrug 1,7 Wochen (Bereich 0,4 – 34,1 Wochen). Bei der Mehrzahl der Patienten kam es zu einem Ereignis von Grad 1 oder 2 (23 von 39 Patienten), wobei Ereignisse von Grad 3 bei 8 % (16 von 39) der mit Crovalimab behandelten Patienten auftraten, die von Eculizumab oder Ravulizumab umgestellt worden waren. Die meisten Ereignisse klangen ohne Veränderung der Studienbehandlung mit Crovalimab ab.
In der COMPOSER-Studie berichteten 2 von 26 Patienten, die von Eculizumab auf Crovalimab umgestellt wurden, über eine Nebenwirkung in Form einer Typ-III-Immunkomplex-Reaktion. Diese Ereignisse waren leicht/mäßig und nicht schwerwiegend. Ein weiterer Patient entwickelte eine leichte Typ-III-Immunkomplex-Reaktion, nachdem er Crovalimab abgesetzt hatte und auf einen anderen C5‑Inhibitor umgestellt wurde.
Immunogenität
In zwei randomisierten Phase-III-Studien (COMMODORE 1 und COMMODORE 2) und einer einarmigen Phase-III-Studie (COMMODORE 3) war der ADA-Status bei 392 Patienten evaluierbar. Von diesen 392 Patienten waren 118 (30,1 %) ADA-positiv. Zwischen ADA-positiven und ADA-negativen Patienten wurden keine Unterschiede in der Häufigkeit von Nebenwirkungen beobachtet, die typischerweise mit Immunogenität assoziiert sind (z. B. Reaktionen im Zusammenhang mit einer Infusion, Reaktionen an der Injektionsstelle oder Überempfindlichkeit) (siehe Abschnitt 5.1).
Immunogenität mit daraus resultierendem Verlust der Exposition und Wirksamkeit
Bei Patienten, die ADA entwickeln, kann infolgedessen die Crovalimab-Exposition beeinträchtigt sein bzw. werden. Bei 23 von 392 Patienten (5,9 %), die hinsichtlich des ADA-Status untersucht wurden, wurde ein teilweiser oder vollständiger Verlust der Exposition im Zusammenhang mit dem Auftreten von ADA beobachtet; bei 17 (4,3 %) von ihnen trat ein Verlust der pharmakologischen Aktivität auf, der mit einem Verlust der Exposition zusammenfiel und einen Verlust der Wirksamkeit, der sich bei 7 Patienten (1,8 %) als erlittener Verlust der Hämolysekontrolle manifestierte.
Im Falle klinischer Anzeichen eines Wirksamkeitsverlusts hat eine sofortige Beurteilung durch medizinisches Fachpersonal zu erfolgen (siehe Abschnitt 4.4).
Reaktionen im Zusammenhang mit einer Infusion und injektionsbedingte Reaktionen
In allen Phase-III-Studien kam es bei 10,2 % der Patienten, die mit Crovalimab behandelt wurden, zu einer Reaktion im Zusammenhang mit einer Infusion. Die häufigsten Anzeichen und Symptome, die berichtet wurden, waren Kopfschmerzen (7,1 %), Ausschlag (0,8 %), Schwindelgefühl (0,8 %), Abdominalschmerzen (0,5 %), Erythem (0,5 %), Übelkeit (0,5 %), Fieber (0,5 %) und Parästhesie (0,3 %). Alle berichteten Ereignisse waren von Grad 1 bis 2.
In allen Phase-III-Studien traten bei 8,4 % der Patienten, die mit Crovalimab behandelt wurden, injektionsbedingte Reaktionen auf. Die häufigsten Anzeichen und Symptome, die berichtet wurden, waren Kopfschmerzen (2,5 %), Erythem an der Injektionstelle (1,0 %), Schmerzen an der Injektionsstelle (1,0 %), und Ausschlag an der Injektionsstelle (1,0 %). Die Mehrzahl der Ereignisse war von Grad 1 bis 2.
Infektionen mit verkapselten Bakterien
Aufgrund seines Wirkmechanismus kann die Anwendung von Crovalimab möglicherweise das Risiko von Infektionen erhöhen, insbesondere Infektionen, die durch verkapselte Bakterien verursacht werden, einschließlich Streptococcus pneumoniae, Neisseria meningitidis der Typen A, C, W, Y und B sowie Haemophilus influenzae (siehe Abschnitt 4.4).
In allen Phase-III-Studien wurden Infektionen mit verkapselten Bakterien Klebsiella pneumoniae, Klebsiella (nicht anderweitig spezifiziert), Haemophilus influenzae und Neisseria subflava berichtet, wobei letztere bei einem Patienten ein unerwünschtes Ereignis einer Bakteriämie verursachte.
Kinder und Jugendliche
Bei 12 pädiatrischen PNH-Patienten mit einem Körpergewicht von ≥ 40 kg (im Alter von 13 bis 17 Jahren), die an den Studien COMMODORE 1, COMMODORE 2 und COMMODORE 3 teilnahmen, zeigte sich ein ähnliches Sicherheitsprofil wie bei erwachsenen PNH-Patienten. Die mit Crovalimab assoziierten Nebenwirkungen, die bei pädiatrischen PNH-Patienten berichtet wurden, sind Infektionen der oberen Atemwege (16,7 %), Harnwegsinfektionen (16,7 %), Ermüdung (Fatigue) (16,7 %), Fieber (16,7 %), Kopfschmerzen (8,3 %), Reaktionen im Zusammenhang mit einer Infusion (8,3 %) und injektionsbedingte Reaktionen (8,3 %).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51‑59
63225 Langen
Tel: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
anzuzeigen.
Im Fall einer Überdosierung sind die Patienten engmaschig auf Anzeichen oder Symptome von Nebenwirkungen zu überwachen und eine geeignete symptomatische Behandlung ist einzuleiten.
Pharmakotherapeutische Gruppe: Immunsuppressiva, Komplementinhibitoren, ATC-Code: L04AJ07
Wirkmechanismus
Crovalimab ist ein rekombinanter humanisierter monoklonaler Antikörper auf der Basis von Immunglobulin G1 (IgG1), der mit hoher Affinität spezifisch an das Komplementprotein C5 des Komplementsystems bindet, dessen Spaltung in C5a und C5b hemmt und so die Bildung des Membranangriffskomplexes (MAC, membrane attack complex) verhindert. Crovalimab verursacht eine Hemmung der terminalen Komplementaktivität. Bei Patienten mit PNH hemmt Crovalimab die terminale komplementvermittelte intravaskuläre Hämolyse.
Pharmakodynamische Wirkungen
In klinischen Studien mit PNH-Patienten wurde nach der Behandlung mit Crovalimab eine konzentrationsabhängige Hemmung der terminalen Komplementaktivität beobachtet. Die Inhibition der terminalen Komplementaktivität (CH50, gemessen mittels Liposomen-Immunoassay [LIA]) wurde unmittelbar nach Ende der initialen Crovalimab-Infusion erreicht und blieb im Allgemeinen über die Dauer der Crovalimab-Behandlung erhalten. Ebenso sanken die mittleren freien C5-Konzentrationen im Vergleich zum Ausgangswert auf niedrige Werte (< 0,0001 g/l) und blieben während der gesamten Behandlungsphase niedrig.
Die Spiegel von freiem C5 und CH50 waren bei pädiatrischen und erwachsenen Patienten, die mit Crovalimab behandelt wurden, ähnlich.
Klinische Wirksamkeit und Sicherheit
Die Sicherheit und Wirksamkeit von Crovalimab bei Patienten mit PNH wurden in einer Nicht-Unterlegenheitsstudie der Phase III (COMMODORE 2, BO42162) untersucht und durch klinische Nachweise aus zwei weiteren Phase-III-Studien (COMMODORE 3, YO42311 und COMMODORE 1, BO42161) gestützt.
In allen Phase-III-Studien mussten die Patienten entweder innerhalb von 3 Jahren vor Behandlungsbeginn oder innerhalb von 7 Tagen nach Behandlungsbeginn mit Crovalimab gegen Neisseria meningitidis geimpft werden. Patienten, die innerhalb von 2 Wochen vor Beginn der Crovalimab-Therapie oder nach Beginn der Prüfbehandlung geimpft wurden, erhielten prophylaktisch geeignete Antibiotika ab dem Zeitpunkt der ersten Anwendung von Piasky bis mindestens 2 Wochen nach der Impfung (siehe Abschnitt 4.4 zu Warnhinweisen und Vorsichtsmaßnahmen im Zusammenhang mit einer schwerwiegenden Meningokokken-Infektion). Patienten mit einer Neisseria meningitidis-Infektion in der Vorgeschichte in den 6 Monaten vor dem Screening und bis zur ersten Anwendung des Prüfpräparats wurden ausgeschlossen.
Patienten wurden ebenfalls ausgeschlossen, wenn eine allogene Knochenmarktransplantation in der Vorgeschichte vorlag.
Crovalimab wurde in den Phase-III-Studien in der in Abschnitt 4.2 empfohlenen Dosierung angewendet. Notfalldosen von 340 mg intravenös verabreichtem Crovalimab waren nach Einschätzung der Prüfärzte zulässig, wenn bei einem Patienten Anzeichen und Symptome einer PNH auftraten. Diese Studien waren jedoch nicht darauf ausgelegt, die Auswirkungen einer Notfalldosierung auf die Wirksamkeit von Crovalimab zu untersuchen. Eculizumab wurde gemäß lokaler Verschreibungsinformation verabreicht bzw. wurden in einem Land ohne Zugang zu kommerziell erhältlichem Eculizumab (COMMODORE 2) in den ersten 4 Wochen 600 mg Eculizumab einmal wöchentlich intravenös verabreicht, gefolgt von alle 2 Wochen 900 mg. Notfalldosen von Eculizumab waren in der Studie nicht erlaubt.
Die Phase-III-Studien bestanden aus einer primären Behandlungsphase von 24 Wochen, nach der die Patienten die Option hatten, mit einer Verlängerungsphase fortzufahren/ auf Crovalimab zu wechseln.
Studie bei Komplementinhibitor-naiven Patienten mit PNH
COMMODORE 2 (Studie BO42162)
COMMODORE 2 war eine randomisierte, offene, aktiv kontrollierte, multizentrische Phase-III-Studie zur Beurteilung der Wirksamkeit und Sicherheit von Crovalimab im Vergleich zu Eculizumab bei Patienten mit PNH, die zuvor nicht mit einem C5-Inhibitor behandelt worden waren. 204 Patienten (Körpergewicht ≥ 40 kg) wurden im Verhältnis 2:1 randomisiert und erhielten entweder Crovalimab (n = 135) oder Eculizumab (n = 69). In die Studie wurden zusätzlich 6 pädiatrische Patienten (im Alter von < 18 Jahren und mit einem Körpergewicht von ≥ 40 kg) in einen deskriptiven Studienarm zur Behandlung mit Crovalimab eingeschlossen (siehe Abschnitt 5.1). Geeignete Patienten hatten beim Screening eine hohe Krankheitsaktivität, nachgewiesen durch einen LDH-Wert von ≥ 2 x des oberen Normalwertes (ULN, upper limit of normal) und durch das Vorliegen eines oder mehrerer Anzeichen oder Symptome in Zusammenhang mit PNH in den letzten 3 Monaten: Ermüdung (Fatigue), Hämoglobinurie, Abdominalschmerzen, Kurzatmigkeit (Dyspnoe), Anämie (Hämoglobin < 10 g/dl), schweres unerwünschtes vaskuläres Ereignis (einschließlich Thrombose) in der Vorgeschichte, Dysphagie oder erektile Dysfunktion; oder Erythrozytenkonzentrat (packed red blood cells, pRBC)-Transfusion aufgrund von PNH in der Vorgeschichte.
Die Randomisierung wurde nach dem jüngsten LDH-Wert (≥ 2 bis ≤ 4 x ULN oder > 4 x ULN) und nach der Transfusionsanamnese (0, > 0 bis ≤ 6 oder > 6 Einheiten (pRBC), die innerhalb von 6 Monaten vor der Randomisierung verabreicht wurden) stratifiziert; die jeweiligen Stratifizierungskategorien waren zwischen den Behandlungsarmen ausgewogen.
Die demographischen Daten und Charakteristika der randomisierten Studienpopulation zu Studienbeginn waren zwischen den Behandlungsarmen im Allgemeinen ausgewogen und werden in Tabelle 3 dargestellt.
Tabelle 3: Demographische Merkmale und Ausgangscharakteristika für COMMODORE 2 (randomisierte Population)
Parameter |
Crovalimab |
Eculizumab |
Alter (Jahre) bei PNH-Diagnose |
||
Mittelwert (SD) |
35,8 (15,5) |
37,4 (16,4) |
Alter (Jahre) bei der ersten Verabreichung der Studienbehandlung* |
||
Mittelwert (SD) |
40,5 (15,2) |
41,9 (16,0) |
< 18 Jahre (n, %) |
0 |
2 (2,9 %) |
Gewicht |
131 (97,0 %) |
66 (95,7 %) |
Geschlecht |
||
Männlich, (n, %) |
77 (57,0 %) |
35 (50,7 %) |
LDH-Spiegel bei Baseline (x ULN) |
||
Median (Bereich) |
7,0 (2,0 – 16,3) |
7,7 (2,0 – 20,3) |
Transfusionsbedarf in den 12 Monaten vor dem Screening in der Vorgeschichte |
103 (77,4 %) |
50 (73,5 %) |
Einheiten von pRBC in den 12 Monaten vor dem Screening |
||
Median (Bereich) |
3,8 (0 – 43,5) |
3,0 (0 – 41,0) |
PNH-Granulozyten-Gesamtklongröße (%) |
91,4 (5,8 – 100) |
93,6 (6,8 – 99,9) |
PNH-Monozyten-Gesamtklongröße (%) |
90,9 (42,5 – 99,9) |
95,1 (41,5 – 99,9) |
PNH-Erythrozyten-Gesamtklongröße (%) |
25,3 (3,5 – 96,0) |
44,6 (0,1 – 88,9) |
Hämoglobinwerte bei Baseline (g/l) |
85,0 (77,0 – 93,0) |
87,0 (81,0 – 97,0) |
Aplastische Anämie in der Vorgeschichte |
||
Ja (n, %) |
53 (39,3 %) |
26 (37,7 %) |
Myelodysplastisches Syndrom in der Vorgeschichte |
||
Ja (n, %) |
6 (4,4 %) |
6 (8,7 %) |
Schweres unerwünschtes vaskuläres Ereignis in der Vorgeschichte (MAVE) |
||
Ja (n, %) |
21 (15,6 %) |
10 (14,5 %) |
Arzneimittel zur Baseline** |
35 (25,9 %) |
17 (24,6 %) |
PNH-bezogene Anzeichen oder Symptome innerhalb von 3 Monaten vor dem Screening |
21 (15,6 %) |
11 (15,9 %) |
Hinweis: IQR = Interquartilbereich (interquartile range).
* Zwei jugendliche Patienten (beide 17 Jahre alt) wurden vor der Eröffnung des separaten deskriptiven pädiatrischen Arms in den Eculizumab-Arm randomisiert. Beide Patienten wechselten in der Verlängerungsphase nach Abschluss der Primärbehandlungsphase zu Crovalimab; ein Patient war noch < 18 Jahre alt, während der andere Patient zum Zeitpunkt der ersten Crovalimab-Behandlung 18 Jahre alt geworden war. Siehe unten „Kinder und Jugendliche“.
** Umfasst Arzneimittel, die vor Beginn der Studienbehandlung begonnen wurden und entweder vor Beginn der Studienbehandlung abgesetzt wurden oder zum Zeitpunkt der Einleitung der Studienbehandlung noch im Einsatz waren.
Das primäre Ziel der Studie war die Beurteilung der Wirksamkeit von Crovalimab im Vergleich zu Eculizumab, basierend auf der Beurteilung der Nicht-Unterlegenheit (NI) der folgenden co-primären Endpunkte: Hämolyse-Kontrolle, gemessen anhand des mittleren Anteils von Patienten mit LDH ≤ 1,5 x oberer Normalwert (ULN) von Woche 5 bis Woche 25; und der Anteil von Patienten, bei denen eine Transfusion vermieden werden konnte, definiert als Patienten, die von Baseline bis Woche 25 frei von pRBC-Transfusionen waren. Sekundäre Wirksamkeitsendpunkte waren der Anteil an Patienten mit Durchbruchhämolyse, der Anteil an Patienten mit stabilisiertem Hämoglobin und die Veränderung der Ermüdung (Fatigue) (gemessen anhand der FACIT [Functional Assessment of Chronic Illness Therapy]-Fatigue-Skala ) von Baseline bis Woche 25.
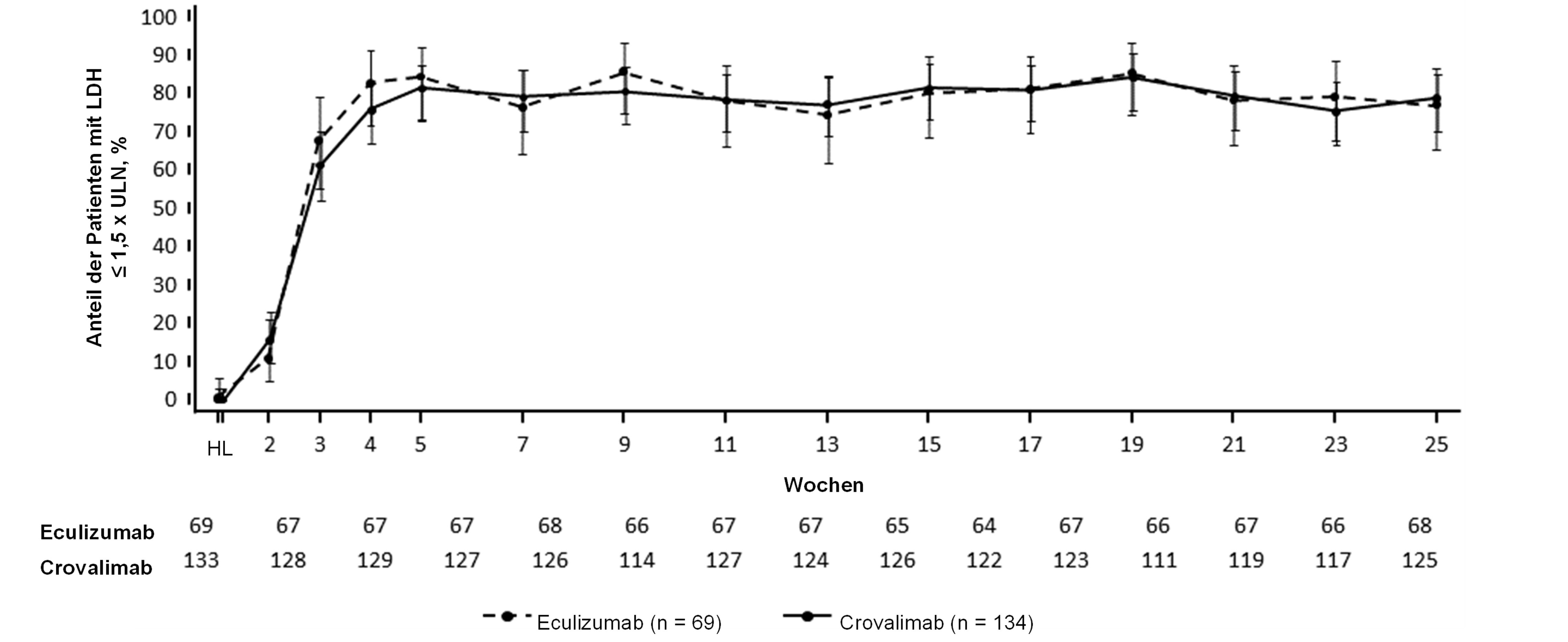
Crovalimab war im Vergleich zu Eculizumab hinsichtlich der beiden co-primären Endpunkte Hämolysekontrolle und Vermeidung einer Transfusion sowie hinsichtlich der sekundären Endpunkte Hämoglobinstabilisierung und Durchbruchhämolyse nicht unterlegen (Abbildung 1). Abbildung 2 zeigt den Anteil der Patienten mit einem LDH-Wert ≤ 1,5 × ULN von Studienbeginn (Baseline) bis Woche 25.
Abbildung 1: Ergebnisse für co-primäre und sekundäre Endpunkte in (COMMODORE 2, Population der Primäranalyse)
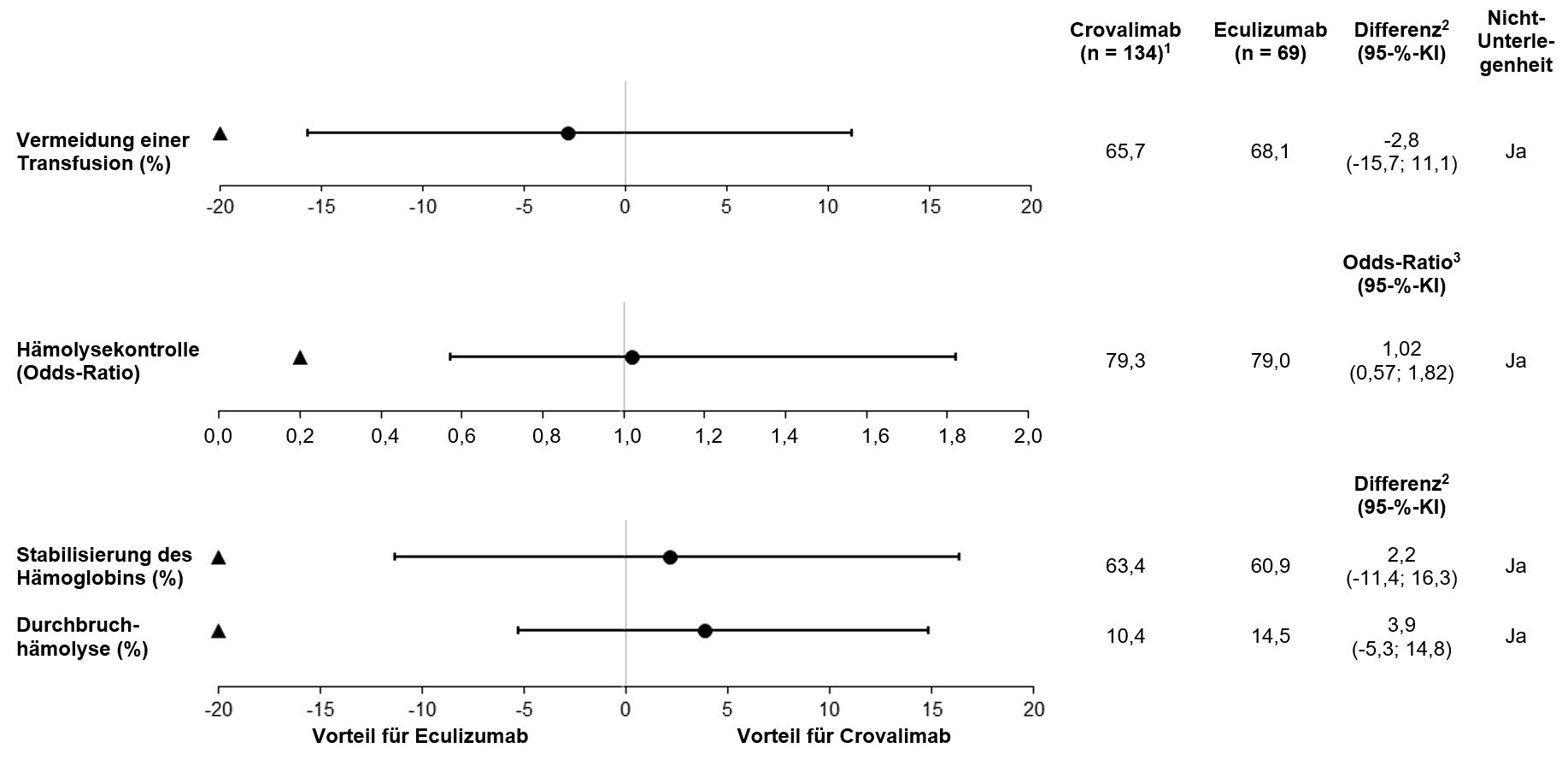
Hinweis: Die Dreiecke zeigen die Nicht-Unterlegenheitsgrenzen an, die Kreise zeigen Punktschätzungen an. KI = Konfidenzintervall.
1 Ein in die Crovalimab-Gruppe randomisierter Patient hatte keine Post-Baseline-LDH und wurde nicht in die primäre Wirksamkeitsanalyse eingeschlossen
2 Für die Vermeidung von Transfusionen und die Hämoglobinstabilisierung wird die Differenz als gewichtete Differenz von Crovalimab minus Eculizumab berechnet. Für die Durchbruchhämolyse wird die Differenz als gewichtete Differenz von Eculizumab minus Crovalimab berechnet.
3 Odds-Ratio berechnet als Odds für Crovalimab dividiert durch Odds für Eculizumab
Abbildung 2: Anteil der Patienten mit LDH ≤ 1,5 x ULN von Baseline bis Woche 25, mit 95‑%‑KI (COMMODORE 2, Population der Primäranalyse)
Studie bei PNH-Patienten, die zuvor mit einem C5-Komplementinhibitor behandelt wurden
COMMODORE 1 (Studie BO42161) randomisierte Eculizumab-Wechsel-Patienten
COMMODORE 1 war eine randomisierte, offene, multizentrische Phase-III-Studie mit aktiver Kontrolle zur Untersuchung der Sicherheit, Pharmakodynamik, Pharmakokinetik und explorativen Wirksamkeit von Crovalimab bei Patienten, die von einer Therapie mit einem anderen C5-Inhibitor umgestellt wurden. Das primäre Ziel der Studie war die Bewertung der Sicherheit (siehe Abschnitt 4.8). Neunundachtzig Patienten wurden im Verhältnis 1:1 randomisiert und erhielten entweder Crovalimab (n = 45) oder Eculizumab (n = 44). Die Patienten waren für die Aufnahme in die randomisierten Studienarme geeignet, wenn sie von den zugelassenen Eculizumab-Dosen umgestellt wurden und beim Screening eine Hämolysekontrolle aufwiesen, definiert durch einen LDH-Wert ≤ 1,5 x ULN. Patienten wurden ausgeschlossen, wenn bei ihnen innerhalb der 6 Monate vor der ersten Verabreichung des Prüfpräparats ein schweres unerwünschtes vaskuläres Ereignis (Major Adverse Vascular Event, MAVE) aufgetreten war. Die Randomisierung erfolgte stratifiziert nach Transfusionshistorie des Patienten (ob ein Patient innerhalb von 12 Monaten vor der Randomisierung eine Transfusion von pRBCs erhalten hatte).
Die demographischen Merkmale und Ausgangscharakteristika der randomisierten Studienpopulation waren zwischen den Behandlungsarmen ausgeglichen. Der mediane LDH-Wert bei Baseline betrug 1,01 x ULN (Bereich: 0,6 – 1,7) für Crovalimab und 0,96 x ULN (Bereich: 0,7 – 1,9) für Eculizumab. Der Anteil an Patienten mit einer Vorgeschichte von Transfusionen in den 12 Monaten vor dem Screening betrug 22,7 % im Crovalimab-Arm und 25 % im Eculizumab-Arm, mit einem Mittelwert (SD) von 1,6 (3,7) bzw. 2,3 (5,4) Einheiten transfundierter pRBC im Crovalimab- bzw. Eculizumab‑Arm. Der Baseline-Mittelwert (Bereich) der PNH-Klongrößen für die Gesamtzahl der Erythrozyten, Monozyten und Granulozyten im Crovalimab-Arm ist im Vergleich zum Eculizumab jeweils wie folgt: 44,6 % (2,6 – 100) gegenüber 54,2 % (1,3 – 100), 88,6 % (13,8 – 100) gegenüber 96,4 % (7,6 – 99,9) und 88,1 % (5,2 – 100) gegenüber 95,7 % (7,9 – 99.9).
Bei 76 von 89 randomisierten Patienten (n = 39 für Crovalimab und n = 37 für Eculizumab), die mindestens 24 Wochen vor dem Cut-Off-Datum der Primäranalyse in die Studie aufgenommen wurden, wurde die Wirksamkeit exploratorisch untersucht. Insgesamt zeigten die Ergebnisse der explorativen Wirksamkeitsendpunkte, dass bei Patienten, die von Eculizumab auf Crovalimab umgestellt wurden, die Krankheitskontrolle aufrechterhalten wurde. Der mittlere Anteil der Patienten, die von Studienbeginn bis Woche 25 eine Kontrolle der Hämolyse beibehielten, betrug 92,9 % [95‑%‑KI: 86,6; 96,4] bei Patienten, die in die Crovalimab-Gruppe randomisiert wurden, und 93,7 % [95‑%‑KI: 87,3; 97,0] bei Patienten, die in die Eculizumab-Gruppe randomisiert wurden. Eine Vermeidung der Transfusion wurde bei 79,5 % [95‑%‑KI: 63,1; 90,1] der Patienten, die zu Crovalimab randomisiert wurden, und bei 78,4 % [95‑%‑KI: 61,3; 89,6] der Patienten, die zu Eculizumab randomisiert wurden, beobachtet.
COMMODORE 1 (Studie BO42161) und COMMODORE 2 (Studie BO42162) - klinisch stabile Wechsel-Patienten
Unterstützende Daten bei klinisch stabilen Patienten, die auf Eculizumab umgestellt wurden, wurden von Patienten in COMMODORE 1 (25 Patienten, deren Wirksamkeit ausgewertet werden konnte) und COMMODORE 2 (29 Patienten, deren Wirksamkeit ausgewertet werden konnte) berichtet, die in der Primärbehandlung mindestens 24 Wochen lang mit Eculizumab behandelt wurden und bei der Umstellung auf Crovalimab einen LDH-Wert ≤ 1,5 x ULN aufwiesen.
Die Wirksamkeit wurde bei den Patienten bewertet, die mindestens 24 Wochen lang mit Crovalimab behandelt worden waren (oder die Behandlung vor Ablauf der 24 Wochen abgebrochen hatten). Der mittlere Anteil der klinisch stabilen Wechsel-Patienten, bei denen die Hämolyse vom Beginn der Wechsel-Behandlung bis zum Wechsel in Woche 25 unter Kontrolle blieb, betrug bei COMMODORE 1 und COMMODORE 2 98,7 % [95-%-KI: 96,2; 99,5] bzw. 95,3 % [95-%-KI: 89,5; 97,9]. Bei 80,0 % [95-%-KI: 58,70; 92,39] bzw. 86,2 % [95-%-KI: 67,43; 95,49] der klinisch stabilen Wechsel-Patienten wurde eine Transfusionsvermeidung beobachtet. Diese Ergebnisse bei klinisch stabilen Eculizumab-Wechsel-Patienten stimmten mit den Ergebnissen bei den randomisierten Eculizumab-Wechsel- Patienten während des primären Behandlungszeitraums von COMMODORE 1 überein.
Des Weiteren konnte im nicht-randomisierten Arm von COMMODORE 1, von den 19 klinisch stabilen Patienten, die von Ravulizumab umgestellt wurden, bei 95,8% [95-%-KI: 89,11; 98,43] die Hämolyse unter Kontrolle gehalten werden. Bei 57,9% [95-%-KI: 33,97; 78,88] der Patienten konnten von Studienbeginn bis Woche 25 Transfusionen vermieden werden.
Immunogenität
Wie bei allen therapeutischen Proteinen besteht die Möglichkeit einer Immunantwort auf Crovalimab.
Die Ergebnisse von Immunogenitätstests hängen stark von mehreren Faktoren ab, darunter Assay-Sensitivität und -Spezifität, Assay-Methodik, Probenhandhabung, Zeitpunkt der Probenentnahme, Begleitmedikationen und Grunderkrankung. Aus diesen Gründen kann ein Vergleich der Inzidenz von Antikörpern gegen Crovalimab mit der Inzidenz von Antikörpern gegen andere Produkte irreführend sein.
In der Phase-III-Studie COMMODORE 2 wurden bei 35,0 % (49/140) der behandlungsnaiven Patienten, die Crovalimab erhielten, und bei 38,2 % (26/68) der Patienten, die von einer Behandlung mit einem anderen C5-Inhibitor zu Crovalimab wechselten, behandlungsbedingte Anti-Wirkstoff-Antikörper (antidrug antibodies, ADAs) beobachtet. Die mediane Zeit bis zur Entwicklung der ersten ADA nach Baseline betrug bei den nicht vorbehandelten Patienten 16,1 Wochen (Bereich: 1,1 bis 72,3 Wochen) bzw. 16,6 Wochen (Bereich: 2,1 bis 36,3 Wochen) bei den Patienten, die zuvor mit einem anderen C5‑Inhibitor behandelt worden waren. In allen Phase-III-Studien betrug die Inzidenz behandlungsbedingter ADA bei nicht vorbehandelten Patienten 35,1 % (67 von 191 Patienten) bzw. 25,4 % (51 von 201 Patienten) bei Patienten, die von einer Behandlung mit einem anderen C5‑Inhibitor auf Crovalimab umgestellt wurden.
In allen Phase-III-Studien waren die medianen Konzentrations-Zeit-Verläufe bei ADA-positiven Patienten etwas geringer als bei ADA-negativen Patienten. Trotz dieser Wirkung blieben die Konzentrationen bei mehr als 80 % der ADA-positiven Patienten über 100 µg/ml (dem Grenzwert für die vollständige terminale Komplementinhibition). Das Vorhandensein von ADA war bei den meisten Patienten nicht mit klinisch relevanten Auswirkungen auf die Pharmakokinetik, Pharmakodynamik und Wirksamkeit verbunden. Bei 23 von 392 auf den ADA-Status untersuchten Patienten (5,9 %) wurde jedoch ein teilweiser oder vollständiger Verlust der mit dem Auftreten von ADA verbundenen Exposition beobachtet; von diesen zeigten 17 (4,3 %) Patienten mit ADA-positivem Ansprechen einen Verlust der pharmakologischen Aktivität (basierend auf CH50 oder freiem C5), der mit einem Verlust der Exposition und einem Verlust der Wirksamkeit zusammenfiel, der sich bei 7 Patienten (1,8 %) als erlittener Verlust der Hämolysekontrolle manifestierte. Es gab keine Hinweise auf klinische Auswirkungen des ADA-Status auf das Sicherheitsprofil von Piasky (siehe Abschnitte 4.4 und 4.8).
Kinder und Jugendliche
Zehn pädiatrische Patienten (mit einem Körpergewicht von ≥ 40 kg), die in COMMODORE 2 (n = 7; 13 – 17 Jahre alt) und COMMODORE 3 (n = 3; 15 – 17 Jahre alt) mit Crovalimab behandelt wurden, waren hinsichtlich der Wirksamkeit auswertbar.
Neun Patienten waren behandlungsnaiv und ein Patient wechselte in der Verlängerungsphase von Eculizumab zu Crovalimab. Alle pädiatrischen Patienten erhielten, basierend auf dem Körpergewicht, die gleiche Dosierung wie erwachsene Patienten. Alle 9 behandlungsnaiven Patienten erreichten bis Woche 4 eine Hämolysekontrolle (definiert als LDH ≤ 1,5 x oberer Normalwert), und diese wurde bei 7 Patienten bei jedem Besuch von Baseline bis Woche 25 aufrechterhalten; der Patient, der von Eculizumab zu Crovalimab wechselte, behielt die Hämolysekontrolle über 24 Behandlungswochen in der Verlängerungsphase bei. Bei sieben der zehn pädiatrischen Patienten wurde eine Vermeidung der Transfusion und eine Hämoglobinstabilisierung erreicht, und kein Patient erlitt während der 24-wöchigen Behandlungsphase eine Durchbruchhämolyse.
Insgesamt war der Behandlungseffekt von Crovalimab bei pädiatrischen PNH-Patienten vergleichbar mit dem, der bei erwachsenen PNH-Patienten beobachtet wurde.
Die Europäische Arzneimittel-Agentur hat für Piasky eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen mit PNH gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Pharmakokinetik von Crovalimab wurde sowohl bei gesunden Probanden als auch bei Patienten mit PNH untersucht. Die Pharmakokinetik wurde anhand von nichtlinearen gemischten pharmakokinetischen Analysemethoden bestimmt, die auf einer gepoolten Datenbasis von 9 gesunden Probanden sowie 210 bzw. 211 behandlungsnaiven Patienten und Patienten, die von einer vorherigen Behandlung mit einem anderen C5-Inhibitor auf Crovalimab umgestellt wurden, basieren.
Der Konzentrations-Zeit-Verlauf von Crovalimab lässt sich am besten anhand eines offenen Zwei-Kompartimentmodells mit Elimination erster Ordnung und einer subkutanen Resorptionskonstante erster Ordnung beschreiben. Zur Beschreibung des vorübergehenden Anstiegs der Clearance aufgrund der Bildung von Immunkomplexen, der bei Patienten beobachtet wurde, die von der Behandlung mit einem anderen C5-Inhibitor auf Crovalimab umgestellt wurden, wurde ein zusätzlicher zeitvariabler Clearance-Parameter hinzugefügt, der mit der Zeit exponentiell abnimmt. Im Steady-State ist zu erwarten, dass die Exposition zwischen nicht vorbehandelten Patienten und Patienten, die auf eine andere Behandlung umgestellt werden, vergleichbar ist.
Resorption
Die Resorptionsratenkonstante wurde auf 0,126 Tag-1 [CV %: 38,3] geschätzt. Nach subkutaner Anwendung wurde die Bioverfügbarkeit auf 83,0 % [CV %: 116] geschätzt.
Verteilung
Das zentrale Verteilungsvolumen wurde auf 3,23 l [CV %: 22,4] und das periphere Verteilungsvolumen auf 2,32 l [CV %: 70,6] geschätzt.
Das geringe Verteilungsvolumen deutet darauf hin, dass Crovalimab wahrscheinlich hauptsächlich im Serum und/oder in gefäßreichem Gewebe verteilt wird.
Biotransformation
Der Metabolismus von Crovalimab wurde nicht direkt untersucht. IgG-Antikörper werden hauptsächlich durch lysosomale Proteolyse katabolisiert und dann vom Körper ausgeschieden oder wiederverwendet.
Elimination
Die Clearance wurde auf 0,0791 l/Tag [CV %: 20,6] geschätzt. Die terminale Halbwertszeit von Crovalimab wurde auf 53,1 Tage [CV %: 39,9] geschätzt, was im Vergleich zu anderen humanisierten IgG-Antikörpern länger ist. Diese lange Halbwertszeit wird durch die Recycling-Eigenschaften von Crovalimab erreicht.
Besondere Patientengruppen
Es wurden keine pharmakokinetischen Studien mit Crovalimab bei besonderen Patientengruppen durchgeführt. Das Körpergewicht ergab sich als signifikante Kovariante, wobei die Clearance und das Verteilungsvolumen zunahmen und die Crovalimab-Exposition mit zunehmendem Körpergewicht abnahm. Daher basiert die Dosierung von Crovalimab auf dem Körpergewicht des Patienten (siehe Abschnitt 4.2).
Nach Einbeziehung des Körpergewichts in das Modell zeigten populationspharmakokinetische Analysen bei Patienten mit PNH, dass Alter (13 – 85 Jahre) und Geschlecht die Pharmakokinetik von Crovalimab nicht wesentlich beeinflussten. Eine weitere Dosisanpassung ist nicht erforderlich.
Es konnte auch gezeigt werden, dass die ethnische Zugehörigkeit keinen Einfluss auf die Pharmakokinetik von Crovalimab hat; die Daten für Patienten mit dunkler Hautfarbe sind jedoch begrenzt und werden daher für diese Population nicht als ausreichend erachtet.
Ältere Patienten
Es wurden keine speziellen Studien zur Untersuchung der Pharmakokinetik von Crovalimab bei Patienten im Alter von ≥ 65 Jahren durchgeführt, jedoch wurden 46 (10,9 %) ältere PNH-Patienten in klinische Studien eingeschlossen, darunter 35 Patienten im Alter von 65 – 74 Jahren, 10 Patienten im Alter von 75 – 84 Jahren und 1 Patient im Alter von ≥ 85 Jahren. Die in klinischen Studien zu PNH erhaltenen Daten weisen darauf hin, dass die Exposition bei Patienten im Alter von ≥ 65 Jahren mit der von jüngeren Patienten in anderen Altersgruppen vergleichbar ist. Aufgrund der begrenzten Daten bei Patienten im Alter von ≥ 85 Jahren ist die Pharmakokinetik von Crovalimab bei diesen Patienten jedoch nicht bekannt.
Nierenfunktionsstörung
Es wurden keine speziellen Studien zur Untersuchung der Pharmakokinetik von Crovalimab bei Patienten mit Nierenfunktionsstörung durchgeführt. Die in klinischen PNH-Studien erhobenen Daten (62 [14,7 %] Patienten mit leichter Nierenfunktionsstörung, 38 [9 %] Patienten mit mäßiger Nierenfunktionsstörung und 4 [1 %] Patienten mit schwerer Nierenfunktionsstörung) weisen jedoch darauf hin, dass die Exposition bei Patienten mit leichter, mäßiger oder schwerer Nierenfunktionsstörung vergleichbar ist mit der von Patienten ohne Nierenfunktionsstörung. Für Patienten mit schwerer Nierenfunktionsstörung wurden in klinischen Studien zu PNH jedoch nur begrenzte Daten erhoben.
Leberfunktionsstörung
Es wurden keine speziellen Studien bei Patienten mit Leberfunktionsstörung durchgeführt, jedoch deuten Daten aus klinischen Studien zu PNH darauf hin, dass die Exposition bei Patienten mit leichter Leberfunktionsstörung (46 [11 %]), gestaffelt nach dem Alanin-Aminotransferase-Spiegel, vergleichbar ist mit der von Patienten ohne Leberfunktionsstörung. Es lagen nur begrenzte pharmakokinetische Daten bei PNH-Patienten mit mäßiger (0 [0 %]) bis schwerer (1 [0,23 %]) Leberfunktionsstörung vor, daher sind die Auswirkungen einer mäßigen oder schweren Leberfunktionsstörung auf die Pharmakokinetik von Crovalimab nicht bekannt, und es kann keine Dosisempfehlung gegeben werden (siehe Abschnitt 4.2).
Kinder und Jugendliche
Die Daten, die bei 12 pädiatrischen Patienten (13 – 17 Jahre) in den klinischen PNH-Studien gewonnen wurde, zeigen, dass die Exposition bei pädiatrischen Patienten ab 12 Jahren mit einem Gewicht von 40 kg und mehr mit der von erwachsenen Patienten vergleichbar war.
Basierend auf den konventionellen Studien zur Toxizität bei wiederholter Gabe, sowie Reproduktions- und Entwicklungstoxizität lassen die präklinischen Daten keine besonderen Gefahren im Zusammenhang mit einer Crovalimab-Behandlung für den Menschen erkennen.
Genotoxizität
Es wurden keine spezifischen Studien zum Nachweis des genotoxischen Potenzials von Crovalimab durchgeführt.
Es wird nicht erwartet, dass monoklonale Antikörper direkt mit DNA oder anderem Chromosomenmaterial interagieren.
Kanzerogenität
Es wurden keine Studien zum Nachweis des kanzerogenen Potenzials von Crovalimab durchgeführt. Die Bewertung der verfügbaren Evidenz in Bezug auf pharmakodynamische Wirkungen und toxikologische Daten bei Tieren deutet nicht auf ein kanzerogenes Potenzial von Crovalimab hin.
Reproduktions- und Entwicklungstoxizität
Die wiederholte Gabe von Crovalimab an trächtige Cynomolgus-Affen während der Trächtigkeit verursachte keine maternale Toxizität und hatte keinen Einfluss auf den Ausgang der Schwangerschaft. Während der 6-monatigen postnatalen Phase wurden keine Auswirkungen auf die Lebensfähigkeit, das Wachstum und die Entwicklung der Jungen beobachtet.
Fertilität
Bei Cynomolgus-Affen wurden nach wiederholter Gabe von Crovalimab über einen Zeitraum von bis zu 6 Monaten keine Auswirkungen auf die weiblichen oder männlichen Fortpflanzungsorgane beobachtet. Separate tierexperimentelle Fertilitätsstudien wurden mit Crovalimab nicht durchgeführt.
Histidin
Aspartinsäure
Argininhydrochlorid
Poloxamer 188
Wasser für Injektionszwecke
Das Arzneimittel darf, außer mit den unter Abschnitt 6.6 aufgeführten, nicht mit anderen Arzneimitteln gemischt werden.
Ungeöffnete Durchstechflasche
3 Jahre
Vor der Verabreichung können ungeöffnete Durchstechflaschen von Piasky bei Bedarf außerhalb des Kühlschranks bei Raumtemperatur gelagert und dann wieder gekühlt werden. Bei Temperaturabweichungen außerhalb von 2 °C bis 8 °C, kann die ungeöffnete Durchstechflasche im Umkarton für einen kumulativen Zeitraum bis zu 7 Tage bei Raumtemperatur (bei bis zu 30 °C) aufbewahrt werden. Entsorgen Sie sie, wenn sie länger als 7 Tage außerhalb des Kühlschranks bei Raumtemperatur aufbewahrt wurde.
Verdünnte Lösung zur intravenösen Infusion
Unter mikrobiologischen Gesichtspunkten muss die verdünnte Lösung zur intravenösen Infusion sofort verwendet werden, es sei denn, die Verdünnungsmethode schließt das Risiko einer mikrobiellen Kontamination aus. Wird es nicht sofort verwendet, liegt die Verantwortung für die Aufbewahrungszeit und die Lagerungsbedingungen beim Anwender.
Wenn die verdünnte Lösung unter kontrollierten und validierten aseptischen Bedingungen zubereitet wird, kann das Arzneimittel im Kühlschrank bei 2 °C bis 8 °C und bei Raumtemperatur (bis zu 30 °C) gelagert werden. Detaillierte Lagerungsbedingungen der zubereiteten Infusionslösung, abhängig von der Art des verwendeten Infusionsbeutels, sind in Tabelle 4 aufgeführt.
Tabelle 4: Aufbewahrungsbedingungen für die unter aseptischen Bedingungen zubereitete Infusionslösung
Infusionsbeutel |
Lagerungsbedingungen |
PO/PE/PP |
Bis zu 30 Tage bei 2 °C bis 8 °C lichtgeschützt und bis zu 24 Stunden bei Raumtemperatur (bis 30 °C) unter Umgebungslichtbedingungen. |
PVC |
Bis zu 12 Stunden bei 2 °C bis 8 °C lichtgeschützt und bis zu 12 Stunden bei Raumtemperatur (bis 30 °C) unter Umgebungslichtbedingungen. |
Polyolefine (PO), Polyethylen (PE), Polypropylen (PP), Polyvinylchlorid (PVC)
Unverdünnte Lösung zur subkutanen Injektion
Unter mikrobiologischen Gesichtspunkten muss das Arzneimittel sofort verwendet werden. Wenn es nicht sofort verwendet wird, liegen die Lagerungszeiten und -bedingungen vor der Verwendung in der Verantwortung des Anwenders und sollten normalerweise nicht länger als 24 Stunden bei 2 °C bis 8 °C betragen, es sei denn, die Zubereitung erfolgte unter kontrollierten und validierten aseptischen Bedingungen.
Wenn Piasky unter kontrollierten und validierten aseptischen Bedingungen aus der Durchstechflasche in die Spritze aufgezogen wird, kann das Arzneimittel in der verschlossenen Spritze im Kühlschrank bei 2 °C bis 8 °C für bis zu 14 Tage lichtgeschützt und bei Raumtemperatur (bis zu 30 °C) für bis zu 24 Stunden bei Umgebungslicht aufbewahrt werden.
Die Lösung von Piasky muss vor direkter Sonneneinstrahlung geschützt werden.
Ungeöffnete Durchstechflasche
Im Kühlschrank lagern (2 °C – 8 °C).
Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen der verdünnten Lösung zur intravenösen Infusion und der unverdünnten Lösung zur subkutanen Injektion, siehe Abschnitt 6.3.
Injektionslösung in einer 2-ml-Durchstechflasche (Typ-I-Glas) zum einmaligen Gebrauch mit einem Stopfen (Gummi) und einer Versiegelung (Aluminium).
Jede Packung enthält 1 Durchstechflasche.
Die Durchstechflasche mit Piasky ist nur zum einmaligen Gebrauch bestimmt.
Piasky wird verdünnt zur intravenösen Infusion oder unverdünnt zur subkutanen Injektion angewendet.
Piasky muss vor der Anwendung visuell überprüft werden, um sicherzustellen, dass keine Partikel oder Verfärbungen vorhanden sind. Piasky ist eine klare bis stark opaleszente und nahezu farblose bis bräunlich-gelbe Lösung. Piasky muss entsorgt werden, wenn das Arzneimittel trüb oder verfärbt aussieht oder Partikel enthält.
Intravenöse Verabreichung
Piasky muss von medizinischem Fachpersonal unter aseptischer Technik zubereitet werden. Die Lösung von Piasky muss vor der Anwendung mit Natriumchlorid-Infusionslösung 9 mg/ml (0,9 %) verdünnt werden. Während der Anwendung muss ein 0,2-μm-Inline-Filter mit dem Infusionsset verwendet werden.
Während der intravenösen Anwendung muss ein dafür vorgesehener Infusionsschlauch verwendet werden.
Verdünnung
1. Mit einer sterilen Spritze das erforderliche Volumen von Piasky aus der Durchstechflasche entnehmen (siehe Tabelle 5) und im Infusionsbeutel verdünnen. Es müssen mehrere Durchstechflaschen verwendet werden, um die erforderliche Menge an Piasky zu erhalten, die dem Infusionsbeutel hinzugefügt werden muss. Verwerfen Sie in der Durchstechflasche übrig gebliebene rekonstituierte Lösung.
Die Verdünnung von Piasky in Infusionsbeuteln, die Natriumchlorid-Infusionslösung 9 mg/ml (0,9 %) enthalten, muss im Bereich von 4 – 15 mg/ml (Endkonzentration nach Verdünnung) liegen.
Intravenöse Infusionsbeutel mit einem Volumen von 100 ml oder 250 ml können verwendet werden.
Tabelle 5: Dosisbeispiel Volumenbestimmung
Dosis |
Konzentration im Beutel |
Volumen von Piasky in 0,9 %iger Natriumchloridlösung* (ml) |
Größe der Infusionsbeutel |
1 000 |
4 |
5,9 |
250 |
1 500 |
6 |
8,8 |
250 |
1 000 |
10 |
5,9 |
100 |
1 500 |
15 |
8,8 |
100 |
* Jede 340 mg-Durchstechflasche enthält ein nominales Füllvolumen von 2,0 ml.
Den Infusionsbeutel durch langsames Umdrehen vorsichtig mischen. Nicht schütteln.
Den Infusionsbeutel auf Partikel prüfen und falls vorhanden entsorgen.
Das Spülen des Infusionsschlauchs ist erforderlich, um die vollständige Gabe der Gesamtdosis sicherzustellen.
Zwischen Piasky und intravenösen Infusionsbeuteln mit produktberührenden Materialien aus Polyvinylchlorid (PVC) oder Polyolefinen (PO) wie Polyethylen (PE) und Polypropylen (PP) wurden keine Inkompatibilitäten festgestellt. Darüber hinaus wurden keine Inkompatibilitäten mit Infusionssets oder Infusionshilfsmitteln mit produktberührenden Materialien aus PVC, PE, Polyurethan (PU), Polybutadien (PBD), Acrylnitril-Butadien-Styrol (ABS), Polycarbonat (PC) oder Polytetrafluorethylen (PTFE) beobachtet.
Lagerungsbedingungen der Infusionsbeutel, siehe Abschnitt 6.3.
Subkutane Verabreichung
Piasky ist unverdünnt anzuwenden und muss mit aseptischer Technik zubereitet werden. Es werden eine Spritze, eine Transfernadel und eine Injektionsnadel benötigt, um die Piasky Lösung aus der Durchstechflasche aufzuziehen und subkutan zu injizieren.
Jede Injektion hat ein Volumen von 2 ml, entsprechend 340 mg. Für jede Injektion ist eine 2-ml- oder 3-ml-Spritze zu verwenden. Eine Dosis von 680 mg wird durch zwei aufeinanderfolgende subkutane Injektionen von 340 mg erreicht. Eine Dosis von 1 020 mg wird durch drei aufeinanderfolgende subkutane Injektionen von 340 mg erreicht.
2-ml- oder 3-ml-Spritze
Kriterien: durchsichtige Spritze aus Polypropylen oder Polycarbonat mit Luer-Lock™-Spitze (falls nicht lokal verfügbar, kann eine Spritze mit Luer-Slip™-Spitze verwendet werden), steril, zum einmaligen Gebrauch, latexfrei und pyrogenfrei.
Transfernadel
Kriterien: Edelstahl, steril, vorzugsweise Gauge 18 G mit einfacher Abschrägung bei ca. 45 Grad, um das Risiko einer Nadelstichverletzung zu reduzieren, oder Gauge 21 G Standard-Nadel als alternative Einwegnadel, latexfrei und pyrogenfrei. Eine Transfernadel ohne Filter wird empfohlen.
Injektionsnadel
Kriterien: Subkutannadel, Edelstahl, steril, Stärke 25 G, 26 G oder 27 G, Länge 9 bis 13 mm, zum einmaligen Gebrauch, latexfrei und pyrogenfrei, vorzugsweise einschließlich Nadelschutzsystem.
Weitere Informationen zur Anwendung siehe Abschnitt 4.2.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Die folgenden Punkte müssen beim Umgang mit Spritzen und anderen spitzen oder scharfen Medizinutensilien sowie deren Entsorgung strikte Beachtung finden:
Nadeln und Spritzen dürfen niemals erneut oder gleichzeitig bei anderen Personen verwendet werden.
Alle verwendeten Nadeln und Spritzen in einem Entsorgungsbehälter für scharfe und spitze Gegenstände entsorgen.
Roche Registration GmbH
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Deutschland
EU/1/24/1848/001
Datum der Erteilung der Zulassung: 22. August 2024
Oktober 2025
Verschreibungspflichtig
Roche Pharma AG
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Telefon (07624) 14-0
Telefax (07624) 1019
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.