
Erbitux 5 mg/ml Infusionslösung
1 ml Infusionslösung enthält 5 mg Cetuximab.
Eine Durchstechflasche mit 20 ml enthält 100 mg Cetuximab.
Eine Durchstechflasche mit 100 ml enthält 500 mg Cetuximab.
Cetuximab ist ein mittels rekombinanter DNA-Technologie aus einer Säugerzelllinie (Sp2/0) gewonnener chimärer monoklonaler IgG1-Antikörper.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Infusionslösung.
Farblose Lösung.
Erbitux wird angewendet zur Behandlung des metastasierenden, EGFR (epidermalen Wachstumsfaktor-Rezeptor) exprimierenden Kolorektalkarzinoms mit RAS-Wildtyp
in Kombination mit einer Irinotecan-basierten Chemotherapie,
als Erstlinienbehandlung in Kombination mit FOLFOX,
als Monotherapie bei Patienten, bei denen die Therapie mit Oxaliplatin und Irinotecan versagt hat und die Irinotecan nicht vertragen.
Einzelheiten siehe Abschnitt 5.1.
Erbitux wird angewendet zur Behandlung von erwachsenen Patienten mit metastasierendem Kolorektalkarzinom mit BRAF-V600E-Mutation
in Kombination mit Encorafenib und FOLFOX als Erstlinienbehandlung,
in Kombination mit Encorafenib bei Patienten, die zuvor eine systemische Therapie erhalten haben.
Einzelheiten siehe Abschnitt 5.1.; für die biomarkerbasierte Patientenwahl siehe Abschnitt 4.2.
Erbitux wird angewendet zur Behandlung von Patienten mit Plattenepithelkarzinom im Kopf- und Halsbereich
in Kombination mit einer Strahlentherapie für eine lokal fortgeschrittene Erkrankung,
in Kombination mit einer platin-basierten Chemotherapie für eine rezidivierende und/oder metastasierende Erkrankung.
Die Verabreichung von Erbitux muss stets unter Aufsicht eines in der Anwendung von antineoplastischen Arzneimitteln erfahrenen Arztes erfolgen. Während der Infusion und mindestens eine Stunde über deren Ende hinaus ist der Patient engmaschig zu überwachen. Die notwendige Ausrüstung zur Durchführung von Notfallmaßnahmen muss einsatzbereit sein.
Dosierung
Vor der ersten Infusion müssen die Patienten mindestens 1 Stunde vor der Verabreichung von Cetuximab mit einem Antihistaminikum und einem Kortikosteroid vorbehandelt werden. Diese Prämedikation empfiehlt sich auch vor allen weiteren Infusionen.
Kolorektalkarzinom
Metastasierendes Kolorektalkarzinom mit RAS-Wildtyp
Bei Patienten mit metastasierendem Kolorektalkarzinom mit RAS‑Wildtyp wird Cetuximab in Kombination mit einer Chemotherapie oder als Monotherapie angewendet (siehe Abschnitt 5.1). Der Nachweis des RAS‑Wildtyp‑Status (K‑RAS und N‑RAS) ist vor Beginn der Behandlung mit Erbitux erforderlich. Der Mutationsstatus sollte durch ein erfahrenes Labor unter Einsatz validierter Prüfmethoden zum Nachweis von K‑RAS (Exons 2, 3 und 4)- und N‑RAS (Exons 2, 3 und 4)‑Mutationen bestimmt werden (siehe Abschnitt 4.4 und 5.1).
Erbitux kann einmal wöchentlich oder einmal alle zwei Wochen verabreicht werden.
Wöchentliches Dosierungsschema
Erbitux wird einmal wöchentlich verabreicht. Die Initialdosis beträgt 400 mg Cetuximab/m2 Körperoberfläche. Danach werden einmal wöchentlich 250 mg/m2 verabreicht.
Zweiwöchentliches Dosierungsschema
Erbitux wird einmal alle zwei Wochen verabreicht. Jede Dosis beträgt 500 mg Cetuximab/m2 Körperoberfläche.
Metastasierendes Kolorektalkarzinom mit BRAF-V600E-Mutation
Bei Patienten mit BRAF-V600E-mutiertem metastasierendem Kolorektalkarzinom wird Cetuximab in Kombination mit Encorafenib und FOLFOX oder mit Encorafenib angewendet (siehe Abschnitt 5.1). Der Nachweis des BRAF-V600E‑Mutationsstatus ist vor Beginn der Behandlung mit Erbitux erforderlich. Der Mutationsstatus sollte mittels eines CE‑gekennzeichneten In‑vitro‑Diagnostikums (IVD) mit dem entsprechenden Verwendungszweck bestimmt werden. Steht kein CE‑gekennzeichnetes IVD zur Verfügung, ist ein alternativer validierter Test anzuwenden.
In Kombination mit Encorafenib bei Patienten, die zuvor eine systemische Therapie erhalten haben
Erbitux wird einmal wöchentlich verabreicht. Die erste Dosis beträgt 400 mg Cetuximab/m2 Körperoberfläche. Danach werden einmal wöchentlich 250 mg/m2 verabreicht.
In Kombination mit Encorafenib und FOLFOX
Erbitux wird einmal alle zwei Wochen verabreicht. Jede Dosis beträgt 500 mg Cetuximab/m2 Körperoberfläche.
Angaben zur Dosierung sowie zu empfohlenen Dosisänderungen von gleichzeitig angewandten Chemotherapeutika und Encorafenib sind den Produktinformationen dieser Arzneimittel zu entnehmen. Zwischen dem Ende der Cetuximab-Infusion und der Gabe der Chemotherapeutika muss ein Mindestabstand von einer Stunde eingehalten werden. Encorafenib sollte vor Beginn der Cetuximab-Infusion angewendet werden.
Es wird empfohlen, die Behandlung mit Cetuximab bis zum Fortschreiten der Grunderkrankung fortzusetzen.
Plattenepithelkarzinom im Kopf- und Halsbereich
In Kombination mit einer Strahlentherapie
Zur Behandlung von Patienten mit lokal fortgeschrittenem Plattenepithelkarzinom im Kopf- und Halsbereich wird Cetuximab in Kombination mit Bestrahlung angewendet. Es wird empfohlen, die Behandlung mit Cetuximab eine Woche vor der Bestrahlung zu beginnen und bis zum Ende des Bestrahlungszeitraumes fortzusetzen.
Erbitux wird einmal wöchentlich verabreicht. Die Initialdosis beträgt 400 mg Cetuximab/m2 Körperoberfläche. Danach werden einmal wöchentlich 250 mg/m2 verabreicht.
In Kombination mit einer platin-basierten Chemotherapie
Bei Patienten mit rezidivierendem und/oder metastasierendem Plattenepithelkarzinom im Kopf- und Halsbereich wird Cetuximab in Kombination mit einer platin-basierten Chemotherapie angewendet, gefolgt von Cetuximab als Erhaltungstherapie bis zur Progression der Erkrankung (siehe Abschnitt 5.1). Zwischen dem Ende der Cetuximab-Infusion und der Chemotherapie muss ein Mindestabstand von einer Stunde eingehalten werden.
Erbitux kann einmal wöchentlich oder einmal alle zwei Wochen verabreicht werden.
Wöchentliches Dosierungsschema
Erbitux wird einmal wöchentlich verabreicht. Die Initialdosis beträgt 400 mg Cetuximab/m2 Körperoberfläche. Danach werden einmal wöchentlich 250 mg/m2 verabreicht.
Zweiwöchentliches Dosierungsschema
Erbitux wird einmal alle zwei Wochen verabreicht. Jede Dosis beträgt 500 mg Cetuximab/m2 Körperoberfläche.
Spezielle Patientengruppen
Cetuximab wurde bei Patienten mit vorbestehenden hämatologischen Erkrankungen nicht untersucht (siehe Abschnitt 4.4).
Bei älteren Patienten ist keine Dosisanpassung notwendig, allerdings liegen Erfahrungen mit Patienten im Alter von ≥75 Jahren nur in beschränktem Umfang vor.
Bisher wurden Patienten mit leichter (CRCL ≥ 60 und < 90 ml/min) und mäßiger (CRCL ≥ 30 und < 60 ml/min) Nierenfunktionsstörung sowie leichter Leberfunktionsstörung gemäß den Kriterien der National Cancer Institute Organ Dysfunction Working Group (NCI‑ODWG) untersucht. Der Einfluss einer mäßigen oder schweren Leberfunktionsstörung (gemäß NCI‑ODWG‑Kriterien) auf die Pharmakokinetik von Cetuximab wurde nicht untersucht. Die Pharmakokinetik von Cetuximab bei Patienten mit schwerer (CRCL ≥ 15 und < 30 ml/min) Nierenfunktionsstörung oder terminaler Niereninsuffizienz wurde nicht evaluiert. Siehe Abschnitt 5.2.
Kinder und Jugendliche
Es gibt in den zugelassenen Anwendungsgebieten keinen relevanten Nutzen von Cetuximab bei Kindern und Jugendlichen.
Art der Anwendung
Erbitux 5 mg/ml wird intravenös entweder mit einer Infusionspumpe oder als Tropfinfusion oder mit einem Perfusor verabreicht (Hinweise für die Handhabung siehe Abschnitt 6.6).
Die Initialdosis sollte langsam gegeben werden, um das Risiko für infusionsbedingte Reaktionen zu minimieren (siehe Abschnitt 4.4). Die empfohlene Infusionsdauer beträgt 120 Minuten. Bei nachfolgenden Gaben von Cetuximab darf die Infusionsgeschwindigkeit 10 mg/min nicht überschreiten. Wenn die Erstinfusion gut vertragen wird, beträgt die empfohlene Infusionsdauer beim wöchentlichen Dosierungsschema mit 250 mg/m2 60 Minuten und beim zweiwöchentlichen Dosierungsschema mit 500 mg/m2 120 Minuten.
Erbitux ist kontraindiziert bei Patienten mit bekannten schweren Überempfindlichkeitsreaktionen (Grad 3 oder 4) gegen Cetuximab.
Die Kombination von Erbitux mit Oxaliplatin-haltiger Chemotherapie ist kontraindiziert bei Patienten mit metastasierendem Kolorektalkarzinom mit RAS-Mutation oder unbekanntem RAS-Mutationsstatus (siehe auch Abschnitt 4.4).
Vor Beginn einer Kombinationsbehandlung sind die Gegenanzeigen für die gleichzeitig angewandten Chemotherapeutika oder für eine Strahlentherapie zu beachten.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Infusionsbedingte Reaktionen einschließlich anaphylaktischer Reaktionen
Häufig können schwere infusionsbedingte Reaktionen einschließlich anaphylaktischer Reaktionen auftreten, die in einigen Fällen zum Tode führen. Das Auftreten einer schweren infusionsbedingten Reaktion macht den sofortigen und dauerhaften Abbruch der Behandlung mit Cetuximab erforderlich. Gegebenenfalls sind Notfallmaßnahmen zu ergreifen. Einige dieser Reaktionen können anaphylaktischer oder anaphylaktoider Natur sein oder ein Zytokinfreisetzungssyndrom (CRS) darstellen. Die Symptome können während der ersten Infusion und auch noch mehrere Stunden danach oder bei nachfolgenden Infusionen auftreten. Es wird empfohlen, die Patienten über die Möglichkeit eines derartig verspäteten Einsetzens aufzuklären und ihnen nahezulegen, bei Symptomen oder Anzeichen von infusionsbedingten Reaktionen ihren Arzt zu kontaktieren. Die Symptome können Bronchospasmen, Urtikaria, Blutdruckanstieg oder -abfall, Bewusstlosigkeit oder Schock umfassen. In seltenen Fällen wurden Angina pectoris, Myokardinfarkt oder Herzstillstand beobachtet.
Anaphylaktische Reaktionen können bereits innerhalb weniger Minuten während der ersten Infusion auftreten, z. B. wenn es zu einer Kreuzreaktion zwischen bereits gebildeten IgE-Antikörpern und Cetuximab kommt. Diese Reaktionen gehen häufig mit Bronchospasmus und Urtikaria einher und können trotz Gabe einer Prämedikation auftreten.
Das Risiko für anaphylaktische Reaktionen ist stark erhöht bei Patienten mit einer bekannten Allergie gegen rotes Fleisch oder Zeckenbisse oder positiven Ergebnissen bei Tests auf IgE‑Antikörper gegen Cetuximab (α‑1‑3‑Galactose). Bei diesen Patienten sollte Cetuximab nur verabreicht werden nach sorgfältiger Abwägung des Nutzen‑Risiko‑Verhältnisses einschließlich alternativer Behandlungen und nur unter engmaschiger Überwachung durch gut geschultes Personal mit Zugang zu einsatzbereiter Ausrüstung zur Durchführung von Reanimationen.
Die erste Dosis sollte langsam gegeben werden. Dabei müssen alle Vitalparameter mindestens zwei Stunden lang engmaschig kontrolliert werden. Wenn bei der ersten Infusion innerhalb der ersten 15 Minuten eine infusionsbedingte Reaktion auftritt, sollte die Infusion abgebrochen werden. Vor einer erneuten Infusion sollte eine sorgfältige Abwägung des Nutzen‑Risiko‑Verhältnisses unter Berücksichtigung möglicher, beim Patienten bereits gebildeter, IgE‑Antikörper erfolgen.
Kommt es im späteren Verlauf der Infusion oder bei einer nachfolgenden Infusion zu einer infusionsbedingten Reaktion, ist die weitere Behandlung von der Schwere der Reaktion abhängig:
a) Grad 1 Langsame Infusion unter engmaschiger Überwachung fortsetzen
b) Grad 2 Langsame Infusion fortsetzen und unverzüglich die Symptome behandeln
c) Grad 3 und 4 Infusion sofort abbrechen und Symptome intensiv behandeln. Die weitere Anwendung von Cetuximab ist kontraindiziert.
Ein Zytokinfreisetzungssyndrom (CRS) tritt typischerweise innerhalb einer Stunde nach Infusion auf und geht weniger häufig mit Bronchospasmus und Urtikaria einher. Ein CRS ist normalerweise im Rahmen der ersten Infusion am stärksten ausgeprägt.
Sehr häufig sind leichte oder mittelschwere infusionsbedingte Reaktionen mit Symptomen wie Fieber, Schüttelfrost, Schwindel oder Atemnot, die in engem zeitlichem Zusammenhang vor allem zur ersten Infusion von Cetuximab stehen. Wenn beim Patienten leichte oder mittelschwere infusionsbedingte Reaktionen auftreten, kann die Infusionsgeschwindigkeit reduziert werden. Es wird empfohlen, auch alle nachfolgenden Infusionen mit der langsameren Infusionsgeschwindigkeit durchzuführen.
Eine engmaschige Beobachtung der Patienten ist erforderlich, vor allem während der ersten Anwendung. Bei Patienten mit reduziertem Allgemeinzustand und bestehenden Herz-Lungen-Erkrankungen ist besondere Vorsicht angezeigt.
Erkrankungen der Atemwege
Es traten Fälle einer interstitiellen Lungenerkrankung (ILD), auch mit tödlichem Ausgang, auf, wobei die meisten Patienten japanischer Herkunft waren.
Bei den tödlich verlaufenen Fällen lagen häufig Stör- oder Einflussfaktoren vor, z. B. eine gleichzeitige Chemotherapie, die bekanntlich mit ILD assoziiert ist, oder vorbestehende Lungenerkrankungen. Solche Patienten sollten engmaschig überwacht werden. Im Falle von Symptomen (wie z. B. Atemnot, Husten oder Fieber) oder radiologischen Befunden, die auf eine ILD hindeuten, sollte umgehend eine diagnostische Abklärung erfolgen.
Wenn eine interstitielle Lungenerkrankung diagnostiziert wird, muss die Behandlung mit Cetuximab abgebrochen und der Patient angemessen behandelt werden.
Hautreaktionen
Nebenwirkungen von Cetuximab sind vorwiegend Hautreaktionen, die insbesondere in Kombination mit einer Chemotherapie einen schweren Verlauf nehmen können. Das Risiko von Sekundärinfektionen (hauptsächlich bakteriell) ist erhöht und es wurden Fälle von staphylogenem Lyell-Syndrom, nekrotisierender Fasziitis und Sepsis, in manchen Fällen mit tödlichem Ausgang, berichtet (siehe Abschnitt 4.8).
Hautreaktionen sind sehr häufig und können unter Umständen eine Unterbrechung oder Beendigung der Behandlung erfordern. Entsprechend den klinischen Praxisleitlinien sollte die prophylaktische Gabe von oralen Tetrazyklinen (6‑8 Wochen) und die topische Anwendung einer feuchtigkeitsspendenden 1%igen Hydrocortisoncreme erwogen werden. Zur Behandlung von Hautreaktionen wurden mäßig bis stark wirksame topische Kortikosteroide oder orale Tetrazykline eingesetzt.
Beim Auftreten von nicht tolerierbaren oder schweren Hautreaktionen (≥Grad 3; Common Terminology Criteria for Adverse Events, CTCAE) muss die Behandlung mit Cetuximab unterbrochen werden. Die Behandlung darf erst wieder aufgenommen werden, wenn sich die Hautreaktion auf Grad 2 zurückgebildet hat.
Ist diese schwere Hautreaktion zum ersten Mal aufgetreten, kann die Behandlung ohne Dosisanpassung wieder aufgenommen werden.
Treten die schweren Hautreaktionen ein zweites oder drittes Mal auf, muss die Cetuximabtherapie erneut abgebrochen werden.
Wenn sich die Reaktion auf Grad 2 zurückgebildet hat, darf die Behandlung nach dem zweiten Auftreten nur mit einer um 20 % niedrigeren Dosis (200 mg/m2 Körperoberfläche beim wöchentlichen Dosierungsschema, 400 mg/m2 Körperoberfläche beim zweiwöchentlichen Dosierungsschema) und nach dem dritten Auftreten nur mit einer um 40 % niedrigeren Dosis (150 mg/m2 Körperoberfläche beim wöchentlichen Dosierungsschema, 300 mg/m2 Körperoberfläche beim zweiwöchentlichen Dosierungsschema) wieder aufgenommen werden.
Wenn eine schwere Hautreaktion zum vierten Mal auftritt oder sich während der Therapieunterbrechung nicht auf Grad 2 zurückbildet, muss die Behandlung mit Cetuximab endgültig abgebrochen werden.
Elektrolytstörungen
Häufig tritt ein fortschreitender Abfall des Magnesium-Serumspiegels auf, der zu schwerer Hypomagnesiämie führen kann. Die Hypomagnesiämie ist nach Absetzen von Cetuximab reversibel. Zusätzlich kann es infolge einer Diarrhö zu einer Hypokaliämie kommen. Eine Hypokalzämie kann ebenfalls auftreten; vor allem in Kombination mit einer platin-basierten Chemotherapie kann die Häufigkeit einer schweren Hypokalzämie erhöht sein.
Vor und in regelmäßigen Abständen während der Behandlung mit Cetuximab wird eine Bestimmung der Elektrolytwerte im Serum empfohlen. Falls erforderlich wird ein Elektrolytersatz empfohlen.
Neutropenie und damit verbundene infektiöse Komplikationen
Für Patienten, die Cetuximab in Kombination mit einer platin-basierten Chemotherapie erhalten, besteht ein erhöhtes Risiko für das Auftreten einer schweren Neutropenie, die zu nachfolgenden infektiösen Komplikationen wie febriler Neutropenie, Pneumonie oder Sepsis führen kann. Diese Patienten sollten sorgfältig überwacht werden, insbesondere jene, die Hautläsionen, Mukositis oder eine Diarrhö entwickeln (was möglicherweise das Auftreten von Infektionen erleichtert) (siehe Abschnitt 4.8).
Kardiovaskuläre Erkrankungen
Bei der Behandlung von nicht-kleinzelligen Lungenkarzinomen, Plattenepithelkarzinomen des Kopf- und Halsbereichs und Kolorektalkarzinomen wurde eine erhöhte Häufigkeit schwerer und gelegentlich tödlich verlaufender kardiovaskulärer Ereignisse und behandlungsbedingter Todesfälle beobachtet. In manchen Studien wurde eine Assoziation mit einem Alter ≥ 65 Jahren oder dem Allgemeinzustand beobachtet. Bei der Verschreibung von Cetuximab sollte der kardiovaskuläre Status und der Allgemeinzustand der Patienten und eine begleitende Verabreichung kardiotoxischer Substanzen wie z. B. Fluoropyrimidine berücksichtigt werden.
Augenerkrankungen
Patienten, bei denen Anzeichen und Symptome vorliegen, die auf eine Keratitis hindeuten, wie akute oder sich verschlechternde Entzündung des Auges, Tränensekretion, Lichtempfindlichkeit, verschwommenes Sehen, Schmerzen im Auge und/oder gerötete Augen, sollten umgehend einen Augenarzt aufsuchen.
Bei Bestätigung der Diagnose einer ulzerativen Keratitis sollte die Behandlung mit Cetuximab unterbrochen oder abgebrochen werden. Wenn eine Keratitis diagnostiziert wurde, sollten der Nutzen und die Risiken einer Weiterbehandlung sorgfältig abgewogen werden.
Cetuximab sollte bei Patienten mit einer Vorgeschichte von Keratitis, ulzerativer Keratitis oder schwerer Form eines trockenen Auges mit Vorsicht angewendet werden. Die Verwendung von Kontaktlinsen ist auch ein Risikofaktor für Keratitis und Ulzeration.
Patienten mit RAS-mutiertem Kolorektalkarzinom
Cetuximab sollte bei Patienten mit Kolorektalkarzinom, deren Tumoren RAS-Mutationen aufweisen oder bei denen der RAS-Tumorstatus unbekannt ist, nicht angewendet werden. Ergebnisse klinischer Studien zeigen für Tumoren mit RAS-Mutationen ein negatives Nutzen-Risiko-Verhältnis. Insbesondere zeigte sich bei diesen Patienten bei Verwendung von Cetuximab als Zusatzbehandlung zu FOLFOX4 ein negativer Effekt auf die progressionsfreie Überlebenszeit (PFS) und die Gesamtüberlebenszeit (OS) (siehe Abschnitt 5.1).
Ähnliche Befunde wurden auch von der Gabe von Cetuximab als Zusatzbehandlung zu XELOX in Kombination mit Bevacizumab (CAIRO2) berichtet. In dieser Studie gelang es jedoch auch nicht, positive Effekte auf die PFS oder OS bei Patienten mit K-RAS-Wildtyp-Tumoren zu zeigen.
Spezielle Patientengruppen
Es liegen keine Erfahrungen mit Cetuximab bei Patienten mit einem oder mehreren der folgenden Laborwerte vor:
Hämoglobin < 9 g/dl
Leukozyten < 3 000/mm³
absolute Neutrophilen < 1 500/mm³
Thrombozyten < 100 000/mm³
Erfahrungen mit dem Einsatz von Cetuximab in Kombination mit Bestrahlung zur Behandlung von Kolorektalkarzinomen liegen nur in beschränktem Umfang vor.
Bisher wurden Patienten mit leichter (CRCL ≥ 60 und < 90 ml/min) und mäßiger (CRCL ≥ 30 und < 60 ml/min) Nierenfunktionsstörung sowie leichter Leberfunktionsstörung gemäß den NCI‑ODWG‑Kriterien untersucht. Der Einfluss einer mäßigen oder schweren Leberfunktionsstörung (gemäß NCI‑ODWG‑Kriterien) auf die Pharmakokinetik von Cetuximab wurde nicht untersucht. Die Pharmakokinetik von Cetuximab bei Patienten mit schwerer (CRCL ≥ 15 und < 30 ml/min) Nierenfunktionsstörung oder terminaler Niereninsuffizienz wurde nicht evaluiert. Siehe Abschnitt 5.2.
Kinder und Jugendliche
Die Wirksamkeit von Cetuximab bei Kindern und Jugendlichen unter 18 Jahren ist nicht erwiesen. Aus einer Phase-I-Studie gingen keine neuen sicherheitsrelevanten Hinweise bei Kindern und Jugendlichen hervor.
In Kombination mit einer platin-basierten Chemotherapie kann die Häufigkeit einer schweren Leukopenie oder einer schweren Neutropenie erhöht sein. Dies führt - im Vergleich zu einer alleinigen platin-basierten Chemotherapie - möglicherweise zu einer höheren Inzidenz infektiöser Komplikationen wie febriler Neutropenie, Pneumonie oder Sepsis (siehe Abschnitt 4.4).
Bei einer Kombinationstherapie mit Fluoropyrimidinen traten häufiger kardiovaskuläre Ischämien (einschließlich Herzinfarkt und kongestive Herzinsuffizienz) sowie häufiger ein Hand-Fuß-Syndrom (palmar-plantare Erythrodysästhesie) auf als unter Gabe von Fluoropyrimidinen.
In Kombination mit Capecitabin und Oxaliplatin (XELOX) kann die Häufigkeit einer schweren Diarrhö erhöht sein.
In einer formalen Wechselwirkungsstudie blieben die pharmakokinetischen Parameter von Cetuximab nach gleichzeitiger Verabreichung einer Einzeldosis Irinotecan (350 mg/m² Körperoberfläche) unverändert. Ebenso blieb auch die Pharmakokinetik von Irinotecan bei gleichzeitiger Applikation von Cetuximab unbeeinflusst.
Mit Cetuximab wurden beim Menschen bisher keine weiteren formalen Studien zur Erfassung von Wechselwirkungen durchgeführt.
Schwangerschaft
EGFR ist an der fetalen Entwicklung beteiligt. Begrenzte Beobachtungen an Tieren deuten darauf hin, dass Cetuximab die Plazenta passiert, und auch von anderen IgG1-Antikörpern ist bekannt, dass sie plazentagängig sind. Tierexperimentelle Daten ergaben keine Hinweise auf Teratogenität. Es kam jedoch zu einem dosisabhängigen Anstieg der Abortrate (siehe Abschnitt 5.3). Es liegen keine ausreichenden Daten von schwangeren Frauen oder stillenden Müttern vor.
Es wird dringend empfohlen, Erbitux bei Schwangeren sowie bei allen Frauen, die keine zuverlässige Empfängnisverhütung betreiben, nur dann anzuwenden, wenn der potenzielle Nutzen für die Mutter das mögliche Risiko für den Fetus rechtfertigt.
Stillzeit
Da nicht bekannt ist, ob Cetuximab in die Muttermilch übergeht, wird empfohlen, während der Therapie und mindestens bis zu zwei Monate nach der letzten Gabe nicht zu stillen.
Fertilität
Es liegen keine Daten zur Wirkung von Cetuximab auf die Fertilität beim Menschen vor. Die Wirkungen auf die männliche und weibliche Fertilität sind nicht im Rahmen formaler Tierstudien beurteilt worden (siehe Abschnitt 5.3).
Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Kommt es bei Patienten behandlungsbedingt zu Symptomen, die das Konzentrations- und Reaktionsvermögen beeinträchtigen, empfiehlt es sich, bis zum Abklingen solcher Wirkungen vom Führen eines Kraftfahrzeugs bzw. vom Bedienen von Maschinen abzusehen.
Die wesentlichen Nebenwirkungen von Cetuximab sind Hautreaktionen (bei über 80 % der Patienten), Hypomagnesiämie (bei über 10 % der Patienten) und infusionsbedingte Reaktionen (mit leichten bis mittelschweren Symptomen bei über 10 % der Patienten, mit schweren Symptomen bei über 1 % der Patienten).
Die Sicherheit von Cetuximab in Kombination mit Encorafenib (300 mg oral einmal täglich) (Dosierung gemäß Fachinformation) wurde im Rahmen der Phase‑III‑Studie ARRAY-818-302 an 216 Patienten mit BRAF-V600E-mutiertem metastasierendem Kolorektalkarzinom untersucht. Die in dieser Population am häufigsten berichteten unerwünschten Arzneimittelwirkungen (> 25 %) waren: Fatigue, Übelkeit, Diarrhö, Akneiforme Dermatitis, Abdominalschmerz, Arthralgie/Schmerzen des Muskel- und Skelettsystems, Appetit vermindert, Ausschlag und Erbrechen.
Die Abbruchrate aller Studienmedikamente aufgrund von Nebenwirkungen betrug 1,9 % bei Patienten, die mit Cetuximab in Kombination mit Encorafenib behandelt wurden.
Die Sicherheit von Cetuximab in Kombination mit Encorafenib (300 mg oral einmal täglich) und FOLFOX wurde im Rahmen der Studie C4221015 – BREAKWATER an 259 Patienten mit BRAF-V600E-mutiertem metastasierendem Kolorektalkarzinom untersucht (im Folgenden als die gepoolte EC+FOLFOX-Population bezeichnet, d. h. alle Patienten, die in den verschiedenen Studienabschnitten Encorafenib und Cetuximab [EC]+FOLFOX erhielten). Die in dieser Population am häufigsten berichteten unerwünschten Arzneimittelwirkungen (> 25 %) waren: periphere Neuropathie, Neutropenie, Übelkeit, Fatigue, Anämie, Diarrhö, Erbrechen, Appetit vermindert, Ausschlag, Thrombozytopenie, Abdominalschmerz, Blutungen, Arthralgie/Schmerzen des Muskel- und Skelettsystems, Fieber, Obstipation, Schleimhautentzündung und Infektion.
Die Abbruchrate aller Studienmedikamente aufgrund von Nebenwirkungen betrug 3,5 % bei Patienten, die mit Cetuximab in Kombination mit Encorafenib und FOLFOX behandelt wurden.
Die folgenden Definitionen beziehen sich auf die nachstehend verwendeten Häufigkeitsangaben:
Sehr häufig (≥ 1/10)
Häufig (≥ 1/100, < 1/10)
Gelegentlich (≥ 1/1 000, < 1/100)
Selten (≥ 1/10 000, < 1/1 000)
Sehr selten (< 1/10 000)
Häufigkeit nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Tabelle: Nebenwirkungen
Cetuximab |
Cetuximab in Kombination mit Encorafenib |
Cetuximab in Kombination mit Encorafenib und FOLFOX |
|
Erkrankungen des Blutes und des Lymphsystems | |||
Sehr häufig |
Neutropenie* |
||
Herzerkrankungen | |||
Häufig |
Tachykardie supraventrikulära |
Tachykardie supraventrikulära |
|
Augenerkrankungen | |||
Häufig |
Konjunktivitis |
||
Gelegentlich |
Blepharitis |
||
Erkrankungen des Gastrointestinaltrakts | |||
Sehr häufig |
Übelkeit |
Übelkeit |
|
Häufig |
Diarrhö |
||
Gelegentlich |
Pankreatitis* |
Pankreatitis* |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |||
Sehr häufig |
Leichte oder mittelschwere infusionsbedingte Reaktion (siehe Abschnitt 4.4) |
Ermüdung* |
Ermüdung* |
Häufig |
Schwere infusionsbedingte Reaktion, in einigen Fällen mit tödlichem Verlauf (siehe Abschnitt 4.4) |
||
Leber- und Gallenerkrankungen | |||
Sehr häufig |
Anstieg der Leberenzymwerte (ASAT, ALAT, AP) |
||
Erkrankungen des Immunsystems | |||
Häufig |
Überempfindlichkeitb |
||
Sehr häufig |
Überempfindlichkeitb |
||
Infektionen und parasitäre Erkrankungen | |||
Sehr häufig |
Infektionen |
||
Häufig |
Infektion der oberen Atemwege* |
||
Untersuchungen | |||
Sehr häufig |
Lipase erhöht* |
||
Häufig |
Kreatinin im Blut erhöht* |
Kreatinin im Blut erhöht* |
|
Gelegentlich |
Amylase erhöht |
||
Stoffwechsel- und Ernährungsstörungen | |||
Sehr häufig |
Hypomagnesiämie (siehe Abschnitt 4.4). |
Appetit vermindert |
Appetit vermindert |
Häufig |
Dehydratation, insbesondere infolge Diarrhö oder Mukositis |
||
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |||
Sehr häufig |
Arthralgie/Schmerzen des Muskel- und Skelettsystems* |
Arthralgie/Schmerzen des Muskel- und Skelettsystems* |
|
Häufig |
Schmerz in einer Extremität* |
||
Gutartige, bösartige und nicht spezifizierte Neubildungen | |||
Sehr häufig |
Melanozytischer Nävus |
||
Häufig |
cuSCCe |
Melanozytischer Nävus |
|
Gelegentlich |
Basalzellkarzinom* |
Neues primäres Melanom* |
|
Erkrankungen des Nervensystems | |||
Sehr häufig |
periphere Neuropathie* |
periphere Neuropathief |
|
Häufig |
Kopfschmerzen |
Schwindelgefühl* |
Schwindelgefühl* |
Nicht bekannt |
Aseptische Meningitis |
||
Psychiatrische Erkrankungen | |||
Sehr häufig |
Schlaflosigkeit |
Schlaflosigkeit* |
|
Erkrankungen der Nieren und Harnwege | |||
Häufig |
Nierenversagen* |
Nierenversagen* |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |||
Gelegentlich |
Lungenembolie |
||
Erkrankungen der Haut und des Unterhautgewebes | |||
Sehr häufig |
Hautreaktionen* |
Akneiforme Dermatitis* |
Ausschlag* |
Häufig |
Hyperpigmentierung der Haut |
Erythem |
|
Gelegentlich |
Exfoliation der Hauth |
||
Sehr selten |
Stevens-Johnson-Syndrom/Epidermolysis acuta toxica |
||
Nicht bekannt |
Superinfektion von Hautläsionen |
||
Gefäßerkrankungen | |||
Gelegentlich |
Tiefe Venenthrombose |
||
Sehr häufig |
Blutungi |
Blutungi |
|
*Zusammengesetzte Begriffe, die mehrere bevorzugte Begriffe (Preferred Terms) enthalten
a umfasst unter anderem Extrasystolen, supraventrikuläre Tachykardie, Vorhoftachykardie und Sinustachykardie
b umfasst unter anderem Angioödem, anaphylaktische Reaktion, Arzneimittelüberempfindlichkeit, Überempfindlichkeit, Hypersensitivitätsvaskulitis und Urtikaria
c umfasst unter anderem Harnwegsinfektion, Gastroenteritis, Peritonitis, Zellulitis, abdominelle Infektion, gastrointestinale Infektion, Infektion, Infektion der unteren Atemwege, virale und bakterielle Pneumonie, Virusinfektion
d umfasst unter anderem Myalgie, Muskelschwäche, Muskelkrämpfe, Muskelverletzung, Myopathie, Myositis
e umfasst Keratoakanthom, Plattenepithelkarzinom und Plattenepithelkarzinom der Haut
f umfasst unter anderem Kälte-Dysästhesie, Dysästhesie, Hyperästhesie, Hypoästhesie, Neuralgie, periphere Neuropathie, Neurotoxizität, Parästhesie, periphere sensorische Neuropathie und Polyneuropathie
g umfasst Erythem, generalisiertes Erythem und Plantarerythem
h umfasst Dermatitis exfoliativa, Exfoliation der Haut und exfoliativen Hautausschlag
i umfasst Blutungen an verschiedenen Stellen, darunter unter anderem Hirnblutung, intrakranielle Blutung, vaginale Blutung, starke Menstruationsblutung, Zwischenblutung, Hämatochezie, Epistaxis, Hämoptyse, Hämothorax, gastrointestinale Blutung und Hämaturie
Beschreibung ausgewählter Nebenwirkungen
Hautreaktionen
Hautreaktionen können bei über 80 % der Patienten auftreten und äußern sich vor allem in akneartigem Hautausschlag und/oder weniger häufig in Pruritus, Hauttrockenheit, Hautabschuppung, Hypertrichose oder Nagelstörungen (z. B. Paronychie). Etwa 15 % der Hautreaktionen sind schwer, darunter einzelne Fälle von Hautnekrosen. Die meisten Hautreaktionen entwickeln sich innerhalb der ersten drei Behandlungswochen. In der Regel bilden sie sich nach Therapieende im Laufe der Zeit ohne Folgeerscheinungen zurück, sofern die empfohlenen Dosierungsanpassungen eingehalten werden (siehe Abschnitt 4.4).
Superinfektion von Hautläsionen
Patienten mit durch Cetuximab hervorgerufenen Hautläsionen sind möglicherweise für Superinfektionen (z. B. mit S. aureus) prädisponiert, was Komplikationen wie Cellulitis, Erysipel oder - eventuell mit tödlichem Ausgang - staphylogenes Lyell-Syndrom (Staphylococcal scalded skin syndrome), nekrotisierende Fasziitis oder eine Sepsis nach sich ziehen kann.
Kombinationstherapie
Bei Anwendung von Cetuximab in Kombination mit Chemotherapeutika sind auch deren Produktinformationen zu beachten.
In Kombination mit einer platin-basierten Chemotherapie kann die Häufigkeit einer schweren Leukopenie oder einer schweren Neutropenie erhöht sein. Dies führt - im Vergleich zu einer alleinigen platin-basierten Chemotherapie - möglicherweise zu einer höheren Inzidenz infektiöser Komplikationen wie febriler Neutropenie, Pneumonie oder Sepsis (siehe Abschnitt 4.4).
Bei einer Kombinationstherapie mit Fluoropyrimidinen traten häufiger kardiovaskuläre Ischämien (einschließlich Herzinfarkt und kongestive Herzinsuffizienz) sowie häufiger ein Hand-Fuß-Syndrom (palmar-plantare Erythrodysästhesie) auf als unter Gabe von Fluoropyrimidinen.
In Kombination mit Encorafenib und FOLFOX oder mit Encorafenib wurden keine neuen Nebenwirkungen beobachtet, die nicht bereits unter Cetuximab, Encorafenib oder FOLFOX bekannt sind. Es gab keine relevante Zunahme der Häufigkeit oder des Schweregrades bekannter Nebenwirkungen. Bitte die Produktinformationen zu Encorafenib und den einzelnen Bestandteilen von FOLFOX beachten.
In Verbindung mit einer lokalen Bestrahlung des Kopf- und Halsbereiches traten zusätzlich die für eine Strahlentherapie typischen Nebenwirkungen auf (wie Mukositis, Strahlendermatitis, Dysphagie oder Leukopenie, hauptsächlich in Form einer Lymphozytopenie). In einer randomisierten, kontrollierten klinischen Studie an 424 Patienten traten eine schwere akute Strahlendermatitis und Mukositis sowie verzögerte bestrahlungsbedingte Nebenwirkungen etwas häufiger bei Patienten auf, die eine Bestrahlung in Kombination mit Cetuximab erhielten, als bei Patienten, die nur eine Strahlentherapie erhielten.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen‑Risiko‑Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung
dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: http://www.pei.de anzuzeigen.
Zur Anwendung von wöchentlichen Dosen über 250 mg/m² Körperoberfläche oder zweiwöchentlichen Dosen über 500 mg/m² Körperoberfläche liegen nur begrenzte Erfahrungen vor. In klinischen Studien mit Dosen von bis zu 700 mg/m² alle zwei Wochen stimmte das Sicherheitsprofil mit dem in Abschnitt 4.8 beschriebenen überein.
Pharmakotherapeutische Gruppe: Antineoplastische Mittel, monoklonale Antikörper, ATC‑Code: L01FE01
Wirkmechanismus
Cetuximab ist ein chimärer monoklonaler IgG1-Antikörper, der spezifisch gegen den epidermalen Wachstumsfaktor-Rezeptor (EGFR) gerichtet ist.
EGFR-Signalwege sind an der Steuerung der Überlebensfähigkeit von Zellen, des Ablaufs des Zellzyklus, der Angiogenese, Zellmigration sowie der zellulären Invasion bzw. Metastasierung beteiligt.
Cetuximab bindet an den EGFR mit einer ungefähr 5- bis 10fach höheren Affinität als endogene Liganden. Cetuximab blockiert die Bindung endogener EGFR-Liganden und hemmt dadurch die Funktion des Rezeptors. Es induziert die Internalisierung des EGFR und kann somit zu dessen Downregulierung führen. Außerdem kann Cetuximab gezielt zytotoxische Effektorzellen des Immunsystems auf die EGFR-exprimierenden Tumorzellen lenken und auf diese Weise die Antikörper-abhängige zelluläre Zytotoxizität (ADCC) vermitteln.
Cetuximab bindet nicht an andere Rezeptoren der HER-Familie.
Das vom Protoonkogen RAS (Rat Sarcoma) codierte Protein spielt eine zentrale, nachgeschaltete Rolle in der Signaltransduktionskette von EGFR. In Tumoren trägt die Aktivierung von RAS durch EGFR zur EGFR-vermittelten gesteigerten Proliferation, zum Zellüberleben sowie zur Produktion angiogenesefördernder Faktoren bei.
RAS ist eine der am häufigsten aktivierten Onkogenfamilien bei Krebserkrankungen des Menschen. Mutationen der RAS-Gene an bestimmten „Hotspots“ auf den Exons 2, 3 und 4 bewirken die konstitutive Aktivierung der RAS-Proteine, unabhängig von einer Signalwirkung durch EGFR.
Pharmakodynamische Wirkungen
Sowohl in vitro als auch in vivo hemmt Cetuximab die Proliferation und induziert die Apoptose EGFR-exprimierender humaner Tumorzellen. In vitro hemmt Cetuximab die Produktion von Angiogenesefaktoren durch Tumorzellen und blockiert die endotheliale Zellmigration. In vivo hemmt Cetuximab die Exprimierung von Angiogenesefaktoren durch Tumorzellen und reduziert die Neuvaskularisierung und Metastasierung von Tumoren.
Beim BRAF‑mutierten Kolorektalkarzinom wurde die Induktion der EGFR‑vermittelten MAPK‑Signalwegaktivierung als Resistenzmechanismus gegen BRAF‑Inhibitoren identifiziert. Kombinationen eines BRAF‑Inhibitors mit EGFR‑gerichteten Wirkstoffen zeigten in präklinischen Modellen eine verbesserte Antitumorwirkung.
Immunogenität
Die Bildung humaner antichimärer Antikörper (HACA) ist eine klassenspezifische Wirkung monoklonaler chimärer Antikörper. Derzeit liegen nur begrenzt Daten zur Entwicklung von HACAs vor. Insgesamt wurden bei 3,4 % der untersuchten Patienten messbare HACA-Titer festgestellt. Die Inzidenz variierte in den Zielindikationsstudien zwischen 0 % und 9,6 %. Zur neutralisierenden Wirkung von HACAs auf Cetuximab liegen bislang keine aussagekräftigen Erkenntnisse vor. Das Auftreten von HACA korrelierte nicht mit dem Auftreten von Überempfindlichkeitsreaktionen oder einer anderen Nebenwirkung von Cetuximab.
Kolorektalkarzinom
Metastasierendes Kolorektalkarzinom mit RAS‑Wildtyp
Der immunhistochemische Nachweis der EGFR-Exprimierung in Tumormaterial erfolgte mit einem diagnostischen Test (EGFR PharmDx). Ein Tumor galt als EGFR-exprimierend, wenn eine einzige gefärbte Zelle nachweisbar war. Ca. 75% der im Rahmen der klinischen Prüfungen für eine eventuelle Studienaufnahme untersuchten Patienten mit einem metastasierenden kolorektalen Karzinom hatten einen EGFR-exprimierenden Tumor und kamen daher für eine Behandlung mit Cetuximab in Frage. Für Patienten mit Tumoren, in denen sich EGFR nicht nachweisen ließ, ist die Wirksamkeit und Sicherheit von Cetuximab nicht dokumentiert.
Studiendaten belegen, dass es höchst unwahrscheinlich ist, dass Patienten mit einem metastasierenden Kolorektalkarzinom und aktivierenden RAS-Mutationen von einer Therapie mit Cetuximab oder einer Kombinationsbehandlung mit Cetuximab und einer Chemotherapie profitieren, und bei Verwendung als Zusatzbehandlung zu FOLFOX4 zeigte sich ein signifikant negativer Effekt auf die progressionsfreie Überlebenszeit (PFS).
Cetuximab in Monotherapie oder in Kombination mit einer Chemotherapie wurde in fünf randomisierten, kontrollierten klinischen Studien sowie in mehreren ergänzenden Studien untersucht. An den fünf randomisierten Studien nahmen insgesamt 3 734 Patienten mit EGFR-exprimierendem, metastasierendem Kolorektalkarzinom und einem ECOG-Performance-Status von ≤ 2 teil. Die Mehrzahl der eingeschlossenen Patienten hatte einen ECOG-Performance-Status von ≤ 1. In allen Studien wurde Cetuximab verabreicht, wie in Abschnitt 4.2 beschrieben.
In vier der randomisierten, kontrollierten Studien (EMR 62 202‑013, EMR 62 202‑047, CA225006 und CA225025) wurde der K-RAS-Exon-2-Status als prädikativer Faktor für den Cetuximab-Behandlungserfolg ermittelt. Von 2 072 Patienten lag der K-RAS-Mutationsstatus vor. In den Studien EMR 62 202‑013 und EMR 62 202‑047 wurden zusätzliche Post‑hoc‑Analysen durchgeführt, bei denen auch andere Mutationen in RAS‑Genen (N‑RAS und K‑RAS) als im K‑RAS‑Exon 2 untersucht wurden. Lediglich in Studie EMR 62 202-007 war eine Post‑hoc‑Analyse nicht möglich.
Cetuximab wurde darüber hinaus in Kombination mit einer Chemotherapie in einer prüferinitiierten randomisierten kontrollierten Phase‑III‑Studie (COIN, COntinuous chemotherapy plus cetuximab or INtermittent chemotherapy) untersucht. Die EGFR-Expression stellte in dieser Studie kein Einschlusskriterium dar. Tumorproben von etwa 81 % der Patienten wurden retrospektiv auf K-RAS-Expression untersucht.
In der prüfergesponserten klinischen Phase‑III‑Studie FIRE‑3 wurde die Behandlung mit FOLFIRI entweder in Kombination mit Cetuximab oder Bevacizumab in der Erstlinienbehandlung von Patienten mit metastasierendem Kolorektalkarzinom mit K‑RAS‑Exon‑2‑Wildtyp verglichen. Weitere Post‑hoc‑Analysen von Mutationen in RAS‑Genen außer denen im K‑RAS‑Exon 2 wurden vorgenommen.
Cetuximab in Kombination mit Chemotherapie
EMR 62 202-013:
In dieser randomisierten Studie bei Patienten mit metastasierendem Kolorektalkarzinom ohne Vorbehandlung der metastasierenden Erkrankung wurde die Kombination von Cetuximab und Irinotecan plus 5-Fluorouracil-/Folinsäure-Infusionen (FOLFIRI) (599 Patienten) mit der entsprechenden Chemotherapie allein (599 Patienten) verglichen. Innerhalb der für den K-RAS-Status auswertbaren Patientengruppe betrug der Anteil der Patienten mit K-RAS-Wildtyp-Tumoren 63 %. Für die Beurteilung des RAS‑Status wurden für alle auswertbaren Tumorproben der K‑RAS‑Exon‑2‑Wildtyp‑Population (65 %) andere als in Exon 2 des K‑RAS‑Gens liegende Mutationen bestimmt. Die Population mit RAS‑Mutationen besteht aus Patienten mit bekannten K‑RAS‑Exon‑2‑Mutationen sowie zusätzlich identifizierten RAS‑Mutationen.
Die in dieser klinischen Studie erhobenen Daten zur Wirksamkeit sind in der nachfolgenden Tabelle zusammengefasst:
|
Population mit RAS-Wildtyp |
Population mit RAS-Mutation |
||
Variable/Statistik |
Cetuximab plus FOLFIRI |
FOLFIRI |
Cetuximab plus FOLFIRI |
FOLFIRI |
|
(N=178) |
(N=189) |
(N=246) |
(N=214) |
OS |
|
|||
Monate, Median |
28,4 |
20,2 |
16,4 |
17,7 |
(95 % KI) |
(24,7; 31,6) |
(17,0; 24,5) |
(14,9; 18,4) |
(15,4; 19,6) |
Hazard Ratio (95 % KI) |
0,69 (0,54; 0,88) |
1,05 (0,86; 1,28) |
||
p-Wert |
0,0024 |
0,6355 |
||
PFS |
||||
Monate, Median |
11,4 |
8,4 |
7,4 |
7,5 |
(95 % KI) |
(10,0; 14,6) |
(7,4; 9,4) |
(6,4; 8,0) |
(7,2; 8,5) |
Hazard Ratio (95 % KI) |
0,56 (0,41; 0,76) |
1,10 (0,85; 1,42) |
||
p-Wert |
0,0002 |
0,4696 |
||
ORR |
||||
% |
66,3 |
38,6 |
31,7 |
36,0 |
(95 % KI) |
(58,8; 73,2) |
(31,7; 46,0) |
(25,9; 37,9) |
(29,6; 42,8) |
Odds Ratio (95 % KI) |
3,1145 (2,0279; 4,7835) |
0,8478 (0,5767; 1,2462) |
||
p-Wert |
<0,0001 |
0,3970 |
||
KI = Konfidenzintervall, FOLFIRI = Irinotecan plus 5-FU/FA-Infusion, ORR = objektive Ansprech- bzw. Remissionsrate (Patienten mit Voll- oder Teilremission), OS = Gesamtüberlebenszeit, PFS = progressionsfreie Überlebenszeit
EMR 62 202-047:
In dieser randomisierten Studie bei Patienten mit metastasierendem Kolorektalkarzinom ohne Vorbehandlung der metastasierenden Erkrankung wurde die Kombination von Cetuximab und Oxaliplatin plus 5-Fluorouracil-/Folinsäure-Dauerinfusionen (FOLFOX4) (169 Patienten) mit der entsprechenden Chemotherapie allein (168 Patienten) verglichen. Innerhalb der für den K-RAS-Status auswertbaren Patientengruppe betrug der Anteil der Patienten mit K-RAS-Wildtyp-Tumoren 57 %. Für die Beurteilung des RAS‑Status wurden für alle auswertbaren Tumorproben der K‑RAS‑Exon‑2‑Wildtyp‑Population andere als in Exon 2 des K‑RAS‑Gens liegende Mutationen bestimmt. Die Population mit RAS‑Mutationen besteht aus Patienten mit bekannten K‑RAS‑Mutationen in Exon 2 sowie zusätzlich identifizierten RAS‑Mutationen.
Die in dieser klinischen Studie erhobenen Daten zur Wirksamkeit sind in der nachfolgenden Tabelle zusammengefasst:
|
Population mit RAS-Wildtyp |
Population mit RAS-Mutation |
||
Variable/Statistik |
Cetuximab plus FOLFOX4 |
FOLFOX4 |
Cetuximab plus FOLFOX4 |
FOLFOX4 |
|
(N=38) |
(N=49) |
(N=92) |
(N=75) |
OS |
|
|||
Monate, Median |
19,8 |
17,8 |
13,5 |
17,8 |
(95 % KI) |
(16,6; 25,4) |
(13,8; 23,9) |
(12,1; 17,7) |
(15,9; 23,6) |
Hazard Ratio (95 % KI) |
0,94 (0,56; 1,56) |
1,29 (0,91; 1,84) |
||
p-Wert |
0,8002 |
0,1573 |
||
PFS |
||||
Monate, Median |
12,0 |
5,8 |
5,6 |
7,8 |
(95 % KI) |
(5,8; NA) |
(4,7; 7,9) |
(4,4; 7,5) |
(6,7; 9,3) |
Hazard Ratio (95 % KI) |
0,53 (0,27; 1,04) |
1,54 (1,04; 2,29) |
||
p-Wert |
0,0615 |
0,0309 |
||
ORR |
||||
% |
57,9 |
28,6 |
37,0 |
50,7 |
(95 % KI) |
(40,8; 73,7) |
(16,6; 43,3) |
(27,1; 47,7) |
(38,9; 62,4) |
Odds Ratio (95 % KI) |
3,3302 (1,375; 8,172) |
0,580 (0,311; 1,080) |
||
p-Wert |
0,0084 |
0,0865 |
||
KI = Konfidenzintervall, FOLFOX4 = Oxaliplatin plus 5-FU/FA-Dauerinfusion, ORR = objektive Ansprech- bzw. Remissionsrate (Patienten mit Voll- oder Teilremission), OS = Gesamtüberlebenszeit, PFS = progressionsfreie Überlebenszeit, NA = nicht abschätzbar
Im Besonderen wurde in der Population mit RAS-Mutation ein negativer Effekt der zusätzlichen Cetuximab-Therapie beobachtet.
COIN:
In dieser offenen, 3-armigen, randomisierten Studie an 2 445 Patienten mit inoperablem metastasierendem oder lokoregionärem Kolorektalkarzinom ohne Vorbehandlung der metastasierenden Erkrankung, wurde die Kombination von Cetuximab und Oxaliplatin plus Fluoropyrimidine (5-Fluorouracil-/Folinsäure-Infusionen [OxMdG] oder Capecitabin [XELOX]) mit der entsprechenden Chemotherapie allein verglichen. Im dritten experimentellen Arm wurde ein intermittierendes OxMdG- oder XELOX-Regime ohne Cetuximab angewendet. Die Daten für das XELOX-Regime und den dritten experimentellen Arm sind nicht aufgeführt.
Tumorproben von etwa 81 % der Patienten wurden retrospektiv auf K-RAS-Expression untersucht; 55 % davon erwiesen sich als K-RAS-Wildtyp. Davon erhielten 362 Patienten Cetuximab und Oxaliplatin plus Fluoropyrimidine (117 Patienten OxMdG und 245 Patienten XELOX) und 367 Patienten erhielten Oxaliplatin plus Fluoropyrimidine allein (127 Patienten OxMdG und 240 Patienten XELOX). Von der Population mit K-RAS-Mutation erhielten 297 Cetuximab und Oxaliplatin plus Fluoropyrimidine (101 Patienten OxMdG und 196 Patienten XELOX) und 268 Patienten erhielten Oxaliplatin plus Fluoropyrimidine allein (78 Patienten OxMdG und 190 Patienten XELOX).
Die in dieser Studie erhobenen Daten zur Wirksamkeit des OxMdG-Regimes sind in der nachfolgenden Tabelle zusammengefasst:
Population mit K-RAS-Wildtyp |
Population mit K-RAS-Mutation |
|||
Variable/Statistik |
Cetuximab |
OxMdG |
Cetuximab |
OxMdG |
(N=117) |
(N=127) |
(N=101) |
(N=78) |
|
OS |
||||
Monate, Median |
16,3 |
18,2 |
13,1 |
14,6 |
(95 % KI) |
(10,3; 32,2) |
(9,8; 27,5) |
(8,0; 23,9) |
(9,5; 22,0) |
Hazard Ratio (95 % KI) |
0,93 (0,72; 1,19) |
0,99 (0,75; 1,30) |
||
p-Wert |
0,617 |
0,931 |
||
PFS |
||||
Monate, Median |
9,0 |
9,2 |
6,8 |
8,5 |
(95 % KI) |
(5,8; 15,5) |
(5,8; 12,7) |
(5,0; 10,7) |
(3,4; 10,8) |
Hazard Ratio (95 % KI) |
0,77 (0,59; 1,01) |
1,05 (0,77; 1,41) |
||
p-Wert |
0,056 |
0,78 |
||
Beste Gesamtansprechrate |
||||
% |
68 |
59 |
47 |
51 |
(95 % KI) |
(58; 76) |
(50; 68) |
(37; 57) |
(40; 63) |
Odds Ratio (95 % KI) |
1,44 (0,85; 2,43) |
0,83 (0,46; 1,49) |
||
p-Wert |
0,171 |
0,529 |
||
KI = Konfidenzintervall, OxMdG = Oxaliplatin plus 5‑FU/FA-Infusionen, OS = Gesamtüberlebenszeit, PFS = progressionsfreie Überlebenszeit
Bei den zeitbezogenen Endpunkten konnten für Patienten, die Cetuximab in Kombination mit dem XELOX-Regime erhielten, keine Trends aufgezeigt werden, die auf einen klinischen Nutzen hindeuten würden.
Hauptsächlich infolge einer größeren Häufigkeit von Diarrhö in den Cetuximab-Armen kam es zu signifikanten Dosisminderungen und Aufschüben der Capecitabin- bzw. Oxaliplatin-Verabreichung. Darüber hinaus erhielten signifikant weniger mit Cetuximab behandelte Patienten eine Zweitlinientherapie.
FIRE‑3 (Erstlinienkombination von Cetuximab mit FOLFIRI):
Bei der FIRE‑3‑Studie handelte es sich um eine multizentrische, randomisierte Phase‑III‑Studie, in der bei Patienten mit metastasierendem Kolorektalkarzinom mit K‑RAS‑Exon‑2‑Wildtyp ein Direktvergleich von 5‑FU, Folinsäure und Irinotecan (FOLFIRI) in Kombination mit entweder Cetuximab oder Bevacizumab erfolgte. Der RAS‑Status war in Tumorproben von 407 Patienten mit K‑RAS‑Exon‑2‑Wildtyp auswertbar, was einem Anteil von 69 % der Gesamtpopulation von Patienten mit K‑RAS‑Exon‑2‑Wildtyp entspricht (592 Patienten). Von diesen hatten 342 Patienten RAS‑Wildtyp‑Tumoren, während bei 65 Patienten RAS‑Mutationen nachgewiesen wurden. Die Population mit RAS-Mutationen bestand aus diesen 65 Patienten plus 113 Patienten mit Tumoren mit K‑RAS‑Exon 2‑Mutation, die behandelt worden waren, bevor die Aufnahme in die Studie auf Patienten mit metastasierendem Kolorektalkarzinom mit K‑RAS‑Exon‑2‑Wildtyp begrenzt wurde.
Die in dieser klinischen Studie erhobenen Daten zur Wirksamkeit sind in der nachfolgenden Tabelle zusammengefasst:
Population mit RAS-Wildtyp |
Population mit RAS-Mutation |
|||
Variable/Statistik |
Cetuximab |
Bevacizumab |
Cetuximab |
Bevacizumab |
(N=171) |
(N=171) |
(N=92) |
(N=86) |
|
OS |
||||
Monate, Median |
33,1 |
25,6 |
20,3 |
20,6 |
(95 % KI) |
(24,5; 39,4) |
(22,7; 28,6) |
(16,4; 23,4) |
(17,0; 26,7) |
Hazard Ratio (95 % KI) |
0,70 (0,53; 0,92) |
1,09 (0,78; 1,52) |
||
p‑Wert |
0,011 |
0,60 |
||
PFS |
||||
Monate, Median |
10,4 |
10,2 |
7,5 |
10,1 |
(95 % KI) |
(9,5; 12,2) |
(9,3; 11,5) |
(6,1; 9,0) |
(8,9; 12,2) |
Hazard Ratio (95 % KI) |
0,93 (0,74; 1,17) |
1,31 (0,96; 1,78) |
||
p‑Wert |
0,54 |
0,085 |
||
ORR |
||||
% |
65,5 |
59,6 |
38,0 |
51,2 |
(95 % KI) |
(57,9; 72,6) |
(51,9; 67,1) |
(28,1; 48,8) |
(40,1; 62,1) |
Odds Ratio (95 % KI) |
1,28 (0,83; 1,99) |
0,59 (0,32; 1,06) |
||
p‑Wert |
0,32 |
0,097 |
||
KI = Konfidenzintervall, FOLFIRI = Irinotecan plus 5‑FU/FA-Infusion, ORR = objektive Ansprech- bzw. Remissionsrate (Patienten mit Voll- oder Teilremission), OS = Gesamtüberlebenszeit, PFS = progressionsfreie Überlebenszeit
In der K‑RAS‑Wildtyp-Population der Studie CALGB/SWOG 80405 (n=1.137) wurde die Überlegenheit von Cetuximab plus Chemotherapie gegenüber Bevacizumab plus Chemotherapie nicht gezeigt.
CA225006:
In dieser randomisierten Studie bei Patienten mit metastasierendem Kolorektalkarzinom, die initial bereits eine Kombinationstherapie aus Oxaliplatin plus Fluoropyrimidin gegen die metastasierende Erkrankung erhalten hatten, wurde die Kombination von Cetuximab und Irinotecan (648 Patienten) mit der Irinotecan-Monotherapie (650 Patienten) verglichen. Nach Krankheitsprogression wurde bei 50 % der Patienten aus der Irinotecan-Monotherapiegruppe eine spezifisch gegen den EGFR gerichtete Therapie eingeleitet.
In der Gesamtpopulation wurden unabhängig vom K-RAS-Status für Cetuximab plus Irinotecan (648 Patienten) im Vergleich zur Irinotecan-Monotherapie (650 Patienten) folgende Ergebnisse berichtet: Median der Gesamtüberlebenszeit (OS) 10,71 vs. 9,99 Monate (HR 0,98); Median der progressionsfreien Überlebenszeit (PFS) 4,0 vs. 2,6 Monate (HR 0,69); objektive Ansprechrate (ORR) 16,4 % vs. 4,2 %.
Im Hinblick auf den K-RAS-Status standen lediglich von 23 % der Patienten (300 von 1 298) Tumorproben zur Verfügung. In der auf den K-RAS-Status hin ausgewerteten Population wiesen 64 % der Patienten (192) K-RAS-Wildtyp-Tumoren und 108 Patienten K-RAS-Mutationen auf. Auf der Grundlage dieser Daten, und da keine unabhängige Auswertung der Bildgebungsdaten erfolgt ist, werden die Ergebnisse im Hinblick auf den Mutationsstatus als nicht interpretierbar angesehen.
EMR 62 202-007:
In dieser randomisierten Studie bei Patienten mit metastasierendem Kolorektalkarzinom, die als letzte Therapie vor Studienbeginn eine Irinotecan-haltige Chemotherapie gegen die metastasierende Erkrankung erhalten hatten und darauf nicht mehr ansprachen, wurde die Kombination von Cetuximab und Irinotecan (218 Patienten) mit der Cetuximab-Monotherapie (111 Patienten) verglichen.
Die Kombination von Cetuximab und Irinotecan verringerte im Vergleich zu Cetuximab allein das Gesamt-Progressionsrisiko um 46 % und bewirkte eine signifikante Steigerung der objektiven Ansprechrate. Hinsichtlich der Verbesserung der Gesamtüberlebenszeit wurde in der randomisierten Studie keine statistische Signifikanz erreicht; allerdings erhielten im weiteren Behandlungsverlauf knapp 50 % der Patienten aus dem Cetuximab-Monotherapiearm nach einer Krankheitsprogression noch eine Kombinationstherapie aus Cetuximab und Irinotecan, was die Gesamtüberlebenszeit beeinflusst haben könnte.
Cetuximab in Monotherapie
CA225025:
In dieser randomisierten Studie wurde bei Patienten mit metastasierendem Kolorektalkarzinom, die zuvor bereits Oxaliplatin-, Irinotecan- und Fluoropyrimidin-haltige Therapieregime gegen die metastasierende Erkrankung erhalten hatten, die Gabe von Cetuximab in Monotherapie zusätzlich zu optimalen unterstützenden Maßnahmen (Best Supportive Care, BSC) (287 Patienten) mit den optimalen unterstützenden Maßnahmen allein (285 Patienten) verglichen. Innerhalb der hinsichtlich des K-RAS-Status auswertbaren Patientengruppe betrug der Anteil der Patienten mit K-RAS-Wildtyp-Tumoren 58 %.
Die in dieser klinischen Studie erhobenen Daten zur Wirksamkeit sind in der nachfolgenden Tabelle zusammengefasst:
|
Population mit K-RAS-Wildtyp |
Population mit K-RAS-Mutation |
||
Variable/Statistik |
Cetuximab plus BSC |
BSC |
Cetuximab plus BSC |
BSC |
|
(N=117) |
(N=113) |
(N=81) |
(N=83) |
OS |
|
|||
Monate, Median |
9,5 |
4,8 |
4,5 |
4,6 |
(95 % KI) |
(7,7; 10,3) |
(4,2; 5,5) |
(3,8; 5,6) |
(3,6; 5,5) |
Hazard Ratio (95 % KI) |
0,552 (0,408; 0,748) |
0,990 (0,705; 1,389) |
||
p-Wert |
<0,0001 |
0,9522 |
||
PFS |
|
|
||
Monate, Median |
3,7 |
1,9 |
1,8 |
1,8 |
(95 % KI) |
(3,1; 5,1) |
(1,8; 2,0) |
(1,7; 1,8) |
(1,7; 1,8) |
Hazard Ratio (95 % KI) |
0,401 (0,299; 0,536) |
1,002 (0,732; 1,371) |
||
p-Wert |
<0,0001 |
0,9895 |
||
ORR |
|
|||
% |
12,8 |
0 |
1,2 |
0 |
(95 % KI) |
(7,4; 20,3) |
(-) |
(0,0; 6,7) |
(-) |
p-Wert |
<0,001 |
0,314 |
||
BSC = optimale unterstützende Maßnahmen, KI = Konfidenzintervall, ORR = objektive Ansprech- bzw. Remissionsrate (Patienten mit Voll- oder Teilremission), OS = Gesamtüberlebenszeit, PFS = progressionsfreie Überlebenszeit
Metastasierendes Kolorektalkarzinom mit BRAF-V600E-Mutation
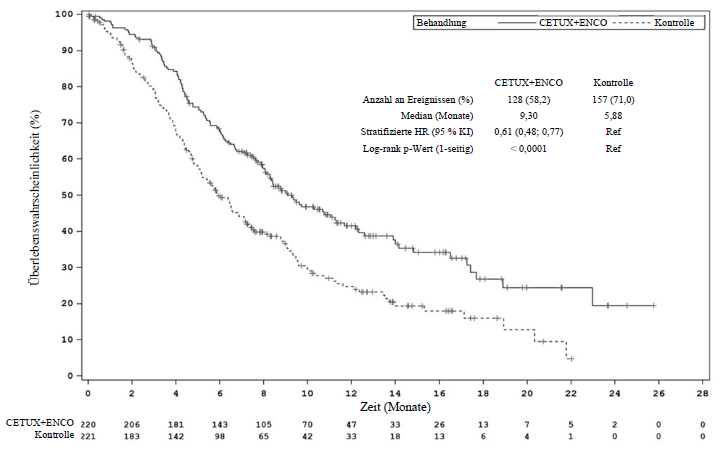
Cetuximab in Kombination mit Encorafenib – Studie ARRAY-818-302 (BEACON)
Cetuximab in Kombination mit Encorafenib wurde in einer randomisierten, aktiv kontrollierten, unverblindeten, multizentrischen Studie (ARRAY-818-302 BEACON CRC) untersucht. Teilnahmeberechtigt waren Patienten mit BRAF-V600E-mutiertem metastasierendem Kolorektalkarzinom, deren Erkrankung nach 1 oder 2 Vortherapien fortgeschritten war. Die aufgenommenen Patienten konnten Cetuximab gemäß der lokal zugelassenen Fachinformation in Abhängigkeit vom RAS‑Status des Tumors erhalten. Die vorherige Anwendung von RAF‑Inhibitoren, MEK‑Inhibitoren oder EGFR‑Inhibitoren war ausgeschlossen. Die Randomisierung erfolgte stratifiziert nach ECOG-Performance-Status (Eastern Cooperative Oncology Group), vorheriger Anwendung von Irinotecan und der verwendeten Cetuximab-Formulierung.
Insgesamt wurden 665 Patienten im Verhältnis 1:1:1 randomisiert und erhielten entweder Cetuximab in Kombination mit 300 mg Encorafenib täglich oral (n = 220) oder Cetuximab in Kombination mit 300 mg Encorafenib täglich oral und 45 mg Binimetinib zweimal täglich oral (n = 224) oder eine Kontrolltherapie (Cetuximab mit Irinotecan oder Cetuximab mit Irinotecan/5‑Fluorouracil/Folinsäure [FOLFIRI], n = 221). Die Behandlung wurde bis zum Fortschreiten der Erkrankung oder dem Auftreten inakzeptabler Toxizität fortgesetzt.
Die Wirksamkeitsendpunkte waren das Gesamtüberleben (OS) und die objektive Ansprechrate (ORR), beurteilt von einem verblindeten, unabhängigen zentralen Review Committee (blinded independent central review committee, BIRC) bei dem Vergleich von Cetuximab in Kombination mit 300 mg Encorafenib mit der Kontrolltherapie. Weitere Wirksamkeitsendpunkte sind in der untenstehenden Tabelle zusammengefasst.
Das mediane Alter der Patienten betrug 61 Jahre (Spanne: 26‑91 Jahre), 47 % waren männlich und 83 % kaukasischer Herkunft. 51 % der Patienten hatten zu Studienbeginn einen ECOG‑Performance-Status von 0, und 51 % hatten zuvor Irinotecan erhalten. Bei 46,8 % der Patienten waren zu Studienbeginn mindestens 3 Organe vom Tumor befallen.
Die mediane Expositionsdauer betrug 3,2 Monate bei Patienten, die mit Cetuximab in Kombination mit 300 mg Encorafenib behandelt wurden, und 1,4 Monate bei Patienten, die mit Cetuximab/Irinotecan oder Cetuximab/FOLFIRI (Kontrollarm) behandelt wurden. Bei Patienten, die mit der Kombination aus Cetuximab und 300 mg Encorafenib behandelt wurden, betrug die mediane relative Dosisintensität (RDI) 98 % für Encorafenib und 93,5 % für Cetuximab. Im Kontrollarm lag die mediane RDI bei 85,4 % für Cetuximab, 75,7 % für Irinotecan und in der Subgruppe der Patienten, die Folinsäure und 5‑FU erhielten, bei 75,2 % bzw. 75 %.
Die Kombination von Cetuximab mit 300 mg Encorafenib führte im Vergleich zur Kontrolltherapie zu einer statistisch signifikanten Verbesserung des OS, der ORR und des PFS. Die Wirksamkeitsergebnisse sind in der folgenden Tabelle und den Abbildungen zusammengefasst.
Studie ARRAY-818-302 BEACON CRC: Wirksamkeitsdaten
Cetuximab mit Encorafenib |
Cetuximab mit Irinotecan oder Cetuximab mit FOLFIRI (Kontrollarm) |
|
Datenstichtag: 11. Februar 2019 (Primäranalyse) | ||
ORR (über BIRC) | ||
Anzahl an Patientene |
113 |
107 |
ORR n (%) |
23 (20,4) |
2 (1,9) |
p-Wertb,d,g |
< 0,0001 |
|
PFS (über BIRC) | ||
Anzahl an Patientena |
220 |
221 |
Anzahl an Ereignissen (%) |
133 (60,5) |
128 (57,9) |
Medianes PFS, Monate (95 % KI) |
4,2 (3,7; 5,4) |
1,5 (1,5; 1,7) |
HR (95 % KI)b,c |
0,40 (0,30; 0,55) |
|
Aktualisierte Analyse, Datenstichtag: 15. August 2019 | ||
OS | ||
Anzahl an Patientena |
220 |
221 |
Anzahl an Ereignissen (%) |
128 (58,2) |
157 (71,0) |
Median, Monate (95 % KI) |
9,3 (8,0; 11,3) |
5,9 (5,1; 7,1) |
HR (95 % KI)b (vs. Kontrolle) |
0,61 (0,48; 0,77) |
|
Mediane Dauer der Nachbeobachtung, Monate |
12,3 (11,1; 14,1) |
12,9 (10,9; 14,6) |
KI = Konfidenzintervall; HR = Hazard Ratio; ORR = Objektive Ansprechrate; OS = Gesamtüberlebenszeit
a Randomisierte Phase 3, Full-Analysis-Set
b Stratifiziert nach ECOG PS, Quelle anhand Cetuximab und vorheriger Irinotecan-Anwendung zum Zeitpunkt der Randomisierung
c Wiederholte KI, abgeleitet unter Verwendung der Lan DeMets O’Brien-Fleming-Grenzen, die mit dem beobachteten Informationsanteil bei der Zwischenanalyse verknüpft sind
d 1‑seitig
e Unter den ersten 331 randomisierten Patienten
f Clopper-Pearson-Methode
g Cochran-Mantel-Haenszel-Test
h Nominaler p-Wert
Studie ARRAY-818-302 BEACON CRC: Kaplan-Meier-Kurve des Gesamtüberlebens (Datenstichtag: 15. August 2019)
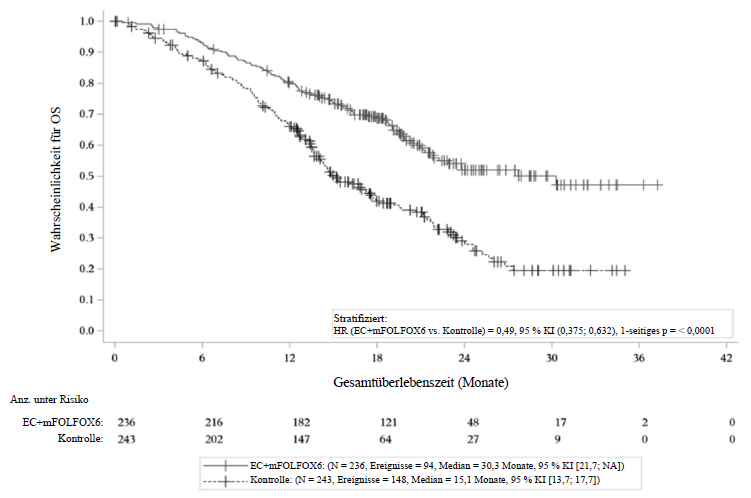
Cetuximab in Kombination mit Encorafenib und FOLFOX – Studie C4221015 (BREAKWATER)
Cetuximab in Kombination mit Encorafenib und mFOLFOX6 wurde in einer randomisierten, aktiv kontrollierten, unverblindeten, multizentrischen Studie (BREAKWATER) untersucht. Geeignet waren Patienten mit metastasierendem, zuvor unbehandeltem Kolorektalkarzinom mit MSS/pMMR (außer Patienten mit bestätigtem dMMR- oder MSI-H-Status, die keine Immuncheckpoint-Inhibitoren erhalten konnten), RAS-Wildtyp und BRAF-V600E-Mutation. Weitere wichtige Eignungskriterien waren: keine vorherige systemische Therapie im metastasierten Stadium, keine Vorbehandlung mit selektiven BRAF- oder EGFR‑Inhibitoren, kein Tumor mit hochgradiger Mikrosatelliteninstabilität (MSI‑H) oder Mismatch-Reparatur-Defizienz (dMMR) (es sei denn, der Patient war nicht für Immuncheckpoint-Inhibitoren geeignet), kein RAS‑mutierter Tumor oder unbekannter RAS‑Mutationsstatus sowie ein Performance-Status nach Eastern Cooperative Oncology Group (ECOG) von 0‑1. Die Randomisierung erfolgte stratifiziert nach ECOG‑Performance-Status (0 versus 1) und Region (USA/Kanada versus Europa versus Rest der Welt).
Eingeschlossene Patienten mit bestätigtem RAS‑Wildtyp konnten Cetuximab und mFOLFOX6 erhalten. Die vorherige Anwendung von BRAF- oder EGFR‑Inhibitoren war ausgeschlossen. Die Randomisierung erfolgte stratifiziert nach ECOG‑Performance-Status und Region.
Die Patienten wurden zunächst im Verhältnis 1:1:1 für einen der folgenden Behandlungsarme randomisiert und anschließend im Verhältnis 1:1 nach Abbruch der Rekrutierung für den Cetuximab+Encorafenib-Arm (158 Patienten):
Cetuximab 500 mg/m2 als intravenöse Infusion alle 2 Wochen in Kombination mit Encorafenib 300 mg oral einmal täglich (Cetuximab+Encorafenib-Arm)
Cetuximab 500 mg/m2 als intravenöse Infusion alle 2 Wochen in Kombination mit Encorafenib 300 mg oral einmal täglich und mFOLFOX6 alle 2 Wochen (Cetuximab+Encorafenib+mFOLFOX6-Arm)
mFOLFOX6 (alle 2 Wochen) oder FOLFOXIRI (alle 2 Wochen) oder CAPOX (alle 3 Wochen), jeweils mit oder ohne Bevacizumab (Anwendung gemäß ärztlicher Verordnung)
mFOLFOX6 bestand aus Oxaliplatin 85 mg/m2, Leucovorin 400 mg/m2, 5‑FU 400 mg/m2 als intravenöse Bolusgabe, gefolgt von einer intravenösen Dauerinfusion von 5‑FU 2 400 mg/m2 über 46‑48 Stunden; CAPOX bestand aus Oxaliplatin 130 mg/m2 als intravenöse Infusion und Capecitabin 1 000 mg/m2 als Tablette zum Einnehmen zweimal täglich an den Tagen 1‑14; FOLFOXIRI bestand aus Irinotecan 165 mg/m2, Oxaliplatin 85 mg/m2, Leucovorin 400 mg/m2, 5‑FU 2 400 oder 3 200 mg/m2 (gemäß lokalem Behandlungsstandard) als intravenöse Dauerinfusion über 46‑48 Stunden.
Die Behandlung wurde bis zum Fortschreiten der Erkrankung, dem Auftreten inakzeptabler Toxizität, dem Widerruf der Einwilligung, Lost-to-Follow-up oder dem Tod fortgesetzt. Im Folgenden werden ausschließlich die Ergebnisse des zugelassenen Behandlungsregimes (Cetuximab in Kombination mit Encorafenib und mFOLFOX6) beschrieben.
Die primären Wirksamkeitsendpunkte waren das PFS und die ORR, beurteilt durch das BICR. Weitere Wirksamkeitsendpunkte waren das OS und die Dauer des Ansprechens (duration of response, DoR), ebenfalls beurteilt durch das BICR. OS und PFS wurden bei allen randomisierten Patienten beurteilt. ORR und DoR wurden in der Subgruppe der ersten 110 Patienten jedes Studienarms beurteilt.
Insgesamt wurden 236 Patienten dem Cetuximab+Encorafenib+mFOLFOX6-Arm und 243 dem Kontrollarm randomisiert zugeteilt. Das mediane Alter dieser Patienten betrug 61 Jahre, 50 % waren weiblich, 60 % kaukasischer Herkunft, 37 % asiatischer Herkunft, 0,4 % hatten mehrere ethnische Zugehörigkeiten, 0,2 % afrikanischer/afroamerikanischer Herkunft und bei 2,5 % fehlten die Angaben. 12 % waren hispanischer oder lateinamerikanischer Herkunft, 81 % waren nicht hispanischer oder lateinamerikanischer Herkunft und bei 7 % fehlten die Angaben. 54 % hatten zu Studienbeginn einen ECOG‑Performance-Status von 0.
Die Kombination von Cetuximab mit Encorafenib und mFOLFOX6 zeigte im Vergleich zum aktiven Vergleichspräparat eine statistisch signifikante Verbesserung von PFS, ORR und OS. Die Wirksamkeitsergebnisse sind in der folgenden Tabelle und den Abbildungen zusammengefasst.
Studie C4221015: Ergebnisse zur Wirksamkeit
Cetuximab mit Encorafenib und mFOLFOX6 |
mFOLFOX6, FOLFOXIRI oder CAPOX; je mit oder ohne Bevacizumab (Kontrollarm) |
|
Datenstichtag: 22. Dezember 2023 (ORR Primäranalyse) | ||
ORR (über BIRC) | ||
Anzahl an Patientena |
110 |
110 |
ORR n (%) |
67 (60,9) |
44 (40,0) |
p-Wertb,c,d |
0,0008 |
|
Datenstichtag: 06. Januar 2025 (PFS Primäranalyse) | ||
PFS (über BIRC) | ||
Anzahl an Patientene |
236 |
243 |
Anzahl an Ereignissen (%) |
122 (51,7) |
132 (54,3) |
Medianes PFS, Monate (95 % KI) |
12,8 (11,2; 15,9) |
7,1 (6,8; 8,5) |
HR (95 % KI)c,d, f, g |
0,53 (0,41; 0,68) |
|
OS |
||
Anzahl an Patientene |
236 |
243 |
Anzahl an Ereignissen (%) |
94 (39,8) |
148 (60,9) |
Medianes OS, Monate (95 % KI) |
30,3 (21,7; NA) |
15,1 (13,7; 17,7) |
HR (95 % KI)c,d, f, g |
0,49 (0,38; 0,63) |
|
Mediane Dauer der Nachbeobachtung, Monate |
21,8 (20,4; 23,4) |
22,2 (18,9; 23,5) |
a ORR-Subsgruppe Full-Analysis-Set
b p-Wert, basierend auf Cochran-Mantel-Haenszel(CMH)-Test
c Randomisierung stratifiziert nach ECOG‑PS und geografischer Region
d 1-seitig
e Randomisierte Phase 3, Full-Analysis-Set
f Hazard Ratio basierend auf dem Cox-Proportional-Hazards-Modell; bei proportionalen Hazardraten deutet eine Hazard Ratio < 1 auf eine Reduktion der Hazardrate zugunsten von EC+mFOLFOX6 im Vergleich zur Kontrolle hin
g p-Wert aus dem Log-Rank-Test
BIRC = Verblindetes, unabhängiges zentrales Review Committee; KI = Konfidenzintervall; CR = Komplettes Ansprechen; HR = Hazard Ratio; NA = Nicht abschätzbar; ORR = Objektive Ansprechrate; OS = Gesamtüberleben; PFS = Progressionsfreies Überleben; PR = Partielles Ansprechen; SD = Stabile Erkrankung
Studie C4221015: Kaplan-Meier-Kurve des Gesamtüberlebens (Datenstichtag: 06. Januar 2025)
Plattenepithelkarzinom im Kopf- und Halsbereich
Ein immunhistochemischer Nachweis der EGFR-Exprimierung erfolgte nicht, da mehr als 90 % der Patienten mit Plattenepithelkarzinom im Kopf- und Halsbereich EGFR-exprimierende Tumore aufweisen.
Cetuximab in Kombination mit einer Strahlentherapie für eine lokal fortgeschrittene Erkrankung
EMR 62 202-006:
In dieser randomisierten Studie wurde bei Patienten mit lokal fortgeschrittenem Plattenepithelkarzinom im Kopf- und Halsbereich die Kombination von Cetuximab und einer Strahlentherapie (211 Patienten) verglichen mit einer alleinigen Strahlentherapie (213 Patienten). Die Behandlung mit Cetuximab wurde eine Woche vor der Bestrahlung begonnen und bis zum Ende des Bestrahlungszeitraumes in der in Abschnitt 4.2 beschriebenen Dosierung verabreicht.
Die in dieser klinischen Studie erhobenen Daten zur Wirksamkeit sind in der nachfolgenden Tabelle zusammengefasst:
Variable/Statistik |
Bestrahlung plus Cetuximab |
Bestrahlung alleine |
||
|
(N=211) |
(N=213) |
||
Lokoregionäre Kontrolle |
|
|
|
|
Monate, Median (95 % KI) |
24,4 |
(15,7; 45,1) |
14,9 |
(11,8; 19,9) |
Hazard Ratio (95 % KI) |
0,68 (0,52; 0,89) |
|||
p-Wert |
0,005 |
|||
OS |
|
|
|
|
Monate, Median (95 % KI) |
49,0 |
(32,8; 69,5+) |
29,3 |
(20,6; 41,4) |
Hazard Ratio (95 % KI) |
0,73 (0,56; 0,95) |
|||
p-Wert |
0,018 |
|||
mediane Nachbeobachtungsdauer, Monate |
60,0 |
60,1 |
||
1-Jahres-Überlebensrate, % (95 % KI) |
77,6 (71,4; 82,7) |
73,8 (67,3; 79,2) |
||
2-Jahres-Überlebensrate, % (95 % KI) |
62,2 (55,2; 68,4) |
55,2 (48,2; 61,7) |
||
3-Jahres-Überlebensrate, % (95 % KI) |
54,7 (47,7; 61,2) |
45,2 (38,3; 51,9) |
||
5-Jahres-Überlebensrate, % (95 % KI) |
45,6 (38,5; 52,4) |
36,4 (29,7; 43,1) |
||
KI = Konfidenzintervall, OS = Gesamtüberlebenszeit, ein „+“ bedeutet, dass die Obergrenze bei Beendigung nicht erreicht worden war.
Patienten mit einer guten Prognose, angezeigt durch Tumor-Klassifizierung, Karnofsky Performance Status (KPS) und Alter, hatten einen stärker ausgeprägten Vorteil, wenn Cetuximab zusätzlich zur Bestrahlung angewendet wurde. Kein klinischer Vorteil konnte für Patienten ≥ 65 Jahre mit einem KPS ≤ 80 gezeigt werden.
Die Anwendung von Cetuximab in Kombination mit einer Chemo-Strahlentherapie wurde bislang nicht ausreichend untersucht. Daher wurde das Nutzen/Risiko-Verhältnis für diese Kombination bisher nicht ermittelt.
Cetuximab in Kombination mit einer platin-basierten Chemotherapie für eine rezidivierende und/oder metastasierende Erkrankung
EMR 62 202-002:
In dieser randomisierten Studie wurde, bei chemotherapeutisch nicht-vorbehandelten Patienten mit rezidivierendem und/oder metastasierendem Plattenepithelkarzinom im Kopf- und Halsbereich, die Kombination von Cetuximab und Cisplatin oder Carboplatin plus 5-Fluorouracil-Infusionen (222 Patienten) verglichen mit der entsprechenden Chemotherapie allein (220 Patienten). Die Behandlung im Cetuximab-Arm bestand aus bis zu 6 Zyklen einer platin-basierten Chemotherapie in Kombination mit Cetuximab, gefolgt von Cetuximab als Erhaltungstherapie bis zur Progression der Erkrankung.
Die in dieser klinischen Studie erhobenen Daten zur Wirksamkeit sind in der nachfolgenden Tabelle zusammengefasst:
Variable/ Statistik |
Cetuximab plus CTX |
CTX |
|
(N=222) |
(N=220) |
OS |
|
|
Monate, Median (95 % KI) |
10,1 (8,6; 11,2) |
7,4 (6,4; 8,3) |
Hazard Ratio (95 % KI) |
0,797 (0,644; 0,986) |
|
p-Wert |
0,0362 |
|
PFS |
|
|
Monate, Median (95 % KI) |
5,6 (5,0; 6,0) |
3,3 (2,9; 4,3) |
Hazard Ratio (95 % KI) |
0,538 (0,431; 0,672) |
|
p-Wert |
<0,0001 |
|
ORR |
|
|
% (95 % KI) |
35,6 (29,3; 42,3) |
19,5 (14,5; 25,4) |
p-Wert |
0,0001 |
|
KI = Konfidenzintervall, CTX = Platin-basierte Chemotherapie, ORR = objektive Ansprech- bzw. Remissionsrate, OS = Gesamtüberlebenszeit, PFS = progressionsfreie Überlebenszeit
Patienten mit einer guten Prognose, angezeigt durch Tumor-Klassifizierung, Karnofsky Performance Status (KPS) und Alter, hatten einen stärker ausgeprägten Vorteil, wenn Cetuximab zusätzlich zu einer platin-basierten Chemotherapie angewendet wurde. Im Gegensatz zur progressionsfreien Überlebenszeit konnte für die Gesamtüberlebenszeit kein Vorteil für Patienten ≥ 65 Jahre mit einem KPS ≤ 80 gezeigt werden.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Cetuximab eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in folgenden Anwendungsgebieten gewährt: Adenokarzinom des Kolons und Rektums und Epithelkarzinome des Oropharynx, des Larynx oder der Nase (ausgenommen Nasopharynxkarzinom oder Lymphoepitheliom, siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Pharmakokinetik von Cetuximab wurde im Rahmen klinischer Prüfungen sowohl bei monotherapeutischer Anwendung als auch bei Kombination mit Chemo- oder Strahlentherapie untersucht. Dabei zeigte Cetuximab bei einmal wöchentlicher intravenöser Infusion in Dosierungen von 5 - 500 mg/m² Körperoberfläche eine dosisabhängige Kinetik.
Bei Verabreichung von Cetuximab in einer Initialdosis von 400 mg/m² Körperoberfläche entsprach das mittlere Verteilungsvolumen in etwa dem Intravasalraum (2,9 l/m², Streubereich 1,5 - 6,2 l/m²). Die Cmax lag im Mittel (± Standardabweichung) bei 185±55 Mikrogramm pro ml. Die mittlere Clearance betrug 0,022 l/h pro m² Körperoberfläche. Cetuximab hat eine lange Eliminationshalbwertszeit von 70 - 100 Stunden.
Bei Verabreichung von Cetuximab in einem wöchentlichen Dosierungsschema (400 mg/m² Aufsättigungsdosis gefolgt von 250 mg/m² wöchentlich) wurden Steady-State-Serumkonzentrationen nach dreiwöchiger Cetuximab-Monotherapie erreicht. Die Cmax lag in der 3. Woche bei durchschnittlich 155,8 Mikrogramm pro ml und in der 8. Woche bei 151,6 Mikrogramm pro ml, während die entsprechenden Cmin-Werte 41,3 bzw. 55,4 Mikrogramm pro ml betrugen. In einer Studie, in der Cetuximab in Kombination mit Irinotecan geprüft wurde, lag der mittlere Talspiegel von Cetuximab in der 12. Woche bei 50,0 Mikrogramm pro ml und in der 36. Woche bei 49,4 Mikrogramm pro ml.
Bei Verabreichung von Cetuximab in einem zweiwöchentlichen Dosierungsschema (500 mg/m² alle zwei Wochen) wurden Steady-State-Serumkonzentrationen nach fünfwöchiger Cetuximab-Monotherapie erreicht. Die Cmax lag in der 5. Woche bei durchschnittlich 297 Mikrogramm pro ml, der entsprechende Cmin-Wert betrug 31,0 Mikrogramm pro ml.
Es wurden verschiedene Stoffwechselwege beschrieben, die an der Metabolisierung von Antikörpern beteiligt sein können. Allen gemein ist der Abbau des Antikörpers im Organismus in kleinere Moleküle, also in kleine Peptide oder Aminosäuren.
Pharmakokinetik in speziellen Patientengruppen
Populationspharmakokinetische Analysen zeigten, dass ethnische Zugehörigkeit, Alter, Geschlecht, Nieren- oder Leberfunktion keinen klinisch relevanten Einfluss auf die pharmakokinetischen Eigenschaften von Cetuximab haben. Es wurde zwar ein Zusammenhang zwischen Geschlecht und Arzneimittelexposition beobachtet, wobei Frauen eine höhere Exposition aufwiesen, dieser Zusammenhang wurde jedoch als klinisch nicht relevant eingestuft.
Basierend auf einer populationspharmakokinetischen Analyse hatte eine leichte (CRCL ≥ 60 und < 90 ml/min) und eine mäßige (CRCL ≥ 30 und < 60 ml/min) Nierenfunktionsstörung im Vergleich zu Patienten mit normaler Nierenfunktion (CRCL ≥ 90 ml/min) keinen klinisch relevanten Einfluss auf die Pharmakokinetik von Cetuximab. Für Patienten mit leichter oder mäßiger Nierenfunktionsstörung wird keine Dosisanpassung empfohlen. Die Pharmakokinetik von Cetuximab bei Patienten mit schwerer (CRCL ≥ 15 und < 30 ml/min) Nierenfunktionsstörung oder terminaler Niereninsuffizienz wurde nicht untersucht.
Basierend auf einer populationspharmakokinetischen Analyse hatte eine leichte Leberfunktionsstörung gemäß Definition der NCI‑ODWG im Vergleich zu Patienten mit normaler Leberfunktion keinen klinisch relevanten Einfluss auf die Pharmakokinetik von Cetuximab. Für Patienten mit leichter Leberfunktionsstörung wird keine Dosisanpassung empfohlen. Der Einfluss einer mäßigen oder schweren Leberfunktionsstörung, wie sie nach den NCI‑ODWG‑Kriterien definiert sind, auf die Pharmakokinetik von Cetuximab wurde nicht untersucht.
Kinder und Jugendliche
In einer Phase-I-Studie an Kindern und Jugendlichen (1-18 Jahre) mit refraktären soliden Tumoren wurde Cetuximab in Kombination mit Irinotecan verabreicht. Die pharmakokinetischen Ergebnisse waren vergleichbar mit denen bei Erwachsenen.
Dosisabhängige Hautveränderungen - ab einem Dosisspiegel, der dem Gebrauch im Humanbereich entspricht - waren die Hauptbefunde der an Cynomolgusaffen durchgeführten Toxizitätsstudien (eine Studie zur chronischen Toxizität bei wiederholter Verabreichung und eine Studie zur embryonalen und fötalen Entwicklung).
Eine Toxizitätsstudie zur embryonalen und fötalen Entwicklung an Cynomolgus-Affen ergab keine Anzeichen für Teratogenität. Jedoch war ein dosisabhängiger Anstieg der Abortrate zu beobachten.
Präklinische Daten zur Gentoxizität und zur lokalen Verträglichkeit einschließlich des unbeabsichtigten Gebrauchs über nicht vorgesehene Verabreichungswege ließen keine besonderen Gefahren für den Menschen erkennen.
Zum kanzerogenen Potenzial von Cetuximab sowie zur Abklärung einer eventuellen Beeinträchtigung der männlichen und weiblichen Fertilität durch die Substanz wurden keine formalen Tierstudien durchgeführt.
Es wurden keine Toxizitätsstudien mit der Kombination von Cetuximab und Chemotherapeutika durchgeführt.
Zum Einfluss von Cetuximab auf die Wundheilung liegen keine präklinischen Daten vor. In präklinischen Wundheilungsmodellen verzögerte die Gabe von EGFR-selektiven Tyrosinkinaseinhibitoren jedoch die Wundheilung.
Natriumchlorid
Glycin
Polysorbat 80
Citronensäure-Monohydrat
Natriumhydroxid
Wasser für Injektionszwecke
Das Arzneimittel darf, außer mit den unter Abschnitt 6.6 aufgeführten, nicht mit anderen Arzneimitteln gemischt werden.
4 Jahre.
Bei 25°C ist die chemische und physikalische Stabilität der angebrochenen Erbitux 5 mg/ml Infusionslösung über 48 Stunden belegt, wenn die Lösung, wie in Abschnitt 6.6 beschrieben, zubereitet wurde.
Erbitux enthält weder ein antimikrobiell wirksames Konservierungsmittel noch ein Bakteriostatikum. Unter mikrobiologischen Gesichtspunkten soll das Produkt direkt nach Anbruch verwendet werden. Wird das Produkt nicht sofort verwendet, so obliegen die Lagerzeiten und -bedingungen der angebrochenen Lösung der Verantwortung des Anwenders. In der Regel soll ein Zeitraum von 24 Stunden bei 2 ‑ 8°C nicht überschritten werden, sofern das Produkt nicht unter kontrollierten und validierten aseptischen Bedingungen geöffnet wurde.
Im Kühlschrank lagern (2°C ‑ 8°C).
Aufbewahrungsbedingungen nach Anbruch, siehe Abschnitt 6.3.
20 ml oder 100 ml Lösung in Durchstechflaschen aus Glas Typ 1 mit Stopfen aus Halogenbutylgummi und Aluminium/Polypropylen-Siegel.
Eine Durchstechflasche pro Packung.
Es werden möglicherweise nicht alle Größen der Durchstechflaschen in den Verkehr gebracht.
Die Verabreichung von Erbitux kann entweder als Tropfinfusion, mit einer Infusionspumpe oder einem Perfusor erfolgen. Für die Infusion von Erbitux ist ein separates Infusionsset zu verwenden, und der Infusionsschlauch ist am Ende der Infusion mit steriler 0,9%iger Kochsalzlösung (9 mg/ml) zu spülen.
Erbitux 5 mg/ml ist mit folgenden Materialien kompatibel:
Infusionsbeutel aus Polyethylen (PE), Ethylvinylacetat (EVA) oder Polyvinylchlorid (PVC),
Infusionssets aus Polyethylen (PE), Polyurethan (PUR), Ethylvinylacetat (EVA), Polyolefin-Thermoplast (TP) oder Polyvinylchlorid (PVC),
Spritzen aus Polypropylen (PP) für einen Perfusor.
Bei der Vorbereitung der Infusion ist auf aseptische Bedingungen zu achten.
Die Erbitux 5 mg/ml Infusionslösung ist wie folgt vorzubereiten:
Für die Verabreichung mittels Infusionspumpe oder Tropfinfusion (verdünnt mit steriler 0,9%iger Kochsalzlösung [9 mg Natriumchlorid/ml]): Einen Infusionsbeutel mit steriler 0,9%iger Kochsalzlösung (9 mg Natriumchlorid/ml) in geeigneter Größe wählen und das benötigte Volumen Erbitux berechnen. Ein adäquates Volumen der Kochsalzlösung mittels einer geeigneten sterilen Spritze mit passender Kanüle aus dem Infusionsbeutel entnehmen. Auf eine geeignete sterile Spritze eine passende Kanüle aufsetzen. Die benötigte Menge Erbitux aus der Durchstechflasche aufziehen. Erbitux in den vorbereiteten Infusionsbeutel geben. Diesen Vorgang wiederholen, bis das errechnete Volumen erreicht ist. Die Infusionsleitung anschließen und vor dem Start der Infusion mit dem verdünnten Erbitux spülen. Mittels Tropfinfusion oder Infusionspumpe verabreichen. Die Einstellung bzw. Regulierung der Infusionsgeschwindigkeit erfolgt wie in Abschnitt 4.2 erläutert.
Für die Verabreichung mittels Infusionspumpe oder Tropfinfusion (unverdünnt): Das benötigte Volumen Erbitux berechnen. Auf eine geeignete sterile Spritze (min. 50 ml) eine passende Kanüle aufsetzen. Die benötigte Menge Erbitux aus der Durchstechflasche aufziehen. Erbitux in einen sterilen, luftleeren Behälter oder Infusionsbeutel geben. Diesen Vorgang wiederholen, bis das errechnete Volumen erreicht ist. Die Infusionsleitung anschließen und vor dem Start der Infusion mit Erbitux spülen. Die Einstellung bzw. Regulierung der Infusionsgeschwindigkeit erfolgt wie in Abschnitt 4.2 erläutert.
Für die Verabreichung mittels Perfusor: Das benötigte Volumen Erbitux berechnen. Auf eine geeignete sterile Spritze eine passende Kanüle aufsetzen. Die benötigte Menge Erbitux aus der Durchstechflasche aufziehen. Die Kanüle entfernen und die Spritze in den Perfusorschlitten einsetzen. Anschließend die Infusionsleitung an die Spritze anschließen und die Infusionsgeschwindigkeit wie in Abschnitt 4.2 beschrieben einstellen bzw. regulieren. Infusionsschlauch mit Erbitux oder steriler 0,9%iger Kochsalzlösung (9 mg Natriumchlorid/ml) spülen und Infusion starten. Diesen Vorgang gegebenenfalls so lange wiederholen, bis das berechnete Volumen infundiert ist.
Merck Europe B.V.
Gustav Mahlerplein 102
1082 MA Amsterdam
Niederlande
EU/1/04/281/003
EU/1/04/281/005
Datum der Erteilung der Zulassung: 29. Juni 2004
Datum der letzten Verlängerung der Zulassung: 17. Juni 2009
Juni 2026
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig