
Evrysdi® 0,75 mg/ml Pulver zur Herstellung einer Lösung zum Einnehmen
Jede Flasche enthält 60 mg Risdiplam in 2 g Pulver zur Herstellung einer Lösung zum Einnehmen.
Jeder ml der rekonstituierten Lösung enthält 0,75 mg Risdiplam.
Sonstige Bestandteile mit bekannter Wirkung
Jeder ml enthält 0,38 mg Natriumbenzoat (E 211) und 2,97 mg Isomalt (Ph.Eur.) (E 953).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Pulver zur Herstellung einer Lösung zum Einnehmen.
Hellgelbes, gelbes, grau-gelbes, grün-gelbes oder hellgrünes Pulver.
Evrysdi wird angewendet zur Behandlung der 5q-assoziierten spinalen Muskelatrophie (SMA) bei Patienten mit einer klinisch diagnostizierten Typ 1-, Typ 2- oder Typ 3-SMA oder mit einer bis vier Kopien des SMN2-Gens.
Eine Therapie mit Risdiplam ist von einem Arzt mit Erfahrung in der Behandlung der SMA einzuleiten.
Dosierung
Die empfohlene einmal tägliche Dosis von Risdiplam wird nach Alter und Körpergewicht bestimmt (siehe Tabelle 1).
Tabelle 1: Evrysdi Pulver zur Herstellung einer Lösung zum Einnehmen; Dosierungsschema nach Alter und Körpergewicht
Alter* und Körpergewicht |
Empfohlene tägliche Dosis |
< 2 Monate |
0,15 mg/kg |
2 Monate bis < 2 Jahre |
0,20 mg/kg |
≥ 2 Jahre (< 20 kg) |
0,25 mg/kg |
≥ 2 Jahre (≥ 20 kg) |
5 mg |
* für Frühgeborene gilt das korrigierte Alter | |
Für Patienten im Alter von ≥ 2 Jahren und mit einem Gewicht von ≥ 20 kg steht eine Filmtablette als alternative Darreichungsform zur Verfügung. Die Zusammenfassung der Merkmale des Arzneimittels (Fachinformation) für Evrysdi Filmtabletten ist zu beachten.
Der Arzt muss die geeignete Darreichungsform entsprechend der erforderlichen Dosis und den Bedürfnissen des Patienten, einschließlich seiner Fähigkeit zu schlucken, verordnen. Für Patienten, die Schwierigkeiten haben, eine ganze Tablette zu schlucken, oder bei denen die Verabreichung über eine Nasogastralsonde oder Gastrostomiesonde erfolgen muss, kann die Filmtablette in Wasser aufgelöst oder das Pulver zur Herstellung einer Lösung zum Einnehmen verschrieben werden.
Eine Behandlung mit einer täglichen Dosis von mehr als 5 mg wurde nicht untersucht.
Verspätete oder versäumte Dosen
Wurde eine geplante Dosis versäumt, ist die Einnahme so früh wie möglich nachzuholen, wenn seit dem geplanten Einnahmezeitpunkt nicht mehr als 6 Stunden vergangen sind. Andernfalls ist die versäumte Dosis zu überspringen und die nächste Dosis ist zum regulär geplanten Zeitpunkt am nächsten Tag einzunehmen.
Wenn eine Dosis nicht vollständig geschluckt wurde oder wenn es nach der Einnahme einer Dosis Risdiplam zu Erbrechen kommt, darf keine weitere Dosis eingenommen werden, um die unvollständige Dosis auszugleichen. Die nächste Einnahme ist zum regulär geplanten Zeitpunkt vorzunehmen.
Ältere Patienten
Bei älteren Patienten ist, basierend auf der begrenzten Datenlage zu Personen im Alter ab 65 Jahren, keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Patienten mit Nierenfunktionsstörung
Risdiplam wurde in dieser Patientengruppe nicht untersucht. Es wird nicht erwartet, dass bei Patienten mit Nierenfunktionsstörung eine Dosisanpassung erforderlich ist (siehe Abschnitt 5.2).
Patienten mit Leberfunktionsstörung
Bei Patienten mit leichter oder mäßiger Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Patienten mit schwerer Leberfunktionsstörung wurden nicht untersucht. Bei ihnen kann die Risdiplam-Exposition erhöht sein (siehe Abschnitte 5.1 und 5.2).
Kinder und Jugendliche
Es liegen keine Daten zur Pharmakokinetik von Risdiplam bei Patienten im Alter von weniger als 16 Tagen vor.
Art der Anwendung
Zum Einnehmen.
Evrysdi Pulver zur Herstellung einer Lösung zum Einnehmen muss vor der Abgabe von medizinischem Fachpersonal (z. B. Apotheker) rekonstituiert werden.
Es wird empfohlen, dass medizinisches Fachpersonal vor der ersten Einnahme des Arzneimittels die Vorbereitung der verordneten täglichen Dosis mit dem Patienten oder der Pflegeperson bespricht.
Evrysdi wird einmal täglich, etwa zur gleichen Uhrzeit, mit oder ohne Nahrung unter Verwendung der mitgelieferten wiederverwendbaren Applikationsspritze für Zubereitungen zum Einnehmen eingenommen. Evrysdi darf nicht mit Milch oder Formulamilch gemischt werden.
Evrysdi sollte sofort nach dem Aufziehen in die Applikationsspritze eingenommen werden. Wenn es nicht innerhalb von 5 Minuten eingenommen wird, sollte der Inhalt der Applikationsspritze verworfen und eine neue Dosis zubereitet werden. Wenn Evrysdi verschüttet wird oder auf die Haut gelangt, ist der Bereich mit Wasser und Seife zu waschen.
Nach der Einnahme von Evrysdi sollte der Patient Wasser trinken, um sicherzustellen, dass das Arzneimittel vollständig geschluckt wurde. Wenn der Patient nicht schlucken kann und eine Nasogastralsonde oder eine Gastrostomiesonde in situ hat, kann Evrysdi Pulver zur Herstellung einer Lösung zum Einnehmen über die Sonde verabreicht werden. Nach der Verabreichung von Evrysdi muss die Sonde mit Wasser gespült werden.
Auswahl der Applikationsspritze für die verordnete tägliche Dosis:
Spritzengröße |
Dosierungsvolumen |
Spritzenmarkierungen |
1 ml |
0,3 ml bis 1 ml |
0,01 ml |
6 ml |
1 ml bis 6 ml |
0,1 ml |
12 ml |
6,2 ml bis 6,6 ml |
0,2 ml |
Bei der Berechnung des Dosierungsvolumens müssen die Markierungen der Applikationsspritze berücksichtigt werden. Das Dosierungsvolumen sollte auf den am nächsten liegenden Teilstrich der ausgewählten Applikationsspritze gerundet werden.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Potenzielle embryofetale Toxizität
In tierexperimentellen Studien wurde embryofetale Toxizität beobachtet (siehe Abschnitt 5.3). Patienten im reproduktionsfähigen Alter müssen über die Risiken aufgeklärt werden und müssen während der Behandlung und nach der letzten Einnahme im Fall von weiblichen Patienten mindestens 1 Monat lang und im Fall von männlichen Patienten mindestens 4 Monate lang eine hoch zuverlässige Verhütungsmethode anwenden. Vor Beginn der Behandlung mit Risdiplam sollte der Schwangerschaftsstatus von fortpflanzungsfähigen Patientinnen geprüft werden (siehe Abschnitt 4.6).
Potenzielle Auswirkungen auf die männliche Fertilität
Aufgrund von Beobachtungen in tierexperimentellen Studien, sollten männliche Patienten während der Behandlung sowie für einen Zeitraum von 4 Monaten nach der letzten Einnahme von Risdiplam keinen Samen spenden. Vor Beginn der Behandlung sollten mit männlichen Patienten im reproduktionsfähigen Alter Strategien zur Erhaltung der Fertilität besprochen werden (siehe Abschnitte 4.6 und 5.3). Die Auswirkungen von Risdiplam auf die männliche Fertilität wurden bei Menschen nicht untersucht.
Sonstige Bestandteile
Isomalt
Evrysdi enthält Isomalt (2,97 mg pro ml). Patienten mit einer seltenen hereditären Fructose-Intoleranz sollten dieses Arzneimittel nicht einnehmen.
Natrium
Dieses Arzneimittel enthält 0,375 mg Natriumbenzoat pro ml Lösung. Natriumbenzoat kann Gelbsucht (Gelbfärbung von Haut und Augen) bei Neugeborenen (im Alter bis zu 4 Wochen) verstärken.
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro 5-mg-Dosis, d. h. es ist nahezu „natriumfrei“.
Wirkungen anderer Arzneimittel auf Risdiplam
Die gleichzeitige, zweimal tägliche Einnahme von 200 mg Itraconazol, einem starken CYP3A-Inhibitor, mit einer oralen Einzeldosis von 6 mg Risdiplam zeigte keine klinisch relevante Wirkung auf die pharmakokinetischen Parameter von Risdiplam (11 % Zunahme der AUC, 9 % Abnahme der Cmax). Bei gleichzeitiger Anwendung von Risdiplam mit einem CYP3A-Inhibitor ist keine Dosisanpassung erforderlich.
Arzneimittelwechselwirkungen über den FMO1- und FMO3-Signalweg sind nicht zu erwarten.
Wirkungen von Risdiplam auf andere Arzneimittel
Risdiplam ist ein schwacher Inhibitor von CYP3A. Bei gesunden erwachsenen Probanden führte die Einnahme von Risdiplam einmal täglich über 2 Wochen zu einer leichten Erhöhung der Exposition gegenüber Midazolam, einem sensitiven CYP3A-Substrat (11 % Anstieg der AUC; 16 % Anstieg der Cmax). Das Ausmaß der Wechselwirkung wird nicht als klinisch relevant angesehen, weshalb für CYP3A-Substrate keine Dosisanpassung erforderlich ist.
In-vitro-Untersuchungen haben gezeigt, dass Risdiplam und sein Hauptmetabolit M1 keine wesentlichen Inhibitoren des humanen MDR1, der organic anion transporting polypeptide (OATP) 1B1 und 1B3 sowie der organic anion transporter 1 und 3 (OAT 1 und 3) sind. Risdiplam und sein Metabolit inhibieren jedoch in vitro den human organic cation transporter 2 (OCT2) und die multidrug and toxin extrusion transporter 1 und 2-K (MATE1 und MATE2-K). Bei therapeutischen Arzneimittelkonzentrationen ist keine Wechselwirkung mit OCT2-Substraten zu erwarten. Die Wirkung der gleichzeitigen Anwendung von Risdiplam auf die Pharmakokinetik von MATE1- und MATE2-K-Substraten beim Menschen ist nicht bekannt. Basierend auf In-vitro-Daten kann Risdiplam die Plasmakonzentrationen von Arzneimitteln, die über MATE1 oder MATE2-K eliminiert werden, wie z. B. Metformin, erhöhen. Wenn sich eine gleichzeitige Anwendung nicht vermeiden lässt, sollten arzneimittelbezogene Toxizitäten überwacht und bei Bedarf eine Dosisreduzierung des gleichzeitig angewendeten Arzneimittels in Betracht gezogen werden.
Es liegen keine Daten zur Wirksamkeit oder Sicherheit vor, die die gleichzeitige Anwendung von Risdiplam und Nusinersen stützen.
Patienten im reproduktionsfähigen Alter
Verhütung bei männlichen und weiblichen Patienten
Männliche und weibliche Patienten im reproduktionsfähigen Alter müssen die folgenden Maßnahmen zur Verhütung ergreifen:
Patientinnen im gebärfähigen Alter müssen während der Behandlung und für mindestens 1 Monat nach der letzten Dosis eine hoch zuverlässige Verhütungsmethode anwenden.
Männliche Patienten und deren Partnerinnen im gebärfähigen Alter müssen beide sicherstellen, dass während der Behandlung und für mindestens 4 Monate nach der letzten Dosis eine hoch zuverlässige Verhütungsmethode angewendet wird.
Schwangerschaftstest
Vor der Einleitung einer Therapie mit Risdiplam sollte bei Patientinnen im gebärfähigen Alter der Schwangerschaftsstatus festgestellt werden. Schwangere Frauen müssen eindeutig auf das mögliche Risiko für den Fötus hingewiesen werden.
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Risdiplam bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3).
Die Anwendung von Risdiplam während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen (siehe Abschnitt 4.4).
Stillzeit
Es ist nicht bekannt, ob Risdiplam in die menschliche Muttermilch übergeht. Studien an Ratten haben gezeigt, dass Risdiplam in die Milch ausgeschieden wird (siehe Abschnitt 5.3). Da über eine potenzielle gesundheitsschädigende Wirkung für den gestillten Säugling nichts bekannt ist, wird empfohlen, während der Behandlung nicht zu stillen.
Fertilität
Männliche Patienten
Wie nicht-klinische Befunde zeigen, kann die männliche Fertilität während der Behandlung beeinträchtigt sein. In den Reproduktionsorganen von Ratten und Affen wurden eine Degeneration des Spermas und reduzierte Spermienzahlen beobachtet (siehe Abschnitt 5.3). Aufgrund von Beobachtungen aus tierexperimentellen Studien ist zu erwarten, dass die Auswirkungen auf die Spermien nach Absetzen von Risdiplam reversibel sind.
Männliche Patienten können eine Spermakonservierung vor Beginn der Behandlung oder nach einem behandlungsfreien Zeitraum von mindestens 4 Monaten in Betracht ziehen. Männliche Patienten, die ein Kind zeugen möchten, sollten die Behandlung mindestens 4 Monate lang unterbrechen. Nach der Empfängnis kann die Behandlung wieder aufgenommen werden.
Weibliche Patienten
Basierend auf nicht-klinischen Daten (siehe Abschnitt 5.3) ist eine Beeinträchtigung der weiblichen Fertilität durch Risdiplam nicht zu erwarten.
Risdiplam hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Bei Patienten mit infantiler SMA sind die Nebenwirkungen, die in klinischen Studien mit Risdiplam am häufigsten beobachtet wurden, Fieber (54,8 %), Ausschlag (29,0 %) und Diarrhö (19,4 %).
Bei Patienten mit später einsetzender SMA sind die Nebenwirkungen, die in klinischen Studien mit Risdiplam am häufigsten beobachtet wurden, Fieber (21,7 %), Kopfschmerzen (20,0 %), Diarrhö (16,7 %) und Ausschlag (16,7 %).
Die oben genannten Nebenwirkungen traten ohne identifizierbares klinisches Muster oder Zeitmuster auf und klangen trotz fortgesetzter Behandlung bei Patienten mit infantiler und Patienten mit später einsetzender SMA im Allgemeinen wieder ab.
Tabellarische Auflistung der Nebenwirkungen
Den jeweiligen Häufigkeitskategorien für die einzelnen Nebenwirkungen liegt folgende Konvention zugrunde: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Die in klinischen Studien aufgetretenen Nebenwirkungen (Tabelle 2) sind nach MedDRA-Systemorganklassen aufgelistet.
Tabelle 2: Nebenwirkungen bei Patienten mit infantiler und später einsetzender SMA auf der Grundlage klinischer Studien zu Risdiplam und Erfahrungen nach der Markteinführung
Systemorganklasse |
Infantile SMA |
Später einsetzende SMA |
Infektionen und parasitäre Erkrankungen | ||
Harnwegsinfektion |
Häufig |
Häufig |
Erkrankungen des Nervensystems | ||
Kopfschmerzen |
Nicht zutreffend |
Sehr häufig |
Erkrankungen des Gastrointestinaltrakts | ||
Diarrhö |
Sehr häufig |
Sehr häufig |
Übelkeit |
Nicht zutreffend |
Häufig |
Mundulzeration und aphthöse Geschwüre |
Häufig |
Häufig |
Erkrankungen der Haut und des Unterhautgewebes | ||
Ausschlag* |
Sehr häufig |
Sehr häufig |
Kutane Vaskulitis** |
Nicht bekannt |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | ||
Arthralgie |
Nicht zutreffend |
Häufig |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | ||
Fieber (einschließlich Hyperpyrexie) |
Sehr häufig |
Sehr häufig |
*Umfasst Dermatitis, akneiforme Dermatitis, allergische Dermatitis, Erythem, Follikulitis, Ausschlag, erythematösen Ausschlag, makulo-papulösen Ausschlag, papulösen Ausschlag
**Nach der Markteinführung wurde über kutane Vaskulitis berichtet. Nach dauerhaftem Absetzen von Risdiplam klangen die Symptome ab. Die Häufigkeit ist auf Grundlage der verfügbaren Daten nicht abschätzbar.
Sicherheitsprofil bei präsymptomatischen Patienten
Auf Grundlage der Primäranalyse von RAINBOWFISH stimmt das Sicherheitsprofil von Evrysdi bei präsymptomatischen Patienten mit dem Sicherheitsprofil symptomatischer Patienten mit infantiler SMA und Patienten mit später einsetzender SMA überein. In die Studie RAINBOWFISH waren 26 Patienten mit präsymptomatischer SMA aufgenommen worden, die zum Zeitpunkt der ersten Dosis zwischen 16 und 41 Tage alt waren (Gewichtsspanne 3,1 bis 5,7 kg). Die mediane Expositionsdauer betrug 20,4 Monate (Bereich: 10,6 bis 41,9 Monate). Für Neugeborene im Alter von < 20 Tagen liegen nur begrenzte Daten nach der Markteinführung vor.
Sicherheitsprofil bei Patienten, die zuvor mit anderen SMA-modifizierenden Therapien behandelt wurden
Das Sicherheitsprofil von Risdiplam bei vorbehandelten SMA-Patienten (einschließlich derjenigen, die zuvor mit Nusinersen oder mit Onasemnogen-Abeparvovec behandelt wurden), stimmt mit dem Sicherheitsprofil bei nicht vorbehandelten SMA-Patienten überein, die in den klinischen Studien FIREFISH, SUNFISH und RAINBOWFISH mit Risdiplam behandelt wurden (siehe Abschnitt 5.1).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
53175 Bonn
Website: http://www.bfarm.de
anzuzeigen.
Es gibt kein bekanntes Antidot bei Überdosierung mit Risdiplam. Im Falle einer Überdosierung muss der Patient engmaschig überwacht und es müssen unterstützende Maßnahmen eingeleitet werden.
Pharmakotherapeutische Gruppe: Andere Mittel gegen Störungen des Muskel- und Skelettsystems
ATC-Code: M09AX10
Wirkmechanismus
Risdiplam ist ein Spleiß-Modifikator der Survival of Motor Neuron 2 (SMN2)-Prä-mRNA zur Behandlung von SMA, die durch Mutationen im SMN1-Gen im Chromosom 5q verursacht wird, welche zu einem Mangel an SMN-Protein führen. Eine funktionelle SMN-Defizienz ist direkt mit der SMA-Pathophysiologie verbunden, die einen fortschreitenden Verlust von Motoneuronen und Muskelschwäche zur Folge hat. Risdiplam korrigiert das Spleißen von SMN2, um das Gleichgewicht vom Exon-7-Ausschluss hin zu dessen Einschluss in das mRNA-Transkript zu verschieben. Dies führt zu einer erhöhten Produktion von funktionellem und stabilem SMN-Protein. Risdiplam wirkt daher bei SMA über eine Erhöhung und Aufrechterhaltung von funktionellen SMN-Proteinspiegeln.
Pharmakodynamische Wirkungen
In den Studien FIREFISH (Patienten im Alter von 2 bis 7 Monaten bei Aufnahme in die Studie), SUNFISH (Patienten im Alter von 2 bis 25 Jahren bei Aufnahme in die Studie) und JEWELFISH (Patienten im Alter von 1 bis 60 Jahren bei Aufnahme in die Studie) bei Patienten mit infantiler sowie Patienten mit später einsetzender SMA führte Risdiplam bei allen untersuchten SMA-Typen zu einem Anstieg des SMN-Proteins im Blut, mit einer mehr als 2‑fachen medianen Veränderung gegenüber dem Ausgangswert innerhalb von 4 Wochen nach Behandlungsbeginn. Dieser Anstieg blieb während der gesamten Behandlungszeit (von mindestens 24 Monaten) erhalten.
Kardiale Elektrophysiologie
Die Wirkung von Risdiplam auf das QTc-Intervall wurde in einer Studie mit 47 gesunden erwachsenen Probanden untersucht. Bei therapeutischer Exposition verlängerte Risdiplam das QTc-Intervall nicht.
Klinische Wirksamkeit und Sicherheit
Die Wirksamkeit von Risdiplam bei der Behandlung von SMA-Patienten mit infantiler (SMA Typ 1) und später einsetzender SMA (SMA Typ 2 und 3) wurde in 2 klinischen Zulassungsstudien, FIREFISH und SUNFISH, untersucht. Wirksamkeitsdaten von Risdiplam bei der Behandlung von präsymptomatischen SMA-Patienten wurden in der klinischen Studie RAINBOWFISH untersucht. Patienten mit der klinischen Diagnose einer SMA Typ 4 wurden nicht in klinischen Studien untersucht.
Infantile SMA
Die Studie BP39056 (FIREFISH) ist eine offene, zweiteilige Studie zur Untersuchung der Wirksamkeit, Sicherheit, Pharmakokinetik und Pharmakodynamik von Risdiplam bei symptomatischen Patienten mit SMA Typ 1. (Bei allen Patienten lag eine genetisch bestätigte Erkrankung mit 2 Kopien des SMN2-Gens vor.) Teil 1 von FIREFISH war als Dosisfindungsphase der Studie konzipiert. Im konfirmatorischen Teil 2 von FIREFISH wurde die Wirksamkeit von Risdiplam beurteilt. Die Patienten von Teil 1 nahmen nicht an Teil 2 teil.
Der primäre Wirksamkeitsendpunkt war die Fähigkeit, nach 12 Behandlungsmonaten mindestens 5 Sekunden lang ohne Unterstützung zu sitzen, gemessen anhand von Testelement 22 der Grobmotorik-Skala der Bayley Scales of Infant and Toddler Development – Dritte Ausgabe (BSID-III).
FIREFISH Teil 2
In die Studie FIREFISH Teil 2 wurden 41 Patienten mit SMA Typ 1 eingeschlossen. Das mediane Alter bei Einsetzen der klinischen Anzeichen und Symptome der SMA Typ 1 betrug 1,5 Monate (Bereich: 1,0 – 3,0 Monate); 54 % der Patienten waren weiblich, 54 % kaukasischer und 34 % asiatischer Herkunft. Das mediane Alter bei Aufnahme in die Studie betrug 5,3 Monate (Bereich: 2,2 – 6,9 Monate) und der mediane Zeitraum zwischen dem Einsetzen der Symptome und der ersten Dosis betrug 3,4 Monate (Bereich: 1,0 – 6,0 Monate). Zum Ausgangszeitpunkt lag die mediane Children's Hospital of Philadelphia Infant Test for Neuromuscular Disease- (CHOP-INTEND-) Punktzahl bei 22,0 Punkten (Bereich: 8,0 – 37,0) und die mediane Hammersmith Infant Neurological Examination Module 2 (HINE-2)-Punktzahl bei 1,0 (Bereich: 0,0 – 5,0).
Der primäre Endpunkt war der Anteil der Patienten mit der Fähigkeit, nach 12 Behandlungsmonaten mindestens 5 Sekunden lang ohne Unterstützung zu sitzen (BSID-III-Grobmotorik-Skala, Punkt 22). Die wichtigsten Wirksamkeitsendpunkte der mit Risdiplam behandelten Patienten sind in Tabelle 3 dargestellt.
Tabelle 3: Zusammenfassung der wichtigsten Wirksamkeitsergebnisse nach 12 und 24 Monaten (FIREFISH Teil 2)
Wirksamkeitsendpunkte |
Anteil der Patienten |
|
Monat 12 |
Monat 24 |
|
Motorische Funktion und Entwicklungsmeilensteine | ||
BSID-III: Sitzen ohne Unterstützung für mindestens 5 Sekunden |
29,3 % |
61,0 % |
CHOP-INTEND: Gesamtpunktzahl von mindestens 40 |
56,1 % |
75,6 % |
CHOP-INTEND: Erhöhung um ≥ 4 Punkte gegenüber dem Ausgangswert |
90,2 % |
90,2 % |
HINE-2: Motorik-Meilenstein Responderb |
78,0 % |
85,4 % |
HINE-2: Sitzen ohne Unterstützungc |
24,4 % |
53,7 % |
Überleben und ereignisfreies Überleben | ||
Ereignisfreies Überlebend |
85,4 % |
82,9 % |
Am Leben |
92,7 % |
92,7 % |
Nahrungsaufnahme | ||
Fähigkeit zur oralen Nahrungsaufnahmee |
82,9 % |
85,4 % |
Abkürzungen: CHOP‑INTEND = Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; HINE‑2 = Module 2 of the Hammersmith Infant Neurological Examination.
a Der p-Wert wurde mit einem einseitigen, exakten Binomialtest ermittelt. Das Ergebnis wird mit einem Grenzwert von 5 % verglichen.
b Gemäß HINE-2: Ein Anstieg um ≥ 2 Punkte [oder die höchste Punktzahl] bei der Fähigkeit zu treten ODER ein Anstieg um ≥ 1 Punkt bei den motorischen Meilensteinen Kopfkontrolle, Auf-die-Seite-Rollen, Sitzen, Krabbeln, Stehen oder Gehen UND Verbesserungen in mehr Kategorien von motorischen Meilensteinen als Verschlechterungen ist für die Zwecke dieser Analyse als Ansprechen definiert.
c Sitzen ohne Unterstützung beinhaltet Patienten, die „stabiles Sitzen“ (24 %, 10/41) und „Schwenken (Drehen)“ (29 %, 12/41) erreicht haben, wie anhand von HINE-2 in Monat 24 beurteilt.
d Ein Ereignis erfüllt die Kriterien des Endpunkts „dauerhafte Beatmung“, definiert als Tracheotomie oder ≥ 16 Stunden nicht-invasive Beatmung pro Tag oder Intubation an > 21 aufeinanderfolgenden Tagen in Abwesenheit von oder im Anschluss an das Abklingen eines akuten reversiblen Ereignisses. Drei Patienten verstarben innerhalb der ersten 3 Monate nach Studieneinschluss und 4 Patienten erreichten den Endpunkt „dauerhafte Beatmung“ vor Monat 24. Bei diesen 4 Patienten war die CHOP-INTEND-Punktzahl gegenüber dem Ausgangswert um mindestens 4 Punkte angestiegen.
e Schließt Patienten ein, die in Monat 24 ausschließlich oral ernährt wurden (insgesamt 29 Patienten) sowie diejenigen, die oral in Kombination mit einer Ernährungssonde ernährt wurden (insgesamt 6 Patienten).
Im Monat 24 konnten 44 % der Patienten 30 Sekunden lang ohne Unterstützung sitzen (BSID-III, Item 26). Die Patienten erreichten weiterhin zusätzliche motorische Meilensteine, die mit dem HINE-2 gemessen wurden. 80,5 % konnten sich rollen und 27 % der Patienten erreichten einen Stehversuch (12 % konnten ihr Gewicht tragen und 15 % standen mit Unterstützung).
Unbehandelte Patienten mit infantiler SMA wären nie in der Lage, ohne Unterstützung zu sitzen, und bei nur 25 % wäre ein Überleben ohne dauerhafte Beatmung über ein Alter von 14 Monaten hinaus zu erwarten.
Abbildung 1: Kaplan-Meier-Kurve des ereignisfreien Überlebens (FIREFISH Teil 1 und Teil 2)
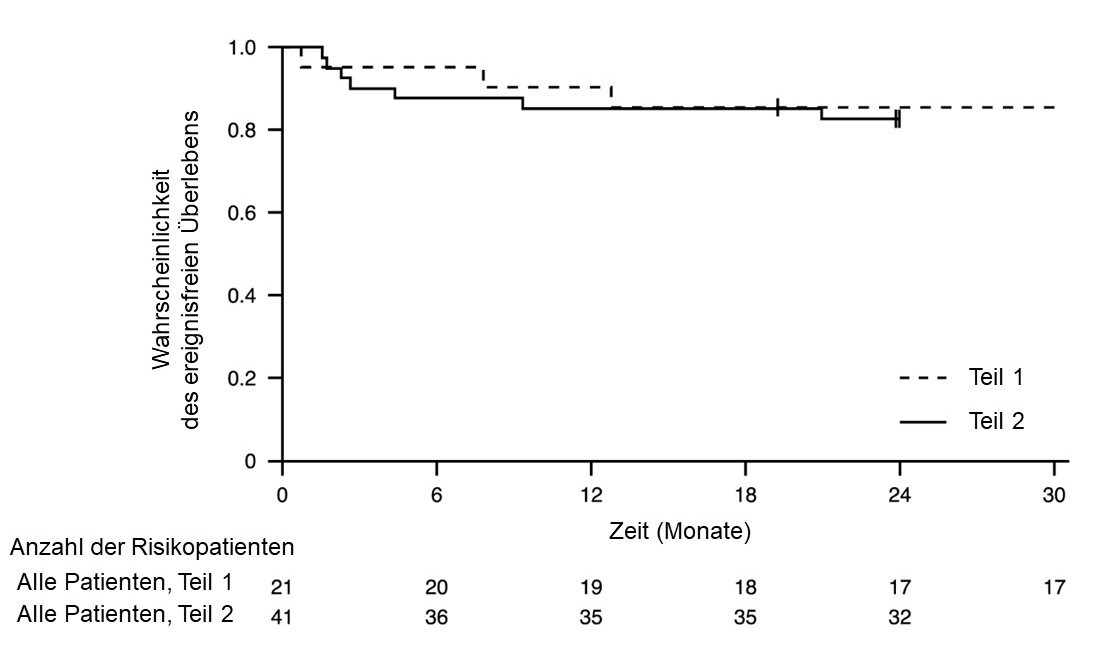
+ Zensiert: Zwei Patienten in Teil 2 wurden wegen zu frühem Erscheinen zum Termin in Monat 24 zensiert. Ein Patient in Teil 1 wurde zensiert, nachdem er die Behandlung abgebrochen hatte, und verstarb 3,5 Monate später.
Abbildung 2: Mittlere Veränderung der CHOP-INTEND-Gesamtpunktzahl gegenüber dem Ausgangswert (FIREFISH Teil 2)
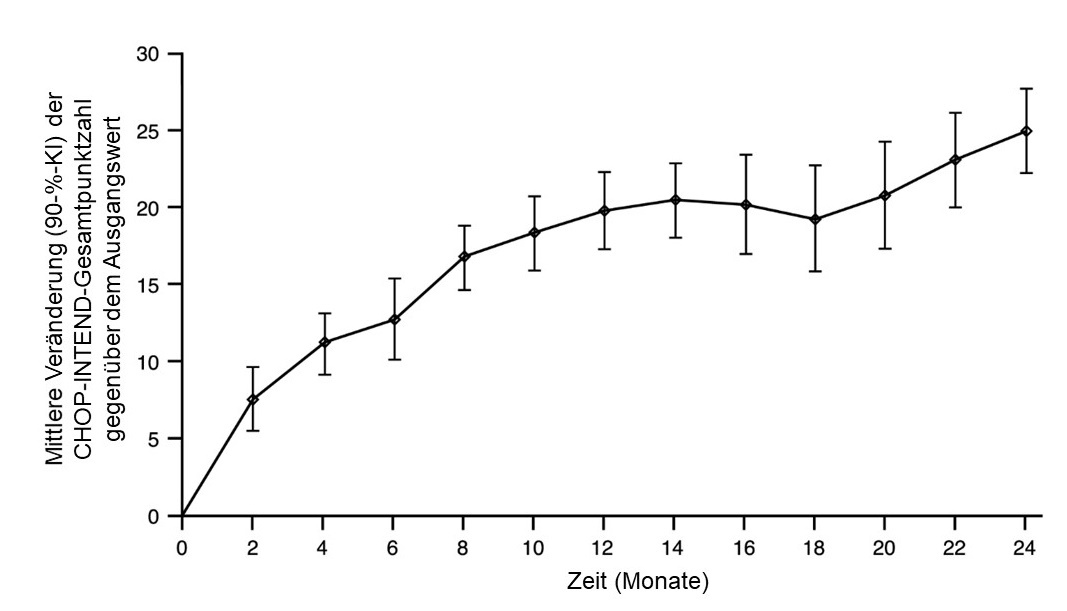
FIREFISH Teil 1
Die Wirksamkeit von Risdiplam bei Patienten mit SMA Typ 1 wird auch durch Ergebnisse der Studie FIREFISH Teil 1 unterstützt. Bei den 21 in Teil 1 eingeschlossenen Patienten entsprachen die Basischarakteristika denen symptomatischer Patienten mit SMA Typ 1. Das mediane Alter bei Aufnahme in die Studie betrug 6,7 Monate (Bereich: 3,3 – 6,9 Monate). Der mediane Zeitraum zwischen dem Einsetzen der Symptome und der ersten Behandlung betrug 4,0 Monate (Bereich: 2,0 – 5,8 Monate).
Insgesamt 17 Patienten erhielten die therapeutische Dosis von Risdiplam (für Teil 2 ausgewählte Dosis). Nach 12 Monaten Behandlung waren 41 % (7/17) dieser Patienten in der Lage, mindestens 5 Sekunden lang selbstständig zu sitzen (BSID-III, Punkt 22). Nach 24 Monaten Behandlung waren 3 weitere Patienten, die die therapeutische Dosis erhielten, in der Lage, mindestens 5 Sekunden lang selbstständig zu sitzen, sodass insgesamt 10 Patienten (59 %) diesen motorischen Meilenstein erreichten.
Nach 12 Monaten Behandlung waren 90 % (19/21) der Patienten am Leben und ereignisfrei (ohne dauerhafte Beatmung) und erreichten ein Alter von 15 Monaten oder mehr. Nach mindestens 33 Monaten Behandlung waren 81 % (17/21) der Patienten am Leben und ereignisfrei und erreichten ein Alter von 37 Monaten oder mehr (Median 41 Monate; Bereich 37 - 53 Monate), siehe Abbildung 1. Drei Patienten starben während der Behandlung und ein Patient starb 3,5 Monate nach Absetzen der Behandlung.
Später einsetzende SMA
Die Studie BP39055 (SUNFISH) ist eine zweiteilige, multizentrische Studie zur Untersuchung der Wirksamkeit, Sicherheit, Pharmakokinetik und Pharmakodynamik von Risdiplam bei Patienten mit SMA Typ 2 oder Typ 3 im Alter von 2 – 25 Jahren. Teil 1 war die explorative Dosisfindungsphase und Teil 2 die randomisierte, doppelblinde, placebokontrollierte Bestätigungsphase. Die Patienten von Teil 1 nahmen nicht an Teil 2 teil.
Der primäre Endpunkt war die Veränderung der Motor Function Measure-32-Gesamtpunktzahl (MFM-32) gegenüber dem Ausgangswert in Monat 12. Die Skala MFM-32 bietet die Möglichkeit, eine Vielzahl unterschiedlicher motorischer Funktionen bei einem breiten Spektrum von SMA-Patienten zu beurteilen. Die MFM-32-Gesamtpunktzahl wird ausgedrückt als der Prozentsatz (Bereich: 0 – 100) der höchstmöglichen Punktzahl, wobei höhere Punktzahlen eine bessere motorische Funktion anzeigen.
SUNFISH Teil 2
SUNFISH Teil 2 ist die randomisierte, doppelblinde, placebokontrollierte Phase der Studie SUNFISH mit 180 nicht gehfähigen Patienten mit SMA Typ 2 (71 %) oder Typ 3 (29 %). Die Patienten wurden im Verhältnis 2:1 entweder der therapeutischen Dosis von Risdiplam (siehe Abschnitt 4.2) oder Placebo zugeteilt. Die Randomisierung erfolgte nach Altersgruppen stratifiziert (2 bis 5, 6 bis 11, 12 bis 17, 18 bis 25 Jahre alt).
Das mediane Alter der Patienten zu Beginn der Behandlung betrug 9,0 Jahre (Bereich 2 – 25 Jahre), der mediane Zeitraum zwischen dem Einsetzen der SMA-Symptome und der ersten Behandlung betrug 102,6 (1 – 275) Monate. Insgesamt waren 30 % bei Aufnahme in die Studie 2 bis 5 Jahre alt, 32 % waren 6 bis 11 Jahre alt, 26 % waren 12 bis 17 Jahre alt und 12 % waren 18 bis 25 Jahre alt. Von den 180 in die Studie aufgenommenen Patienten waren 51 % weiblich, 67 % kaukasischer und 19 % asiatischer Herkunft. Zum Ausgangszeitpunkt litten 67 % der Patienten an Skoliose (32 % an schwerer Skoliose). Die Patienten hatten zum Ausgangszeitpunkt eine mittlere MFM-32-Punktzahl von 46,1 und eine Punktzahl von 20,1 im Revised Upper Limb Module (RULM). Die demographischen Charakteristika bei Studienbeginn waren zwischen dem Arm mit Risdiplam und dem Placebo-Arm ausgeglichen, mit Ausnahme von Skoliose (63 % der Patienten im Arm mit Risdiplam und 73 % der Patienten im Placebo-Kontrollarm).
Die Primäranalyse von SUNFISH Teil 2 ergab hinsichtlich der Änderung der MFM-32-Gesamtpunktzahl in Monat 12 gegenüber dem Ausgangswert einen klinisch bedeutsamen und statistisch signifikanten Unterschied zwischen den mit Risdiplam und den mit Placebo behandelten Patienten. Die Ergebnisse der Primäranalyse und wichtige sekundäre Endpunkte sind in Tabelle 4, Abbildung 3 und Abbildung 4 zusammengefasst.
Tabelle 4: Zusammenfassung der Wirksamkeitsdaten für Patienten mit später einsetzender SMA in Behandlungsmonat 12 (SUNFISH Teil 2)
Endpunkt |
Risdiplam |
Placebo |
Primärer Endpunkt: |
||
Änderung der MFM-32-Gesamtpunktzahl1 in Monat 12 gegenüber dem Ausgangswert |
1,36 |
-0,19 |
Unterschied gegenüber Placebo |
1,55 |
|
Sekundäre Endpunkte: |
||
Anteil der Patienten mit einer Änderung der MFM-32-Gesamtpunktzahl1 von mindestens 3 Punkten gegenüber dem Ausgangswert (95-%-KI) in Monat 121 |
38,3 % |
23,7 % |
Odds-Ratio für Gesamtansprechen (95-%-KI) |
2,35 (1,01; 5,44) |
|
Änderung der RULM-Gesamtpunktzahl5 in Monat 12 gegenüber dem Ausgangswert |
1,61 |
0,02 |
Unterschied gegenüber Placebo, Schätzwert (95-%-KI) |
1,59 (0,55; 2,62) |
|
LS = Least Squares (kleinste Fehlerquadrate)
1 Entsprechend der Regel zu fehlenden Daten für MFM-32 wurden 6 Patienten von der Analyse ausgeschlossen (Risdiplam n = 115; Placebokontrolle n = 59).
2 Die Daten wurden mithilfe eines gemischten Modells für wiederholte Messungen analysiert. Bestimmt wurden Gesamtpunktzahl bei Studienbeginn, Behandlungsarm, Visite, Alterskategorie, Behandlung nach Visite (Treatment-by-Visit) und Ausgangswert nach Visite (Baseline-by-Visit).
3 Die Daten wurden mittels logistischer Regression mit den Zielvariablen Gesamtpunktzahl zum Ausgangszeitpunkt, Behandlung und Altersgruppe analysiert.
4 Der bereinigte p-Wert wurde für die in die hierarchischen Tests einbezogenen Endpunkte ermittelt und auf der Grundlage aller p-Werte von Endpunkten in der Reihenfolge der Hierarchie bis zum aktuellen Endpunkt abgeleitet.
5 Entsprechend der Regel zu fehlenden Daten für RULM wurden 3 Patienten von der Analyse ausgeschlossen (Risdiplam n = 119; Placebo n = 58).
Nach Abschluss der 12-monatigen Behandlung erhielten 117 Patienten weiterhin Risdiplam. Zum Zeitpunkt der 24-Monats-Analyse zeigten diese Patienten, die 24 Monate lang mit Risdiplam behandelt wurden, insgesamt eine anhaltende Verbesserung der motorischen Funktion zwischen Monat 12 und Monat 24. Die mittlere Veränderung gegenüber dem Ausgangswert für MFM-32 betrug 1,83 (95-%-KI: 0,74; 2,92) und für RULM 2,79 (95-%-KI: 1,94; 3,64).
Abbildung 3: Mittlere Änderung der MFM-32-Gesamtpunktzahl gegenüber dem Ausgangswert im Verlauf von 12 Monaten in der SUNFISH-Studie Teil 21
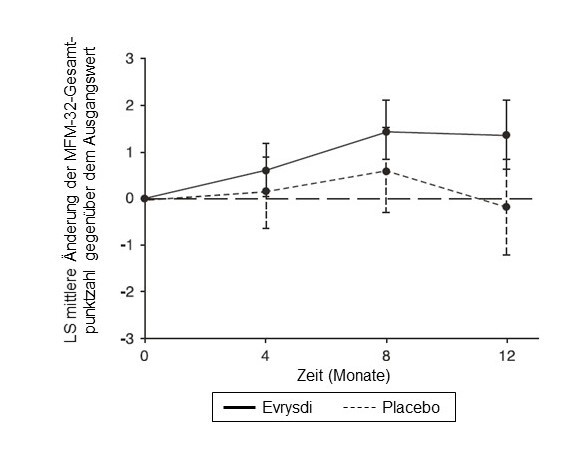
1 Differenz der Mittelwerte der kleinsten Fehlerquadrate (LS) für die Änderung der MFM-32-Punktzahl gegenüber dem Ausgangswert [95-%-KI]
Abbildung 4: Mittlere Änderung der RULM-Gesamtpunktzahl gegenüber dem Ausgangswert im Verlauf von 12 Monaten in der SUNFISH-Studie Teil 21
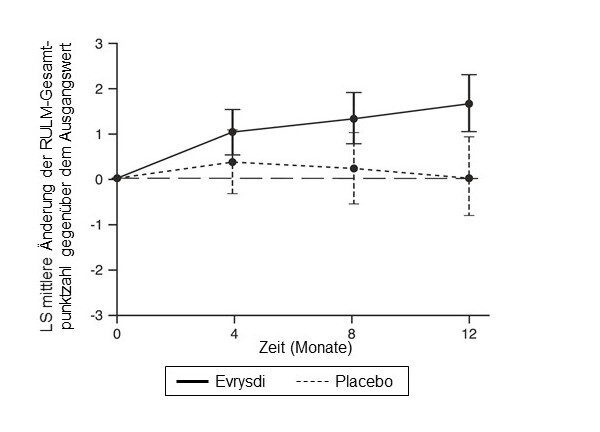
1 Differenz der Mittelwerte der kleinsten Fehlerquadrate (LS) für die Änderung der RULM-Punktzahl gegenüber dem Ausgangswert [95-%-KI]
SUNFISH Teil 1
Die Wirksamkeit bei Patienten mit später einsetzender SMA wird außerdem durch die Ergebnisse von Teil 1, der Dosisfindungsphase der SUNFISH-Studie, untermauert. In Teil 1 wurden 51 Patienten mit SMA Typ 2 und Typ 3 (einschließlich 7 gehfähiger Patienten) im Alter von 2 – 25 Jahren aufgenommen. Nach einem Jahr Behandlung zeigte die anhand von MFM-32 gemessene motorische Funktion eine klinisch bedeutsame Verbesserung. Die mittlere Änderung gegenüber dem Ausgangswert betrug 2,7 Punkte (95-%-KI: 1,5; 3,8). Die Verbesserung der MFM-32-Punktzahl blieb unter Behandlung bis zu 2 Jahre erhalten (mittlere Änderung um 2,7 Punkte [95-%-KI: 1,2; 4,2]).
Anwendung bei Patienten, die zuvor mit anderen SMA-modifizierenden Therapien behandelt wurden (JEWELFISH)
Bei der Studie BP39054 (JEWELFISH, n = 174) handelt es sich um eine einarmige, offene Studie zur Untersuchung der Sicherheit, Verträglichkeit, PK und PD von Risdiplam bei Patienten mit infantiler und später einsetzender SMA (medianes Alter 14 Jahre [Spanne 1 – 60 Jahre]), die zuvor mit anderen zugelassenen (Nusinersen n = 76, Onasemnogen-Abeparvovec n = 14) oder in der Erprobung befindlichen SMA-modifizierenden Therapien behandelt wurden. Bei Studienbeginn hatten von den 168 Patienten im Alter von 2 bis 60 Jahren 83 % eine Skoliose und 63 % eine Hammersmith Functional Motor Scale Expanded-(HFMSE)-Punktzahl < 10 Punkte.
Bei der Analyse im Monat 24 der Behandlung zeigte sich bei den Patienten im Alter von 2 bis 60 Jahren eine allgemeine Stabilisierung der motorischen Funktion im MFM-32 und RULM (n = 137 bzw. n = 133). Patienten unter 2 Jahren (n = 6) konnten motorische Meilensteine wie Kopfkontrolle, Rollen und selbstständiges Sitzen beibehalten oder erreichen. Alle gehfähigen Patienten (im Alter von 5 bis 46 Jahren, n = 15) behielten ihre Gehfähigkeit.
Präsymptomatische SMA (RAINBOWFISH)
Die Studie BN40703 (RAINBOWFISH) ist eine unverblindete, einarmige, multizentrische klinische Studie zur Untersuchung der Wirksamkeit, Sicherheit, Pharmakokinetik und Pharmakodynamik von Risdiplam bei Säuglingen von der Geburt bis zum Alter von 6 Wochen (bei der ersten Dosis), bei denen genetisch eine SMA diagnostiziert wurde, die aber noch keine Symptome aufweisen.
Die Wirksamkeit bei präsymptomatischen SMA-Patienten wurde in Monat 12 bei 26 Patienten [Intention‑to‑treat(ITT)-Population] untersucht, die mit Risdiplam behandelt worden waren: Acht Patienten, 13 Patienten und 5 Patienten hatten jeweils 2, 3 und ≥ 4 Kopien des SMN2‑Gens. Bei diesen Patienten lag das mediane Alter bei Gabe der ersten Dosis bei 25 Tagen (Bereich: 16 bis 41 Tage), 62 % waren weiblich und 85 % waren kaukasischer Herkunft. Bei Studienbeginn betrug die mediane CHOP-INTEND-Punktzahl 51,5 (Bereich: 35,0 bis 62,0), die mediane HINE‑2-Punktzahl 2,5 (Bereich: 0 bis 6,0) und die mediane Amplitude des zusammengesetzten Muskelaktionspotentials (CMAP, compound muscle action potential) des Nervus ulnaris 3,6 mV (Bereich: 0,5 bis 6,7 mV).
Die primäre Wirksamkeitspopulation (n = 5) umfasste Patienten mit 2 SMN2-Kopien und einer CMAP-Amplitude ≥ 1,5 mV bei Studienbeginn. Bei diesen Patienten betrug die mediane CHOP-INTEND-Punktzahl 48,0 (Bereich: 36,0 bis 52,0), die mediane HINE-2-Punktzahl 2,0 (Bereich: 1,0 bis 3,0) und die mediane CMAP-Amplitude 2,6 mV (Bereich: 1,6 bis 3,8 mV) bei Studienbeginn.
Der primäre Endpunkt war der Anteil der Patienten in der primären Wirksamkeitspopulation, die nach 12 Monaten mindestens 5 Sekunden lang ohne Unterstützung sitzen konnten (BSID-III Grobmotorik-Skala, Testelement 22); im Vergleich zum vorgegebenen Leistungskriterium von 5 % erreichte ein statistisch signifikanter und klinisch bedeutsamer Anteil der Patienten diesen Meilenstein.
Die wichtigsten Wirksamkeitsendpunkte der mit Risdiplam behandelten Patienten sind in den Tabellen 5 und 6 sowie in Abbildung 5 dargestellt.
Tabelle 5: Sitzfähigkeit gemäß BSID-III-Testelement 22 für präsymptomatische Patienten in Monat 12
Wirksamkeitsendpunkt |
Population |
||
Primäre Wirksamkeit (n = 5) |
Patienten mit 2 SMN2-Kopiena (n = 8) |
ITT |
|
Anteil der Patienten, die mindestens 5 Sekunden lang ohne Unterstützung sitzen (BSID-III, Testelement 22); (90-%-KI) |
80 % |
87,5 % |
96,2 % |
Abkürzungen: BSID-III = Bayley Scales of Infant and Toddler Development – 3. Edition; KI = Konfidenzintervall; ITT = Intention-to-Treat.
a Patienten mit 2 SMN2-Kopien hatten bei Studienbeginn eine mediane CMAP-Amplitude von 2,0 (Bereich 0,5 - 3,8).
b Der p-Wert wurde mit einem einseitigen, exakten Binomialtest ermittelt. Das Ergebnis wird mit einem Grenzwert von 5 % verglichen.
Zusätzlich erreichten 80 % (4/5) der primären Wirksamkeitspopulation, 87,5 % (7/8) der Patienten mit 2 SMN2-Kopien und 80,8 % (21/26) der Patienten in der ITT-Population für eine Dauer von 30 Sekunden das Sitzen ohne Unterstützung (BSID-III, Testelement 26).
Die Patienten in der ITT-Population erreichten auch motorische Meilensteine, gemessen mit dem HINE-2 in Monat 12 (n = 25). In dieser Population konnten 96,0 % der Patienten sitzen [1 Patient (1/8 Patienten mit 2 SMN2-Kopien) konnte stabil sitzen und 23 Patienten (6/8, 13/13, 4/4 der Patienten mit 2, 3 bzw. ≥ 4 SMN2-Kopien) konnten sich drehen/wenden]. Darüber hinaus konnten 84 % der Patienten stehen; 32 % (n = 8) der Patienten konnten mit Unterstützung stehen (3/8, 3/13 bzw. 2/4 Patienten mit 2, 3 bzw. ≥ 4 SMN2-Kopien) und 52 % (n = 13) der Patienten konnten ohne Unterstützung stehen (1/8, 10/13 bzw. 2/4 der Patienten mit 2, 3 bzw. ≥ 4 SMN2-Kopien). Des Weiteren konnten 72 % der Patienten hüpfen, mit Unterstützung gehen oder laufen; 8 % (n = 2) der Patienten konnten hüpfen (2/8 Patienten mit 2 SMN2-Kopien), 16 % (n = 4) konnten mit Unterstützung gehen (3/13 bzw. 1/4 Patienten mit 3 bzw. ≥ 4 SMN2-Kopien) und 48 % (n = 12) konnten eigenständig laufen (1/8, 9/13 bzw. 2/4 Patienten mit 2, 3 bzw. ≥ 4 SMN2-Kopien). Sieben Patienten wurden in Monat 12 nicht auf Gehfähigkeit getestet.
Tabelle 6: Zusammenfassung der wichtigsten Wirksamkeitsendpunkte für präsymptomatische Patienten in Monat 12
Wirksamkeitsendpunkte |
ITT-Population (n = 26) |
Motorische Funktion | |
Anteil der Patienten, die im CHOP-INTEND eine Gesamtpunktzahl von 50 oder mehr erreichen (90‑%‑KI) |
92 %a |
Anteil der Patienten, die im CHOP-INTEND eine Gesamtpunktzahl von 60 oder mehr erreichen (90‑%‑KI) |
80 %a |
Nahrungsaufnahme | |
Anteil der Patienten mit der Fähigkeit zur oralen Nahrungsaufnahme (90‑%‑KI) |
96,2 %b |
Inanspruchnahme einer Pflegeperson | |
Anteil der Patienten ohne Krankenhausaufenthaltec (90‑KI‑%) |
92,3 % |
Ereignisfreies Überlebend | |
Anteil der Patienten mit ereignisfreiem Überleben (90‑%‑KI) |
100 % |
Abkürzungen: CHOP‑INTEND = Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; KI = Konfidenzintervall | |
Abbildung 5: Mediane CHOP-INTEND-Gesamtpunktzahl pro Untersuchung und Anzahl der SMN2‑Kopien (ITT-Population)
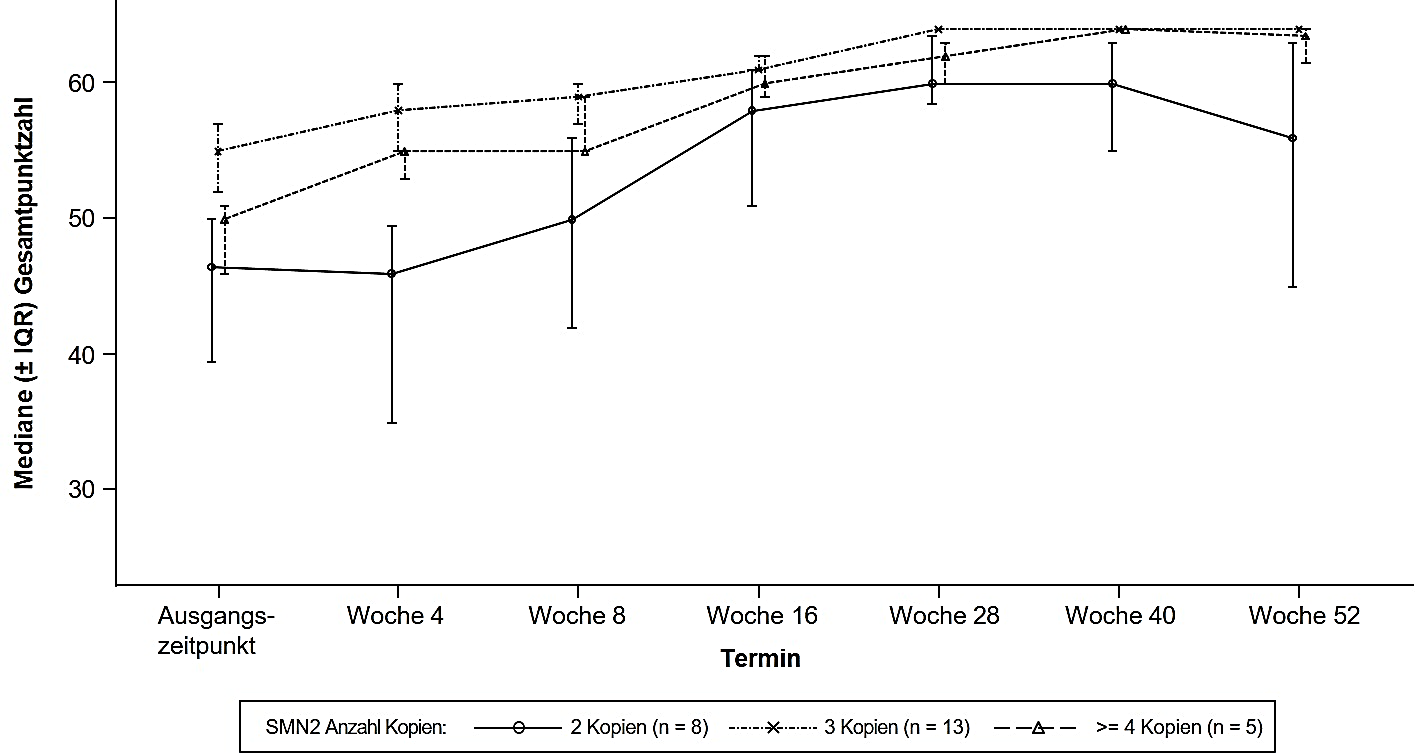
Abkürzungen: IQR = Interquartilsabstand (interquartile range); SMN2 = survival of motor neuron 2.
Die pharmakokinetischen Parameter wurden bei gesunden erwachsenen Probanden und bei Patienten mit SMA bestimmt.
Nach Anwendung des Arzneimittels in Form einer Lösung zum Einnehmen war die Pharmakokinetik von Risdiplam zwischen 0,6 und 18 mg annähernd dosislinear. Die Pharmakokinetik von Risdiplam ließ sich am besten durch ein Populations-Pharmakokinetik-Modell mit Resorption in drei Transitkompartimenten, Disposition in zwei Kompartimenten und Elimination mit einer Kinetik erster Ordnung beschreiben. Es konnte gezeigt werden, dass Körpergewicht und Alter einen signifikanten Einfluss auf die Pharmakokinetik haben.
Die geschätzte Exposition (mittlere AUC0-24h) bei Patienten mit infantiler SMA (Alter 2 – 7 Monate zum Zeitpunkt des Studieneinschlusses) betrug 1 930 ng h/ml bei der therapeutischen Dosis von 0,2 mg/kg einmal täglich. Die geschätzte mittlere Exposition bei präsymptomatischen Säuglingen (im Alter von 16 Tagen bis < 2 Monaten) in der Studie RAINBOWFISH betrug 2 020 ng h/ml bei 0,15 mg/kg nach zweiwöchiger einmal täglicher Gabe. Die geschätzte Exposition bei Patienten mit später einsetzender SMA (Alter 2 – 25 Jahre zum Zeitpunkt des Studieneinschlusses) in der Studie SUNFISH (Teil 2) betrug 2 070 ng h/ml nach einem Jahr Behandlung und 1 940 ng h/ml nach 5 Jahren Behandlung bei der therapeutischen Dosis (0,25 mg/kg einmal täglich bei Patienten mit einem Körpergewicht < 20 kg; 5 mg einmal täglich bei Patienten mit einem Körpergewicht ≥ 20 kg). Die geschätzte Exposition (mittlere AUC0-24h) für vorbehandelte SMA-Patienten (Alter 1 – 60 Jahre bei Studieneinschluss) betrug 1 700 ng h/ml bei der therapeutischen Dosis von 0,25 mg/kg oder 5 mg. Die beobachtete maximale Konzentration (mittlere Cmax) betrug 194 ng/ml bei 0,2 mg/kg in der Studie FIREFISH, 140 ng/ml in der Studie SUNFISH Teil 2, 129 ng/ml in der Studie JEWELFISH, und die geschätzte maximale Konzentration bei 0,15 mg/kg in der Studie RAINBOWFISH lag bei 111 ng/ml.
Resorption
Risdiplam wurde im nüchternen Zustand schnell resorbiert, wobei die Plasma-tmax nach der Einnahme des rekonstituierten Pulvers zur Herstellung einer Lösung zum Einnehmen zwischen 1 und 5 Stunden lag. Auf der Basis von Daten von 47 gesunden Probanden hatte Nahrung (kalorienreiches Frühstück mit hohem Fettgehalt) keinen wesentlichen Einfluss auf die Exposition gegenüber Risdiplam. In den klinischen Studien wurde Risdiplam mit einer Morgenmahlzeit oder nach dem Stillen verabreicht.
Verteilung
Risdiplam wird durch Überwinden der Blut-Hirn-Schranke gleichmäßig in allen Körperteilen verteilt, einschließlich des Zentralnervensystems (ZNS), und führt somit zu erhöhten SMN-Protein-Spiegeln im ZNS und im gesamten Körper. Die Konzentration von Risdiplam im Plasma und von SMN-Protein im Blut spiegelt die Verteilung von Risdiplam und dessen pharmakodynamische Wirkungen in Geweben wie dem Gehirn und den Muskeln wider.
Die geschätzten Parameter der Populations-Pharmakokinetik waren 98 l für das scheinbare Verteilungsvolumen im zentralen Kompartiment, 93 l für das periphere Volumen und 0,68 l/h für die Interkompartiment-Clearance.
Risdiplam bindet überwiegend an Serumalbumin, nicht dagegen an saures Alpha-1-Glykoprotein. Der ungebundene Anteil beträgt 11 %.
Biotransformation
Risdiplam wird hauptsächlich von FMO1 und FMO3 sowie auch von CYP 1A1, 2J2, 3A4 und 3A7 metabolisiert.
Die gleichzeitige, zweimal tägliche Anwendung von 200 mg Itraconazol, einem starken CYP3A-Inhibitor, mit einer oralen Einzeldosis von 6 mg Risdiplam zeigte keine klinisch relevante Auswirkung auf die Pharmakokinetik von Risdiplam (11 % Zunahme der AUC, 9 % Abnahme der Cmax).
Elimination
In populationspharmakokinetischen Analysen wurde die scheinbare Clearance (CL/F) für Risdiplam auf 2,6 l/h geschätzt.
Die effektive Halbwertszeit von Risdiplam bei SMA-Patienten betrug etwa 50 Stunden.
Risdiplam ist kein Substrat des humanen Multidrug Resistance Proteins 1 (MDR1).
Etwa 53 % der Dosis (14 % unverändertes Risdiplam) wurden über den Stuhl und 28 % (8 % unverändertes Risdiplam) über den Urin ausgeschieden. Die Ausgangssubstanz war die Hauptkomponente im Plasma und machte 83 % der Stoffwechselprodukte des Arzneimittels im Kreislauf aus. Der pharmakologisch inaktive Metabolit M1 wurde als der wichtigste zirkulierende Metabolit identifiziert.
Pharmakokinetik bei speziellen Patientengruppen
Kinder und Jugendliche
Körpergewicht und Alter wurden bei der populationspharmakokinetischen Analyse als Kovariate identifiziert. Auf der Basis dieses Modells wird die Dosis daher entsprechend dem Alter (jünger und älter als 2 Monate bzw. 2 Jahre) und Körpergewicht (bis 20 kg) angepasst, um über den gesamten Alters- und Körpergewichtsbereich eine ähnliche Exposition zu erzielen. Für Patienten im Alter von unter 20 Tagen liegen nur begrenzte Daten zur Pharmakokinetik vor, da nur ein 16 Tage altes Neugeborenes in klinischen Studien Risdiplam in einer niedrigeren Dosis (0,04 mg/kg) erhielt.
Ältere Patienten
Spezielle Studien zur Untersuchung der Pharmakokinetik bei Patienten mit SMA im Alter über 60 Jahre wurden nicht durchgeführt. Studienteilnehmer ohne SMA im Alter bis 69 Jahre waren in die klinischen Pharmakokinetik-Studien eingeschlossen. Diese zeigten, dass für Patienten im Alter bis 69 Jahre keine Dosisanpassung erforderlich ist.
Patienten mit eingeschränkter Nierenfunktion
Es wurden keine Studien zur Untersuchung der Pharmakokinetik von Risdiplam bei Patienten mit eingeschränkter Nierenfunktion durchgeführt. Die Elimination von Risdiplam durch renale Ausscheidung der unveränderten Substanz ist gering (8 %).
Patienten mit eingeschränkter Leberfunktion
Leichte und mäßige Leberfunktionsstörungen hatten keinen signifikanten Einfluss auf die Pharmakokinetik von Risdiplam. Nach Einnahme einer einzelnen oralen Dosis von 5 mg Risdiplam betrug der mittlere Quotient für Cmax und AUC 0,95 bzw. 0,80 bei Probanden mit leichten (n = 8) und 1,20 bzw. 1,08 bei Studienteilnehmern mit mäßig schweren Leberfunktionsstörungen (n = 8) gegenüber einer gesunden Kontrollgruppe (n = 10). Die Sicherheit und Pharmakokinetik wurden bei Patienten mit schweren Leberfunktionsstörungen nicht untersucht.
Ethnizität
Die Pharmakokinetik von Risdiplam unterscheidet sich bei japanischen und kaukasischen Probanden nicht.
Beeinträchtigung der Fertilität
Bei Ratten und Affen wurde ein Zusammenhang der Behandlung mit Risdiplam mit dem Arrest der männlichen Keimzellen beobachtet, ohne dass eine Sicherheitsspanne auf der Grundlage von systemischen Expositionen beim No Observed Adverse Effect Level (NOAEL) bestand. Dieser Effekt führte zu degenerierten Spermatozyten, Degeneration/Nekrose des seminiferösen Epithels und Oligospermie/Aspermie in den Nebenhoden. Effekte von Risdiplam auf Samenzellen gehen wahrscheinlich auf eine Störung des Zellzyklus sich teilender Zellen durch Risdiplam zurück und sind stadienspezifisch und voraussichtlich reversibel. Effekte auf die weiblichen Reproduktionsorgane bei Ratten und Affen nach Behandlung mit Risdiplam wurden nicht beobachtet.
Es wurden keine Studien zur Fertilität und frühen Embryonalentwicklung bei gleichzeitiger Verabreichung von Risdiplam durchgeführt, da bereits bei der Behandlung von Ratten und Affen in anderen Toxizitätsstudien ein Spermienstillstand und ein embryotoxisches Potenzial unter der Behandlung festgestellt wurde. In zwei Studien, in denen Ratten gepaart wurden, wurde keine Beeinträchtigung der männlichen oder weiblichen Fertilität beobachtet. Die Auswertung erfolgte entweder nach Abschluss einer 13-wöchigen Behandlungsperiode, die mit dem Absetzen begann, oder 8 Wochen nach Abschluss einer 4-wöchigen Behandlungsperiode, die im Alter von 4 Tagen begann.
Wirkung auf die Struktur der Retina
Die chronische Behandlung von Affen mit Risdiplam ergab Hinweise auf einen Effekt auf die Retina in Form einer Degeneration der Photorezeptoren, die an der Peripherie der Retina beginnt. Nach Beendigung der Behandlung waren die Effekte im Retinogramm teilweise reversibel, die Degeneration der Photorezeptoren ging jedoch nicht zurück. Die Effekte wurden durch optische Kohärenztomographie (OCT) und Elektroretinographie (ERG) überwacht. Die Effekte wurden bei Expositionen beobachtet, die mehr als das Doppelte der Exposition beim Menschen bei Einnahme einer therapeutischen Dosis betrugen, ohne Sicherheitsspanne auf der Grundlage von systemischen Expositionen beim NOAEL. Keine derartigen Befunde wurden bei Albino- oder pigmentierten Ratten beobachtet, wenn diese chronisch mit Risdiplam bei Expositionen, die über denen des Affen lagen, dosiert wurden. Derartige Befunde wurden in klinischen Studien mit SMA-Patienten mit regelmäßiger ophthalmologischer Überwachung [einschließlich Spectral-Domain-optische-Kohärenztomographie (SD-OCT) und Beurteilung der visuellen Funktion] nicht beobachtet.
Wirkung auf Epithelgewebe
Bei mit Risdiplam behandelten Ratten und Affen waren Auswirkungen auf die Histologie der Haut, des Kehlkopfs und des Augenlids sowie auf den Gastrointestinaltrakt erkennbar. Die Veränderungen waren erstmals nach 2 oder mehr Behandlungswochen mit hohen Dosen erkennbar. Bei chronischer Behandlung von Affen über 39 Wochen lag der NOAEL bei einer Exposition, die mehr als dem Zweifachen der durchschnittlichen Exposition bei Menschen bei Einnahme einer therapeutischen Dosis entsprach.
Wirkung auf hämatologische Parameter
Beim akuten Knochenmark-Mikronukleus-Test an Ratten wurde bei hoher Dosisstufe (Exposition, die mehr als 15‑fach höher war als die durchschnittliche Exposition beim Menschen unter der therapeutischen Dosis) eine Verringerung des Verhältnisses von polychromatischen (jungen) zu normochromatischen (reifen) Erythrozyten um mehr als 50 % beobachtet. Dies deutet auf eine erhebliche Knochenmarkstoxizität hin. Bei längerer Behandlung von Ratten über 26 Wochen lagen die Expositionsgrenzen zum NOAEL etwa beim 4-Fachen der durchschnittlichen Exposition beim Menschen in der therapeutischen Dosis.
Genotoxizität
Risdiplam zeigt bei einem Rückmutationstest an Bakterien keine Mutagenität. Bei Säugetierzellen in vitro und im Knochenmark von Ratten erhöht Risdiplam die Häufigkeit von Zellen mit Mikronuklei. In verschiedenen Toxizitätsstudien an Ratten (adulte und juvenile Tiere) wurde eine Mikronukleus-Induktion im Knochenmark beobachtet. Der NOAEL ist bei sämtlichen Studien mit einer Exposition assoziiert, die etwa dem 1,5-Fachen der Exposition von Menschen bei der therapeutischen Dosis entspricht. Daten zeigen, dass dieser Effekt indirekt ist und eine Folge der Störung des Zellzyklus sich teilender Zellen durch Risdiplam darstellt. Eine direkte Schädigung der DNA durch Risdiplam erfolgt nicht.
Reproduktionstoxizität
In Studien an trächtigen Ratten, die mit Risdiplam behandelt wurden, zeigte sich embryofötale Toxizität durch ein geringeres Gewicht und eine verzögerte Entwicklung der Föten. Der NOAEL für diesen Effekt war etwa doppelt so hoch wie die Expositionsniveaus, die mit der therapeutischen Dosis von Risdiplam bei Patienten erreicht werden. In Studien an trächtigen Kaninchen wurden dysmorphogene Effekte bei Expositionen beobachtet, die auch mit maternaler Toxizität verbunden waren. Dabei handelte es sich um vier Föten (4 %) aus vier Würfen (22 %) mit Hydrocephalus. Der NOAEL für diesen Effekt war etwa viermal so hoch wie die Expositionsniveaus, die mit der therapeutischen Dosis von Risdiplam bei Patienten erreicht werden. In einer prä- und postnatalen Entwicklungsstudie an Ratten, die täglich mit Risdiplam behandelt wurden, verursachte Risdiplam eine leichte Verlängerung der Trächtigkeitsdauer. Untersuchungen an trächtigen und säugenden Ratten zeigten, dass Risdiplam die Plazenta passiert und in die Muttermilch übergeht.
Karzinogenität
In einer Studie an transgenen rasH2-Mäusen mit 6-monatiger Behandlungsdauer und in einer 2-jährigen Studie an Ratten mit Expositionen, die denen der empfohlenen Höchstdosis beim Menschen (MRHD, maximum recommended human dose) entsprachen, ergaben sich keine Hinweise auf ein karzinogenes Potenzial von Risdiplam. Eine signifikante Zunahme von Tumoren der Präputialdrüse bei männlichen Ratten und der Klitorisdrüse bei weiblichen Ratten, die bei der 4‑fachen MRHD-Exposition beobachtet wurde, hat keine Relevanz für den Menschen, da es sich bei beiden um nagetierspezifische Organe handelt.
Studien mit Jungtieren
Daten von Jungtieren lassen keine besondere Gefahr für den Menschen erkennen.
Mannitol (Ph.Eur.) (E 421)
Isomalt (Ph.Eur.) (E 953)
Erdbeer-Aroma: natürliche(r) Aromastoff(e), Aromaextrakt(e), Maltodextrin aus Mais, modifizierte Wachsmaisstärke (E 1450)
Weinsäure (Ph.Eur.) (E 334)
Natriumbenzoat (E 211)
Macrogol 6000 (E 1521)
Sucralose
Ascorbinsäure (E 300)
Natriumedetat (Ph.Eur.)
Nicht zutreffend.
Pulver zur Herstellung einer Lösung zum Einnehmen
2 Jahre
Rekonstituierte Lösung zum Einnehmen
64 Tage bei Lagerung im Kühlschrank (2 °C bis 8 °C).
Falls erforderlich, kann der Patient oder seine Pflegeperson die Lösung zum Einnehmen bei Raumtemperatur (unter 40 °C) aufbewahren, jedoch nicht länger als insgesamt 120 Stunden (5 Tage).
Die Lösung zum Einnehmen soll wieder in den Kühlschrank gestellt werden, wenn es nicht mehr notwendig ist, die Flasche bei Raumtemperatur aufzubewahren. Die Gesamtdauer der Aufbewahrung außerhalb des Kühlschranks (unter 40 °C) soll überwacht werden.
Die Lösung zum Einnehmen soll verworfen werden, wenn sie länger als insgesamt 120 Stunden (5 Tage) bei Raumtemperatur (unter 40 °C) oder über einen beliebigen Zeitraum bei über 40 °C gelagert wurde.
Pulver zur Herstellung einer Lösung zum Einnehmen
Nicht über 25 °C lagern.
Die Flasche fest verschlossen halten, um den Inhalt vor Feuchtigkeit zu schützen.
Rekonstituierte Lösung zum Einnehmen
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
Die Lösung zum Einnehmen in der Original-Braunglas-Flasche aufbewahren, um den Inhalt vor Licht zu schützen. Die Flasche stets aufrecht mit fest verschlossenem Schraubdeckel lagern.
Braunglas-Flasche Typ III, mit fälschungssicherem, kindergesichertem Schraubdeckel.
Jeder Umkarton enthält 1 Flasche, 1 Einpress-Flaschenadapter, je zwei braune, wiederverwendbare Applikationsspritzen für Zubereitungen zum Einnehmen mit Graduierung zu 1 ml und 6 ml sowie eine zu 12 ml.
Das Risdiplam Pulver muss vor der Abgabe an den Patienten von einem Angehörigen der Gesundheitsberufe (z. B. Apotheker) zu einer Lösung zum Einnehmen rekonstituiert werden.
Vorbereitung
Evrysdi Pulver zur Herstellung einer Lösung zum Einnehmen ist mit Vorsicht zu handhaben (siehe Abschnitt 4.4). Inhalation und direkter Kontakt mit der Haut und den Schleimhäuten mit dem Trockenpulver und der rekonstituierten Lösung sind zu vermeiden.
Während der Rekonstitution und beim Abwischen der Außenfläche der Flasche und des Deckels sowie beim Reinigen der Arbeitsfläche nach der Rekonstitution sind Einmalhandschuhe zu tragen. Bei Kontakt gründlich mit Wasser und Seife waschen; Augen mit Wasser spülen.
Anleitung zur Rekonstitution:
Klopfen Sie vorsichtig gegen den Flaschenboden, um das Pulver zu lösen.
Nehmen Sie den Deckel ab. Werfen Sie den Deckel nicht weg.
Füllen Sie vorsichtig 79 ml gereinigtes Wasser oder Wasser für Injektionszwecke in die Risdiplam Flasche, um die Lösung zum Einnehmen mit 0,75 mg/ml zu erhalten.
Halten Sie die Arzneimittelflasche mit einer Hand auf einer ebenen Oberfläche fest. Setzen Sie den Flaschenadapter in die Öffnung ein, indem Sie ihn mit der anderen Hand hineindrücken. Achten Sie darauf, dass der Adapter vollständig in die Flasche eingedrückt ist.
Setzen Sie den Deckel wieder auf die Flasche und verschließen Sie die Flasche fest. Stellen Sie sicher, dass die Flasche vollständig geschlossen ist und schütteln Sie sie dann kräftig 15 Sekunden lang. Warten Sie 10 Minuten. Die Lösung sollte nun klar sein. Anschließend die Flasche erneut kräftig 15 Sekunden lang schütteln.
Schreiben Sie das Haltbarkeitsdatum der Lösung zum Einnehmen auf das Flaschenetikett und den Umkarton. (Das Haltbarkeitsdatum wird als 64 Tage nach der Rekonstitution berechnet, wobei der Tag der Rekonstitution als Tag 0 gezählt wird.) Stellen Sie die Flasche wieder zurück in den Originalkarton mit den Applikationsspritzen (einzeln verpackt), der Packungsbeilage und der Broschüre „Gebrauchsanweisung“. Lagern Sie den Originalkarton im Kühlschrank (2 °C bis 8 °C).
Nicht verbrauchte Lösung ist 64 Tage nach der Rekonstitution zu entsorgen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Roche Registration GmbH
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Deutschland
EU/1/21/1531/001
Datum der Erteilung der Zulassung: 26. März 2021
Datum der letzten Verlängerung der Zulassung: 08. Januar 2026
Februar 2026
Verschreibungspflichtig
Roche Pharma AG
Emil-Barell-Straße 1
79639 Grenzach-Wyhlen
Telefon (07624) 14-0
Telefax (07624) 1019
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.