
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Itovebi® 3 mg Filmtabletten
Itovebi® 9 mg Filmtabletten
Itovebi 3 mg Filmtabletten
Jede Filmtablette enthält 3 mg Inavolisib.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 22 mg Lactose.
Itovebi 9 mg Filmtabletten
Jede Filmtablette enthält 9 mg Inavolisib.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Filmtablette enthält 66 mg Lactose.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette (Tablette).
Itovebi 3 mg Filmtabletten
Rote, runde, konvexe Filmtablette mit der Prägung „INA 3“ auf einer Seite. Ungefährer Durchmesser: 6 mm.
Itovebi 9 mg Filmtabletten
Pinkfarbene, ovale Filmtablette mit der Prägung „INA 9“ auf einer Seite. Ungefähre Größe: 13 mm (Länge), 6 mm (Breite).
Itovebi wird in Kombination mit Palbociclib und Fulvestrant zur Behandlung von erwachsenen Patienten mit PIK3CA-mutiertem, Östrogenrezeptor (ER)-positivem, HER2-negativem, lokal fortgeschrittenem oder metastasiertem Brustkrebs angewendet, wenn während einer adjuvanten endokrinen Behandlung oder innerhalb von 12 Monaten nach Abschluss einer adjuvanten endokrinen Behandlung ein Rezidiv auftritt (siehe Abschnitt 5.1).
Bei Patienten, die zuvor im Rahmen der (neo)adjuvanten Therapie mit einem CDK4/6-Inhibitor behandelt wurden, sollte zwischen dem Absetzen des CDK4/6-Inhibitors und dem Nachweis des Rezidivs ein Intervall von mindestens 12 Monaten liegen.
Bei prä-/perimenopausalen Frauen und bei Männern ist die endokrine Therapie mit einem LHRH-Agonisten (LHRH = luteinising hormone-releasing hormone) zu kombinieren.
Die Behandlung mit Itovebi ist von einem Arzt einzuleiten, der Erfahrung in der Anwendung von antineoplastischen Arzneimitteln hat.
Für die Behandlung mit Itovebi sind Patienten mit ER-positivem, HER2-negativem, lokal fortgeschrittenem oder metastasiertem Brustkrebs basierend auf dem Vorhandensein einer oder mehrerer PIK3CA-Mutationen in einer Tumor- oder Plasmaprobe auszuwählen. Der Nachweis der PIK3CA-Mutation(en) ist unter Verwendung eines CE-gekennzeichneten In-vitro-Diagnostikums (IVD) mit dem entsprechenden Verwendungszweck durchzuführen (siehe Abschnitt 5.1). Wenn kein CE-gekennzeichnetes IVD verfügbar ist, muss ein alternativer validierter Test verwendet werden. Wenn bei einem Probenmaterial keine Mutation festgestellt wird, könnte bei dem anderen Probentyp, falls verfügbar, eine Mutation festgestellt werden.
Dosierung
Die empfohlene Dosis Itovebi beträgt 9 mg oral einmal täglich mit oder ohne Nahrung.
Itovebi ist in Kombination mit Palbociclib und Fulvestrant zu verabreichen. Die empfohlene Dosis von Palbociclib beträgt 125 mg, die oral einmal täglich an 21 aufeinander folgenden Tagen eingenommen wird, gefolgt von 7 Tagen ohne Einnahme, dies ergibt einen vollständigen Zyklus von 28 Tagen. Die empfohlene Dosis von Fulvestrant beträgt 500 mg, intramuskulär verabreicht an den Tagen 1, 15 und 29, danach einmal monatlich. Weitere Informationen sind der Zusammenfassung der Merkmale des Arzneimittels (Fachinformation) von Palbociclib und Fulvestrant zu entnehmen.
Die Behandlung von prä-/perimenopausalen Frauen und Männern mit Itovebi soll gemäß der lokalen klinischen Praxis auch einen LHRH-Agonisten umfassen.
Dauer der Behandlung
Es wird empfohlen, Patienten bis zur Krankheitsprogression oder bis zum Auftreten einer inakzeptablen Toxizität mit Itovebi zu behandeln.
Verspätete oder versäumte Dosen
Die Patienten sind aufzufordern, ihre Dosis jeden Tag ungefähr zur gleichen Zeit einzunehmen. Wenn eine Einnahme von Itovebi vergessen wurde, kann diese innerhalb von 9 Stunden nach der gewohnten Einnahmezeit nachgeholt werden. Nach mehr als 9 Stunden ist die Dosis für diesen Tag auszulassen. Am nächsten Tag ist Itovebi zur üblichen Zeit einzunehmen. Wenn der Patient nach der Einnahme der Itovebi Dosis erbricht, soll der Patient an diesem Tag keine zusätzliche Dosis einnehmen und am nächsten Tag zum gewohnten Zeitpunkt mit dem üblichen Dosierungsschema fortfahren.
Dosisanpassungen
Die Behandlung von Nebenwirkungen kann eine vorübergehende Unterbrechung, Dosisreduktion oder einen Abbruch der Behandlung mit Itovebi erfordern. Die empfohlenen Dosisreduktionen bei Nebenwirkungen sind in Tabelle 1 aufgeführt.
Tabelle 1: Empfehlungen zur Dosisreduktion bei Nebenwirkungen
Dosisstufe |
Dosis und Zeitplan |
Anfangsdosis |
9 mg täglich |
Erste Dosisreduktion |
6 mg täglich |
Zweite Dosisreduktion |
3 mg täglicha |
a Die Behandlung mit Itovebi soll dauerhaft abgebrochen werden, wenn die Patienten die tägliche Dosis von 3 mg nicht vertragen.. | |
Basierend auf der klinischen Beurteilung des Patienten durch den behandelnden Arzt kann die Dosis von Itovebi wieder bis auf eine maximale Tagesdosis von 9 mg erhöht werden. Empfehlungen zur Dosisanpassung bei spezifischen Nebenwirkungen sind in den Tabellen 2 – 4 dargestellt.
Hyperglykämie
Tabelle 2: Dosisanpassung und Behandlung von Hyperglykämie
Nüchtern-Glukosespiegela |
Empfehlung |
> ULN bis 160 mg/dl |
|
> 160 bis 250 mg/dl |
|
> 250 bis 500 mg/dl |
|
> 500 mg/dl |
|
ULN = obere Normalgrenze (upper limit of normal) | |
Stomatitis
Tabelle 3: Dosisanpassung und Behandlung von Stomatitis
Grada |
Empfehlung |
Grad 1 |
|
Grad 2 |
|
Grad 3 |
|
Grad 4 |
|
a Basierend auf CTCAE Version 5.0. | |
Andere Nebenwirkungen
Tabelle 4: Dosisanpassung und Behandlung von anderen Nebenwirkungen
Grada |
Empfehlung |
Für alle Grade: Unterstützende Therapie einleiten und, falls klinisch angezeigt, überwachen. | |
Grad 1 |
|
Grad 2 |
|
Grad 3, erstes Ereignis |
|
Grad 3, wiederauftretend |
|
Grad 4, lebensbedrohlich |
|
a Basierend auf CTCAE Version 5.0. | |
Besondere Patientengruppen
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Itovebi bei Kindern und Jugendlichen im Alter von 0 bis 17 Jahren sind nicht erwiesen. Es liegen keine Daten vor.
Ältere Patienten
Auf Grundlage einer pharmakokinetischen Populationsanalyse ist bei Patienten ≥ 65 Jahren keine Dosisanpassung von Itovebi erforderlich. Bei Patienten ≥ 65 Jahren liegen nur begrenzte Daten vor (siehe Abschnitt 5.2).
Nierenfunktionsstörung
Die empfohlene Anfangsdosis von Itovebi bei Patienten mit mittelschwerer Nierenfunktionsstörung (eGFR 30 bis < 60 ml/min nach CKD-EPI-Formel) beträgt 6 mg oral einmal täglich und bei Patienten mit schwerer Nierenfunktionsstörung (eGFR < 30 ml/min nach CKD-EPI-Formel) 3 mg oral einmal täglich. Bei Patienten mit leichter Nierenfunktionsstörung (eGFR 60 bis < 90 ml/min) ist keine Dosisanpassung erforderlich.
Leberfunktionsstörung
Bei Patienten mit leichter Leberfunktionsstörung (Gesamtbilirubin > ULN bis ≤ 1,5 ULN oder AST > ULN und Gesamtbilirubin ≤ ULN) ist keine Dosisanpassung erforderlich. Die Sicherheit und Wirksamkeit von Itovebi wurden bei Patienten mit mittelschwerer bis schwerer Leberfunktionsstörung nicht untersucht (siehe Abschnitt 5.2).
Art der Anwendung
Itovebi ist zum Einnehmen. Die Tabletten können mit oder ohne Nahrung eingenommen werden. Die Tabletten sind im Ganzen zu schlucken und dürfen nicht gekaut, zerdrückt, aufgelöst oder geteilt werden.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Hyperglykämie
Die Sicherheit und Wirksamkeit von Itovebi bei Patienten mit Typ-1-Diabetes mellitus oder Typ-2-Diabetes mellitus, die eine laufende antihyperglykämische Therapie benötigen, wurden nicht untersucht, da diese Patienten von der INAVO120 Studie ausgeschlossen waren. Nur ein Patient mit Typ-2-Diabetes mellitus war in den Itovebi Arm der INAVO120 Studie eingeschlossen. Dies sollte berücksichtigt werden, wenn Itovebi Patienten mit Diabetes mellitus verschrieben wird. Patienten mit Diabetes mellitus in der Vorgeschichte benötigen möglicherweise eine intensivierte antihyperglykämische Behandlung und häufigere Nüchtern-Glukosetests während der Behandlung mit Itovebi. Die Behandlung mit Itovebi sollte erst begonnen werden, wenn der Nüchtern-Glukosespiegel optimiert ist. Vor Beginn der Behandlung mit Itovebi sollte die Konsultation eines in der Behandlung von Hyperglykämie erfahrenen Arztes in Betracht gezogen werden.
Hyperglykämie wurde häufig berichtet bei Patienten, die mit Itovebi behandelt wurden. Schwere Fälle von Hyperglykämie, einschließlich Ketoazidose mit tödlichen Komplikationen, sind aufgetreten.
In der INAVO120-Studie wurde Hyperglykämie durch eine antihyperglykämische Behandlung und eine klinisch indizierte Dosisanpassung von Itovebi behandelt (siehe Abschnitt 4.8). Eine kurzfristige Gabe von Insulin kann als Rescue-Therapie bei Hyperglykämie eingesetzt werden. Es liegen nur begrenzte Erfahrungen zu Patienten vor, die Insulin erhalten haben, während sie mit Itovebi behandelt wurden. Bei der Behandlung von Hyperglykämie mit antihyperglykämischen Arzneimitteln (z. B. Insulin, Sulfonylharnstoffe) muss das mögliche Auftreten von Hypoglykämien in Betracht gezogen werden, wenn die Behandlung mit Itovebi unterbrochen oder abgesetzt wird.
Vor Beginn der Behandlung mit Itovebi sind die Patienten über die Anzeichen und Symptome einer Hyperglykämie (z. B. übermäßiger Durst, häufigeres Wasserlassen, verschwommenes Sehen, geistige Verwirrung, Atembeschwerden oder gesteigerter Appetit bei Gewichtsverlust) aufzuklären und darüber zu informieren, sich unverzüglich an einen Arzt zu wenden, wenn diese Symptome auftreten. Vor und während der Behandlung ist eine optimale Hydratation aufrechtzuerhalten.
Bei den Patienten sind vor der Behandlung mit Itovebi und in regelmäßigen Abständen während der Behandlung der Nüchtern-Glukosespiegel (FPG, fasting plasma glucose oder FBG, fasting blood glucose) und HbA1C (glykosyliertes Hämoglobin) zu testen (siehe Tabelle 5). Bei Patienten, die Risikofaktoren für eine Hyperglykämie aufweisen oder bei denen eine Hyperglykämie auftritt, ist die Einleitung einer häuslichen Nüchtern-Glukoseüberwachung in Betracht zu ziehen. Eine Metformin Prämedikation kann bei Patienten mit Risikofaktoren für Hyperglykämie in Betracht gezogen werden. Alle Patienten sollten dazu angehalten werden, ihren Lebensstil (z. B. Ernährungsumstellungen, körperliche Aktivität) zu ändern.
Tabelle 5: Zeitplan für die Überwachung der Nüchtern-Glukose- und HbA1C-Spiegel
Empfohlener Zeitplan für die Überwachung der Nüchtern-Glukose- und HbA1C-Spiegel bei allen mit Itovebi behandelten Patienten |
|
Beim Screening, vor Beginn der Behandlung mit Itovebi |
Bei den Patienten sind der Nüchtern-Glukosespiegel (FPG oder FBG) und der HbA1C-Spiegel zu testen und ihr Blutglukosespiegel ist zu optimieren (siehe Tabelle 2). |
Nach dem Behandlungsbeginn mit Itovebi |
Eine Überwachung/Selbstüberwachung der Nüchtern-Glukose soll in der ersten Woche (Tag 1 bis 7) einmal alle 3 Tage, dann in den nächsten 3 Wochen (Tag 8 bis 28) einmal wöchentlich, in den nächsten 8 Wochen einmal alle 2 Wochen, danach einmal alle 4 Wochen und wie klinisch indiziert erfolgen.* Eine Überwachung/Selbstüberwachung des Nüchtern-Glukosespiegels soll gemäß klinischer Indikation häufiger in Betracht gezogen werden* bei Patienten mit Risikofaktoren für Hyperglykämie, einschließlich, aber nicht beschränkt auf (Prä-)Diabetes, HbA1C ≥ 5,7 %, BMI ≥ 30 kg/m2, Alter ≥ 45 Jahre, Schwangerschaftsdiabetes in der Anamnese und Diabetes mellitus in der Familienanamnese. Bei Patienten mit gleichzeitiger Anwendung von Corticosteroiden, interkurrenten Infektionen oder anderen Erkrankungen, die ein intensiviertes Blutglukosemanagement erfordern können, sind häufigere Nüchtern-Glukosetests erforderlich, um eine Verschlechterung des gestörten Glukosestoffwechsels und potenzielle Komplikationen, einschließlich diabetischer Ketoazidose, zu verhindern. Bei diesen Patienten wird eine Überwachung von HbA1C und Ketonen (vorzugsweise im Blut) zusätzlich zur Nüchtern-Glukose empfohlen. Bei Bedarf ist eine antihyperglykämische Behandlung einzuleiten oder anzupassen (siehe Abschnitt 4.2). |
HbA1C soll alle 3 Monate kontrolliert werden. | |
Wenn sich nach Beginn der Behandlung mit Itovebi eine Hyperglykämie entwickelt |
Nüchtern-Glukose engmaschiger überwachen, wie klinisch angezeigt.* |
Während einer antihyperglykämischen Behandlung sollte der Nüchtern-Glukosespiegel weiterhin mindestens einmal wöchentlich über einen Zeitraum von 8 Wochen, danach einmal alle 2 Wochen und, wenn klinisch indiziert, kontrolliert werden.* | |
* Jegliche Glukosemessungen sollen nach ärztlichem Ermessen gemäß klinischer Indikation durchgeführt werden. | |
Stomatitis
Bei Patienten, die mit Itovebi behandelt wurden, wurden Fälle von Stomatitis berichtet (siehe Abschnitt 4.8). Je nach Schweregrad der Stomatitis kann die Behandlung mit Itovebi unterbrochen, reduziert oder dauerhaft abgesetzt werden (siehe Tabelle 3).
In der Studie INAVO120 wurde zur Prophylaxe von Stomatitis eine corticosteroidhaltige Mundspülung empfohlen. Bei den Patienten, die Itovebi in Kombination mit Palbociclib und Fulvestrant erhielten, wurde bei 19,1 % bzw. 1,2 % der Patienten eine Prophylaxe mit Dexamethason oder Triamcinolon angewendet.
Die Patienten sind darauf hinzuweisen, beim ersten Anzeichen einer Stomatitis mit der Anwendung einer alkoholfreien Mundspülung mit Corticosteroiden zu beginnen und alkoholische oder peroxidhaltige Mundspülungen zu vermeiden, da diese den Zustand verschlimmern können (siehe Abschnitt 4.8). Diätetische Anpassungen (z. B. Verzicht auf scharfe Speisen) sind in Betracht zu ziehen.
Anwendung bei Patienten, die zuvor einen CDK4/6-Inhibitor erhalten haben
Bei Patienten, die zuvor im Rahmen einer neodadjuvanten oder adjuvanten Behandlung einen CDK4/6-Inhibitor erhalten haben, liegen nur sehr begrenzte Informationen zur Wirksamkeit der Kombination von Itovebi, Palbociclib und Fulvestrant vor. Die Wirksamkeit kann bei diesen Patienten geringer sein.
Lactose
Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactase-Mangel oder einer Glukose-Galactose-Malabsorption sollten dieses Arzneimittel nicht anwenden.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Filmtablette, d. h. es ist nahezu „natriumfrei“.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
CYP-Inhibitoren und -Induktoren
Die Ergebnisse klinischer Studien weisen darauf hin, dass die Hauptmetaboliten von Inavolisib nicht durch CYP-Enzyme metabolisiert werden und Hydrolyse der wichtigste Stoffwechselweg ist. Dies deutet auf eine geringe Wahrscheinlichkeit für klinisch relevante Wechselwirkungen zwischen Inavolisib und CYP-Inhibitoren oder -Induktoren hin.
CYP-Substrate
Inavolisib induziert CYP3A und ist ein zeitabhängiger Inhibitor von CYP3A in vitro. Daher ist Inavolisib in Kombination mit sensitiven CYP3A4-Substraten mit geringer therapeutischer Breite (z. B. Alfentanil, Astemizol, Cisaprid, Cyclosporin, Chinidin, Sirolimus, Tacrolimus) mit Vorsicht anzuwenden, da Inavolisib die systemische Exposition dieser Substrate erhöhen oder verringern kann.
Außerdem induziert Inavolisib CYP2B6, CYP2C8, CYP2C9 und CYP2C19 in vitro. Daher sollte Inavolisib mit Vorsicht zusammen mit sensitiven Substraten dieser Enzyme mit geringer therapeutischer Breite (z. B. Paclitaxel, Warfarin, Phenytoin, S‑Mephenytoin) angewendet werden, da Inavolisib die systemische Exposition dieser Substrate verringern und folglich zu einer verminderten Wirksamkeit führen kann.
Frauen im gebärfähigen Alter/Verhütung bei Männern und Frauen
Frauen
Die Patientinnen sind anzuweisen, während der Behandlung mit Itovebi und für 1 Woche nach der letzten Dosis von Itovebi eine wirksame nicht-hormonelle Verhütungsmethode anzuwenden.
Männer
Es ist nicht bekannt, ob Inavolisib in das Sperma gelangt. Um eine potenzielle fetale Exposition während der Schwangerschaft zu vermeiden, müssen männliche Patienten mit Partnerinnen im gebärfähigen Alter oder schwangeren Partnerinnen während der Behandlung mit Itovebi und für 1 Woche nach der letzten Dosis von Itovebi ein Kondom verwenden.
Schwangerschaft
Vor dem Beginn einer Therapie mit Itovebi ist bei Frauen im gebärfähigen Alter der Schwangerschaftsstatus festzustellen. Schwangere Frauen müssen klar auf das mögliche Risiko für den Fötus hingewiesen werden.
Bisher liegen keine oder nur sehr begrenzte Erfahrungen mit der Anwendung von Inavolisib bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Die Anwendung von Itovebi während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen.
Stillzeit
Es ist nicht bekannt, ob Inavolisib/Metabolite in die Muttermilch übergehen. Ein Risiko für das Neugeborene/den Säugling kann nicht ausgeschlossen werden. Das Stillen soll während der Behandlung mit Itovebi und für 1 Woche nach der letzten Dosis von Itovebi unterbrochen werden.
Fertilität
Es liegen keine Daten zur Wirkung von Inavolisib auf die Fertilität von Menschen vor. Tierexperimentelle Studien haben gezeigt, dass Inavolisib die Fertilität von fortpflanzungsfähigen Frauen und Männern beeinträchtigen könnte (siehe Abschnitt 5.3).
Itovebi hat geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen, da während der Behandlung mit Itovebi Ermüdung (Fatigue) berichtet wurde.
Zusammenfassung des Sicherheitsprofils
Die häufigsten Nebenwirkungen bei Patienten, die Itovebi erhielten, waren Hyperglykämie (59,9 %), Stomatitis (51,2 %), Diarrhö (48,1 %), Thrombozytopenie (48,1 %), Ermüdung (Fatigue) (37,7 %), Anämie (37 %), Übelkeit (27,8 %), verminderter Appetit (23,5 %), Ausschlag (22,8 %), Kopfschmerzen (21 %), Gewichtsabnahme (17,3 %), Erbrechen (14,8 %) und Harnwegsinfektionen (13 %).
Die häufigsten schwerwiegenden Nebenwirkungen bei Patienten, die mit Itovebi behandelt wurden, waren Anämie (1,9 %), Diarrhö (1,2 %) und Harnwegsinfektion (1,2 %).
Bei 3,1 % der Patienten wurde Itovebi aufgrund einer Nebenwirkung dauerhaft abgesetzt. Die Nebenwirkungen, die zu einem dauerhaften Absetzen von Itovebi führten, waren Hyperglykämie (1,2 %), Stomatitis (0,6 %), erhöhte Alanintransaminase (ALT) (0,6 %) und Gewichtsverlust (0,6 %).
Tabellarische Auflistung der Nebenwirkungen
Die Nebenwirkungen, die auf den Daten von 162 Patienten mit lokal fortgeschrittenem oder metastasiertem Brustkrebs basieren, die mit Itovebi in Kombination mit Palbociclib und Fulvestrant in der randomisierten Phase-3-Studie INAVO120 behandelt wurden und auf Daten aus der Überwachung nach der Markteinführung basieren, sind in Tabelle 6 nach MedDRA-Systemorganklasse aufgeführt. Die mediane Behandlungszeit betrug zum Zeitpunkt der primären Auswertung 9,2 Monate (Bereich: 0 bis 38,8 Monate).
Innerhalb jeder Systemorganklasse sind die Nebenwirkungen nach Häufigkeit geordnet, wobei die häufigsten Nebenwirkungen zuerst aufgeführt sind. Die entsprechenden Häufigkeitskategorien der einzelnen Nebenwirkungen sind wie folgt definiert: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Die Nebenwirkungen werden für jede Häufigkeitsgruppe nach abnehmendem Schweregrad aufgeführt.
Tabelle 6: Beobachtete Nebenwirkungen bei Patienten, die mit Itovebi behandelt wurden
Systemorganklasse |
Itovebi + Palbociclib + Fulvestrant |
||||
Häufigkeitskategorie |
Alle Grade (%) |
Grad 3 – 4 (%) |
|||
Infektionen und parasitäre Erkrankungen | |||||
Harnwegsinfektion |
Sehr häufig |
13 |
1,2* |
||
Erkrankungen des Bluts und des Lymphsystems | |||||
Thrombozytopenie |
Sehr häufig |
48,1 |
14,2 |
||
Anämie |
Sehr häufig |
37 |
6,2* |
||
Stoffwechsel- und Ernährungsstörungen | |||||
Hyperglykämiea |
Sehr häufig |
59,9 |
5,6* |
||
Appetit vermindert |
Sehr häufig |
23,5 |
0 |
||
Hypokaliämie |
Sehr häufig |
16 |
2,5 |
||
Hypokalzämie |
Häufig |
8,6 |
1,2* |
||
Ketoazidose |
Gelegentlichb |
- |
- |
||
Erkrankungen des Nervensystems | |||||
Kopfschmerzen |
Sehr häufig |
21 |
0 |
||
Augenerkrankungen | |||||
Trockenes Auge |
Häufig |
8,6 |
0 |
||
Erkrankungen des Gastrointestinaltrakts | |||||
Stomatitisc |
Sehr häufig |
51,2 |
5,6* |
||
Diarrhö |
Sehr häufig |
48,1 |
3,7* |
||
Übelkeit |
Sehr häufig |
27,8 |
0,6* |
||
Abdominalschmerz |
Sehr häufig |
15,4 |
0,6* |
||
Erbrechen |
Sehr häufig |
14,8 |
0,6* |
||
Dysgeusie |
Häufig |
8,6 |
0 |
||
Dyspepsie |
Häufig |
8 |
0 |
||
Erkrankungen der Haut und des Unterhautgewebes | |||||
Ausschlagd |
Sehr häufig |
22,8 |
0 |
||
Alopezie |
Sehr häufig |
18,5 |
0 |
||
Trockene Haute |
Sehr häufig |
13 |
0 |
||
Dermatitisf |
Häufig |
2,5 |
0 |
||
Follikulitis |
Häufig |
1,2 |
0 |
||
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |||||
Ermüdung (Fatigue) |
Sehr häufig |
37,7 |
1,9* |
||
Untersuchungen | |||||
Alanin-Aminotransferase erhöht |
Sehr häufig |
17,3 |
3,7* |
||
Gewicht erniedrigt |
Sehr häufig |
17,3 |
3,7* |
||
Insulin im Blut erhöht |
Häufig |
6,2 |
0 |
||
Einstufung gemäß CTCAE Version 5.0. | |||||
Beschreibung ausgewählter Nebenwirkungen
Hyperglykämie
In der INAVO120-Studie wurde bei 59,9 % der Patienten, die mit Itovebi in Kombination mit Palbociclib und Fulvestrant behandelt wurden, über Hyperglykämie jeden Grades berichtet; das Auftreten von Grad 2 und Grad 3 wurde bei 38,3 % bzw. 5,6 % der Patienten berichtet (basierend auf CTCAE Version 5.0). Bei den Patienten, bei denen eine Hyperglykämie auftrat, war die Rate neu auftretender Hyperglykämien während der ersten zwei Monate der Behandlung am höchsten, mit einer medianen Zeit bis zum ersten Auftreten von 7 Tagen (Bereich: 2 bis 955 Tage).
Von den 97 Patienten, die Itovebi in Kombination mit Palbociclib und Fulvestrant erhielten und bei denen es zu Hyperglykämie kam, erhielten 74,2 % (72/97) antihyperglykämische Arzneimittel, einschließlich SGLT2-Inhibitoren, Thiazolidindione und DPP-4-Inhibitoren zur Prophylaxe oder Behandlung von Hyperglykämie. Alle Patienten, die antihyperglykämische Arzneimittel bekamen, erhielten Metformin als Monotherapie oder in Kombination mit anderen antihyperglykämischen Arzneimitteln (d. h. Insulin, DPP-4-Inhibitoren und Sulfonylharnstoffe); und 11,3 % (11/97) erhielten Insulin (siehe Abschnitt 4.4).
Bei Patienten mit einem Nüchtern-Glukosespiegel > 160 mg/dl (> 8,9 mmol/l) und einer Verbesserung des Nüchtern-Blutglukosespiegels um mindestens eine Stufe (siehe Tabelle 2) (n = 52) betrug die mediane Zeit bis zur Verbesserung 8 Tage (Bereich: 2 bis 43 Tage).
Eine Hyperglykämie führte bei 27,8 % der Patienten zur Unterbrechung der Behandlung mit Itovebi, bei 2,5 % zur Dosisreduktion von Itovebi und bei 1,2 % der Patienten zum Absetzen von Itovebi.
Stomatitis
Stomatitis wurde bei 51,2 % der Patienten berichtet, die mit Itovebi in Kombination mit Palbociclib und Fulvestrant behandelt wurden; Ereignisse von Grad 1 wurden bei 32,1 % der Patienten, Ereignisse von Grad 2 bei 13,6 % der Patienten und Ereignisse von Grad 3 bei 5,6 % der Patienten berichtet. Bei den Patienten, bei denen Stomatitis auftrat, betrug die mediane Zeit bis zum ersten Auftreten 13 Tage (Spanne: 1 bis 610 Tage).
Stomatitis führte bei 9,9 % der Patienten zur Unterbrechung der Behandlung mit Itovebi, bei 3,7 % zur Dosisreduktion von Itovebi und bei 0,6 % der Patienten zum Absetzen von Itovebi.
Von den Patienten, die Itovebi in Kombination mit Palbociclib und Fulvestrant erhielten, wendeten 24,1 % eine Mundspülung mit Dexamethason zur Behandlung der Stomatitis an (siehe Abschnitt 4.4).
Diarrhö
Diarrhö wurde bei 48,1 % der Patienten berichtet, die mit Itovebi in Kombination mit Palbociclib und Fulvestrant behandelt wurden; Ereignisse von Grad 1 wurden bei 27,8 % der Patienten, Ereignisse von Grad 2 bei 16,7 % der Patienten und Ereignisse von Grad 3 bei 3,7 % der Patienten berichtet. Bei den Patienten, bei denen Diarrhö auftrat, betrug die mediane Zeit bis zum ersten Auftreten 15 Tage (Bereich: 2 bis 602 Tage).
Diarrhö führte bei 6,8 % zu einer Unterbrechung der Behandlung mit Itovebi, bei 1,2 % zu einer Dosisreduktion von Itovebi und bei keinem Patienten zum Absetzen von Itovebi.
Antidiarrhoika (z. B. Loperamid) wurden bei 28,4 % der Patienten, die Itovebi in Kombination mit Palbociclib und Fulvestrant erhielten, zur Behandlung der Symptome angewendet.
Ältere Patienten
Eine Analyse der Sicherheit von Itovebi, bei der Patienten im Alter von ≥ 65 Jahren (14,8 %) mit jüngeren Patienten (85,2 %) verglichen wurden, deutet auf eine höhere Inzidenz von Dosisanpassungen/-unterbrechungen von Itovebi hin (79,2 % gegenüber 68,1 %).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
53175 Bonn
Website: http://www.bfarm.de
anzuzeigen.
Die höchste Dosis von Itovebi, die in der INAVO120-Studie verabreicht wurde, betrug 18 mg bei einem Patienten. Dieses Ereignis einer versehentlichen Überdosierung klang innerhalb eines Tages ab und erforderte keine Behandlung oder führte zu einer Dosisanpassung der Studienmedikamente.
Patienten, bei denen es zu einer Überdosierung gekommen ist, sind engmaschig zu überwachen und unterstützende Maßnahmen sind einzuleiten. Es gibt kein bekanntes Antidot für Itovebi.
Pharmakotherapeutische Gruppe: Antineoplastische Mittel, PI3K-Inhibitoren, ATC-Code: L01EM06
Wirkmechanismus
Inavolisib ist ein Inhibitor der katalytischen Untereinheit der Phosphatidylinositol-4,5-Bisphosphat-3-Kinase (PI3K) Alpha-Isoform (p110α; codiert durch das PIK3CA-Gen). Darüber hinaus fördert Inavolisib den Abbau von mutiertem p110α (mutant degrader). Der PI3K-Signalweg ist bei HR-positivem Brustkrebs häufig fehlreguliert, oft aufgrund aktivierender PIK3CA-Mutationen. Mit seinem dualen Wirkmechanismus hemmt Inavolisib die Aktivität nachgeordneter Ziele des PI3K-Signalweges einschließlich AKT, was zu einer verringerten zellulären Proliferation und zur Apoptoseinduktion in PIK3CA-mutierten Brustkrebszelllinien führt.
Klinische Wirksamkeit und Sicherheit
Lokal fortgeschrittener oder metastasierender Brustkrebs
Basierend auf den Daten aus der INAVO120-Studie sind die Patienten in diesem Setting definiert als endokrin resistente Patienten (Rezidiv der Erkrankung während einer adjuvanten endokrinen Behandlung oder innerhalb von 12 Monaten nach Abschluss einer adjuvanten endokrinen Behandlung), die keine vorherige Behandlung für ihre lokal fortgeschrittene oder metastasierte Erkrankung erhalten haben.
INAVO120
Die Wirksamkeit von Itovebi in Kombination mit Palbociclib und Fulvestrant wurde in einer randomisierten, doppelblinden, placebokontrollierten Phase-3-Studie bei erwachsenen Patienten mit PIK3CA-mutiertem, HR-positivem, HER2-negativem, lokal fortgeschrittenem oder metastasiertem Brustkrebs untersucht, deren Erkrankung während einer adjuvanten endokrinen Behandlung oder innerhalb von 12 Monaten nach Abschluss einer adjuvanten endokrinen Therapie (endokrin-resistent) fortschritt, und die davor keine systemische Therapie für ihre lokal fortgeschrittene oder metastasierte Erkrankung erhalten hatten. In die Studie wurden Patienten eingeschlossen, die zuvor eine (neo)adjuvante endokrine Therapie einschließlich eines CDK4/6-Inhibitors erhalten hatten, wenn das Progressionsereignis > 12 Monate nach Abschluss des CDK4/6-Inhibitor-Anteils der (neo)adjuvanten Therapie eingetreten war, und die ein HbA1C < 6% und einen Nüchtern-Glukosespiegel von < 126 mg/dl aufwiesen. Von der Studie ausgeschlossen waren Patienten mit Typ-1-Diabetes mellitus oder Typ-2-Diabetes mellitus, die zu Beginn der Studienbehandlung eine laufende antihyperglykämische Therapie benötigten, Patienten, die zuvor mit Fulvestrant behandelt wurden (außer als Teil einer neoadjuvanten Therapie mit einer Behandlungsdauer von ≤ 6 Monaten) und Patienten mit bekannten und unbehandelten oder aktiven ZNS-Metastasen (fortschreitend oder die Antikonvulsiva oder Corticosteroide zur symptomatischen Kontrolle benötigten).
Der PIK3CA-Mutationsstatus wurde prospektiv durch das Testen von aus Plasma gewonnener zirkulierender Tumor-DNA (ctDNA) unter Verwendung eines Next-Generation-Sequencing (NGS)-Assays (FoundationOne® Liquid CDx Assay oder PredicineCARETM) in einem Zentrallabor (87,4 %) oder in lokalen Labors (12,6 %) unter Verwendung verschiedener validierter Polymerase-Kettenreaktion (PCR)- oder NGS-Assays an Tumorgewebe oder Plasma bestimmt. Die folgenden PIK3CA-Mutationen an den angegebenen Aminosäurepositionen waren für den Einschluss geeignet: H1047D/I/L/N/P/Q/R/T/Y, G1049A/C/D/R/S, E545A/D/G/K/L/Q/R/V, E453A/D/G/K/Q/V, E542A/D/G/K/Q/R/V, K111N/R/E, Q546E/H/K/L/P/R, G106A/D/R/S/V, N345D/H/I/K/S/T/Y, G118D, C420R, R88Q und M1043I/T/V. In allen Proben der eingeschlossenen Patienten wurde mindestens eine relevante PIK3CA-Mutation an mindestens einer dieser Aminosäurepositionen identifiziert. Basierend auf den Ergebnissen des zentralen FoundationOne® Liquid CDx-Assays waren die häufigsten PIK3CA-Mutationen kurze Varianten an den Aminosäuren H1047 (n = 115, 42,6 %), E 545 (n = 58, 21,5 %) und E 542 (n = 39, 14,4 %). Bei 25 Patienten wiesen die Proben mehr als eine PIK3CA-Mutation (d. h. multiple PIK3CA-Mutationen) auf, bei 33 Patienten traten seltenere PIK3CA-Mutationen auf.
Insgesamt wurden 325 Patienten im Verhältnis 1:1 randomisiert und erhielten entweder Itovebi 9 mg (n = 161) oder Placebo (n = 164) oral einmal täglich in Kombination mit Palbociclib und Fulvestrant bis zur Krankheitsprogression oder bis zum Auftreten einer inakzeptablen Toxizität. Darüber hinaus erhielten prä-/perimenopausale Frauen und Männer während der gesamten Therapie einen LHRH-Agonisten. Die Randomisierung wurde nach Vorhandensein einer viszeralen Erkrankung (ja oder nein), endokriner Resistenz (primär oder sekundär) und geografischer Region (Nordamerika/Westeuropa, Asien, andere) stratifiziert.
Die Charakteristika bezüglich Demographie und Krankheitsbild bei Behandlungsbeginn waren: medianes Alter 54 Jahre (Bereich: 27 bis 79 Jahre, 18,2 % waren ≥ 65 Jahre alt); 98,2 % weiblich; 38,2 % prä-/perimenopausal; 58,8 % weiß, 38,2 % asiatisch, 2,5 % unbekannt, 0,6 % schwarz oder afroamerikanisch; 6,2 % hispanisch oder lateinamerikanisch; und Leistungsstatus gemäß der ECOG (Eastern Cooperative Oncology Group) von 0 (63,4 %) oder 1 (36,3 %). Tamoxifen (56,9 %) und Aromatasehemmer (50,2 %) waren die am häufigsten angewendeten adjuvanten endokrinen Therapien. Drei (0,9 %) Patienten hatten zuvor eine Therapie mit einem CDK4/6-Inhibitor erhalten. Die demographischen Merkmale und die Krankheitsmerkmale zu Studienbeginn waren zwischen den Studienarmen ausgeglichen und vergleichbar.
Der primäre Wirksamkeitsendpunkt war das vom Prüfarzt (INV, investigator) beurteilte progressionsfreie Überleben (PFS, progression‑free survival) gemäß den Response Evaluation Criteria in Solid Tumors (RECIST) Version 1.1. Die sekundären Wirksamkeitsendpunkte umfassten das Gesamtüberleben (OS, overall survival), die objektive Ansprechrate (ORR, objective response rate), die beste Gesamtansprechrate (BOR, best overall response), die klinische Benefitrate (CBR, clinical benefit rate), die Dauer des Ansprechens (DOR, duration of response) und die Zeit bis zur bestätigten Verschlechterung (TTCD, time to confirmed deterioration) bei Schmerzen, körperlicher Funktion, Rollenfunktion und den allgemeinen Gesundheitszustand/die gesundheitsbezogene Lebensqualität (HRQoL, health‑related quality of life).
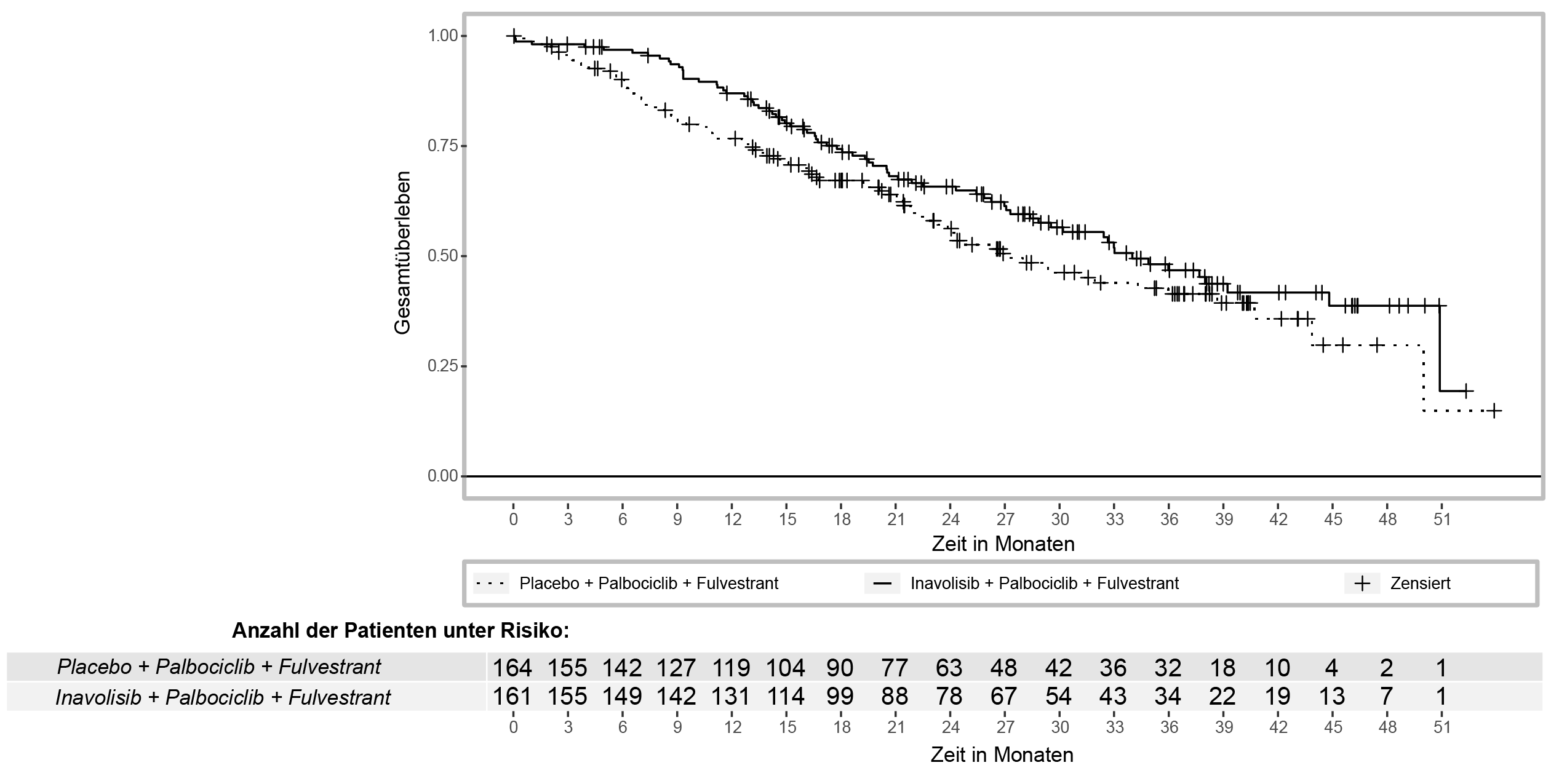
Die Ergebnisse zur Wirksamkeit sind in Tabelle 7, Abbildung 1 und Abbildung 2 zusammengefasst. INV-bewertete PFS-Ergebnisse wurden durch konsistente Ergebnisse der verblindeten, unabhängigen, zentralen Überprüfung (BICR, blinded independent central review) gestützt.
Tabelle 7: Ergebnisse zur Wirksamkeit bei Patienten mit lokal fortgeschrittenem oder metastasiertem Brustkrebs in der Studie INAVO120
Wirksamkeitsendpunkt |
Itovebi+ Palbociclib + Fulvestrant |
Placebo + Palbociclib + Fulvestrant |
INV-beurteiltes progressionsfreies Überlebena | ||
Patienten mit Ereignis, n (%) |
82 (50,9) |
113 (68,9) |
Median, Monate (95-%-KI) |
15 (11,3; 20,5) |
7,3 (5,6; 9,3) |
Hazard-Ratio (95-%-KI) |
0,43 (0,32; 0,59) |
|
p-Wert |
< 0,0001 |
|
Gesamtüberlebenb,c | ||
Patienten mit Ereignis, n (%) |
72 (44,7) |
82 (50) |
Median, Monate (95-%-KI) |
34 (28,4; 44,8) |
27 (22,8; 38,7) |
Hazard-Ratio (95-%-KI) |
0,67 (0,48; 0,94) |
|
p-Wert |
0,0190 |
|
Objektive Ansprechrateb,d | ||
Patienten mit CR oder PR, n (%) |
101 (62,7) |
46 (28) |
95-%-KI |
(54,8; 70,2) |
(21,3; 35,6) |
p-Wert |
< 0,0001 |
|
Ansprechdauer | ||
Mediane DOR, Monate (95‑%‑KI) |
19,2 (14,7; 28,3) |
11,1 (8,5; 20,2) |
KI = Konfidenzintervall (confidence interval); CR = vollständiges Ansprechen (complete response); PR = partielles Ansprechen (partial response) | ||
Abbildung 1: Vom Prüfarzt beurteiltes progressionsfreies Überleben bei Patienten mit lokal fortgeschrittenem oder metastasiertem Mammakarzinom in der INAVO120
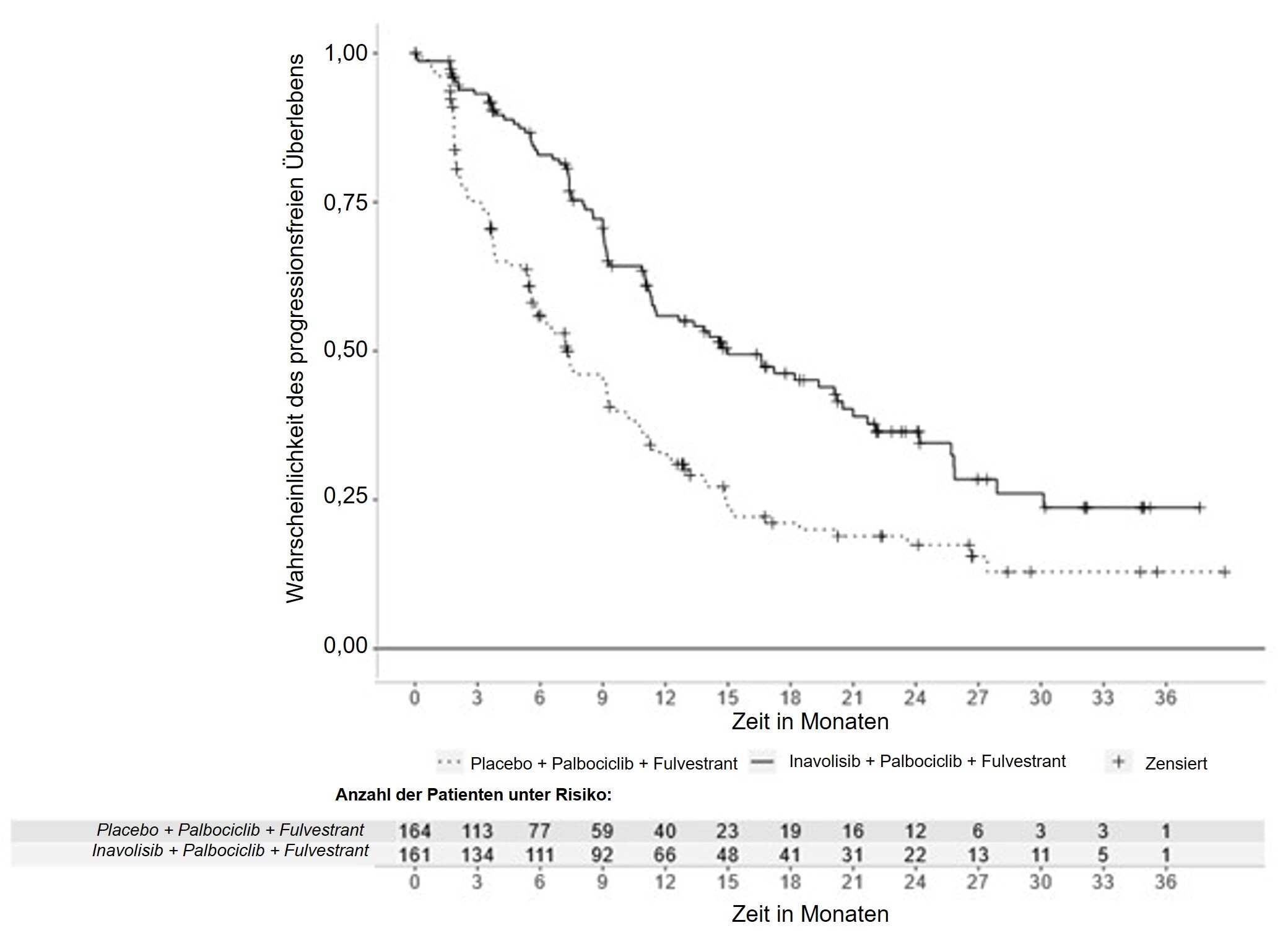
Abbildung 2: Gesamtüberleben bei Patienten mit lokal fortgeschrittenem oder metastasiertem Brustkrebs in der Studie INAVO120
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Itovebi eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen bei Brustkrebs gewährt (für Informationen zur pädiatrischen Anwendung siehe Abschnitt 4.2).
Die Pharmakokinetik von Inavolisib wurde bei gesunden Probanden und bei Patienten mit lokal fortgeschrittenen oder metastasierten PIK3CA-mutierten soliden Tumoren, einschließlich Brustkrebs, unter einem oralen Dosierungsschema im Bereich von 6 mg bis 12 mg täglich und bei gesunden Probanden bei einer Einzeldosis von 9 mg charakterisiert.
Sofern nicht anders angegeben, wird die Pharmakokinetik von Inavolisib als geometrischer Mittelwert (geometrischer Variationskoeffizient [geo CV] %) nach Verabreichung der zugelassenen empfohlenen Dosierung angegeben. Basierend auf einer populationspharmakokinetischen Analyse betrug die AUC im Steady-State von Inavolisib 1 019 h*ng/ml (29 %) und die Cmax 67 ng/ml (28 %). Steady-State-Konzentrationen sollen gemäß Vorhersage bis Tag 5 erreicht sein.
Bei einer Dosierung von 9 mg einmal täglich betrug das geometrische Mittel des Akkumulationsverhältnisses ungefähr das 2-Fache.
Resorption
Die Zeit bis zur maximalen Plasmakonzentration (Tmax) wurde nach einer täglichen Gabe von 9 mg Inavolisib unter Nüchternbedingungen im Median nach 3 Stunden (Bereich: 0,5 bis 4 Stunden) im Steady State erreicht.
Die absolute orale Bioverfügbarkeit von Inavolisib betrug 76 %.
Wechselwirkung mit Nahrung
Es wurde keine klinisch signifikante Wirkung von Nahrung auf die Bioverfügbarkeit von Inavolisib beobachtet. Das Verhältnis der geometrischen Mittelwerte (GMR, geometric mean ratio) (90-%-KI) für AUC0-24 beim Vergleich von nicht nüchternem und nüchternem Zustand betrug 0,895 (0,737 – 1,09) nach einer Einzeldosis und 0,876 (0,701 – 1,09) im Steady State. Die GMR (90-%-KI) für C max im Vergleich von nicht nüchternem zu nüchternem Zustand betrug 0,925 (0,748 – 1,14) nach einer Einzeldosis und 0,910 (0,712 – 1,16) im Steady State.
Verteilung
Die Plasmaproteinbindung von Inavolisib beim Menschen beträgt 37 % und schien über den getesteten Konzentrationsbereich nicht konzentrationsabhängig zu sein (0,1 – 10 μM). Beim Menschen beträgt das auf Basis einer populationspharmakokinetischen Analyse geschätzte orale Verteilungsvolumen im Steady State 155 l (26 %).
Biotransformation
Nach oraler Gabe einer radioaktiv markierten Einzeldosis von 9 mg Inavolisib an gesunde Probanden war die Muttersubstanz die am häufigsten vorkommende wirkstoffbezogene Verbindung im Plasma und im Urin. Hydrolyse war der hauptsächliche Stoffwechselweg. Es wurden keine spezifischen Hydrolyseenzyme identifiziert, die am Stoffwechsel von Inavolisib beteiligt sind.
Elimination
Nach oraler Gabe einer radioaktiv markierten Einzeldosis von 9 mg Inavolisib an gesunde Probanden wurden 48,5 % der verabreichten Dosis im Urin (40,4 % unverändert) und 48 % in den Fäzes (10,8 % unverändert) wiedergefunden.
In klinischen Studien, basierend auf einer populationspharmakokinetischen Analyse, betrug das geometrische Mittel der Schätzungen der individuellen Halbwertszeit für Inavolisib 15 Stunden (24 %) nach einer Einzeldosis von 9 mg. Die geschätzte Gesamtclearance von Inavolisib beträgt 8,8 l/h (29 %).
Linearität/Nicht-Linearität
Begrenzte Daten deuten auf eine Dosisproportionalität innerhalb des getesteten Dosisbereichs (6 bis 12 mg) für die Einzeldosis-Cmax, die AUC0-24 und die Steady-State-AUC0-24 hin; für die Steady-State-Cmax deuten die Daten jedoch auf eine Nichtproportionalität hin.
Arzneimittelwechselwirkungen
Die Ergebnisse klinischer Studien weisen darauf hin, dass die Hauptmetaboliten von Inavolisib nicht durch CYP-Enzyme metabolisiert werden. Dies deutet auf eine geringe Wahrscheinlichkeit für klinisch relevante Wechselwirkungen zwischen Inavolisib und CYP-Inhibitoren oder -Induktoren hin. Darüber hinaus zeigten in‑vitro-Ergebnisse, dass Inavolisib die Enzyme CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 oder CYP2D6 nicht hemmt.
In-vitro- Studien weisen darauf hin, dass Inavolisib nicht das Potential hat, einen der getesteten Arzneimitteltransporter zu hemmen. Darüber hinaus ist Inavolisib in vitro ein Substrat von P-Glykoprotein (P-gp) und Breast Cancer Resistance Protein (BCRP). Aufgrund der pharmakokinetischen Gesamtcharakteristika von Inavolisib ist aber nicht zu erwarten, dass Inhibitoren oder Induktoren von P-gp und/oder BCRP mit Inavolisib klinisch relevante Arzneimittelwechselwirkungen hervorrufen.
Besondere Patientengruppen
Ältere Patienten
Basierend auf der populationspharmakokinetischen Analyse wurden keine klinisch relevanten Unterschiede in der Pharmakokinetik von Inavolisib zwischen Patienten, die 65 Jahre oder älter waren, und Patienten, die jünger als 65 Jahre waren, festgestellt. Von den 162 Patienten, die Itovebi in der INAVO120 Studie erhielten, waren 24 Patienten ≥ 65 Jahre alt.
Nierenfunktionsstörung
Populationspharmakokinetische Analysen zeigten, dass eine leichte Nierenfunktionsstörung keine klinisch relevante Kovariate der Inavolisib-Exposition ist. Die Pharmakokinetik von Inavolisib bei Patienten mit leichter Nierenfunktionsstörung (eGFR 60 bis < 90 ml/min) war ähnlich wie bei Patienten mit normaler Nierenfunktion. Die AUC und Cmax von Inavolisib waren bei Patienten mit mittelschwerer Nierenfunktionsstörung (eGFR 30 bis < 60 ml/min) um 73 % bzw. 11 % höher und bei Patienten mit schwerer Nierenfunktionsstörung (eGFR < 30 ml/min) um 123 % bzw. 33 % höher als bei Patienten mit normaler Nierenfunktion (eGFR ≥ 90 ml/min).
Leberfunktionsstörung
Populationspharmakokinetische Analysen zeigten, dass eine leichte Leberfunktionsstörung keine klinisch relevante Kovariate der Inavolisib Exposition ist. Die Pharmakokinetik von Inavolisib bei Patienten mit leichter Leberfunktionsstörung (Gesamtbilirubin > ULN bis ≤ 1,5 × ULN oder AST > ULN und Gesamtbilirubin ≤ ULN) war ähnlich wie bei Patienten mit normaler Leberfunktion. Die Auswirkungen einer mittelschweren bis schwerwiegenden Leberfunktionsstörung auf die Pharmakokinetik von Inavolisib wurden nicht untersucht.
Genotoxizität
Inavolisib war im bakteriellen Mutagenitätstest nicht mutagen.
Inavolisib zeigte In vitro-Klastogenität; es gab jedoch keine Hinweise auf eine durch Inavolisib induzierte In-vivo-Genotoxizität (Klastogenität, Aneugenität oder DNA-Schädigung) in der Mikronukleus- und Comet-Studie an Ratten bei Dosen bis zu einer maximal tolerierten Dosis (MTD, maximum tolerated dose) des 16-Fachen der Exposition bei einer klinischen Dosis von 9 mg.
Karzinogenität
Es wurden keine Karzinogenitätsstudien mit Inavolisib durchgeführt.
Entwicklungstoxizität
In einer embryofetalen Entwicklungsstudie an Sprague-Dawley-Ratten wurden dosisabhängige Wirkungen von Inavolisib auf die embryofetale Entwicklung festgestellt, die eine Abnahme des fetalen Körpergewichts und des Plazentagewichts, einen Postimplantationsverlust, eine geringere Lebensfähigkeit des Fetus und Teratogenität (fetale äußere, viszerale und skelettale Fehlbildungen) umfassten, wobei die maternale Exposition beim No-Observed-Adverse-Effect Level (NOAEL) das 0,2-Fache der Exposition bei einer klinischen Dosis von 9 mg betrug.
Fertilität
Es wurden keine speziellen Fertilitätsstudien mit Inavolisib durchgeführt.
Bei männlichen Ratten wurden eine dosisabhängige Atrophie der Prostata und der Samenbläschen sowie verminderte Organgewichte ohne mikroskopische Korrelate in den Nebenhoden und Hoden beobachtet (bei einem ΝΟΑΕL ≥ der 0,4-Fachen Exposition bei einer klinischen Dosis von 9 mg). Diese Befunde waren reversibel. Bei männlichen Hunden wurden nach 4-wöchiger Dosierung (bei dem ≥ 2-Fachen der Exposition bei einer klinischen Dosis von 9 mg) fokale Eindickungen des Inhalts der Samenkanälchen und mehrkernige Spermatiden in den Hoden sowie eine Epitheldegeneration/-nekrose in den Nebenhoden beobachtet. Nach 3-monatiger Behandlung mit Dosen, die der Exposition bis zu dem 1,2-Fachen der Exposition bei einer klinischen Dosis von 9 mg entsprachen, wurde eine reversible Abnahme der Gesamtspermienzahl mit einem NOAEL der 0,4-Fachen Exposition bei einer klinischen Dosis von 9 mg beobachtet. Es ergaben sich aber keine durch Inavolisib bedingten mikroskopischen Befunde in den Hoden oder Nebenhoden oder Auswirkungen auf die Spermienkonzentration, -motilität oder -morphologie.
Bei weiblichen Ratten wurden eine minimale bis leichte Atrophie in Uterus und Vagina, eine Abnahme der Ovarialfollikel und Befunde, die auf eine Unterbrechung/Veränderung des Östruszyklus hindeuten (bei dem ≥ 1,2-Fachen der Exposition bei einer klinischen Dosis von 9 mg), beobachtet, mit einem NOAEL des 0,5-Fachen der Exposition bei einer klinischen Dosis von 9 mg. Diese Befunde wurden in der 4-wöchigen Toxizitätsstudie nach der Erholungsphase nicht beobachtet. Die Erholung wurde in der 3-monatigen Studie an Ratten nicht untersucht.
Andere
Nebenwirkungen, die nicht in klinischen Studien beobachtet wurden, aber bei Tieren nach Exposition im humantherapeutischen Bereich auftraten und als möglicherweise relevant für die klinische Anwendung zu bewerten sind, umfassten Entzündungen bei Hunden und eine Degeneration der Augenlinse bei Ratten. Die Entzündung steht im Einklang mit den erwarteten pharmakologischen Wirkungen der PI3K-Hemmung, war im Allgemeinen dosisabhängig und reversibel. Eine bei einigen Ratten beobachtete minimale Linsenfaserdegeneration (bei dem ≥ 3,6-Fachen der Exposition bei einer klinischen Dosis von 9 mg) wurde als irreversibel angesehen.
Itovebi 3 mg und 9 mg Tablettenkern
Lactose-Monohydrat
Magnesiumstearat (E 470b)
Mikrokristalline Cellulose (E 460)
Carboxymethylstärke-Natrium
Itovebi 3 mg Filmüberzug
Polyvinylalkohol, partiell hydrolysiert
Titandioxid (E 171)
Macrogol
Talkum (E 553b)
Eisen(III)-oxid (E 172)
Itovebi 9 mg Filmüberzug
Polyvinylalkohol, partiell hydrolysiert
Titandioxid (E 171)
Macrogol
Talkum (E 553b)
Eisen(III)-oxid (E 172)
Eisen(III)-hydroxid-oxid-Hydrat (E 172)
Nicht zutreffend.
3 Jahre
Für dieses Arzneimittel sind bezüglich der Temperatur keine besonderen Lagerungsbedingungen erforderlich. In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Perforierte Alu/Alu (Aluminium/Aluminium) Einzeldosis-Blisterpackungen in Umkartons mit 28 × 1 Filmtabletten.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Roche Registration GmbH
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Deutschland
EU/1/25/1942/001
EU/1/25/1942/002
Datum der Erteilung der Zulassung: 18. Juli 2025
April 2026
Verschreibungspflichtig
Roche Pharma AG
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Telefon (07624) 14-0
Telefax (07624) 1019
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.