
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
VYDURA® 75 mg Lyophilisat zum Einnehmen
Jedes Lyophilisat zum Einnehmen enthält Rimegepanthemisulfat-Sesquihydrat, entsprechend 75 mg Rimegepant.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1
Lyophilisat zum Einnehmen
Das Lyophilisat zum Einnehmen ist weiß bis gebrochen weiß, rund, hat einen Durchmesser von 14 mm und trägt das eingeprägte Symbol ![]() .
.
VYDURA wird angewendet zur
Akuttherapie der Migräne mit oder ohne Aura bei Erwachsenen.
präventiven Behandlung von episodischer Migräne bei Erwachsenen, die mindestens 4 Migräneattacken pro Monat haben.
Dosierung
Akuttherapie der Migräne
Die empfohlene Dosis beträgt 75 mg Rimegepant, bei Bedarf, einmal täglich.
Prophylaxe der Migräne
Die empfohlene Dosis beträgt 75 mg Rimegepant jeden zweiten Tag.
Die Höchstdosis pro Tag beträgt 75 mg Rimegepant.
VYDURA kann zu einer Mahlzeit oder unabhängig von den Mahlzeiten eingenommen werden.
Gleichzeitig angewendete Arzneimittel
Bei gleichzeitiger Anwendung von mittelstarken CYP3A4-Inhibitoren oder starken P‑gp-Inhibitoren ist die Einnahme einer weiteren Dosis Rimegepant innerhalb von 48 Stunden zu vermeiden (siehe Abschnitt 4.5).
Besondere Patientengruppen
Ältere Patienten (ab 65 Jahren)
Es liegen nur begrenzte Erfahrungen mit der Anwendung von Rimegepant bei Patienten ab 65 Jahren vor. Eine Dosisanpassung ist nicht erforderlich, da die Pharmakokinetik von Rimegepant vom Alter nicht beeinflusst wird (siehe Abschnitt 5.2).
Eingeschränkte Nierenfunktion
Bei Patienten mit leicht, mittelstark oder stark eingeschränkter Nierenfunktion ist eine Dosisanpassung nicht erforderlich. Eine stark eingeschränkte Nierenfunktion führte zu einem > 2‑fachen Anstieg der AUC des ungebundenen Wirkstoffs, aber nur zu einem Anstieg der Gesamt-AUC um weniger als 50 % (siehe Abschnitt 5.2). Bei häufiger Anwendung ist bei Patienten mit stark eingeschränkter Nierenfunktion Vorsicht geboten. Rimegepant wurde bei Patienten mit terminaler Niereninsuffizienz und bei Dialysepatienten nicht untersucht. Die Anwendung von Rimegepant bei Patienten mit terminaler Niereninsuffizienz (CLcr < 15 ml/min) sollte vermieden werden.
Eingeschränkte Leberfunktion
Bei Patienten mit leicht (Child-Pugh A) oder mittelstark (Child-Pugh B) eingeschränkter Leberfunktion ist eine Dosisanpassung nicht erforderlich. Bei Patienten mit stark eingeschränkter Leberfunktion (Child-Pugh C) waren die Plasmakonzentrationen von Rimegepant (AUC des ungebundenen Wirkstoffs) signifikant höher (siehe Abschnitt 5.2). Die Anwendung von Rimegepant bei Patienten mit stark eingeschränkter Leberfunktion sollte vermieden werden.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von VYDURA bei Kindern und Jugendlichen (< 18 Jahren) ist nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
VYDURA ist zum Einnehmen.
Das Lyophilisat zum Einnehmen wird auf oder unter die Zunge gelegt. Es löst sich im Mund auf und kann ohne Flüssigkeit eingenommen werden.
Die Patienten müssen darauf hingewiesen werden, dass sie zum Öffnen der Blisterpackung trockene Hände haben müssen. Zwecks vollständiger Anwendungshinweise ist auf die Packungsbeilage zu verweisen.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Überempfindlichkeitsreaktionen, wie Dyspnoe und Ausschlag, traten bei weniger als 1 % der Patienten auf, die im Rahmen von klinischen Studien mit Rimegepant behandelt wurden (siehe Abschnitt 4.8). Überempfindlichkeitsreaktionen, einschließlich schwerwiegender Überempfindlichkeit wie anaphylaktische Reaktionen, wurden im klinischen Anwendungsbereich und nach dem Inverkehrbringen berichtet (siehe Abschnitt 4.8). Einige Überempfindlichkeitsreaktionen können auch noch Tage nach der Einnahme auftreten. Wenn eine Überempfindlichkeitsreaktion auftritt, ist Rimegepant abzusetzen und eine geeignete Therapie einzuleiten.
VYDURA wird nicht empfohlen:
bei Patienten mit stark eingeschränkter Leberfunktion (siehe Abschnitt 4.2);
bei Patienten mit terminaler Niereninsuffizienz (CLcr < 15 ml/min) (siehe Abschnitt 4.2);
bei gleichzeitiger Anwendung mit starken CYP3A4-Inhibitoren (siehe Abschnitt 4.5);
bei gleichzeitiger Anwendung mit starken oder mittelstarken CYP3A4-Induktoren (siehe Abschnitt 4.5).
Kopfschmerzen durch Medikamentenübergebrauch (MOH, medication overuse headache)
Ein übermäßiger Gebrauch von Schmerzmitteln jeglicher Art bei Kopfschmerzen kann diese verschlimmern. Wenn dies der Fall ist oder ein solcher Verdacht besteht, sollte ärztlicher Rat eingeholt und die Behandlung abgebrochen werden. Die Diagnose MOH sollte bei Patienten naheliegend sein, die trotz (oder wegen) der regelmäßigen Einnahme von Schmerzmitteln bei akuten Kopfschmerzen häufig oder täglich Kopfschmerzen haben.
Rimegepant ist ein Substrat von CYP3A4 sowie der Effluxtransporter P-Glycoprotein (P‑gp) und Brust-Krebs-Resistenzprotein (BCRP) (siehe Abschnitt 5.2).
CYP3A4-Inhibitoren
CYP3A4-Inhibitoren erhöhen die Plasmakonzentrationen von Rimegepant. Die gleichzeitige Anwendung von Rimegepant mit starken CYP3A4-Inhibitoren (z. B. Clarithromycin, Itraconazol, Ritonavir) wird nicht empfohlen (siehe Abschnitt 4.4). Die gleichzeitige Anwendung von Rimegepant mit Itraconazol führte zu einem signifikanten Anstieg der Rimegepant-Exposition (AUC um das 4-Fache und Cmax um das 1,5-Fache).
Die gleichzeitige Anwendung von Rimegepant mit Arzneimitteln, die mittelstarke CYP3A4-Hemmer sind (z. B. Diltiazem, Erythromycin, Fluconazol), kann die Exposition gegenüber Rimegepant erhöhen. Die gleichzeitige Anwendung von Rimegepant mit Fluconazol führte zu erhöhten Rimegepant-Expositionen (AUC um das 1,8-Fache), jedoch ohne relevanten Einfluss auf die Cmax. Bei gleichzeitiger Anwendung von mittelstarken CYP3A4-Inhibitoren (z. B. Fluconazol) ist die Einnahme einer weiteren Dosis Rimegepant innerhalb von 48 Stunden zu vermeiden (siehe Abschnitt 4.2).
CYP3A4-Induktoren
CYP3A4-Induktoren verringern die Plasmakonzentrationen von Rimegepant. Die gleichzeitige Anwendung von VYDURA mit starken CYP3A4-Induktoren (z. B. Phenobarbital, Rifampicin, Johanniskraut (Hypericum perforatum)) oder mit mittelstarken CYP3A4-Induktoren (z. B. Bosentan, Efavirenz, Modafinil) wird nicht empfohlen (siehe Abschnitt 4.4). Die Wirkung der CYP3A4-Induktion kann nach dem Absetzen des starken oder mittelstarken CYP3A4-Induktors für bis zu 2 Wochen anhalten. Die gleichzeitige Anwendung von Rimegepant mit Rifampicin führte zu einer signifikanten Abnahme der Rimegepant-Exposition (Abnahme der AUC um 80 % und der Cmax um 64 %), die eventuell zu einem Wirksamkeitsverlust führen kann.
Inhibitoren, die ausschließlich P‑gp und BCRP hemmen
Inhibitoren der Effluxtransporter P‑gp und BCRP können die Plasmakonzentrationen von Rimegepant erhöhen. Bei gleichzeitiger Anwendung von starken P‑gp-Inhibitoren (z. B. Ciclosporin, Verapamil, Chinidin) ist die Einnahme einer weiteren Dosis Rimegepant innerhalb von 48 Stunden zu vermeiden (siehe Abschnitt 4.2). Die gleichzeitige Anwendung von Rimegepant mit Ciclosporin (einem potenten P‑gp- und BCRP-Inhibitor) oder mit Chinidin (einem selektiven P‑gp-Inhibitor) führte zu einem signifikanten Anstieg der Rimegepant-Exposition ähnlichen Ausmaßes (AUC und Cmax um > 50 %, aber um weniger als das 2-Fache).
Schwangerschaft
Es liegen nur begrenzte Erfahrungen mit der Anwendung von Rimegepant bei Schwangeren vor. Tierexperimentelle Studien haben gezeigt, dass Rimegepant nicht embryozid ist und in klinisch relevanten Expositionen wurde kein teratogenes Potenzial beobachtet. Unerwünschte Wirkungen auf die embryo-fetale Entwicklung (vermindertes Körpergewicht der Feten und vermehrte Skelettveränderungen bei Ratten) nach Gabe von Rimegepant während der Schwangerschaft wurden nur bei Expositionen beobachtet, die zu einer Toxizität bei den Muttertieren führten (etwa 200 Mal höher als die klinischen Expositionen) (siehe Abschnitt 5.3). Aus Vorsichtsgründen soll eine Anwendung von VYDURA während der Schwangerschaft vermieden werden.
Stillzeit
In einer monozentrischen klinischen Studie an 12 stillenden Frauen, die mit einer einmaligen Dosis von 75 mg Rimegepant behandelt wurden, fand man minimale Konzentrationen von Rimegepant in der Muttermilch. Der relative Prozentsatz einer maternalen Dosis, der das Kind erreicht, wird auf weniger als 1 % geschätzt. Es liegen keine Daten über die Auswirkungen auf die Milchproduktion vor. Die entwicklungs- und gesundheitsfördernden Wirkungen des Stillens sollten zusammen mit dem klinischen Bedarf der Mutter an VYDURA und möglichen unerwünschten Wirkungen von Rimegepant oder der Grunderkrankung der Mutter auf den gestillten Säugling berücksichtigt werden.
Fertilität
Tierexperimentelle Studien zeigten keine klinisch relevanten Auswirkungen auf die weibliche und männliche Fertilität (siehe Abschnitt 5.3).
VYDURA hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Nebenwirkungsprofils
Die häufigste Nebenwirkung in der Akuttherapie und in der Migräneprophylaxe war Übelkeit (1,2 % bzw. 1,4 %). Die meisten Nebenwirkungen waren leicht oder mittelschwer. Überempfindlichkeit, einschließlich Dyspnoe und starkem Ausschlag, traten bei weniger als 1 % der behandelten Patienten auf.
Tabellarische Auflistung der Nebenwirkungen
Die Nebenwirkungen sind in Tabelle 1 nach MedDRA-Systemorganklasse aufgelistet. Die entsprechende Häufigkeitskategorie für die einzelnen Nebenwirkungen basiert auf folgender Konvention (CIOMS III): sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1.000, <1/100); selten (≥1/10.000, <1/1.000); sehr selten (<1/10.000).
Tabelle 1 Auflistung der Nebenwirkungen
Systemorganklasse |
Nebenwirkung |
Häufigkeit |
Akuttherapie | ||
Erkrankungen des Immunsystems |
Anaphylaktische Reaktiona |
Gelegentlich |
Erkrankungen des Gastrointestinaltrakts |
Übelkeit |
Häufig |
Prophylaxe | ||
Erkrankungen des Immunsystems |
Anaphylaktische Reaktiona |
Nicht bekannt |
Erkrankungen des Gastrointestinaltrakts |
Übelkeit |
Häufig |
a Nach dem Inverkehrbringen identifizierte unerwünschte Arzneimittelwirkungen (UAW)
Langzeitsicherheit
Die Langzeitsicherheit von Rimegepant wurde in zwei einjährigen unverblindeten Verlängerungsstudien untersucht. 1662 Patienten erhielten Rimegepant für mindestens 6 Monate und 740 Patienten erhielten Rimegepant für 12 Monate als Akuttherapie oder als Prophylaxe.
Beschreibung ausgewählter Nebenwirkungen
Überempfindlichkeitsreaktionen
Überempfindlichkeit, einschließlich Dyspnoe und schwerem Ausschlag, traten bei weniger als 1 % der in klinischen Studien behandelten Patienten auf. Überempfindlichkeitsreaktionen können auch noch Tage nach der Anwendung auftreten und es kam auch zu verzögert aufgetretenen schwerwiegenden Überempfindlichkeitsreaktionen.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das aufgeführte nationale Meldesystem anzuzeigen.
Deutschland |
Österreich |
Es liegen nur begrenzte klinische Erfahrungen mit einer Überdosierung von Rimegepant vor. Es liegen keine Berichte über Symptome unter Überdosierung vor. Die Behandlung einer Überdosierung von Rimegepant sollte aus allgemeinen unterstützenden Maßnahmen bestehen, einschließlich Überwachung der Vitalparameter und Beobachtung des klinischen Zustandes des Patienten. Ein spezifisches Antidot zur Behandlung einer Überdosierung mit Rimegepant ist nicht verfügbar. Aufgrund der hohen Serumproteinbindung ist es unwahrscheinlich, das Rimegepant durch Dialyse in signifikantem Umfang aus dem Körper entfernt werden kann.
Pharmakotherapeutische Gruppe: Analgetika, Calcitonin-Gene-Related-Peptide (CGRP)-Antagonisten,
ATC-Code: N02CD06
Wirkmechanismus
Rimegepant bindet selektiv mit hoher Affinität an den humanen Calcitonin-Gene-Related-Peptide (CGRP)-Rezeptor und antagonisiert dessen Funktion.
Der Zusammenhang zwischen der pharmakodynamischen Aktivität und dem Mechanismus bzw. den Mechanismen, durch die Rimegepant seine klinischen Wirkungen entfaltet, ist nicht bekannt.
Klinische Wirksamkeit: Akuttherapie
Die Wirksamkeit von VYDURA in der Akuttherapie der Migräne mit und ohne Aura bei Erwachsenen wurde in drei randomisierten, doppelblinden, placebokontrollierten klinischen Studien (Studie 1‑3) untersucht. Die Patienten wurden angewiesen, eine Migräne mit mittelstarker bis starker Kopfschmerzintensität zu behandeln. Notfallarzneimittel (d. h. NSAR, Paracetamol und/oder ein Antiemetikum) waren 2 Stunden nach der ersten Behandlung erlaubt. Andere Arten von Notfallarzneimitteln wie Triptane waren innerhalb von 48 Stunden nach der ersten Behandlung nicht erlaubt. Etwa 14 % der Patienten nahmen zu Studienbeginn prophylaktische Migränemittel ein. Keiner der Patienten in Studie 1 nahm gleichzeitig Prophylaxearzneimittel ein, die auf den Calcitonin-Gene-Related-Peptid-Signalweg wirken.
Die primären Wirksamkeitsanalysen wurden bei Patienten durchgeführt, die eine Migräne mit mittelstarken bis starken Kopfschmerzen behandelten. Schmerzfreiheit war definiert als ein Rückgang der mittelstarken oder starken Kopfschmerzen bis auf keine Kopfschmerzen, und die Freiheit von dem belastendsten Symptom (MBS, most bothersome symptom) war definiert als das Fehlen des selbst angegebenen MBS (d. h. Photophobie, Phonophobie oder Übelkeit). Die Patienten, die ein MBS angaben, nannten Photophobie als häufigstes Symptom (54 %), gefolgt von Übelkeit (28 %) und Phonophobie (15 %).
In Studie 1 war der Prozentsatz von Patienten, die 2 Stunden nach einer Einzeldosis Kopfschmerzfreiheit und MBS-Freiheit erreichten, bei den Patienten, die VYDURA erhielten, statistisch signifikant höher als bei den Patienten, die das Placebo erhielten (Tabelle 2). Außerdem wurden statistisch signifikante Wirkungen von VYDURA im Vergleich zu Placebo für die zusätzlichen Wirksamkeitsendpunkte Schmerzlinderung nach 2 Stunden, anhaltende Schmerzfreiheit von 2 bis 48 Stunden, Anwendung von Notfallarzneimitteln innerhalb von 24 Stunden und Fähigkeit, 2 Stunden nach der Einnahme normal zu funktionieren, nachgewiesen. Schmerzlinderung war definiert als ein Rückgang der Migräneschmerzen von mittelstarken oder starken Schmerzen auf leichte oder gar keine Schmerzen. Die zulassungsrelevanten doppelblinden, placebokontrollierten Studien 2 und 3 zur Untersuchung von einzelnen Migräneattacken wurden an Migränepatienten durchgeführt, die eine bioäquivalente Darreichungsform von 75 mg Rimegepant erhielten.
Tabelle 2: Migräne-Wirksamkeitsendpunkte in Studien zur Akuttherapie
Studie 1 |
Studie 2 |
Studie 3 |
||||
VYDURA 75 mg |
Placebo |
Rimegepant 75 mg |
Placebo |
Rimegepant 75 mg |
Placebo |
|
Kopfschmerzfreiheit nach 2 Stunden |
||||||
n/N* |
142/669 |
74/682 |
105/537 |
64/535 |
104/543 |
77/541 |
% Responder |
21,2 |
10,9 |
19,6 |
12,0 |
19,2 |
14,2 |
Unterschied im Vergleich zu Placebo (%) |
10,3 |
7,6 |
4,9 |
|||
p-Wert |
<0,0001 a |
0,0006a |
0,0298 a |
|||
MBS-Freiheit nach 2 Stunden |
||||||
n/N* |
235/669 |
183/682 |
202/537 |
135/535 |
199/543 |
150/541 |
% Responder |
35,1 |
26,8 |
37,6 |
25,2 |
36,6 |
27,7 |
Unterschied im Vergleich zu Placebo (%) |
8,3 |
12,4 |
8,9 |
|||
p-Wert |
0,0009 a |
<0,0001 a |
0,0016 a |
|||
Schmerzlinderung nach 2 Stunden |
||||||
n/N* |
397/669 |
295/682 |
312/537 |
229/535 |
304/543 |
247/541 |
% Responder |
59,3 |
43,3 |
58,1 |
42,8 |
56,0 |
45,7 |
Unterschied im Vergleich zu Placebo |
16,1 |
15,3 |
10,3 |
|||
p-Wert |
<0,0001a |
<0,0001a |
0,0006a |
|||
Anhaltende Kopfschmerzfreiheit über 2 bis 48 Stunden |
||||||
n/N* |
90/669 |
37/682 |
53/537 |
32/535 |
63/543 |
39/541 |
% Responder |
13,5 |
5,4 |
9,9 |
6,0 |
11,6 |
7,2 |
Unterschied im Vergleich zu Placebo (%) |
8.0 |
3.9 |
4.4 |
|||
p-Wert |
<0,0001a |
0,0181b |
0,0130b |
|||
**n=Anzahl Responder/N=Anzahl Patienten in der betreffenden Behandlungsgruppe
a Signifikanter p-Wert im hierarchischen Test
b Nominaler p-Wert im hierarchischen Test
MBS: belastendstes Symptom
Abbildung 1 zeigt den Prozentsatz von Patienten in Studie 1, die innerhalb von 2 Stunden nach der Behandlung Migränefreiheit erreichten.
Abbildung 1: Prozentsatz von Patienten in Studie 1, die innerhalb von 2 Stunden Kopfschmerzfreiheit erreichten
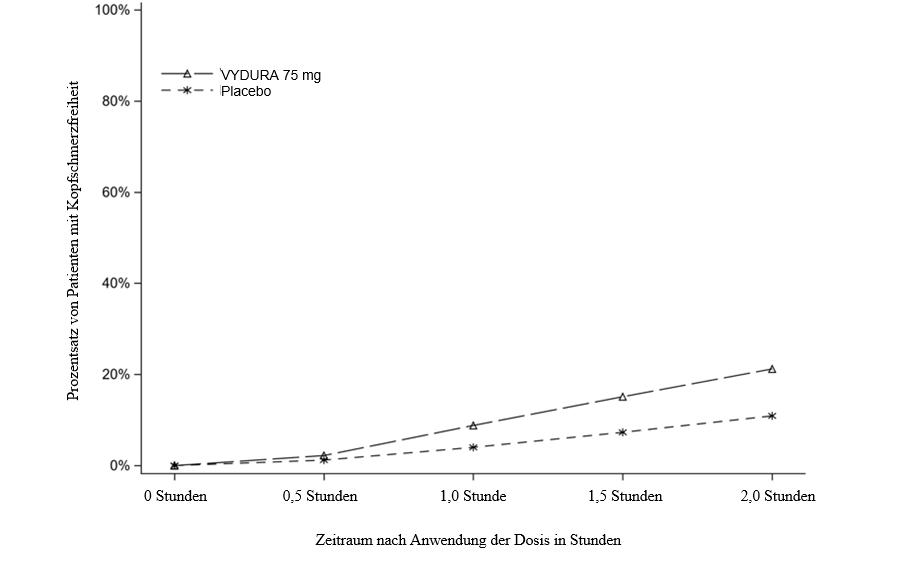
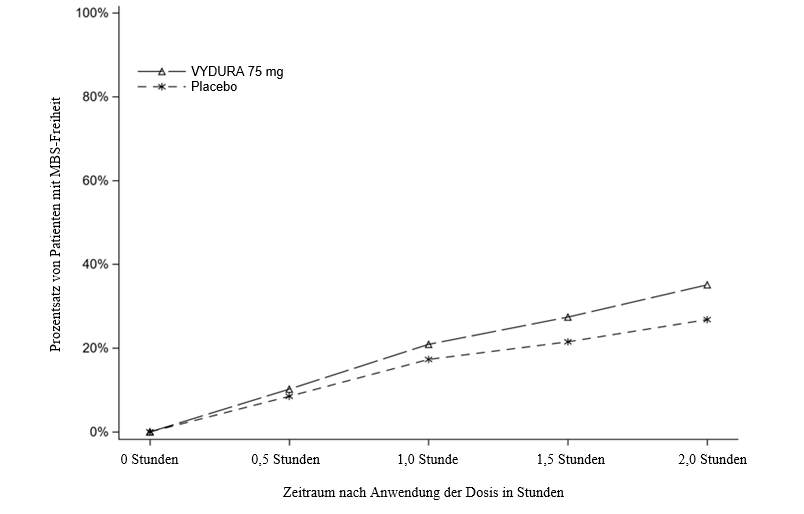
Abbildung 2 zeigt den Prozentsatz von Patienten in Studie 1, die innerhalb von 2 Stunden nach der Behandlung MBS-Freiheit erreichten.
Abbildung 2: Prozentsatz von Patienten in Studie 1, die innerhalb von 2 Stunden MBS-Freiheit erreichten
In allen 3 Studien war die Inzidenz von Photophobie und Phonophobie 2 Stunden nach der Einnahme von VYDURA 75 mg im Vergleich zu Placebo reduziert.
Klinische Wirksamkeit: Prophylaxe
Die Wirksamkeit von Rimegepant als prophylaktische Migränebehandlung wurde in einer randomisierten, doppelblinden, placebokontrollierten Studie (Studie 4) untersucht.
Studie 4 schloss männliche und weibliche Erwachsene mit einer mindestens 1-jährigen Migräneanamnese (mit oder ohne Aura) ein. Die Patienten hatten eine Vorgeschichte mit 4 bis 18 Migräneattacken mit mittelstarker bis starker Kopfschmerzintensität pro 4-Wochen-Zeitraum in den 12 Wochen vor dem Screeningbesuch. Die Patienten hatten während des 28-tägigen Beobachtungszeitraums vor der Randomisierung zur Studie durchschnittlich 10,9 Kopfschmerztage, darunter im Durchschnitt 10,2 Migränetage. In der Studie wurden die Patienten zu einer Behandlung mit Rimegepant 75 mg (N=373) oder Placebo (N=374) für bis zu 12 Wochen randomisiert. Die Patienten wurden angewiesen, die ihnen zugeteilte Behandlung während des 12-wöchigen Behandlungszeitraums einmal jeden zweiten Tag (EOD, every other day) einzunehmen. Die Patienten durften bei Bedarf andere Akuttherapien der Migräne (z. B. Triptane, NSAR, Paracetamol, Antiemetika) einnehmen. Zu Studienbeginn nahmen etwa 22 % der Patienten Prophylaxearzneimittel gegen Migräne ein. Die Patienten hatten die Möglichkeit, für weitere 12 Monate an einer unverblindeten Verlängerungsstudie teilzunehmen.
Der primäre Wirksamkeitsendpunkt von Studie 4 war die Veränderung der durchschnittlichen Anzahl von Migränetagen pro Monat (MMDs, monthly migraine days) gegenüber dem Ausgangswert in Woche 9 bis 12 der doppelblinden Behandlungsphase. Sekundäre Endpunkte waren das Erreichen einer Reduktion der monatlichen Migränetage mit mittelstarken bis starken Kopfschmerzen um ≥ 50 % gegenüber dem Ausgangswert.
Die Behandlung mit Rimegepant 75 mg jeden zweiten Tag zeigte statistisch signifikante Verbesserungen bei den wichtigen Wirksamkeitsendpunkten im Vergleich zu Placebo. Die Ergebnisse sind in Tabelle 3 zusammengestellt und in Abbildung 3 grafisch veranschaulicht.
Tabelle 3: Wichtige Wirksamkeitsendpunkte in Studie 4
Rimegepant |
Placebo |
|
Monatliche Migränetage (MMD) in Woche 9 bis 12 |
N=348 |
N=347 |
Veränderung gegenüber dem Ausgangswert |
-4,3 |
-3,5 |
Veränderung im Vergleich zu Placebo |
-0,8 |
|
p-Wert |
0,010a |
|
Abnahme der MMDs mit mittelstarken bis starken Kopfschmerzen um ≥ 50 % in Woche 9 bis 12 |
N=348 |
N=347 |
% Responder |
49,1 |
41,5 |
Unterschied im Vergleich zu Placebo |
7,6 |
|
p-Wert |
0,044a |
|
a Signifikanter p-Wert bei hierarchischen Tests | ||
Abbildung 3: Veränderung der monatlichen Migränetage gegenüber dem Ausgangswert in Studie 4
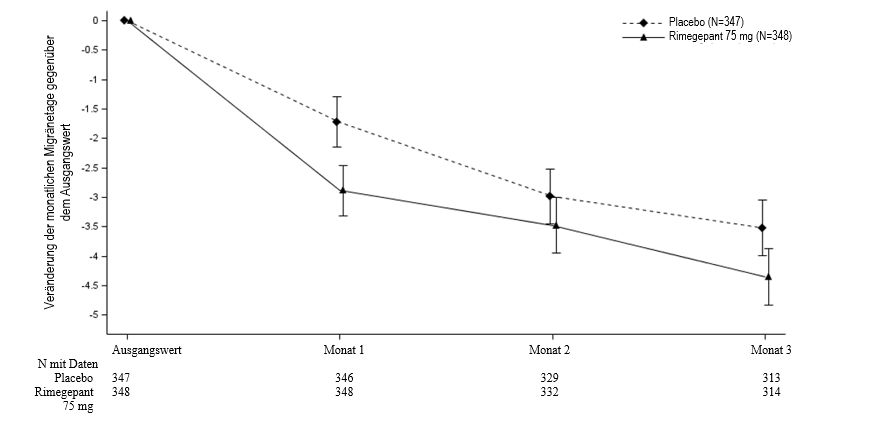
Langfristige Wirksamkeit
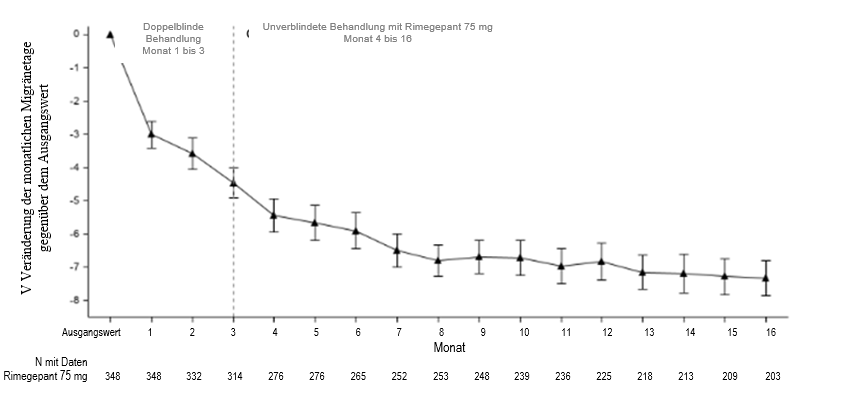
Die an Studie 4 teilnehmenden Patienten hatten die Möglichkeit, für weitere 12 Monate an einer offenen Verlängerungsstudie teilzunehmen. In der offenen Verlängerungsstudie, in der Patienten Rimegepant 75 mg jeden zweiten Tag erhielten sowie je nach Bedarf an nicht geplanten Behandlungstagen, wurde die Wirksamkeit für bis zu 1 Jahr aufrechterhalten (Abbildung 4). Ein Teil der Teilnehmer, bestehend aus 203 Patienten, die Rimegepant zugewiesen waren, beendete den insgesamt 16-monatigen Behandlungszeitraum. Bei diesen Patienten betrug die durchschnittliche Gesamtabnahme der Anzahl der MMDs gegenüber dem Ausgangswert im Verlauf des 16-monatigen Behandlungszeitraums durchschnittlich 6,2 Tage.
Abbildung 4: Längsschnittkurve der Veränderung der durchschnittlichen Anzahl von MMDs gegenüber dem Beobachtungszeitraum im Laufe der Zeit während der doppelblinden Behandlung (Monat 1 bis 3) und während der unverblindeten Behandlung mit Rimegepant (Monat 4 bis 16)
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für VYDURA eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in der prophylaktischen Behandlung von Migränekopfschmerzen gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Europäische Arzneimittel-Agentur hat für VYDURA eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in der Akuttherapie der Migräne gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Resorption
Nach oraler Anwendung wird Rimegepant unter Erreichen der Höchstkonzentration nach 1,5 Stunden resorbiert. Nach einer supratherapeutischen Dosis von 300 mg betrug die absolute orale Bioverfügbarkeit von Rimegepant etwa 64 %.
Einfluss von Nahrungsmitteln
Nach Einnahme von Rimegepant nach dem Verzehr einer fettreichen oder fettarmen Mahlzeit war die Tmax um 1 bis 1,5 Stunden verzögert. Eine fettreiche Mahlzeit reduzierte die Cmax um 41 bis 53 % und die AUC um 32 bis 38 %. Eine fettarme Mahlzeit reduzierte die Cmax um 36 % und die AUC um 28 %. In den klinischen Sicherheits- und Wirksamkeitsstudien wurde Rimegepant unabhängig von den Mahlzeiten angewendet.
Verteilung
Das Verteilungsvolumen von Rimegepant im Steady-State beträgt 120 l. Die Plasmaproteinbindung von Rimegepant beträgt etwa 96 %.
Biotransformation
Rimegepant wird in erster Linie durch CYP3A4 und in geringerem Umfang durch CYP2C9 metabolisiert. Rimegepant ist die Hauptform (~77 %), wobei keine wesentlichen Metaboliten (d. h. > 10 %) im Plasma nachgewiesen wurden.
Basierend auf In-vitro-Studien ist Rimegepant kein Inhibitor von CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 oder UGT1A1 in klinisch relevanten Konzentrationen. Allerdings ist Rimegepant ein schwacher Inhibitor von CYP3A4 mit zeitabhängiger Hemmung. Rimegepant ist kein Induktor von CYP1A2, CYP2B6 oder CYP3A4 in klinisch relevanten Konzentrationen.
Elimination
Die Eliminationshalbwertszeit von Rimegepant beträgt bei gesunden Probanden etwa 11 Stunden. Nach oraler Anwendung von [14C]-Rimegepant bei gesunden männlichen Probanden wurden 78 % der Gesamtradioaktivität in den Fäzes und 24 % im Urin wiedergefunden. Unverändertes Rimegepant ist die wichtigste Einzelkomponente in den ausgeschiedenen Fäzes (42 %) und im Urin (51 %).
Transporter
In vitro ist Rimegepant ein Substrat der Effluxtransporter P‑gp und BCRP. Inhibitoren der P‑gp- und BCRP-Effluxtransporter können die Plasmakonzentration von Rimegepant erhöhen (siehe Abschnitt 4.5).
Rimegepant ist kein Substrat von OATP1B1 oder OATP1B3. In Anbetracht seiner geringen renalen Clearance wurde Rimegepant nicht als Substrat von OAT1, OAT3, OCT2 , MATE1 oder MATE2-K eingestuft.
Rimegepant ist kein Inhibitor von P‑gp, BCRP, OAT1 oder MATE2-K in klinisch relevanten Konzentrationen. Es ist ein schwacher Inhibitor von OATP1B1 und OAT3.
Rimegepant ist ein Inhibitor von OATP1B3, OCT2 und MATE1. Die gleichzeitige Anwendung von Rimegepant mit Metformin, einem MATE1-Transportersubstrat, führte zu keinen klinisch signifikanten Auswirkungen auf die Pharmakokinetik von Metformin oder auf die Glukoseverwertung. Für Rimegepant sind mit OATP1B3 oder OCT2 in klinisch relevanten Konzentrationen keine klinischen Arzneimittelwechselwirkungen zu erwarten.
Linearität/Nicht-Linearität
Rimegepant zeigt nach einmaliger oraler Gabe einen überproportionalen Anstieg der Exposition auf, der offenbar mit einer dosisabhängigen Zunahme der Bioverfügbarkeit zusammenhängt.
Alter, Geschlecht, Körpergewicht, Ethnie
Es wurden keine klinisch signifikanten Unterschiede in der Pharmakokinetik von Rimegepant aufgrund von Alter, Geschlecht, Ethnie, Körpergewicht, Migränestatus oder CYP2C9-Genotyp beobachtet.
Eingeschränkte Nierenfunktion
In einer speziellen klinischen Studie zum Vergleich der Pharmakokinetik von Rimegepant bei Patienten mit leicht (geschätzte Kreatinin-Clearance [CLcr] 60-89 ml/min), mittelschwer (CLcr 30-59 ml/min) und schwer (CLcr 15-29 ml/min) eingeschränkter Nierenfunktion mit der von nierengesunden Probanden (gepoolte gesunde Kontrolle) wurde nach einer Einzeldosis von 75 mg ein Anstieg der Gesamtexposition von Rimegepant um weniger als 50 % beobachtet. Die AUC von ungebundenem Rimegepant war bei Patienten mit schwer eingeschränkter Nierenfunktion um das 2,57-Fache erhöht. VYDURA wurde nicht bei Patienten mit terminaler Niereninsuffizienz (CLcr < 15 ml/min) untersucht.
Eingeschränkte Leberfunktion
In einer speziellen klinischen Studie zum Vergleich der Pharmakokinetik von Rimegepant bei Patienten mit leicht, mittelschwer oder schwer eingeschränkter Leberfunktion mit der von lebergesunden Probanden (gesunde merkmalsgleiche Kontrolle) war die Exposition von Rimegepant (AUC des ungebundenen Wirkstoffs) nach einer Einzeldosis von 75 mg bei Patienten mit schwer eingeschränkter Leberfunktion (Child-Pugh Klasse C) um das 3,89-Fache höher. Es bestanden keine klinisch bedeutsamen Unterschiede in der Exposition von Rimegepant bei Patienten mit leicht (Child-Pugh Klasse A) und mittelstark eingeschränkter Leberfunktion (Child-Pugh Klasse B) im Vergleich zu Probanden mit normaler Leberfunktion.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität, Phototoxizität, Reproduktions- und Entwicklungstoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Rimegepant-bedingte Wirkungen bei höheren Dosen in Studien mit wiederholter Gabe umfassten hepatische Lipidose bei Mäusen und Ratten, intravaskuläre Hämolyse bei Ratten und Affen sowie Erbrechen bei Affen. Diese Befunde wurden nur bei Expositionen beobachtet, die als ausreichend über der maximalen Exposition beim Menschen liegend angesehen wurden, was auf eine geringe Relevanz für die klinische Anwendung hindeutet (≥ 12-mal [Mäuse] und ≥ 49-mal [Ratten] für hepatische Lipidose, ≥ 95-mal [Ratten] und ≥ 9-mal [Affen] für intravaskuläre Hämolyse und ≥ 37-mal für Erbrechen [Affen]).
In einer Fertilitätsstudie an Ratten wurden Rimegepant-bedingte Wirkungen nur bei der hohen Dosis von 150 mg/kg/Tag festgestellt (verminderte Fertilität und vermehrte Präimplantationsverluste), die zu maternaler Toxizität und systemischen Expositionen führte, die ≥ 95-mal höher waren als die maximale Exposition beim Menschen. Die orale Anwendung von Rimegepant während der Organogenese führte bei Ratten, nicht aber bei Kaninchen, zu Auswirkungen auf die Föten. Bei Ratten wurden ein vermindertes Körpergewicht der Föten sowie eine erhöhte Inzidenz fötaler Veränderungen nur bei der höchsten Dosis von 300 mg/kg/Tag beobachtet, die eine maternale Toxizität bei einer Exposition bewirkte, die etwa dem 200-Fachen der maximalen Exposition beim Menschen entsprach. Darüber hinaus hatte Rimegepant in Dosen bis zu 60 mg/kg/Tag (das ≥ 24-Fache der maximalen Exposition beim Menschen) keine Auswirkungen auf die prä- und postnatale Entwicklung bei Ratten oder auf Wachstum, Entwicklung oder Reproduktionsleistung von juvenilen Ratten in Dosen bis zu 45 mg/kg/Tag (das ≥ 14-Fache der maximalen Exposition beim Menschen).
Gelatine
Mannitol (Ph.Eur.) (E421)
Minz-Aroma
Sucralose
Nicht zutreffend.
4 Jahre
Nicht über 30 °C lagern.
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Einzeldosis-Blisterpackung aus Polyvinylchlorid (PVC), orientiertem Polyamid (OPA) und Aluminiumfolie, versiegelt mit einer abziehbaren Aluminiumfolie.
Packungsgrößen:
Einzeldosis mit 2 x 1 Lyophilisat zum Einnehmen.
Einzeldosis mit 8 x 1 Lyophilisat zum Einnehmen.
Einzeldosis mit 16 x 1 Lyophilisat zum Einnehmen.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Keine besonderen Anforderungen für die Beseitigung.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Brüssel
Belgien
EU/1/22/1645/001
EU/1/22/1645/002
EU/1/22/1645/003
Datum der Erteilung der Zulassung: 25. April 2022
Januar 2026

Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
VERKAUFSABGRENZUNG IN DEUTSCHLAND
Verschreibungspflichtig
REZEPTPFLICHT/ APOTHEKENPFLICHT IN ÖSTERREICH
Rezept- und apothekenpflichtig, wiederholte Abgabe verboten
PACKUNGSGRÖSSEN IN DEUTSCHLAND
75 mg: Blisterpackung mit 16 x 1 Lyophilisat zum Einnehmen
PACKUNGSGRÖSSEN IN ÖSTERREICH
75 mg: Blisterpackung mit 2 x 1 Lyophilisat zum Einnehmen
Blisterpackung mit 8 x 1 Lyophilisat zum Einnehmen
Blisterpackung mit 16 x 1 Lyophilisat zum Einnehmen
REPRÄSENTANT IN DEUTSCHLAND
PFIZER PHARMA GmbH
Friedrichstr. 110
10117 Berlin
Tel.: 030 550055-51000
Fax: 030 550054-10000
REPRÄSENTANT IN ÖSTERREICH
Pfizer Corporation Austria Ges.m.b.H.
Floridsdorfer Hauptstraße 1
A-1210 Wien
Tel.: +43 (0)1 521 15-0