
Pedmarqsi 80 mg/ml Infusionslösung
Jede Durchstechflasche mit 100 ml enthält 8 g Natriumthiosulfat als wasserfreies Salz.
Jeder ml Infusionslösung enthält 80 mg Natriumthiosulfat.
Sonstige(r) Bestandteil(e) mit bekannter Wirkung:
Jeder ml Infusionslösung enthält 0,25 mg Borsäure und 23 mg Natrium.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Infusionslösung
Die Infusionslösung ist klar, farblos und nahezu feststofffrei mit einem pH-Wert von 7,7–9,0 und einer Osmolalität von 980-1200 mOsm/kg.
Pedmarqsi ist angezeigt für die Vorbeugung einer durch eine Cisplatin-Chemotherapie induzierten Ototoxizität bei Patienten im Alter von 1 Monat bis < 18 Jahren mit lokalisierten, nicht metastasierten, soliden Tumoren.
Pedmarqsi ist nur zur Anwendung im Krankenhaus unter fachärztlicher Aufsicht bestimmt.
Dosierung
Die empfohlene Dosis von Natriumthiosulfat zur Prävention einer Cisplatin-induzierten Ototoxizität richtet sich nach dem Körpergewicht und wird gemäß der nachstehenden Tabelle mit der Körperoberfläche normalisiert:
Tabelle 1 Empfohlene Dosis von Natriumthiosulfat zur intravenösen Infusion
Körpergewicht |
Dosis |
Menge |
> 10 kg |
12,8 g/m2 |
160 ml/m2 |
5 bis 10 kg |
9,6 g/m2 |
120 ml/m2 |
< 5 kg |
6,4 g/m2 |
80 ml/m2 |
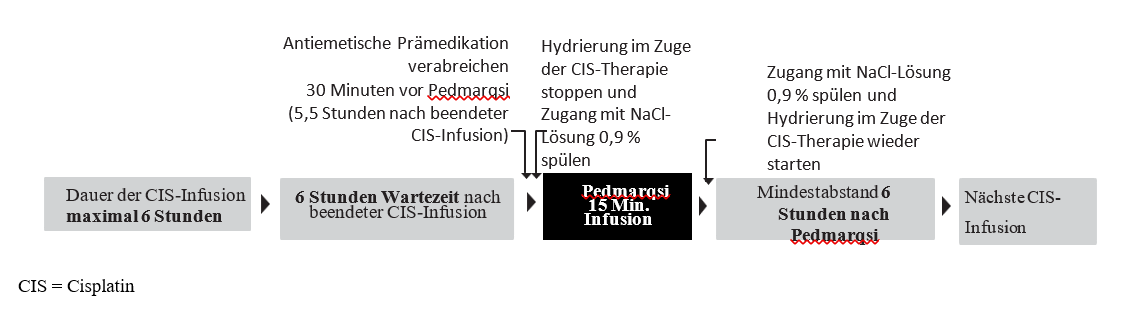
Natriumthiosulfat sollte als 15-minütige intravenöse Infusion 6 Stunden nach Abschluss jeder Cisplatin-Gabe verabreicht werden, wenn die Cisplatin-Infusion nicht länger als 6 Stunden dauert (siehe Art der Anwendung).
Eine Vorbehandlung mit Antiemetika wird empfohlen, um das Auftreten von Übelkeit und Erbrechen zu verringern (siehe Abschnitt 4.4).
Besondere Patientengruppen
Frühgeborene und Neugeborene bis zu einem Alter von weniger als 1 Monat
Natriumthiosulfat ist bei Frühgeborenen und Neugeborenen bis zu einem Alter von weniger als 1 Monat kontraindiziert (siehe Abschnitte 4.3 und 4.4).
Nierenfunktionsstörung
Bei Patienten mit Nierenfunktionsstörung wird keine Dosisanpassung empfohlen (siehe Abschnitt 5.2). Aufgrund des Natriumgehalts von Natriumthiosulfat besteht bei Patienten mit eingeschränkter Nierenfunktion ein erhöhtes Risiko für Nebenwirkungen (siehe Abschnitt 4.4).
Leberfunktionsstörung
Bei Patienten mit Leberfunktionsstörung wird keine Dosisanpassung empfohlen (siehe Abschnitt 5.2).
Art der Anwendung
Zur intravenösen Anwendung.
Aufgrund der hypertonen Formulierung wird eine zentralvenöse Verabreichung empfohlen.
Natriumthiosulfat wird als 15-minütige Infusion verabreicht.
Jede Durchstechflasche ist nur zur einmaligen Anwendung bestimmt.
Zeitpunkt der Verabreichung im zeitlichen Verhältnis zur Gabe von Cisplatin
Der Zeitpunkt der Verabreichung von Natriumthiosulfat im zeitlichen Verhältnis zur Durchführung der Cisplatin-Chemotherapie ist entscheidend.
Die Verabreichung von Natriumthiosulfat:
Weniger als 6 Stunden nach Beendigung der Cisplatin-Infusion: kann die tumorhemmende Wirkung von Cisplatin verringern
Mehr als 6 Stunden nach Beendigung der Cisplatin-Infusion: zeigt für die Vorbeugung von Ototoxizität möglicherweise keine Wirkung
Natriumthiosulfat darf nur nach Cisplatin-Infusionen mit einer Dauer von bis zu 6 Stunden angewendet werden. Natriumthiosulfat darf nicht angewendet werden, wenn
die Cisplatin-Infusion länger als 6 Stunden dauert oder
innerhalb der nächsten 6 Stunden eine weitere Cisplatin-Infusion geplant ist.
Wenn Cisplatin an aufeinanderfolgenden Tagen verabreicht wird, ist nach der Natriumthiosulfat-Infusion ein Mindestabstand von 6 Stunden vor Gabe der nächsten Cisplatin-Infusion einzuhalten.
Nach Beendigung der Cisplatin-Infusion:
30 Minuten vor der Gabe von Natriumthiosulfat ist eine hochwirksame kombinierte intravenöse antiemtische Therapie zu verabreichen, d. h. 5,5 Stunden nach Abschluss der Cisplatin-Infusion (siehe Abschnitt 4.4).
Dieses Arzneimittel ist eine gebrauchsfertige Infusionslösung.
Überprüfen Sie den Inhalt der Durchstechflasche(n) vor der Verabreichung visuell auf Partikel und Verfärbungen.
Die erforderliche Dosis Natriumthiosulfat aus der (den) Durchstechflasche(n) in einer Spritze aufziehen oder in einen leeren, sterilen Infusionsbeutel geben.
Hydrierung im Zuge der Cisplatin-Therapie stoppen und Zugang mit 0,9 %iger NaCl-Lösung spülen.
Natriumthiosulfat über einen Zeitraum von 15 Minuten (6 Stunden nach Abschluss der Cisplatin-Infusion) infundieren.
Zugang mit 0,9 %iger NaCl-Lösung spülen und Hydrierung im Zuge der Cisplatin-Therapie unmittelbar im Anschluss fortsetzen.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Neugeborene unter 1 Monat, aufgrund des Risikos von Hypernatriämie (siehe Abschnitt 4.4).
Überempfindlichkeit
In klinischen Studien nach Verabreichung von Natriumthiosulfat (siehe Abschnitt 4.8) wurden Überempfindlichkeitsreaktionen gemeldet. Zu den Symptomen zählten Hautausschlag, Tachykardie, Schüttelfrost und Dyspnoe.
Natriumthiosulfat kann Spuren von Natriumsulfit enthalten. Es kann in seltenen Fällen verschiedene Überempfindlichkeitsreaktionen und Bronchospasmen hervorrufen. Sulfitempfindlichkeit tritt bei asthmatischen Patienten häufiger auf als bei nicht-asthmatischen Patienten.
Antihistamine (z. B. Diphenhydramin und Steroide) sollten im Falle einer allergischen Reaktion, sofern klinisch indiziert, sofort verfügbar sein. Sollte die Überempfindlichkeitsreaktion des Patienten so ausfallen, dass die Gabe von Natriumthiosulfat nach der nächsten Cisplatin-Infusion fortgesetzt werden kann, sollte eine Prämedikation mit Antihistaminika und Steroiden verabreicht und der Patient sorgfältig beobachtet werden.
Störung des Elektrolytgleichgewichtes
Eine Dosis von 12,8 g/m2 führt zu einer Natriumbeladung von 162 mmol/m2, eine Dosis von 9,6 g/m2 zu einer Natriumbeladung von 121 mmol/m2 und eine Dosis von 6,4 g/m2 zu einer Natriumbeladung von 81 mmol/m2. Elektrolythaushalt und Blutdruck sollten sorgfältig überwacht werden, und Natriumthiosulfat sollte nicht gegeben werden, wenn das Serumnatrium zu Behandlungsbeginn vor der Gabe von Natriumthiosulfat innerhalb eines Behandlungszyklus > 145 mmol/Liter beträgt.
Patienten im Alter von < 1 Monat haben eine weniger gut entwickelte Natriumhomöostase; daher ist Natriumthiosulfat bei Neugeborenen kontraindiziert (siehe Abschnitt 4.3).
Die Magnesium-, Kalium- und Phosphatspiegel im Serum sollten ebenfalls überwacht und bei Bedarf sollten Ergänzungen vorgenommen werden, da die Kombination aus Flüssigkeitsbeladung in Verbindung mit einer Chemotherapie auf Cisplatin-Basis und der Verabreichung von Natriumthiosulfat vorübergehende Elektrolytstörungen verursachen kann.
Übelkeit und Erbrechen
Vorübergehende Zunahmen der Inzidenz und Schwere von Übelkeit und Erbrechen können bei Natriumthiosulfat-Infusion aufgrund der über einen kurzen Zeitraum verabreichten hohen Natriumkonzentrationen beobachtet werden (siehe Abschnitt 4.8). Zusätzlich zu allen prophylaktischen Antiemetika, die vor der Verabreichung von Cisplatin verabreicht werden, sollten in den 30 Minuten vor der Verabreichung von Natriumthiosulfat weitere Antiemetika kombiniert verabreicht werden. Übelkeit und Erbrechen enden tendenziell kurz nach Abschluss der Natriumthiosulfat-Infusion.
Nierenfunktionsstörung
Es ist bekannt, dass Natriumthiosulfat im Wesentlichen über die Nieren ausgeschieden wird (siehe Abschnitt 5.2), und bei Patienten mit eingeschränkter Nierenfunktion kann das Nebenwirkungsrisiko von Natriumthiosulfat höher sein. Da eine Cisplatin-Chemotherapie mit Nierentoxizität einhergeht, sollte die Nierenfunktion überwacht und der Elektrolythaushalt genau beobachtet werden, wenn die glomeruläre Filtrationsrate (GFR) unter 60 ml/min/1,73 m2 fällt.
Sonstige Bestandteile mit bekannter Wirkung
Dieses Arzneimittel enthält 0,25 mg/ml Borsäure als Puffer. Borsäure kann die Fertilität beeinträchtigen, wenn sie dauerhaft in Dosen über 0,2 mg/kg/Tag verabreicht wird. Dieses Arzneimittel wird über einen Zeitraum von 6 Monaten zwischen 6 und 30 Mal intermittierend in Verbindung mit einer Cisplatin-Chemotherapie verabreicht. Zusammen mit der im Trinkwasser enthaltenen Borsäure summiert sich dies je nach Alter und Größe des Kindes auf 0,17–0,22 mg/kg/Tag.
Dieses Arzneimittel enthält 23 mg Natrium pro ml, entsprechend 1,15 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahmemenge mit der Nahrung von 2 g. Dies entspricht auch 1,15–2,1 % der von der Europäischen Behörde für Lebensmittelsicherheit (EFSA) für Kinder im Alter von 1 bis 17 Jahren festgelegten sicheren täglichen Höchstmenge von 1,1–2 g Natrium und 11,5 % der von der EFSA festgelegten sicheren täglichen Höchstmenge von 0,2 g für Säuglinge im Alter von 7 bis 11 Monaten. Dies ist bei Patienten, die eine natriumarme Diät einhalten müssen, zu berücksichtigen.
Natriumthiosulfat sollte frühestens 6 Stunden nach Beendigung einer Cisplatin-Infusion verabreicht werden. Natriumthiosulfat sollte nicht verabreicht werden, wenn die Infusion mit Cisplatin länger als 6 Stunden dauert oder wenn innerhalb der nächsten 6 Stunden eine weitere Infusion mit Cisplatin geplant ist (siehe Abschnitt 4.2). Die verzögerte Verabreichung verhindert eine potenzielle Beeinflussung der tumorhemmenden Wirkung der Cisplatin-Chemotherapie.
Es wurden keine weiteren Studien zur Erfassung von Arzneimittelwechselwirkungen durchgeführt. Relevante pharmakokinetische Wechselwirkungen sind unwahrscheinlich, da Natriumthiosulfat nur selten und nur in Verbindung mit Cisplatin verabreicht wird und Natriumthiosulfat binnen weniger Stunden nach der Verabreichung eliminiert wird. Natriumthiosulfat kann CYP2B6 induzieren (siehe Abschnitt 5.2).
Schwangerschaft
Es liegen keine oder nur eingeschränkte Daten aus der Verwendung von Natriumthiosulfat bei Schwangeren vor. Es liegen keine ausreichenden tierexperimentellen Studien zur Reproduktionstoxizität bei intravenöser Infusion von Natriumthiosulfat vor (siehe Abschnitt 5.3). Aus Vorsichtsgründen wird empfohlen, die Anwendung von Natriumthiosulfat während der Schwangerschaft möglichst zu vermeiden.
Natriumthiosulfat ist nur zur Verabreichung in Verbindung mit einer Cisplatin-Chemotherapie bestimmt. Cisplatin wird während der Schwangerschaft nicht angewendet, es sei denn, der Arzt hält das Risiko bei einer einzelnen Patientin für klinisch gerechtfertigt. Patienten, die Cisplatin erhalten, werden darauf hingewiesen, dass während der Behandlung und für 6 Monate nach der Behandlung mit Cisplatin eine angemessene Kontrazeption erforderlich ist, da Cisplatin embryotoxisch und fetotoxisch ist.
Stillzeit
Es ist nicht bekannt, ob Natriumthiosulfat/Metaboliten in menschlicher Milch ausgeschieden werden. Ein Risiko für Neugeborene/Säuglinge kann nicht ausgeschlossen werden. Aus Vorsichtsgründen wird empfohlen, die Anwendung von Natriumthiosulfat während des Stillens zu vermeiden.
Natriumthiosulfat ist nur zur Verabreichung in Verbindung mit einer Cisplatin-Chemotherapie bestimmt, bei der das Stillen oder Füttern mit Muttermilch kontraindiziert ist.
Fertilität
Zu den Auswirkungen von Natriumthiosulfat auf die Fertilität liegen keine klinischen Daten vor. Es liegen keine ausreichenden Informationen aus tierexperimentellen Studien vor, um die Auswirkungen einer intravenösen Infusion von Natriumthiosulfat auf die Fertilität zu beurteilen.
Natriumthiosulfat ist nur zur Verabreichung in Verbindung mit einer Cisplatin-Chemotherapie bestimmt. Es ist bekannt, dass die Behandlung mit Cisplatin die Fertilität beeinträchtigt.
Dieses Arzneimittel enthält 0,25 mg/ml Borsäure, die bei dauerhafter Verabreichung in Dosen von mehr als 0,2 mg/kg/Tag die Fertilität beeinträchtigen kann (siehe Abschnitt 4.4).
Natriumthiosulfat hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrsfähigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die am häufigsten berichteten Nebenwirkungen mit einer Häufigkeit von ≥ 1 Fall pro 10 Patienten sind Erbrechen (44 %), Übelkeit (23 %), Hypokaliämie (21%), Hypernatriämie (19 %), Hypophosphatämie (18 %) und Überempfindlichkeit (11 %).
Ein schwerwiegender Fall einer Überempfindlichkeit, der zu einem Behandlungsabbruch führte, wurde in einer der klinischen Studien (SIOPEL 6 Studie - Behandlungsarm) beobachtet.
Tabellarische Auflistung der Nebenwirkungen
Die nachstehendeTabelle 2 richtet sich nach der Organklassifikation gemäß MedDRA System Organ Classification (SOC), Preferred Term (PT) Level und der Häufigkeit. Dabei wurden die Häufigkeiten wie folgt aufgeschlüsselt: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1.000, < 1/100), selten (≥ 1/10.000, < 1/1.000), sehr selten (< 1/10.000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb der einzelnen Häufigkeitsgruppen sind die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 2. Nebenwirkungen (aus klinischen Studien) | ||
Systemorganklasse |
Nebenwirkung |
Häufigkeit |
Erkrankungen des Immunsystems |
Überempfindlichkeit |
Sehr häufig (11 %) |
Stoffwechsel- und Ernährungsstörungen |
Hypokaliämie |
Sehr häufig (21 %) |
Hypernaträmie |
Sehr häufig (19 %) |
|
Hypophosphatämie |
Sehr häufig (18 %) |
|
Metabolische Azidose |
Häufig (3 %) |
|
Hypokalzämie |
Häufig (7 %) |
|
Gefäßerkrankungen |
Hypertonie |
Häufig (2 %) |
Hypotonie |
Häufig (2 %) |
|
Erkrankungen des Gastrointestinaltrakts |
Erbrechen |
Sehr häufig (44 %) |
Übelkeit |
Sehr häufig (23 %) |
|
Beschreibung ausgewählter Nebenwirkungen
Übelkeit und Erbrechen
Die Verabreichung von Natriumthiosulfat geht mit einer hohen Inzidenz von Übelkeit und Erbrechen einher. Übelkeit und Erbrechen enden tendenziell kurz nach Beendigung der Natriumthiosulfat-Infusion (siehe Abschnitt 4.4).
Hypernaträmie
Eine Dosis von 12,8 g/m2 führt zu einer Natriumbeladung von 162 mmol/m2, eine Dosis von 9,6 g/m2 zu einer Natriumbeladung von 121 mmol/m2 und eine Dosis von 6,4 g/m2 zu einer Natriumbeladung von 81 mmol/m2. In klinischen Studien führten vergleichbare Natriumthiosulfat-Dosen zu einem leichten, vorübergehenden Anstieg der Natriumspiegel im Serum, unabhängig von Alter, Körperoberfläche, Körpergewicht, Gesamt-Tages-Natriumthiosulfat-Dosis oder Cisplatin-Zyklus. Die Natriumspiegel sinken 18 Stunden bzw. 24 Stunden nach der Verabreichung wieder auf die Ausgangswerte.
Störung des Elektrolytgleichgewichtes
Hypophosphatämie und Hypokaliämie treten nach einer Behandlung mit Natriumthiosulfat sehr häufig auf. Elektrolythaushalt und Blutdruck sollten sorgfältig überwacht werden (siehe Abschnitt 4.4).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das,
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: http://www.bfarm.de
Es kann davon ausgegangen werden, dass zu hohe Dosen von Natriumthiosulfat zu schwerer Übelkeit und Erbrechen sowie zu Elektrolytstörungen, Blutdruckveränderungen und Azidose führen. Die Behandlung einer Überdosierung sollte aus allgemeinen unterstützenden Maßnahmen bestehen, einschließlich der Verabreichung von Flüssigkeiten und der Beobachtung des klinischen Zustands des Patienten. Für eine Überdosierung mit Natriumthiosulfat gibt es kein spezifisches Gegenmittel.
Pharmakotherapeutische Gruppe: Antidote, ATC-Code: V03AB06
Wirkmechanismus
Wie Natriumthiosulfat gegen Ototoxizität schützt, ist noch nicht vollständig geklärt, der Mechanismus kann jedoch steigende Konzentrationen endogener Antioxidantien, die Hemmung des intrazellulären oxidativen Stresses und direkte Wechselwirkungen zwischen Cisplatin und der Thiol-Gruppe in Natriumthiosulfat zur Bildung inaktiver Platinspezies umfassen.
Die gleichzeitige Inkubation von Natriumthiosulfat und Cisplatin verringerte in vitro die Zytotoxizität von Cisplatin auf Tumorzellen; eine verzögerte Zugabe von Natriumthiosulfat zu diesen Kulturen verhinderte diese Schutzwirkung.
Pharmakodynamische Wirkungen
Es sind keine klinischen pharmakodynamischen Informationen verfügbar, die über die Angaben im Abschnitt „Wirkmechanismus“ hinausgehen.
Klinische Wirksamkeit und Sicherheit
Die Wirksamkeit von Natriumthiosulfat (STS) bei der Vorbeugung einer durch Cisplatin (CIS) induzierten Ototoxizität wurde in zwei multizentrischen Studien untersucht, in denen 112 pädiatrische Patienten mit verschiedenen soliden Tumorarten nach jeder Verabreichung von CIS mit STS behandelt wurden. Die Sicherheit wurde durch Gabe von 1 bis 5 Dosen Natriumthiosulfat pro Chemotherapiezyklus ermittelt, wobei die Dosierschemata von einer Dosis CIS+STS pro Zyklus bis zu 5 Dosen CIS+STS pro Zyklus variierten.
Studie 1(SIOPEL 6) – Schlüsselstudie
Studie 1 war eine multizentrische, randomisierte, kontrollierte, unverblindete („open-label“) Studie zur Beurteilung der Wirksamkeit und Sicherheit von STS bei der Verringerung von Ototoxizität bei Kindern, die eine CIS-Chemotherapie für das Standardrisiko Hepatoblastom (SR-HB) erhielten. In die Studie aufgenommen wurden Kinder im Alter von 1 Monat bis 18 Jahren mit histologisch bestätigtem, neu diagnostiziertem HB. Die Kinder wurden im Verhältnis 1:1 randomisiert und erhielten nach jeder CIS-Dosis STS (CIS+STS-Gruppe) oder nur CIS.
Das CIS wurde als 6-stündige intravenöse Infusion verabreicht. Vier CIS-Gaben wurden vor der Operation und zwei weitere Gaben nach der ‑Operation verabreicht.
Der CIS+STS-Gruppe wurde die intravenöse STS-Infusion über einen Zeitraum von 15 Minuten verabreicht, und zwar 6 Stunden nach Beendigung der einzelnen CIS-Infusionen. Die STS-Dosierung wurde wie folgt an das Körpergewicht der Kinder angepasst: Kinder > 10 kg erhielten ein Äquivalent von 12,8 g/m2 STS, Kinder ≥ 5 bis ≤ 10 kg erhielten ein Äquivalent von 9,6 g/m2 STS und Kinder < 5 kg erhielten ein Äquivalent von 6,4 g/m2 STS.
Insgesamt wurden 129 Kinder registriert und 114 Kinder wurden in der Studie randomisiert (61 Patienten in der CIS+STS-Gruppe und 53 Patienten in der Nur-CIS-Gruppe). Von den 114 randomisierten Patienten zogen 5 Patienten ihre Teilnahme vor Aufnahme der Behandlung zurück: 2 Patienten aufgrund des Widerrufs der elterlichen Einwilligung, 2 Patienten aufgrund einer Neueinstufung als Hochrisiko-HB und 1 Patient aufgrund von Nichterfüllung der Teilnahmekriterien.
Als Hörverlust wurde ein Brock-Grad ≥ 1 definiert, der mittels audiologischer Auswertung nach Ende der Studienbehandlung gemessen wurde bzw. im Alter von mindestens 3,5 Jahren, sobald ein zuverlässiges Ergebnis erzielt werden konnte, je nachdem, welcher Zeitpunkt später eintrat. Der Anteil der Kinder mit Hörverlust im Alter von ≥ 3,5 Jahren in der CIS+STS-Gruppe (20 Kinder [35,1 %]) lag im Vergleich mit der Nur-CIS Gruppe (35 Kinder [67,3 %]) nur ungefähr halb so‑ hoch (Tabelle 2). Die ereignisfreie Überlebenszeit (EFS) und die Gesamtüberlebenszeit (OS) wurden ebenfalls untersucht.
Tabelle 3: Zusammenfassung Patientenpopulation und Hörverlust in Studie 1 | ||
Nur CIS |
CIS + STS |
|
Patientenpopulation | ||
N (Intent-to-Treat-Population) |
||
Alter (Jahre), Median (min; max) |
1,1 (0,3; 5,9) |
1,1 (0,1; 8,2) |
Gewicht (kg) (Mittelwert, SD) |
10,25 (3,26) |
10,23 (3,76) |
N (behandelte Population) |
||
Anzahl der CIS-Zyklen (Mittelwert, SD) |
5,8 (1,0) |
5,9 (0,6) |
Kumulative CIS-Dosis (mg/m2) (Mittelwert, SD) |
362,851 (98,871) |
363,860 (96,607) |
Kumulative STS-Dosis (g/m2) (Mittelwert, SD) |
-- |
85,149 (24,390) |
Patienten, bei denen ein Hörverlust aufgetreten ist | ||
N (Intent-to-Treat-Population) |
||
Ja, n (%) |
35 (67,3) |
20 (35,1) |
Nein, n (%) |
17 (32,7) |
37 (64,9) |
Relatives Risiko (95 %-KI) |
0,521 (0,349; 0,778) |
|
p-Wert |
<0,001 |
|
Das Risiko eines Hörverlusts lag in der CIS+STS-Gruppe statistisch deutlich niedriger als in der Nur-CIS-Gruppe, was einem klinisch relevanten, um 48 % geringeren Risiko nach der STS-Behandlung entspricht.
Bei einem Median von 4,27 Jahren Nachbeobachtung betrug der Risikoquotient zwischen den beiden Gruppen in der ereignisfreien Überlebenszeit (EFS) ([CIS+STS vs. Nur-CIS]: 0,96; 95 %-KI: 0,42; 2.23) und in der Gesamtüberlebenszeit (OS) (Risikoquotient: 0,48; 95 %-KI: 0,09; 2,61).
Studie 2 (COG ACCL0431) – unterstützende Studie
Bei Studie 2 handelte es sich um eine multizentrische, randomisierte, kontrollierte, unverblindete („open label“) Studie zur Bewertung der Wirksamkeit und Sicherheit von STS zur Vorbeugung von Hörverlusten bei Kindern, die eine CIS-Chemotherapie zur Behandlung von neu diagnostizierten Keimzelltumoren (25,6 %), Hepatoblastomen (5,6 %), Medulloblastomen (20,8 %), Neuroblastomen (20,8 %), Osteosarkomen (23,2 %), atypischen Teratoid/Rhabdoid-Tumoren (1,6 %), Choroid-Plexus-Karzinomen (0,8 %) und anaplastischen Astrocytoma (0,8 %) oder anderen maligner Erkrankungen erhielten, die mit CIS behandelt wurden; 7,5 % hatten zuvor Schädelbestrahlung erhalten. In die Studie aufgenommen wurden Kinder im Alter zwischen 1 und 18 Jahren, die ein Chemotherapieschema mit einer kumulativen CIS-Dosis von ≥ 200 mg/m2 erhielten, wobei einzelne CIS-Dosen über einen Zeitraum von ≤ 6 Stunden infundiert wurden. Die Kinder wurden im Verhältnis 1:1 randomisiert und erhielten entweder 6 Stunden nach jeder CIS-Dosis STS verabreicht (CIS+STS) oder eine Chemotherapie mit CIS ohne nachfolgende Gabe von STS (Nur-CIS).
Das CIS wurde gemäß den zu diesem Zeitpunkt gültigen krankheitsspezifischen Krebsbehandlungsprotokollen der teilnehmenden Zentren verabreicht. Wenn mehrere Tagesdosen CIS geplant waren, sah das Protokoll einen mindestens 10-‑stündigen Abstand zwischen einer STS-Infusion und dem Beginn der CIS-Infusion am darauffolgenden Tag vor.
In der CIS+STS-Gruppe wurden sechs Stunden nach Beendigung jeder CIS-Infusion 10,2 g/m2 STS mittels intravenöser Infusion über einen Zeitraum von 15 Minuten verabreicht. Für Kinder, deren therapeutisches Protokoll aufgrund des jungen Alters oder des niedrigen Körpergewichts eine CIS-Gabe in Abhängigkeit vom Körpergewicht vorsah, wurde eine Dosisreduzierung vorgenommen, d. h. 341 mg/kg STS.
Der primäre Endpunkt der Studie lag in der Bewertung der proportionalen Inzidenz von Hörverlust in der CIS+STS-Gruppe gegenüber der Nur-CIS-Gruppe, definiert durch Vergleich der Kriterien der American Speech-language-Hearing Association (ASHA), die zu Studienbeginn und 4 Wochen nach der letzten Gabe von Cisplatin bewertet wurden. Die EFS, d. h. das Auftreten bzw. Nichtauftreten einer Tumorprogression oder eines Rezidivs oder die anschließende Entwicklung eines malignen Neoplasmas, und die OS wurden ebenfalls untersucht.
Insgesamt wurden 131 Kinder registriert und 125 Kinder wurden in der Studie randomisiert (61 Patienten in der CIS+STS-Gruppe und 64 Patienten in der Nur-CIS-Gruppe). Von den 125 randomisierten Patienten zogen 2 Patienten ihre Teilnahme vor Beginn der Behandlung zurück: 1 Patient aufgrund des Widerrufs der elterlichen Einwilligung und 1 Patient aufgrund einer Entscheidung des Prüfers.
Unter den 104 Patienten, bei denen eine Hörbewertung zu Studienbeginn sowie im Anschluss über einen Zeitraum von 4 Wochen vorgenommen wurde, lag der Anteil der Kinder mit Hörverlust in der CIS+STS-Gruppe (14 Patienten [28,6 %]) etwa halb so hoch wie in der Nur-CIS-Gruppe (31 Patienten [56,4 %]) (Tabelle 4).
Tabelle 4: Zusammenfassung Patientenpopulation und Hörverlust in Studie 2 | ||
Nur CIS |
CIS + STS |
|
Patientenpopulation | ||
N (Intent-to-Treat-Population) |
||
Alter (Jahre), Median (min; max) |
8,3 (1, 18) |
10,7 (1, 18) |
N (Intent-to-Treat-Population) |
||
Gewicht (kg) (Mittelwert, SD) |
37,3 (24,9) |
39,1 (28,3) |
N (Sicherheitspopulation) |
||
Anzahl der CIS-Zyklen (Mittelwert, SD) |
3,8 (1,5) |
3,1 (1,4) |
Kumulative CIS-Dosis (mg/m2) (Mittelwert, SD) |
391,47 (98,40) |
337,57 (118,33) |
Kumulative STS-Dosis (g/m2) (Mittelwert, SD) |
-- |
108,23 (80,24) |
Patienten, bei denen ein Hörverlust aufgetreten ist | ||
N (Wirksamkeitspopulation) |
||
Ja, n (%) |
31 (56,4) |
14 (28,6) |
Nein, n (%) |
24 (43,6) |
35 (71,4) |
Relatives Risiko (95 %-KI) |
0,516 (0,318; 0,839) |
|
p-Wert |
0,0040 |
|
Das Risiko eines Hörverlusts lag in der CIS+STS-Gruppe statistisch deutlich niedriger als in der Nur-CIS-Gruppe, was einem klinisch relevanten, um 48 % geringeren Risiko nach der STS-Behandlung entspricht.
Bei einem Median von 5,33 Jahren Nachbeobachtung betrug der Risikoquotient zwischen den beiden Gruppen in der ereignisfreien Überlebenszeit (EFS) ([CIS+STS vs. Nur-CIS]: 1,27; 95 %-KI: 0,73; 2,18). Es wurde eine Disparität in der OS festgestellt (Risikoquotient: 1,79; 95 %-KI: 0,86; 3,72). Bei Patienten, die post-hoc als Patienten mit lokalisierter Erkrankung eingestuft wurden, betrug der Risikoquotient zwischen den beiden Gruppen in der EFS (1,02; 95 %-KI: 0,49; 2,15 und in der OS (Risikoquotient: 1,23; 95 %-KI: 0,41; 3,66)).
Resorption
Natriumthiosulfat wird nach oraler Verabreichung schlecht resorbiert und muss intravenös verabreicht werden. Am Ende einer intravenösen Natriumthiosulfat-Infusion liegen die Plasmaspiegel von Natriumthiosulfat hoch und sinken danach rasch wieder ab, mit einer terminalen Eliminationshalbwertszeit von etwa 50 Minuten. Innerhalb von 3 bis 6 Stunden nach der Infusion werden die Vordosenwerte wieder erreicht. Mehr als 95 % des Natriumthiosulfats werden innerhalb der ersten 4 Stunden nach der Anwendung im Urin ausgeschieden. Daher gibt es keine Plasmaakkumulation, wenn Natriumthiosulfat an zwei aufeinanderfolgenden Tagen verabreicht wird.
Bei Kindern und Erwachsenen lag der Höchstgehalt an Natriumthiosulfat im Plasma nach 15-minütiger Infusion einer Dosis von 12,8 g/m² bei etwa 13 mM. Der Plasmaspiegel von Thiosulfat ändert sich im Verhältnis zur verabreichten Dosis. Das Alter schien keinen Einfluss auf die maximalen Plasmakonzentrationen von Natriumthiosulfat bzw. auf die anschließende Abnahme zu haben. Ein populationspharmakokinetisches Modell, das Wachstums- und Reifungsvariablen für die pädiatrische Population beinhaltete, zeigte, dass die prognostizierten Plasmakonzentrationen von Natriumthiosulfat am Ende der Infusion über die empfohlenen Dosierungen für die angegebenen Alters- und Körpergewichtsbereiche hinweg konsistent waren.
Verteilung
Natriumthiosulfat bindet sich nicht an menschliche Plasmaproteine. Natriumthiosulfat ist ein anorganisches Salz, und Thiosulfatanionen sind nicht ohne Weiteres zellgängig. Daher scheint sich das Verteilungsvolumen weitgehend auf extrazelluläre Räume zu beschränken und wird bei Erwachsenen auf 0,23 l/kg geschätzt. Bei Tieren wurde festgestellt, dass sich Natriumthiosulfat entlang der Cochlea verteilt. Eine Überwindung der Blut-Hirn-Schranke oder Verteilung über die Plazenta scheint nicht oder nur eingeschränkt möglich. Thiosulfat ist eine endogene Verbindung, die ubiquitär in allen Zellen und Organen vorkommt. Die endogenen Thiosulfatspiegel im Serum betrugen bei erwachsenen Probanden 5,5 ± 1,8 µM.
Biotransformation
Die Metaboliten von Natriumthiosulfat wurden nicht im Rahmen klinischer Studien bestimmt. Thiosulfat ist ein endogenes Zwischenprodukt des schwefelhaltigen Aminosäurestoffwechsels. Der Thiosulfat-Stoffwechsel beinhaltet keine CYP-Enzyme; er wird durch Thiosulfat-Schwefeltransferase und Thiosulfat-Reduktase-Aktivität zu Sulfit metabolisiert, das rasch zu Sulfat oxidiert wird.
Elimination
Natriumthiosulfat (Thiosulfat) wird durch glomeruläre Filtration ausgeschieden. Nach der Verabreichung sind die Thiosulfatspiegel im Urin hoch, und etwa die Hälfte der Natriumthiosulfat-Dosis wird unverändert im Urin ausgeschieden; nahezu die gesamte Dosis wird innerhalb der ersten 4 Stunden nach der Verabreichung ausgeschieden. Die renale Clearance von Thiosulfat als Maß für die GFR war gut mit Inulin-Clearance vergleichbar.
Die Ausscheidung von endogen produziertem Thiosulfat in die Galle war sehr gering und stieg nach Verabreichung von Natriumthiosulfat nicht an. Es wurden keine Studien zur Massenbilanz durchgeführt, es ist jedoch zu erwarten, dass eine nicht-renale‑ Clearance hauptsächlich zur renalen Ausscheidung von Sulfaten führt. Ein geringer Teil des Sulfan-Schwefels in Natriumthiosulfat kann in den endogenen zellulären Schwefelstoffwechsel Eingang finden.
Nierenfunktionsstörung
Bei Hämodialyse-Patienten lag die Gesamtclearance von Natriumthiosulfat bei 2,04 ± 0,72 ml/min/kg (vor/nach der Dialyse), verglichen mit 4,11 ± 0,77 ml/min/kg bei gesunden Freiwilligen. Diese Clearance war im Wesentlichen vergleichbar mit der nicht-renalen‑ Clearance, die bei gesunden Probanden beobachtet wurde (1,86 ml/min/kg ± 0,45 ml/min/kg). In Ermangelung einer glomerulären Filtration bei Hämodialysepatienten führte dies nur zu einem Anstieg der maximalen Thiosulfatplasmaspiegel um ca. 25 % und einem etwa 2‑fachen Anstieg der Gesamtexposition. Die Plasmakonzentration von Thiosulfat gilt als wichtigster Parameter im Zusammenhang mit der Wirksamkeit des Produkts. Darüber hinaus wird davon ausgegangen, dass die häufigsten Nebenwirkungen mit der Natriumbelastung bei Verabreichung von Natriumthiosulfat und gleichzeitigen Elektrolytenungleichgewichten zusammenhängen (siehe Abschnitt 4.4). Nichtklinische Studien wiesen darauf hin, dass die dosislimitierenden akuten Wirkungen mit der Natriumaufnahme in Zusammenhang standen. Natriumthiosulfat ist nur zur Verabreichung in Verbindung mit einer Cisplatin-Chemotherapie bestimmt. Cisplatin ist bei Patienten mit vorbestehender Nierenfunktionsstörung kontraindiziert, weshalb Natriumthiosulfat ohne die Gabe von Cisplatin nicht verabreicht werden würde.
Leberfunktionsstörung
Für die Anwendung von Natriumthiosulfat bei Patienten mit Leberfunktionsstörung liegen keine Informationen vor. Thiosulfat-Sulfurtransferase/-reduktase-Aktivität ist jedoch allgegenwärtig und umfasst auch Gewebe wie rote Blutkörperchen, Leber, Niere, Darm, Muskel und Gehirn. Daher sind die Veränderungen der Pharmakokinetik von Thiosulfat bei Patienten mit Leberfunktionsstörung wahrscheinlich begrenzt und ohne klinische Bedeutung.
Studien zu Wechselwirkungen
Natriumthiosulfat bindet sich nicht an menschliche Plasmaproteine. Die chemischen Eigenschaften von Natriumthiosulfat sowie Beobachtungen, dass Natriumthiosulfat nicht ohne Weiteres Membranbarrieren überwindet und durch glomeruläre Filtration ausgeschieden wird, machen eine Interaktion mit Membrantransportern unwahrscheinlich.
In-vitro-Studien
Cytochrom-P450-Enzyme
Natriumthiosulfat ist ein Induktor von CYP2B6, jedoch nicht von CYP1A2 oder CYP3A4. Natriumthiosulfat ist in klinisch relevanten Konzentrationen kein Inhibitor von CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 und CYP3A4.
Genotoxizität
Natriumthiosulfat zeigte in vitro in einem bakteriellen Rückmutationstest (Ames-Test) mit oder ohne Stoffwechselaktivierung keine genotoxische Wirkung und war in vitro in einem Säugerzelltest (Seterchromatidaustausch) unter Verwendung menschlicher peripherer Lymphozyten nicht klastogen.
Karzinogenität
Es wurden keine Langzeitstudien an Tieren durchgeführt, um die potenzielle Karzinogenität von Natriumthiosulfat zu bewerten.
Beeinträchtigung der Fertilität
Es liegen keine ausreichenden Informationen aus tierexperimentellen Studien vor, um die Auswirkungen einer intravenösen Infusion von Natriumthiosulfat auf die Fertilität zu beurteilen.
Entwicklungstoxizität
Es liegen keine ausreichenden Daten aus Tierstudien vor, um die Entwicklungsrisiken bei intravenöser Infusion von Natriumthiosulfat zu bewerten.
Borsäure
Wasser für Injektionszwecke
Salzsäure (zur pH-Einstellung)
Natriumhydroxid (zur pH-Wert-Einstellung)
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
3 Jahre
Aus mikrobiologischer Sicht sollte das Arzneimittel unmittelbar nach dem Öffnen angewendet werden. Wenn es nicht sofort verwendet wird, liegen die Lagerzeiten und -bedingungen vor der Verwendung in der Verantwortung des Anwenders und belaufen sich normalerweise bei 2 °C – 8 °C auf nicht länger als 24 Stunden.
Für das Produkt, das in Infusionsbeuteln aus Polyvinylchlorid, Ethylenvinylacetat und Polyolephin zur intravenösen Infusion aufbewahrt wird, wurde eine chemische und physikalische Stabilität bei kontrollierter Raumtemperatur für 24 Stunden nachgewiesen.
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Aufbewahrungsbedingungen nach Anbruch des Arzneimittels, siehe Abschnitt 6.3.
Durchstechflaschen aus Klarglas Typ I, 100 ml, verschlossen mit einem chlorierten Butylgummistopfen und einer Flip-off-Aluminiumkappe. Jede Durchstechflasche enthält 100 ml Infusionslösung.
Die Durchstechflaschen sind in Kartons mit jeweils 1 Durchstechflasche erhältlich.
Dieses Arzneimittel ist eine sterile und gebrauchsfertige Infusionslösung.
Jede Durchstechflasche ist nur zur einmaligen Anwendung bestimmt, und nicht verwendete Lösungen sollten verworfen werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Norgine B.V.
Antonio Vivaldistraat 150
1083 HP Amsterdam
Niederlande
EU/1/23/1734/001
/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 26. Mai 2023
05.2025